

Abstract
Cisplatin resistance is a major therapeutic challenge in advanced head and neck squamous cell carcinoma (HNSCC). Here, we aimed to investigate the key signaling pathway for cisplatin resistance in HNSCC cells. Vomeronasal type-1 receptor 5 (VN1R5) was identified as a cisplatin resistance-related protein and was highly expressed in cisplatin-resistant HNSCC cells and tissues. The long noncoding RNA (lncRNA) lnc-POP1-1 was confirmed to be a downstream target induced by VN1R5. VN1R5 transcriptionally regulated lnc-POP1-1 expression by activating the specificity protein 1 (Sp1) transcription factor via the cyclic AMP (cAMP)/protein kinase A (PKA) pathway. VN1R5 promoted cisplatin resistance in HNSCC cells in a lnc-POP1-1-dependent manner. Mechanistically, lnc-POP1-1 bound to the minichromosome maintenance deficient 5 (MCM5) protein directly and decelerated MCM5 degradation by inhibiting ubiquitination of the MCM5 protein, which facilitated the repair of DNA damage caused by cisplatin. In summary, we identified the cisplatin resistance-related protein VN1R5 and its downstream target lnc-POP1-1. Upon upregulation by VN1R5, lnc-POP1-1 promotes DNA repair in HNSCC cells through interaction with MCM5 and deceleration of its degradation.
Keywords: long noncoding RNA, lnc-POP1-1, vomeronasal type-1 receptor 5, minichromosome maintenance deficient 5, specificity protein 1, cisplatin resistance, DNA repair, ubiquitination, head and neck squamous cell carcinoma
Graphical abstract
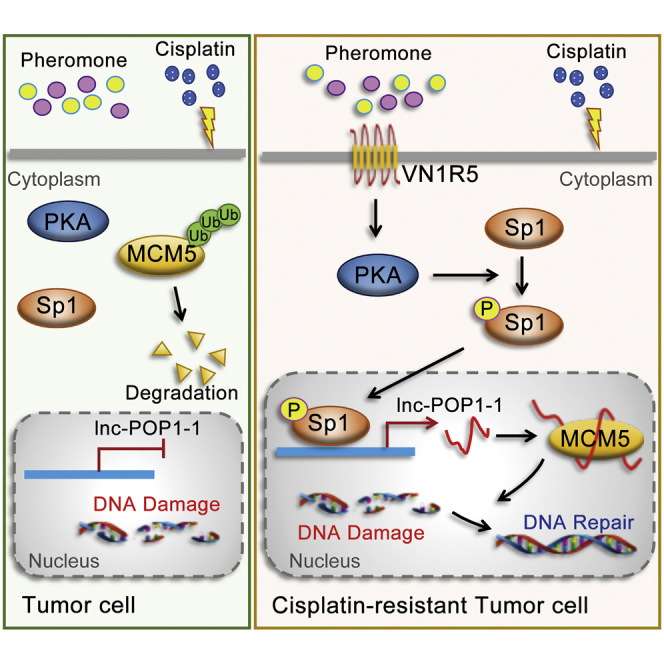
VN1R5 was identified to be a key cisplatin resistance-related protein. VN1R5 transcriptionally regulated lnc-POP1-1 expression by activating the Sp1 transcription factor. lnc-POP1-1 bound to the MCM5 protein directly and decelerated MCM5 degradation by inhibiting ubiquitination of MCM5 protein, which facilitated the repair of DNA damage caused by cisplatin.
Introduction
Head and neck squamous cell carcinoma (HNSCC) is one of the most common malignant tumors in the oral and maxillofacial regions. More than 600,000 new cases are diagnosed annually worldwide.1, 2, 3 Because of its high local recurrence rate, high metastasis rate, and poor prognosis, HNSCC gravely affects human health and quality of life. According to the National Comprehensive Cancer Network (NCCN) Clinical Practice Guidelines in Oncology (NCCN Guidelines), concurrent radiotherapy, and chemotherapy are the standard treatments for locally advanced HNSCC that cannot be surgically removed.4 Although technological advances in surgery, radiotherapy, and chemotherapy have improved local control of HNSCC, the 5-year survival rate of HNSCC patients has remained at approximately 60%; furthermore, patients who are not candidates for surgery have a 5-year survival rate of only approximately 20%.5. The 5-year survival rate of HNSCC patients has remained low because the incidence of treatment failure is high, with recurrence and lymph node metastasis frequently observed.5
Currently, cisplatin (DDP) is the most widely used chemotherapeutic drug for HNSCC.6,7 However, some patients develop cisplatin resistance, leading to cancer recurrence; such acquired resistance is the main cause of cisplatin treatment failure in HNSCC patients.8 Therefore, investigating the key regulatory factors that lead to cisplatin resistance and exploring the molecular mechanism of cisplatin resistance in HNSCC are very important for developing effective treatment methods to enhance the clinical efficacy of HNSCC therapy.
Long noncoding RNAs (lncRNAs) are RNA transcription products that are more than 200 nucleotides in length but lack protein-coding potential. lncRNAs are important regulatory factors in cell biology and participate in DNA replication, RNA transcription, protein translation, cell development, and cell differentiation.9 Many previous studies, including those by our group, have confirmed that lncRNAs are closely related to the occurrence and development of HNSCC and that their abnormal expression affects the growth, recurrence, metastasis, and other behaviors of HNSCC.10, 11, 12, 13 In addition, the regulatory roles of lncRNAs in tumor cisplatin resistance have been gradually recognized.14 A few studies have addressed these roles, most frequently with regard to changes in drug import or efflux, intracellular detoxification, apoptosis, autophagy, DNA repair, and other processes.15 lncRNAs are also known to play regulatory roles in cisplatin resistance in HNSCC. Specifically, the expression levels of lncRNAs such as miRNA processing-related lncRNA (MPRL),16 urothelial cancer associated 1 (UCA1),17 KCNQ1 overlapping transcript 1 (KCNQ1OT1),18 HOX transcript antisense RNA (HOTAIR),19 and HOMEOBOX A11 antisense RNA (HOXA11-AS)20 have been found to be related to the cisplatin response in HNSCC.21 However, there is still an urgent need to identify key functional lncRNAs and clarify their molecular mechanisms promoting cisplatin resistance to further improve the efficacy of HNSCC therapy.
In this study, we identified a key cisplatin resistance-related protein, vomeronasal type-1 receptor 5 (VN1R5), and its downstream lncRNA, lnc-POP1-1. We also explored the functions and molecular mechanisms of these molecules involved in mediating cisplatin resistance in HNSCC cells, aiming to provide a putative prediagnostic marker for cisplatin resistance and a potential therapeutic target for reversing cisplatin resistance in HNSCC cells.
Results
VN1R5 facilitated the acquisition of cisplatin resistance in HNSCC cells
HNSCC cell lines that were sensitive (HN4 and HN30) or resistant (HN4/DDP and HN30/DDP) to cisplatin were used for this study. The survival rates of cisplatin-resistant HNSCC cells were greater than those of the corresponding cisplatin-sensitive cells treated with the same cisplatin dose (Figures S1A and S1B). Moreover, cisplatin-resistant human melanoma cell lines (A375/DDP) and human lung cancer cell lines (A549/DDP) were also established (Figures S1C and S1D). To identify the roles of proteins in the acquisition of cisplatin resistance, we analyzed abnormally expressed proteins via protein mass spectrometry methods (isobaric tags for relative and absolute quantitation, iTRAQ) in cisplatin-sensitive and cisplatin-resistant cancer cells and found that VN1R5 was highly expressed in cisplatin-resistant cells (Figure 1A; Figures S1E and S1F). The upregulation of VN1R5 in cisplatin-resistant cells was confirmed by qPCR and western blotting (Figures 1B and 1C; Figures S1G and S1H). Moreover, VN1R5 was highly expressed in docetaxel+cisplatin+5-fluororacil (TPF) regimen-resistant HNSCC tissues compared to TPF regimen-sensitive tissues (Figure 1D; Table S2). The 5-year survival rate of the VN1R5 high-expression group was significantly lower than that of the VN1R5 low-expression group (Figure 1E).
Figure 1.

Upregulated VN1R5 expression was related to cisplatin resistance in HNSCC cells
(A) Microarray analysis of cisplatin-sensitive and -resistant HNSCC cells. VN1R5 was upregulated in cisplatin-resistant HNSCC cells. (B) VN1R5 expression was measured by qPCR in cisplatin-sensitive and -resistant HN4 and HN30 cells. (C) VN1R5 expression was measured by western blotting in cisplatin-sensitive and -resistant HN4 and HN30 cells. (D) VN1R5 expression was measured by qPCR in TPF-sensitive and -resistant HNSCC tissues. (E) 5-year overall survival rates of HNSCC patients with different VN1R5 expression levels. (F) Compared with NC-transfected cells (black line), HN30/DDP cells with VN1R5 downregulation were sensitized to cisplatin (blue line). The IC50 values are shown on the right. (G) Compared with NC-transfected cells (black line), HN30 cells with VN1R5 upregulation exhibited cisplatin resistance (red line). The IC50 values are shown on the right. (H) Under intraperitoneal injection with 5 mg/kg cisplatin every 3 days 5 times, the volumes and weights of tumors in nude mice subcutaneously inoculated with KO-VN1R5 HN30/DDP cells at the end of the experiment are shown (n = 6/group). (I) The tumor volumes of the KO-VN1R5/KO-NC groups were calculated every 5 days. The mice were intraperitoneally injected with 5 mg/kg cisplatin at days 6, 9, 12, 15, and 18 after tumor inoculation. (J) Under intraperitoneal injection with 5 mg/kg cisplatin every 3 days 5 times, the volumes and weights of tumors in nude mice subcutaneously inoculated with HN30 cells stably transfected with LV-VN1R5 at the end of the experiment are shown (n = 6/group). (K) The tumor volumes of the LV-VN1R5/LV-NC groups were calculated every 5 days. The mice were intraperitoneally injected with 5 mg/kg cisplatin at days 6, 9, 12, 15, and 18 after tumor inoculation. ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001, ∗∗∗∗p < 0.0001. Error bars, means ± SDs. (TPF, docetaxel+cisplatin+5-fluororacil; NC, negative control; LV, lentiviral vector; KO, knockout; i.p., intraperitoneal.)
To examine the effect of VN1R5 on cisplatin resistance in HNSCC cells, we determined the half-maximal inhibitory concentration (IC50) of cisplatin and assessed colony formation ability in cancer cells with VN1R5 knockout (KO) or overexpression. The VN1R5 gene was knocked out in HNSCC cells using CRISPR-Cas9 technology (Figures S2A to S2D). In cisplatin-resistant HN4/DDP and HN30/DDP cells, VN1R5 KO resulted in significant decreases in the IC50 value of cisplatin (Figure S2E; Figure 1F) and in colony formation ability under exposure to cisplatin (Figure S2F). In contrast, VN1R5 overexpression (Figures S2G and S2H) dramatically increased the IC50 value of cisplatin (Figure S2I; Figure 1G) and facilitated colony formation in HN4 and HN30 cells treated with cisplatin (Figure S2J). In animal experiments, under treatment with cisplatin, tumor volumes and weights were significantly lower in the KO-VN1R5 group than in the control group (Figures 1H and 1I), and the results of hematoxylin and eosin (H&E) staining and immunohistochemistry (IHC) of Ki-67 confirmed the alterations in tumor formation (Figure S2K).
Moreover, under treatment with cisplatin, tumor volumes and weights were significantly higher in the VN1R5-overexpression group than in the control group (Figures 1J and 1K), and the results of H&E and Ki-67 staining further confirmed the alterations in tumor formation (Figure S2L).
These results demonstrated that VN1R5 facilitated the acquisition of cisplatin resistance in HNSCC cells.
lnc-POP1-1 was upregulated by VN1R5 and facilitated the acquisition of cisplatin resistance in HNSCC cells
To explore the downstream targets regulated by VN1R5, we performed gene microarray analysis on HNSCC cells with VN1R5 KO or overexpression. The lncRNA lnc-POP1-1 was identified as a downstream target responsive to VN1R5 (Figure 2A; Figures S3A and S3B). The genomic locus of lnc-POP1-1 in humans is chr8:98176523–98180044 (https://lncipedia.org/db/script/lnc-POP1-1:1; Figure 2B).
Figure 2.

lnc-POP1-1 expression was regulated by VN1R5 and positively associated with VN1R5 expression
(A) Venn diagram of the microarray analysis results. lnc-POP1-1 was the downstream gene regulated by VN1R5. (B) Schematic annotation of the lnc-POP1-1 genomic locus on chr8:98176523–98180044 in humans. The blue rectangles represent the exons of lnc-POP1-1. (C) lnc-POP1-1 expression was measured by qPCR in cisplatin-resistant HNSCC cells with VN1R5 KO. (D) lnc-POP1-1 expression was measured by qPCR in cisplatin-sensitive HNSCC cells overexpressing VN1R5. (E) lnc-POP1-1 expression was measured by qPCR in cisplatin-sensitive and -resistant HN4 and HN30 cells. (F) lnc-POP1-1 expression was measured by qPCR in TPF-sensitive and -resistant HNSCC tissues. (G) Pearson correlation analysis of the expression of lnc-POP1-1 and VN1R5 in HNSCC tissue. r2 = 0.7025, n = 70. (H) Compared with NC-transfected cells (black line), HN30/DDP cells with lnc-POP1-1 downregulation were sensitized to cisplatin (blue line). (I) Under intraperitoneal injection with 5 mg/kg cisplatin every 3 days 5 times, the tumor volumes and weights of the mice subcutaneously inoculated with HN30 cells treated with ASO-lnc-POP1-1/ASO-NC are shown (n = 6/group). (J) The tumor volumes of the ASO-lnc-POP1-1 and ASO-NC groups were calculated every 5 days. The mice were intraperitoneally injected with 5 mg/kg cisplatin at days 6, 9, 12, 15, and 18 after tumor inoculation. (K) Compared with NC-transfected cells (black line), HN30 cells with lnc-POP1-1 upregulation exhibited resistance to cisplatin (red line). (L) Under intraperitoneal injection with 5 mg/kg cisplatin every 3 days 5 times, the volumes and weights of tumors in nude mice subcutaneously inoculated with HN30 cells stably transfected with LV-lnc-POP1-1 at the end of the experiment are shown (n = 6/group). (M) The tumor volumes of the LV-lnc-POP1-1 and LV-NC groups were calculated every 5 days. The mice were intraperitoneally injected with 5 mg/kg cisplatin at days 6, 9, 12, 15, and 18 after tumor inoculation. ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001, ∗∗∗∗p < 0.0001. Error bars, means ± SDs. (SS, Smart Silencer; ASO, antisense oligonucleotide.)
lnc-POP1-1 expression was positively associated with VN1R5 expression in HNSCC cells (Figures 2C and 2D). The expression levels of lnc-POP1-1 were significantly higher in cisplatin-resistant HN4/DDP and HN30/DDP cells than in the corresponding cisplatin-sensitive HN4 and HN30 cells (Figure 2E). Moreover, lnc-POP1-1 was highly expressed in TPF regimen-resistant HNSCC tissues compared to TPF regimen-sensitive tissues (Figure 2F; Table S3).
lnc-POP1-1 expression was positively associated with VN1R5 expression in HNSCC tissues (Figure 2G). lnc-POP1-1 was found to be distributed mainly in the nucleus in HNSCC cells by cytoplasmic/nuclear fractionation and fluorescence in situ hybridization (FISH) assays (Figures S3C to S3E).
To examine the effect of lnc-POP1-1 on cisplatin resistance in HNSCC cells, we knocked down lnc-POP1-1 in HN4/DDP and HN30/DDP cells by transfection with Smart Silencer (SS-lnc-POP1-1) (Figure S4A). Silencing of lnc-POP1-1 resulted in a significant decrease in the IC50 of cisplatin in HN4/DDP and HN30/DDP cells (Figure S4B; Figure 2H) and inhibited colony formation in HN4/DDP and HN30/DDP cells treated with cisplatin (Figure S4C). In animal experiments, lnc-POP1-1 was silenced with antisense oligonucleotides (ASOs). The qPCR results showed that ASO-1 was the most effective in knocking down lnc-POP1-1 expression in both HN30 and HN30/DDP cells; thus, this ASO was used in animal experiments (Figure S4D). Tumor volumes and weights were significantly lower in the lnc-POP1-1-knockdown group than in the control group under cisplatin treatment (Figures 2I and 2J), and the results from H&E staining and IHC of Ki-67 confirmed the alterations in tumor formation (Figure S4E).
Conversely, lnc-POP1-1 was overexpressed in HN4 and HN30 cells via transduction of a lentiviral lnc-POP1-1 expression vector (LV-lnc-POP1-1; Figure S4F). Overexpression of lnc-POP1-1 increased the IC50 of cisplatin (Figure S4G; Figure 2K) and facilitated colony formation in HN4 and HN30 cells treated with cisplatin (Figure S4H). Moreover, tumor volumes and weights were significantly higher in the lnc-POP1-1-overexpression group than in the control group under treatment with cisplatin (Figures 2L and 2M), and the results of H&E and Ki-67 staining further confirmed the alterations in tumor formation (Figure S4I). These results demonstrated that lnc-POP1-1 was a downstream target responsive to VN1R5 and facilitated the acquisition of cisplatin resistance in HNSCC cells.
lnc-POP1-1 mediated the promoting effect of VN1R5 on cisplatin resistance in HNSCC cells
Flow cytometry revealed that the apoptosis rates of HN4/DDP and HN30/DDP cells were obviously lower than those of HN4 and HN30 cells (Figure S5A). When VN1R5 was knocked out in HN4/DDP and HN30/DDP cells, the number of apoptotic cells was obviously increased (Figure S5B), consistent with the results of the lnc-POP1-1-knockdown experiment (Figure S5C). In addition, the expression levels of cleaved caspase-3 in different treatment groups were also analyzed to detect cell apoptosis, and the results were consistent with those of flow cytometry (Figures S5D to S5F).
Silencing of lnc-POP1-1 using SS-lnc-POP1-1 in HN4 and HN30 cells significantly blocked the ability of VN1R5 to promote cisplatin resistance in HNSCC cells (Figure S6A; Figure 3A). In contrast, overexpression of lnc-POP1-1 using LV-lnc-POP1-1 restored cisplatin resistance in KO-VN1R5 HNSCC cells (Figure S6B; Figure 3B). In VN1R5-overexpressing HNSCC cells, the number of apoptotic cells was dramatically increased when lnc-POP1-1 was simultaneously knocked down (Figures 3C and 3D). However, VN1R5 KO did not dramatically increase the apoptosis rate in HNSCC cells overexpressing lnc-POP1-1 (Figures S6C and S6D). Moreover, the results of cleaved caspase-3 also confirmed the effect of lnc-POP1-1 on VN1R5 promoting cisplatin resistance (Figures S6E and S6F).
Figure 3.

lnc-POP1-1 was regulated by VN1R5 to affect cisplatin resistance in HNSCC cells
(A) Cell viability was detected by CCK-8 assays when lnc-POP1-1 was knocked down in HN30 cells stably transfected with LV-VN1R5. The IC50 values are shown on the right. (B) Cell viability was detected by CCK-8 assays when lnc-POP1-1 was overexpressed in KO-VN1R5 HN30/DDP cells. The IC50 values are shown on the right. (C and D) Apoptosis was detected using flow cytometry in VN1R5-overexpressing HN4 and HN30 cells treated with 0 or 5 μM cisplatin after knockdown of lnc-POP1-1. (E) Under intraperitoneal injection with 5 mg/kg cisplatin every 3 days 5 times, the tumor volumes and weights of the mice subcutaneously inoculated with LV-VN1R5 HN30 cells treated with ASO-lnc-POP1-1/ASO-NC are shown (n = 6/group). (F) The tumor volumes of the four groups were calculated every 5 days. Cisplatin was injected intraperitoneally every 3 days 5 times. (G) Under intraperitoneal injection with 5 mg/kg cisplatin every 3 days 5 times, the tumor volume and weight from the mice subcutaneously inoculated with KO-VN1R5 HN30/DDP cells treated with LV-lnc-POP1-1 and LV-NC are shown (n = 6/group). (H) The tumor volumes of the four groups were calculated every 5 days. Cisplatin was injected intraperitoneally every 3 days 5 times. ∗p < 0.05, ∗∗p < 0.01, ∗∗∗∗p < 0.0001. Error bars, means ± SDs.
In animal experiments, under conditions of exposure to a certain concentration of cisplatin, the significant increases in tumor volumes and weights in mice subcutaneously injected with stable VN1R5-expressing HN30 cells were reversed by knockdown of lnc-POP1-1 (Figures 3E and 3F). In contrast, the VN1R5 KO-induced decreases in tumor growth were significantly reversed by overexpression of lnc-POP1-1 under treatment with cisplatin (Figures 3G and 3H).
These results showed that VN1R5 affected cisplatin resistance in HNSCC cells in a lnc-POP1-1-dependent manner.
VN1R5 activated lnc-POP1-1 expression through the transcription factor (TF) specificity protein 1 (Sp1)
To understand the mechanism by which VN1R5 regulates lnc-POP1-1, we predicted TFs occupying the lnc-POP1-1 promoter region. The AliBaba2.1 prediction results (Figure S7A) and JASPAR 2020 prediction results22 revealed five TFs (Sp1, nuclear factor-κB [NF-κB], c-Fos, TBP, and apurinic/apyrimidinic-1 [AP-1]) predicted to bind to the promoter of lnc-POP1-1 (Figure S7B). When the predicted TFs were knocked down individually in HNSCC cells, Sp1 knockdown (Figures S7C and S7D) was found to downregulate lnc-POP1-1 expression in HN4/DDP and HN30/DDP cells (Figure 4A). Conversely, Sp1 overexpression (Figures S7E and S7F) upregulated lnc-POP1-1 expression in HN4 and HN30 cells (Figure 4B). Moreover, Sp1 knockdown reversed lnc-POP1-1 expression in VN1R5-overexpressing HN4 and HN30 cells (Figure 4C), while Sp1 overexpression reversed lnc-POP1-1 expression in KO-VN1R5 HN4/DDP and HN30/DDP cells (Figure 4D). In VN1R5-overexpressing or KO-VN1R5 HNSCC cells, the expression level of phosphorylated Sp1 (p-Sp1) was positively associated with that of VN1R5 (Figures S7G and S7H). However, VN1R5 did not regulate the transcription of Sp1 (Figures S7I and S7J), although the silencing of Sp1 resulted in a significant decrease in the IC50 of cisplatin in HN4/DDP and HN30/DDP cells (Figure S7K).
Figure 4.

VN1R5 regulated the promoter activity of Sp1 to affect lnc-POP1-1 expression
(A) lnc-POP1-1 expression was analyzed by qPCR in HN4/DDP and HN30/DDP cells with Sp1 knockdown. (B) lnc-POP1-1 expression was analyzed by qPCR in HN4 and HN30 cells overexpressing Sp1. (C) lnc-POP1-1 expression were analyzed by qPCR in VN1R5-overexpressing HN4 and HN30 cells with Sp1 knockdown. (D) lnc-POP1-1 expression were analyzed by qPCR in VN1R5-knockout HN4/DDP and HN30/DDP cells overexpressing Sp1. (E) The Sp1 binding motifs and predicted binding sites in the lnc-POP1-1 promoter were determined with AliBaba2.1 and JASPAR. (F) Primers specific for the promoter region including predicted TF binding sites were designed for ChIP assays. (G) ChIP-qPCR analysis of Sp1 genomic occupancy of the lnc-POP1-1 promoter in HN4/DDP and HN30/DDP cells. (H) Mutation constructs for Sp1 binding sites C, D, and E of the lnc-POP1-1 promoter were constructed. (I) The relative luciferase activity of mutation constructs for the lnc-POP1-1 promoter was measured. (J) The relative luciferase activity of the lnc-POP1-1 promoter was affected by VN1R5 and Sp1. ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001, ∗∗∗∗p < 0.0001. Error bars, means ± SDs. (ns, no significance; si, siRNA; EV, empty vector; TSS, transcription start site; WT, wild-type; Mut, mutation.)
Collectively, the results of the AliBaba2.1 and JASPAR 2020 analyses revealed three binding motifs and five binding sites for Sp1 in the lnc-POP1-1 promoter (Figure 4E). To verify the binding of Sp1 to the lnc-POP1-1 promoter and to further determine the motifs of the binding sites, we performed chromatin immunoprecipitation (ChIP) and luciferase assays. Primers specific for the promoter of lnc-POP1-1 containing the predicted Sp1 binding sites were designed (Figure 4F), and the ChIP assay results indicated that Sp1 was enriched on the lnc-POP1-1 promoter fragment at base pairs −2,000/–1,801 (Figure 4G). To further clarify the Sp1 transcriptional regulatory sites in the promoter region of lnc-POP1-1, we constructed a series of promoter-deletion luciferase reporters (Figure S7L). Luciferase reporters (including Sp1 binding sites D and E) were significantly activated under wild-type (WT) Sp1 expression, but silencing Sp1 expression dramatically inhibited luciferase reporter activity in HNSCC cells (Figure S7M). To verify which motifs mediated the transcriptional activity of the lnc-POP1-1 promoter, deletion mutations of the predicted Sp1 binding sites C, D, and E were constructed (Figure 4H). The mutant constructs were transfected into HNSCC cells, and the Mut D site (the promoter fragment at base pairs −1,927/–1,918) showed significantly lower transcriptional activity than the WT site (Figure 4I). The effects of VN1R5 and Sp1 on lnc-POP1-1 promoter activity were assessed, and VN1R5 was found to regulate lnc-POP1-1 promoter activity driven by Sp1 (Figure 4J).
Since lnc-POP1-1 transcription was regulated mainly by VN1R5 and Sp1, we deeply investigated the pathways activated by VN1R5 that regulate Sp1 activity in HNSCC cells. Various signaling pathways, including the cyclic AMP/protein kinase A (cAMP/PKA), phosphoinositide 3-kinase (PI3K), and p38 mitogen-activated protein kinase (MAPK) pathways, have been linked to Sp1 expression in multiple cancers (Figure S8A).23, 24, 25, 26 Therefore, inhibitors of PKA (H-89 dihydrochloride, 2 μM),27 inhibitors of PI3K (LY294002, 50 μM),28,29 and inhibitors of p38 MAPK (SB202190, 10 μM)30 were used to characterize the potential role of VN1R5 in mediating Sp1 phosphorylation. The luciferase assay results showed that H-89 dihydrochloride or LY294002 might inhibit lnc-POP1-1 promoter activity in HNSCC cells (Figure S8B). However, lnc-POP1-1 expression was appreciably decreased with H-89 dihydrochloride treatment but not with LY294002 or SB202190 treatment (Figure S8C). Blocking the PKA pathway with H-89 dihydrochloride decreased the levels of phosphorylated Sp1 and lnc-POP1-1 in VN1R5-overexpressing HNSCC cells (Figures S8D and S8E). Knocking down the expression of PKA downregulated the expression of phosphorylated Sp1 and lnc-POP1-1 in HN4/DDP and HN30/DDP cells (Figures S8F and S8G). In addition, PKA overexpression in KO-VN1R5 HNSCC cells significantly increased the levels of both phosphorylated Sp1 and lnc-POP1-1 (Figures S8H and S8I). These results showed that VN1R5 regulated the transcriptional activity of Sp1 to affect lnc-POP1-1 expression via the cAMP/PKA pathway.
lnc-POP1-1 promoted the DNA repair process and bound to minichromosome maintenance deficient 5 (MCM5)
To deeply investigate the mechanism by which lnc-POP1-1 affects cisplatin resistance in HNSCC cells, we used RNA pull-down assays followed by mass spectrometry to explore the putative RNA-binding proteins (RBPs) interacting with lnc-POP1-1. Pathway analysis demonstrated that putative RBPs are mainly involved in several DNA repair pathways in humans (Figure S9A). Cisplatin-resistant HN4/DDP and HN30/DDP cells exhibited lower expression of γH2AX (a DNA damage marker) than cisplatin-sensitive HN4 and HN30 cells (Figure S9B). Moreover, HN4/DDP and HN30/DDP cells exhibited enhanced DNA repair, as verified by comet assays (Figure S9C) and apurinic/apyrimidinic (AP) site counting assays (Figures S9D and S9E). In other words, DNA repair pathways were evident in cisplatin-resistant HN4/DDP and HN30/DDP cells. Gene ontology (GO) analysis also demonstrated that the RBPs played important roles in DNA repair, DNA replication, double-strand break (DSB) repair, the cellular response to DNA damage stimulation, etc. (Figure S9F). The possible binding sites between the putative RBPs and lnc-POP1-1 were predicted with the Protein-RNA Interaction predictor (PRIdictor, http://bclab.inha.ac.kr/pridictor).31 When biological processes and binding sites were considered together, five RBPs (PRKDC, MCM5, SUPT16H, KDM2A, and UBA52) were predicted for lnc-POP1-1 (Figure S9G). Western blot analysis was performed following the RNA pull-down assays to confirm that MCM5 was an RBP binding with lnc-POP1-1 (Figure 5A; Figure S9H). The interaction between MCM5 and lnc-POP1-1 was confirmed in HN4 and HN30 cells by RNA immunoprecipitation (RIP) assays (Figures 5B and 5C). Immunofluorescence (IF) showed that MCM5 was located in the nucleus (Figure S10A), and the colocalization of MCM5 and lnc-POP1-1 was detected by double FISH assays (Figure 5D). In addition, there was a weak positive correlation between the expression of lnc-POP1-1 and MCM5 in HNSCC tissues (Figure S10B). The possible binding sites between MCM5 and lnc-POP1-1 were predicted by PRIdictor.31 A possible binding site on MCM5 for lnc-POP1-1 was located at the arginine (R) residue at amino acid position 724 (Figure S10C), while the nucleotides at base pairs 600–609 were putative MCM5 binding sites on the sequence of lnc-POP1-1 (Figure S10D). An MCM5 mutant vector with the R at amino acid 724 mutated to alanine (A) was constructed based on the predicted lnc-POP1-1 binding sites (Figures S10E and S10F). The ability of MCM5 to bind lnc-POP1-1 was significantly weakened, suggesting that the R at amino acid 724 of MCM5 was important for the interaction with lnc-POP1-1 (Figure 5E). Based on the results obtained with the RIP primers, we speculate that the nucleotides at base pairs 600–609 of lnc-POP1-1 might be responsible for binding with MCM5. After preparation of a biotin-labeled lnc-POP1-1 probe with deletion of base pairs 600–609, RNA pull-down assays were performed on HN4 and HN30 cells. Minimal MCM5 was pulled down by the lnc-POP1-1 deletion probe, suggesting that the nucleotides at base pairs 600–609 of lnc-POP1-1 may be responsible for binding with MCM5 (Figure 5F).
Figure 5.

lnc-POP1-1 bound to MCM5 and participated in DNA repair pathways
(A) RNA pull-down plus western blot analyses showed that MCM5 was pulled down by lnc-POP1-1 probes in HN4 and HN30 cells transfected with LV-lnc-POP1-1. (B and C) qPCR analysis of lnc-POP1-1 enriched with anti-MCM5 in HN4 (B) and HN30 (C) cells transfected with LV-lnc-POP1-1 in RIP assays (six primers were detected). (D) The colocalization of lnc-POP1-1 and MCM5 in HNSCC cells was detected by double FISH analysis. Scale bar, 10 μm. (E) qPCR analysis of lnc-POP1-1 enriched with anti-FLAG in HN4 and HN30 cells transfected with an MCM5-FLAG vector and an MCM5-FLAG mutant vector in which arginine (R) was changed to alanine (A) at site 724 in RIP assays. (F) Western blot of MCM5 following RNA pull-down assays retrieved by the lnc-POP1-1 probe and base pair 600–609 deletion probes in HN4 and HN30 cells transfected with LV-lnc-POP1-1. (G) MCM5 and γH2AX expression was measured by western blotting in cisplatin-resistant or -sensitive HNSCC cells with lnc-POP1-1 knockdown or overexpression. (H) Representative images of alkaline comet assays used to analyze DNA damage in HN4 and HN30 cells overexpressing lnc-POP1-1. Scale bar, 20 μm. (I) Following exposure to 0 μM or 5 μM cisplatin for 24 h, the AP sites of HN4 and HN30 cells overexpressing lnc-POP1-1 were quantified using an AP site counting kit to analyze DNA damage. (J) Compared with the WT plasmid, the lnc-POP1-1 mutant plasmid sensitized HN4/DDP and HN30/DDP cells with KO-VN1R5 to cisplatin. The IC50 values are shown. ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001, ∗∗∗∗p < 0.0001. Error bars, means ± SDs.
Then, the effects of lnc-POP1-1 on MCM5 and DNA damage repair in HNSCC cells were examined. lnc-POP1-1 overexpression upregulated the expression of MCM5 and downregulated the expression of γH2AX in HN4 and HN30 cells, while lnc-POP1-1 knockdown downregulated the expression of MCM5 and upregulated the expression of γH2AX in HN4/DDP and HN30/DDP cells (Figure 5G). The results suggested that lnc-POP1-1 maintained the stability of the MCM5 protein and contributed to DNA damage repair. Moreover, lnc-POP1-1 enhanced DNA repair, as verified by comet assays (Figure 5H) and AP site counting assays (Figure 5I). In other words, when lnc-POP1-1 was overexpressed, DNA damage and the number of AP sites were decreased. Then, MCM5 was overexpressed in HN4 and HN30 cells via the transduction of a lentiviral MCM5 overexpression vector (LV-MCM5; Figure S10G), and γH2AX expression was decreased when MCM5 was overexpressed (Figure S10H), which suggested that MCM5 decreased DNA damage. This result was consistent with those of comet assays (Figure S10I) and AP site counting assays (Figure S10J). A lnc-POP1-1 mutant plasmid with deletion of base pairs 600–609 (lnc-POP1-1-Mut) was constructed to study the effect of the predicted binding site on the cisplatin resistance of HN4/DDP and HN30/DDP cells. The results showed that cisplatin resistance in HNSCC cells transfected with the lnc-POP1-1 mutant plasmid was lower than that in HNSCC cells transfected with the WT plasmid (Figure S10K; Figure 5J).
lnc-POP1-1 prevented the degradation of MCM5 through the proteasome pathway by reducing the ubiquitination of MCM5
HN4 and HN30 cells were treated with 200 μM cycloheximide (CHX) and/or 10 μM MG132 for 0–16 h, and proteins were isolated every 4 h and analyzed by western blotting. Compared with that in the non-MG132-treated group, the level of MCM5 protein in the MG132-treated group was significantly lower, indicating that the MCM5 degradation was partly related to the proteasome (Figure S11A; Figure 6A). Moreover, the proteins of HN4 and HN30 cells overexpressing lnc-POP1-1 in the presence of 200 μM CHX for 0–16 h were isolated every 4 h and analyzed by western blotting. The results showed that lnc-POP1-1 overexpression resulted in slower decay of the MCM5 protein (Figure 6B). Importantly, we observed that MCM5 levels were decreased in the nucleus when lnc-POP1-1 was knocked down (Figure 6C) and that MCM5 levels were increased in the nucleus when lnc-POP1-1 was overexpressed (Figure 6D), suggesting that lnc-POP1-1 contributed to MCM5 retention in the nucleus. We then examined whether lnc-POP1-1-dependent degradation of MCM5 was mediated by MCM5 ubiquitination. After MCM5 was immunoprecipitated in HN4/DDP and HN30/DDP cells, obviously higher ubiquitin signals of MCM5 protein were detected in cells with lnc-POP1-1 silencing than in control cells (Figure 6E). Consistent with these findings, MCM5 ubiquitination was lower in cells overexpressing lnc-POP1-1 than in control cells (Figure 6F).
Figure 6.

lnc-POP1-1 bound to MCM5 to inhibit the ubiquitination of MCM5
(A) The grayscale values of MCM5 protein expression affected by MG132 and the relative intensity are shown. (B) Western blot of the effect of lnc-POP1-1 on MCM5 protein degradation in the presence of 200 μM CHX. (C) The protein levels of MCM5 in nuclear and cytoplasmic fractions were analyzed by western blotting of HN4/DDP and HN30/DDP cells transfected with SS-lnc-POP1-1. (D) The protein levels of MCM5 in nuclear and cytoplasmic fractions were analyzed by western blotting of HN4 and HN30 cells transfected with LV-lnc-POP1-1. (E) Western blot analysis of the ubiquitination of MCM5 in HN4/DDP and HN30/DDP cells transfected with SS-lnc-POP1-1 and treated with 10 μM MG132 for 8 h. (F) Western blot analysis of the ubiquitination of MCM5 in HN4 and HN30 cells overexpressing lnc-POP1-1 and treated with 10 μM MG132 for 8 h. ∗p < 0.05, ∗∗p < 0.01, ∗∗∗∗p < 0.0001. Error bars, means ± SDs. (Ub, ubiquitin; IP, immunoprecipitation; IB, immunoblotting.)
lnc-POP1-1 affected cisplatin resistance in HNSCC cells by interacting with MCM5
MCM5 overexpression in HN4 and HN30 cells increased the IC50 of cisplatin (Figure S11B; Figure 7A). Downregulation of MCM5 (Figure S11C) resulted in a significant decrease in the IC50 of cisplatin in HN4/DDP and HN30/DDP cells (Figure S11D; Figure 7B).
Figure 7.

lnc-POP1-1 affected the cisplatin resistance of HNSCC cells by interacting with MCM5
(A) Compared with NC-transfected cells (black line), HN30 cells with MCM5 upregulation exhibited resistance to cisplatin (red line). The IC50 values are shown on the right. (B) Compared with NC-transfected cells (black line), HN30/DDP cells with MCM5 downregulation were sensitized to cisplatin (blue line). The IC50 values are shown on the right. (C) Cell viability was detected by CCK-8 assay when lnc-POP1-1 was knocked down in HN30 cells stably transfected with LV-MCM5. The IC50 values are shown on the right. (D) Cell viability was detected by CCK-8 assay when lnc-POP1-1 and MCM5 were both knocked down in HN30/DDP cells. The IC50 values are shown on the right. (E) Under intraperitoneal injection with 5 mg/kg cisplatin every 3 days 5 times, the tumor volumes and weights of the mice subcutaneously inoculated with LV-MCM5 HN30 cells treated with ASO-lnc-POP1-1/ASO-NC are shown (n = 6/group). (F) The tumor volumes of the four groups were calculated every 5 days. Cisplatin was injected intraperitoneally every 3 days 5 times. (G) Under intraperitoneal injection with 5 mg/kg cisplatin every 3 days 5 times, the tumor volumes and weights of the mice subcutaneously inoculated with HN30/DDP cells and then treated with ASO-lnc-POP1-1 and/or si-MCM5 are shown (n = 6/group). (H) The tumor volumes of the four groups were calculated every 5 days. Cisplatin was injected intraperitoneally every 3 days 5 times. ∗p < 0.05, ∗∗∗p < 0.001, ∗∗∗∗p < 0.0001. Error bars, means ± SDs.
Silencing of lnc-POP1-1 using SS-lnc-POP1-1 significantly blocked the ability of MCM5 to promote cisplatin resistance in HN4 and HN30 cells (Figure S11E; Figure 7C). Silencing of lnc-POP1-1 also dramatically coordinated the ability of MCM5 knockdown to promote cisplatin sensitivity in HNSCC cells (Figure S11F; Figure 7D).
In animal experiments, under conditions of exposure to a certain concentration of cisplatin, the significant increases in tumor volumes and weights in mice subcutaneously injected with HN30 cells stably expressing MCM5 were reversed by the knockdown of lnc-POP1-1, as shown by the comparison of the knockdown group with the control group (Figures 7E and 7F). In contrast, under treatment with cisplatin, the significant MCM5 knockdown-mediated decreases in tumor volumes and weights were aggravated by silencing lnc-POP1-1, as shown by the comparison of the silenced group with the control group (Figures 7G and 7H).
These results showed that lnc-POP1-1 affected cisplatin resistance in HNSCC cells by interacting with MCM5.
Discussion
Cisplatin resistance and recurrence are the major factors leading to cisplatin-based therapeutic failure in HNSCC patients.8 In patients with locally advanced HNSCC, the initial response rate to cisplatin-based chemotherapy is up to 50%; however, most patients develop cisplatin resistance, which induces cancer recurrence.32 Understanding the mechanism of cisplatin resistance may enable the development of strategies for overcoming chemoresistance and improving clinical outcomes in HNSCC.
Cisplatin resistance is driven by three main mechanisms: (1) a mechanism involving alterations in cisplatin transport and metabolism, decreases in drug uptake or increases in drug efflux, and increases in detoxification ability; (2) a mechanism involving increases in DNA repair ability or DNA damage tolerance; and (3) a mechanism involving abnormalities in cell signal transduction pathways and blockage of apoptosis.8 Multiple mechanisms can be active simultaneously in cisplatin-resistant tumor cells, but the predominant mechanism differs in different cell states.33,34 Our previous studies have confirmed that excessively enhanced DNA repair activity is one of the dominant mechanisms of cisplatin resistance in HNSCC.35
Cisplatin-induced DNA adducts block the processes of transcription and DNA synthesis, thus triggering complex intracellular signal transduction cascades. Cells must remove or tolerate these lesions to resist cisplatin treatment, and the cell cycle is arrested to provide adequate time for DNA repair mechanisms to remove the lesions. If this process is not completed, impaired repair or excessive damage initiates cell death programs, including apoptosis. DNA damage induced by endogenous and environmental DNA-damaging agents can be repaired via DNA repair pathways, thus protecting the genome. The main DNA repair pathways are nucleotide excision repair (NER), homologous recombination (HR), mismatch repair (MMR), base excision repair (BER), nonhomologous end joining (NHEJ), and Fanconi anemia (FA)-pathway repair.36 Most of the major DNA repair systems help repair the many different DNA lesions caused by cisplatin. Cisplatin-induced intrastrand chain crosslinking can lead to distortion of the DNA double helix, thus primarily activating the NER pathway, while the formation of interstrand crosslinks (ICLs) depends on the interaction of the translation/synthesis, HR, NER, translesion synthesis (TLS), and FA pathways.37
Recent studies have evaluated noncoding RNAs involved in DNA damage repair.38, 39, 40 Accumulating evidence has shown that lncRNAs are involved in the regulation of chemoresistance and has elucidated specific mechanisms by which they contribute to DNA repair pathways that lead to pathological conditions such as cancer. lncRNAs also play important roles in cisplatin resistance in HNSCC. For example, MPRL can inhibit the production of miR-483-5p and upregulate the expression of FIS1 to enhance the cisplatin sensitivity of tongue cancer cells.16 In addition, KCNQ1OT1 regulates the cisplatin resistance of tongue cancer cells by specifically sponging miR-211-5p and mediating the Ezrin/Fak/Src signaling pathway.18 HOTAIR can increase the cisplatin resistance of oral squamous cell carcinoma (OSCC) cells by accelerating autophagy and reducing apoptosis.19 The UCA1/miR-184/SF1 axis17 and HOXA11-AS/miR-214-3p/PIM1 axis20 can also regulate the cisplatin resistance of OSCC cells. In our study, nuclear lnc-POP1-1 was found to mediate cisplatin resistance through the DNA repair system and to interact with DNA repair proteins.
lnc-POP1-1 is highly expressed in cisplatin-resistant HNSCC cells, and its abnormal expression can promote the resistance of HNSCC cells to cisplatin. Clarification of the molecular regulatory pathway that causes abnormal expression could thus help explain the mechanism of cisplatin resistance in HNSCC. TF-mediated regulation, histone modification, and DNA methylation are common causes of abnormal lncRNA expression and play regulatory roles in tumor development. In our study, Sp1 was indeed confirmed to regulate the expression of lnc-POP1-1, and 5 predicted binding sites for Sp1 in the lnc-POP1-1 promoter were revealed by AliBaba2 and JASPAR. The Sp1 binding sites in the promoter fragment at base pairs −1,927/–1,918 were ultimately confirmed with a dual-luciferase reporter gene system and ChIP assays.
Sp1 is a tissue-specific TF that regulates gene expression by binding to a GC/GT box in a gene promoter region. Sp1 has been found to be directly involved in the transcriptional regulation of miRNA92b,41 Annexin A2,42 and other genes, promoting the development of OSCC. Moreover, Sp1 participates in transcriptional regulation of chemoresistance-related genes and is related to chemoresistance in lung cancer,43 leukemia,25 breast cancer,44 and other malignancies. Sp1 can be phosphorylated by a number of kinases, such as PKA and PKC, and the phosphorylation of Sp1 has been implicated in increased binding of the TF to consensus sites within various promoters. In our study, VN1R5 controlled the expression of lnc-POP1-1 via Sp1. VN1R5, a cell membrane-localized G protein-coupled receptor (GPCR), is a member of the vomeronasal receptor (VN1R) family and has pheromone receptor activity.45 Previous studies have confirmed that GPCRs can activate intracellular signaling pathways when stimulated by specific ligands; these pathways include the cAMP, Ca2+, MAPK/ERK, p38 MAPK, PI3K, Rho, and other signaling pathways,46 which affect cell biological properties and activate TFs for gene regulation.47 Moreover, the p38 MAPK,25 PI3K/Akt,26 cAMP/PKA,23,24 and other signaling pathways can induce the transcriptional activity of Sp1 to regulate downstream gene expression. VN1R5 could regulate Sp1 by activating these intracellular signaling pathways. Our study results showed that VN1R5 regulated lnc-POP1-1 expression via the PKA-Sp1 pathway.
Moreover, we discovered that MCM5 was an RBP of lnc-POP1-1 and directly interacted with lnc-POP1-1 to affect DNA repair and cisplatin resistance in HNSCC cells. MCM5, a DNA replication regulatory protein, is a member of the minichromosome maintenance family and a component of the MCM2-7 complex (MCM complex), which is the putative replicative helicase essential for “once-per-cell-cycle” DNA replication initiation and elongation in eukaryotic cells.48 MCM5, a subunit of the MCM hexamer, plays an important role in the formation of the MCM dihexamer (in which MCM2 and MCM7 are connected head-on),49,50 the assembly of the CMG complex (MCM2/5 gating function),51,52 and DNA binding (double-stranded DNA is directly connected with the MCM5 helix in the region from isoleucine 526 to lysine 53453). The predicted binding sites for lnc-POP1-1 and MCM5 are listed in the PRIdictor database. When the predicted binding sites (R at amino acid 724 of MCM5 or nucleotide positions 600–609 of lnc-POP1-1) were mutated, the binding capacity between MCM5 and lnc-POP1-1 decreased, suggesting that the predicted binding sites played important roles in maintaining the binding of MCM5 and lnc-POP1-1. Moreover, we found that MCM5 was degraded more slowly and that MCM5 was retained in the nucleus in lnc-POP1-1-overexpressing HNSCC cells. Ubiquitination is an important protein posttranslational modification (PTM) that regulates many key cellular processes.54,55 Ubiquitylation and degradation involve a series of regulatory molecules.56 In our study, lnc-POP1-1 bound to MCM5 to retain MCM5 in the nucleus and thus prevent its ubiquitination.
Our results confirmed that VN1R5 upregulated lnc-POP1-1 expression by promoting the transcriptional activity of Sp1 via the cAMP/PKA pathway, while lnc-POP1-1 was found to bind to the DNA repair protein MCM5 and decelerate its degradation to participate in DNA repair pathways and thus promote the cisplatin resistance of HNSCC cells. VN1R5 and lnc-POP1-1 might be used as predictive markers of cisplatin resistance and therapeutic targets for reversing cisplatin resistance. Therapeutic strategies and methods targeting VN1R5 and lnc-POP1-1 may benefit HNSCC patients with cisplatin resistance.
Conclusions
VN1R5 and lnc-POP1-1 are upregulated in cisplatin-resistant HNSCC cells and tissues and are associated with cisplatin resistance in HNSCC cells. VN1R5 transcriptionally regulates lnc-POP1-1 expression by activating the TF Sp1 via the cAMP/PKA pathway. lnc-POP1-1 facilitates the repair of DNA damage through interaction with MCM5 protein and deceleration of its degradation.
Materials and methods
Specimens
All HNSCC tissue samples were collected from the Department of Oral and Maxillofacial-Head and Neck Oncology, Ninth People’s Hospital, Shanghai Jiao Tong University School of Medicine (Shanghai, China). 70 HNSCC tissues were collected to analyze the association between the expression of VN1R5/lnc-POP1-1 or lnc-POP1-1/MCM5. In addition, 83 tissue samples from HNSCC patients who received the TPF regimen (docetaxel+cisplatin+5-fluorouracil) were tested. According to the efficacy of the TPF regimen, complete remission (CR), and partial remission (PR) were defined as indicators of drug sensitivity, while no progression (stable disease, SD) and progression (PD) were defined as indicators of drug resistance. The clinical characteristics (such as age, sex, smoking and drinking habits, pathological grade, tumor-lymph node-metastasis (TNM) stage, lymph node metastasis, local invasion, etc.) were collected. Regular follow-up was conducted by checking the original medical records, telephone interviews, and outpatient reviews. The follow-up time was 5 years (up to April 2021).
Cell culture and drug treatments
The human HNSCC cell lines HN4 (RRID: CVCL_5515) and HN30 (RRID: CVCL_5525) were from the Shanghai Jiao Tong University School of Medicine, while the human skin melanoma cell line A375 (ATCC cat# 300110/p852_A-375, RRID: CVCL_0132) and the human lung adenocarcinoma cell line A549 (ATCC cat# A549, RRID: CVCL_0023) were purchased from the Cell Bank of the Chinese Academy of Sciences, Shanghai, China. The cells were cultured in Dulbecco’s modified Eagle’s medium (DMEM; GIBCO, NY, USA) supplemented with 10% fetal bovine serum (FBS; GIBCO-BRL, NY, USA) and 1% penicillin-streptomycin. The cells were cultured at 37°C in a humidified 5% CO2 atmosphere.
HNSCC cells (HN4, HN30), A375 cells, and A549 cells were exposed to gradually increasing doses of cisplatin (Sigma, MO, USA) over a 6-month period to establish cisplatin-resistant cells (HN4/DDP, HN30/DDP, A375/DDP, and A549/DDP) that were resistant to treatment with a certain concentration of cisplatin.
The cells were incubated in culture medium containing 200 μM CHX (Sigma, MO, USA) for 0, 4, 8, 12, and 16 h, and western blot analysis was performed.
iTRAQ analysis
The abnormally expressed proteins associated with cisplatin resistance were analyzed in cisplatin-resistant cells (HN4/DDP, HN30/DDP, A375/DDP, and A549/DDP) with iTRAQ (isobaric tags for relative and absolute quantitation, OE Biotechnology, Shanghai, China).
RNA extraction and qPCR
Total RNA from cultured cells was isolated with TRIzol reagent (TaKaRa, Tokyo, Japan) according to the manufacturer’s instructions. A PrimeScript RT Reagent Kit (TaKaRa, Tokyo, Japan) was used for reverse transcription of cDNA. A TB Green Premix Ex Taq Reagent Kit (TaKaRa, Tokyo, Japan) was used for qPCR analysis of mRNA. All qPCRs were performed in an ABI StepOne Real-Time PCR System (Life Technologies, MD, USA). The mRNA levels were normalized to those of glyceraldehyde-3-phosphate dehydrogenase (GAPDH) and/or U6 using the 2–ΔΔCt method. The sequences of the qPCR primers used for mRNA analysis are listed in Table S1.
Western blot analysis
Western blot analysis was performed as previously described.11 The primary antibodies used included the following: VN1R5 antibody (1:1,000, cat# orb165310) from Biorbyt (Cambridge, UK); MCM5 (1:3,000, cat# 11703-1-AP, RRID:AB_2235162); caspase-3 (1:1,000, cat# 19677-1-AP, RRID:AB_10733244) and DYKDDDDK tag (FLAG, 1:500, cat# 20543-1-AP, RRID:AB_11232216) antibodies from Proteintech (Chicago, IL, USA); PKA C-α (PKA, 1:1,000, cat# 4782, RRID:AB_2170170), phospho-histone H2A.X (Ser139; γH2AX, 1:1,000, cat# 9718, RRID:AB_2118009), ubiquitin (Ub, 1:1,000, cat# 3936, RRID:AB_331292), and histone H3 (1:2,000, cat#4499, RRID:AB_10544537) antibodies from Cell Signaling Technology (MA, USA); and Sp1 (1:1,000, cat# ab231778), Sp1 (phospho-T453, 1:1,000, cat# ab37707, RRID:AB_1524434) and GAPDH (1:1,000, cat# ab181602, RRID:AB_2630358) antibodies from Abcam (MA, USA). Moreover, goat anti-rabbit and mouse horseradish peroxidase (HRP)-labeled secondary antibodies (1:10,000, cat# ab205718, RRID:AB_2819160; cat# ab205719, RRID:AB_2755049; Abcam) were used. The signals were visualized with ECLUltra (New Cell and Molecular Biotech, Suzhou, China).
Transfection of smart silencer/siRNAs or plasmids
The SS and small interfering RNA (siRNA) used in our study were designed and synthesized by Guangzhou RiboBio (Guangzhou, China). The sequences of the SS and siRNAs are listed in Tables S4 and S5. Sp1, PKA catalytic subunit (PKA-Cα; PKA), or MCM5-FLAG plasmids were constructed by HanYin Biotechnology (Shanghai, China). Transfection was performed using Lipofectamine 3000 (Thermo Fisher Scientific, MA, USA) following the manufacturer’s instructions.
KO of VN1R5 (KO-VN1R5) via CRISPR-Cas9 genome editing
The CRISPR-Cas9 lentiviral vectors were constructed by HanYin Biotechnology (Shanghai, China). The Hu6-SgRNA-EF1A-hspCas9-Flag-puro-WPRE (PHY-701) plasmid was used as a delivery system. Single guide RNA (sgRNA) sequences targeting VN1R5 and a scrambled sgRNA (Table S6) were cloned into the BsmBI restriction site of the PHY-701 plasmid (HanYin Biotechnology, Shanghai, China). The plasmids were stably transfected into HNSCC cells, and the cells were screened with puromycin (10 μg/mL) for 2 weeks. After sequence validation by DNA sequencing and qPCR analyses, the stable cell lines were diluted and cultured, and clones grown from single cells were selected and expanded to obtain specific stable cell lines.
Lentiviral transduction and screening of stable strains
Lentiviral overexpression vectors for VN1R5 (LV-VN1R5), lnc-POP1-1 (LV-lnc-POP1-1), and MCM5 (LV-MCM5) were constructed by HanYin Biotechnology (Shanghai, China). Lentiviral transduction was performed according to the manufacturer’s instructions. After incubation for approximately 72 h, the cells were cultured with puromycin (10 μg/mL, for LV-VN1R5) or blasticidin (100 μg/mL, for LV-lnc-POP1-1 or LV-MCM5) and were passaged 2–3 times; then, the stably stained cells were screened.
Agilent microarray analysis
An Agilent SurePrint G3 Human Gene Expression v3 8x60K Microarray (Design ID: 072363) was used in this experiment and data analysis of the 8 samples was conducted by OE Biotechnology (Shanghai, China). Total RNA was quantified with a NanoDrop ND-2000 (Thermo Scientific), and RNA integrity was assessed using an Agilent Bioanalyzer 2100 (Agilent Technologies). Sample labeling, microarray hybridization, and washing were performed based on the manufacturer’s standard protocols. Briefly, total RNA was reverse-transcribed into double-stranded cDNA, which was used to synthesize cRNA. The cRNA was labeled with Cyanine-3-CTP. The labeled cRNA was hybridized onto the microarray. After washing, the arrays were scanned with an Agilent Scanner G2505C (Agilent Technologies). Feature Extraction software (version 10.7.1.1, Agilent Technologies) was used to analyze the array images to obtain raw data. GeneSpring (version 14.8, Agilent Technologies) was employed to finish the basic analysis with the raw data.
Cell viability
The viability of HNSCC cells was evaluated by measuring the IC50 value using a cell counting kit-8 (CCK-8) assay. To evaluate the cisplatin response of HNSCC cells, we seeded a total of 3,000 cells in each well of 96-well plates in sextuplicate and treated them with increasing concentrations of cisplatin. After 72 h, viability was assessed by adding 10 μL of CCK-8 solution (Dojindo, Kumamoto, Japan) to each well. The cells were subsequently incubated for 2 h at 37°C, and the optical density at 450 nm was measured in a microplate reader (SpectraMax i3, Molecular Devices, USA). Survival at each concentration was plotted as a percentage of survival in drug-free medium.
Colony formation assay
Approximately 1,000 cells were cultured in 6-well plates and treated with cisplatin in a specific concentration gradient for 2 weeks. The colonies were fixed with 4% paraformaldehyde and stained with crystal violet. Colony formation ability was evaluated according to the size and density of the colonies.
Apoptosis analyses
Cells were seeded in 6-well plates for 24 h and then cultured in FBS-free DMEM (GIBCO, NY, USA) for 24 h. The cells were exposed to normal medium with 5 μM cisplatin for 48 h (the control group was exposed to normal medium without cisplatin).
For apoptosis analysis, cells were harvested, washed, and stained with a fluorescein isothiocyanate (FITC)/Annexin V Apoptosis Detection Kit I (BD PharMingen, CA, USA) according to the instructions. The stained cells were detected by flow cytometry.
IF
Cells were seeded onto coverslips in 24-well plates for 24 h, fixed with 4% paraformaldehyde for 20 min and permeabilized with 0.1% Triton X-100 for 10 min. After being blocked in 3% BSA for 30 min, the cells were incubated with the MCM5 antibody (1:100, Cat# 67049-1-Ig, RRID:AB_2882362; Proteintech) overnight at 4°C, washed with PBST, and then incubated with CoraLite488-conjugated goat anti-mouse immunoglobulin G (IgG; H+L; 1:200, Cat# SA00013-1, Proteintech) for 1 h at room temperature in the dark. The cells were costained with 4′, 6-diamidino-2-phenylindole (DAPI, Beyotime) for 5 min for detection of nuclei and then observed and imaged under a Leica TCS-SP2 laser-scanning confocal microscope (Leica Microsystems, Germany).
Isolation of nuclear and cytoplasmic RNA
Nuclear RNA, cytoplasmic RNA, and total RNA were isolated using a PARIS kit (Invitrogen, CA, USA) following the manufacturer’s instructions. The data were normalized with respect to total RNA with the following equation: % of Input = 100 × [2ˆ (Ct total RNA – Ct RNA fraction)]. U6 was used as the nuclear marker, while GAPDH was used as the cytoplasmic marker. The primer sequences used are listed in Table S1.
FISH and double FISH
Fluorescence-labeled probes for lnc-POP1-1, 18S rRNA, and U6 RNA were designed and synthesized, and FISH experiments were performed using a Ribo Fluorescent In Situ Hybridization Kit (RiboBio). For double FISH, primary and secondary antibodies were added, and hybridization was performed. Images were acquired on a TCS-SP2 laser-scanning confocal microscope (Leica Microsystems, Germany).
Prediction of TF binding sites in the lnc-POP1-1 promoter
The sequence of the lnc-POP1-1 promoter region (base pairs −2,000 to 0) was downloaded from the University of California, Santa Cruz (UCSC) Genome Browser (http://genome.ucsc.edu/). Potential TFs and binding sites were predicted using the AliBaba2.1 program (http://gene-regulation.com/pub/programs/alibaba2/index.html) and the JASPAR database (http://jaspar.genereg.net/).
ChIP assay
ChIP assays were performed as previously described.11,16 In brief, HN4/DDP cells, HN30/DDP cells, or HN4-LV-lnc-POP1-1 cells (5 × 106) were washed with 1 × phosphate-buffered saline (PBS) and incubated for 10 min with 1% formaldehyde at room temperature. Crosslinking was terminated by treatment with 0.1 M glycine for 5 min. The cells were washed twice with 1 × PBS, lysed for 1 h at 4°C in lysis buffer, and sonicated into chromatin fragments with an average length of 500–800 bp, as assessed via agarose gel electrophoresis. The samples were precleared with protein A agarose (Beyotime, Beijing, China) for 1 h at 4°C on a rocking platform. Then, 5 μg of specific antibodies was added, and the samples were incubated overnight at 4°C with shaking. Immunoprecipitated DNA was purified using a QIAquick PCR purification kit (QIAGEN, Hilden, Germany) according to the manufacturer’s protocol. The final ChIP DNA was then used as a template for qPCR with the primers listed in Table S7. ChIP-grade antibodies (anti-Sp1, cat# ab231778; anti-IgG, cat# ab171870, RRID:AB_2687657; Abcam) were used in this study.
Luciferase assay
The plasmids were constructed by HanYin Biotechnology (Shanghai, China). We cloned the potential lnc-POP1-1 promoter region located at base pair −2,000 with respect to the transcription start site (TSS) of lnc-POP1-1 into the pGL3-Basic plasmid upstream of the luciferase reporter gene (pro-2000). According to the predicted Sp1 binding sites in pro-2000, truncated promoter fragments were selected to construct lnc-POP1-1 promoter-deletion plasmids. After initial determination of the binding sites, the binding sites were mutated to construct promoter-mutation plasmids. Luciferase activity assays were performed as described previously.12 In brief, 1 × 105 cells were seeded in 24-well plates for 24 h. Then, HN4 and HN30 cells with stable expression of LV-VN1R5, KO-VN1R5, or the corresponding NC construct were cotransfected with pGL3-lnc-POP1-1-wt or pGL3-lnc-POP1-1-mut plasmids and si-Sp1 (si-NC as the control) using Lipofectamine 3000 (Thermo Fisher Scientific, MA, USA). A constant amount of transfected DNA was maintained across transfections. After 24 h of transfection, cells were collected, and the assay was performed following the manufacturer’s instructions. The data from each luciferase assay were analyzed based on the Renilla/firefly luciferase ratio. Luciferase activity was measured with a Dual-Luciferase Reporter Assay System (Promega, WI, USA).
RNA pull-down assays followed by liquid chromatography-tandem mass spectrometry (LC-MS/MS)
RNA pull-down assays were performed according to the instructions of a Target RNA Purification Kit (ZEHENG Biotech, Shanghai, China).11, 12, 13,57 In brief, HN4-LV-lnc-POP1-1 cells were crosslinked with 1% formaldehyde for 10 min, equilibrated in glycine buffer for 5 min, and washed with cold PBS three times. Then the nuclear extract was isolated, lysed, and sonicated according to the instructions. After 50 μL of the supernatant was preserved for input analysis, the remaining supernatant was incubated with rotation with lnc-POP1-1 biotin probes (or with negative probes as controls) and streptavidin magnetic beads.
After 10% of the bead-sample mixture was collected to verify the efficiency of target RNA purification, the remaining 90% was separated into two parts for protein purification and DNA recovery. After subsequent washing and elution, the precipitated proteins were analyzed with LC-MS/MS by OE Biotech (Shanghai, China). Subsequently, the proteins were verified by immunoblot analysis after RNA pulldown assays.
RIP assay
RIP was performed using an EZ-Magna RIP RNA-Binding Protein Immunoprecipitation Kit (Millipore, MA, USA) according to the manufacturer’s instructions, as previously described.11 After cell lysis with RIP lysis buffer, 100 μL of the lysate was incubated with RIP buffer containing magnetic beads conjugated to a human MCM5 (4 μg, cat# 11703-1-AP) or FLAG antibody (4 μg, cat# 20543-1-AP; Proteintech, Chicago, IL, USA) and normal rabbit IgG (Millipore, MA, USA). IgG was used as the NC. Proteinase K buffer was then added to the samples. Finally, the target RNA was extracted and purified for further analysis by qPCR. The primers used for PCR are listed in Table S8.
Xenograft formation assay
4-week-old BALB/c nude mice (Shanghai Laboratory Animal Center, Shanghai, China) were used in all the in vivo experiments, which were conducted in accordance with the appropriate ethical standards and national guidelines. Tumor cells (1 × 107) were mixed with 100 μL of serum-free DMEM and subcutaneously injected into the right flanks of mice. HN30 or HN30/DDP cells stably transfected with LV-VN1R5, KO-VN1R5, LV-lnc-POP1-1, or LV-MCM5 were used for overexpression in vivo experiments, while cholesterol-conjugated ASO-lnc-POP1-1 (sequences shown in Table S5) and MCM5 siRNA (si-MCM5) from RiboBio (Guangzhou, China) were used for in vivo ASO or siRNA delivery. When the size of the tumors reached approximately 5 mm × 5 mm, ASO or siRNA (10 nmol in 0.1 mL of saline buffer per tumor nodule) was injected into the tumor mass once every 3 days 5 times. The mice were intraperitoneally injected with 5 mg/kg cisplatin at days 6, 9, 12, 15, and 18 after tumor inoculation. Each group consisted of 6 animals. Tumor volume measurements (volume in mm3 = L × W2/2, where L is the length in mm and W is the width in mm) began on day 5 and continued every 5 days until the end of the study. At the end of the experiment, the mice were sacrificed. Their primary tumors were carefully removed, imaged, weighed, and subjected to H&E staining and IHC analysis.
IHC analysis
For IHC, xenograft tissues were fixed, dehydrated, paraffin-embedded, and sectioned. Then, the sections were incubated with a Ki-67 antibody (1:600, Abcam cat# ab15580, RRID:AB_443209) and then incubated with goat anti-rabbit IgG H&L (HRP; 1:20,000, #ab205718, Abcam, MA, USA). Images were captured at 100× and 400× magnification with a light microscope (Olympus, Tokyo, Japan) and quantified with Image-Pro Plus.
Comet assay
A single-cell gel electrophoresis (comet assay) kit was employed to evaluate DNA damage following the manufacturer’s instructions (Abcam, Cambridge, MA, USA). The appropriate treatment was applied, and the cells were collected and washed with prechilled 1 × PBS. Next, 1 × 105 cells/mL were combined with molten comet agarose (at 37°C) at a 1:10 ratio (v/v), and the mixture (75 μL) was immediately pipetted onto the top of the comet agarose base layer. After gentle cell lysis, the samples were treated with alkaline electrophoresis buffer to unwind and denature the DNA and hydrolyze associated sites of damage. The samples were electrophoresed following the manufacturer’s instructions, stained with diluted Vista Green DNA Dye, and visualized on slides with epifluorescence microscopy (Carl Zeiss, Göttingen, Germany) using a FITC filter.
Genomic DNA AP site counts
After treatment of HN4 and HN30 cells with LV-lnc-POP1-1, genomic DNA was purified using a DNA isolation kit (TIANGEN, Beijing, China). The AP sites in DNA were counted using a DNA damage quantification (AP site counting) kit according to the manufacturer’s instructions (Dojindo, Kumamoto, Japan). Briefly, 10 μL of purified genomic DNA solution (100 μg/mL) and 10 μL of ARP solution were mixed and incubated at 37°C for 1 h. Then, 60 μL of the diluted ARP-labeled genomic DNA solution and 100 μL of the DNA binding solution were added to each well, and the plate was incubated at room temperature overnight. The next day, the DNA binding solution was discarded, and the wells were washed 5 times with washing buffer. Then, 150 μL of diluted HRP-streptavidin solution was added to each well, and the plate was incubated at 37°C for 1 h. After washing the plate five times with washing buffer, 100 μL of substrate solution was added to each well, and the plate was incubated for 1 h at 37°C. The absorbance of the samples was analyzed using a SpectraMax i3 microplate reader (Molecular Devices, CA, USA) with a 650-nm filter.
Isolation of nuclear and cytoplasmic protein
Cytoplasmic and nuclear extracts were separated and prepared using NE-PER Nuclear and Cytoplasmic Extraction Reagents (Thermo Fisher Scientific, MA, USA) according to the manufacturer’s instructions and a previous study.11,58 Then, western blot analysis was performed to detect MCM5 expression in the nucleus and cytoplasm in HNSCC cells.
Ubiquitination immunoprecipitation assay
Cells were treated with 10 μM MG132 for 8 h and lysed in 500 μL of ice cold radioimmunoprecipitation assay (RIPA) lysis buffer (Beyotime, Beijing, China) containing a protease inhibitor cocktail (MedChemExpress, NJ, USA) and a phosphatase inhibitor (New Cell and Molecular Biotech, Suzhou, China) before being harvested. After 30 min at 4°C, the cell lysates were centrifuged at 10,000 rpm for 15 min. The precipitates were lysed and denatured with 300 μL of whole-cell lysis buffer for 15 min at 105°C, diluted in a 6-fold volume of RIPA lysis buffer, and incubated with Protein A/G MagBeads (Yeasen, Shanghai, China) binding an MCM5 antibody (4 μg, cat# 11703-1-AP, Proteintech, Chicago, IL, USA) overnight at 4°C. The next day, the beads were washed, and the proteins were separated from the beads using 1× immunoblot loading buffer (Beyotime, Beijing, China) for 10 min at 105°C. The supernatants were collected for subsequent immunoblot analysis with a ubiquitin antibody.
Statistical analysis
All statistical analyses were performed using Statistical Package for Social Sciences software version 16.0 (SPSS 16.0) and GraphPad Prism 7.0. For all experiments, the data in bar graphs are presented as the mean ± SD values from three independent experiments. Significant differences between means were determined using Student’s t test. The association between the expression of VN1R5/lnc-POP1-1 or lnc-POP1-1/MCM5 was analyzed with the Pearson correlation coefficient. Analyses of associations between the expression levels of VN1R5 or lnc-POP1-1 and clinical features were performed using the Mann-Whitney U-test. Kaplan-Meier survival analysis was used to analyze the effects of VN1R5 on the 5-year overall survival rate of HNSCC patients. The IC50 values were determined using a curve fitting model with a four-parameter logistic equation model in GraphPad Prism software. A p value of < 0.05 was considered to indicate significance in all of our analyses.
Ethics approval and consent to participate
The Ethics Committee of Shanghai Jiao Tong University approved our study. Written informed consent was provided by the participants prior to enrollment. All experimental methods abided by the Helsinki Declaration.
Availability of data and material
Data supporting the findings of this study are available within the paper and its Supplemental informationfiles. The iTRAQ and mass spectrometry proteomics data have been deposited to the ProteomeXchange Consortium (http://proteomecentral.proteomexchange.org) via the iProX partner repository59 with the dataset identifier PXD022799. The gene microarray data have been deposited in the NCBI GEO database: GSE161935.
Acknowledgments
The authors would like to thank Shanghai LuMing Biological Technology Co., Ltd. (Shanghai, China) for providing proteomics services. We also thank Dr. Qidong Zu (OE Biotech, Inc., Shanghai, China, https://www.oebiotech.com/) for assistance with the bioinformatic analyses of the iTRAQ protein profile data, Agilent RNA microarray, and LC-MS/MS data. This study was supported by grants from the National Natural Science Foundation of China (81972573, 81772933, 82103008, 82073036), Shandong Provincial Natural Science Foundation (ZR2020MH192), and Shanghai Science and Technology Commission (18JC1413700). The authors have no other relevant affiliations or financial involvement with any organization or entity with a financial interest in or financial conflict with the subject matter or materials discussed in the manuscript apart from those disclosed.
Author contributions
J.Z. and W.C. designed the research. Y.J. and H.G. carried out the molecular biology studies and wrote the manuscript. X.W. performed the statistical analysis. J.Z. and W.C. interpreted the data. T.T., F.X., and X.Q. provided administrative, technical, or material support. J.Z. and W.C. helped to draft the manuscript. The study supervisors are W.C. and J.Z. All authors read and approved the final manuscript.
Declaration of interests
The authors declare no competing interests.
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.ymthe.2021.06.006.
Contributor Information
Wantao Chen, Email: chenwantao196323@sjtu.edu.cn.
Jianjun Zhang, Email: zjjshuobo@163.com.
Supplemental information
References
- 1.Chen W., Zheng R., Baade P.D., Zhang S., Zeng H., Bray F., Jemal A., Yu X.Q., He J. Cancer statistics in China, 2015. CA Cancer J. Clin. 2016;66:115–132. doi: 10.3322/caac.21338. [DOI] [PubMed] [Google Scholar]
- 2.Siegel R.L., Miller K.D., Jemal A. Cancer statistics, 2020. CA Cancer J. Clin. 2020;70:7–30. doi: 10.3322/caac.21590. [DOI] [PubMed] [Google Scholar]
- 3.Chow L.Q.M. Head and Neck Cancer. N. Engl. J. Med. 2020;382:60–72. doi: 10.1056/NEJMra1715715. [DOI] [PubMed] [Google Scholar]
- 4.Adelstein D., Gillison M.L., Pfister D.G., Spencer S., Adkins D., Brizel D.M., Burtness B., Busse P.M., Caudell J.J., Cmelak A.J., et al. NCCN Guidelines Insights: Head and Neck Cancers, Version 2.2017. J. Natl. Compr. Canc. Netw. 2017;15:761–770. doi: 10.6004/jnccn.2017.0101. [DOI] [PubMed] [Google Scholar]
- 5.Hedberg M.L., Goh G., Chiosea S.I., Bauman J.E., Freilino M.L., Zeng Y., Wang L., Diergaarde B.B., Gooding W.E., Lui V.W., et al. Genetic landscape of metastatic and recurrent head and neck squamous cell carcinoma. J. Clin. Invest. 2016;126:169–180. doi: 10.1172/JCI82066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Gao Y., Liu D. The roles of excision repair cross-complementation group1 in objective response after cisplatin-based concurrent chemoradiotherapy and survival in head and neck cancers: a systematic review and meta-analysis. Oral Oncol. 2015;51:570–577. doi: 10.1016/j.oraloncology.2015.03.009. [DOI] [PubMed] [Google Scholar]
- 7.López-Verdín S., Lavalle-Carrasco J., Carreón-Burciaga R.G., Serafín-Higuera N., Molina-Frechero N., González-González R., Bologna-Molina R. Molecular Markers of Anticancer Drug Resistance in Head and Neck Squamous Cell Carcinoma: A Literature Review. Cancers (Basel) 2018;10:376. doi: 10.3390/cancers10100376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Galluzzi L., Senovilla L., Vitale I., Michels J., Martins I., Kepp O., Castedo M., Kroemer G. Molecular mechanisms of cisplatin resistance. Oncogene. 2012;31:1869–1883. doi: 10.1038/onc.2011.384. [DOI] [PubMed] [Google Scholar]
- 9.Lee J.T. Epigenetic regulation by long noncoding RNAs. Science. 2012;338:1435–1439. doi: 10.1126/science.1231776. [DOI] [PubMed] [Google Scholar]
- 10.Dhamija S., Diederichs S. From junk to master regulators of invasion: lncRNA functions in migration, EMT and metastasis. Int. J. Cancer. 2016;139:269–280. doi: 10.1002/ijc.30039. [DOI] [PubMed] [Google Scholar]
- 11.Jiang Y., Cao W., Wu K., Qin X., Wang X., Li Y., Yu B., Zhang Z., Wang X., Yan M., et al. LncRNA LINC00460 promotes EMT in head and neck squamous cell carcinoma by facilitating peroxiredoxin-1 into the nucleus. J. Exp. Clin. Cancer Res. 2019;38:365. doi: 10.1186/s13046-019-1364-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Jiang Y., Wu K., Cao W., Xu Q., Wang X., Qin X., Wang X., Li Y., Zhang J., Chen W. Long noncoding RNA KTN1-AS1 promotes head and neck squamous cell carcinoma cell epithelial-mesenchymal transition by targeting miR-153-3p. Epigenomics. 2020;12:487–505. doi: 10.2217/epi-2019-0173. [DOI] [PubMed] [Google Scholar]
- 13.Wu K., Jiang Y., Zhou W., Zhang B., Li Y., Xie F., Zhang J., Wang X., Yan M., Xu Q., et al. Long Noncoding RNA RC3H2 Facilitates Cell Proliferation and Invasion by Targeting MicroRNA-101-3p/EZH2 Axis in OSCC. Mol. Ther. Nucleic Acids. 2020;20:97–110. doi: 10.1016/j.omtn.2020.02.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Vera O., Rodriguez-Antolin C., de Castro J., Karreth F.A., Sellers T.A., Ibanez de Caceres I. An epigenomic approach to identifying differential overlapping and cis-acting lncRNAs in cisplatin-resistant cancer cells. Epigenetics. 2018;13:251–263. doi: 10.1080/15592294.2018.1436364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hu Y., Zhu Q.N., Deng J.L., Li Z.X., Wang G., Zhu Y.S. Emerging role of long non-coding RNAs in cisplatin resistance. OncoTargets Ther. 2018;11:3185–3194. doi: 10.2147/OTT.S158104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Tian T., Lv X., Pan G., Lu Y., Chen W., He W., Lei X., Zhang H., Liu M., Sun S., et al. Long Noncoding RNA MPRL Promotes Mitochondrial Fission and Cisplatin Chemosensitivity via Disruption of Pre-miRNA Processing. Clin. Cancer Res. 2019;25:3673–3688. doi: 10.1158/1078-0432.CCR-18-2739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Fang Z., Zhao J., Xie W., Sun Q., Wang H., Qiao B. LncRNA UCA1 promotes proliferation and cisplatin resistance of oral squamous cell carcinoma by sunppressing miR-184 expression. Cancer Med. 2017;6:2897–2908. doi: 10.1002/cam4.1253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Zhang S., Ma H., Zhang D., Xie S., Wang W., Li Q., Lin Z., Wang Y. LncRNA KCNQ1OT1 regulates proliferation and cisplatin resistance in tongue cancer via miR-211-5p mediated Ezrin/Fak/Src signaling. Cell Death Dis. 2018;9:742. doi: 10.1038/s41419-018-0793-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Wang X., Liu W., Wang P., Li S. RNA interference of long noncoding RNA HOTAIR suppresses autophagy and promotes apoptosis and sensitivity to cisplatin in oral squamous cell carcinoma. J. Oral Pathol. Med. 2018;47:930–937. doi: 10.1111/jop.12769. [DOI] [PubMed] [Google Scholar]
- 20.Wang X., Li H., Shi J. LncRNA HOXA11-AS Promotes Proliferation and Cisplatin Resistance of Oral Squamous Cell Carcinoma by Suppression of miR-214-3p Expression. BioMed Res. Int. 2019;2019:8645153. doi: 10.1155/2019/8645153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ghafouri-Fard S., Mohammad-Rahimi H., Jazaeri M., Taheri M. Expression and function of long non-coding RNAs in head and neck squamous cell carcinoma. Exp. Mol. Pathol. 2020;112:104353. doi: 10.1016/j.yexmp.2019.104353. [DOI] [PubMed] [Google Scholar]
- 22.Fornes O., Castro-Mondragon J.A., Khan A., van der Lee R., Zhang X., Richmond P.A., Modi B.P., Correard S., Gheorghe M., Baranašić D., et al. JASPAR 2020: update of the open-access database of transcription factor binding profiles. Nucleic Acids Res. 2020;48:D87–D92. doi: 10.1093/nar/gkz1001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Zhu P., Wang W., Zuo R., Sun K. Mechanisms for establishment of the placental glucocorticoid barrier, a guard for life. Cell. Mol. Life Sci. 2019;76:13–26. doi: 10.1007/s00018-018-2918-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Gupta V., Khan A.A., Sasi B.K., Mahapatra N.R. Molecular mechanism of monoamine oxidase A gene regulation under inflammation and ischemia-like conditions: key roles of the transcription factors GATA2, Sp1 and TBP. J. Neurochem. 2015;134:21–38. doi: 10.1111/jnc.13099. [DOI] [PubMed] [Google Scholar]
- 25.Zhang Y., Chen H.X., Zhou S.Y., Wang S.X., Zheng K., Xu D.D., Liu Y.T., Wang X.Y., Wang X., Yan H.Z., et al. Sp1 and c-Myc modulate drug resistance of leukemia stem cells by regulating survivin expression through the ERK-MSK MAPK signaling pathway. Mol. Cancer. 2015;14:56. doi: 10.1186/s12943-015-0326-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Yin P., Zhao C., Li Z., Mei C., Yao W., Liu Y., Li N., Qi J., Wang L., Shi Y., et al. Sp1 is involved in regulation of cystathionine γ-lyase gene expression and biological function by PI3K/Akt pathway in human hepatocellular carcinoma cell lines. Cell. Signal. 2012;24:1229–1240. doi: 10.1016/j.cellsig.2012.02.003. [DOI] [PubMed] [Google Scholar]
- 27.Kurowski P., Gawlak M., Szulczyk P. Muscarinic receptor control of pyramidal neuron membrane potential in the medial prefrontal cortex (mPFC) in rats. Neuroscience. 2015;303:474–488. doi: 10.1016/j.neuroscience.2015.07.023. [DOI] [PubMed] [Google Scholar]
- 28.Lin J., Guan Z., Wang C., Feng L., Zheng Y., Caicedo E., Bearth E., Peng J.R., Gaffney P., Ondrey F.G. Inhibitor of differentiation 1 contributes to head and neck squamous cell carcinoma survival via the NF-kappaB/survivin and phosphoinositide 3-kinase/Akt signaling pathways. Clin. Cancer Res. 2010;16:77–87. doi: 10.1158/1078-0432.CCR-08-2362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Maseki S., Ijichi K., Tanaka H., Fujii M., Hasegawa Y., Ogawa T., Murakami S., Kondo E., Nakanishi H. Acquisition of EMT phenotype in the gefitinib-resistant cells of a head and neck squamous cell carcinoma cell line through Akt/GSK-3β/snail signalling pathway. Br. J. Cancer. 2012;106:1196–1204. doi: 10.1038/bjc.2012.24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Zhong Y., Naito Y., Cope L., Naranjo-Suarez S., Saunders T., Hong S.M., Goggins M.G., Herman J.M., Wolfgang C.L., Iacobuzio-Donahue C.A. Functional p38 MAPK identified by biomarker profiling of pancreatic cancer restrains growth through JNK inhibition and correlates with improved survival. Clin. Cancer Res. 2014;20:6200–6211. doi: 10.1158/1078-0432.CCR-13-2823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Tuvshinjargal N., Lee W., Park B., Han K. PRIdictor: Protein-RNA Interaction predictor. Biosystems. 2016;139:17–22. doi: 10.1016/j.biosystems.2015.10.004. [DOI] [PubMed] [Google Scholar]
- 32.Chen D., Wu M., Li Y., Chang I., Yuan Q., Ekimyan-Salvo M., Deng P., Yu B., Yu Y., Dong J., et al. Targeting BMI1+ Cancer Stem Cells Overcomes Chemoresistance and Inhibits Metastases in Squamous Cell Carcinoma. Cell Stem Cell. 2017;20:621–634.e6. doi: 10.1016/j.stem.2017.02.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Chen S.H., Chang J.Y. New Insights into Mechanisms of Cisplatin Resistance: From Tumor Cell to Microenvironment. Int. J. Mol. Sci. 2019;20:4136. doi: 10.3390/ijms20174136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Amable L. Cisplatin resistance and opportunities for precision medicine. Pharmacol. Res. 2016;106:27–36. doi: 10.1016/j.phrs.2016.01.001. [DOI] [PubMed] [Google Scholar]
- 35.Qin X., Guo H., Wang X., Zhu X., Yan M., Wang X., Xu Q., Shi J., Lu E., Chen W., Zhang J. Exosomal miR-196a derived from cancer-associated fibroblasts confers cisplatin resistance in head and neck cancer through targeting CDKN1B and ING5. Genome Biol. 2019;20:12. doi: 10.1186/s13059-018-1604-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Curtin N.J. DNA repair dysregulation from cancer driver to therapeutic target. Nat. Rev. Cancer. 2012;12:801–817. doi: 10.1038/nrc3399. [DOI] [PubMed] [Google Scholar]
- 37.Ceccaldi R., O’Connor K.W., Mouw K.W., Li A.Y., Matulonis U.A., D’Andrea A.D., Konstantinopoulos P.A. A unique subset of epithelial ovarian cancers with platinum sensitivity and PARP inhibitor resistance. Cancer Res. 2015;75:628–634. doi: 10.1158/0008-5472.CAN-14-2593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Khanduja J.S., Calvo I.A., Joh R.I., Hill I.T., Motamedi M. Nuclear Noncoding RNAs and Genome Stability. Mol. Cell. 2016;63:7–20. doi: 10.1016/j.molcel.2016.06.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Zhang C., Peng G. Non-coding RNAs: an emerging player in DNA damage response. Mutat. Res. Rev. Mutat. Res. 2015;763:202–211. doi: 10.1016/j.mrrev.2014.11.003. [DOI] [PubMed] [Google Scholar]
- 40.Majidinia M., Yousefi B. Long non-coding RNAs in cancer drug resistance development. DNA Repair (Amst.) 2016;45:25–33. doi: 10.1016/j.dnarep.2016.06.003. [DOI] [PubMed] [Google Scholar]
- 41.Fang H., Pang P., Liu F.Y., Sun C.F. [Role of specificity protein 1 in transcription regulation of microRNA-92b in head and neck squamous cell carcinoma] Zhonghua Kou Qiang Yi Xue Za Zhi. 2017;52:563–568. doi: 10.3760/cma.j.issn.1002-0098.2017.09.011. [DOI] [PubMed] [Google Scholar]
- 42.Liu X.B., Wang J., Li K., Fan X.N. Sp1 promotes cell migration and invasion in oral squamous cell carcinoma by upregulating Annexin A2 transcription. Mol. Cell. Probes. 2019;46:101417. doi: 10.1016/j.mcp.2019.06.007. [DOI] [PubMed] [Google Scholar]
- 43.Li J., Ao J., Li K., Zhang J., Li Y., Zhang L., Wei Y., Gong D., Gao J., Tan W., et al. ZNF32 contributes to the induction of multidrug resistance by regulating TGF-β receptor 2 signaling in lung adenocarcinoma. Cell Death Dis. 2016;7:e2428. doi: 10.1038/cddis.2016.328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Dong H., Wang W., Mo S., Chen R., Zou K., Han J., Zhang F., Hu J. SP1-induced lncRNA AGAP2-AS1 expression promotes chemoresistance of breast cancer by epigenetic regulation of MyD88. J. Exp. Clin. Cancer Res. 2018;37:202. doi: 10.1186/s13046-018-0875-3. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 45.Wallrabenstein I., Gerber J., Rasche S., Croy I., Kurtenbach S., Hummel T., Hatt H. The smelling of Hedione results in sex-differentiated human brain activity. Neuroimage. 2015;113:365–373. doi: 10.1016/j.neuroimage.2015.03.029. [DOI] [PubMed] [Google Scholar]
- 46.Morri M., Sanchez-Romero I., Tichy A.M., Kainrath S., Gerrard E.J., Hirschfeld P.P., Schwarz J., Janovjak H. Optical functionalization of human Class A orphan G-protein-coupled receptors. Nat. Commun. 2018;9:1950. doi: 10.1038/s41467-018-04342-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Cvicek V., Goddard W.A., 3rd, Abrol R. Structure-Based Sequence Alignment of the Transmembrane Domains of All Human GPCRs: Phylogenetic, Structural and Functional Implications. PLoS Comput. Biol. 2016;12:e1004805. doi: 10.1371/journal.pcbi.1004805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Simonetti G., Padella A., do Valle I.F., Fontana M.C., Fonzi E., Bruno S., Baldazzi C., Guadagnuolo V., Manfrini M., Ferrari A., et al. Aneuploid acute myeloid leukemia exhibits a signature of genomic alterations in the cell cycle and protein degradation machinery. Cancer. 2019;125:712–725. doi: 10.1002/cncr.31837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Li N., Zhai Y., Zhang Y., Li W., Yang M., Lei J., et al. Structure of the eukaryotic MCM complex at 3.8 A. Nature. 2015;524:186–191. doi: 10.1038/nature14685. [DOI] [PubMed] [Google Scholar]
- 50.Zhai Y., Li N., Jiang H., Huang X., Gao N., Tye B.K. Unique Roles of the Non-identical MCM Subunits in DNA Replication Licensing. Mol. Cell. 2017;67:168–179. doi: 10.1016/j.molcel.2017.06.016. [DOI] [PubMed] [Google Scholar]
- 51.Zhai Y., Cheng E., Wu H., Li N., Yung P.Y., Gao N., Tye B.K. Open-ringed structure of the Cdt1-Mcm2-7 complex as a precursor of the MCM double hexamer. Nat. Struct. Mol. Biol. 2017;24:300–308. doi: 10.1038/nsmb.3374. [DOI] [PubMed] [Google Scholar]
- 52.Wang Y., Chen H., Zhang J., Cheng A.S.L., Yu J., To K.F., Kang W. MCM family in gastrointestinal cancer and other malignancies: From functional characterization to clinical implication. Biochim. Biophys. Acta Rev. Cancer. 2020;1874:188415. doi: 10.1016/j.bbcan.2020.188415. [DOI] [PubMed] [Google Scholar]
- 53.Yuan Z., Schneider S., Dodd T., Riera A., Bai L., Yan C., Magdalou I., Ivanov I., Stillman B., Li H., Speck C. Structural mechanism of helicase loading onto replication origin DNA by ORC-Cdc6. Proc. Natl. Acad. Sci. USA. 2020;117:17747–17756. doi: 10.1073/pnas.2006231117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Nakayama K.I., Nakayama K. Ubiquitin ligases: cell-cycle control and cancer. Nat. Rev. Cancer. 2006;6:369–381. doi: 10.1038/nrc1881. [DOI] [PubMed] [Google Scholar]
- 55.Ma A., Malynn B.A. A20: linking a complex regulator of ubiquitylation to immunity and human disease. Nat. Rev. Immunol. 2012;12:774–785. doi: 10.1038/nri3313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Swatek K.N., Komander D. Ubiquitin modifications. Cell Res. 2016;26:399–422. doi: 10.1038/cr.2016.39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Su X., Wang H., Ge W., Yang M., Hou J., Chen T., Li N., Cao X. An In Vivo Method to Identify microRNA Targets Not Predicted by Computation Algorithms: p21 Targeting by miR-92a in Cancer. Cancer Res. 2015;75:2875–2885. doi: 10.1158/0008-5472.CAN-14-2218. [DOI] [PubMed] [Google Scholar]
- 58.Qin X., Yan M., Wang X., Xu Q., Wang X., Zhu X., Shi J., Li Z., Zhang J., Chen W. Cancer-associated Fibroblast-derived IL-6 Promotes Head and Neck Cancer Progression via the Osteopontin-NF-kappa B Signaling Pathway. Theranostics. 2018;8:921–940. doi: 10.7150/thno.22182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Ma J., Chen T., Wu S., Yang C., Bai M., Shu K., Li K., Zhang G., Jin Z., He F., et al. iProX: an integrated proteome resource. Nucleic Acids Res. 2019;47:D1211–D1217. doi: 10.1093/nar/gky869. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Data supporting the findings of this study are available within the paper and its Supplemental informationfiles. The iTRAQ and mass spectrometry proteomics data have been deposited to the ProteomeXchange Consortium (http://proteomecentral.proteomexchange.org) via the iProX partner repository59 with the dataset identifier PXD022799. The gene microarray data have been deposited in the NCBI GEO database: GSE161935.