

Abstract
Activating mutations in c-KIT are associated with the mast cell (MC) clonal disorders cutaneous mastocytosis and systemic mastocytosis and its variants, including aggressive systemic mastocytosis, MC leukemia, and MC sarcoma. Currently, therapies inhibiting KIT signaling are a leading strategy to treat MC proliferative disorders. However, these approaches may have off-target effects, and in some patients, complete remission or improved survival time cannot be achieved. These limitations led us to develop an approach using chemically stable exon skipping oligonucleotides (ESOs) that induce exon skipping of precursor (pre-)mRNA to alter gene splicing and introduce a frameshift into mature KIT mRNA transcripts. The result of this alternate approach results in marked downregulation of KIT expression, diminished KIT signaling, inhibition of MC proliferation, and rapid induction of apoptosis in neoplastic HMC-1.2 MCs. We demonstrate that in vivo administration of KIT targeting ESOs significantly inhibits tumor growth and systemic organ infiltration using both an allograft mastocytosis model and a humanized xenograft MC tumor model. We propose that our innovative approach, which employs well-tolerated, chemically stable oligonucleotides to target KIT expression through unconventional pathways, has potential as a KIT-targeted therapeutic alone, or in combination with agents that target KIT signaling, in the treatment of KIT-associated malignancies.
Keywords: mastocytosis, mast cell leukemia, mast cell sarcoma, therapeutic oligonucleotide, exon skipping, receptor tyrosine kinase
Graphical abstract
Cruse and colleagues describe an application of exon skipping oligonucleotides to introduce a frameshift into mRNA transcripts for the growth factor receptor KIT, resulting in induction of apoptosis and inhibition of proliferation of neoplastic mast cells in vitro and in vivo. The study highlights potential therapeutic application for KIT-associated neoplasia.
Introduction
c-KIT is a proto-oncogene that is highly conserved between species and encodes the receptor tyrosine kinase, KIT (CD117). KIT is critically required for the proliferation, survival, and differentiation of bone marrow-derived hematopoietic stem cells including mast cell (MC) progenitors.1 Expression of KIT in most hematopoietic cells is lost during differentiation, but MCs retain KIT expression throughout their lifespan, and KIT signaling remains essential for mature MC survival and proliferation.2, 3, 4, 5 The oncogenic potential of c-KIT was initially realized when its viral counterpart, v-KIT, was found to be responsible for the transforming activity of the Hardy-Zuckerman IV feline sarcoma virus.6 Since then, activating gain-of-function c-KIT mutations have been associated with the initiation and progression of several human malignancies, but there is a particularly high occurrence in gastrointestinal stromal tumors (GISTs) and MC proliferative disorders, which include MC leukemia (MCL) and MC sarcoma.7
Mastocytosis is an unusual neoplastic condition with a number of variants, all of which demonstrate aberrant expansion and accumulation of MCs.8 The disease can be difficult to treat in part due to the extensive biological heterogeneity of mastocytosis subtypes, which exhibit distinct clinical manifestations and require therapy adjusted to the disease variant.8 Patients with non-advanced mastocytosis generally do not require cytoreductive or KIT-targeting therapies, while patients with aggressive disease require intervention. Current frontline treatments for advanced systemic mastocytosis (ASM) include cladribine and KIT-targeting tyrosine kinase inhibitors (TKIs).9,10 In some cases, interferon-alpha is prescribed, and in rapidly progressing ASM and MCL, poly-chemotherapy and stem cell transplantation may be considered. However, these treatments are not always effective in advanced disease, in which complete remission is infrequent and improvement in prognosis is variable. Moreover, due to the non-specific nature of chemotherapeutics and TKIs, there is a risk of off-target toxicity, including bone marrow suppression, immunological dysfunction, and cardiotoxicity.11 Although hematopoietic stem cell transplantation is possible for some patients, there remains an unmet clinical need for additional and novel therapeutic approaches that will selectively target and induce apoptosis of neoplastic MCs.
With a strong connection to cancer development and aberrant MC growth, KIT represents a desirable but challenging therapeutic target. Human c-KIT is located on chromosome 4q12 and contains 21 exons.12 KIT-stem cell factor (SCF) interactions are necessary for the survival, proliferation, and differentiation of MCs, but in the presence of activating mutations, SCF dependence is less important and oncogenic KIT signaling drives neoplastic MC growth.7,13 Although mutations can occur throughout the gene, many cluster in hotspots, which are associated with specific diseases.7 For example, mutations in exon 11, which encodes the juxtamembrane domain of KIT, commonly occur in GISTs,14,15 while the D816V point mutation in exon 17,16 encoding the second kinase domain of KIT, is present in most patients with systemic mastocytosis.13 Identifying these mutations within patients is important for assessing disease prognosis and determining treatment approaches, as the type of mutation and the occurrence of additional mutations in part dictate the success of therapies for KIT-associated malignancies. In particular, the KIT D816V mutation found in >80% of adult systemic mastocytosis cases13,17 causes a conformational change in KIT that renders it resistant to some TKIs such as imatinib mesylate (Gleevec)18,19 that target the inactive ATP-binding site conformation of KIT. Therefore, in these patient populations, there is an unmet need for therapeutics. Recent and emerging compounds that can more selectively target KIT, and inhibit KIT with the D816V mutation, such as avapritinib, hold promise,20 but a critical need for selective therapeutics that target KIT remains.
Due to the heterogeneity of c-KIT mutations in mastocytosis, and the occurrence of mutations that confer resistance to TKIs, we proposed that targeting KIT expression would prove an effective additional approach to therapy. Conventionally, the therapeutic development of oligonucleotides to target gene expression within many diseases has followed either classic antisense or small interfering RNA (siRNA) approaches that rely on RNase H or RNA-induced silencing complex (RISC)-mediated pathways of transcript degradation (reviewed in Lundin et al.21). These approaches have had some success in early stage clinical trials for treatment of some diseases such as age-related macular degeneration.22 This has contributed to increased attention for antisense oligonucleotide (ASO) therapy (reviewed in Lundin et al.21 and Potaczek et al.23). In recent decades, several ASOs have been approved by both the US Food and Drug Administration (FDA) and European Medicines Agency (EMA) for various applications.24,25 Most recently, two vaccines containing oligonucleotides of RNA with modified nucleobases designed to introduce non-native gene expression of viral spike protein have received worldwide attention as vaccination against SARS CoV-2 CoVID-19.26,27 These examples highlight the potential for therapeutic oligonucleotides to temporarily alter protein expression in a variety of settings and demonstrate that they are a well-tolerated and viable therapeutic approach.
Splice switching oligonucleotides (SSOs) are a type of oligonucleotide that are emerging as promising therapeutics in personalized medicine. The short synthetic single strands of nucleic acids that are typically less than 50 nt long provide a targeted approach to gene modification by manipulating RNA splicing. Compared to classic ASO-mediated mRNA transcript degradation, SSOs comprise a different type of ASO therapy that alters normal splicing of the targeted transcript, which may prove more versatile and exhibit high sequence specificity (reviewed in Havens and Hastings28). SSOs are a class of oligonucleotides that have altered backbone chemistry. Some SSO chemistries, such as phosphorodiamidate morpholino oligonucleotides (PMOs), are substantially different from DNA and RNA backbones, with a neutral charge that increases stability and apparent evasion of recognition by pattern recognition receptors, such as Toll-like receptors in the immune system.29, 30, 31, 32 SSOs can be used to promote inclusion of exons or to induce exon skipping. With respect to exon skipping, SSOs can prevent the inclusion of a specific exon in mature mRNA by binding to specific splice sites in precursor (pre-)mRNA molecules, which has the effect of sterically shielding the splice site from the spliceosome machinery. These specific types of SSOs are termed exon skipping oligonucleotides (ESOs). Importantly, chemical modifications of SSOs prevent the degradation of pre-mRNA-SSO complexes by RNase H, allowing transcription of an altered mRNA sequence to continue (reviewed in Havens and Hastings28). In genetic diseases in particular, this has important clinical implications, since exon exclusion or inclusion can correct an aberrant splicing pattern, or skip an exon containing a frameshift mutation to restore expression of a functional protein. For instance, therapeutic splice switching in Duchenne muscular dystrophy (DMD) aims to reestablish the correct reading frame to permit production of a truncated but partially functional dystrophin protein, which reduces the clinical severity of the DMD phenotype (reviewed in Nakamura and Takeda33 and Nakamura 34). A number of other genetic diseases may also benefit from SSO-based therapy, as earlier reviewed.21,23
We thus employed chemically stable antisense oligonucleotides that induce exon skipping of pre-mRNA to alter gene splicing. These ESOs contain various chemical modifications to their backbones that prevent recognition by RNA degradation pathways. Targeting pre-mRNA with ESOs thus provides a more versatile and chemically stable approach to gene targeting compared to conventional siRNA approaches. Rather than induce RNA degradation through energetic enzymatic pathways, ESOs prevent inclusion of a specific exon in mature mRNA by binding to splice sites in pre-mRNA molecules, which has the effect of sterically shielding the splice site from the spliceosome machinery.28 Therefore, transcription is not perturbed, but rather an altered mRNA sequence is translated to produce an alternative splice variant. In genetic diseases in particular, this has important clinical implications, since exon exclusion can partially correct aberrant mRNA caused by a frameshift mutation.35,36
We utilized this exon skipping approach to induce a frameshift into mature KIT mRNA transcripts. Our ESO, which we abbreviate in this report as KitStop, results in an immediate stop codon and loss-of-function culminating in rapid neoplastic MC death in vitro and in vivo, and inhibition of tumor growth with systemic injection in both an allograft model of mastocytosis and a humanized xenograft MC tumor model. These data act as a proof of principle for the therapeutic utility of a frameshifting KitStop ESO in KIT-associated malignancies. Given the recent approval of the ESO eteplirsen by the FDA for the treatment of DMD,35,36 we propose that our KitStop approach shows similar promise for a novel KIT-targeted therapeutic.
Results
Exon skipping of c-KIT exon 4 in HMC-1.2 cells
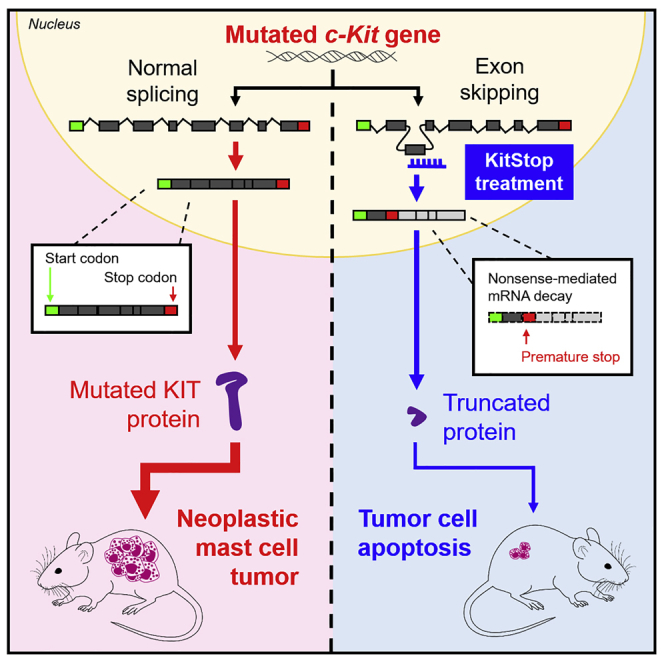
We previously reported >95% transfection efficiency of ESOs for FcεRIβ in human and mouse MCs with no evidence of cytotoxicity, as determined by propidium iodide (PI) and Live/Dead staining.37 We similarly designed a stable 25-mer morpholino ESO to target the donor splice site of exon 4 in c-KIT pre-mRNA that we termed KitStop, with the prediction that ESO-induced skipping of exon 4 would introduce a frameshift into the mature mRNA open reading frame and a premature termination (or stop) codon. We selected exon 4 of c-KIT based on the gene sequence and mRNA product after analysis of the sequence revealed that exon 4 was the first exon that would result in a frameshift when skipped, and this frameshift would introduce an immediate premature stop codon that should trigger a nonsense-mediated mRNA decay (NMD) response to degrade transcripts. Exon 4 is very early in the transcript, and so if transcripts evade NMD, or if saturation of NMD occur, the protein product would most likely be non-functional. Thus, frameshifting with ESOs would either induce early termination of KIT mRNA translation, resulting in a severely truncated protein, or induce NMD of mRNA transcripts (Figure 1A). Either outcome should eliminate expression of the KIT receptor.
Figure 1.

ESO-mediated alternative splicing of exon 4 in c-KIT pre-mRNA
(A) KitStop ESO was designed to target the donor splice site of exon 4, which led to exclusion of exon 4 by the spliceosome. This is predicted to introduce a premature stop codon due to a frameshift in the open reading frame of the mRNA transcript, even in the presence of G560V and D816V activating mutations, which are located downstream of the target site of KitStop. The resulting premature stop codon was predicted to induce degradation of transcripts by NMD. If the NMD pathway becomes saturated, it was predicted that a truncated mRNA transcript encoding a non-functional protein would be produced. Yellow boxes represent exons; thick black bar represents introns. FL-c-KIT, full-length c-KIT; t-c-KIT, truncated c-KIT. (B) Gel electrophoresis data demonstrating splice switching of c-KIT by KitStop ESO in comparison to standard control (Stndcon) in HMC-1.2 as assessed by analysis of total RNA by RT-PCR. Black arrow indicates full-length c-KIT; orange arrow indicates alternatively spliced truncated c-KIT mRNA. Each lane of the gel represents a paired replicate for Stndcon- and KitStop-treated cells in sequence. Gel electrophoresis RT-PCR images were acquired using a LI-COR Odyssey Fc imaging system. (C) qRT-PCR of c-KIT transcripts in HMC-1.2 cells demonstrates a significant reduction in KIT mRNA transcripts with KitStop relative to Stndcon-treated cells after correction against the housekeeping gene β-actin. (D) Gel electrophoresis data demonstrating splice switching of wild-type c-KIT mRNA by KitStop ESO in comparison to Stndcon ASO in LAD2 cells. (E) qRT-PCR of c-KIT transcripts in LAD2 cells demonstrates a significant reduction with KitStop relative to Stndcon cells. Data include mean ± SEM from three independent experiments. ∗p < 0.05, one-sample t test against hypothetical mean of 1.
To test this predicted outcome, we transfected HMC-1.2 cells exhibiting V560G and D816V mutations with KitStop or an oligonucleotide control. KitStop induced exon skipping as indicated by the appearance of an additional, truncated band of KIT-mRNA by RT-PCR, compared with cells transfected with an equivalent 25-mer standard control antisense oligonucleotide (Figure 1B). Exon 4 is 137 bp long. RT-PCR products were designed with primers spanning exons 3–6, and PCR product from the full-length transcript should be 577 bp, and 440 bp for exon 4-skipped KIT mRNA. On occasion with exon skipping, more than one exon can be skipped. Our primer design would detect transcripts with exon 4 and exon 5 removed, but we did not detect any of these transcripts. In addition, quantitative RT-PCR targeting exons upstream of exon 4 indicated that total c-KIT mRNA was reduced by about 30% in HMC-1.2 cells transfected with KitStop (Figure 1C). In order to establish whether efficacy of KitStop was comparable in slowly dividing human MCs with non-mutant KIT, we performed the same experiments in the human MC line LAD-2, which is a transformed cell line that more closely represents “normal” MCs.38 We found comparable results in exon skipping in the LAD-2 cells with evidence of skipped transcripts (Figure 1D) and a reduction in total mRNA transcript number (Figure 1E). Because introduction of a stop codon early in an exon can induce NMD,39,40 our data are consistent with the conclusion that a proportion of KIT transcripts are degraded by NMD. However, even if the NMD pathway becomes saturated, or transcripts evade NMD, the produced truncated transcript of the first three exons would not be functional as a receptor as it would code only for a segment of the extracellular domain (Figure 1A).
Loss of KIT expression and downstream signaling with KitStop exon skipping
We then used flow cytometry to measure KIT expression in HMC-1.2 cells transfected with KitStop. MCs express KIT on their cell surface, but KIT is rapidly internalized following activation by its ligand, SCF. After endocytosis, KIT signals within intracellular compartments before degradation.41 In neoplastic MCs such as the HMC-1.2 cell line, oncogenic KIT signaling occurs within intracellular compartments, independently of SCF.7,42 Consequently, we measured total KIT expression in HMC-1.2 cells (for gating strategies, see Figure S1) and found that it was significantly reduced by KitStop (Figures 2A–2C). The reduction in KIT protein expression was also demonstrated by western blot (Figure 2D). Unlike KIT, KitStop ESO did not affect the expression of the kinase extracellular signal-regulated kinase (ERK) or the cytoskeletal protein β-actin (Figure 2D). However, transfection of the KitStop ESO reduced by comparable amounts the phosphorylation of both KIT (Figure 2E) and ERK (Figure 2F), which are constitutively phosphorylated in HMC-1.2 cells (Figure 2D). Thus, this reduction in phosphorylation of KIT and the downstream signaling protein ERK are likely a direct result of reduced expression of the constitutively active KIT protein. Taken together, these data are consistent with the conclusion that KitStop frameshifting ESOs efficiently deplete KIT expression by reducing mRNA expression, and thus silence constitutive KIT signaling.
Figure 2.

KitStop ESO reduces KIT expression and loss of function in human MCs
(A) Flow cytometry histograms of total KIT expression in HMC-1.2 cells at 24 h following transfection with KitStop ESO. (B) Mean flow cytometry data for total KIT expression calculated from the geometric mean fluorescence intensity (MFI) (B) and expressed as a percentage of Stndcon ASO (C). KIT expression data were taken from only the viable cells, and the total number of viable cells in the KitStop-treated HMC-1.2 cells was low. (D) Immunoblots of constitutive phosphorylation of KIT and ERK in HMC-1.2 cells after 24 h of KitStop treatment from three independent experiments shown from left to right. (E and F) Combined phosphorylation data for phosphorylated (p)KIT (E) and pERK (F) after correction against β-actin or total ERK, respectively, and normalized to phosphorylation in control cells. (G) Combined data from flow cytometry assessing annexin V surface staining expressed as geometric MFI. (H) Combined geometric MFI of Live/Dead staining in HMC-1.2 cells at each time point. (I) Total number of viable HMC-1.2 cells cultured under normal conditions assessed by trypan blue counts. Graphical representation of data includes mean ± SEM from three independent experiments. ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001, ∗∗∗∗p < 0.0001, using ANOVA with Sidak’s post-test (B, C, and G–I) or Student’s paired t test (E and F).
Frameshifting KIT mRNA induces neoplastic MC death
In neoplastic MC diseases and cell lines such as HMC-1.2, constitutive KIT activation enables continuous SCF-independent MC survival and growth. Therefore, we examined whether KitStop treatment can eliminate the pro-survival effects of imatinib-insensitive activating c-KIT mutations in neoplastic HMC-1.2 cells.
First, we assessed HMC-1.2 cell apoptosis and cell death using fluorescein isothiocyanate (FITC)-conjugated annexin V and PI co-staining (Figures S2A and S3). In comparison to cells transfected with the standard control oligonucleotide, KitStop-treated cells exhibited increased annexin V-FITC staining during the course of 72 h (Figure 2G; Figures S2A and S2B). KitStop reduced the number of non-apoptotic cells during 72 h (Figure S2C) with a corresponding increase in early (Figure S2D) and late (Figure S2E) apoptotic cells as assessed by annexin V and PI staining. To examine cell viability, HMC-1.2 cells stained with PI were analyzed (Figures S4A and S5). In contrast to the standard control oligonucleotide, KitStop caused increased PI staining during 72 h (Figures S4A–S4C). A similar pattern of staining was also seen with Live/Dead staining (Figure 2H; Figures S4D, S4E, and S6) with an increase in dead cells at each time point measured. Taken together, these findings demonstrate that ESO-mediated frameshifting of the KIT mRNA transcript rapidly induces apoptosis and cell death in neoplastic MCs.
Frameshifting c-KIT prevents neoplastic MC proliferation
We next examined the effects of KIT mRNA frameshifting on neoplastic MC proliferation, given that the activating c-KIT mutations expressed by HMC-1.2 cells facilitate SCF-independent proliferation.43,44 In contrast to cells transfected with the standard control antisense oligonucleotide that increased in numbers during 72 h, KitStop prevented HMC-1.2 growth, as demonstrated by a marked decrease in viable cell number with trypan blue cell counts (Figure 2I). In addition to total viable cell counts, we performed a proliferation assay using CellTrace dilution and Live/Dead staining. Similar to the cell counts, the population of viable KitStop-treated cells, as determined by Live/Dead staining, retained the CellTrace dye, whereas CellTrace fluorescence in standard control oligonucleotide-treated cells was increasingly diluted at each time point (Figures S7A–S7C). The reduced cell proliferation is consistent with reduced KIT expression and the decrease in ERK phosphorylation, a pathway activated by KIT with a critical role in proliferation. KitStop treatment also increased the number of non-viable cells, which retained the CellTrace dye and thus had not proliferated (Figures S7B and S7D). Longer time courses were not evaluated in detail due to lack of viable cells in culture, which died off by 5–7 days. Taken together, these findings indicate that the ESO-induced frameshifting of KIT mRNA markedly suppresses mutant KIT-mediated neoplastic cell proliferation and survival. Consequently, frameshifting oligonucleotides targeting KIT could represent an efficient approach to reduce MC burden in vivo.
Frameshifting c-KIT markedly reduces KIT expression in slowly dividing and primary MCs
To examine the effects of KitStop ESO on an MC line with less proliferative capacity than HMC-1.2 cells, we employed the human LAD-2 MC line that is SCF responsive and expresses non-mutated KIT.38 KitStop ESO rapidly reduced both surface and total KIT expression in LAD-2 cells with >95% reduction by 2 days, which lasted for at least 7 days (Figure S8). Functionally, the almost complete loss of KIT expression had no effect on apoptosis or cell death by 2 days, but it resulted in a trend toward induction of apoptosis and cell death by 7 days (Figures S9A–S9D). These data suggest that slowly dividing MCs with non-mutated KIT are slower to respond to KitStop and loss of KIT expression compared to rapidly dividing mutant KIT cells. To further explore the delayed effects of KitStop in these cells, we examined the effects of removing and washing out SCF in LAD-2 cells and staining the cells with Live/Dead stain. In agreement with the KitStop data, LAD-2 cells were resistant to removal of SCF during 7 days (Figures S9E and S9F). We then examined the effects of KitStop on proliferation of LAD-2 cells, which have a slower doubling time.38 We found that KitStop appeared to inhibit proliferation as evidenced by retention of CellTrace dye (Figure S9G), but given the slow division of these cells, the difference did not reach significance (Figure S9H).
Finally, we also examined the efficacy of KitStop in primary MCs from mouse bone marrow-derived MCs (BMMCs) to establish whether KitStop reduced Kit expression in mouse cells to enable in vivo assessment. To target mouse Kit, we examined the sequence of c-Kit and Kit mRNA, and compared it to human c-KIT and KIT mRNA, to establish that in both species, exon 4 was the first exon in the transcript to result in a frameshift in mRNA when the exon was skipped and an immediate stop codon was introduced in both species. Therefore, we designed a comparable mouse KitStop ESO and found that the mouse version resulted in exon skipping and markedly reduced Kit expression in mouse BMMCs (Figure S10). Taken together, these results suggest that KitStop functions comparably in both human and mouse MCs, and that the effects of KitStop are conserved across cell types. However, neoplastic MCs may be more rapidly affected by KitStop and more susceptible to apoptosis.
KitStop systemic administration inhibited human neoplastic MC growth in vivo in a humanized xenograft MC neoplasia model
In order to establish efficacy in vivo, we utilized Vivo-Morpholinos for KitStop ESO. Vivo-Morpholinos have an octa-guanidine dendrimer around a triazine core to act as a cell-penetrating delivery moiety for in vivo applications.45 We based our dosing protocol on knowing the maximum suggested dose of 12.5 mg/kg daily from the supplier, and the efficacy of in vitro testing on HMC-1.2 cells during 72 h, which suggested that administration every 3 days would be effective. Using a conservative treatment protocol of 8.3 mg/kg every 3 days (Figure 3A) and groups of five mice per cohort, we established that systemic intravenous (i.v.) administration of KitStop significantly reduced tumor growth, as measured by assessing tumor volume after inoculation of NSG female mice with 1 × 106 human neoplastic HMC-1.2 cells into a primary site at the right flank, subcutaneously (Figures 3B and 3C). After 14 days of treatment, mice were euthanized and the tumors were excised and weighed (Figures 3D and 3E). The tumors from the KitStop i.v. group were significantly smaller than those of the vehicle control group. The spleens showed a trend for reduced size in the KitStop i.v. group (Figure 3F), but liver weight (Figure 3G) and mouse weight during the course of the experiment were not different (Figure 3H), suggesting it is well tolerated. Taken together, these data support the conclusion that systemic (i.e., i.v.) administration of KitStop significantly inhibits tumor growth in an aggressive humanized MC neoplasia model.
Figure 3.

Systemic delivery of human KitStop ESO inhibits tumor growth in a humanized xenograft MC neoplasia model
(A) Schematic representation of the protocol design for the humanized xenograft model used. (B) Examples of tumors in NSG female mice that were injected with 1 × 106 HMC-1.2 cells into a primary site at the right flank subcutaneously. The red arrows indicate the tumor and the green arrows show a secondary growth caused by drag of the needle during inoculation. (C) Measurements of tumor volume over time during treatment (n = 5 mice per group). Arrows indicate days that KitStop or vehicle control was administered. (D) Photograph of tumors after mice were euthanized at day 14. Measurements are in centimeters. (E) Weight of excised tumors after mice were euthanized on day 14. (F) Spleen weight at the conclusion of the experiment. (G) Liver weight at the conclusion of the experiment. (H) Weight of mice was monitored during the course of the experiment. No significant differences were observed during the course of the experiment. Data are the mean ± SEM from five mice per group. ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001, using ANOVA with Dunnett’s post-test (B) or an unpaired t test (D and E).
We found evidence of reduced MC infiltrates in the livers of the mice with almost no lesions in the liver of KitStop i.v.-treated mice (Figures 4A and 4C). Immunohistochemistry (IHC) for KIT receptor confirmed that these lesions were human KIT+ MCs (Figures 4B and 4D). Histopathological assessment of neoplastic liver infiltration as a percentage of tissue (Figure 4E) and as foci size (Figure 4F) both demonstrated a significantly reduced neoplastic burden in the liver. Taken together, these results suggest that KitStop treatment to reduce MC burden in vivo effectively targets MC proliferation and survival in different tissues.
Figure 4.

KitStop ESOs reduce liver infiltration of neoplastic MCs in a humanized xenograft model of MC neoplasia
(A–D) Examples of liver sections from NSG mice showing neoplastic MC infiltrates with H&E (A) and IHC for human KIT (B) in the vehicle control liver and almost no infiltrates in the KitStop-treated livers (C and D). (E) Histopathological assessment of % of neoplastic infiltration in liver. (F) Histopathological assessment of neoplastic foci size in livers. Graphical representation of data includes boxplots where middle bar represents median and whiskers represent minimum and maximum. All data are derived from five NSG female mice per group. The p value was determined by a Fisher’s least significant difference (LSD) test with ordinary one-way ANOVA or an uncorrected Dunn’s test with a Kruskal-Wallis one-way ANOVA. ∗p ≤ 0.05. Scale bars, 2 mm.
KitStop ESO reduced primary tumor growth in an aggressive isograft model of MC neoplasia
We have established that KitStop is effective in vitro and in vivo using HMC-1.2 cells (Figures 1, 2, and 3). We first tested a mouse version of KitStop to reduce KIT expression using mouse BMMCs and found that KitStop was very effective in mouse cells (Figure S10). We next sought to further characterize KitStop efficacy in vivo using an established isograft model of aggressive SM and MCL.46,47 The DBA/2 P815 model produces a solid but diffuse tumor with ill-defined borders, making measurements of tumor volume difficult. Therefore, we stably transduced P815 cells with luciferase to enable measurement of bioluminescence using i.v. injection of d-luciferin and an in vivo imaging system (IVIS).
Using the P815 model of aggressive SM and MCL (Figure 5A), DBA/2J female mice were injected with 5 × 105 bioluminescent P815 cells into a primary site at the right flank. Vehicle control or KitStop ESO was administered every 3 days for 24 days to groups of six mice per treatment cohort. We observed that vehicle control animals had marked signs of primary tumor growth as measured by IVIS (Figures 5B and 5C). Bioluminescence from P815 neoplastic cells was significantly reduced by both intratumoral (i.t.) (p = 0.0007) (Figure 5D) and i.v. (p = 0.0220) (Figure 5E) injection of KitStop ESO when compared to controls. Tumor mass was significantly reduced for i.t. (p = 0.0132) and a trend for reduction in the i.v. (p = 0.0674) KitStop ESO treatment group compared to controls (Figure 5F). Tumor volume was measured with calipers after tumors were excised post-mortem, and these measurements were also reduced for i.t. (p = 0.0188) and i.v. (p = 0.0530) KitStop ESO treatment groups (Figure 5G). Tumor depth, measured by caliper measurements of excised tumor, was significantly reduced for i.t. (p < 0.0001) and i.v. (p = 0.0008) KitStop ESO treatment groups compared to controls (Figure 5H). Finally, tumor necrosis was assessed and expressed as the percent of area of tissue affected by morphological changes characteristic of cell death as assessed by a veterinary pathologist (D.B.S.), and was increased for i.t. (p = 0.0434) and i.v. (p = 0.0588) use with KitStop ESO treatment groups (Figure 5I). Taken together, these data demonstrate that KitStop effectively reduces MC burden in vivo, demonstrating an inverse relationship between tumor size and necrosis induced by KitStop treatment.
Figure 5.

KitStop ESO administration reduced primary tumor growth in an isograft model of MC neoplasia carrying D814Y auto-activating mutation of the c-Kit proto-oncogene
P815 cells, which have the mouse equivalent of the D816V mutation, D814Y, were transduced to make intracellular firefly luciferase providing a bioluminescent reporter that increased relative to neoplastic cell proliferation. (A) DBA/2J female mice (n = 6 per treatment group) were injected with 5 × 105 bioluminescent P815 cells into a primary site at the right flank followed by IVIS to confirm tumor size and subsequent euthanasia for tissue collection at day 24. Vehicle control or KitStop ESO was administered every 3 days for 24 days. (B) Bioluminescence from P815 neoplastic cells was significantly reduced in the i.t. (p = 0.0007) and i.v. (p = 0.0220) KitStop ESO treatment groups compared to controls. (C–E) IVIS images overlay primary site tumor where bioluminescent P815 neoplastic cells were detected. Representative images show equal-sized regions of interest (ROIs) over the primary tumor site standardized area for photon flux (total counts) detected from all mice in the study. (F–I) Tumor weight (F), volume (G), and depth (H) collectively represent decreased primary site tumor burden (F and G) and penetration into underlying structures (H) for the i.t. and i.v. KitStop treatment groups compared to controls. (F) Tumor mass was reduced for the i.t. (p = 0.0132) and i.v. (p = 0.0674) KitStop ESO treatment groups compared to controls. (G) Tumor volume was reduced for the i.t. (p = 0.0188) and i.v. (p = 0.0530) KitStop ESO treatment groups compared to controls. (H) Tumor depth was significantly reduced for the i.t. (p < 0.0001) and i.v. (p = 0.0008) KitStop ESO treatment groups compared to controls. (G) Tumor necrosis was increased for the i.t. (p = 0.0434) and i.v. (p = 0.0588) KitStop ESO treatment groups compared to controls. (B and F–H) Graphical representation of data includes boxplots where middle bar represents median and whiskers represent minimum and maximum. The p value was determined by a Fisher’s LSD test with an ordinary one-way ANOVA or uncorrected Dunn’s test with a Kruskal-Wallis one-way ANOVA. ∗p ≤ 0.05, ∗∗∗p ≤ 0.001, ∗∗∗∗p ≤ 0.0001.
KitStop ESO administration reduced hepatosplenomegaly
Having established that KitStop ESO administration reduces the primary tumor growth in the DBA model, we next assessed neoplastic MC burden in organs. IVIS imaging of organs after necropsy demonstrated that luciferase-positive P815 cells spread from the primary inoculation site to infiltrate organs commonly involved in advanced cases of systemic mastocytosis (Figures 6A–6F). Bioluminescence from P815 neoplastic cells revealed that liver infiltration was significantly reduced by i.t. KitStop ESO compared to controls (Figures 6A, 6B, and 6G). However, the i.v. treated group did not show statistical significance for overall liver luminescence (Figures 6A, 6C, and 6G). Hepatic mass was significantly reduced for i.t. KitStop ESO treatment group compared to controls, which could be related to neoplastic cell infiltration (Figure 6H). Comparable results were found for spleen bioluminescence (Figures 6D–6F and 6I) and weight (Figure 6J).
Figure 6.

KitStop ESO administration reduced hepatosplenomegaly and infiltration in an isograft model of MC neoplasia carrying D814Y auto-activating mutation of the c-Kit proto-oncogene
Luciferase-positive P815 cells spread from the primary injection site to infiltrate organs commonly involved in advanced cases of systemic mastocytosis. (A–F) IVIS images overlay bioluminescence detected from luciferase-positive P815 neoplastic cells. Representative images show equal-sized ROIs over the liver and spleen to standardize the area of photon flux (total counts) detected. (G) Bioluminescence from P815 neoplastic cells infiltrating the liver was significantly reduced by the i.t. (p = 0.0330) KitStop ESO treatment group compared to controls. (H) Hepatic mass was significantly reduced for the i.t. (p = 0.0317) KitStop ESO treatment group compared to controls. (I) Bioluminescence from P815 neoplastic cells infiltrating the spleen was reduced by the i.t. (p = 0.0116) and i.v. (p = 0.0716) KitStop ESO treatment groups compared to controls. (J) Splenic mass was reduced for the i.t. (p = 0.0019) and i.v. (p = 0.0570) KitStop ESO treatment groups compared to controls. (A–F) Luminescence scale is 5–25 counts. (G–J) Graphical representation of data includes boxplots where middle bar represents median and whiskers represent minimum and maximum. The p value was determined by a Fisher’s LSD test with ordinary one-way ANOVA or an uncorrected Dunn’s test with a Kruskal-Wallis one-way ANOVA. ∗p ≤ 0.05, ∗∗p ≤ 0.01. Scale bars, 0.5 cm.
Analysis of multiple tissues at necropsy confirmed that cells in multicentric neoplasia spread from the primary tumor site to infiltrate organs commonly involved in advanced cases of systemic mastocytosis, including liver, spleen, the gastrointestinal tract, lymph nodes, and occasionally other tissues. Grossly detectable neoplastic MC infiltration replaced hepatic parenchyma (Figures 7A–7C, inset, arrowheads) in a pale cream-colored multifocal to coalescing distribution (i.e., irregular areas) (Figure 7A) and discrete nodules (Figure 7C). Quantification revealed that the percent of liver surface replaced by neoplastic MCs was significantly reduced for the i.t. (p = 0.0153) KitStop ESO treatment group compared to controls. Histopathology sections of liver showed infiltration by neoplastic cells in multifocal to coalescing distribution (Figure 7E) and multiple discrete variable-sized foci (Figures 7F and 7G), with occasional neoplastic MCs evident within blood vessels. Infiltrates were significantly reduced for i.t. (p = 0.0067) and i.v. (p = 0.0042) KitStop ESO treatment groups compared to controls (Figure 7H). Histopathology sections of spleen revealed infiltration by neoplastic cells as individualized cells and multifocal coalescing distribution infiltrating red (Figures 7I–7K) and white (Figures 7I and 7K) pulp with occasional neoplastic MCs within vessels (Figure 7I, inset). The infiltration of neoplastic MCs into spleen was significantly reduced for the i.t. (p = 0.0201) KitStop ESO treatment group compared to controls (Figure 7L).
Figure 7.

KitStop ESO administration reduced multicentric spread to organs and tissues in an isograft model of mast cell neoplasia carrying D814Y auto-activating mutation of the c-Kit proto-oncogene
Multicentric neoplasia cells spread from the primary tumor site to infiltrate organs commonly involved in advanced cases of systemic mastocytosis, including liver, spleen, gastrointestinal tract, lymph nodes, and occasionally other tissues. (A–C) Representative images show grossly detectable neoplastic mast cell infiltration-replaced hepatic parenchyma (inset, arrowheads) as pale cream-colored irregular zones and nodules. (D) Percent of liver surface replaced by neoplastic mast cells was reduced for the i.t. (p = 0.0153) KitStop ESO treatment group compared to controls. (E–G) Histopathology sections of liver show infiltration by neoplastic cells in multifocal coalescing distribution in vehicle mice (E) and with KitStop treatment had multiple discrete foci (F and G). (H) Infiltration of neoplastic mast cells into liver was reduced for the i.t. (p = 0.0067) and i.v. (p = 0.0042) KitStop ESO treatment groups compared to controls. (I–K) Histopathology sections of spleen had infiltration by neoplastic cells as individualized cells and multifocal coalescing distribution infiltrating red (I–K) and white (I and K) pulp. (L) Infiltration of neoplastic mast cells into spleen was reduced for the i.t. (p = 0.0201) KitStop ESO treatment group compared to controls. (D, H, and L) Graphical representation of data includes boxplots where middle bar represents median and whiskers represent minimum and maximum. The p value was determined by a Fisher’s LSD test with an ordinary one-way ANOVA or uncorrected Dunn’s test with a Kruskal-Wallis one-way ANOVA. ∗p ≤ 0.05, ∗∗p ≤ 0.01. Scale bars, 0.5 cm.
KitStop ESO administration reduced MCL
Next, we examined the P815 neoplastic MC involvement in bone marrow (Figures 8A–8D). We identified marked infiltration of neoplastic MCs within the bone marrow, and the percent of bone marrow infiltrated by P815 neoplastic cells was significantly reduced in the i.t. (p = 0.0149) KitStop ESO treatment group compared to controls. High numbers of neoplastic MCs infiltrated, replaced, and effaced bone marrow for the vehicle control (Figure 8B) compared to less apparent infiltration for i.t. (Figure 8C) and i.v. (Figure 8D) KitStop ESO administration. Displacement of hematopoietic bone marrow tissue (myelophthisis) by neoplastic MCs was reduced for i.t. (p = 0.0095) and i.v. (p = 0.0333) KitStop ESO treatment groups compared to controls (Figure 8E).
Figure 8.

KitStop ESO administration reduced cardinal signs of mast cell leukemia in an isograft model of mutant mast cell neoplasia
(A) Percent of bone marrow infiltrated by P815 neoplastic cells. (B) Representative micrograph of bone marrow from vehicle controls (B) compared to i.t. (C) and i.v. (D) Kitstop ESO administration. (E) Displacement of hematopoietic bone marrow tissue (myelophthisis) by neoplastic mast cells. (F) Packed red blood cell volume (hematocrit). Some animals from each group had a hematocrit below the lower 2SD reference interval (dotted line) for female DBA/2J mice. (G) Bone marrow hematopoiesis as measured by the myeloid progenitor-to-erythroid progenitor (M:E) ratio. (H) Circulating neoplastic mast cells on differential counts. (I) Blood smear of neoplastic mast cells with Romanowsky stain (representative image shown from vehicle control animal blood). (J) H&E histopathology assessment of neoplastic emboli and circulating neoplastic cells in organs. (K–L) Emboli of neoplastic mast cells in pulmonary vessels from H&E histopathology sections from control (K) and i.v. KitStop ESO (L) treatment groups. (A and F–H) Graphical representation of data includes boxplots where middle bar represents median and whiskers represent minimum and maximum. The p value was determined by a Fisher’s LSD test with an ordinary one-way ANOVA or uncorrected Dunn’s test with a Kruskal-Wallis one-way ANOVA. (E and J) Graphical representation of data includes mean and SEM. The p values were determined independently by a Mann-Whitney test. ∗p ≤ 0.05, ∗∗p ≤ 0.01. Scale bars, 100 μm.
Because c-KIT is involved in hematopoiesis and erythropoiesis,48 we assessed basic factors that represent these processes. Packed red blood cell volume (hematocrit) was not significantly different for i.t. or i.t. treatment groups compared to controls (Figure 8F). Bone marrow hematopoiesis as measured by the myeloid progenitor-to-erythroid progenitor (M:E) ratio was within reference intervals and not significantly different between groups (Figure 8G).
We also determined that the blood and MC numbers exceeding the World Health Organization criteria for diagnosis of MCL in humans were present in the vehicle control group (Figure 8H). Circulating neoplastic MCs on differential counts were significantly reduced by i.t. KitStop ESO treatment compared to controls (Figure 8H). Neoplastic emboli, neoplasm encroachment on vessels, and circulating neoplastic MC prevalence was reduced on H&E histopathology sections of organs for the i.t. (p = 0.0238) and i.v. (p = 0.0022) KitStop ESO treatment groups compared to controls (Figures 8I and 8K). Intravascular clusters and individual neoplastic MCs and emboli filling the vascular lumen in pulmonary vessels were evident in H&E histopathology sections from vehicle control lungs (Figure 8K), and lymphatics carried neoplastic MCs to the subcapsular sinus (arrowheads) of draining lymph nodes (Figure 8L). These features were rarely observed in KitStop-treated mice (Figure 8J).
Discussion
This study presents and summarizes an innovative approach to treat MC neoplastic diseases by targeting KIT mRNA splicing with an ESO we term KitStop, markedly reducing KIT protein levels and function in neoplastic MCs. By employing chemically stable ESOs that introduce a frameshift into mature mRNA, we propose that we induce both degradation of mRNA by NMD and production of a truncated mRNA variant that encodes a small non-functional peptide. Thus, if saturation of mRNA degradation does occur, or if the transcripts evade premature termination codon-dependent NMD, it would not reduce efficacy because the result would be production of non-functional mRNA and protein. We established that KitStop is effective in vitro and reduces neoplastic MC numbers in vivo in two separate MC tumor models without obvious associated pathologies or defects in hematopoiesis.
Using the P815 model of aggressive SM and MCL, we observed vehicle control animals that had multiple measurable signs of primary tumor proliferation and proliferation of neoplastic MCs at distant sites including liver, spleen, gastrointestinal tract, lymph nodes, and other tissues. Vehicle control mice had consistently larger tumors at the primary site with increased tumor bioluminescence, increased hepatic bioluminescence, measurable hepatomegaly, increased neoplastic cell infiltration into liver, increased splenic bioluminescence, measurable splenomegaly, increased neoplastic cell infiltration into spleen, and infiltration into multiple organs, when compared to KitStop-treated mice. Notably, vehicle controls had MCL when measured by World Health Organization (WHO) criteria for human samples, including the bone marrow and blood smears. Additionally, histopathology slides from multiple tissues had neoplastic MCs in vessels and lymph node subcapsular sinus confirming neoplastic MC circulation in afferent lymphatic fluid-draining tissues. These histopathological features of this model were all improved with KitStop treatment.
MC proliferative diseases are heterogeneous and difficult to treat relating to variation in age of onset, patient age, organ involvement, co-morbidities, and disease aggressiveness.8 While conservative treatment is appropriate for cutaneous mastocytosis, increasingly specific treatments are necessary for aggressive forms of mastocytosis and are generally directed at inhibiting common mutations found within KIT. However, the diversity of c-KIT mutations that underlie oncogenic KIT signaling makes targeting such mutations a challenge. Among these mutations, KIT D816V, which is present in most systemic mastocytosis patients, confers resistance to the TKI imatinib mesylate (Gleevec) by inducing a conformational change in the tyrosine kinase domain of KIT.49,50 This has led to the development of additional TKIs that target KIT D816V. One such TKI approved for use is midostaurin, which exhibits high response rates. However, there remains risk of disease progression and transformation into leukemia even after treatment with midostaurin.8,10 An emerging therapeutic that may also provide promise is avapritinib, which has been demonstrated to more selectively target KIT, can inhibit KIT with the D816V mutation, and resulted in a reduction of >50% neoplastic colony formation in 64% of patients with ASM.20 However, 36% of the cohort were resistant to avapritinib.
Other neoplastic disorders such as GISTs, in which somatic gain-of-function mutations in c-KIT are common and predictive of the risk of metastasis,51,52 can be treated with imatinib. However, similarly to mastocytosis, imatinib sensitivity can be lost over time with the acquisition of additional c-KIT mutations.51,53 Combinations of TKIs can have synergistic inhibitory effects on neoplastic MC growth and could have therapeutic benefit. However, the risk of acquired resistance to TKIs and off-target toxicity remain.54,55 Consequently, given the strong association between aberrant MC proliferation and survival with activating c-KIT mutations, another and additional desirable approach for treatment of these diseases would be one capable of specifically targeting KIT expression with high efficacy, regardless of the many possible mutations and conformations of the receptor.
For selective targeting of KIT expression, ESO technology represents a promising approach if the challenge of efficient delivery with therapeutic ESOs can be overcome. Although further development is needed for ASO delivery, progress with new generations of chemical modifications that improve oligonucleotide stability, bioavailability, efficacy, and delivery are exciting advances in ESO technology.56, 57, 58 Antisense oligonucleotides, including ESOs, are versatile and, depending on their design, are capable of performing a wide range of gene-manipulating applications. For example, some applications of antisense oligonucleotides are to block protein translation, promote exon inclusion, or induce exon skipping of mature mRNA. In this study, we used ESOs to achieve the latter and utilized this unconventional approach to cause a frameshift in the mRNA and introduce a premature stop codon. In doing so, we have not only prohibited KIT expression, but also significantly inhibited KIT-dependent neoplastic MC growth and survival in vitro and in vivo. In neoplastic HMC-1.2 cells, KitStop downregulated intracellular KIT expression, which is particularly significant because the D816V mutation eliminates the need for surface expression of KIT. Instead, oncogenic KIT signaling occurs intracellularly and independently of ligand binding,7,42 and thus lack of mutant KIT expression at the surface negates antibody-based therapeutic approaches. In addition to our in vitro findings, we established that i.t. and i.v. administration of KitStop reduced tumor growth and, in the case of intratumoral administration, could actually eliminate the tumors. Furthermore, we established that systemic administration of KitStop significantly reduced tumor development in a humanized xenograft MC neoplasia model, suggesting that the approach would translate to humans. Oligonucleotides have some weaknesses in distributing to tissues. While the i.v. administration did not eliminate the tumor, i.t. delivery did eliminate the tumor and shows that with effective delivery of the ESOs to MCs, we could further improve efficacy.
Two key reasons for choosing to modify pre-mRNA splicing, rather than target KIT mRNA using conventional siRNA approaches are, first, the sheer abundance of KIT transcripts expressed by MCs and, second, the inefficacy of siRNA approaches on MCs, particularly in vivo. Although KitStop targeting of KIT mRNA did not result in a full switch into skipped transcripts in human and mouse MCs when measured by standard RT-PCR, this level of exon skipping was sufficient to reduce by 30% KIT mRNA species (including skipped and truncated mRNA transcripts), as well as a markedly reduced KIT protein expression, loss of proliferation, and induction of apoptosis and cell death. These effects on reduction of KIT mRNA levels and protein expression were even more marked in slowly dividing MCs, as evidenced in LAD-2 cells. While it is not yet clear whether KitStop results in production of a truncated protein, RT-PCR data suggest that there is production of truncated mRNA transcripts, and only partial reduction in total transcripts despite a marked reduction in protein expression, possibly after NMD is saturated or transcripts evade NMD. Therefore, if the splicing-dependent NMD pathway is evaded or saturated, KitStop may then result in production of a severely truncated C-terminal protein that would be difficult to detect, similar to what has been observed in CRISPR-Cas systems that introduce a frameshift into transcripts.59 With high copy number non-desirable transcripts, such as mutant KIT, this phenomenon could be critical for therapeutic success.
Further reasoning for our choice to use ESOs to target gene expression rather than conventional gene silencing approaches are the therapeutic and safety profile and prior success of therapeutic SSOs for genetic diseases such as DMD. Multiple relevant examples to our choice of PMO chemical backbone for KitStop have been recently approved by the FDA as new drugs. Eteplirsen (Exondys 51),35,36 golodirsen (Vyondys 53),60 casimersen (Amondys 45),61 and viltolarsen (Viltepso 53)62 are all PMOs that cause exon skipping of various target exons in the dystrophin gene to treat specific exon mutations in DMD. The basis of these drugs is to utilize splice switching and exon skipping to partially restore protein function in the dystrophin gene mutated in DMD by restoring the reading frame in the mRNA. While these examples provide strong support for therapeutic utility of PMOs and i.v. administration, they also highlight the burden on development of the classic personalized medicine approach with ESOs, where each exon mutation requires a different drug. Our unconventional use of ESOs reported in this study to introduce a frameshift, rather than correct a frameshift, would be less personalized and thus developing the approach would be more direct.
In summary, our in vitro and in vivo data are a proof of concept for developing ESO-induced frameshifting of KIT mRNA as an additional therapeutic approach for the treatment for MC neoplastic diseases. As we have discussed,37 ESOs represent reversible gene-targeting therapies, thereby avoiding irreversible alteration of the genome. The nonsense frameshift induced by KitStop exemplifies a promising additional approach for treatment of MC neoplasia and possibly GISTs. This KIT-specific therapeutic strategy has translational potential for clinical applications. Furthermore, c-KIT plays a role in other types of cancer, including those that are KIT driven, such as certain melanomas, and those that acquire secondary c-KIT mutations, such as small cell lung cancer and brain tumors, in which c-KIT mutations subsequently contribute to tumor progression.1 Further study is required to optimize dosing, establish whether resistant clones can be developed after longer treatment regimens, and determine the effects of longer treatment on hematopoiesis. However, this study provides proof-of-concept data for the application of KitStop ESOs as a therapeutic approach. The therapeutic potential of ESOs used to alter mRNA splice variants and protein isoforms have already been highlighted in diseases such as hypercholesterolemia and cardiovascular disease,63 breast cancer,64 and allergy.37 Although improvements can be made toward specific delivery platforms for ESOs, the recent approval of the ESO for the treatment of DMD (eteplirsen)35,36 demonstrates the increased likelihood that other ESOs will emerge as personalized and targeted therapeutics.
Materials and methods
Animal studies
Mice were housed in American Association for the Accreditation of Laboratory Animal Care (AAALAC)-accredited National Institute of Allergy and Infectious Diseases (NIAID) or North Carolina State University animal facilities, and experimental protocols with live mice were performed under an animal study proposal (LAD2E) approved by the NIAID Division of Intramural Research (DIR) Animal Care and Use Committee under the guidance of the Office of Animal Care and Use of the National Institutes of Health and North Carolina State University laboratory animal care protocol (17-108-B) approved by an Institutional Animal Care and Use Committee (IACUC).
Transfection of MCs with ESOs
The HMC-1.2 human MC line was purchased from MilliporeSigma (Burlington, MA, USA) and cultured according to the manufacturer’s instructions. For transfection of MCs with ESOs, 2 × 106 HMC-1.2 cells were used for each transfection. The human MC line LAD2 was obtained from Dr. A.S. Kirshenbaum from the Laboratory of Allergic Diseases (NIAID, NIH) and cultured as described.38 BMMCs were obtained as described.65 Transfection was performed as described,66 and efficiency was determined using FITC-conjugated ESOs (Gene Tools). Cell viability was monitored during experiments, and there was no evidence of cytotoxicity with control (non-targeting) ESO transfection.
ESO design
KitStop ESO was designed to target exon 4 of human c-KIT (GenBank: NM_000222.2). A region within the splicing donor site was targeted with the following sequence for human c-KIT: 5′-GAATGAAGCGATTCACTCACCTGAC-3′. For murine c-Kit, the following ESO was used: 5′-AGGACTTAAACAGCACTCACCTGAG-3′. All oligonucleotides sequences were BLAST searched for specificity and were purchased from Gene Tools. The unconjugated standard control ESO provided by Gene Tools had the following sequence: 5′-CCTCTTACCTCAGTTACAATTTATA-3′. For in vivo studies, morpholino ESOs linked through the terminal 3′-N to an octaguanidinium dendrimer (Vivo-Morpholino) were purchased from Gene Tools.
RT-PCR
Total RNA was harvested using the RNeasy plus mini-kit (QIAGEN) according to the manufacturer’s instructions with inclusion of the QIAshredder step. The primers used were designed to amplify exon 4 and the surrounding exons and spanned multiple exons. For human c-KIT mRNA, the following primers were used: forward, 5′-GAAGCCTCTTCCCAAGGACT-3′, reverse, 5′-GTGTTCAGGTTTGGGGAATG-3′. For murine c-Kit mRNA, the following primers were used: forward, 5′-TCATCGAGTGTGATGGGAAA-3′, reverse, 5′-TCACAGGGGAGATGTTGATG-3′.
Receptor expression
For human MCs, flow cytometry for total KIT expression was performed at each time point with allophycocyanin (APC)-conjugated anti-human CD117 monoclonal antibodies (clone 104D2) alongside APC-conjugated mouse IgG1, κ, isotype control antibodies (BioLegend, San Diego, CA, USA).
Apoptosis and viability assays
At each time point, cells were pelleted and analyzed for apoptosis with FITC-annexin V (eBioscience). To assess viability, cells were stained with Live/Dead green dead cell stain (Invitrogen), according to the manufacturer’s instructions, or PI. Flow cytometry was performed on a CytoFLEX flow cytometer (Beckman Coulter).
Proliferation assays
Cells were stained with 1 μM CellTrace Far Red (Invitrogen) at a concentration of 1 × 106 cells/mL, according to the manufacturer’s instructions. Briefly, cells were stained for 20 min at 37°C, followed by an addition of 5× vol of culture media for 5 min. Cells were pelleted, then resuspended in complete medium and incubated for 10 min at 37°C to allow the CellTrace reagent to undergo acetate hydrolysis. Stained cells were transfected with standard control ASO or KitStop ESO. At each designated time point, cells were stained with Live/Dead green dead cell stain. Proliferating cells were identified with an additional gate set for the single-cell population. Flow cytometry was performed on a CytoFLEX flow cytometer. Trypan blue was also used routinely to assess viable cell counts across different time points.
Bioluminescence assay in isograft model of aggressive SM and MCL
The neoplastic mouse MC line derived from DBA/2 mouse liver, P815, was selected for the isograft model because these cells contain a c-KitD814Y mutation. The mutation at c-Kit codon 814 is the mouse equivalent of human c-KIT codon 816 mutation, and both result in constitutive signaling of the Kit/KIT receptor present in the overwhelming majority of clinical cases of SM and nearly 100% of aggressive SM/MCL. P815 cells were transduced to make intracellular firefly luciferase, providing a bioluminescent reporter that increased relative to neoplastic cell proliferation. Cytomegalovirus (CMV)-firefly luciferase lentivirus with neomycin selection marker (Cellomics Technology) was applied to rapidly dividing P815 cells cultured to 5 × 105 to 1 × 106 cells/mL of routine culture media composed of DMEM and 10% fetal bovine serum (FBS) (v/v) in the presence of 2 μg/mL Polybrene for 12 h. Cells were pelleted by gentle centrifugation at 100 × g for 5 min at room temperature and subsequently cultured without antibiotics for 48 h in routine culture media composed of DMEM and 10% FBS (v/v). The G418 antibiotic kill curve was established by culturing non-transduced cells in media ranging from 0.5 to 1.5 mg/mL to select for transduced cells with complete killing of all non-transduced cells, including controls (1.4 mg/mL G418 in this case). After transduction, cells were maintained in 1 mg/mL G418 added to DMEM-based culture media that included 10% FBS (v/v) along with glutamine, pyruvate, and essential amino acids.
DBA/2J female mice were injected with 5 × 105 bioluminescent P815 cells into a primary site at the right flank. Vehicle control or KitStop ESO was administered every 3 days for 24 days. More specifically, 12.5 mg/kg KitStop ESO was administered by either i.v. injection or i.t. injection for mice in each respective treatment group. The hair was gently removed with clippers over the primary tumor site to allow visualization and subsequent photon flux measurements. Bioluminescence from P815 neoplastic cells was measured by IVIS (PerkinElmer), an imaging system that measures photon flux, 15 min after i.v. injection with 100 μL of 100 mg/mL solution containing d-luciferin (Pierce, Thermo Fisher Scientific) and produces bioluminescence measurements low in background compared to other in vivo visualization modalities.
IVIS images confirmed tumor size at 16 days and organ infiltration at 24 days. After bioluminescence detection, mice were euthanized for tissue collection at day 24 to include organ weights and preparation of tissues for histopathology.
Xenograft model of aggressive SM
HMC-1.2 cells were selected because the cell line contains the c-KitD816V mutation that leads to auto-activation of KIT receptor present in the overwhelming majority of clinical cases of SM and nearly 100% of aggressive SM/MCL. NSG female mice were injected with 1 × 106 HMC-1.2 cells into a primary site at the right flank subcutaneously. Once the tumor volume reached 50 mm3 (day 0), KitStop was administered by i.v. injection of 8.3 mg/kg once a day every 3 days and compared to vehicle control. Tumor volumes were measured daily. Mice were euthanized for tissue collection at day 14 to include organ weights and preparation of tissues for histopathology.
Acknowledgments
We thank Alicia G. Schubert (NCSU, CVM) for technical assistance. This work was supported by Department of Molecular Biomedical Sciences, College of Veterinary Medicine, start-up funds (to G.C.), Institutional National Research Service Award from the Office of the Director of National Institutes of Health (NIH) T32OD011130 (fellowship to D.B.S.), and by the Division of Intramural Research of NIAID within the National Institutes of Health (to G.H.F., A.O., and D.D.M.).
Author contributions
D.B.S. performed and designed experiments, analyzed data, prepared micrographs, and contributed to manuscript; G.K.A. performed and designed experiments, analyzed data, prepared figures, and wrote the manuscript; G.H.F. performed experiments, analyzed data, and prepared figures; A.O. supervised studies, designed experiments, assisted with experiments, and edited the manuscript; L.C.E.-H performed experiments and analyzed data; E.S. and C.S. assisted with experiments; D.D.M. supervised studies and edited the manuscript; and G.C. conceived and designed the studies, directed and supervised the research, and wrote the manuscript.
Declaration of interests
G.C. has filed a patent application related to the research reported in this study. G.C. has research support from Hoth Therapeutics for a project not directly related to the research reported in this publication and also serves on their Scientific Advisory Board. The research findings included in this publication were not funded by Hoth Therapeutics, but a technology licensing agreement has been granted to Hoth Therapeutics for this technology after submission and while this study was under review. The terms of this arrangement have been reviewed and approved by North Carolina State University in accordance with its policy on objectivity in research. The remaining authors declare no competing interests.
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.ymthe.2021.08.009.
Supplemental information
References
- 1.Pittoni P., Piconese S., Tripodo C., Colombo M.P. Tumor-intrinsic and -extrinsic roles of c-Kit: Mast cells as the primary off-target of tyrosine kinase inhibitors. Oncogene. 2011;30:757–769. doi: 10.1038/onc.2010.494. [DOI] [PubMed] [Google Scholar]
- 2.Tsai M., Takeishi T., Thompson H., Langley K.E., Zsebo K.M., Metcalfe D.D., Geissler E.N., Galli S.J. Induction of mast cell proliferation, maturation, and heparin synthesis by the rat c-kit ligand, stem cell factor. Proc. Natl. Acad. Sci. USA. 1991;88:6382–6386. doi: 10.1073/pnas.88.14.6382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Mekori Y.A., Oh C.K., Metcalfe D.D. IL-3-dependent murine mast cells undergo apoptosis on removal of IL-3. Prevention of apoptosis by c-kit ligand. J. Immunol. 1993;151:3775–3784. [PubMed] [Google Scholar]
- 4.Iemura A., Tsai M., Ando A., Wershil B.K., Galli S.J. The c-kit ligand, stem cell factor, promotes mast cell survival by suppressing apoptosis. Am. J. Pathol. 1994;144:321–328. [PMC free article] [PubMed] [Google Scholar]
- 5.Galli S.J., Tsai M., Wershil B.K., Tam S.Y., Costa J.J. Regulation of mouse and human mast cell development, survival and function by stem cell factor, the ligand for the c-kit receptor. Int. Arch. Allergy Immunol. 1995;107:51–53. doi: 10.1159/000236928. [DOI] [PubMed] [Google Scholar]
- 6.Besmer P., Murphy J.E., George P.C., Qiu F.H., Bergold P.J., Lederman L., Snyder H.W., Jr., Brodeur D., Zuckerman E.E., Hardy W.D. A new acute transforming feline retrovirus and relationship of its oncogene v-kit with the protein kinase gene family. Nature. 1986;320:415–421. doi: 10.1038/320415a0. [DOI] [PubMed] [Google Scholar]
- 7.Cruse G., Metcalfe D.D., Olivera A. Functional deregulation of KIT: Link to mast cell proliferative diseases and other neoplasms. Immunol. Allergy Clin. North Am. 2014;34:219–237. doi: 10.1016/j.iac.2014.01.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Valent P., Akin C., Hartmann K., Nilsson G., Reiter A., Hermine O., Sotlar K., Sperr W.R., Escribano L., George T.I., et al. Advances in the classification and treatment of mastocytosis: Current status and outlook toward the future. Cancer Res. 2017;77:1261–1270. doi: 10.1158/0008-5472.CAN-16-2234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Gleixner K.V., Mayerhofer M., Aichberger K.J., Derdak S., Sonneck K., Böhm A., Gruze A., Samorapoompichit P., Manley P.W., Fabbro D., et al. PKC412 inhibits in vitro growth of neoplastic human mast cells expressing the D816V-mutated variant of KIT: comparison with AMN107, imatinib, and cladribine (2CdA) and evaluation of cooperative drug effects. Blood. 2006;107:752–759. doi: 10.1182/blood-2005-07-3022. [DOI] [PubMed] [Google Scholar]
- 10.Gotlib J., Kluin-Nelemans H.C., George T.I., Akin C., Sotlar K., Hermine O., Awan F.T., Hexner E., Mauro M.J., Sternberg D.W., et al. Efficacy and safety of midostaurin in advanced systemic mastocytosis. N. Engl. J. Med. 2016;374:2530–2541. doi: 10.1056/NEJMoa1513098. [DOI] [PubMed] [Google Scholar]
- 11.Jensen B.M., Akin C., Gilfillan A.M. Pharmacological targeting of the KIT growth factor receptor: A therapeutic consideration for mast cell disorders. Br. J. Pharmacol. 2008;154:1572–1582. doi: 10.1038/bjp.2008.204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Giebel L.B., Strunk K.M., Holmes S.A., Spritz R.A. Organization and nucleotide sequence of the human KIT (mast/stem cell growth factor receptor) proto-oncogene. Oncogene. 1992;7:2207–2217. [PubMed] [Google Scholar]
- 13.Arock M., Sotlar K., Akin C., Broesby-Olsen S., Hoermann G., Escribano L., Kristensen T.K., Kluin-Nelemans H.C., Hermine O., Dubreuil P., et al. KIT mutation analysis in mast cell neoplasms: Recommendations of the European Competence Network on Mastocytosis. Leukemia. 2015;29:1223–1232. doi: 10.1038/leu.2015.24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Taniguchi M., Nishida T., Hirota S., Isozaki K., Ito T., Nomura T., Matsuda H., Kitamura Y. Effect of c-kit mutation on prognosis of gastrointestinal stromal tumors. Cancer Res. 1999;59:4297–4300. [PubMed] [Google Scholar]
- 15.Miettinen M., Majidi M., Lasota J. Pathology and diagnostic criteria of gastrointestinal stromal tumors (GISTs): A review. Eur. J. Cancer. 2002;38(Suppl 5):S39–S51. doi: 10.1016/s0959-8049(02)80602-5. [DOI] [PubMed] [Google Scholar]
- 16.Nagata H., Worobec A.S., Oh C.K., Chowdhury B.A., Tannenbaum S., Suzuki Y., Metcalfe D.D. Identification of a point mutation in the catalytic domain of the protooncogene c-kit in peripheral blood mononuclear cells of patients who have mastocytosis with an associated hematologic disorder. Proc. Natl. Acad. Sci. USA. 1995;92:10560–10564. doi: 10.1073/pnas.92.23.10560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Garcia-Montero A.C., Jara-Acevedo M., Teodosio C., Sanchez M.L., Nunez R., Prados A., Aldanondo I., Sanchez L., Dominguez M., Botana L.M., et al. KIT mutation in mast cells and other bone marrow hematopoietic cell lineages in systemic mast cell disorders: a prospective study of the Spanish Network on Mastocytosis (REMA) in a series of 113 patients. Blood. 2006;108:2366–2372. doi: 10.1182/blood-2006-04-015545. [DOI] [PubMed] [Google Scholar]
- 18.Ma Y., Zeng S., Metcalfe D.D., Akin C., Dimitrijevic S., Butterfield J.H., McMahon G., Longley B.J. The c-KIT mutation causing human mastocytosis is resistant to STI571 and other KIT kinase inhibitors; kinases with enzymatic site mutations show different inhibitor sensitivity profiles than wild-type kinases and those with regulatory-type mutations. Blood. 2002;99:1741–1744. doi: 10.1182/blood.v99.5.1741. [DOI] [PubMed] [Google Scholar]
- 19.Pardanani A., Elliott M., Reeder T., Li C.Y., Baxter E.J., Cross N.C., Tefferi A. Imatinib for systemic mast-cell disease. Lancet. 2003;362:535–536. doi: 10.1016/s0140-6736(03)14115-3. [DOI] [PubMed] [Google Scholar]
- 20.Lübke J., Naumann N., Kluger S., Schwaab J., Metzgeroth G., Evans E., Gardino A.K., Lengauer C., Hofmann W.K., Fabarius A., et al. Inhibitory effects of midostaurin and avapritinib on myeloid progenitors derived from patients with KIT D816V positive advanced systemic mastocytosis. Leukemia. 2019;33:1195–1205. doi: 10.1038/s41375-019-0450-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Lundin K.E., Gissberg O., Smith C.I. Oligonucleotide therapies: The past and the present. Hum. Gene Ther. 2015;26:475–485. doi: 10.1089/hum.2015.070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Singerman L. Combination therapy using the small interfering RNA bevasiranib. Retina. 2009;29(6, Suppl):S49–S50. doi: 10.1097/IAE.0b013e3181ad2341. [DOI] [PubMed] [Google Scholar]
- 23.Potaczek D.P., Garn H., Unger S.D., Renz H. Antisense molecules: A new class of drugs. J. Allergy Clin. Immunol. 2016;137:1334–1346. doi: 10.1016/j.jaci.2015.12.1344. [DOI] [PubMed] [Google Scholar]
- 24.Crooke S.T., Liang X.H., Baker B.F., Crooke R.M. Antisense technology: A review. J. Biol. Chem. 2021;296:100416. doi: 10.1016/j.jbc.2021.100416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Crooke S.T., Baker B.F., Crooke R.M., Liang X.H. Antisense technology: An overview and prospectus. Nat. Rev. Drug Discov. 2021;20:427–453. doi: 10.1038/s41573-021-00162-z. [DOI] [PubMed] [Google Scholar]
- 26.Verbeke R., Lentacker I., De Smedt S.C., Dewitte H. The dawn of mRNA vaccines: The COVID-19 case. J. Control. Release. 2021;333:511–520. doi: 10.1016/j.jconrel.2021.03.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Lamb Y.N. BNT162b2 mRNA COVID-19 vaccine: First approval. Drugs. 2021;81:495–501. doi: 10.1007/s40265-021-01480-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Havens M.A., Hastings M.L. Splice-switching antisense oligonucleotides as therapeutic drugs. Nucleic Acids Res. 2016;44:6549–6563. doi: 10.1093/nar/gkw533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Zhang A., Uaesoontrachoon K., Shaughnessy C., Das J.R., Rayavarapu S., Brown K.J., Ray P.E., Nagaraju K., van den Anker J.N., Hoffman E.P., Hathout Y. The use of urinary and kidney SILAM proteomics to monitor kidney response to high dose morpholino oligonucleotides in the mdx mouse. Toxicol. Rep. 2015;2:838–849. doi: 10.1016/j.toxrep.2015.05.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Sazani P., Weller D.L., Shrewsbury S.B. Safety pharmacology and genotoxicity evaluation of AVI-4658. Int. J. Toxicol. 2010;29:143–156. doi: 10.1177/1091581809359206. [DOI] [PubMed] [Google Scholar]
- 31.Sazani P., Ness K.P., Weller D.L., Poage D., Nelson K., Shrewsbury A.S. Chemical and mechanistic toxicology evaluation of exon skipping phosphorodiamidate morpholino oligomers in mdx mice. Int. J. Toxicol. 2011;30:322–333. doi: 10.1177/1091581811403504. [DOI] [PubMed] [Google Scholar]
- 32.Sazani P., Ness K.P., Weller D.L., Poage D.W., Palyada K., Shrewsbury S.B. Repeat-dose toxicology evaluation in cynomolgus monkeys of AVI-4658, a phosphorodiamidate morpholino oligomer (PMO) drug for the treatment of duchenne muscular dystrophy. Int. J. Toxicol. 2011;30:313–321. doi: 10.1177/1091581811403505. [DOI] [PubMed] [Google Scholar]
- 33.Nakamura A., Takeda S. Exon-skipping therapy for Duchenne muscular dystrophy. Neuropathology. 2009;29:494–501. doi: 10.1111/j.1440-1789.2009.01028.x. [DOI] [PubMed] [Google Scholar]
- 34.Nakamura A. Moving towards successful exon-skipping therapy for Duchenne muscular dystrophy. J. Hum. Genet. 2017;62:871–876. doi: 10.1038/jhg.2017.57. [DOI] [PubMed] [Google Scholar]
- 35.Syed Y.Y. Eteplirsen: First global approval. Drugs. 2016;76:1699–1704. doi: 10.1007/s40265-016-0657-1. [DOI] [PubMed] [Google Scholar]
- 36.Dowling J.J. Eteplirsen therapy for Duchenne muscular dystrophy: Skipping to the front of the line. Nat. Rev. Neurol. 2016;12:675–676. doi: 10.1038/nrneurol.2016.180. [DOI] [PubMed] [Google Scholar]
- 37.Cruse G., Yin Y., Fukuyama T., Desai A., Arthur G.K., Bäumer W., Beaven M.A., Metcalfe D.D. Exon skipping of FcεRIβ eliminates expression of the high-affinity IgE receptor in mast cells with therapeutic potential for allergy. Proc. Natl. Acad. Sci. USA. 2016;113:14115–14120. doi: 10.1073/pnas.1608520113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Kirshenbaum A.S., Akin C., Wu Y., Rottem M., Goff J.P., Beaven M.A., Rao V.K., Metcalfe D.D. Characterization of novel stem cell factor responsive human mast cell lines LAD 1 and 2 established from a patient with mast cell sarcoma/leukemia; activation following aggregation of FcεRI or FcγRI. Leuk. Res. 2003;27:677–682. doi: 10.1016/s0145-2126(02)00343-0. [DOI] [PubMed] [Google Scholar]
- 39.Lykke-Andersen J., Bennett E.J. Protecting the proteome: Eukaryotic cotranslational quality control pathways. J. Cell Biol. 2014;204:467–476. doi: 10.1083/jcb.201311103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Mugridge J.S., Coller J., Gross J.D. Structural and molecular mechanisms for the control of eukaryotic 5′-3′ mRNA decay. Nat. Struct. Mol. Biol. 2018;25:1077–1085. doi: 10.1038/s41594-018-0164-z. [DOI] [PubMed] [Google Scholar]
- 41.Wiley H.S., Burke P.M. Regulation of receptor tyrosine kinase signaling by endocytic trafficking. Traffic. 2001;2:12–18. doi: 10.1034/j.1600-0854.2001.020103.x. [DOI] [PubMed] [Google Scholar]
- 42.Obata Y., Toyoshima S., Wakamatsu E., Suzuki S., Ogawa S., Esumi H., Abe R. Oncogenic Kit signals on endolysosomes and endoplasmic reticulum are essential for neoplastic mast cell proliferation. Nat. Commun. 2014;5:5715. doi: 10.1038/ncomms6715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Longley B.J., Jr., Metcalfe D.D., Tharp M., Wang X., Tyrrell L., Lu S.Z., Heitjan D., Ma Y. Activating and dominant inactivating c-KIT catalytic domain mutations in distinct clinical forms of human mastocytosis. Proc. Natl. Acad. Sci. USA. 1999;96:1609–1614. doi: 10.1073/pnas.96.4.1609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Chan I.J., Kasprowicz S., Tharp M.D. Distinct signalling pathways for mutated KIT(V560G) and KIT(D816V) in mastocytosis. Clin. Exp. Dermatol. 2013;38:538–544. doi: 10.1111/ced.12000. [DOI] [PubMed] [Google Scholar]
- 45.Morcos P.A., Li Y., Jiang S. Vivo-Morpholinos: A non-peptide transporter delivers morpholinos into a wide array of mouse tissues. Biotechniques. 2008;45:613–614, 616, 618 passim. doi: 10.2144/000113005. [DOI] [PubMed] [Google Scholar]
- 46.Demehri S., Corbin A., Loriaux M., Druker B.J., Deininger M.W. Establishment of a murine model of aggressive systemic mastocytosis/mast cell leukemia. Exp. Hematol. 2006;34:284–288. doi: 10.1016/j.exphem.2005.11.015. [DOI] [PubMed] [Google Scholar]
- 47.Evans E.K., Gardino A.K., Kim J.L., Hodous B.L., Shutes A., Davis A., Zhu X.J., Schmidt-Kittler O., Wilson D., Wilson K., et al. A precision therapy against cancers driven by KIT/PDGFRA mutations. Sci. Transl. Med. 2017;9:eaao1690. doi: 10.1126/scitranslmed.aao1690. [DOI] [PubMed] [Google Scholar]
- 48.Broudy V.C. Stem cell factor and hematopoiesis. Blood. 1997;90:1345–1364. [PubMed] [Google Scholar]
- 49.Theoharides T.C., Valent P., Akin C. Mast cells, mastocytosis, and related disorders. N. Engl. J. Med. 2015;373:1885–1886. doi: 10.1056/NEJMc1510021. [DOI] [PubMed] [Google Scholar]
- 50.Antonescu C.R., Besmer P., Guo T., Arkun K., Hom G., Koryotowski B., Leversha M.A., Jeffrey P.D., Desantis D., Singer S., et al. Acquired resistance to imatinib in gastrointestinal stromal tumor occurs through secondary gene mutation. Clin. Cancer Res. 2005;11:4182–4190. doi: 10.1158/1078-0432.CCR-04-2245. [DOI] [PubMed] [Google Scholar]
- 51.Heinrich M.C., Corless C.L., Blanke C.D., Demetri G.D., Joensuu H., Roberts P.J., Eisenberg B.L., von Mehren M., Fletcher C.D., Sandau K., et al. Molecular correlates of imatinib resistance in gastrointestinal stromal tumors. J. Clin. Oncol. 2006;24:4764–4774. doi: 10.1200/JCO.2006.06.2265. [DOI] [PubMed] [Google Scholar]
- 52.Ito T., Yamamura M., Hirai T., Ishikawa T., Kanda T., Nakai T., Ohkouchi M., Hashikura Y., Isozaki K., Hirota S. Gastrointestinal stromal tumors with exon 8 c-kit gene mutation might occur at extragastric sites and have metastasis-prone nature. Int. J. Clin. Exp. Pathol. 2014;7:8024–8031. [PMC free article] [PubMed] [Google Scholar]
- 53.Tamborini E., Pricl S., Negri T., Lagonigro M.S., Miselli F., Greco A., Gronchi A., Casali P.G., Ferrone M., Fermeglia M., et al. Functional analyses and molecular modeling of two c-Kit mutations responsible for imatinib secondary resistance in GIST patients. Oncogene. 2006;25:6140–6146. doi: 10.1038/sj.onc.1209639. [DOI] [PubMed] [Google Scholar]
- 54.Gleixner K.V., Mayerhofer M., Sonneck K., Gruze A., Samorapoompichit P., Baumgartner C., Lee F.Y., Aichberger K.J., Manley P.W., Fabbro D., et al. Synergistic growth-inhibitory effects of two tyrosine kinase inhibitors, dasatinib and PKC412, on neoplastic mast cells expressing the D816V-mutated oncogenic variant of KIT. Haematologica. 2007;92:1451–1459. doi: 10.3324/haematol.11339. [DOI] [PubMed] [Google Scholar]
- 55.Gallogly M.M., Lazarus H.M., Cooper B.W. Midostaurin: A novel therapeutic agent for patients with FLT3-mutated acute myeloid leukemia and systemic mastocytosis. Ther. Adv. Hematol. 2017;8:245–261. doi: 10.1177/2040620717721459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Juliano R.L. The delivery of therapeutic oligonucleotides. Nucleic Acids Res. 2016;44:6518–6548. doi: 10.1093/nar/gkw236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Godfrey C., Desviat L.R., Smedsrød B., Piétri-Rouxel F., Denti M.A., Disterer P., Lorain S., Nogales-Gadea G., Sardone V., Anwar R., et al. Delivery is key: Lessons learnt from developing splice-switching antisense therapies. EMBO Mol. Med. 2017;9:545–557. doi: 10.15252/emmm.201607199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.McClorey G., Banerjee S. Cell-penetrating peptides to enhance delivery of oligonucleotide-based therapeutics. Biomedicines. 2018;6:E51. doi: 10.3390/biomedicines6020051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Reber S., Mechtersheimer J., Nasif S., Benitez J.A., Colombo M., Domanski M., Jutzi D., Hedlund E., Ruepp M.D. CRISPR-Trap: A clean approach for the generation of gene knockouts and gene replacements in human cells. Mol. Biol. Cell. 2018;29:75–83. doi: 10.1091/mbc.E17-05-0288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Heo Y.A. Golodirsen: First approval. Drugs. 2020;80:329–333. doi: 10.1007/s40265-020-01267-2. [DOI] [PubMed] [Google Scholar]
- 61.Shirley M. Casimersen: First approval. Drugs. 2021;81:875–879. doi: 10.1007/s40265-021-01512-2. [DOI] [PubMed] [Google Scholar]
- 62.Dhillon S. Viltolarsen: First approval. Drugs. 2020;80:1027–1031. doi: 10.1007/s40265-020-01339-3. [DOI] [PubMed] [Google Scholar]
- 63.Disterer P., Al-Shawi R., Ellmerich S., Waddington S.N., Owen J.S., Simons J.P., Khoo B. Exon skipping of hepatic APOB pre-mRNA with splice-switching oligonucleotides reduces LDL cholesterol in vivo. Mol. Ther. 2013;21:602–609. doi: 10.1038/mt.2012.264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Wan J., Sazani P., Kole R. Modification of HER2 pre-mRNA alternative splicing and its effects on breast cancer cells. Int. J. Cancer. 2009;124:772–777. doi: 10.1002/ijc.24052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Jensen B.M., Swindle E.J., Iwaki S., Gilfillan A.M. Generation, isolation, and maintenance of rodent mast cells and mast cell lines. Curr. Protoc. Immunol. 2006;Chapter 3:Unit 3.23. doi: 10.1002/0471142735.im0323s74. [DOI] [PubMed] [Google Scholar]
- 66.Cruse G., Beaven M.A., Ashmole I., Bradding P., Gilfillan A.M., Metcalfe D.D. A truncated splice-variant of the FcεRIβ receptor subunit is critical for microtubule formation and degranulation in mast cells. Immunity. 2013;38:906–917. doi: 10.1016/j.immuni.2013.04.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.