

Summary
CRISPR/Cas has revolutionized genome engineering in plants. However, the use of anti‐CRISPR proteins as tools to prevent CRISPR/Cas‐mediated gene editing and gene activation in plants has not been explored yet. This study describes the characterization of two anti‐CRISPR proteins, AcrIIA4 and AcrVA1, in Nicotiana benthamiana. Our results demonstrate that AcrIIA4 prevents site‐directed mutagenesis in leaves when transiently co‐expressed with CRISPR/Cas9. In a similar way, AcrVA1 is able to prevent CRISPR/Cas12a‐mediated gene editing. Moreover, using a N. benthamiana line constitutively expressing Cas9, we show that the viral delivery of AcrIIA4 using Tobacco etch virus is able to completely abolish the high editing levels obtained when the guide RNA is delivered with a virus, in this case Potato virus X. We also show that AcrIIA4 and AcrVA1 repress CRISPR/dCas‐based transcriptional activation of reporter genes. In the case of AcrIIA4, this repression occurs in a highly efficient, dose‐dependent manner. Furthermore, the fusion of an auxin degron to AcrIIA4 results in auxin‐regulated activation of a downstream reporter gene. The strong anti‐Cas activity of AcrIIA4 and AcrVA1 reported here opens new possibilities for customized control of gene editing and gene expression in plants.
Keywords: anti‐CRISPR/Cas9, anti‐CRISPR/Cas12a, AcrIIA4, AcrVA1, gene expression regulation, Nicotiana benthamiana
Introduction
The systems based on the clustered regularly interspaced short palindromic repeats (CRISPR) and their CRISPR associated (Cas) proteins have been used as the site‐directed nucleases of choice for plant genome engineering due to their simplicity and specificity (Baltes and Voytas, 2015). In addition to gene editing through site‐specific DNA cleavage and subsequent repair by the host machinery, CRISPR/Cas‐based platforms provide new ways of engineering other catalytic and non‐catalytic genome‐associated functions, such as DNA base modifiers, epigenetic effectors or programmable transcription factors (TFs); the latter enabling new control strategies for gene expression known as CRISPR activation (CRISPRa) or interference (CRISPRi; Adli, 2018; Lo and Qi, 2017). This is achieved by coupling nuclease‐deactivated versions of Cas (dCas) to transcriptional activator or repressor domains, which then act as functionally customized RNA‐guided DNA‐binding complexes (Lo and Qi, 2017; Qi et al., 2013).
Although different CRISPR‐based architectures in plants have been shown to induce both efficient mutagenesis and powerful transcriptional activation when expressed constitutively (Lowder and Paul, 2017; Moradpour and Abdulah, 2020; Zhan et al., 2020), the spatial–temporal control on such activities would considerably expand the range of their potential applications. To this date, the control of CRISPR/Cas activity in plants has been attempted mainly at the transcriptional level using inducible promoters controlling expression of Cas transcripts (Barone et al., 2020; Ochoa‐Fernandez et al., 2020; Wang et al., 2020). However, in practical terms, the control of effector function at the transcriptional level alone is only partially effective due to inherent leakiness and noise. Furthermore, transcriptional regulation imposes a lag in the responses due to the requirement for de novo protein synthesis. Conversely, many biological switches make use of post‐translational regulatory strategies such as enzymatic modification or protein–protein interactions as relay mechanisms. In this context, the recent discovery of phage‐derived proteins with Cas‐inhibitory activities, named collectively as anti‐CRISPR (Acr) proteins, offers exciting new possibilities to control Cas activity at the post‐translational level. Acr proteins constitute a collective arsenal of naturally evolved CRISPR/Cas antagonists that inhibit CRISPR/Cas immune activity at various stages (Borges and Davidson, 2017; Marino et al., 2020; Stanley and Maxwell, 2018). To date, 45 non‐homologous Acr proteins (24 for class I CRISPR/Cas and 21 for class II) have been discovered, comprising different mechanisms and structures (Dong et al., 2017; Knott et al., 2019; Trasanidou et al., 2019). Acr proteins can inhibit CRISPR/Cas function (i) by directly interacting with a Cas protein to prevent DNA binding (e.g. the anti‐Cas9 protein AcrIIA4), CRISPR RNA (crRNA) loading or DNA cleavage, or (ii) by interfering with effector‐complex formation for example cleaving the crRNA when bound to the Cas (e.g. the anti‐Cas12a protein AcrVA1; Marino et al., 2020). Whereas Acrs have been successfully tested in mammalian and bacterial cells, no evidence of their functionality in plants exists. Here, we study the ability of two Acr proteins, AcrIIA4 from a Listeria monocytogenes prophage and AcrVA1 from Moraxella bovoculi, to inhibit the activity of Streptococcus pyogenes Cas9 (Cas9) and Lachnospiraceae bacterium Cas12a (Cas12a) proteins, respectively, in plants. We found that Acrs are highly active in Nicotiana benthamiana and up to the point of virtually abolishing the highest levels of Cas9 editing activity obtained with a viral delivery system. Similarly, we found that Acrs work as efficient and tunable CRISPRa inhibitors, thus opening the way for post‐translational regulation of CRISPR/Cas systems in plants.
Results
AcrIIA4 and AcrVA1 prevent editing and CRISPRa in N. benthamiana
In yeast and mammalian cells, AcrIIA4 binds the Cas9‐gRNA complex preventing its binding to the target genomic DNA and blocking DNA cleavage or dCas9‐mediated CRISPRa (Figure 1a). To study its use in plants, we first analysed the ability of AcrIIA4 to prevent Cas9 site‐specific mutagenesis in N. benthamiana using three previously well‐characterized targets: the xylosyltransferase gene (XT2) and two different genes of the Squamosa‐promoter binding protein‐like family (Table S1). As shown in Figure 1b, transient expression of specific gRNAs along with the Cas9 resulted in average editing efficiencies of 6%, 8% and 10% for each target, respectively, at 5 days post‐infiltration (dpi), whereas co‐infiltrations of the same editing constructs with a transcriptional unit (TU) expressing a nuclear‐localized AcrIIA4 under the constitutive CaMV 35S promoter reduced the editing efficiencies to undetectable levels for all three targets (Table S2). Next, as a stress test of AcrIIA4 functionality, we studied its ability to prevent virus‐induced genome editing (VIGE). In a recent work, we reported that delivery of a gRNA using a Potato virus X (PVX)‐based vector into N. benthamiana plants constitutively expressing Cas9 results in high editing efficiencies (~80%; Uranga, Aragonés, et al., 2021). Here, we confirmed these results by agro‐inoculating a PVX‐based vector carrying a gRNA targeting two homeologs of the Phytoene desaturase 3 (PDS3) gene (Niben101Scf01283g02002.1 and Niben101Scf14708g00023.1) in Cas9 plants. This construct includes the 5′ end (102 nt) of the Arabidopsis thaliana Flowering locus T (FT) coding sequence (truncated FT, tFT) fused to the 3′ end of the gRNA (PVX::gPDS3ii‐tFT). The tFT RNA element promotes the mobility of the gRNA, so it can access and edit the germline cells (Ellison et al., 2020). As anticipated, at 7 dpi, we obtained edition levels of 71.7% and 77% for each of the targeted PDS3 genes respectively. Remarkably, when AcrIIA4 was delivered simultaneously using a Tobacco etch virus (TEV)‐based vector (TEV::AcrIIA4), editing levels at 7 dpi dropped dramatically to undetectable levels (Figure 1c; Table S3). These results confirmed the high efficiency of AcrIIA4 in preventing Cas9‐mediated mutagenesis in plants.
Figure 1.
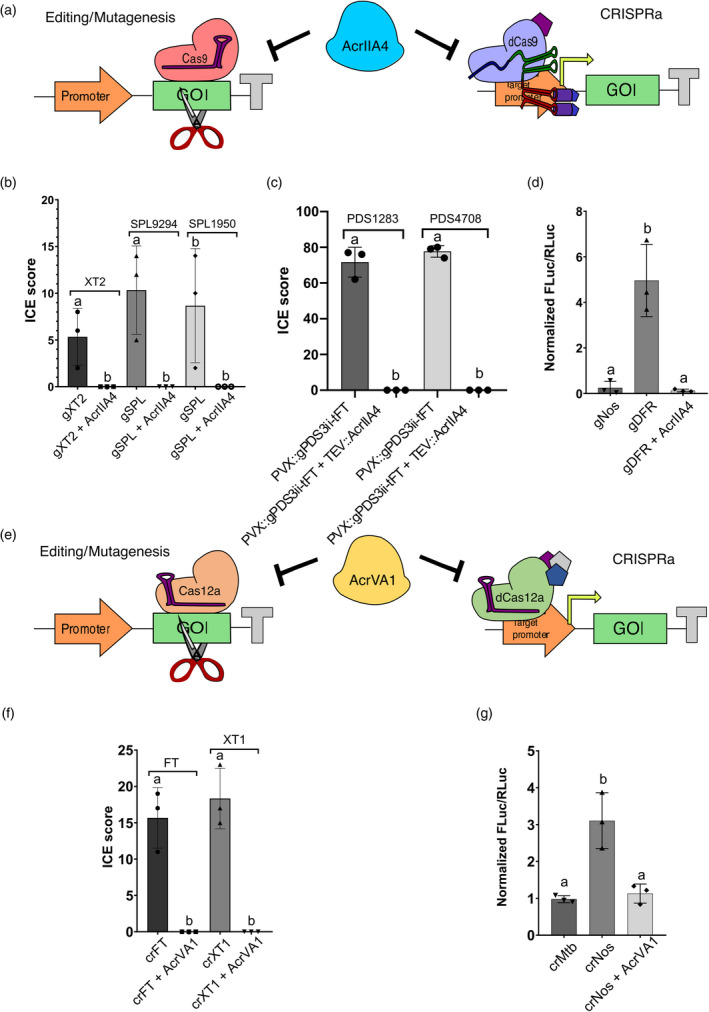
AcrIIA4 and AcrVA1 prevent Cas‐mediated gene editing and activation in Nicotiana benthamiana. (a) Schematic representation of dCasEV2.1 and the corresponding specific or unspecific gRNAs, with or without AcrIIA4 anti‐Cas9 activity. (b) Editing efficiency of NbXT2 and NbSPL in the absence and in the presence of NLS‐AcrIIA4. (c) Editing efficiency of NbPDS3 in Cas9 transgenic plants after agroinoculation of PVX::gRNA in the absence and in the presence of TEV::AcrIIA4. (d) Normalized FLuc/RLuc ratios of N. benthamiana leaves expressing a pDFR:Luc reporter in combination t AcrIIA4. (e) Schematic representation of AcrVA1 anti‐Cas12a activity. (f) Editing efficiency of NbXT1 and NbFT in the absence and in the presence of NLS‐AcrVA1. (g) Normalized FLuc/RLuc ratios of N. benthamiana leaves expressing a pNos:Luc reporter in combination with dCas12a:VPR and the corresponding specific or unspecific crRNAs, with or without AcrVA1. For (b) and (f) N. benthamiana leaves were agroinfiltrated with a Cas9 or Cas12a TU, a P19 TU and the corresponding gRNA/crRNA, with or without the NLS‐AcrIIA4 or NLS‐AcrVA1 TU. Editing efficiencies displayed in (b), (c) and (f) were determined using Synthego. For (d) and (g), FLuc/RLuc ratios were normalized using the FLuc/RLuc values obtained for a pNos:Luc reporter construct. Error bars indicate SD (n = 3). Statistical analyses were performed using unpaired t‐test (P ≤ 0.05). Variables within the same statistical groups are marked with the same letters.
Next, we wanted to test to which extent AcrIIA4 could also inhibit other Cas9 engineered functions that make use of its nuclease‐deployed dCas9 versions. One of the most widely used are the so‐called dCas9‐based programmable transcriptional activators (PTAs), which are becoming increasingly employed in plants for customized up‐regulation of endogenous gene expression (CRISPRa). In our laboratory, we previously developed a collection of dCas9‐based PTAs for plants, selecting the so‐called dCasEV2.1 as the strongest activator complex in our hands (Selma et al., 2019). dCasEV2.1 comprises three elements, each of them encoded in a different TU: (i) a modified gRNA scaffold, which includes anchoring sites for the phage MS2 coat protein (MCP; Konermann et al., 2015); (ii) a dCas9 fused to the EDLL plant activator domain and (iii) a MCP fused to the synthetic VP64‐p65‐Rta (VPR) activator domain. First, we validated the ability of AcrIIA4 to prevent dCasEV2.1‐based gene activation in plants using a transient assay based on a luciferase reporter. The reporter system comprised a firefly luciferase (FLuc) driven by the Solanum lycopersicum dihydroflavonol reductase (DFR) promoter (pSlDFR) and a dCasEV2.1 complex targeted to position −147 of the pSlDFR. Luminescence assays showed that the relative transcription activity (RTA) conferred by pSlDFR changed from 0.25 ± 0.28 in its basal state (with dCasEV2.1 loaded with an unrelated gRNA, gNos) to 4.96 ± 1.59 in the activated stage (with dCasEV2.1 loaded with a specific gRNA at −147 position of pSlDFR, gDFR) (Figure 1d) that correspond approximately to a 20‐fold activation. Remarkably, co‐expression of AcrIIA4 along with dCasEV2.1 and gDFR completely abolished dCasEV2.1 reducing RTA to basal levels of 0.13 ± 0.26 (Figure 1d).
Additionally, we decided to test another anti‐CRISPR protein targeting the second most used CRISPR system, CRISPR/Cas12a. The combination of two different CRISPR/Cas systems having different PAM requirements (e.g. Cas9 and Cas12) has often been considered as a requisite for creating complex regulatory circuits. This allows simultaneous gene activation and repression by targeting each of the Cas enzymes, one optimized for CRISPRa and the other for CRISPRi, to a different subset of genes. Therefore, development of anti‐Cas12a activities in planta can offer new possibilities for gene regulation. Anti‐Cas12a protein AcrVA1 has been described to cleave the crRNA when bound to Cas12a preventing editing and CRISPRa in yeast and mammalian cells (Kempton et al., 2020; Yu and Marchisio, 2021; Figure 1e). Following the same procedure as described for AcrIIA4, we selected two targets previously characterized for Cas12a: XT1 and FT genes (Bernabé‐Orts et al., 2019). Transient expression of specific crRNAs targeting these genes along with the Cas12a TU resulted in average editing efficiencies of 15% and 18% respectively (Figure 1f; Table S4). However, co‐infiltration of the same crRNAs and the Cas12a TU resulted in undetectable editing levels when the AcrVA1 was also co‐infiltrated. These results demonstrate that AcrVA1 can prevent CRISPR/Cas12a editing in N. benthamiana.
Next, we tested the ability of AcrVA1 to inhibit CRISPRa in N. benthamiana. Plant dCas12a‐based PTAs are not as potent activators as dCasEV2.1, but earlier comparative analyses showed that a fusion of dCas12a with the VP64 activation domain provided good activation rates when targeting the Agrobacterium tumefaciens nopaline synthase promoter (pNos; Vazquez‐Vilar et al., 2020). As previously described for AcrIIA4, we also used a reporter system to evaluate the performance of AcrVA1 preventing dCas12a‐based CRISPRa. In this example, the reporter system comprised the FLuc CDS driven by the pNos and the dCas12a PTA was targeted to positions −133, −33 and +26 of the pNos. As PTA for this experiment, we used a fusion of dCas12a with the VPR activation domain (dCas12a:VPR). Co‐infiltration of the reporter construct with the dCas12a:VPR TU and an unrelated crRNA (crMtb) resulted in basal RTA values of 0.97 ± 0.09 measured as normalized FLuc/RLuc ratios. When the reporter construct was co‐expressed with dCas12a:VPR and the three crRNAs targeting pNos, RTA went up to 3.11 ± 0.75 (>threefold induction). As in the case of AcrIIA4, the co‐expression of AcrVA1 along with the reporter construct, dCas12a:VPR and the pNos specific crRNAs reduced the promoter RTA to its basal levels 1.12 ± 0.26 (Figure 1g), demonstrating the inhibitory activity of AcrVA1.
AcrIIA4 prevents CRISPRa in N. benthamiana in a regulable fashion
Next, we wanted to further explore the behaviour of anti‐CRISPR proteins for regulating CRISPRa in N. benthamiana. Since dCasEV2.1 results in a higher activation range than dCas12a:VPR in our experimental conditions, we continued this work using the combination of dCasEV2.1 and AcrIIA4 for this purpose. It has been suggested that AcrIIA4 inhibits Cas9 activity by a binding mimicking mechanism, occupying the PAM DNA‐binding site with higher affinity than the DNA substrate (Yang and Patel, 2017). According to this, AcrIIA4 anti‐Cas activity is expected to be dependent on AcrIIA4 concentration, thus opening the way to establish dose‐dependent control systems. To test this hypothesis, we performed a dose–response experiment by infiltrating decreasing amounts of the A. tumefaciens culture that drives expression of AcrIIA4 together with the reporter/dCasEV2.1/gDFR mix. An enhanced GFP (EGFP) construct infiltrated in the same experimental conditions showed a linear relation between the optical densities (OD600) of the Agrobacterium culture and the levels of GFP fluorescence (Figure S1), thus validating the use of this culture dilution approach for dose–response experiments in the range of OD600 assayed. As expected, AcrIIA4 anti‐Cas9 activity showed a strong dose‐dependent effect (Figure 2a). The two highest OD600 assayed, namely 0.1 and 0.05, resulted in the complete inhibition of the dCasEV2.1‐mediated activation, with RTA values of ~0.09. The lowest OD600 that resulted in a significant reduction of the dCasEV2.1‐mediated activation was 0.005. This is a bacterial density 20 times lower than that used for transferring the dCasEV2.1 construct. Considering that all TUs in the assay are under the control of the CaMV 35S promoter, these results suggest a strong anti‐Cas9 activity of AcrIIA4 in plants, a very efficient expression of AcrIIA4, or both. To further confirm the dose‐dependent activity of AcrIIA4, we evaluated anti‐Cas activity when AcrIIA4 expression is driven by different promoters at a constant OD600. In addition to the CaMV 35S promoter, we selected three additional promoters with lower strengths, namely the nopaline synthase promoter (pNos) and two promoters from Nicotiana tabacum cv. K326 corresponding to gene IDs Nitab4.5_0000037g0360.1 (named as pNtA1) and Nitab4.5_0006225g0030.1 (named as pNtA11). Luminiscence assays showed that RTA values for CaMV 35S, pNtA1 and pNtA11 were ~11.67, 0.15 and 0.08, respectively, when compared to pNos (Figure S1). The infiltration of N. benthamiana leaves with A. tumefaciens culture mixes carrying the reporter/dCasEV2.1/gDFR cultures and the AcrIIA4 driven by the different promoters at an OD600 of 0.05 resulted in different degrees of inhibition of the CRISPRa activity. The AcrIIA4 expressed under the Nos promoter, resulted in a significant fourfold reduction of the dCasEV2.1‐mediated activation, while under pNtA1 and pNtA11, RTA values of ~2.8 and 4.3 were obtained respectively (Figure 2b). Thus, the expression of AcrIIA4 under a very week promoter resulted in a dCas‐driven activation similar to that obtained in the absence of AcrIIA4. These results strongly indicate again that AcrIIA4 activity could be successfully modulated in a dose‐dependent manner.
Figure 2.
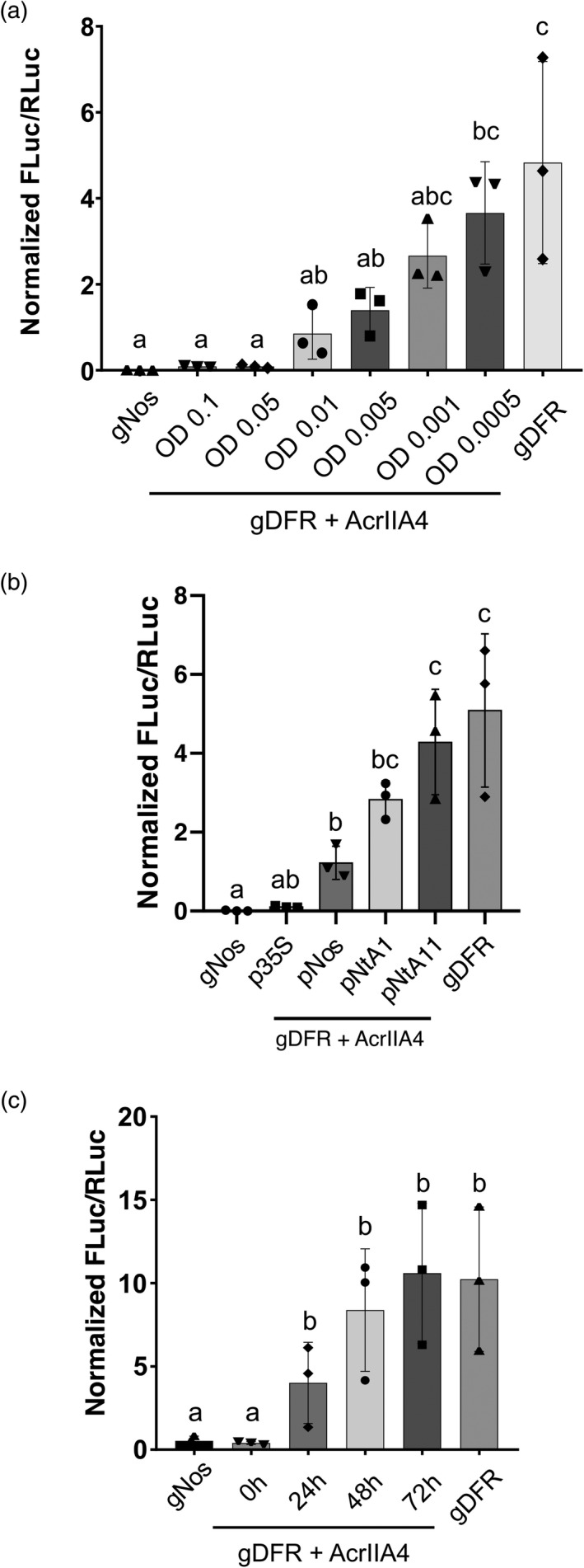
AcrIIA4 prevents dCas9‐based activation of a reporter gene in Nicotiana benthamiana in a regulable manner. (a) Normalized FLuc/RLuc ratios of N. benthamiana leaves expressing dCasEV2.1 and the corresponding specific or unspecific gRNAs, with AcrIIA4 infiltrated at different OD600 or (b) under different promoters. (c) Normalized FLuc/RLuc ratios of N. benthamiana leaves expressing dCasEV2.1 and the corresponding specific or unspecific gRNAs, with AcrIIA4 infiltrated at different time points. Error bars indicate SD (n = 3). Statistical analyses were performed using one‐way ANOVA (Tukey's multiple comparisons test, P ≤ 0.05). Variables within the same statistical groups are marked with the same letters.
A key factor to consider for gene regulation is time‐course dependency. Therefore, we decided to test the ability of AcrIIA4 to suppress an ongoing dCasEV2.1 activation. For this analysis, an AcrIIA4 construct was infiltrated simultaneously, or with a delay of 24, 48 and 72 h after the infiltration of the reporter/dCasEV2.1/gDFR mix, and all tissue samples were subsequently collected 96 h after the initial infiltration. In a parallel experiment, an A. tumefaciens culture driving the simultaneous expression of AcrIIA4 and EGFP was infiltrated at the different timepoints in the same experimental conditions to validate that re‐infiltration of previously agroinfiltrated tissue does not affect the gene expression capacity (Figure S1). As shown in Figure 2b, AcrIIA4 treatment is partially effective up to 24 h post‐initial infiltration, but this efficiency is completely lost at longer time points. These results suggest that displacement of dCas9‐DNA activation complexes by AcrIIA4 is a relatively slow or inefficient process and that PTA inhibition by AcrIIA4 is more successfully achieved by early formation of inhibitory complexes rather than by binding competition.
The repression of a TF is often used as a key control mechanism in the cell. In many cases, TF repressors are regulated through selective proteolytic degradation, circumventing lengthy transcriptional signalling. To emulate this regulatory theme using a fully synthetic approach, we attempted to engineer a de‐repression mechanism based on degron technology (Prozzillo et al., 2020). In a first attempt to modulate anti‐CRISPR activity in this way, we created and assayed an N‐terminal fusion of the AcrIIA4 with the K2 degron, which was earlier described to induce temperature‐dependent protein degradation when translationally fused to other proteins of interest (Faden et al., 2016; Figure 3a). We assayed two K2 configurations with or without the SV40 nuclear localization signal (NLS). As shown in Figure 3b, both degron fusions produced a shift in AcrIIA4 repression activities when moved from permissive (14 °C) to restrictive (28 °C) temperature, although only the K2:AcrIIA4 configuration was found statistically significant. However, even at permissive temperature, both K2‐AcrIIA4 fusions showed a reduced ability to repress dCasEV2.1‐dependent luciferase activation when compared with a native repressor, thus limiting its ability to function as an effective regulatory system.
Figure 3.
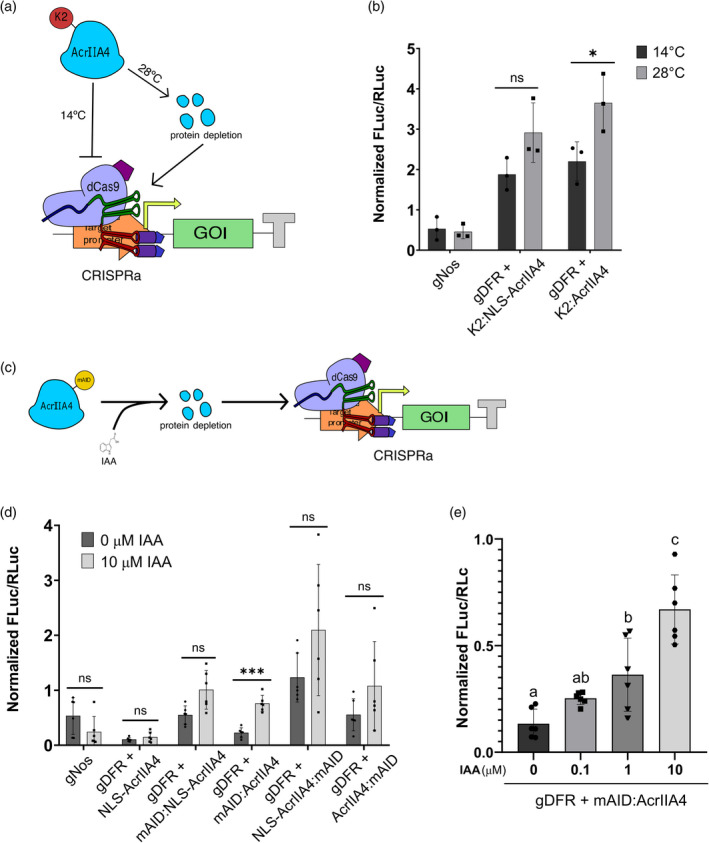
Post‐translational degradation of AcrIIA4 confers tunable dCas9‐based activation control in Nicotiana benthamiana. (a) Schematic representation of high temperature mediated AcrIIA4 protein depletion. (b) Changes in transcriptional de‐activation measured as normalized FLuc/RLuc ratios of N. benthamiana leaves expressing the low temperature degron K2‐AcrII4 fusions in response to low (14°C) and high (28°C) temperature treatments. (c) Schematic representation of IAA mediated AcrIIA4 protein depletion. (d) Changes in transcriptional de‐activation measured as normalized FLuc/RLuc ratios of N. benthamiana leaves expressing dCasEV2.1, the corresponding specific or unspecific gRNAs and four different versions of mAID‐AcrIIA4 fusions in response to IAA treatments. (e) Normalized FLuc/RLuc ratios of N. benthamiana leaves expressing dCasEV2.1, the corresponding specific or unspecific gRNAs and mAID:AcrIIA4 treated with different IAA concentrations. Error bars indicate SD: n = 3 for (b); n = 6 for (d) and (e). Statistical analyses were performed using one‐way ANOVA (Tukey's multiple comparisons test, P ≤ 0.05) or unpaired t‐test for two samples comparisons (*P ≤ 0.05 and ***P ≤ 0.0005).
To explore alternative regulatory options with wider dynamic range, we next analysed N‐ and C‐termini fusions of the AcrIIA4 to the minimal auxin inducible degron (AID) mAID47 (Brosh et al., 2016), again with or without the SV40 NLS signal, as schematically depicted in Figure 3c. AIDs were previously used for reprogramming A. thaliana development when coupled to dCas9‐based repressors (Khakhar et al., 2018), as well as for inducing protein depletion in other eukaryotic systems (Brosh et al., 2016; Nishimura et al., 2009; Zhang et al., 2015). Results in Figure 3d show that most of the mAID47 fusions assayed partially retained the anti‐Cas9 activity of native AcrIIA4, with some configurations showing almost full activity as it was the case for the mAID47 N‐terminal fusion without NLS (mAID‐AcrIIA4). Upon auxin treatment, all protein fusions showed a clear de‐repression trend indicative of auxin‐mediated degradation. However, the increase in reporter activity was only statistically significant for the above‐mentioned mAID‐AcrIIA4 fusion (fourfold activation). This is still an incomplete de‐repression when compared with the luminescence levels obtained when AcrIIA4 is not present, thus indicating that this degron fusion needs further optimization to reach its full potential.
To further confirm the auxin‐dependency of the observed de‐repression, we conducted a dose–response analysis, spraying agroinfiltrated leaves with increasing IAA concentrations. As shown in Figure 3e, luciferase expression is dependent on hormone concentration. Despite the low inducibility range because of the aforementioned incomplete de‐repression, it is clear that mAID‐AcrIIA4 degradation rises with increasing IAA concentrations, and thus results in higher luminescence. These results indicate that mAID‐AcrIIA4 has certain ability to function as a quantitative IAA sensor, taking IAA concentrations as input and translating them into a proportional transcriptional output.
AcrIIA4 prevents CRISPRa of endogenous genes in N. benthamiana
Finally, we analysed the ability of AcrIIA4 to prevent dCasEV2.1‐mediated gene activation of two endogenous N. benthamiana genes, a DFR (NbDFR; Niben101Scf00305g05035), and a MYB TF involved in phenylpropanoid biosynthesis (NbAN2; Niben101Scf00156g02004). Both genes were earlier shown to be efficiently activated with the dCasEV2.1 system (Selma et al., 2019). N. benthamiana leaves were agroinfiltrated with the dCasEV2.1 system targeting NbDFR or NbAN2 promoters with or without AcrIIA4. At 4 dpi, leaf samples were collected and reverse transcription‐quantitative PCR (RT‐qPCR) assays were performed for each gene. To estimate gene expression levels, agroinfiltrated NbDFR samples were used as negative control for NbAN2 samples and vice versa. Infiltration of dCasEV2.1 targeting NbDFR or NbAN2 promoter regions conferred strong activation reaching up to 400‐ and 30‐fold, respectively, although the absolute levels were highly dependent on the age of the leaf (Figure 4a,b). Remarkably, co‐infiltration of AcrIIA4 strongly repressed CRISPRa in both genes, resulting in a significant reduction of the relative gene expression values in all samples assayed.
Figure 4.
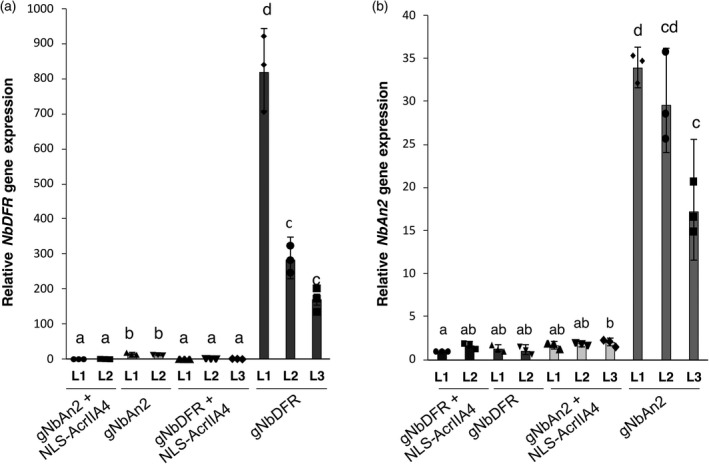
AcrIIA4 prevents dCas9‐based activation of NbDFR and NbAN2 endogenous genes in Nicotiana benthamiana. N. benthamiana leaves expressing dCas9EV2.1 and the corresponding specific or unspecific gRNAs for the activation of NbDFR or NbAN2, with or without NLS‐AcrIIA4. L1, L2 and L3 indicate independent agroinfiltrated leaves. Error bars indicate SD (n = 3, technical replicates). Statistical analyses were performed using one‐way ANOVA (Tukey's multiple comparisons test, P ≤ 0.05).
Discussion
The rapid expansion of CRISPR/Cas for genome engineering in plants, and its increasing use for programmable gene regulation have unveiled the need of generating control mechanisms for the different types of CRISPR‐derived activities. In this regard, Acr proteins represent an unexplored control tool in plant biotechnology. In plants, CRISPR/Cas9 and CRISPR/Cas12a are the most popular tools for genome engineering; therefore, we chose to characterize Acr proteins inhibiting these systems. Previous studies in other organisms, including bacteria, yeast and mammalian cells, already demonstrated the potent anti‐Cas9 effect of AcrIIA4 (Li et al., 2018; Nakamura et al., 2019; Rauch et al., 2017). Recently, Yu and Marchisio (2021) also reported the high anti‐Cas12a inhibitory capacity of AcrVA1 in yeast.
In this work, we demonstrated that transient expression of AcrIIA4 and AcrVA1 in N. benthamiana leaves reduces Cas9‐ and Cas12a‐mediated editing to undetectable levels for all seven tested loci. Notably, we showed that the high levels obtained with VIGE using a PVX vector for gRNA delivery can be completely counteracted with TEV‐delivered AcrIIA4. Altogether, our results confirmed the potent ability of AcrIIA4 and AcrVA1 to prevent CRISPR/Cas9 and CRISPR/Cas12a‐mediated mutagenesis. Among other potential uses, off‐switch editing tools as those shown here will be important for the control of gene drive systems in plants, as previously shown in other organisms (Basgall et al., 2018). CRISPR/Cas‐based gene drives, which have been proposed for weed control and for the conservation of endangered plant populations (Barrett et al., 2019), were recently demonstrated in A. thaliana (Zhang et al., 2021), but require effective control systems that safeguard the extension of the drive to non‐target populations.
Many of the potential applications of Acr proteins rely on their ability to regulate CRISPRa and CRISPRi‐based genetic circuits. Acr proteins offer an additional control point for implementing, for example spatio‐temporal regulation in this type of circuits. Owing to its simplicity and its strong activity, Acr can result advantageous for circuit engineering in plants as compared to other dCas‐off‐switch methods such as those that rely on degron fusions to dCas (Kleinjan et al., 2017), or on catalytically inactivating gRNAs (Rose et al., 2020). One major issue when implementing inducible genetic circuits is transcriptional read‐through or ‘leaky’ expression. On this regard, Acr proteins could act as ‘safeguards’ preventing the activity of small amounts of Cas protein generated by leaky expression in CRISPRa and CRISPRi‐based genetic circuits. Moreover, Acr proteins are small in size (87 and 170 aa for AcrIIA4 and AcrVA1 respectively), facilitating their cloning and in situ delivery via viral vectors, which usually impose size constraints (Hefferon, 2017; Marino et al., 2020). Despite their small size, they could remain functional upon fusions of tags both on their N‐ and C‐terminus (Basgall et al., 2018). However, the incorporation of Acr proteins to synthetic genetic circuits relies in the proper characterization of the effectiveness and the dynamics of the inhibition. In this work, we showed that, when transiently expressed, AcrIIA4 and AcrVA1 are capable of totally blocking the strong dCasEV2.1‐ and dCas12a‐driven CRISPRa of reporter genes in N. benthamiana, consistent with previous studies in other organisms. However, the delivery of AcrIIA4 twenty‐four hours after the delivery of dCasEV2.1 already resulted in a non‐significant reduction of CRISPRa‐mediated activation. These results are consistent with those previously reported (Yang and Patel, 2017), suggesting that AcrIIA4 cannot access and replace the Cas9‐bound dsDNA substrate.
Genetic circuits design also relies on the possibility of incorporating sensors or ligand‐responsive gene switches. Previous works in eukaryotic cells demonstrated that destabilization of anti‐CRISPR proteins either by the fusion of the Avena sativa LOV2 domain or the Shield‐1‐responsive destabilization domain caused the misfold of the anti‐CRISPR protein upon photoexcitation and Shield‐1 application, respectively, and resulted in the loss of its affinity to the Cas (Bubeck et al., 2018; Nakamura et al., 2019). In order to investigate to what extent the inhibitory activity of AcrIIA4 could be tunable with exogenous signals in plants, we tested two degrons, one being temperature‐based (i.e. K2) and another chemical‐based (i.e. mAID). While the K2 degron conferred only partial AcrIIA4 depletion, the mAID degron displayed a wide range of protein degradation depending on the protein configuration. The AcrIIA4 version carrying an N‐terminal mAID and lacking the NLS was the best of the tested configurations. mAID:AcrIIA4 was able to inhibit CRISPRa as efficiently as NLS‐AcrIIA4 and its activity can be modulated in an auxin dose‐based manner. However, the maximum activation levels obtained upon auxin treatment were far below those conferred by dCasEV2.1 in the absence of AcrIIA4. These results may suggest that mAID:AcrIIA4 depletion is only partial or that mAID:AcrIIA4 cannot be targeted to degradation once it is bound to the dCasEV2.1. Given the relatively high doses of auxins reached in the dose–response experiment (Yesbolatova and Tominari, 2019), the most likely explanation is that mAID:AcrIIA4‐dCasEV2.1 complexes are formed immediately after protein folding impeding the degradation of the dCasEV2.1‐bound mAID:AcrIIA4 via the ubiquitin‐proteasome pathway.
In conclusion, this work demonstrates that Acr proteins AcrIIA4 and AcrVA1 are effective suppressors of CRISPR/Cas‐mediated editing and CRISPRa in N. benthamiana. It also represents a first step towards the incorporation of Acr proteins to CRISPRa‐based genetic switches in plants. Remarkably, AcrIIA4 shows a great potential as a ‘safeguard’ component of these circuits. The discovery of more Acr proteins and the elucidation of their mechanism of action will provide new insights for circuit design. In particular, the anti‐Cas12a protein AcrVA4, which was recently reported to dislodge dsDNA bound to dCas12a, has a great potential for circuits in which the turnover of the inhibition is required (Knott et al., 2019).
Experimental procedures
GoldenBraid cloning
DNA constructs used in this work were assembled using GoldenBraid (Sarrion‐Perdigones et al., 2013). AcrIIA4 protein sequence, as originally reported (Rauch et al., 2017) fused to SV40 NLS and mAID47 sequence, as reported (Brosh et al., 2016) (modified as N‐tag or C‐tag), were codon optimized for N. benthamiana using IDT Codon Optimization Tool (https://eu.idtdna.com/CodonOpt) and subsequently domesticated at https://gbcloning.upv.es/do/synthesis/. Additionally, a N. benthamiana codon optimized AcrIIA4 protein version without SV40‐NLS, B3‐B4 version was domesticated using GoldenBraid GB Domesticator tool (https://gbcloning.upv.es/do/domestication/). Level 1 assemblies of TUs from individual Level 0 parts were performed through Golden Gate‐like multipartite BsaI reactions. All Level 0 and Level 1 assemblies were performed as previously reported (Vazquez‐Vilar et al., 2017). Level 0 assemblies were confirmed by restriction analysis and Sanger sequencing, and Level 1 assemblies were verified by restriction analysis. An exhaustive list of all plasmids used in this work is listed in Table S5.
Viral vectors assembly
Cloning of the gRNA targeting both NbPDS3 homeologs in a PVX‐based vector was previously described (Uranga, Aragonés, et al., 2021). The viral clone TEV::AcrIIA4, which includes AcrIIA4 between the NIb and CP cistron, was assembled as previously described by (Uranga, Vazquez‐Vilar, et al., 2021), using primers listed in Table S6 for AcrIIA4 amplification.
N. benthamiana transient expression
For transient expression assays, plasmids were transferred to A. tumefaciens strain C58C1. Five to six weeks old N. benthamiana plants grown at 24 °C and 16 h (light)/20 °C and 8 h (darkness) conditions were used. Agroinfiltration was carried out as previously reported (Orzaez et al., 2009). Briefly, overnight A. tumefaciens cultures were pelleted and resuspended in agroinfiltration buffer (10 mM MES, pH 5.6, 10 mM MgCl2 and 200 µM acetosyringone) to an optical density of 0.1 at 600 nm (OD600). Bacterial suspensions were incubated for 2 h at room temperature on a horizontal rolling mixer and then mixed for co‐expression experiments, in which more than one GB element was used. Finally, agroinfiltrations were carried out through the abaxial surface of the three youngest fully expanded leaves of each plant with a 1‐ml needle‐free syringe. For some experiments, agroinfiltrations were carried out with small modifications from the general procedure described above. For dose–response assays, SV40‐AcrIIA4 TU (GB3344) was resuspended in agroinfiltration solution to an OD600 of 0.1 and subsequently diluted to 0.05, 0.01, 0.005 and 0.001 using a culture carrying an empty vector to maintain the final OD600 at 0.1. For the time‐course assay, the strain carrying the SV40‐AcrIIA4 TU (GB3344) was agroinfiltrated at OD600 of 0.05 at 0, 24, 48 and 72 h after infiltration of the dCasEV2.1 with the DFR gRNA (GB2513) and the pSlDFR reporter construct (GB1160). Detailed information of the experimental design can be found in the Methods S1 section.
N. benthamiana agroinoculation
For agroinoculation experiments, plasmids PVX::gPDS3ii‐tFT and TEV::AcrIIA4 were transferred to A. tumefaciens strain C58C1, containing the pCLEAN‐S48 helper plasmid (https://pubmed.ncbi.nlm.nih.gov/17932303/) in the case of TEV::AcrIIA4. Five to six weeks old Cas9‐expressing N. benthamiana plants grown at 25 °C under a 16 h day/8 h night cycle were used. For agroinoculation of PVX::gPDS3ii‐tFT either in combination with TEV::AcrIIA4 or with TEV::GFP (used as control), the procedure previously described (Bedoya et al., 2010; Uranga, Aragonés, et al., 2021) was followed. Briefly, single colonies were grown at 28 °C for about 24 h, pelleted and resuspended in agroinfiltration buffer to an OD600 of 0.5. Resuspended cells were incubated for 2 h at 28 °C in darkness, and bacteria suspensions of PVX::gPDS3ii‐tFT and TEV::AcrIIA4 or TEV::GFP were mixed in a 1:1 ratio. Agroinfiltrations were carried out through the abaxial surface of the two oldest fully expanded leaves of each plant with a 1‐ml needle‐free syringe. Three independent plants were used as biological replicates. Following agroinfiltration, plants were transferred to a growth chamber at 25 °C under a 12‐h day/12‐h night cycle.
Luciferase activity and determination of relative transcriptional activity
Leaf samples were collected at 4 dpi. For the determination of the FLuc/RLuc activity, one 0.8 cm diameter disc per agroinfiltrated leaf was excised. Leaf discs were frozen in liquid nitrogen and subsequently homogenized with 180 µl of Passive Lysis buffer, followed by 15 min of centrifugation at 14 000× g at 4°C. Crude extract (10 µl) was mixed with 40 µl of LARII, and FLuc activity was determined using a GloMax 96 microplate luminometer (Promega) with a 2‐s delay and a 10‐s measurement time. After the measurements, 40 µl of Stop&Glo reagent was added per sample and Renilla luciferase (RLuc) activity was determined using the same protocol. Sample FLuc/RLuc ratios were calculated as the mean value of the three independent agroinfiltrated leaves. Relative transcriptional activities were calculated as the FLuc/RLuc ratios of the pSlDFR reporter in each sample normalized with the FLuc/RLuc ratios produced by a pNos reporter (GB1116) assayed in parallel and expressed in relative promoter units (Vazquez‐Vilar et al., 2017).
Fluorescence activity assay
Leaf samples were collected at 4 dpi. For the determination of fluorescence, one 0.5 cm diameter disc per agroinfiltrated leaf was excised and transferred to a black 96‐well microplate. Subsequently, fluorescence was determined using EX 490, EM 510‐570 filters in a GloMax 96 microplate luminometer (Promega) following the manufacturer instructions.
Determination of Cas9‐mediated editing activity
For determination of editing efficiencies on plants transiently expressing AcrIIA4 and their corresponding controls, approximately 150 mg of leaf tissue was collected from each of the three infiltrated leaves at 5 dpi. For determination of editing efficiencies on Cas9‐expressing plants agroinoculated with TEV::AcrIIA4 and their corresponding controls, approximately 150 mg of leaf tissue was collected at 7 dpi from the first symptomatic systemic leaf for each plant. Genomic DNA was extracted from leaf tissue following the CTAB protocol (Murray and Thompson, 1980). The DNA was used as template for PCR amplification of the targeted sites with primers listed on Table S6 and using MyTaq™ DNA Polymerase (Bioline). Subsequently, PCR products were analysed in 1% agarose gel electrophoresis, purified with the ExoSAP‐IT™ PCR Product Cleanup Reagent (Applied Biosystems™) following manufacturer instructions and Sanger‐sequenced. Finally, sequencing results were analysed using Synthego CRISPR Performance Analysis (https://ice.synthego.com/#/) to determine the ICE score.
RNA isolation and RT‐qPCR
Leaf samples from infiltrated plants were harvested at 4 dpi, and 100 mg of tissue was used for total RNA isolation using the Thermo Scientific™ GeneJET RNA Purification Kit. Total RNA was treated with recombinant DNase I (RNase‐free) (Takara) following manufacturer´s instructions. Aliquots of 1 µg of the treated RNA were used for cDNA synthesis with oligo dT using PrimeScript™ RT‐PCR kit (Takara). cDNAs (0.4 µl) were used to determine the expression levels for each gene in triplicated 25 µl reactions with the SYBR® Premix Ex Taq (Takara) using the Applied biosystem 7500 Fast Real Time PCR system. N. benthamiana F‐BOX gene was used as internal reference (Liu et al., 2012). Calculations of each sample were carried out according the comparative ΔΔCT method (Livak and Schmittgen, 2001). Primers used for qRT‐PCR reactions are listed in Table S6.
Conflict of interest
The authors declare no conflict of interest.
Author contributions
D.O., J.A.D., C.C., M.V‐V. and M.U. designed the experiments. C.C., M.V‐V., S.S., M.U. and A.F‐d‐C. conducted the experiments. C.C., M.V‐V. and D.O. drafted the manuscript. All the authors discussed and revised the manuscript.
Supporting information
Figure S1. AcrIIA4 prevents dCas9‐based activation of a reporter gene in N. benthamiana in a regulable manner.
Table S1. Targeted genes and gRNA sequences of the editing experiments.
Table S2. Summary of Synthego's ICE results for the Cas9‐mediated editing assay.
Table S3. Summary of Synthego's ICE results for the VIGE assay.
Table S4. Summary of Synthego's ICE results for the Cas12a‐mediated editing assay.
Table S5. List of GoldenBraid plasmids used in this work. Sequence information is available at https://gbcloning.upv.es/search/ by entering the GB Number.
Table S6. List of primers used in this work. Primers used for Sanger sequencing are underlined.
Method S1. List of tables including the Agrobacterium cultures co‐infiltrated in the same mix for each experiment.
Acknowledgements
This work has been funded by grants PID2019‐108203RB‐100, PID2020‐114691RB‐I00 and BIO2017‐83184‐R from the Spanish Ministerio de Ciencia e Innovación, through the Agencia Estatal de Investigación (co‐financed European Regional Development Fund). Calvache, C. is recipient of FPI‐UPV (PAID‐01‐20) fellowship from Universitat Politècnica de València. Vazquez‐Vilar, M. is recipient of APOSTD/2020/096 (Generalitat Valenciana and Fondo Social Europeo post‐doctoral grant). Selma, S. and Uranga, M. are recipients of FPI (BIO2016‐78601‐R) H2020 Research Program: 760331 Newcotiana and FPU (FPU17/05503) fellowships, respectively, from Ministerio de Ciencia e Innovacion (Spain).
Calvache, C. , Vazquez‐Vilar, M. , Selma, S. , Uranga, M. , Fernández‐del‐Carmen, A. , Daròs, J.‐A. and Orzáez, D. (2022) Strong and tunable anti‐CRISPR/Cas activities in plants. Plant Biotechnol. J., 10.1111/pbi.13723
References
- Adli, M. (2018) The CRISPR tool kit for genome editing and beyond. Nat. Commun., 9, 1911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baltes, N.J. and Voytas, D.F. (2015) Enabling plant synthetic biology through genome engineering. Trends Biotechnol., 33, 120–131. [DOI] [PubMed] [Google Scholar]
- Barone, P. , Wu, E. , Lenderts, B. , Anand, A. , Gordon‐Kamm, W. , Svitashev, S. and Kumar, S. (2020) Efficient gene targeting in maize using inducible CRISPR‐Cas9 and marker‐free donor template. Mol. Plant, 13, 1219–1227. [DOI] [PubMed] [Google Scholar]
- Barrett, L.G. , Legros, M. , Kumaran, N. , Glassop, D. , Raghu, S. and Gardiner, D.M. (2019) Gene drives in plants: opportunities and challenges for weed control and engineered resilience. Proc. R. Soc. B Biol. Sci., 286, 20191515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Basgall, E.M. , Goetting, S.C. , Goeckel, M.E. , Giersch, R.M. , Roggenkamp, E. , Schrock, M.N. , Halloran, M. et al. (2018) Gene drive inhibition by the anti‐CRISPR proteins AcrIIA2 and AcrIIA4 in Saccharomyces cerevisiae. Microbiology, 164, 464–474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bedoya, L. , Martínez, F. , Rubio, L. and Daròs, J.‐A. (2010) Simultaneous equimolar expression of multiple proteins in plants from a disarmed potyvirus vector. J. Biotechnol., 150, 268–275. [DOI] [PubMed] [Google Scholar]
- Bernabé‐Orts, J.M. , Casas‐Rodrigo, I. , Minguet, E.G. , Landolfi, V. , Garcia‐Carpintero, V. , Gianoglio, S. et al. (2019) Assessment of Cas12a‐mediated gene editing efficiency in plants. Plant Biotechnol. J., 17, 1971–1984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Borges, A.L. , Davidson, A.R. and Bondy‐Denomy, J. (2017) The discovery, mechanisms, and evolutionary impact of anti‐CRISPRs. Annu. Rev. Virol., 4, 37–59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brosh, R. , Hrynyk, I. , Shen, J. , Waghray, A. , Zheng, N. and Lemischka, I.R. (2016) A dual molecular analogue tuner for dissecting protein function in mammalian cells. Nat. Commun., 7, 11742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bubeck, F. , Hoffmann, M.D. , Harteveld, Z. , Aschenbrenner, S. , Bietz, A. , Waldhauer, M.C. et al. (2018) Engineered anti‐CRISPR proteins for optogenetic control of CRISPR–Cas9. Nat. Methods, 15, 924–927. [DOI] [PubMed] [Google Scholar]
- Dong, D.E. , Guo, M. , Wang, S. , Zhu, Y. , Wang, S. , Xiong, Z. , Yang, J. et al. (2017) Structural basis of CRISPR‐SpyCas9 inhibition by an anti‐CRISPR protein. Nature, 546, 436–439. [DOI] [PubMed] [Google Scholar]
- Ellison, E.E. , Nagalakshmi, U. , Gamo, M.E. , Huang, P.‐J. , Dinesh‐Kumar, S. and Voytas, D.F. (2020) Multiplexed heritable gene editing using RNA viruses and mobile single guide RNAs. Nat. Plants, 6, 620–624. [DOI] [PubMed] [Google Scholar]
- Faden, F. , Ramezani, T. , Mielke, S. , Almudi, I. , Nairz, K. , Froehlich, M.S. et al. (2016) Phenotypes on demand via switchable target protein degradation in multicellular organisms. Nat. Commun., 7, 12202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hefferon, K. (2017) Plant virus expression vectors: a powerhouse for global health. Biomedicines, 5, 44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kempton, H.R. , Goudy, L.E. , Love, K.S. and Qi, L.S. (2020) Multiple input sensing and signal integration using a split Cas12a system. Mol. Cell, 78, 184–191.e3. [DOI] [PubMed] [Google Scholar]
- Khakhar, A. , Leydon, A.R. , Lemmex, A.C. , Klavins, E. and Nemhauser, J.L. (2018) Synthetic hormone‐responsive transcription factors can monitor and re‐program plant development. eLife, 7, e34702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kleinjan, D.A. , Wardrope, C. , Nga Sou, S. and Rosser, S.J. (2017) Drug‐tunable multidimensional synthetic gene control using inducible degron‐tagged dCas9 effectors. Nat. Commun., 8, 1191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knott, G.J. , Thornton, B.W. , Lobba, M.J. , Liu, J.‐J. , Al‐Shayeb, B. , Watters, K.E. and Doudna, J.A. (2019) Broad‐spectrum enzymatic inhibition of CRISPR‐Cas12a. Nat. Struct. Mol. Biol., 26, 315–321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Konermann, S. , Brigham, M.D. , Trevino, A.E. , Joung, J. , Abudayyeh, O.O. , Barcena, C. , Hsu, P.D. et al. (2015) Genome‐scale transcriptional activation by an engineered CRISPR‐Cas9 complex. Nature, 517, 583–588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li, J. , Xu, Z. , Chupalov, A. and Marchisio, M.A. (2018) Anti‐CRISPR‐based biosensors in the yeast S. cerevisiae . J. Biol. Eng., 12, 11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu, D. , Shi, L. , Han, C. , Yu, J. , Li, D. and Zhang, Y. (2012) Validation of reference genes for gene expression studies in virus‐infected Nicotiana benthamiana using quantitative real‐time PCR. PLoS One, 7, e46451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Livak, K.J. and Schmittgen, T.D. (2001) Analysis of relative gene expression data using real‐time quantitative PCR and the 2−ΔΔCT method. Methods, 25, 402–408. [DOI] [PubMed] [Google Scholar]
- Lo, A. and Qi, L. (2017) Genetic and epigenetic control of gene expression by CRISPR–Cas systems. F1000Research, 6, 747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lowder, L.G. , Paul, J.W. and Qi, Y. (2017) Multiplexed transcriptional activation or repression in plants using CRISPR‐dCas9‐based systems. Methods Mol. Biol., 1629, 167–184. [DOI] [PubMed] [Google Scholar]
- Marino, N.D. , Pinilla‐Redondo, R. , Csörgő, B. and Bondy‐Denomy, J. (2020) Anti‐CRISPR protein applications: natural brakes for CRISPR‐Cas technologies. Nat. Methods, 17, 471–479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moradpour, M. and Abdulah, S.N.A. (2020) CRISPR/dCas9 platforms in plants: strategies and applications beyond genome editing. Plant Biotechnol. J., 18, 32–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murray, M.G. and Thompson, W.F. (1980) Rapid isolation of high molecular weight plant DNA. Nucleic Acids Res., 8, 4321–4325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakamura, M. , Srinivasan, P. , Chavez, M. , Carter, M.A. , Dominguez, A.A. , La Russa, M. et al. (2019) Anti‐CRISPR‐mediated control of gene editing and synthetic circuits in eukaryotic cells. Nat. Commun., 10, 194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nishimura, K. , Fukagawa, T. , Takisawa, H. , Kakimoto, T. and Kanemaki, M. (2009) An auxin‐based degron system for the rapid depletion of proteins in nonplant cells. Nat. Methods, 6, 917–922. [DOI] [PubMed] [Google Scholar]
- Ochoa‐Fernandez, R. , Abel, N.B. , Wieland, F.‐G. , Schlegel, J. , Koch, L.‐A. , Miller, J.B. et al. (2020) Optogenetic control of gene expression in plants in the presence of ambient white light. Nat. Methods, 17, 717–725. [DOI] [PubMed] [Google Scholar]
- Orzaez, D. , Medina, A. , Torre, S. , Fernández‐Moreno, J.P. , Rambla, J.L. , Fernández‐Del‐Carmen, A. et al. (2009) A visual reporter system for virus‐induced gene silencing in tomato fruit based on anthocyanin accumulation. Plant Physiol., 150, 1122–1134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prozzillo, Y. , Fattorini, G. , Santopietro, M.V. , Suglia, L. , Ruggiero, A. , Ferreri, D. and Messina, G. (2020) Targeted protein degradation tools: overview and future perspectives. Biology, 9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qi, L.S. , Larson, M.H. , Gilbert, L.A. , Doudna, J.A. , Weissman, J.S. , Arkin, A.P. and Lim, W.A. (2013) Repurposing CRISPR as an RNA‐guided platform for sequence‐specific control of gene expression. Cell, 152, 1173–1183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rauch, B.J. , Silvis, M.R. , Hultquist, J.F. , Waters, C.S. , McGregor, M.J. , Krogan, N.J. and Bondy‐Denomy, J. (2017) Inhibition of CRISPR‐Cas9 with bacteriophage proteins. Cell, 168, 150–158.e10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rose, J.C. , Popp, N.A. , Richardson, C.D. , Stephany, J.J. , Mathieu, J. , Wei, C.T. et al. (2020) Suppression of unwanted CRISPR‐Cas9 editing by co‐administration of catalytically inactivating truncated guide RNAs. Nat. Commun., 11, 2697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sarrion‐Perdigones, A. , Vazquez‐Vilar, M. , Palaci, J. , Castelijns, B. , Forment, J. , Ziarsolo, P. , Blanca, J. et al. (2013) GoldenBraid 2.0: a comprehensive DNA assembly framework for plant synthetic biology. Plant Physiol., 162, 1618–1631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Selma, S. , Bernabé‐Orts, J.M. , Vazquez‐Vilar, M. , Diego‐Martin, B. , Ajenjo, M. , Garcia‐Carpintero, V. et al. (2019) Strong gene activation in plants with genome‐wide specificity using a new orthogonal CRISPR/Cas9‐based programmable transcriptional activator. Plant Biotechnol. J., 17, 1703–1705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stanley, S.Y. and Maxwell, K.L. (2018) Phage‐encoded anti‐CRISPR defenses. Annu. Rev. Genet., 52, 445–464. [DOI] [PubMed] [Google Scholar]
- Trasanidou, D. , Gerós, A.S. , Mohanraju, P. , Nieuwenweg, A.C. , Nobrega, F.L. and Staals, R.H.J. (2019) Keeping crispr in check: diverse mechanisms of phage‐encoded anti‐crisprs. FEMS Microbiol. Lett., 366(9). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uranga, M. , Aragonés, V. , Selma, S. , Vázquez‐Vilar, M. , Orzáez, D. and Daròs, J.‐A. (2021) Efficient Cas9 multiplex editing using unspaced sgRNA arrays engineering in a Potato virus X vector. The Plant Journal. Cell and Molecular Biology, 106, 555–565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uranga M., Vazquez‐Vilar M., Orzáez D., and Daròs J.‐A., (2021) CRISPR‐Cas12a Genome Editing at the Whole‐Plant Level Using Two Compatible RNA Virus Vectors. The CRISPR Journal. 10.1089/crispr.2021.0049 [DOI] [PubMed] [Google Scholar]
- Vazquez‐Vilar, M. , Garcia‐Carpintero, V. , Selma, S. , Bernabé‐Orts, J.M. , Sanchez‐Vicente, J. , Salazar‐Sarasua, B. , Ressa, A. et al. (2020) Edition of complex gene families in tobacco with GoldenBraid 4.0, a multipurpose web‐based platform for plant genome engineering. bioRxiv. 10.1101/2020.10.06.327841 [DOI] [Google Scholar]
- Vazquez‐Vilar, M. , Quijano‐Rubio, A. , Fernandez‐del‐Carmen, A. , Sarrion‐Perdigones, A. , Ochoa‐Fernandez, R. , Ziarsolo, P. et al. (2017) GB3.0: a platform for plant bio‐design that connects functional DNA elements with associated biological data. Nucleic Acids Res., 45, 2196–2209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang, X. , Ye, L. , Lyu, M. , Ursache, R. , Löytynoja, A. and Mähönen, A.P. (2020) An inducible genome editing system for plants. Nat. Plants, 6, 766–772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang, H. and Patel, D.J. (2017) Inhibition mechanism of an anti‐CRISPR suppressor AcrIIA4 targeting SpyCas9. Mol. Cell, 67, 117–127.e5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yesbolatova, A. , Tominari, Y. and Kanemaki, M.T. (2019) Ligand‐induced genetic degradation as a tool for target validation. Drug Discov. Today Technol., 31, 91–98. [DOI] [PubMed] [Google Scholar]
- Yu, L. and Marchisio, M.A. (2021) Saccharomyces cerevisiae synthetic transcriptional networks harnessing dCas12a and type V‐A anti‐CRISPR proteins. ACS Synth. Biol., 10, 870–883. [DOI] [PubMed] [Google Scholar]
- Zhan, X. , Lu, Y. , Zhu, J.‐K. and Botella, J.R. (2020) Genome editing for plant research and crop improvement. J. Integr. Plant Biol., 63, 3–33. [DOI] [PubMed] [Google Scholar]
- Zhang, L. , Ward, J.D. , Cheng, Z. and Dernburg, A.F. (2015) The auxin‐inducible degradation (AID) system enables versatile conditional protein depletion in C. elegans . Dev. Camb. Engl., 142, 4374–4384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang, T. , Mudgett, M. , Rambabu, R. , Abramson, B. , Dai, X. , Michael, T.P. and Zhao, Y. (2021) Selective inheritance of target genes from only one parent of sexually reproduced F1 progeny in Arabidopsis. Nat. Commun., 12, 3854. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1. AcrIIA4 prevents dCas9‐based activation of a reporter gene in N. benthamiana in a regulable manner.
Table S1. Targeted genes and gRNA sequences of the editing experiments.
Table S2. Summary of Synthego's ICE results for the Cas9‐mediated editing assay.
Table S3. Summary of Synthego's ICE results for the VIGE assay.
Table S4. Summary of Synthego's ICE results for the Cas12a‐mediated editing assay.
Table S5. List of GoldenBraid plasmids used in this work. Sequence information is available at https://gbcloning.upv.es/search/ by entering the GB Number.
Table S6. List of primers used in this work. Primers used for Sanger sequencing are underlined.
Method S1. List of tables including the Agrobacterium cultures co‐infiltrated in the same mix for each experiment.