

Abstract
Exercise training benefits the heart. The knowledge of post-transcription regulation, especially RNA editing, in hearts remain rare. ADAR2 is an enzyme that edits adenosine to inosine nucleotides in double-stranded RNA, and RNA editing is associated with many human diseases. We found that ADAR2 was upregulated in hearts during exercise training. AAV9-mediated cardiac-specific ADAR2 overexpression attenuated acute myocardial infarction (AMI), MI remodeling, and doxorubicin (DOX)-induced cardiotoxicity. In vitro, overexpression of ADAR2 inhibited DOX-induced cardiomyocyte (CM) apoptosis. but it could also induce neonatal rat CM proliferation. Mechanistically, ADAR2 could regulate the abundance of mature miR-34a in CMs. Regulations of miR-34a or its target genes (Sirt1, Cyclin D1, and Bcl2) could affect the pro-proliferation and anti-apoptosis effects of ADAR2 on CMs. These data demonstrated that exercise-induced ADAR2 protects the heart from MI and DOX-induced cardiotoxicity. Our work suggests that ADAR2 overexpression or a post-transcriptional associated RNA editing via ADAR2 may be a promising therapeutic strategy for heart diseases.
Keywords: ADAR2, exercise, myocardial infarction, doxorubicin-induced cardiotoxicity, RNA editing
Graphical abstract
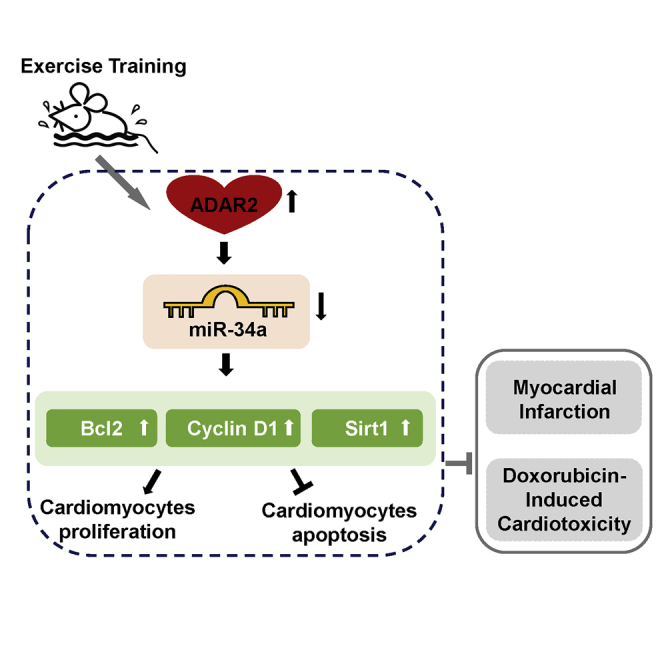
Xiao and colleagues report that exercise-induced ADAR2 can protect the heart from myocardial infarction and doxorubicin-induced cardiotoxicity. Their study suggests that ADAR2 overexpression or a post-transcriptional associated RNA editing via ADAR2 may be a promising therapeutic strategy for heart diseases.
Introduction
Heart disease is a leading cause of mortality worldwide. Cardiomyocyte (CM) death in ischemic heart disease contributes to decreased cardiac function and development toward subsequent heart failure.1 The proliferation and regenerative capacity of adult mammalian CM is limited, and little is known about the essential factors regulating these processes. Clinical studies and animal models support the cardioprotective benefits of exercise via enhancing CM viability and inducing physiological cardiac growth.2,3 However, little is known about the molecular mechanisms underlying exercise-stimulated cardiac functioning. Thus, understanding factors and pathways that enhance the myocardial recovery and endurance for cardiac stresses in response to exercise could provide clinical implications on cardiac protection against heart failure.
Adenosine deaminases acting on RNA (ADARs) are enzymes acting on double-stranded RNAs (dsRNAs), converting adenosine to inosine (A to I). Three ADAR gene family members (ADAR1–3) are known in vertebrates. ADAR1 and ADAR2 are ubiquitously expressed, while ADAR3 is exclusively expressed in the brain.4 ADAR-mediated A-to-I editing is the most frequent type of RNA editing in human, and inosine is interpreted as guanosine by the cellular machinery. The A-to-I modification of RNA at a post-transcription level usually leads to alternative splicing, RNA folding, and RNA sequences.5 Dysregulation of RNA editing is associated with many diseases, including cancers, neurological disorders, and atherosclerosis.6, 7, 8 Very recently, CM-specific deletion of ADAR1 has been reported to lead to severe cardiac dysfunction and eventual embryonic or postnatal lethality.9,10 Although different ADAR mRNAs are found to be significantly regulated in congenital heart disease patients, the critical role of adenosine deaminases in the heart and its underlying regulatory mechanisms remain largely unexplored.11 In particular, the exercise-induced post-transcription adaptions in the heart have not been examined. Therefore, we investigated the regulatory role of adenosine deaminases in exercise-induced cardiac adaptation, myocardial infarction (MI), and doxorubicin (DOX)-induced cardiotoxicity.
The role of microRNAs (miRNAs, miRs) in cardiovascular diseases has been well established.12,13 They have emerged as crucial regulators influencing entire gene expression networks in heart disease.14 MiR-222 regulates cardiac growth and protects against pathological cardiac remodeling.15 MiR-34a inhibition reduces cardiac cell death and improves heart function following MI.16,17 Modification of miRNAs via sequence editing may alter their function, leading to different post-transcriptional regulation of miRNAs. Primary miRNAs (pri-miRNAs) can be A-to-I edited by ADARs.18 The consequence of this editing ranges from the inhibition of mature miRNA processing to the re-direction of miRNA toward an altered set of target genes.19, 20, 21 Therefore, understanding post-transcriptional regulation via editing of miRNAs provides a direction and potential strategy to modulate mature miRNAs levels.
Here, we show that the expression of ADAR2 is increased in exercised hearts. In vitro, ADAR2 promotes CM proliferation and inhibits DOX-induced CM apoptosis. AAV9 (adeno-associated virus serotype 9) carrying the cardiac-specific troponin-T (cTnT) promoter-driven ADAR2 overexpression (AAV9-cTnT-ADAR2) in vivo can attenuate acute myocardial infarction (AMI), MI remodeling, and DOX-induced cardiotoxicity. Next, we identify that ADAR2 negatively regulates mature miR-34a, and the RNA editing activity of ADAR2 contributes to the ADAR2/miR-34a regulation axis in CMs. Moreover, we demonstrate that ADAR2 regulates CM proliferation and apoptosis through miR-34a and its target genes (Sirt1, Cyclin D1, and Bcl2). Our work suggests that ADAR2 overexpression or post-transcriptional associated RNA editing via ADAR2 may be a promising therapeutic strategy for heart diseases.
Results
Exercise-induced ADAR2 promotes CM proliferation and survival
To investigate the functions and regulation of RNA editing in physiological cardiac hypertrophy, we examined the changes in ADAR enzymes in the hearts of swimming mice. We found that the expression of ADAR2 was elevated in mRNA levels, whereas an increased expression level of ADAR1 did not reach significance (Figures 1A and S1A). Consistently, the protein level of ADAR2 was increased in the hearts of swimming mice (Figure 1A), which could also be confirmed, both in the mRNA and protein levels of ADAR2, in the hearts of swimming rats (Figure S1B). Then, we measured the protein level of ADAR2 in mice hearts under pathological stresses, including AMI, 3 weeks post-MI, and DOX-induced cardiotoxicity. Interestingly, the protein levels of ADAR2 were unchanged in all three pathological models, indicating a specific elevation of ADAR2 in response to physiological stimulation (Figures S1C–S1E). We therefore selected ADAR2 for follow-up studies on the role of ADAR2 in the heart.
Figure 1.

Exercise-induced ADAR2 promotes cardiomyocyte proliferation and survival in vitro
(A) Quantitative real-time PCR and representative western blot showing expression change of ADAR2 in swimming-trained mice hearts versus control mice hearts (n = 6, ∗∗p < 0.01). (B) Representative western blot testing expression change of ADAR2 after overexpression (n = 6, ∗∗p < 0.01). (C) Representative western blot testing expression change of ADAR2 after siRNA strand-3 knockdown (n = 6, ∗∗p < 0.01). (D) Analysis of proliferation markers, including EdU (red) and Ki67 (red), after knockdown or overexpression of ADAR2. α-actinin (green) and DAPI (blue) are co-stained (n = 6, ∗∗p < 0.01); scale bar, 50 μm. (E and F) Analysis of apoptosis by TUNEL staining (E) and western blot of Bax, Bcl2, caspase 3, and cleaved-caspase 3 (F) after ADAR2 overexpression or ADAR2 knockdown in doxorubicin (DOX)induced apoptosis model (n = 6, ∗p < 0.05, ∗∗p < 0.01).
To identify cell types that express ADAR2 in the heart, we separated primary neonatal rat cardiac myocytes (NRCMs) and neonatal rat cardiac fibroblasts (NRCFs), and observed that both NRCMs and NRCFs have a similar degree of ADAR2 expression (Figure S2). Next, to determine the function of ADAR2 in NRCFs and NRCMs, an ADAR2 overexpression construct (ADAR2 OE) was generated with its empty vector (EV) used as control (Figure 1B). Three small interfering RNAs (siRNAs) were designed and the most efficient one (strand-3) to reduce the ADAR2 mRNA levels was subsequently used in the following loss-of-function studies in vitro (Figures 1C and S3A). We performed gain- and loss-of-function assays in NRCFs, but observed that ADAR2 affected neither the proliferation of cardiac fibroblasts, via counting Ki67+ nuclei, nor their differentiation into myofibroblasts via the increased presence of α-SMA (Figure S3B). We therefore further explored the role of ADAR2 in NRCMs. As indicated in Figure 1D, overexpression of ADAR2 remarkably increased numbers of 5-ethynyl-2'-deoxyuridine (EdU)+ NRCMs, whereas ADAR2 knockdown significantly decreased that. Similarly, ADAR2 overexpression positively regulated the number of NRCMs Ki67+ nuclei (Figure 1D). Neither ADAR2 OE nor ADAR2 knockdown led to morphological cell size changes (Figure S3C). Moreover, as evidenced by TUNEL (terminal deoxynucleotidyl transferase 2′-deoxyuridine,5′-triphosphate [dUTP] nick end labeling) staining and western blot analyses (Bax/Bcl2 and cleaved-caspase 3/caspase 3), overexpression of ADAR2 inhibited DOX-induced apoptosis in NRCMs, while knockdown of ADAR2 aggravated levels of cell apoptosis (Figures 1E and 1F). These data suggested a protective role of ADAR2 in the heart by promoting CM proliferation and attenuating CM apoptosis.
To further evaluate whether ADAR2 was required for exercise-mediated physiological cardiac hypertrophy, we used the swim-induced cardiac hypertrophy model.2 Mice were tail vein injected with AAV9-cTnT-ADAR2 small hairpin RNA (AAV9-cTnT-ADAR2-shRNA), and mice injected with empty AAV9-cTnT-scramble (AAV9-cTnT-Scr) were used as controls. The cTnT promoter-driven ADAR2 shRNA was generated by using a miR-30d-based shRNA framework as previously reported.22 Cardiac-specific knockdown of ADAR2 was induced in AAV9-cTnT-ADAR2-shRNA-treated mice (Figure S4A). Interestingly, exercise with and without AAV9-cTnT-ADAR2-shRNA treatment both showed an increase in heart weight (HW) and heart weight/tibia length (HW/TL), demonstrating that ADAR2 knockdown did not affect the exercise-induced cardiac growth (Figure S4B). Consistently, wheat germ agglutinin (WGA) staining suggested that no significant difference in CM cell size was observed in exercised mice between AAV9-cTnT-ADAR2-shRNA and AAV9-cTnT-Scr groups (Figure S4C). Hence, although the levels of ADAR2 are successfully changed, our data demonstrate that ADAR2 is not necessary for exercise-induced physiological cardiac hypertrophy in vivo.
Cardiac-specific overexpression ADAR2 protects AMI in mice hearts
Since ADAR2 contributes to CM proliferation and apoptosis in vitro, we proposed that cardiac-specific overexpression of ADAR2 in vivo possibly protects against cardiac injury. AAV9-cTnT-ADAR2 or AAV9-cTnT-ctrl were tail vein injected into C57BL/6 mice, subsequently followed by MI or sham surgery 1 week after the AAV9 injection (Figure 2A). The expression of ADAR2 was significantly increased in mice hearts treated with AAV9-cTnT-ADAR2 (Figure 2B), together with a remarkable decrease in the infarct size as observed by triphenyltetrazolium chloride (TTC) staining (Figure 2C). As shown in Figures 2D and S5A, the percentage of TUNEL+ CM was reduced in mice that received AAV9-cTnT-ADAR2 compared with AAV9-cTnT-ctrl-treated controls. The ameliorated AMI-induced cell apoptosis was further supported by western blot via a reduction in Bax/Bcl2 and cleaved-caspase 3/caspase 3 levels (Figure 2E). Then, lactate dehydrogenase (LDH) assays demonstrated a significant reduction of LDH release in serum from AAV9-cTnT-ADAR2-treated mice after AMI surgery (Figure 2F). Consistently, propidium iodide (PI) staining revealed that the number of PI+ nuclei was reduced in AAV9-cTnT-ADAR2-treated mice hearts after AMI (Figures 2G and S5B). Furthermore, the pro-proliferation effects of ADAR2 overexpression on CMs in vivo in AMI mice were assessed by increased immunofluorescence-stained EDU+, Ki67+, and pHH3+ CMs (Figures 2H, 2I, and S5C–S5E). These data provide in vivo evidence that ADAR2 overexpression in the myocardium can result in a reduction in infarcted size, alleviation of apoptosis and cellular necrosis, and promotion of CM proliferation after AMI.
Figure 2.

Cardiac-specific overexpression of ADAR2 protects AMI in mice hearts
(A) The schedule of virus injection and AMI model establishment. (B) Quantitative real-time PCR testing expression of ADAR2 after overexpression of ADAR2 by injecting AAV9-cTnT-ADAR2 with or without (AMI) surgery (n = 7:8:9:8, ∗∗p < 0.01). (C) Triphenyltetrazolium chloride (TTC) staining of mice hearts treated with AAV9-cTnT-ADAR2 followed by AMI; infarct size is indicated by IS (infarct size)/AAR (area at risk) (n = 7, ∗∗p < 0.01). (D) TUNEL staining (green) together with α-actinin (red) and DAPI (blue) of mice hearts treated with AAV9-cTnT-ADAR2 or AAV9-cTnT-ctrl followed by AMI (n = 6, ∗∗p < 0.01); scale bar, 20 μm. (E) Western blot of Bax, Bcl2, caspase 3, and cleaved-caspase 3 of the mice hearts treated with AAV9-cTnT-ADAR2 followed by AMI (n = 3, ∗∗p < 0.01). (F) Serum levels of LDH in indicated groups (n = 7:10:8:7, ∗∗p < 0.01). (G) Representative images of PI staining and quantification of the PI+ nuclei in the mice hearts treated as indicated (n = 6, ∗∗p < 0.01); scale bar, 20 μm. (H) Representative images of immunofluorescence staining and quantification of the 5-ethynyl-2′-deoxyuridine+ (EdU+) cardiomyocytes in the mice hearts treated as indicated (n = 6, ∗∗p < 0.01); scale bar, 20 μm. (I) Representative images of immunofluorescence staining and quantification of the Ki67+ and pHH3+ cardiomyocytes in the mice hearts treated as indicated (n = 6, ∗∗p < 0.01); scale bar, 20 μm. AMI, acute myocardial infarction; Ctrl, control; LDH, lactate dehydrogenase. ns, non-statistically significant.
Cardiac-specific ADAR2 overexpression improves cardiac function and attenuates cardiac fibrosis during cardiac remodeling post-MI
To investigate the clinical perspective of ADAR2 as a biological therapy, we overexpressed ADAR2 in murine hearts by infecting them with AAV9-cTnT-ADAR2 or AAV9-cTnT-ctrl, and performed MI surgery (Figure 3A). A significant elevation of ADAR2 was observed in the hearts of mice treated with AAV9-cTnT-ADAR2 (Figure S6A). Echocardiography was examined at 3 weeks post-MI surgery. The cardiac function in the AAV9-cTnT-ADAR2 group was partially preserved compared to the AAV9-cTnT-ctrl group, as demonstrated by the increased ejection fraction (EF) and fractional shortening (FS), as well as the decreased left ventricular internal diameters of systole (LVID;s) and left ventricular end-systolic volume (LVes;vol) (Figures 3B and S6B). The cardiac hypertrophic adaptation in response to MI injury in ADAR2 overexpressing mice was improved as indicated by decreases in HW/BW (Figure 3C). MI-induced fibrosis was alleviated by AAV9-cTnT-ADAR2 injection compared with AAV9-cTnT-ctrl, as demonstrated by Masson trichrome staining and the mRNA expression levels of fibrotic factors (Ctgf, Col1a1, and Col3a1) (Figures 3D and 3E). These data suggest that cardiac-specific overexpression ADAR2 can protect the heart from cardiac adverse remodeling, resulting in improved cardiac function and reduced cardiac fibrosis.
Figure 3.

Cardiac-specific overexpression of ADAR2 improves cardiac function in cardiac remodeling after MI
(A) The schedule of virus injection and MI remodeling model establishment. (B) Echocardiography of mice treated with AAV9-cTnT-ADAR2 with or without MI surgery (n = 8:10:8:11, ∗p < 0.05, ∗∗p < 0.01). LVID;s, left ventricular internal diameters of systole. (C) Heart weight/body weight (HW/BW) was calculated to evaluate cardiac adaptation in response to MI remodeling in control and ADAR2 overexpression mice (n = 8:10:8:11, ∗p < 0.05, ∗∗p < 0.01). (D) Masson trichrome staining of the hearts treated with AAV9-cTnT-ADAR2 followed by MI surgery, n = 8:10:8:11; scale bar, 100 μm (∗∗p < 0.01). (E) Quantitative real-time PCR analyzing the fibrotic factors (Ctgf, Col1a1, and Col3a1) in the hearts treated with AAV9-cTnT-ADAR2 followed by MI remodeling (n = 8:9:7:10, ∗∗p < 0.01).
ADAR2 overexpression attenuates DOX-induced cardiotoxicity
Our in vitro study suggested that ADAR2 overexpression would exert beneficial effects in DOX-induced CM apoptosis. Then, we asked whether ADAR2 overexpression could alleviate DOX-induced cardiotoxicity in vivo. Seven days after AAV9-cTnT-ADAR2 or AAV9-cTnT-ctrl injection, DOX was intraperitoneally injected at a dose of 5 mg/kg once per week for 5 consecutive weeks (Figure 4A). Overexpression of ADAR2 was verified by RT-PCR (Figure S7A). Mice with ADAR2 overexpression demonstrated by improved cardiac function, as evidenced by increased EF and FS, declined LVID;s and LVes;vol (Figures 4B and S7B). Moreover, TUNEL staining suggested that ADAR2 overexpression decreased the DOX-induced apoptosis of the myocardium (Figure 4C). Furthermore, the alleviated cell apoptosis was also evidenced by western blot of declined Bax/Bcl2 and cleaved-caspase 3/caspase 3 (Figure 4D). Our data suggest that ADAR2 overexpression attenuates DOX-induced cardiotoxicity via improving cardiac function and attenuating cell apoptosis.
Figure 4.

ADAR2 overexpression attenuates DOX-induced cardiotoxicity
(A) The schedule of virus injection and DOX-induced cardiotoxicity model establishment. (B) Echocardiography of mice treated with AAV9-cTnT-ADAR2 followed by DOX or saline treatment (n = 10:9:10:12, ∗∗p<0.01). (C) TUNEL staining (green) together with α-actinin (red) and DAPI (blue) of mice hearts treated with AAV9-cTnT-ADAR2 or AAV9-cTnT-ctrl followed by DOX or saline treatment (n = 6, ∗∗p < 0.01); scale bar, 20 μm. (D) Western blot of Bax, Bcl2, caspase 3, and cleaved-caspase 3 of mice hearts treated with AAV9-cTnT-ADAR2 followed by DOX or saline treatment (n = 3, ∗p < 0.05, ∗∗p < 0.01).
RNA editing may contribute to the ADAR2/miR-34a regulation in CMs
Previous studies revealed that ADAR2 was involved in miRNA regulation via A-to-I RNA editing of pri-miRNAs.5,20,23 To explore the mechanism underlying the protective effect of ADAR2 in hearts, we focused on miRNAs. To select candidates, we performed an initial screening based on two criteria: (1) miRNAs involved in regulating CM proliferation and (2) miRNAs regulated by ADAR2 alteration. As shown in Figure 5A, ADAR2 overexpression inhibited mature miR-34a levels, whereas ADAR2 knockdown increased miR-34a levels. In addition, the expression of miR-34a was decreased in the exercised hearts, while they were increased in the MI hearts (Figure 5B). Then, we identified three nucleotides change in the sequenced products in the presence of ADAR2 overexpression via TA cloning and Sanger sequencing analysis for pri-miR-34a conducted in 107 clones (Figures 5C and 5D). Major editing sites U44-C and A61-G of pre-miR-34a were identified near its end loop, which is linked to Dicer’s cleavage region. A minor site (A72-G) of pri-miR-34a was found at its antisense strand (Figures 5C and 5D). As the editing ratio we observed in miR-34a was relatively low, we asked whether A-to-I editing is the predominant effect of ADAR2 in CMs and its regulation in the expression of miR-34a. We used the HMMER webserver to locate the two double-stranded RNA-binding domains (dsRBDs) and the deaminase domain.24 Then, we generated the ADAR2-N construct, which did not have the deaminase domain (Figure 5E). As shown in Figures 5F and 5G, we found that overexpression of ADAR2-N without deaminase activity could not promote DNA synthesis and resist to DOX-induced apoptosis in NRCMs. Interestingly, unlike ADAR2 full-length overexpression, ADAR2-N overexpression did not regulate the abundance of mature miR-34a (Figure 5H). Furthermore, to examine whether RNA editing occurs by nucleotide changes in the precursor of miR-34a, we performed high-resolution melting (HRM) analysis. The HRM results showed that, compared with ADAR2-N, ADAR2 full-length overexpression could lead to a RT-PCR product alteration of pri-miR-34a, indicating that nucleotide editing may be present in the transcript of pri-miR-34a (Figure 5I). These observations indicate that RNA editing activity of ADAR2 may contribute to the ADAR2/miR-34a regulation axis in CMs.
Figure 5.

RNA editing activity of ADAR2 contributes to the ADAR2/miR-34a regulation in cardiomyocytes
(A) Screening of miRNAs involved in regulating cardiomyocyte proliferation and regulated by ADAR2 alteration (n = 6, ∗p < 0.05, ∗∗p < 0.01). (B) Quantitative real-time PCR analyzing the expression of miR-34a in the exercised hearts and MI remodeling (3 weeks) hearts (n = 6, ∗p < 0.05, ∗∗p < 0.01). (C and D) Sanger sequencing analysis for pri-miR-34a in 107 clones in the presence of ADAR2 overexpression. (E) Schematic illustration of the generation of the ADAR2-N construct without deaminase activity. (F) Representative images of immunofluorescence staining and quantification of the EdU+ cardiomyocytes treated with or without ADAR2-N overexpression (n = 6); scale bar, 50 μm. (G) Representative images of immunofluorescence staining and quantification of the TUNEL+ cardiomyocytes treated with ADAR2-N or control EV in DOX-induced apoptosis model (n = 6); scale bar, 50 μm. (H) Quantitative real-time PCR analyzing the expression of miR-34a with or without ADAR2-N overexpression (n = 6). (I) High-resolution melting (HRM) analysis showing RT-PCR product alterations of pri-miR-34a after overexpression of ADAR2, ADAR2-N, or EV. ns, non-statistically significant.
ADAR2 regulates CM proliferation and apoptosis through the miR-34a-Sirt1/Cyclin D1/Bcl2 axis
We subsequently investigated whether the above observed editing regulation has functional consequences. Considering the fact that miR-34a has been reported to be involved in regulating cardiac repair and cardiac aging,16,17 we speculated that the ability of ADAR2 to regulate CM proliferation and apoptosis is probably mediated by miR-34a as well. To confirm our hypothesis, we explored the contribution of miR-34a to the regulatory function of ADAR2 in NRCMs. As shown in Figure 6A, miR-34a overexpression (miR-34a mimic) could alleviate the pro-proliferation effects of ADAR2 overexpression in NRCMs, as evidenced by EdU incorporation and Ki67 immunofluorescent staining. Moreover, TUNEL staining and western blot analysis revealed that miR-34a overexpression could abolish the protective effects of ADAR2 overexpression in DOX-induced NRCMs apoptosis (Figures 6B and 6C). These data confirmed that the pro-proliferation and anti-apoptosis effects of ADAR2 were mediated by miR-34a inhibition, and overexpression of miR-34a could block the protective effect of ADAR2 in vitro.
Figure 6.

MiR-34a mimics blunts the pro-proliferation and anti-apoptosis effects of ADAR2 on cardiomyocytes
(A) EdU incorporation and Ki67 immunofluorescent staining of NRCM after ADAR2 overexpression and miR-34a overexpression (miR-34a mimic) (n = 6, ∗∗p < 0.01), scale bar, 50 μm. (B and C) TUNEL staining (scale bar, 50 μm) and western blot analyzing DOX-induced NRCMs apoptosis after ADAR2 overexpression and miR-34a overexpression (miR-34a mimic) (n = 6, ∗p < 0.05, ∗∗p < 0.01).
To obtain more evidence supporting the contribution of miR-34a in the function of ADAR2, the target genes of miR-34a in cardiomyocyte were further checked in NRCMs. Notch1, Pofut1, Sirt1, Cyclin D1, and Bcl2 have been reported to be targets of miR-34a in regulating cardiomyocyte proliferation and death.17,25 Among them, the expression of Sirt1, Cyclin D1, and Bcl2 were elevated after swimming training in mice hearts, while Notch1 and Pofut1 demonstrated no change (Figure S8A). Therefore, we focused on Sirt1, Cyclin D1, and Bcl2. Consistently, we found that miR-34a expression in NRCMs was sufficient to inhibit the expression of all three reported targets (Sirt1, Cyclin D1, and Bcl2), while inhibiting miR-34a increased their expression (Figure S8B). Moreover, upon overexpression of ADAR2, we observed an effective increase in the expression of miR-34a targets (Sirt1, Cyclin D1, and Bcl2), whereas knockdown of ADAR2 demonstrated opposite modulating effects (Figure 7A). Knockdown of either Sirt1 or Cyclin D1 could partially blunt the pro-proliferation capacity of ADAR2 overexpression, and Bcl2 inhibition would partially reverse the anti-apoptosis effect of ADAR2 overexpression in DOX-induced cardiomyocyte apoptosis (Figures S8C–S8E). We further simultaneously inhibited all three targets of miR-34a and found that the ADAR2 overexpression effects (both pro-proliferation and anti-apoptosis) are completely abolished (Figures 7B and 7C). These data indicate that the miR-34a-Sirt1/Cyclin D1/Bcl2 axis may play important roles in mediating the beneficial effects of exercise-induced ADAR2 on cardiomyocyte and hearts.
Figure 7.

ADAR2 regulates cardiomyocyte proliferation and apoptosis through the target genes of miR-34a (Sirt1/Cyclin D1/Bcl2)
(A) Western blot showing the expression of Sirt1, Cyclin D1, and Bcl2 after overexpression or knockdown of ADAR2 (n = 6, ∗p < 0.05, ∗∗p < 0.01). (B) EdU staining of NRCM after ADAR2 overexpression and simultaneous knockdown of Sirt1, Cyclin D1, and Bcl2 (n = 6, ∗∗p < 0.01); scale bar, 50 μm. (C) TUNEL staining analyzing DOX-induced NRCMs apoptosis after ADAR2 overexpression and simultaneous knockdown of Sirt1, Cyclin D1, and Bcl2 (n = 6, ∗∗p < 0.01); scale bar, 50 μm.
C/EBPβ acts as an upstream regulator of ADAR2
Furthermore, we explored what triggers the upregulation of ADAR2 in response to exercise. Transcription factors that may be involved in the regulation of ADAR2 expression at the transcriptional level were predicted by JASPAR.26 Among them, C/EBPβ is of particularly interest as it is a key regulator of exercise-induced physiological cardiac hypertrophy (Figure 8A).2 First, C/EBPβ’s binding to the predicted ADAR2 promoter region was verified by chromatin immunoprecipitation (ChIP) assay (Figure 8B). Second, we used the luciferase assay system to test whether C/EBPβ could regulate the expression of ADAR2. As shown in Figure 8C, the promoter region of ADAR2 exhibited low luciferase activity in the presence of C/EBPβ overexpression, indicating the negative regulation of ADAR2. Also, qRT-PCR suggested that C/EBPβ knockdown in NRCM led to the elevation of ADAR2 (Figure 8D). Furthermore, functional rescue assays demonstrated that the suppression effect of C/EBPβ overexpression on NRCM proliferation was blocked by ADAR2 overexpression (Figure 8E). Moreover, ADAR2 overexpression could blunt the deteriorating effect of C/EBPβ overexpression on DOX-induced NRCM apoptosis (Figure 8F). Our data suggest that a key cardiac adaptor to exercise training, transcription factor C/EBPβ, functions as an upstream regulator of ADAR2, and therefore regulates the upregulation of ADAR2 in response to exercise.
Figure 8.

C/EBPβ acts as an upstream regulator of ADAR2
(A) Schematic illustration of the JASPAR-predicted binding sites of C/EBPβ in the promoter region of Adar2. (B) ChIP-PCR assay showed that C/EBPβ could bind to the ADAR2 promoter region (n = 4, ∗∗p < 0.01). (C) Luciferase assay revealed the negative regulation of C/EBPβ on ADAR2 (n = 6, ∗∗p < 0.01). (D) Quantitative real-time PCR analysis of ADAR2 expression with or without C/EBPβ knockdown (n = 6, ∗∗p < 0.01). (E) Representative images of immunofluorescence staining and quantification of the EdU+ cardiomyocytes displayed the rescue effect of ADAR2 overexpression on C/EBPβ overexpression (n = 6, ∗∗p < 0.01); scale bar, 50 μm. (F) Representative images of immunofluorescence staining and quantification of the TUNEL+ cardiomyocytes displayed the rescue effect of ADAR2 overexpression on C/EBPβ overexpression in DOX-induced NRCMs apoptosis (n = 6, ∗p < 0.05, ∗∗p < 0.01); scale bar, 50 μm.
Discussion
The beneficial effects of exercise on the cardiovascular system have been well accepted, and physical activity contributes to a reduction in deaths from cardiovascular disease.27, 28, 29, 30 However, exercise-induced post-transcriptional adaptions in the heart have not been explored much. Here, we report that exercise-induced ADAR2 mediates pri-miR-34a editing, which is a mechanism that can contribute to the adaptations of the heart to exercise training. To our knowledge, this is the first study to explore the post-transcriptional mechanisms involved in exercise-induced cardiac protection. We show that ADAR2, an important RNA binding protein that converts A-to-I in dsRNAs, is upregulated in endurance exercise both in mice and rat hearts. ADAR2 overexpression can promote NRCM proliferation and increase resistance to apoptosis induced by DOX. Moreover, ADAR2 overexpression can protect mice hearts and lead to a decreased cardiac infarct size via reduced cardiac apoptosis and necrosis, as well as increase CM proliferation. Also, ADAR2 overexpression could improve cardiac function and alleviate cardiac fibrosis after MI remodeling, as well as attenuate DOX-induced cardiotoxicity in vivo. Mechanically, RNA editing activity of ADAR2 contributes to the ADAR2/miR-34a regulation axis in CMs. Overexpression of miR-34a reverses the pro-proliferation and anti-apoptosis of ADAR2 effects on CMs. Furthermore, the previously reported miR-34a targets (Sirt1, Cyclin D1, and Bcl2) also play important roles in mediating the beneficial effects of ADAR2 on cardiomyocytes. A key cardiac adaptor to exercise training, transcription factor C/EBPβ, acts as an upstream regulator of ADAR2 in cardiomyocytes.
Adenosine deamination by ADARs plays an essential role in post-transcriptional regulation, and recent studies indicate that ADARs may contribute to cardiovascular disease and cardiac development.5,9, 10, 11 The biological significance of many A-to-I editing sites remains largely unknown. In this study, we focused on identifying edited target genes of ADAR2 involved in regulating cardiomyocyte proliferation, with particular attention on miRNAs. miRNAs participate in many important biological processes, including cell cycle, differentiation, and apoptosis.31 In the cardiovascular system, miRNAs also attract dramatic attention for their critical regulatory roles as well as potential therapeutic targets.12,32 RNA editing, mediated by ADARs, can affect miRNA expression and processing via either inhibiting Drosha and Dicer cleavage steps or altering the seed sequences of miRNAs.5,23,33 One possible mechanistic explanation about the nucleotide modification of pre-miRNA regulation was that pre-miRNAs in solution adopt multiple conformations and therefore regulate the binding and processing efficiency of the Dicer complex.34,35 In our study, we found that the most edited sites in pri-miR-34a occurred near the Dicer cleavage sites (U44C and A61G) and on its antisense strand (A72G). Among them, A64G and A72G editing change the A:U base pair to the I:U wobble pair; possibly destabilize the dsRNA structure, U44C, at the terminal loop region of pre-miR-34a; and may modulate the RNA conformation shift. Our observations on the nucleotide modification of pre-miR-34a at both the loop and stem regions to inhibit miR-34a may be achieved by reducing the accessibility and processing ability of the Dicer complex to pre-miR34a. However, further investigation into potential allosteric effects between Dicer complexes and edited pre-miR-34a are needed to provide a better understanding of the underlying mechanism.
CM loss and deficiency underlie most causes of heart diseases, including heart failure. Although the regenerative capacity of adult mammalian hearts is limited, tremendous efforts have been made to improve cardiac function by promoting CM proliferation.36,37 Induction of cardiac physiological growth is the most important adaptive response to exercise. Experimental evidence has demonstrated that several exercise-induced factors are required for cardioprotection against myocardial injury and remodeling.2,15,38 Thus, exploring therapeutic targets from physical activity-stimulated factors represents a promising strategy for cardiac diseases.39 However, whether exercise-induced hypertrophy and proliferation mediate the adaptive response independently or cooperatively remains unknown. Different from previously reported critical physiologic growth mediators, such as C/EBPβ, miR-222, and miR-17-3p, our report here shows that exercise-induced ADAR2 did not affect the hypertrophic response to exercise in vivo. Interestingly, ADAR2 demonstrated pro-proliferation and anti-apoptosis effects in NRCMs. In addition, cardiac-specific ADAR2 overexpression in vivo exhibited a protective effect on myocardial injury and remodeling. This finding suggests that, at least to a certain extent, exercise-induced factors in adult hearts contributing to the benefits of exercise are hypertrophy independent. In addition, in the MI 3-week remodeling model, we overexpressed ADAR2 in murine hearts by infecting them with AAV9-cTnT-ADAR2 and performed MI surgery at the same day. As previously reported, several days were needed to achieve the ADAR2 overexpression in the heart.40 Thus, except for the currently investigated pro-proliferation and anti-apoptosis effects in cardiomyocytes, other regulatory mechanisms may also exist in the heart and contribute to ADAR2-mediated cardioprotection. Interestingly, we examined ADAR2 on the proliferation of NRCFs at basal conditions and found that ADAR2 alteration did not affect the proliferation of cardiac fibroblasts. This observed phenomenon that some genes are ubiquitously expressed but are regulated or show distinct function under specific stimulant in a cell-type-specific manner have been reported previously.41,42 In this present study, this may be due to the fact that ADAR2 is an exercise response factor and the regulation of ADAR2 associated with an important transcription factor C/EBPβ alteration, which is closely related to cardiac changes in adaptation to exercise. The well-studied mechanisms underlying the effects of exercise on the heart are cardiomyocyte hypertrophy and growth, vascular adaptions, mitochondrial adaptations, and angiogenesis.27,43 As far as we know, a limited number of studies report the regulation effects of exercise training on fibroblast proliferation, although exercise training has been shown to have antifibrotic properties in heart diseases. This indicates that the antifibrotic properties of exercise might be caused by other reasons such as changes in the collagen profile, crosstalk between other cardiac cells and fibroblasts, other than alter the proliferation of fibroblast. The cell type specificity regulation under specific conditions represents an interesting and active area of investigation; therefore, further studies using spatial transcriptomics and single-cell sequencing to profile the exercised heart with cell type and spatial resolution would contribute to a deep understanding of the cardiac adaptation to exercise and may explain where this cell type specificity came from.
MiR-34a is elevated in the heart and is associated with aging and other cardiac diseases.16,17,25 However, little knowledge is available regarding the role of miR-34a in physiological cardiac growth. In the present study, for the first time, we investigated the role of miR-34a and its targets (Sirt1, Cyclin D1, and Bcl2) involved in regulating the exercise-induced cardiac adaptation. Importantly, exercise-induced ADAR2 regulated miR-34a processing at the post-transcriptional level. The inhibition of miR-34a has been proposed as a promising therapeutic strategy for heart disease. A-to-I RNA editing occurs in over half of the transcriptome, altering the expression and activity of ADARs are the dominant regulation events in response to A-to-I editing regulation.44,45 ADAR2 alteration should lead to global pattern changes in RNA editing in the heart. We cannot exclude the possibility that there are additional mRNAs regulated by ADAR2 that contribute to the protective effects of ADAR2 in the heart. Therefore, it would be important to further explore the role of global RNA editing patterns in the heart. However, with the concept of nucleic acid-based therapeutics targeting transcript-raised and site-directed A-to-I editing system, this editing machinery may indicate an alternative approach to control miR-34a expression with high efficiency and specificity.
Here, we report the functional relationship between ADAR2 and the heart and examine miR-34a, which was one target of ADAR2 in CMs. Although our results demonstrated that ADAR2-N without deaminase activity did not have the pro-survival effects and the regulation ability on miR-34a expression, relatively low miR-34a editing efficiency (only 6 of 107 clones) observed in the present study. We measured the editing effect by TA cloning followed by Sanger sequencing, rather than a discovery-based method of RNA sequencing (RNA-seq), which could not exclude the possibility that the additional editing pattern of RNA transcripts regulated by ADAR2 may also affect the expression of miR-34a. Future studies should return to basic questions regarding the biological role of specific editing transcripts or even genome editing alterations for a deep understanding of the biological significance and the underlying regulatory mechanism about those regulation events would offer significant opportunity to better clarify the coordination between editing activity and the contribution of an individual regulated transcript. A limitation of our study is that we did not address editing anywhere else except miR-34a, which is specifically edited by ADAR2. The low miRNA editing rates have been previously reported.46,47 Those low detected miRNA editing efficiencies have also been found due to either preventing the maturation of miRNA or leading to the degradation of the edited miRNA.20,48 Although the editing efficiency in miR-34a observed in the present study is relatively low, the alteration of ADAR2 significantly regulates the expression fold change of miR-34a (Figure 5A). This expression alteration of mature miR-34a may explain the significant functional effects in CMs. Moreover, further investigations including RNA-seq elucidate the global editing pattern change of ADAR2 alteration, identify other potential targets of ADAR2, and would provide a greater understanding of ADAR2 regulatory mechanisms in the heart.
In summary, our results demonstrate that exercise-induced ADAR2 can protect the heart from MI and DOX-induced cardiotoxicity. Our study suggests that ADAR2 overexpression may be a promising therapeutic strategy for heart diseases.
Materials and methods
Animals and swimming protocol
Male C57BL/6J mice were obtained from the Model Animal Research Center of Nanjing University. The study on animals was in accordance with the Guidelines for the Care and Use of Laboratory Animals published by the National Institutes of Health (no. 85-23, revised 1996). All of the mice were maintained in a specific pathogen-free (SPF) laboratory animal facility of Nanjing Medical University (Nanjing, China). The experimental protocol was approved by the Animal Care and Use Committee of Nanjing Medical University (license no. IACUC-1903016).
To induce physiological cardiac hypertrophy of mice, a 3-week swimming protocol was performed in mice as previously described.38 Briefly, the exercise training began with 10 min swimming twice per day for the first day, and then continued with an increase of 10 min per day until 90 min swimming twice per day was reached. After 3 weeks of swimming, the mice were sacrificed and cardiac tissues were collected.
MI model
For the acute MI model, a corresponding group of C57BL/6J male mice (8–10 weeks) were treated with AAV9 vectors carrying the cTnT promoter-driven ADAR2 overexpression and its control (Hanbio Biotechnology, China) at a dose of 1011 vg/mice via tail vein injection 1 week before the surgery. The MI model was constructed by ligating the left anterior descending coronary artery (LAD) using a 7/0 silk thread. The sham group was treated by the same experimental process, except that the ligation part was not executed. To determine the area at risk (AAR) and the infarct size (IS) of MI, the heart was stained with TTC 24 h after surgery. For TTC straining, 1 mL 1% Evans Blue was injected into the left ventricle before the mice were sacrificed. ImageJ software (NIH, USA) was used to calculate the AAR/left ventricle (LV) weight and IS/AAR. The IS and AAR were measured as previously described.49 The ratio of AAR/LV was determined to evaluate the homogeneity of the surgery, and IS/AAR was calculated to assess the MI severity. Plasma miR-208a and miR-499 have been reported to be biomarkers of myocardial injury.50 To further verify this, cTnT (Roche Diagnostics, Germany), miR-208a, and miR-499 were evaluated in mice plasma to further evaluate the homogeneity of the surgery. Due to ADAR2 overexpression exhibiting a protective effect on AMI (24 h) surgery, we advanced the time point for plasma high-sensitive cTnT, miR-208a, and miR-499 examination at 6 h after MI surgery (Figure S9). For MI remodeling, MI or sham surgery was immediately performed after mice were subjected to tail vein injection, after 3 weeks of remodeling, the cardiac function was detected by echocardiography, and the hearts were collected for further examination. All of the surgeries and analyses were performed by investigators blinded to the treatment.
DOX-induced cardiotoxicity model
The corresponding group of C57BL/6J male mice (8–10 weeks) were treated with AAV9 vectors carrying the cTnT promoter-driven ADAR2 overexpression and its control (Hanbio Biotechnology, China) at a dose of 1011 vg/mice via tail vein injection 1 week before the first DOX injection. To establish DOX -induced cardiotoxicity, mice were administered DOX at a dose of 5 mg/kg weekly for 5 consecutive weeks. The cardiac function was detected by echocardiography and the hearts were collected for further examination.
Echocardiography
Echocardiography measurements was performed by Vevo 2100 (VisualSonics, Canada) with a center frequency of 30 MHz Scan head to detect heart function as previously described.49 M mode echocardiograms were obtained at the papillary muscle level to measure the LVEF and the LV short-axis shortening rate (FS). The baseline cardiac function of the mice before MI surgery (for AMI and MI remodeling) and DOX treatment used in this study are shown in Figure S10.
Quantitative real-time PCRs
Total RNA was isolated using the TRIZOL RNA extraction kit (Invitrogen, USA). The RNA concentration was measured by Nanodrop 2000 (Thermo Fisher, USA), and reverse transcription of 400 ng RNA was performed using iScript reverse transcription supermix (Bio-Rad, USA) according to the manufacturer’s instructions. SYBR Green supermix (Bio-Rad, USA) and the ABI-7900 Real-Time PCR Detection System (Applied Biosystems, USA) were used for quantitative PCR and analysis. Glyceraldehyde 3-phosphate dehydrogenase (GAPDH) or 18 s RNA were used as internal controls for gene expression. The primer sequences used in this study are listed in Table S1. For quantitative miRNA analysis, the Bull-Loop miRNA qPCR primer set (RiboBio, China) was used to determine the expression level of miRNA. U6 was used as an internal control. The relative expression level was calculated using the 2−ΔΔCt method.
Immunofluorescent and TUNEL staining for frozen sections
Heart samples were cut into 5-μm-thick sections. Subsequently, the sections were fixed in 4% paraformaldehyde (PFA), permeabilized with 0.2% Triton X-100, and blocked at room temperature. Next, the sections were incubated with α-actinin antibody (1:200, A7811, Sigma-Aldrich, USA) at 4°C overnight. After washing with PBS, the sections were incubated with Cy3 AffiniPure Goat Anti-Mouse IgG (immunoglobulin G) (H+L) (1:200, 115-165-003, Jackson, USA). To quantify apoptosis, the TUNEL assay was performed using the DeadEnd Fluorometric TUNEL system (Promega, USA) according to the manufacturer’s instructions. DAPI was used for nuclear counterstaining, and the number of TUNEL+ nuclei was counted. Images were captured by fluorescent microscope (Zeiss, Germany).
Masson’s trichrome staining
Heart tissues were fixed in 4% PFA and then embedded in paraffin. Paraffin sections were dewaxed and hydrated. The 5-μm-thick sections were fixed with 4% PFA and then subjected to Masson’s trichrome staining (Servicebio, China) according to the manufacturer’s instructions. Images were taken by a Nikon Eclipse microscope (Nikon, Japan) and the images were analyzed with NIS Elements (Nikon). Image-Pro Plus software (Media Cybernetics, USA) was used to quantify the fibrotic area. The percentage of fibrosis was calculated as fibrosis areas/total myocardial areas.
WGA staining
For WGA staining, paraffin sections were dewaxed and hydrated. The sections were fixed, immersed, and rinsed in PBS. Sections were incubated with WGA dye (Thermo Fisher, W21405) for 30 min at 37°C and washed with PBS. DAPI (Servicebio) was used for nuclear counterstaining. Images were taken by a Nikon Eclipse microscope and the images were analyzed with NIS Elements. ImageJ software was used to measure the cell size.
Statistical analysis
All of the data were analyzed using GraphPad Prism 8 (USA) and presented as means ± SDs. The Shapiro-Wilk test was performed to test the normality of all of the data. For the two independent samples, when the data followed a Gaussian distribution, an unpaired, two-tailed Student’s t test was used; when the data did not follow a Gaussian distribution, we used the Mann-Whitney U test. For the multiple groups data, we performed two-way ANOVA followed by Tukey’s multiple comparisons test. For the data that did not follow a Gaussian distribution, we used robust two-way ANOVA by using R package WRS2.51 The R function pbad2way was used to perform robust two-way ANOVA followed by pairwaiseMeidan test in R package rcompanion for post hoc tests. Differences were considered significant with p < 0.05. An extended methods section is available in the online supplemental information, Supplemental materials and methods section
Acknowledgments
This work was supported by grants from the National Key Research and Development Project (2018YFE0113500 to J.X.), the National Natural Science Foundation of China (81900359 to J.L.; 81730106 and 81670347 to X.L.; 81800358 to L.W.; and 82020108002 and 81911540486 to J.X.), the Innovation Program of Shanghai Municipal Education Commission (2017-01-07-00-09-E00042 to J.X.), the Science and Technology Commission of Shanghai Municipality (20DZ2255400 and 18410722200 to J.X.), the Shanghai Sailing Program (19YF1416400 to J.L.), the “Dawn” Program of Shanghai Education Commission (19SG34 to J.X.), and the Natural Science Foundation of Shanghai (19ZR1474100 to L.W.). X.L. is an Associate Fellow at the Collaborative Innovation Center for Cardiovascular Disease Translational Medicine. J.P.S. is supported by the European Research Council (ERC) ERC-2016-COG EVICARE (No. 725229).
Author contributions
J.X. and X.L. designed the project and supervised the experiments. X.W., L.W., K.W., J.L., R.C., X.W., G.N., and C.L. performed the experiments and analyzed the data. S.D. and J.P.G.S. provided critical advice. J.X. and L.W. wrote the original draft of the manuscript. J.X., X.L., and J.P.G.S. revised and edited the manuscript. All of the authors have read and approved the final manuscript.
Declaration of interests
The authors declare no competing interests.
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.ymthe.2021.07.004.
Contributor Information
Xinli Li, Email: xinli3267_nj@hotmail.com.
Junjie Xiao, Email: junjiexiao@shu.edu.cn.
Supplemental information
References
- 1.Hill J.A., Olson E.N. Cardiac plasticity. N. Engl. J. Med. 2008;358:1370–1380. doi: 10.1056/NEJMra072139. [DOI] [PubMed] [Google Scholar]
- 2.Boström P., Mann N., Wu J., Quintero P.A., Plovie E.R., Panáková D., Gupta R.K., Xiao C., MacRae C.A., Rosenzweig A., Spiegelman B.M. C/EBPβ controls exercise-induced cardiac growth and protects against pathological cardiac remodeling. Cell. 2010;143:1072–1083. doi: 10.1016/j.cell.2010.11.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Young D.R., Reynolds K., Sidell M., Brar S., Ghai N.R., Sternfeld B., Jacobsen S.J., Slezak J.M., Caan B., Quinn V.P. Effects of physical activity and sedentary time on the risk of heart failure. Circ. Heart Fail. 2014;7:21–27. doi: 10.1161/CIRCHEARTFAILURE.113.000529. [DOI] [PubMed] [Google Scholar]
- 4.Hogg M., Paro S., Keegan L.P., O’Connell M.A. RNA editing by mammalian ADARs. Adv. Genet. 2011;73:87–120. doi: 10.1016/B978-0-12-380860-8.00003-3. [DOI] [PubMed] [Google Scholar]
- 5.Nishikura K. Functions and regulation of RNA editing by ADAR deaminases. Annu. Rev. Biochem. 2010;79:321–349. doi: 10.1146/annurev-biochem-060208-105251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Stellos K., Gatsiou A., Stamatelopoulos K., Perisic Matic L., John D., Lunella F.F., Jaé N., Rossbach O., Amrhein C., Sigala F., et al. Adenosine-to-inosine RNA editing controls cathepsin S expression in atherosclerosis by enabling HuR-mediated post-transcriptional regulation. Nat. Med. 2016;22:1140–1150. doi: 10.1038/nm.4172. [DOI] [PubMed] [Google Scholar]
- 7.Shelton P.M., Duran A., Nakanishi Y., Reina-Campos M., Kasashima H., Llado V., Ma L., Campos A., García-Olmo D., García-Arranz M., et al. The Secretion of miR-200s by a PKCζ/ADAR2 Signaling Axis Promotes Liver Metastasis in Colorectal Cancer. Cell Rep. 2018;23:1178–1191. doi: 10.1016/j.celrep.2018.03.118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Tran S.S., Jun H.I., Bahn J.H., Azghadi A., Ramaswami G., Van Nostrand E.L., Nguyen T.B., Hsiao Y.E., Lee C., Pratt G.A., et al. Widespread RNA editing dysregulation in brains from autistic individuals. Nat. Neurosci. 2019;22:25–36. doi: 10.1038/s41593-018-0287-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Moore J.B., 4th, Sadri G., Fischer A.G., Weirick T., Militello G., Wysoczynski M., Gumpert A.M., Braun T., Uchida S. The A-to-I RNA Editing Enzyme Adar1 Is Essential for Normal Embryonic Cardiac Growth and Development. Circ. Res. 2020;127:550–552. doi: 10.1161/CIRCRESAHA.120.316932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.El Azzouzi H., Vilaça A.P., Feyen D.A.M., Gommans W.M., de Weger R.A., Doevendans P.A.F., Sluijter J.P.G. Cardiomyocyte Specific Deletion of ADAR1 Causes Severe Cardiac Dysfunction and Increased Lethality. Front. Cardiovasc. Med. 2020;7:30. doi: 10.3389/fcvm.2020.00030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Altaf F., Vesely C., Sheikh A.M., Munir R., Shah S.T.A., Tariq A. Modulation of ADAR mRNA expression in patients with congenital heart defects. PLoS ONE. 2019;14:e0200968. doi: 10.1371/journal.pone.0200968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Zhou S.S., Jin J.P., Wang J.Q., Zhang Z.G., Freedman J.H., Zheng Y., Cai L. miRNAS in cardiovascular diseases: potential biomarkers, therapeutic targets and challenges. Acta Pharmacol. Sin. 2018;39:1073–1084. doi: 10.1038/aps.2018.30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Lv D., Liu J., Zhao C., Sun Q., Zhou Q., Xu J., Xiao J. Targeting microRNAs in Pathological Hypertrophy and Cardiac Failure. Mini Rev. Med. Chem. 2015;15:475–478. doi: 10.2174/1389557515666150324124751. [DOI] [PubMed] [Google Scholar]
- 14.Small E.M., Olson E.N. Pervasive roles of microRNAs in cardiovascular biology. Nature. 2011;469:336–342. doi: 10.1038/nature09783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Liu X., Xiao J., Zhu H., Wei X., Platt C., Damilano F., Xiao C., Bezzerides V., Boström P., Che L., et al. miR-222 is necessary for exercise-induced cardiac growth and protects against pathological cardiac remodeling. Cell Metab. 2015;21:584–595. doi: 10.1016/j.cmet.2015.02.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Boon R.A., Iekushi K., Lechner S., Seeger T., Fischer A., Heydt S., Kaluza D., Tréguer K., Carmona G., Bonauer A., et al. MicroRNA-34a regulates cardiac ageing and function. Nature. 2013;495:107–110. doi: 10.1038/nature11919. [DOI] [PubMed] [Google Scholar]
- 17.Yang Y., Cheng H.W., Qiu Y., Dupee D., Noonan M., Lin Y.D., Fisch S., Unno K., Sereti K.I., Liao R. MicroRNA-34a Plays a Key Role in Cardiac Repair and Regeneration Following Myocardial Infarction. Circ. Res. 2015;117:450–459. doi: 10.1161/CIRCRESAHA.117.305962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Nishikura K. A-to-I editing of coding and non-coding RNAs by ADARs. Nat. Rev. Mol. Cell Biol. 2016;17:83–96. doi: 10.1038/nrm.2015.4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kawahara Y., Zinshteyn B., Sethupathy P., Iizasa H., Hatzigeorgiou A.G., Nishikura K. Redirection of silencing targets by adenosine-to-inosine editing of miRNAs. Science. 2007;315:1137–1140. doi: 10.1126/science.1138050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Yang W., Chendrimada T.P., Wang Q., Higuchi M., Seeburg P.H., Shiekhattar R., Nishikura K. Modulation of microRNA processing and expression through RNA editing by ADAR deaminases. Nat. Struct. Mol. Biol. 2006;13:13–21. doi: 10.1038/nsmb1041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kawahara Y., Zinshteyn B., Chendrimada T.P., Shiekhattar R., Nishikura K. RNA editing of the microRNA-151 precursor blocks cleavage by the Dicer-TRBP complex. EMBO Rep. 2007;8:763–769. doi: 10.1038/sj.embor.7401011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Chang K., Marran K., Valentine A., Hannon G.J. Creating an miR30-based shRNA vector. Cold Spring Harb. Protoc. 2013;2013:631–635. doi: 10.1101/pdb.prot075853. [DOI] [PubMed] [Google Scholar]
- 23.Cesarini V., Silvestris D.A., Tassinari V., Tomaselli S., Alon S., Eisenberg E., Locatelli F., Gallo A. ADAR2/miR-589-3p axis controls glioblastoma cell migration/invasion. Nucleic Acids Res. 2018;46:2045–2059. doi: 10.1093/nar/gkx1257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Potter S.C., Luciani A., Eddy S.R., Park Y., Lopez R., Finn R.D. HMMER web server: 2018 update. Nucleic Acids Res. 2018;46(W1):W200–W204. doi: 10.1093/nar/gky448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Bernardo B.C., Gao X.M., Winbanks C.E., Boey E.J., Tham Y.K., Kiriazis H., Gregorevic P., Obad S., Kauppinen S., Du X.J., et al. Therapeutic inhibition of the miR-34 family attenuates pathological cardiac remodeling and improves heart function. Proc. Natl. Acad. Sci. USA. 2012;109:17615–17620. doi: 10.1073/pnas.1206432109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Fornes O., Castro-Mondragon J.A., Khan A., van der Lee R., Zhang X., Richmond P.A., Modi B.P., Correard S., Gheorghe M., Baranašić D., et al. JASPAR 2020: update of the open-access database of transcription factor binding profiles. Nucleic Acids Res. 2020;48(D1):D87–D92. doi: 10.1093/nar/gkz1001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Bernardo B.C., Ooi J.Y.Y., Weeks K.L., Patterson N.L., McMullen J.R. Understanding Key Mechanisms of Exercise-Induced Cardiac Protection to Mitigate Disease: Current Knowledge and Emerging Concepts. Physiol. Rev. 2018;98:419–475. doi: 10.1152/physrev.00043.2016. [DOI] [PubMed] [Google Scholar]
- 28.Vega R.B., Konhilas J.P., Kelly D.P., Leinwand L.A. Molecular Mechanisms Underlying Cardiac Adaptation to Exercise. Cell Metab. 2017;25:1012–1026. doi: 10.1016/j.cmet.2017.04.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Wu G., Zhang X., Gao F. The epigenetic landscape of exercise in cardiac health and disease. J. Sport Health Sci. 2020 doi: 10.1016/j.jshs.2020.12.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Wang L., Wang J., Cretoiu D., Li G., Xiao J. Exercise-mediated regulation of autophagy in the cardiovascular system. J. Sport Health Sci. 2020;9:203–210. doi: 10.1016/j.jshs.2019.10.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Bartel D.P. MicroRNAs: genomics, biogenesis, mechanism, and function. Cell. 2004;116:281–297. doi: 10.1016/s0092-8674(04)00045-5. [DOI] [PubMed] [Google Scholar]
- 32.Wang L., Lv Y., Li G., Xiao J. MicroRNAs in heart and circulation during physical exercise. J. Sport Health Sci. 2018;7:433–441. doi: 10.1016/j.jshs.2018.09.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Heale B.S., Keegan L.P., McGurk L., Michlewski G., Brindle J., Stanton C.M., Caceres J.F., O’Connell M.A. Editing independent effects of ADARs on the miRNA/siRNA pathways. EMBO J. 2009;28:3145–3156. doi: 10.1038/emboj.2009.244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Liu Z., Wang J., Cheng H., Ke X., Sun L., Zhang Q.C., Wang H.W. Cryo-EM Structure of Human Dicer and Its Complexes with a Pre-miRNA Substrate. Cell. 2018;173:1191–1203.e12. doi: 10.1016/j.cell.2018.03.080. [DOI] [PubMed] [Google Scholar]
- 35.Gu S., Jin L., Zhang Y., Huang Y., Zhang F., Valdmanis P.N., Kay M.A. The loop position of shRNAs and pre-miRNAs is critical for the accuracy of dicer processing in vivo. Cell. 2012;151:900–911. doi: 10.1016/j.cell.2012.09.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Malek Mohammadi M., Abouissa A., Azizah I., Xie Y., Cordero J., Shirvani A., Gigina A., Engelhardt M., Trogisch F.A., Geffers R., et al. Induction of cardiomyocyte proliferation and angiogenesis protects neonatal mice from pressure overload-associated maladaptation. JCI Insight. 2019;5:e128336. doi: 10.1172/jci.insight.128336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Laflamme M.A., Murry C.E. Heart regeneration. Nature. 2011;473:326–335. doi: 10.1038/nature10147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Shi J., Bei Y., Kong X., Liu X., Lei Z., Xu T., Wang H., Xuan Q., Chen P., Xu J., et al. miR-17-3p Contributes to Exercise-Induced Cardiac Growth and Protects against Myocardial Ischemia-Reperfusion Injury. Theranostics. 2017;7:664–676. doi: 10.7150/thno.15162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Hill J.A. Braking bad hypertrophy. N. Engl. J. Med. 2015;372:2160–2162. doi: 10.1056/NEJMcibr1504187. [DOI] [PubMed] [Google Scholar]
- 40.Zincarelli C., Soltys S., Rengo G., Koch W.J., Rabinowitz J.E. Comparative cardiac gene delivery of adeno-associated virus serotypes 1-9 reveals that AAV6 mediates the most efficient transduction in mouse heart. Clin. Transl. Sci. 2010;3:81–89. doi: 10.1111/j.1752-8062.2010.00190.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Podleśny-Drabiniok A., Marcora E., Goate A.M. Microglial Phagocytosis: A Disease-Associated Process Emerging from Alzheimer’s Disease Genetics. Trends Neurosci. 2020;43:965–979. doi: 10.1016/j.tins.2020.10.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Huang H., Weng H., Chen J. m6A Modification in Coding and Non-coding RNAs: Roles and Therapeutic Implications in Cancer. Cancer Cell. 2020;37:270–288. doi: 10.1016/j.ccell.2020.02.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Moreira J.B.N., Wohlwend M., Wisløff U. Exercise and cardiac health: physiological and molecular insights. Nat. Metab. 2020;2:829–839. doi: 10.1038/s42255-020-0262-1. [DOI] [PubMed] [Google Scholar]
- 44.Bazak L., Haviv A., Barak M., Jacob-Hirsch J., Deng P., Zhang R., Isaacs F.J., Rechavi G., Li J.B., Eisenberg E., Levanon E.Y. A-to-I RNA editing occurs at over a hundred million genomic sites, located in a majority of human genes. Genome Res. 2014;24:365–376. doi: 10.1101/gr.164749.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Jain M., Jantsch M.F., Licht K. The Editor’s I on Disease Development. Trends Genet. 2019;35:903–913. doi: 10.1016/j.tig.2019.09.004. [DOI] [PubMed] [Google Scholar]
- 46.Landgraf P., Rusu M., Sheridan R., Sewer A., Iovino N., Aravin A., Pfeffer S., Rice A., Kamphorst A.O., Landthaler M., et al. A mammalian microRNA expression atlas based on small RNA library sequencing. Cell. 2007;129:1401–1414. doi: 10.1016/j.cell.2007.04.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Chiang H.R., Schoenfeld L.W., Ruby J.G., Auyeung V.C., Spies N., Baek D., Johnston W.K., Russ C., Luo S., Babiarz J.E., et al. Mammalian microRNAs: experimental evaluation of novel and previously annotated genes. Genes Dev. 2010;24:992–1009. doi: 10.1101/gad.1884710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Kawahara Y., Megraw M., Kreider E., Iizasa H., Valente L., Hatzigeorgiou A.G., Nishikura K. Frequency and fate of microRNA editing in human brain. Nucleic Acids Res. 2008;36:5270–5280. doi: 10.1093/nar/gkn479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Tao L., Bei Y., Chen P., Lei Z., Fu S., Zhang H., Xu J., Che L., Chen X., Sluijter J.P., et al. Crucial Role of miR-433 in Regulating Cardiac Fibrosis. Theranostics. 2016;6:2068–2083. doi: 10.7150/thno.15007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Wang G.K., Zhu J.Q., Zhang J.T., Li Q., Li Y., He J., Qin Y.W., Jing Q. Circulating microRNA: a novel potential biomarker for early diagnosis of acute myocardial infarction in humans. Eur. Heart J. 2010;31:659–666. doi: 10.1093/eurheartj/ehq013. [DOI] [PubMed] [Google Scholar]
- 51.Mair P., Wilcox R. Robust statistical methods in R using the WRS2 package. Behav. Res. Methods. 2020;52:464–488. doi: 10.3758/s13428-019-01246-w. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.