

Abstract
Machado-Joseph disease (MJD) is a fatal neurodegenerative disorder clinically characterized by prominent ataxia. It is caused by an expansion of a CAG trinucleotide in ATXN3, translating into an expanded polyglutamine (polyQ) tract in the ATXN3 protein, that becomes prone to misfolding and aggregation. The pathogenesis of the disease has been associated with the dysfunction of several cellular mechanisms, including autophagy and transcription regulation. In this study, we investigated the transcriptional modifications of the autophagy pathway in models of MJD and assessed whether modulating the levels of the affected autophagy-associated transcripts (AATs) would alleviate MJD-associated pathology. Our results show that autophagy is impaired at the transcriptional level in MJD, affecting multiple AATs, including Unc-51 like autophagy activating kinase 1 and 2 (ULK1 and ULK2), two homologs involved in autophagy induction. Reinstating ULK1/2 levels by adeno-associated virus (AAV)-mediated gene transfer significantly improved motor performance while preventing neuropathology in two in vivo models of MJD. Moreover, in vitro studies showed that the observed positive effects may be mainly attributed to ULK1 activity. This study provides strong evidence of the beneficial effect of overexpression of ULK homologs, suggesting these as promising instruments for the treatment of MJD and other neurodegenerative disorders.
Keywords: Machado-Joseph disease, Spinocerebellar Ataxia Type 3, ataxin-3, autophagy, transcriptional dysregulation, Unc-51 like autophagy activating kinase 1 and 2, adeno-associated viral vectors, gene therapy, neuroprotection, motor improvements
Graphical abstract
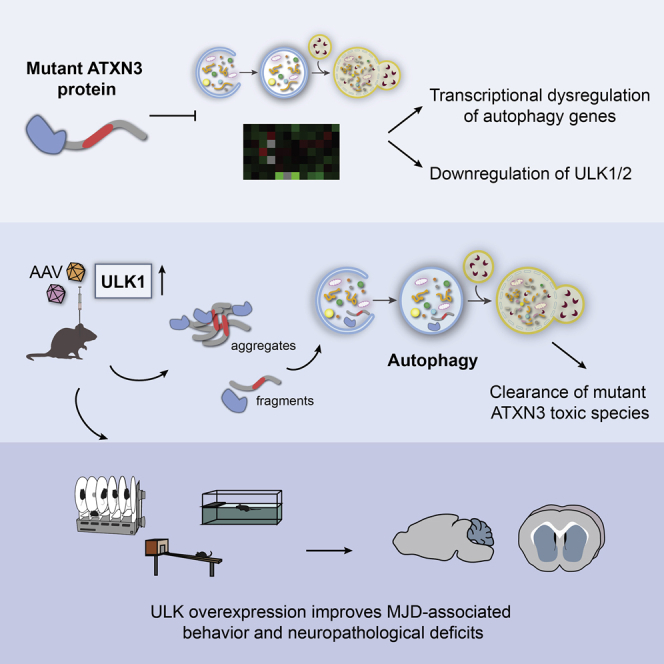
Pereira de Almeida and colleagues show that autophagy is dysregulated at the transcriptional level in SCA3/Machado-Joseph disease, markedly affecting ULK1/2. Viral-mediated ULK overexpression in vitro and in a mouse model reduces mutant ATXN3 toxic species, improves MJD-associated behavioral phenotypes, and prevents cerebellar degeneration.
Introduction
Machado-Joseph disease (MJD), also known as spinocerebellar ataxia type 3 (SCA3), is the most frequent dominantly inherited ataxia worldwide,1 reaching the highest prevalence in the Azorean island of Flores.2 The causative mutation of MJD is mapped to chromosome 14q32.123 within the ataxin-3 (ATXN3) gene and is characterized by the over-repetition of the trinucleotide CAG that translates into an abnormal tract of glutamines within the protein (ATXN3). The accumulation of mutant protein species leads to neurodegeneration in different brain regions, such as the cerebellum, pons, substantia nigra, and striatum.4,5 The onset of MJD typically occurs in the adult age and is characterized by a wide clinical spectrum of symptoms, including progressive cerebellar ataxia, pyramidal signs, peripheral neuropathy, and oculomotor abnormalities.6,7 MJD is an incapacitating and fatal disorder with no current treatment available to prevent or stop disease progression.
Although not fully understood, impairment of macroautophagy (hereafter called autophagy) and transcription dysregulation are considered some of the underlying pathogenic mechanisms. Autophagy is a complex metabolic process, governed by multiple autophagy-related proteins (Atgs), that involves the sequestration of intracellular cargoes by double-membrane autophagosomes, which are then delivered to lysosomes for degradation.8 Initially described as a bulk degradation system, autophagy is a central mechanism for the elimination of insoluble proteins and is highly important for neuronal survival.9, 10, 11 Much evidence suggests the implication of autophagy in the accumulation of mutant ATXN3 (mutATXN3) aggregates and neurodegeneration in MJD. Studies using brain tissue from MJD patients and animal models have identified the presence of ubiquitin and insoluble p62, a classical cargo receptor, in ATXN3-positive inclusions.12, 13, 14 In addition, an abnormal expression of different autophagic markers, the accumulation of autophagosomes, and the decreased levels of the autophagy regulator beclin-1 were also reported, which indicates that autophagy flux is impaired in MJD.13, 14, 15, 16 Furthermore, it is known that mutATXN3 can abnormally interact and recruit specific transcription factors to polyglutamine (polyQ)-rich nuclear inclusions, therefore affecting transcription and contributing to neurotoxicity.17, 18, 19, 20
Despite the compelling evidence of the contribution of autophagy and transcription dysfunction to MJD pathogenesis, it is still unclear whether autophagy is dysregulated at the transcriptional level. Therefore, the first aim of this work was to investigate the transcriptional modifications of the autophagy pathway in postmortem brain tissue from MJD patients and in brain samples from a transgenic mouse model of MJD. Among the multiple dysregulated autophagy-associated transcripts (AATs), Unc-51 like autophagy activating kinase 1 and 2 (ULK1 and ULK2), two orthologs of the yeast Atg1, were found to be drastically reduced in both MJD brain tissue and transgenic mice samples. Given the importance of ULK1/2 activity in the regulation of the autophagy, we hypothesized that the restoration of their levels could allow the normalization of the cellular degradation system, preventing the progression of the disease. Thus, using in vitro and in vivo models of MJD, we provide evidence that ULK overexpression mitigates motor and neuropathological deficits, suggesting that it may constitute a new therapeutic strategy for this condition.
Results
The autophagy pathway is dysregulated at the transcriptional level in MJD
Our previous studies have demonstrated a reduction of the autophagy-associated protein beclin-1, along with an impairment of autophagic clearance in MJD models.13,16 To further evaluate whether autophagy is dysregulated at the transcriptional level in MJD, we took advantage of a qRT-PCR array that allowed the analysis of 84 key genes involved in autophagy and autophagy-related processes (autophagy RT2 Profiler™ PCR array; QIAGEN). For this study, we used (1) postmortem brain tissue from MJD patients, namely the dentate nucleus of the cerebellum (University Medical Center Groningen, Groningen, the Netherlands), and (2) brain samples from 8-week-old transgenic mice expressing a hemagglutinin (HA)-tagged N-terminal truncated fragment of the human ATXN3 with 69 glutamine repeats (Tg-Q69) in the cerebellum (expression driven by the Purkinje cell-specific L7 promoter).21
In the dentate nucleus of MJD patients, we found that 67 out of the 84 AATs were downregulated (Figure 1A). Regarding the cerebellar region of Tg-Q69 mice, 51 AATs were downregulated and 17 were upregulated (Figure 1B), which suggests that the transcriptional levels of autophagy genes are markedly affected in pathological conditions.
Figure 1.

The autophagy pathway is dysregulated at the transcriptional level in MJD
Eighty-four autophagy-associated transcripts (AATs) were analyzed using the RT2 Profiler™ array in samples from MJD patients and Tg-Q69 mice. (A and B) Heatmap depicting fold change of AATs. Green and red colors represent downregulated and upregulated genes, respectively. The list of genes is specified in Materials and methods. (C and D) Scatterplot visualization of the analyzed genes, with a fold change threshold of 1.99, showing top differentially expressed genes in MJD patients and transgenic mice, respectively. Commonly altered genes were detected in both human and mouse samples, including ULK1, ATG16L2, and TNFSF10 Green and red colors represent downregulated and upregulated genes, respectively. (E–J) qPCR (E–G) and western blot (H–J) analysis of ULK1 at 4, 8, and 16 weeks of age. Densitometric quantification of ULK1 levels relative to β-actin. ULK1 mRNA and protein levels are significantly decreased at 8 and 16 weeks of age. Values are expressed as mean ± SEM of n = 5 for WT and transgenic animals. Statistical significance was evaluated with an unpaired Student’s t test (∗∗p < 0.01, ∗∗∗p < 0.001). See also Figure S1.
When analyzing the top differentially expressed AATs, a more extensive list was found in MJD patients, when compared to Tg-Q69 mice. However, some common dysregulated genes were found, such as ULK1, ATG16L2, and TNFS10 (Figures 1C and 1D; Table 1). By the combined analysis of the top 12 downregulated AATs in both MJD patients and Tg-Q69 mice, we identified several genes involved in multiple steps of autophagy, as well as in the co-regulation of the autophagy and autophagy-related processes (Table 1). In fact, genes involved in the autophagosome formation, maturation, and lysosomal function were confirmed to be downregulated in Tg-Q69 mice, when compared to wild-type (WT) animals (Figure S1).
Table 1.
Top 12 differentially expressed AATs in MJD patients and Tg-Q69 mice, combined
| Gene symbol | Name | Human tissue fold regulation | Tg-Q69 mice fold regulation | Protein function (GO molecular function or biological process) |
|---|---|---|---|---|
| ATG16L2 | autophagy-related 16-like 2 | −2.41 | −1.99 | autophagosome formation |
| ATG4D | autophagy-related 4D | −1.85 | −1.90 | autophagosome formation; protein transport |
| ATG7 | autophagy-related 7 | −1.45 | −1.50 | autophagosome formation; protein transport |
| BAX | Bcl2-associated X protein | −1.61 | −1.51 | co-regulation of autophagy and of cell cycle |
| BCL2L1 | Bcl2-like 1 | −1.53 | −1.48 | co-regulation of autophagy, cell death, and cell cycle |
| GABARAPL2 | GABA A receptor-associated protein-like 2 | −1.46 | −1.45 | autophagosome formation/maturation; protein transport |
| HTT | huntingtin | −2.23 | −1.64 | protein transport |
| MAPK14 | mitogen-activated protein kinase 14 | −1.71 | −1.60 | RNA polymerase II transcription factor binding |
| RB1 | retinoblastoma 1 | −1.88 | −1.64 | co-regulation of autophagy and cell cycle |
| TNFSF10 | tumor necrosis factor superfamily, member 10 | −9.22 | −7.37 | co-regulation of autophagy and cell death |
| TRP53 | transformation related protein 53 | −1.67 | −1.45 | co-regulation of autophagy, cell death, and cell cycle |
| ULK1 | Unc-51 like kinase 1 | −1.99 | −2.24 | autophagosome formation; regulation of autophagy |
ULK1, an important factor involved in the initial stages of autophagy, was one of the main affected AATs identified in the qRT-PCR array analysis. To validate this observation, we further evaluated mRNA (Figures 1E–1G) and protein levels (Figures 1H–1J) of ULK1 in 4-, 8-. and 16-week-old Tg-Q69 mice. We observed a significantly altered expression (decrease of about 50%) starting at 8 weeks of age, suggesting that the impairment of ULK1 occurs after the establishment of the disease.
ULK1/2 overexpression improves balance and motor performance in a transgenic mouse model of MJD
The serine/threonine-protein kinases ULK1 and ULK2 are evolutionary conserved orthologs of the yeast Atg1 that are essential for autophagosome formation and for the regulation of autophagy.22, 23, 24 Because of their upstream and redundant functions in the autophagy pathway, and having found that ULK1/2 levels are drastically reduced (Figure 1; Figure S1B), we investigated the therapeutic potential of ULK1/2 overexpression in MJD.
For this purpose, we took advantage of the Tg-Q69 mouse model that exhibits a severe ataxia characterized by the lack of coordination and balance, detected in early age (Figure S2).21,25,26 Six-week-old Tg-Q69 mice and WT littermates were centrally injected into the cerebellar parenchyma with adeno-associated viruses serotype 1/2 (AAV1/2), encoding for green fluorescent protein (GFP) or ULK1 and ULK2 (Figure 2A), and their behavioral performance was accessed before and 3, 6, and 9 weeks post-injection. The injection of AAV1/2 vectors allowed a robust transduction of the cerebellum, as observed by the GFP immunoreactivity and western blot analysis of GFP and ULK1 (Figures 2B and 2C).
Figure 2.

ULK1/2 overexpression improves balance, gait, and motor performance in a transgenic mouse model of MJD
(A) Schematic representation of the stereotaxic injection of AAV1/2 vectors encoding for ULK1 and ULK2 or GFP into cerebellar parenchyma of Tg-Q69 mice. Behavioral assessment was performed before stereotaxic injection (time point 0) and at 3, 6, and 9 weeks post-injection of AAV1/2 vectors. (B and C) Immunofluorescence of GFP (B) and western blot analysis (C) shows that the stereotaxic injection of the viral vectors allowed the overexpression of GFP or ULK1 in the cerebellum of Tg-Q69 mice. Scale bar, 200 μm. (D–G) Behavioral assessment at 9 weeks post-injection reveals an improvement in the performance of ULK1/2-overexpressing transgenic mice, observed in the stationary (D) and accelerated (E) rotarod tests, swimming test (F), and beam walking test (G). (H and I) Behavioral assessment at 6 weeks post-injection reveals an improvement in the footprint test performance of ULK1/2 overexpressing transgenic mice, observed in the overlap (H) and stride length (I) measurements. Values are expressed as mean ± SEM of n = 7–8 males (Tg + GFP) and n = 5–7 males (Tg + ULK1/2). Statistical significance was evaluated with unpaired Student’s t test (∗p < 0.05). See also Figure S2.
Since this transgenic mouse model recapitulates the MJD-associated motor impairments, we used the rotarod, swimming, and beam walking tests to evaluate whether the re-establishment of ULK1/2 levels would improve motor coordination and balance.
Tg-Q69 mice displayed marked behavior deficits, when compared to WT littermates, that were partially rescued by ULK1/2 overexpression (Figure S2). At 9 weeks post-injection, transgenic animals injected with ULK vectors presented a clear improvement in motor performance, when compared to control mice, as evidenced by the significant increase in latency to fall in stationary (Figure 2D, 10.56 ± 2.198 versus 4.367 ± 0.5454 control, Figure S2A) and accelerated rotarod (Figure 2E, 15.45 ± 3.033 versus 7.476 ± 1.551 control). Furthermore, in the swimming (Figure 2F; Figure S2B) and beam walking (Figure 2G; Figure S2C) tests, ULK1/2-overexpressing mice took less time to cross the swimming pool (6.667 ± 0.2041 versus 8.593 ± 0.5368 control) and a squared beam (16.72 ± 1.016 versus 20.85 ± 1.157 control), respectively.
Figure 4.

ULK1/2 overexpression reduces the toxic forms of mutATXN3 in the lentiviral-based mouse model of MJD
(A) Schematic representation of the stereotaxic injection of lentiviral vectors encoding for the human mutATXN3 72Q into the mouse striatum (both hemispheres) and AAV1/2 vectors encoding for GFP (left hemisphere) and ULK1 and ULK2 (right hemisphere). Neuropathological analysis was performed at 4 weeks post-injection of viral vectors. (B and C) Immunofluorescence of GFP (B) and western blot analysis (C) confirm that the stereotaxic injection of the viral vectors mediated the overexpression of GFP and ULK1. Scale bar, 200 μm. (D–F) Western blot analysis shows a significant reduction of high-MW species (D), soluble mutATXN3 (E), and 34-kDa fragment levels (F). Densitometric quantification of mutATXN3 levels relative to β-tubulin is shown. Values are expressed as mean ± SEM of n = 3. Statistical significance was evaluated with a paired Student’s t test (∗p < 0.05, ∗∗p < 0.01). (G–I) Immunohistochemical analysis reveals the reduction of ubiquitin-positive inclusions in MJD mice upon overexpression of ULK1 and ULK2 (H) when compared to mice injected with GFP (G), as quantified in (I). Values are expressed as mean ± SEM of n = 7. Statistical significance was evaluated with a paired Student’s t test (∗p < 0.05). Scale bars, 50 μm. LV, lentiviral vector. See also Figure S4.
In order to evaluate the impact of ULK1/2 overexpression in ataxic gait, we analyzed Tg-Q69 mice footprint patterns. The typical severe ataxic gait observed in this mouse model was partially recovered 6 weeks after AAV delivery, as evidenced by the tendency for a decrease in the footprint overlap distance in Tg + ULK1/2 (Figure 2H). Similarly, a significant increase of the stride length was observed in ULK1/2-overexpressing Tg-Q69 mice in comparison with the control group (Figure 2I and 6.283 ± 0.1195 versus 5.850 ± 0.1376 control, Figure S2D), which suggests an amelioration of the ataxic gait mediated by ULK1/2 overexpression.
Altogether, these results indicate that ULK1/2 overexpression improves the motor performance of MJD transgenic mice.
ULK1/2 overexpression prevents cerebellar neuropathology in transgenic MJD mice
Along with the severe motor impairments, this transgenic mouse model is characterized by a profound cerebellar atrophy, particularly affecting Purkinje cells (Figure S3).21,25,26 Therefore, we further evaluated whether the improvement in behavioral performance was correlated with a preservation of cerebellar structures. For that, we collected brains of ULK1/2-overexpressing and control mice for histological analysis 9 weeks after injection of viral vectors.
Using cresyl violet-stained brain sections, cerebellar volume and layer thickness were measured. Tg-69Q mice exhibited a marked cerebellar neuropathology, when compared to WT littermates, which was attenuated by ULK1/2 overexpression (Figure S3; Figure 3). ULK1/2-overexpressing transgenic mice showed a significantly larger cerebellar volume (normalized to brain volume, Figures 3A–3C, 4.664 ± 0.07376 versus 4.136 ± 0.1383 control, Figure S3D) and a preservation of the molecular layer thickness, when compared to control Tg + GFP mice (Figures 3D–3F, 119.3 ± 2.089 versus 98.58 ± 3.777 control, Figure S3H). In agreement with the previous results, a clear preservation of the number and morphology of Purkinje cells was observed in Tg + ULK1/2 mice (Figure 3H) in comparison with control Tg + GFP mice (Figure 3G), confirmed by the significantly higher number of calbindin-positive Purkinje cells (Figure 3I, 516.1 ± 16.54 versus 427.2 ± 21.41 control).
Figure 3.

ULK1/2 overexpression prevents cerebellar neuropathology in transgenic MJD mice
(A–F) Cresyl violet-stained sections from Tg-Q69 mice. (A–C) ULK1/2 overexpressing transgenic mice (B) presented a larger cerebellar volume, relative to cerebral volume, than did control GFP-expressing transgenic mice (A), as quantified in (C). (D–F) Representative images of molecular layer from Tg + GFP (D) and Tg + ULK1/2 (E) mice show that ULK1/2 overexpression prevented the reduction of molecular layer thickness, confirmed by the respective analysis (F). (G–I) Immunohistochemical analysis, detected by calbindin antibody, indicates a preservation of Purkinje cells in ULK1/2 overexpressing transgenic mice (G), compared with control GFP-expressing transgenic mice (H), which was confirmed by quantification of the number of calbindin-positive Purkinje cells per section (I). Values are expressed as mean ± SEM, n = 5 (Tg + GFP) and 6 (Tg + ULK1/2). Statistical significance was evaluated with an unpaired Student’s t test (∗∗p < 0.01, ∗∗∗p > 0.001). Scale bars, 500 μm (A and B), 100 μm (D and E), and 50 μm (G and H). (J–L) Immunohistochemical analysis of mutATXN3 inclusions, detected by the HA antibody, shows a decrease in the number of HA-positive inclusions in ULK1/2 overexpressing transgenic mice (K), comparing to control GFP-overexpressing transgenic mice (J). Reduction in HA-positive inclusions was confirmed by the quantification of the number of inclusions per section (L). Values are expressed as mean ± SEM, n = 5 (Tg + GFP) and 6 (Tg + ULK1/2). Scale bars, 200 μm (J and K). (M–O) Western blot analysis of mutATXN3 levels confirms a reduction of high-MW species (N) and soluble mutATXN3 levels (O). Densitometric quantification of mutATXN3 levels relative to β-actin is shown. Values are expressed as mean ± SEM, n = 4. Statistical significance was evaluated with an unpaired Student’s t test (∗p < 0.05, ∗∗p > 0.01; ns, not significant). MW, molecular weight; ML, molecular layer. See also Figure S3.
We further evaluated whether ULK1/2 overexpression could reduce mutATXN3 levels. Upon immunohistochemical analysis, we observed a significant decrease of HA-positive inclusions in transgenic mice injected with ULK1/2 vectors, when compared to control Tg + GFP mice (Figures 3J–3L, 4,872 ± 599 versus 9,286 ± 1,700 control). Similarly, western blot analysis revealed a significant reduction of high molecular weight (MW) species (Figures 3M and 3N, −46.18 ± 10.33), while a tendency for a decrease of soluble mutATXN3 species (Figures 3M and 3Q, −31.21 ± 15.88) was observed in Tg + ULK1/2 mice.
Taken together, these results show that ULK1/2 overexpression prevents cerebellar neurodegeneration by promoting mutATXN3 clearance and, thus, preserving Purkinje cells and cerebellar structures.
ULK1/2 overexpression reduces the toxic forms of mutATXN3 in the lentiviral-based mouse model of MJD
To further assess whether ULK1/2 overexpression would alleviate the MJD-associated neuropathology in a different brain region, we took advantage of the striatal lentiviral-based mouse model of MJD.27 Importantly, this strategy allows a localized full-length mutATXN3 expression, which can be useful not only to study the molecular mechanisms of the disease but also to effectively and quantitatively assess the efficacy of a certain therapeutic approach.28, 29, 30
Lentiviral vectors encoding for mutATXN3-72Q were injected in one striatal hemisphere, whereas the contralateral hemisphere, serving as control, was injected with lentiviral vectors encoding for WT ATXN3-27Q (Figure S4). ULK1 levels were initially measured in both hemispheres. As a result of striatal full-length mutATXN3 expression, a tendency toward reduced ULK1 protein levels was observed, when compared to the WT ATXN3 control hemisphere, which is particularly evident at 8 weeks post-injection (p = 0.0592; Figures S4B and S4C).
To further evaluate whether ULK1/2 overexpression would alleviate the neuropathology characteristic of this MJD mouse model,29, 30, 31 6-week-old mice were bilaterally co-injected in the striatum with lentiviral vectors encoding for mutATXN3-72Q and AAV1/2 vectors encoding for GFP (control, left hemisphere) or ULK1 and ULK2 (right hemisphere, Figure 4A) and were sacrificed at 4 weeks post-injection. A large area of striatum was efficiently transduced, as observed by the GFP immunoreactivity (Figure 4B) and the overexpression of GFP or ULK1 by western blot (Figure 4C). The use of AAVs with a single-stranded genome for expression of ULK1/2 allows a delayed sequential expression compared to mutATXN3 by lentiviral vectors.
Similar to other polyQ diseases, mutATXN3 tends to aggregate and its nuclear accumulation in neurons is considered a hallmark of the disease.32 In addition, the expanded protein can suffer proteolytic cleavage, and the generation of ATXN3 fragments has been related to an increase of aggregation and toxicity.31,33, 34, 35 Importantly, ULK1/2 overexpression drastically reduced the toxic forms of mutATXN3, when compared to GFP control hemispheres. ULK1 and ULK2 promoted a significant decrease of high-MW species, soluble mutATXN3, and 34-kDa fragment levels by 67.44% ± 7.21%, 88.45% ± 7.75%, and 68.81% ± 14.59%, respectively (Figures 4C–4F). Furthermore, upon immunohistochemistry, the quantification of the number of ubiquitin-positive inclusions further revealed a similar significant reduction upon ULK1/2 overexpression (37.78% ± 6.50% versus 100% ± 20.99% control, Figures 4G–4I).
These results show that ULK1/2 overexpression prevents the accumulation of mutATXN3 in the striatal lentiviral-based mouse model of MJD.
ULK1 exerts an essential role in autophagy-mediated mutATXN3 clearance in vitro
Although ULK1 and ULK2 display redundant functions in autophagy induction, they may play distinct autophagy-related and non-related roles, which are still not fully understood.36, 37, 38 To investigate the specific contribution of ULK1 and ULK2 in the attenuation of MJD-associated neuropathology, we further evaluated their impact in vitro. Neuro2a cells were transfected with a plasmid encoding for the truncated mutATXN3 with 69Q21 and with GFP, ULK1, ULK2, or ULK1 + ULK2 vectors (Figure 5A). The overexpression of ULK1 and both ULK1 + ULK2 led to the simultaneous reduction of high-MW species by 77.17% ± 5.34% and 61.08% ± 13.37% and of soluble mutATXN3 levels by 52.61% ± 5.052% and 52.38% ± 4.50%, respectively, whereas ULK2 alone had no significant effect on the levels of these species (Figures 5B–5D). Our results suggest that ULK1 plays a major role in the clearance of mutant protein clearance.
Figure 5.

ULK1 exerts an essential role in autophagy-mediated mutATXN3 clearance in vitro
(A) Schematic representation of the experimental conditions. Human ULK1 KO and control HAP1 cell lines were infected with lentivirus encoding for mutATXN3 and ULK1. (B) The stable expression of the transgenes was confirmed by western blot analysis. (C) HAP1 cells were analyzed in the presence or absence of chloroquine (100 μM). Western blot analysis of LC3B shows a significant decrease of LC3BII net flux in the ULK1 KO cell line that trended toward recovery upon ULK1 overexpression. (D) Western blot analysis of ATXN3 shows a significant decrease of mutATXN3 levels in HAP1 KO expressing ULK1. Densitometric quantification of LC3BII levels and mutATNX3 levels relative to β-actin and β-tubulin, respectively. Values are expressed as mean ± SEM of n = 4. A one-way ANOVA with Bonferroni’s post hoc test was used to evaluate statistical significance (∗p < 0.05, ∗∗p < 0.01). (E) Schematic representation of Neuro2a transfection with plasmids encoding for the mutATXN3 with 69Q and GFP, ULK1, or ULK1 + ULK2, using PEI reagent. (F–H) Western blot analysis shows a significant reduction of high-MW species (G) and soluble mutATXN3 levels (H). Densitometric quantification of mutATXN3 levels relative to β-actin. Values are expressed as mean ± SEM of n = 4 and 5. A one-way ANOVA with Dunnett’s post hoc test was used to compare Neuro2a cells expressing ULK constructions to GFP (∗p < 0.05, ∗∗p < 0.01). CTRL NI, non-infected control; CTRL, control; KO, knockout; chQ, chloroquine; PEI, polyethyleneimine; MW, molecular weight
To further explore the role of ULK1 in MJD, we took advantage of a human HAP1 ULK1 knockout (KO) cell line. Control and ULK1 KO HAP1 cells were transduced with lentiviral vectors encoding for mutATXN3. ULK1 KO cells were further infected with lentiviral vectors encoding for ULK1 (Figure 5E). The expression of the transgenes of interest was confirmed by western blot analysis (Figure 5F).
First, to evaluate whether differences of ULK1 expression levels have an impact on autophagy activation, we evaluated the protein levels of the transient autophagosomal membrane-bound LC3BII, commonly used as a marker for autophagosome formation. Autophagic flux was accessed by LC3BII turnover assay, which compares LC3BII levels between samples in the presence and absence of a lysosomal inhibitor (chloroquine).39 We observed a significant decrease in LC3BII net flux in ULK1 KO cells that trended toward recovery in ULK1-overexpressing cells (Figure 5G), suggesting a critical role of ULK1 in the autophagosome formation.
Based on this evidence, we next evaluated the mutATXN3 clearance in the absence or presence of ULK1. When compared to control cells, ULK1 KO cells showed an increase in mutATXN3 levels (p = 0.052), while the overexpression of ULK1 led to a significant decrease of the mutant protein (Figure 5H). These observations support the beneficial effect of ULK1 in autophagy-mediated mutATXN3 clearance.
Discussion
In this study, we provide evidence of the transcriptional dysregulation of key autophagy genes in MJD, including ULK1 and ULK2. We explored their in vivo therapeutic potential, demonstrating that the overexpression of the ULK homologs leads to significant improvements in MJD-associated behavior and neuropathological deficits. Moreover, we also demonstrated a central role of ULK1 in promoting autophagy-mediated mutATXN3 clearance, most likely responsible for the additional positive effects observed in MJD models.
First, we observed a general transcriptional dysregulation of the autophagy-associated genes in two models of MJD, which suggests that, in fact, transcription is markedly affected in pathological conditions, as previously described.17,40,41 The robust transcriptional dysregulation observed might be explained by the severity of the analyzed models. In this study we made use of postmortem tissue of MJD patients, specifically from the dentate nucleus region in the cerebellum, one of the most affected regions in the disease42,43 where, therefore, an exacerbation of the pathogenic mechanisms might be expected. Moreover, the analyzed Tg-Q69 mouse model expresses an N-terminal truncated human ATXN3 driven for expression in Purkinje cells, being characterized by a profound cerebellar atrophy and early motor impairments.21
The impairment of autophagy induction in MJD is well established, given by the observation of decreased beclin-1 levels in several models.13,16,44 Nonetheless, several studies show the accumulation of autophagosomes, autophagy-related proteins (ATG7, ATG12, ATG16L1, ATG16L2), endosomal/lysosomal markers (LAMP-2, RAB7, RAB1A), and the incorporation of ubiquitin-related proteins (p62 and NRB1) into neuronal intranuclear inclusions13, 14, 15 in MJD brains, suggesting that the autophagic substrates are not efficiently delivered to autophagosomes for degradation, and therefore other pathways might be affected. Our results support these observations, since AATs involved in the late steps of autophagy were shown to be dysregulated in the analyzed MJD models. A combined analysis of AATs dysregulation in MJD brain tissue and the Tg-Q69 mouse model led us to the identification of several common transcripts robustly downregulated, such as ATG16L2, ATG4D, ATG7, HTT, GABARAPL2, and ULK1, that are involved in autophagosome formation, maturation, or protein transport.45, 46, 47 Even though further functional studies are needed, the abnormal reduction of expression levels of autophagy genes may affect their normal function, contributing to the impairment of the autophagy pathway in MJD.
One of the main downregulated genes identified in this study was ULK1, the levels of which were drastically decreased in the Tg-69Q mouse model after 8 weeks of age, when neuropathology and robust symptoms are already established. In line with these results, previous studies have found a reduction of beclin-1 levels in a late stage of a MJD transgenic mouse model,13 as well as a decline of beclin-1 in an age-dependent manner in brains of Huntington’s disease patients,48 suggesting that decreased levels of autophagy proteins might influence the disease onset and progression. Furthermore, ULK1 function was shown to be affected in polyQ disease models. ULK1-mediated phosphorylation of ATG14 is impaired in Huntington’s disease, which is possibly caused by the p62-induced sequestration of ULK1.49 In addition, a destabilization of ULK1, possibly due to an increased p53-FIP200 interaction, and subsequent inhibition of autophagy were observed in a SCA7 model.50
Considering that strategies based on autophagy activation can represent promising therapies in diseases characterized by the abnormal accumulation of toxic proteins, it is important to understand and target the mechanism by which mutant proteins disturb the autophagy pathway. By overexpressing ULK1/2 proteins abnormally diminished in the disease, we anticipate a restoration of the cellular degradation system. Moreover, regulating autophagy at its initial step may contribute to a more effective response as compared to more downstream interventions.
ULK1 and ULK2 serine/threonine protein kinases are widely studied factors of the autophagy induction complex, sharing 52% of the overall sequence identity and 76% within their kinase domains.51 The importance of these homologs for autophagy activity is supported by the evidence that kinase-inactivated forms of ULK1 and ULK2 strongly inhibit autophagy.36 Moreover, while ULK1−/− or ULK2−/− mice display a normal development, double KO of ULK1 and ULK2 lead to neonatal lethality.37,52,53 For these reasons, we made the option to overexpress simultaneously both ULK1 and ULK2 in vivo. For this approach, we took advantage of two mouse models vastly used in our previous studies.27,29, 30, 31,54, 55, 56, 57, 58
We first investigated the re-establishment of ULK1 and ULK2 levels in a Tg-Q69 mouse model expressing a truncated form of the human mutATXN3 in the cerebellum.21 This model displays a severe and early phenotype, which is highly relevant for pre-clinical studies. Notably, the marked behavioral deficits observed in these mice were partially prevented by ULK1/2 overexpression. The significant improvements can be explained by the autophagic reduction of mutATXN3 aggregated and soluble species. Nevertheless, since the role of ULK1/2 is not limited to autophagy, other mechanisms might be involved.
Indeed, autophagy and apoptosis are intimately connected, as some regulators can control both processes. Compelling evidence demonstrates that autophagy can be cytoprotective in neurons, by preventing apoptosis, a process where ULK1 has been shown to exert an important function.59,60 In fact, the recovery of motor performance was accompanied by the amelioration of neuropathology, namely the preservation of Purkinje cells and cerebellar morphology, suggesting a reduction of neuronal cell death. Moreover, the neuron-specific functions of ULK1 and ULK2, as well as their involvement in the regulation of autophagy in response to proteotoxic stress, could represent an important advantage of a therapy to be applied in MJD and other neurological disorders.60, 61, 62, 63, 64
To clarify whether ULK1/2 would exert protective effects in a different brain region that is also affected in MJD, we used the striatal lentiviral-based mouse model. This model expresses the full-length human mutATXN3 and allows the assessment of the efficacy of the developed therapy in a short period of time by the precise quantification of the neuropathological deficits. Moreover, ULK1 levels were also found to be reduced in this model, reinforcing the idea of the deleterious impact of mutATXN3 in ULK1 expression and the importance of restoring ULK1 activity. Indeed, 4 weeks after injection of viral vectors, ULK1/2 overexpression promoted the clearance of mutATXN3 toxic forms.
Importantly, autophagy has been shown to be a crucial pathway for the clearance of insoluble protein aggregates, since they cannot be efficiently degraded by the ubiquitin-proteasome system.65 Along with the insoluble protein, a significant reduction of a mutATXN3 fragment was observed, which may suggest, in line with the “toxic fragment hypothesis,” a prevention of the aggregation process until inclusion formation.33,66,67 Independently of the pathophysiological implications of the different polyQ toxic forms, which are still controversial, the overall reduction of mutATXN3 could indicate a retardation of the associated neuropathological mechanisms.
We further evaluated the specific contribution of ULK1 and/or ULK2 in the activation of autophagy and mutATXN3 degradation in in vitro models of the disease. Interestingly, we observed a central role of ULK1 in promoting protein clearance, since ULK1 alone, but not ULK2, strongly reduced mutATXN3 levels in vitro. Moreover, our results show that the absence of ULK1 led to the impairment of the autophagic flux, which was tendentially prevented by reinstating ULK1 levels. This is in accordance with previous evidence showing that only the knockdown of ULK1, but not ULK2, drastically decreases starvation-induced autophagy activity in HEK293T cells and mouse embryonic fibroblasts.23,68
Our results suggest that the neuroprotective effects observed in vivo were more likely due to ULK1 activity; however, further studies are needed to understand the separate function of ULK1 and ULK2 in this context. While being partially redundant, ULK1 and ULK2 may also have important independent functions since they act differently depending on the underlying stimulus or cell type.36,53,69 Indeed, a recent computational analysis that compared the human ULK1 and ULK2 proteins found major differences in their autophagy-related and non-autophagy-related interactors, as well as specific transcriptional and post-translational regulators, suggesting distinct functions for ULK1 and ULK2.38 Therefore, the identification of the specific processes wherein these homologs are involved would result in a better comprehension of the impact of ULK1 and ULK2 imbalance in autophagy and disease pathogenesis.
In conclusion, the present study provides evidence that autophagy is markedly dysregulated at the transcriptional level in MJD, and that the overexpression of the ULK homologs alleviates MJD-associated motor deficits and neuropathology. Thus, gene delivery of AATs, such as ULK1, may be a promising strategy for MJD treatment.
Materials and methods
Vector constructions
For lentiviral ULK1 construction, cloning was performed into XhoI and BamI sites of the LTR-SIN-PGK-ATXN3-LTR vector.27 For AAV ULK1 and ULK2 constructions, cloning was performed into EcoRI/BamH and EcoRI/XhoI sites, respectively, of a single-stranded AAV (ssAAV) backbone under the cytomegalovirus (CMV) promoter.
Viral production, purification, and titer assessment
Lentiviral vectors encoding for human mutATXN3 with 72 glutamines (LV-PGK-ATXN3 72Q)27 and ULK1 (LV-PGK-ULK1) were produced in human embryonic kidney 293 (HEK293T) cells with a four-plasmid system, as previously described.70 Lentiviral particles were suspended in sterile 0.5% bovine serum albumin diluted in phosphate-buffered solution (PBS). The viral particle content of each batch was evaluated by assessing the HIV-1 p24 antigen levels by ELISA (Retro Tek, Gentaur), in accordance with the manufacturer’s instructions.
AAV1/2, encoding for ssAAV-GFP, ssAAV-ULK1, and ssAAV-ULK2, were produced in HEK293T cells as described elsewhere.71 AAV vectors were purified based on the fast protein liquid chromatography (FPLC) technique, using the ÄKTA pure 25 system (GE Healthcare Life Sciences). AAV titer was determined by quantitative real-time PCR using the AAVpro titration kit (for real-time PCR) v2 (Takara Bio), in accordance with the manufacturer’s instructions.
Concentrated viral stocks were stored at −80°C until further use.
HAP1 cell culture and transduction
HAP1 cells (Horizon) are a human adherent cell line, with fibroblast-like morphology, derived from male chronic myelogenous leukemia cell line KBM-7. HAP1 cells are near haploid, having a single copy of nearly every chromosome.72
HAP1 control and ULK1 KO cells were maintained in culture in Iscove’s modified Dulbecco’s medium (IMDM, Sigma-Aldrich) supplemented with 10% fetal bovine serum (FBS, Life Technologies), 1% penicillin-streptomycin (Life Technologies) and kept at 37°C in a 5% CO2 atmosphere incubator.
Both cell lines were infected with lentiviral vectors encoding for mutATXN3 (LV-PGK-ATXN3 72Q), and two passages after infection, HAP1 KO cells were infected with lentiviral vectors encoding for ULK1 at the ratio of 60 ng of p24 antigen/1 × 105 cells of each vector.
After transduction, 0.6 × 104 cells were plated on six-well cell culture-treated multiwell plates (Thermo Fisher Scientific) for western blot analysis. Twenty-four hours later, each line was incubated with 100 μM chloroquine for 3 h prior to collection. Cells without chloroquine incubation were used as control.
Neuro2a cell culture and transfection
A mouse neural crest-derived cell line (neuro2a cells) was obtained from the American Type Culture Collection. The cells were maintained in Dulbecco’s modified Eagle’s medium (DMEM) (Sigma-Aldrich) supplemented with 10% heat-inactivated FBS (Life Technologies) and 1% penicillin- streptomycin (Life Technologies) and cultured at 37°C under a humidified atmosphere containing 5% CO2. Neuro2a cells were transfected with plasmids encoding for truncated mutATXN3 with 69Q21 and (1) GFP (as control), (2) ULK1, (3) ULK2, or (4) ULK1 and ULK2. For that, 1 × 104 cells were plated on 12-well cell culture-treated multiwell plates (Thermo Fisher Scientific) for western blot analysis. After 24 h, cells were transfected with a mixture of DNA/polyethylenimine (PEI) complexes (MW40000, Polysciences). Complex formation was performed by combining 400 ng of each plasmid DNA in 50 μL of serum-free DMEM and 4 μL of PEI (1 mg/mL) per well. This mixture was vortexed for 10 s and incubated at room temperature for 10 min. Afterward, DNA/PEI complexes were diluted in complete DMEM and added to cell culture. At 48 h post-transfection, cells were collected.
Human postmortem tissue
Postmortem human brain tissue from dentate nucleus was obtained from the University Medical Center of Groningen (UMCG), the Netherlands. Tissue from two healthy individuals (male, 68 years old; further details unknown) and two MJD patients (female, 34 years old, 77 CAG repeats; female, 70 years old, 68 CAG repeats) was used. All individuals gave informed consent for clinical data and sample use for research as approved by the Medical Ethical Committee of the UMCG.
Animals
The lentiviral-based MJD mouse model was produced as previously established in our laboratory.27 C57BL/6 mice from Charles River Laboratories were used. The detailed protocol is described in the next section.
The transgenic mouse model used was developed by Hirokazu Hirai (Kanazawa University, Japan),21,73 and a colony of these transgenic mice was established in a licensed animal facility (International Animal Welfare Assurance no. 520.000.000.2006) at the Center for Neuroscience and Cell Biology (CNC) of the University of Coimbra, Portugal. Transgenic mice express an N-terminal truncated human ATXN3 in cerebellum, with 69 CAG repeats together with an N-terminal HA epitope, driven by a L7 promoter. The genotype was confirmed by PCR. Animals were housed in groups (two to five per cage) in plastic cages (365 × 207 × 140 mm) with food and water ad libitum and maintained on a 12-h light/12-h dark cycle at a room with constant temperature (22°C ± 2°C) and humidity (55% ± 15%).
The experiments were carried out in accordance with the European Union Community directive (2010/63/EU) for the care and use of laboratory animals. Researchers received adequate training (Federation of European Laboratory Animal Science Associations (FELASA)-certified course) and certification from Portuguese authorities (Direcção Geral de Alimentação e Veterinária) to perform the experiments.
In vivo injection of viral vectors
Mice with 6 weeks of age were anesthetized by intraperitoneal injection of a mixture of ketamine (80 mg/kg) and xylazine (4 mg/kg).
For experiments using the lentiviral-based MJD mouse model, WT mice were stereotaxically injected into the striatum with concentrated lentiviral vectors containing 400 ng of p24 antigen and encoding for the mutATXN3 with 72Q and 2 × 109 viral genomes of AAV1/2 vectors encoding for GFP or ULK1 + ULK2 in each hemisphere at the following coordinates: +0.6 mm rostral to lambda, −1.8 mm midline, and −3.3 mm ventral to the skull surface, with the mouth bar set at 0.
For experiments using the Tg-Q69 mouse model, transgenic mice and WT littermates were injected into the cerebellum with 2 × 109 viral genomes of AAV1/2 vectors encoding for GFP or ULK1 + ULK2 at the following coordinates: anteroposterior −2.3 mm, lateral 0 mm, and ventral −3.0 mm, relative to lambda. After the injection, the syringe needle was left in place for an additional 5 min to minimize backflow.
Behavioral assessment
Transgenic males were subjected to locomotor tests before and 3, 6, and 9 weeks after injection of viral vectors. Animals were acclimatized for 1 h to a quiet room with controlled temperature and ventilation and dimmed lighting, and handled prior to behavioral testing.
Rotarod
Motor coordination and balance were assessed using the rotarod apparatus (Letica Scientific Instruments, Panlab) at a constant speed (5 rpm) and at an accelerated speed (4–40 rpm), both for a maximum of 5 min. The time during which mice remained walking in the rotation drum was recorded. Mice performed three trials, at each time point, for both constant and accelerated speed rotarod, with at least 20 min of rest between trials. The results were expressed as the average of the trials.
Swimming
Mice were placed in one side of a 100-cm-long rectangular swimming pool made of glass, with a safe platform in the opposite side. The pool was filled with water at 25°C, up to the platform level. Mice performed three trials for each time point, with at least 20 min of rest between trials. The time (in seconds) that mice took to reach the safe platform (with the four paws) was recorded. The results were expressed as the average of the trials.
Beam walking
Motor coordination and balance of mice were assessed by measuring the time (in seconds) to cross a 9-mm squared beam and reach an enclosed safety platform. The beam was placed horizontally and raised to a height of approximately 20 cm, and mice had to traverse 40 cm to reach the safe platform. Three trials were performed in each time point, with at least 20 min of rest between trials. The results were expressed as the average of the trials.
Footprint patterns
Gait was evaluated by performing the footprint analysis, in which the forepaws and hindpaws of the animal were painted with non-toxic dyes of different colors. Mice were allowed to walk along a blank paper runway (100 × 10 × 15 cm). Five consecutive steps were chosen, and the footprint overlap and stride length were measured. The overlap between forepaw and hindpaw placement was measured as the distance between the front and hind footprints on each side. Measurements were performed on the left and right sides and the mean from 10 measurements (5 left, 5 right) was recorded. To measure the stride length, the average distance of the forward movement between each stride was quantified. The results were expressed as the mean value for the forepaw and hindpaw stride length.
mRNA expression analysis
mRNA isolation and integrity assessment
Total RNA of human brain tissue was isolated using a miRCURY RNA isolation kit (Exiqon), while RNA of transgenic mice was extracted with a NucleoSpin RNA kit (Macherey-Nagel), according to the manufacturer’s instructions. For the mouse samples, the protocol was preceded by homogenization of the tissue with TRIzol reagent (Invitrogen) and chloroform (Merck).
RNA integrity, purity, and concentration were determined using an Experion RNA StdSens kit (Bio-Rad) and a NanoDrop 2000 spectrophotometer (Thermo Fisher Scientific). The RNA samples were stored at −80°C until further use.
Reverse transcription and quantitative real-time PCR
For RT2 Profiler™ PCR array autophagy analysis (QIAGEN), cDNA was synthesized by the conversion of 0.5 μg of total DNase I-treated RNA, using an RT2 first-strand kit (QIAGEN), according to the manufacturer’s instructions.
Quantitative real-time PCR (qPCR) was carried out in a 2,700-μL reaction volume containing 102 μL of cDNA synthesis reaction (control or MJD pooled samples) and 1,350 μL of RT2 SYBR Green mastermix (QIAGEN). In each model of the disease, one 96-well plate for control or the MJD group was analyzed. qPCR was performed in the StepOnePlus real-time PCR system (Applied Biosystems), which was initiated with enzyme activation by heating at 95°C during 10 min, followed by 40 cycles of two steps: 95°C for 15 s, and 60°C for 1 min. To verify PCR specificity a melting curve was performed, with the following program: 95°C for 15 s, 60°C for 1 min, and 60°C–95°C with an increment of 0.3°C per 15 s.
For following qPCR assays, cDNA was synthesized by the conversion of 1 μg of the DNase I-treated RNA with an iScript selected cDNA synthesis kit (Bio-Rad), according to the manufacturer’s instructions, and stored at −20°C. qPCR was carried out in 10 μL using the SsoAdvanced SYBR Green supermix (Bio-Rad). Primers (mouse ULK1 forward, 5′-CAGACAGCCTGATGTGCAGT-3′, reverse 5′-TCAATGCGCTGGTAGTTCTG-3′; mouse Gapdh forward 5′-TGGAGAAACCTGCCAAGTATGA-3′, reverse 5′-GTCCTCAGTGTAGCCCAAG-3′) were designed using Primer-BLAST software,74 and their efficiency and specificity were subsequently evaluated. qPCR was performed in the StepOnePlus real-time PCR system (Applied Biosystems) according to the following specifications: 95°C for 30 s, followed by 45 cycles at 95°C for 5 s and 60°C for 15 s. The melting step was performed with slow heating, starting at 65°C with an increment of 0.5°C per 5 s up to 95°C.
Each assay included a non-template control, non-reverse transcription control, and a standard curve for each target gene. The amplification rate was evaluated from the cycle threshold (Ct), which was automatically determined by the software with cDNA dilutions.
Data analysis
For RT2 Profiler™ PCR array autophagy, data analysis was performed in the RT2 Profiler™ data analysis web portal using the ΔΔCt method. Data normalization was performed using the housekeeping genes with a variation of ≤0.5 Ct between samples. The following housekeeping genes were used in qPCR array analysis: ACTB, HPRT1, RPLP0 (human samples) and Actb, Hsp90ab1, Gusb, and Gapdh (mouse samples).
The fold change (FC) value was calculated by converting ΔΔCt from a log2 scale to a linear scale using the following equation: FC = 2−ΔΔCt. For all FC values greater than 1, the fold regulation and FC values are the same. For all FC values less than 1, the fold regulation is the negative inverse of the FC (−1/FC).
The autophagy genes (human and mouse) analyzed in the PCR array are listed in Table 2.
Table 2.
RT2 Profiler™ PCR array human/ mouse autophagy, gene list
| A01 | Aakt1 | B10 | Becn1 | D07 | Gabarap | F04 | Pik3c3 |
| A02 | A mbra1 | B11 | Bid | D08 | Gabarapl1 | F05 | Pik3cg |
| A03 | AApp | B12 | Bnip3 | D09 | Gabarapl2 | F06 | Pik3r4 |
| A04 | Atg10 | C01 | Casp3 | D10 | Hdac1 | F07 | Prkaa1 |
| A05 | Atg12 | C02 | Casp8 | D11 | Hdac6 | F08 | Pten |
| A06 | Atg16l1 | C03 | Cdkn1b | D12 | Hgs | F09 | Rab24 |
| A07 | Atgl2 | C04 | Cdkn2a | E01 | Hsp90aa1 | F10 | Rb1 |
| A08 | Atg3 | C05 | Cln3 | E02 | Hspa8 | F11 | Rgs19 |
| A09 | Atg4a | C06 | Ctsb | E03 | Htt | F12 | Rps6kb1 |
| A10 | Atg4b | C07 | Ctsd | E04 | lfng | G01 | Snca |
| A11 | Atg4c | C08 | Ctss | E05 | Igf1 | G02 | Sqstm1 |
| A12 | Atg4d | C09 | Cxcr4 | E06 | Ins2 | G03 | Tgfb1 |
| B01 | Atg5 | C10 | Dapk1 | E07 | Irgm1 | G04 | Tgm2 |
| B02 | Atg7 | C11 | Dram1 | E08 | Lamp1 | G05 | Tmem74 |
| B03 | Atg9a | C12 | Dram2 | E09 | Map1lc3a | G06 | Tnf |
| B04 | Atg9b | D01 | Eif2ak3 | E10 | Map1lc3b | G07 | Tnfsf10 |
| B05 | Bad | D02 | Eif4g1 | E11 | Mapk14 | G08 | Trp53 |
| B06 | Bak1 | D03 | Esr1 | E12 | Mapk8 | G09 | Ulk1 |
| B07 | Bax | D04 | Fadd | F01 | Mtor | G10 | Ulk2 |
| B08 | Bcl2 | D05 | Fas | F02 | Nfkb1 | G11 | Uvrag |
| B09 | Bcl2l1 | D06 | Gaa | F03 | Npc1 | G12 | Wipi1 |
FC of following qPCR assays was determined using Pfäffl method. To each gene and experiment, a standard curve was performed to assess the efficiency of each set of primers. Data were corrected for Gapdh levels, normalized for the control group, and FC was expressed as percentage.
For Gene Ontology analysis we took advantage of the Gene Ontology Consortium data base.75,76
Protein isolation and western blot analysis
For protein isolation, brain tissue and cells were lysed with radioimmunoprecipitation assay (RIPA) buffer solution (50 mM Tris-base [Fisher Scientific], 150 mM NaCl [Fisher Chemical], 5 mM ethylene glycol tetraacetic acid [Sigma-Aldrich], 1% Triton X-100 [Sigma-Aldrich], 0.5% sodium deoxycholate [Sigma-Aldrich], and 0.1% sodium dodecyl sulfate [SDS, Acros Organics] containing protease inhibitors [Roche Diagnostics] and supplemented with 0.2 mM phenylmethylsulfonyl fluoride [Sigma-Aldrich], 1 mM dithiothreitol [DTT, Sigma-Aldrich], 1 mM sodium orthovanadate [Sigma-Aldrich], and 5 mM sodium fluoride [Sigma-Aldrich]). Tissue and cell lysates were further submitted to two series of 4 s of ultra-sound pulses (1 pulse/s). Protein concentration was determined using the Bradford protein assay (Bio-Rad). Samples were then denatured with 6× sample buffer (9.3% DTT, 10% SDS, 30% glycerol in 0.5 M Tris-HCl/0.4% SDS [pH 6.8] and bromophenol blue [0.012%]) and incubated during 5 min at 95°C. Forty to 60 μg of total protein extract was resolved on SDS-polyacrylamide gels (4% stacking, 10% running). The proteins were transferred onto polyvinylidene fluoride membranes (Millipore) according to standard protocols. The membranes were blocked by incubation in 5% non-fat milk powder in Tris-buffered saline with 0.1% Tween 20 for 1 h at room temperature, and then incubated overnight with the following primary antibodies diluted in blocking buffer, that is, rabbit monoclonal ULK1 (1:1,000, Cell Signaling Technology), mouse monoclonal anti-GFP (1:1,000, #11814460001, Roche), rabbit monoclonal anti-LC3B antibody (1:1,000, Cell Signaling Technology), mouse monoclonal anti-ATXN3 antibody (1H9, 1:5,000, Merck Millipore), mouse monoclonal anti-HA antibody (1:1,000, InvivoGen), mouse monoclonal anti-β-actin (1:5,000, A5316, clone AC74, Sigma-Aldrich), and mouse monoclonal anti-β-tubulin (1:10,000, SAP.4G5, Sigma-Aldrich). Blots were incubated with the corresponding alkaline phosphatase-linked secondary goat anti-mouse or anti-rabbit antibodies (1:10,000, Thermo Fisher Scientific) for 2 h at room temperature. Bands were visualized with enhanced chemifluorescent substrate (GE Healthcare), using chemifluorescence imaging (ChemiDoc imaging system, Bio-Rad). Semiquantitative analysis was carried out based on the optical density of scanned films (Image Lab software version 5.1, Bio-Rad). The specific optical density was then normalized with respect to the amount of β-actin/β-tubulin loaded in the corresponding lane of the same gel and to control group, expressed as a percentage.
Histological staining and immunohistochemistry procedure
Tissue preparation
Mice were sacrificed with an overdose of xylazine/ketamine (8/160 mg/kg body weight, intraperitoneally). Perfusion with PBS and fixation with 4% paraformaldehyde (Sigma-Aldrich) were performed transcardially. Brains were collected and post-fixed in 4% paraformaldehyde for 24 h and were then cryoprotected/dehydrated by immersion in 25% sucrose/PBS for 36–48 h. Dried brains were frozen at −80°C, and 25-μm coronal sections from stereotaxically injected mice were sliced using a cryostat (Leica CM3050 S, Leica Microsystems) at −21°C. Slices were collected in anatomical series and stored in 48-well plates as free-floating sections in PBS supplemented with 0.05% (m/v) sodium azide, at 4°C until immunohistochemical procedures.
Bright-field immunohistochemistry
The immunohistochemical procedure was initiated by endogenous peroxidase quenching with a 30-min incubation at 37°C in a 0.1% phenylhydrazine (Merck)/PBS solution. Sections were then incubated at room temperature for 1 h in blocking solution constituted by PBS with 0.1% Triton X-100 containing 10% normal goat serum (Gibco), and then incubated overnight at 4°C in blocking solution with the primary antibody, that is, rabbit polyclonal anti-ubiquitin antibody (1:1,000, Dako). After washing, sections were incubated for 2 h at room temperature with the respective biotinylated secondary antibody (1:200, Vector Laboratories). Bound antibody was visualized using a Vectastain ABC kit, with 3,3′-diaminobenzidine (DAB) tetrahydrochloride (DAB metal concentrate, Pierce) as substrate. The sections were mounted, dehydrated, and coverslipped with mounting medium (Thermo Fisher Scientific).
Cresyl violet staining
Pre-mounted sagittal sections were stained with cresyl violet (Sigma-Aldrich) for 5 min, differentiated in 70% ethanol, dehydrated by passing through 96% ethanol, 100% ethanol, and xylene solutions, and mounted with mounting medium (Thermo Fisher Scientific).
Immunofluorescence
The immunohistochemical procedure was initiated with 1 h of blocking and permeabilization at room temperature in 0.1% Triton X-100 10% normal goat serum prepared in PBS. Sections were then incubated overnight at 4°C with the following primary antibodies: rabbit polyclonal anti-GFP antibody (1:1000, Thermo Fisher Scientific), rabbit polyclonal anti-HA antibody (1:1,000, BioLegend), or rabbit polyclonal anti-calbindin D-28K antibody (1:1,000, Merck Millipore). Sections were washed and incubated for 2 h at room temperature with the corresponding secondary antibody coupled to fluorophores (1:200, Molecular Probes, Life Technologies) diluted in the respective blocking solution. Nuclei staining was performed with 4′,6-diamidino-2-phenylindole (DAPI, 2 μg/mL in PBS, Invitrogen), after which sections were washed, mounted, and coverslipped with mounting medium (Dako).
Quantification of ubiquitin-positive inclusions
The quantification of ubiquitin-positive inclusions in mouse striata was performed by scanning 12 coronal pre-stained 25-μm-thick sections spread over the anterior-posterior extent of the striatum with an inter-section distance of 200 μm, using a ×20 objective Zeiss Axio Imager 2 microscope (Carl Zeiss Microscopy) with ZEN software. For each animal, the total number of ubiquitin-positive inclusions in the striatum was manually counted in all 12 sections and multiplied by 8 to account for the intermediate sections in 200 μm of tissue.77 Results are expressed as percentage of control.
Quantification of thickness of the cerebellar layers and cerebellar volume
Quantification of neuropathological parameters in the cerebellum of transgenic mice was blindly performed by scanning four sagittal sections with 30-μm thickness, starting in the middle of the cerebellum, with an inter-section distance of 240 μm.
Quantification of cerebellar layer thickness was measured over the sagittal sections stained with cresyl violet in the interception of cerebellar lobules, using a ×20 objective Zeiss Axio Imager 2 microscope (Carl Zeiss Microscopy) with ZEN software. For cerebellar/cerebrum volume evaluation, ×5 objective images were obtained of each section. Cerebellar volume of the selected sections was calculated by quantifying the area of both cerebellum and cerebrum following the formula: volume = d(a1 + a2 + a3 + a4), where d is the distance between serial sections (240 μm) and ax values are cerebellar or cerebral areas for individual sections. The result was expressed as a ratio of cerebellar volume over cerebrum volume.
Quantification of Purkinje cells
Quantification of the number of Purkinje cells was performed after performing calbindin immunohistochemistry and imaging of the selected sections, using a ×20 objective on a Zeiss Cell Observer spinning disk microscope (Carl Zeiss Microscopy) with ZEN software. The number of Purkinje cells was blindly manually counted and expressed as the average of Purkinje cells per section.
Quantification of HA-tagged ATXN3-positive inclusions
For quantification of HA-tagged ATXN3-positive inclusions, serial z stack images (interval = 0.9 μm) were captured by a ×20 objective on confocal microscope (Zeiss Cell Observer spinning disk microscope). HA-positive inclusions were quantified in the axon terminals of Purkinje cells in deep cerebellar nuclei, using semi-automated image analysis software (ImageJ, National Institute of Health). The results are expressed as the average of HA-positive inclusions per section.
Statistical analysis
Statistical analysis was performed with a paired or unpaired Student’s t test and one-way analysis of variance (ANOVA) followed by the adequate post hoc test for multiple comparisons, using GraphPad Prism 6 (version 6.01, GraphPad). For behavior results (Figure S2), a trend analysis was used to compare linear regression slopes using the two-tailed t test hypothesis. Outliers were excluded from analysis using Grubb’s test. Results are expressed as mean ± standard error of the mean (SEM). Significant threshold was set at p < 0.05, as defined in the text.
Acknowledgments
This work was funded by the ERDF through the Regional Operational Program Center 2020, Competitiveness Factors Operational Program (COMPETE 2020), and national funds through FCT (Foundation for Science and Technology)-BrainHealth2020 projects (CENTRO-01-0145-FEDER-000008), UID/NEU/04539/2020, ViraVector (CENTRO-01-0145-FEDER-022095), CortaCAGs (PTDC/NEU-NMC/0084/2014|POCI-01-0145-FEDER-016719), SpreadSilencing POCI-01-0145-FEDER-029716, Imagene POCI-01-0145-FEDER-016807, CancelStem POCI-01-0145-FEDER-016390 and POCI-01-0145-FEDER-032309, as well as SynSpread, ESMI, and ModelPolyQ under the EU Joint Program-Neurodegenerative Disease Research (JPND), with the last two co-funded by the European Union H2020 program, GA no. 643417; E-Rare4/0003/2012 Joint Call for European Research Projects on Rare Diseases (JTC 2012) and FCT; by the National Ataxia Foundation (USA), the American Portuguese Biomedical Research Fund (APBRF), and the Richard Chin and Lily Lock Machado-Joseph Disease Research Fund. A.V.-F. was supported by a PhD fellowship of FCT (SFRH/BD/87804/2012).
Author contributions
Conceptualization, L.P.d.A., A.V.-F., and C.N.; methodology, A.V.-F., D.P., M.M.L., R.F., and S.M.L.; investigation, A.V.-F., I.M.M., and D.L.; writing – original draft, A.V.-F; writing – review & editing, L.P.d.A., I.M.M., D.L., D.P., M.M.L., S.M.L., and D.V.; funding acquisition, L.P.d.A., D.V., and T.S.; resources, L.P.d.A., T.S., and D.V.; supervision, L.P.d.A. and C.N.
Declaration of interests
The authors declare no competing interests.
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.ymthe.2021.07.012.
Supplemental information
References
- 1.Schöls L., Bauer P., Schmidt T., Schulte T., Riess O. Autosomal dominant cerebellar ataxias: Clinical features, genetics, and pathogenesis. Lancet Neurol. 2004;3:291–304. doi: 10.1016/S1474-4422(04)00737-9. [DOI] [PubMed] [Google Scholar]
- 2.Bettencourt C., Santos C., Kay T., Vasconcelos J., Lima M. Analysis of segregation patterns in Machado-Joseph disease pedigrees. J. Hum. Genet. 2008;53:920–923. doi: 10.1007/s10038-008-0330-y. [DOI] [PubMed] [Google Scholar]
- 3.Kawaguchi Y., Okamoto T., Taniwaki M., Aizawa M., Inoue M., Katayama S., Kawakami H., Nakamura S., Nishimura M., Akiguchi I., et al. CAG expansions in a novel gene for Machado-Joseph disease at chromosome 14q32.1. Nat. Genet. 1994;8:221–228. doi: 10.1038/ng1194-221. [DOI] [PubMed] [Google Scholar]
- 4.Klockgether T., Skalej M., Wedekind D., Luft A.R., Welte D., Schulz J.B., Abele M., Bürk K., Laccone F., Brice A., Dichgans J. Autosomal dominant cerebellar ataxia type I. MRI-based volumetry of posterior fossa structures and basal ganglia in spinocerebellar ataxia types 1, 2 and 3. Brain. 1998;121:1687–1693. doi: 10.1093/brain/121.9.1687. [DOI] [PubMed] [Google Scholar]
- 5.Sudarsky L., Coutinho P. Machado-Joseph disease. Clin. Neurosci. 1995;3:17–22. [PubMed] [Google Scholar]
- 6.Maruyama H., Nakamura S., Matsuyama Z., Sakai T., Doyu M., Sobue G., Seto M., Tsujihata M., Oh-i T., Nishio T., et al. Molecular features of the CAG repeats and clinical manifestation of Machado-Joseph disease. Hum. Mol. Genet. 1995;4:807–812. doi: 10.1093/hmg/4.5.807. [DOI] [PubMed] [Google Scholar]
- 7.Rosenberg R.N. Machado-Joseph disease: An autosomal dominant motor system degeneration. Mov. Disord. 1992;7:193–203. doi: 10.1002/mds.870070302. [DOI] [PubMed] [Google Scholar]
- 8.Mizushima N., Yoshimori T., Ohsumi Y. The role of Atg proteins in autophagosome formation. Annu. Rev. Cell Dev. Biol. 2011;27:107–132. doi: 10.1146/annurev-cellbio-092910-154005. [DOI] [PubMed] [Google Scholar]
- 9.Hara T., Nakamura K., Matsui M., Yamamoto A., Nakahara Y., Suzuki-Migishima R., Yokoyama M., Mishima K., Saito I., Okano H., Mizushima N. Suppression of basal autophagy in neural cells causes neurodegenerative disease in mice. Nature. 2006;441:885–889. doi: 10.1038/nature04724. [DOI] [PubMed] [Google Scholar]
- 10.Komatsu M., Waguri S., Chiba T., Murata S., Iwata J., Tanida I., Ueno T., Koike M., Uchiyama Y., Kominami E., Tanaka K. Loss of autophagy in the central nervous system causes neurodegeneration in mice. Nature. 2006;441:880–884. doi: 10.1038/nature04723. [DOI] [PubMed] [Google Scholar]
- 11.Williams A., Jahreiss L., Sarkar S., Saiki S., Menzies F.M., Ravikumar B., Rubinsztein D.C. Aggregate-prone proteins are cleared from the cytosol by autophagy: Therapeutic implications. Curr. Top. Dev. Biol. 2006;76:89–101. doi: 10.1016/S0070-2153(06)76003-3. [DOI] [PubMed] [Google Scholar]
- 12.Hayashi M., Kobayashi K., Furuta H. Immunohistochemical study of neuronal intranuclear and cytoplasmic inclusions in Machado-Joseph disease. Psychiatry Clin. Neurosci. 2003;57:205–213. doi: 10.1046/j.1440-1819.2003.01102.x. [DOI] [PubMed] [Google Scholar]
- 13.Nascimento-Ferreira I., Santos-Ferreira T., Sousa-Ferreira L., Auregan G., Onofre I., Alves S., Dufour N., Colomer Gould V.F., Koeppen A., Déglon N., Pereira de Almeida L. Overexpression of the autophagic beclin-1 protein clears mutant ataxin-3 and alleviates Machado-Joseph disease. Brain. 2011;134:1400–1415. doi: 10.1093/brain/awr047. [DOI] [PubMed] [Google Scholar]
- 14.Sittler A., Muriel M.P., Marinello M., Brice A., den Dunnen W., Alves S. Deregulation of autophagy in postmortem brains of Machado-Joseph disease patients. Neuropathology. 2018;38:113–124. doi: 10.1111/neup.12433. [DOI] [PubMed] [Google Scholar]
- 15.Mori F., Tanji K., Odagiri S., Toyoshima Y., Yoshida M., Kakita A., Takahashi H., Wakabayashi K. Autophagy-related proteins (p62, NBR1 and LC3) in intranuclear inclusions in neurodegenerative diseases. Neurosci. Lett. 2012;522:134–138. doi: 10.1016/j.neulet.2012.06.026. [DOI] [PubMed] [Google Scholar]
- 16.Onofre I., Mendonça N., Lopes S., Nobre R., de Melo J.B., Carreira I.M., Januário C., Gonçalves A.F., de Almeida L.P. Fibroblasts of Machado Joseph disease patients reveal autophagy impairment. Sci. Rep. 2016;6:28220. doi: 10.1038/srep28220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Chou A.H., Yeh T.H., Ouyang P., Chen Y.L., Chen S.Y., Wang H.L. Polyglutamine-expanded ataxin-3 causes cerebellar dysfunction of SCA3 transgenic mice by inducing transcriptional dysregulation. Neurobiol. Dis. 2008;31:89–101. doi: 10.1016/j.nbd.2008.03.011. [DOI] [PubMed] [Google Scholar]
- 18.Evert B.O., Araujo J., Vieira-Saecker A.M., de Vos R.A., Harendza S., Klockgether T., Wüllner U. Ataxin-3 represses transcription via chromatin binding, interaction with histone deacetylase 3, and histone deacetylation. J. Neurosci. 2006;26:11474–11486. doi: 10.1523/JNEUROSCI.2053-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Evert B.O., Vogt I.R., Kindermann C., Ozimek L., de Vos R.A., Brunt E.R., Schmitt I., Klockgether T., Wüllner U. Inflammatory genes are upregulated in expanded ataxin-3-expressing cell lines and spinocerebellar ataxia type 3 brains. J. Neurosci. 2001;21:5389–5396. doi: 10.1523/JNEUROSCI.21-15-05389.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Perez M.K., Paulson H.L., Pendse S.J., Saionz S.J., Bonini N.M., Pittman R.N. Recruitment and the role of nuclear localization in polyglutamine-mediated aggregation. J. Cell Biol. 1998;143:1457–1470. doi: 10.1083/jcb.143.6.1457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Torashima T., Koyama C., Iizuka A., Mitsumura K., Takayama K., Yanagi S., Oue M., Yamaguchi H., Hirai H. Lentivector-mediated rescue from cerebellar ataxia in a mouse model of spinocerebellar ataxia. EMBO Rep. 2008;9:393–399. doi: 10.1038/embor.2008.31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kraft C., Kijanska M., Kalie E., Siergiejuk E., Lee S.S., Semplicio G., Stoffel I., Brezovich A., Verma M., Hansmann I., et al. Binding of the Atg1/ULK1 kinase to the ubiquitin-like protein Atg8 regulates autophagy. EMBO J. 2012;31:3691–3703. doi: 10.1038/emboj.2012.225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ganley I.G., Lam H., Wang J., Ding X., Chen S., Jiang X. ULK1.ATG13.FIP200 complex mediates mTOR signaling and is essential for autophagy. J. Biol. Chem. 2009;284:12297–12305. doi: 10.1074/jbc.M900573200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Russell R.C., Tian Y., Yuan H., Park H.W., Chang Y.Y., Kim J., Kim H., Neufeld T.P., Dillin A., Guan K.L. ULK1 induces autophagy by phosphorylating Beclin-1 and activating VPS34 lipid kinase. Nat. Cell Biol. 2013;15:741–750. doi: 10.1038/ncb2757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Gonçalves N., Simões A.T., Prediger R.D., Hirai H., Cunha R.A., Pereira de Almeida L. Caffeine alleviates progressive motor deficits in a transgenic mouse model of spinocerebellar ataxia. Ann. Neurol. 2017;81:407–418. doi: 10.1002/ana.24867. [DOI] [PubMed] [Google Scholar]
- 26.Nascimento-Ferreira I., Nóbrega C., Vasconcelos-Ferreira A., Onofre I., Albuquerque D., Aveleira C., Hirai H., Déglon N., Pereira de Almeida L. Beclin 1 mitigates motor and neuropathological deficits in genetic mouse models of Machado-Joseph disease. Brain. 2013;136:2173–2188. doi: 10.1093/brain/awt144. [DOI] [PubMed] [Google Scholar]
- 27.Alves S., Régulier E., Nascimento-Ferreira I., Hassig R., Dufour N., Koeppen A., Carvalho A.L., Simões S., de Lima M.C., Brouillet E., et al. Striatal and nigral pathology in a lentiviral rat model of Machado-Joseph disease. Hum. Mol. Genet. 2008;17:2071–2083. doi: 10.1093/hmg/ddn106. [DOI] [PubMed] [Google Scholar]
- 28.Carmona V., Cunha-Santos J., Onofre I., Simões A.T., Vijayakumar U., Davidson B.L., Pereira de Almeida L. Unravelling endogenous microRNA system dysfunction as a new pathophysiological mechanism in Machado-Joseph disease. Mol. Ther. 2017;25:1038–1055. doi: 10.1016/j.ymthe.2017.01.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Duarte-Neves J., Gonçalves N., Cunha-Santos J., Simões A.T., den Dunnen W.F., Hirai H., Kügler S., Cavadas C., Pereira de Almeida L. Neuropeptide Y mitigates neuropathology and motor deficits in mouse models of Machado-Joseph disease. Hum. Mol. Genet. 2015;24:5451–5463. doi: 10.1093/hmg/ddv271. [DOI] [PubMed] [Google Scholar]
- 30.Nóbrega C., Carmo-Silva S., Albuquerque D., Vasconcelos-Ferreira A., Vijayakumar U.G., Mendonça L., Hirai H., de Almeida L.P. Re-establishing ataxin-2 downregulates translation of mutant ataxin-3 and alleviates Machado-Joseph disease. Brain. 2015;138:3537–3554. doi: 10.1093/brain/awv298. [DOI] [PubMed] [Google Scholar]
- 31.Simões A.T., Gonçalves N., Koeppen A., Déglon N., Kügler S., Duarte C.B., Pereira de Almeida L. Calpastatin-mediated inhibition of calpains in the mouse brain prevents mutant ataxin 3 proteolysis, nuclear localization and aggregation, relieving Machado-Joseph disease. Brain. 2012;135:2428–2439. doi: 10.1093/brain/aws177. [DOI] [PubMed] [Google Scholar]
- 32.Paulson H.L., Perez M.K., Trottier Y., Trojanowski J.Q., Subramony S.H., Das S.S., Vig P., Mandel J.L., Fischbeck K.H., Pittman R.N. Intranuclear inclusions of expanded polyglutamine protein in spinocerebellar ataxia type 3. Neuron. 1997;19:333–344. doi: 10.1016/s0896-6273(00)80943-5. [DOI] [PubMed] [Google Scholar]
- 33.Ikeda H., Yamaguchi M., Sugai S., Aze Y., Narumiya S., Kakizuka A. Expanded polyglutamine in the Machado-Joseph disease protein induces cell death in vitro and in vivo. Nat. Genet. 1996;13:196–202. doi: 10.1038/ng0696-196. [DOI] [PubMed] [Google Scholar]
- 34.Paulson H.L., Das S.S., Crino P.B., Perez M.K., Patel S.C., Gotsdiner D., Fischbeck K.H., Pittman R.N. Machado-Joseph disease gene product is a cytoplasmic protein widely expressed in brain. Ann. Neurol. 1997;41:453–462. doi: 10.1002/ana.410410408. [DOI] [PubMed] [Google Scholar]
- 35.Yoshizawa T., Yamagishi Y., Koseki N., Goto J., Yoshida H., Shibasaki F., Shoji S., Kanazawa I. Cell cycle arrest enhances the in vitro cellular toxicity of the truncated Machado-Joseph disease gene product with an expanded polyglutamine stretch. Hum. Mol. Genet. 2000;9:69–78. doi: 10.1093/hmg/9.1.69. [DOI] [PubMed] [Google Scholar]
- 36.Chan E.Y., Longatti A., McKnight N.C., Tooze S.A. Kinase-inactivated ULK proteins inhibit autophagy via their conserved C-terminal domains using an Atg13-independent mechanism. Mol. Cell. Biol. 2009;29:157–171. doi: 10.1128/MCB.01082-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Cheong H., Lindsten T., Wu J., Lu C., Thompson C.B. Ammonia-induced autophagy is independent of ULK1/ULK2 kinases. Proc. Natl. Acad. Sci. USA. 2011;108:11121–11126. doi: 10.1073/pnas.1107969108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Demeter A., Romero-Mulero M.C., Csabai L., Ölbei M., Sudhakar P., Haerty W., Korcsmáros T. ULK1 and ULK2 are less redundant than previously thought: computational analysis uncovers distinct regulation and functions of these autophagy induction proteins. Sci. Rep. 2020;10:10940. doi: 10.1038/s41598-020-67780-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Klionsky D.J., Abdelmohsen K., Abe A., Abedin M.J., Abeliovich H., Acevedo Arozena A., Adachi H., Adams C.M., Adams P.D., Adeli K., et al. Guidelines for the use and interpretation of assays for monitoring autophagy (3rd edition) Autophagy. 2016;12:1–222. doi: 10.1080/15548627.2015.1100356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Evert B.O., Vogt I.R., Vieira-Saecker A.M., Ozimek L., de Vos R.A., Brunt E.R., Klockgether T., Wüllner U. Gene expression profiling in ataxin-3 expressing cell lines reveals distinct effects of normal and mutant ataxin-3. J. Neuropathol. Exp. Neurol. 2003;62:1006–1018. doi: 10.1093/jnen/62.10.1006. [DOI] [PubMed] [Google Scholar]
- 41.Toonen L.J.A., Overzier M., Evers M.M., Leon L.G., van der Zeeuw S.A.J., Mei H., Kielbasa S.M., Goeman J.J., Hettne K.M., Magnusson O.T., et al. Transcriptional profiling and biomarker identification reveal tissue specific effects of expanded ataxin-3 in a spinocerebellar ataxia type 3 mouse model. Mol. Neurodegener. 2018;13:31. doi: 10.1186/s13024-018-0261-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Coutinho P., Guimarães A., Scaravilli F. The pathology of Machado-Joseph disease. Report of a possible homozygous case. Acta Neuropathol. 1982;58:48–54. doi: 10.1007/BF00692697. [DOI] [PubMed] [Google Scholar]
- 43.Koeppen A.H. The pathogenesis of spinocerebellar ataxia. Cerebellum. 2005;4:62–73. doi: 10.1080/14734220510007950. [DOI] [PubMed] [Google Scholar]
- 44.Ashkenazi A., Bento C.F., Ricketts T., Vicinanza M., Siddiqi F., Pavel M., Squitieri F., Hardenberg M.C., Imarisio S., Menzies F.M., Rubinsztein D.C. Polyglutamine tracts regulate beclin 1-dependent autophagy. Nature. 2017;545:108–111. doi: 10.1038/nature22078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Feng Y., He D., Yao Z., Klionsky D.J. The machinery of macroautophagy. Cell Res. 2014;24:24–41. doi: 10.1038/cr.2013.168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Rui Y.N., Xu Z., Patel B., Chen Z., Chen D., Tito A., David G., Sun Y., Stimming E.F., Bellen H.J., et al. Huntingtin functions as a scaffold for selective macroautophagy. Nat. Cell Biol. 2015;17:262–275. doi: 10.1038/ncb3101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Wong Y.C., Holzbaur E.L. The regulation of autophagosome dynamics by huntingtin and HAP1 is disrupted by expression of mutant huntingtin, leading to defective cargo degradation. J. Neurosci. 2014;34:1293–1305. doi: 10.1523/JNEUROSCI.1870-13.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Shibata M., Lu T., Furuya T., Degterev A., Mizushima N., Yoshimori T., MacDonald M., Yankner B., Yuan J. Regulation of intracellular accumulation of mutant Huntingtin by Beclin 1. J. Biol. Chem. 2006;281:14474–14485. doi: 10.1074/jbc.M600364200. [DOI] [PubMed] [Google Scholar]
- 49.Wold M.S., Lim J., Lachance V., Deng Z., Yue Z. ULK1-mediated phosphorylation of ATG14 promotes autophagy and is impaired in Huntington’s disease models. Mol. Neurodegener. 2016;11:76. doi: 10.1186/s13024-016-0141-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Yu X., Muñoz-Alarcón A., Ajayi A., Webling K.E., Steinhof A., Langel Ü., Ström A.L. Inhibition of autophagy via p53-mediated disruption of ULK1 in a SCA7 polyglutamine disease model. J. Mol. Neurosci. 2013;50:586–599. doi: 10.1007/s12031-013-0012-x. [DOI] [PubMed] [Google Scholar]
- 51.Yan J., Kuroyanagi H., Tomemori T., Okazaki N., Asato K., Matsuda Y., Suzuki Y., Ohshima Y., Mitani S., Masuho Y., et al. Mouse ULK2, a novel member of the UNC-51-like protein kinases: unique features of functional domains. Oncogene. 1999;18:5850–5859. doi: 10.1038/sj.onc.1202988. [DOI] [PubMed] [Google Scholar]
- 52.Kuma A., Hatano M., Matsui M., Yamamoto A., Nakaya H., Yoshimori T., Ohsumi Y., Tokuhisa T., Mizushima N. The role of autophagy during the early neonatal starvation period. Nature. 2004;432:1032–1036. doi: 10.1038/nature03029. [DOI] [PubMed] [Google Scholar]
- 53.Kundu M., Lindsten T., Yang C.Y., Wu J., Zhao F., Zhang J., Selak M.A., Ney P.A., Thompson C.B. Ulk1 plays a critical role in the autophagic clearance of mitochondria and ribosomes during reticulocyte maturation. Blood. 2008;112:1493–1502. doi: 10.1182/blood-2008-02-137398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Conceição M., Mendonça L., Nóbrega C., Gomes C., Costa P., Hirai H., Moreira J.N., Lima M.C., Manjunath N., Pereira de Almeida L. Intravenous administration of brain-targeted stable nucleic acid lipid particles alleviates Machado-Joseph disease neurological phenotype. Biomaterials. 2016;82:124–137. doi: 10.1016/j.biomaterials.2015.12.021. [DOI] [PubMed] [Google Scholar]
- 55.Gonçalves N., Simões A.T., Cunha R.A., de Almeida L.P. Caffeine and adenosine A2A receptor inactivation decrease striatal neuropathology in a lentiviral-based model of Machado-Joseph disease. Ann. Neurol. 2013;73:655–666. doi: 10.1002/ana.23866. [DOI] [PubMed] [Google Scholar]
- 56.Mendonça L.S., Nóbrega C., Hirai H., Kaspar B.K., Pereira de Almeida L. Transplantation of cerebellar neural stem cells improves motor coordination and neuropathology in Machado-Joseph disease mice. Brain. 2015;138:320–335. doi: 10.1093/brain/awu352. [DOI] [PubMed] [Google Scholar]
- 57.Nóbrega C., Nascimento-Ferreira I., Onofre I., Albuquerque D., Déglon N., de Almeida L.P. RNA interference mitigates motor and neuropathological deficits in a cerebellar mouse model of Machado-Joseph disease. PLoS ONE. 2014;9:e100086. doi: 10.1371/journal.pone.0100086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Oliveira Miranda C., Marcelo A., Silva T.P., Barata J., Vasconcelos-Ferreira A., Pereira D., Nóbrega C., Duarte S., Barros I., Alves J., et al. Repeated mesenchymal stromal cell treatment sustainably alleviates Machado-Joseph disease. Mol. Ther. 2018;26:2131–2151. doi: 10.1016/j.ymthe.2018.07.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Boya P., González-Polo R.A., Casares N., Perfettini J.L., Dessen P., Larochette N., Métivier D., Meley D., Souquere S., Yoshimori T., et al. Inhibition of macroautophagy triggers apoptosis. Mol. Cell. Biol. 2005;25:1025–1040. doi: 10.1128/MCB.25.3.1025-1040.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Lee E.J., Tournier C. The requirement of uncoordinated 51-like kinase 1 (ULK1) and ULK2 in the regulation of autophagy. Autophagy. 2011;7:689–695. doi: 10.4161/auto.7.7.15450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Li Z., Zhao K., Lv X., Lan Y., Hu S., Shi J., Guan J., Yang Y., Lu H., He H., et al. Ulk1 governs nerve growth factor/TrkA signaling by mediating Rab5 GTPase activation in porcine hemagglutinating encephalomyelitis virus-induced neurodegenerative disorders. J. Virol. 2018;92:e00325-18. doi: 10.1128/JVI.00325-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Lim J., Lachenmayer M.L., Wu S., Liu W., Kundu M., Wang R., Komatsu M., Oh Y.J., Zhao Y., Yue Z. Proteotoxic stress induces phosphorylation of p62/SQSTM1 by ULK1 to regulate selective autophagic clearance of protein aggregates. PLoS Genet. 2015;11:e1004987. doi: 10.1371/journal.pgen.1004987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Tomoda T., Bhatt R.S., Kuroyanagi H., Shirasawa T., Hatten M.E. A mouse serine/threonine kinase homologous to C. elegans UNC51 functions in parallel fiber formation of cerebellar granule neurons. Neuron. 1999;24:833–846. doi: 10.1016/s0896-6273(00)81031-4. [DOI] [PubMed] [Google Scholar]
- 64.Tomoda T., Kim J.H., Zhan C., Hatten M.E. Role of Unc51.1 and its binding partners in CNS axon outgrowth. Genes Dev. 2004;18:541–558. doi: 10.1101/gad.1151204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Verhoef L.G., Lindsten K., Masucci M.G., Dantuma N.P. Aggregate formation inhibits proteasomal degradation of polyglutamine proteins. Hum. Mol. Genet. 2002;11:2689–2700. doi: 10.1093/hmg/11.22.2689. [DOI] [PubMed] [Google Scholar]
- 66.Haacke A., Broadley S.A., Boteva R., Tzvetkov N., Hartl F.U., Breuer P. Proteolytic cleavage of polyglutamine-expanded ataxin-3 is critical for aggregation and sequestration of non-expanded ataxin-3. Hum. Mol. Genet. 2006;15:555–568. doi: 10.1093/hmg/ddi472. [DOI] [PubMed] [Google Scholar]
- 67.Wellington C.L., Ellerby L.M., Gutekunst C.A., Rogers D., Warby S., Graham R.K., Loubser O., van Raamsdonk J., Singaraja R., Yang Y.Z., et al. Caspase cleavage of mutant huntingtin precedes neurodegeneration in Huntington’s disease. J. Neurosci. 2002;22:7862–7872. doi: 10.1523/JNEUROSCI.22-18-07862.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Chan E.Y., Kir S., Tooze S.A. siRNA screening of the kinome identifies ULK1 as a multidomain modulator of autophagy. J. Biol. Chem. 2007;282:25464–25474. doi: 10.1074/jbc.M703663200. [DOI] [PubMed] [Google Scholar]
- 69.Ro S.H., Jung C.H., Hahn W.S., Xu X., Kim Y.M., Yun Y.S., Park J.M., Kim K.H., Seo M., Ha T.Y., et al. Distinct functions of Ulk1 and Ulk2 in the regulation of lipid metabolism in adipocytes. Autophagy. 2013;9:2103–2114. doi: 10.4161/auto.26563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.de Almeida L.P., Zala D., Aebischer P., Déglon N. Neuroprotective effect of a CNTF-expressing lentiviral vector in the quinolinic acid rat model of Huntington’s disease. Neurobiol. Dis. 2001;8:433–446. doi: 10.1006/nbdi.2001.0388. [DOI] [PubMed] [Google Scholar]
- 71.Grieger J.C., Choi V.W., Samulski R.J. Production and characterization of adeno-associated viral vectors. Nat. Protoc. 2006;1:1412–1428. doi: 10.1038/nprot.2006.207. [DOI] [PubMed] [Google Scholar]
- 72.Carette J.E., Raaben M., Wong A.C., Herbert A.S., Obernosterer G., Mulherkar N., Kuehne A.I., Kranzusch P.J., Griffin A.M., Ruthel G., et al. Ebola virus entry requires the cholesterol transporter Niemann-Pick C1. Nature. 2011;477:340–343. doi: 10.1038/nature10348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Oue M., Mitsumura K., Torashima T., Koyama C., Yamaguchi H., Furuya N., Hirai H. Characterization of mutant mice that express polyglutamine in cerebellar Purkinje cells. Brain Res. 2009;1255:9–17. doi: 10.1016/j.brainres.2008.12.014. [DOI] [PubMed] [Google Scholar]
- 74.Ye J., Coulouris G., Zaretskaya I., Cutcutache I., Rozen S., Madden T.L. Primer-BLAST: A tool to design target-specific primers for polymerase chain reaction. BMC Bioinformatics. 2012;13:134. doi: 10.1186/1471-2105-13-134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Ashburner M., Ball C.A., Blake J.A., Botstein D., Butler H., Cherry J.M., Davis A.P., Dolinski K., Dwight S.S., Eppig J.T., et al. The Gene Ontology Consortium Gene ontology: Tool for the unification of biology. Nat. Genet. 2000;25:25–29. doi: 10.1038/75556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.The Gene Ontology Consortium Expansion of the Gene Ontology knowledgebase and resources. Nucleic Acids Res. 2017;45(D1):D331–D338. doi: 10.1093/nar/gkw1108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.de Almeida L.P., Ross C.A., Zala D., Aebischer P., Déglon N. Lentiviral-mediated delivery of mutant huntingtin in the striatum of rats induces a selective neuropathology modulated by polyglutamine repeat size, huntingtin expression levels, and protein length. J. Neurosci. 2002;22:3473–3483. doi: 10.1523/JNEUROSCI.22-09-03473.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.