

Abstract
CTG repeat expansion (CTGexp) is associated with aberrant alternate splicing that contributes to cardiac dysfunction in myotonic dystrophy type 1 (DM1). Excision of this CTGexp repeat using CRISPR-Cas resulted in the disappearance of punctate ribonuclear foci in cardiomyocyte-like cells derived from DM1-induced pluripotent stem cells (iPSCs). This was associated with correction of the underlying spliceopathy as determined by RNA sequencing and alternate splicing analysis. Certain genes were of particular interest due to their role in cardiac development, maturation, and function (TPM4, CYP2J2, DMD, MBNL3, CACNA1H, ROCK2, ACTB) or their association with splicing (SMN2, GCFC2, MBNL3). Moreover, while comparing isogenic CRISPR-Cas9-corrected versus non-corrected DM1 cardiomyocytes, a prominent difference in the splicing pattern for a number of candidate genes was apparent pertaining to genes that are associated with cardiac function (TNNT, TNNT2, TTN, TPM1, SYNE1, CACNA1A, MTMR1, NEBL, TPM1), cellular signaling (NCOR2, CLIP1, LRRFIP2, CLASP1, CAMK2G), and other DM1-related genes (i.e., NUMA1, MBNL2, LDB3) in addition to the disease-causing DMPK gene itself. Subsequent validation using a selected gene subset, including MBNL1, MBNL2, INSR, ADD3, and CRTC2, further confirmed correction of the spliceopathy following CTGexp repeat excision. To our knowledge, the present study provides the first comprehensive unbiased transcriptome-wide analysis of the differential splicing landscape in DM1 patient-derived cardiac cells after excision of the CTGexp repeat using CRISPR-Cas9, showing reversal of the abnormal cardiac spliceopathy in DM1.
Keywords: myotonic dystrophy, induced pluripotent stem cells, CRISPR ribonucleoprotein, alternate splicing, spliceopathy, cardiomyogenic differentiation, transcriptome, ribonuclear foci, gene editing
Graphical abstract
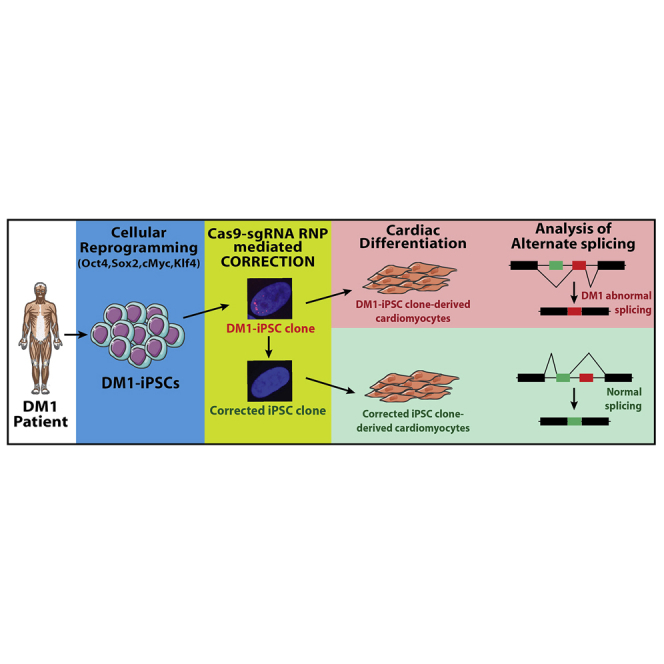
DM1 is due to pathogenic CTG repeats in the DMPK gene that trigger spliceopathy and cardiac dysfunction. Comprehensive transcriptome-wide analysis shows that CRISPR-Cas9-mediated excision of the CTG repeats reverses spliceopathy in DM1 patient-derived cardiomyocyte-like cells. These corrected versus non-corrected isogenic lines provide new molecular insights in DM1 pathology.
Introduction
Myotonic dystrophy type 1 (DM1) is an autosomal dominant multisystemic disorder mainly affecting the skeletal muscles and heart. Apart from muscular atrophy and weakness, a well-established clinical feature in congenital DM1 patients is heart disease, with sudden cardiac arrest being the predominant cause of mortality.1,2 Hence, it is important to validate new therapeutic strategies that can correct the cardiac defects in DM1 patients. DM1 is caused by a trinucleotide repeat expansion (CTGexp) at the 3′ untranslated region (UTR) of the dystrophia myotonica protein kinase (DMPK) gene on chromosome 19.3,4 Transcription of the DMPK gene with a CTGexp repeat leads to expanded DMPK transcripts. These transcripts consequently fold into a supercoiled stem-loop structure that forms complexes with intracellular RNA-binding proteins, resulting in ribonuclear foci, which accumulate in the cell nuclei. The toxic RNA gain-of-function hypothesis is the widely accepted central pathogenesis mechanism for DM1.5, 6, 7, 8 These ribonuclear foci play a central role in the pathogenesis of DM1. In particular, the RNA-binding proteins muscleblind-like 1 (MBNL1) and CUG-binding protein 1 (CUGBP1)4,9,10 are thought to play a role in DM1 pathogenesis. MBNL1 and CUGBP1 are regulators of alternative splicing, exhibit antagonistic effects, and are important in developmental regulation of splicing events.11 During the fetal to adult transition, nuclear levels of MBNL1 are increased and CUGBP1 levels are decreased, leading to a switch from fetal to adult splicing.4,11 Due to the combination of the MBNL1 loss of function and the CUGBP1 gain of function in DM1 pathology, alternative splicing of specific genes is reverted back to the embryonic splicing pattern with retention of fetal-specific exons in precursor (pre-)mRNA of DM1 affected cells.4,11 This abnormal splicing pattern is one of the hallmarks of DM1 and contributes to its pathogenicity.9,12, 13, 14, 15 Consequently, correction of the spliceopathy is a prime target of current therapeutic interventions in DM1 and serves as an important biological marker to analyze its efficacy.
We and others have recently shown that excision of the CTGexp repeat using CRISPR-Cas9 corrects the abnormal splicing defect in DM1 patient-derived myoblast or mesoangioblast-derived myotubes.16,17 However, it is not known whether precise excision of the CTGexp repeat using CRISPR-Cas9 technology is sufficient to completely reverse the underlying spliceopathy in DM1 cardiomyocytes. Moreover, comprehensive genome-wide studies assessing the global impact of the CTGexp repeat on the abnormal cardiac splicing pattern in DM1 are currently lacking. Unfortunately, primary DM1 cardiomyocytes cannot readily be retrieved and expanded in culture, hampering these studies.
In the current study, we therefore aim to overcome this limitation by relying on cardiomyocyte-like cells derived from DM1 patient-specific induced pluripotent stem cells (DM1-iPSCs). This represents an attractive in vitro cellular DM1 disease model to assess the precise molecular consequences of CRISPR-Cas9-mediated gene-specific excision of the pathogenic CTGexp repeat in cardiomyocyte-like cells. To take into account intrinsic interpatient and/or cell line variability in splicing patterns, we used four isogenic pairs of CRISPR-Cas9-corrected and non-corrected iPSC lines that we previously described.16 To our knowledge, the present study provides the first comprehensive transcriptome-wide assessment of the global impact of CRISPR-Cas9-mediated excision of the CTGexp repeat on the splicing landscape in DM1 patient-derived cardiomyocyte-like cells. In our current study, we have broadly looked into two aspects of the transcriptome: the differential expression pattern of genes (DEGs) and their individual transcripts (DETs). Second, we have compared the differentially alternative spliced genes (DASGs) in CRISPR-Cas9 corrected and non-corrected DM1 cardiomyocyte-like cells. The current study reveals new potential disease marker genes and demonstrates that cardiac spliceopathy was effectively reversed using CRISPR-Cas9, supporting its use as a potentially attractive treatment modality to correct the cardiac dysfunction in DM1 patients.
Results
Cardiomyogenic differentiation of CRISPR-Cas9-corrected DM1 patient-specific iPSCs
The human iPSC (hiPSC) clones from healthy donors and CRISPR-Cas9-corrected and non-corrected isogenic iPSC clonal lines from DM1 patients16 (Figures 1A and 1B; Figures S1A, S2A, and S2D) were induced to undergo cardiomyogenic differentiation. The required specific growth and differentiation conditions followed a three-stage process (Figure 1A) based on (1) mesodermal commitment, (2) cardiac mesodermal induction, and (3) cardiomyocyte maturation toward contracting cardiomyocyte-like cells. These contracting cardiomyocyte-like cell clusters emerged around day 14 after initiation of cardiomyogenic differentiation (Figure 1C; Figures S1B and S2B) and were maintained in cardiomyocyte maturation medium between day 14 and day 20 (Figure 1A). A total of 10 iPSC lines sourced from two DM1 patients (i.e., patient FL: FL8 and FL5; patient L: L81 and L23) and two healthy donors (wild-type [WT]: C1 and C7) were established (Table S1). The 10 iPSC lines consisted of four CRISPR-Cas9-corrected DM1-iPSC lines (i.e., FL8-C07, FL5-C12, L81-C10, and L23-C03) (Figure 1B; Figures S1A and S2A), four non-corrected DM1-iPSC lines (i.e., FL8-C35, FL5-C28, L81-C08, and L23-C06) (Figure 1B; Figures S1A and S2A) and two wild-type iPSC lines (i.e., WT-C1 and WT-C7) (Figure 1B; Figure S2A). In order to minimize variation, we obtained isogenic pairs of CRISPR-Cas9-corrected DM1-iPSC lines and their non-corrected counterparts, derived from the same parent DM1-iPSC line (Table S1). For instance, the CRISPR-Cas9-corrected FL8-C07 iPSCs and non-corrected FL8-C35 iPSCs were obtained from the parental DM1 FL8 iPSC line. Prior to DM1-iPSCs to cardiac differentiation, the iPSC clonal lines were quality controlled for any potential off-target mutation caused by transient delivery of the Cas9-single guide RNA (sgRNA) ribonucleoprotein (RNP) complexes.16 Off-target analysis of eight iPSC clonal lines for the five most probable sites based on computational prediction algorithms (which included a site at human splicing factor 3b, subunit 2 [hSF3B2] gene locus with a role in alternate splicing) revealed no off-target activity at those sites (Figures S9A and S9B). This is in line with our previous report where we had detected background level off-target activity between 0.2% and 0.3%, upon constitutive expression of CRISPR-Cas9 components.16
Figure 1.

Cardiomyogenic differentiation of CRISPR-Cas9 RNP-corrected and non-corrected DM1 induced pluripotent stem cells (DM1-iPSCs)
(A) Schematic representation of the various stages of the generation of iPSC-derived cardiomyogenic cells from human iPSCs. The flowchart also depicts the step of magnetic activated cell sorting (MACS) of VCAM+ and/or SIRPA+ cells after differentiation. (B) Representative phase contrast images of DM1-iPSC clonal lines (DM1 patient FL: FL5 C12 and FL5 C28) obtained after CRISPR-Cas9 treatment and clonal isolation. A healthy wild-type (WT) iPSC line (WT-C1) cultured under feeder-free conditions is also shown (scale bars, 250 μm). (C) Representative phase contrast images of DM1 and WT iPSC-derived cardiomyogenic differentiation cells (iPSC-Cardio) obtained after day 14 of differentiation. The cardiomyogenic beating cell clusters are dense multi-layered structures marked with black arrowheads (scale bars, 100 μm). (D) Representative image of DM1 cells and iPSC-Cardio stained for RNA foci by fluorescence in situ hybridization (FISH). Cardiomyogenic cells from CRISPR-Cas9-corrected DM1-iPSC clones (DM1 patient L: L81-C10 and L23-C03) and non-corrected DM1-iPSC clones (DM1 patient L: L81-C08 and L23-C06) were analyzed. Healthy WT iPSC line-derived cardiomyogenic cells (WT-C1) were taken as the negative control for FISH staining. An antisense Cy3-labeled probe was used against trinucleotide CUGexp RNA. Selected nuclei from the non-corrected samples were zoomed into (bottom panel) to show the ribonuclear foci (red) indicated by arrowheads. Upper panel represents stained nuclei at lower magnification (scale bars, 20 μm), and the bottom-most panel represents higher magnification of the selected region. Nuclei were counterstained with DAPI. (E) Graph representing foci per nucleus for cardiomyocytes derived from CRISPR-Cas9-corrected and non-corrected DM1-iPSCs. WT iPSC-derived cardiomyocytes were taken as negative controls. The foci per nucleus were calculated and are represented by each data point in the graph. The data are represented as mean ± SEM. (F) Dual staining for MBNL1 protein and analysis of localization with a DMPK mutated transcript with CTGexp repeats in the ribonuclear foci complex in the corrected FL8-C7 DM1-iPSC- and non-corrected FL8-C35 DM1-iPSC-derived cardiac cells. WT cells (WT-C7) were also stained as a healthy control. Each representative image is a maximum intensity z projection of the z slices. Enlarged views of selected ribonuclear foci negative (NF−) and positive (NF+) nuclei are represented under different filters. Nuclei were counterstained with DAPI.
Characterization of CRISPR-Cas9-corrected DM1-iPSC-derived cardiomyocyte-like cells
One of the main cellular pathological characteristics of DM1 is the accumulation of ribonuclear foci in the nuclei of affected cells. Ribonuclear foci formation was therefore assessed in the DM1-iPSC-derived cardiomyocyte-like cells. Quantification of ribonuclear foci per nucleus of cardiomyocyte-like cells obtained from the CRISPR-Cas9-corrected DM1-iPSC-derived cell lines (i.e., FL8-C07, FL5-C12, L81-C10, and L23-C03) and WT iPSC lines (i.e., WT-C1 and WT-C7) showed an absence of any ribonuclear foci (Figures 1D and 1E; Figure S1C). In contrast, the non-corrected DM1-iPSC (i.e., FL8-C35, FL5-C28, L81-C08, and L23-C06)-derived cardiomyocyte-like cells retained ribonuclear foci (Figures 1D and 1E; Figures S1C). This provides direct evidence that CRISPR-Cas9-mediated excision of the CTGexp repeat in iPSC-derived cardiomyocyte-like cells results in the disappearance of the pathogenic RNP/DMPK mRNA complexes, which is expected to correct its downstream pathological consequences. Co-localization of the MBNL1 protein with the DMPK mutated transcript is one of the hallmarks of DM1 cells. Therefore, in addition to ribonuclear foci, we also examined the MBNL1 delocalization in the corrected cardiomyocyte-like cells. We performed dual immunofluorescence (IF)-fluorescence in situ hybridization (FISH) co-staining to detect MBNL1 protein and DMPK mutated transcripts with CTGexp repeat in CRISPR-Cas9-corrected versus non-corrected DM1-iPSC-derived cardiomyocyte-like cells (Figure 1F; Figure S10). Analysis of the microscopic data shows the presence of MBNL1 punctate staining (marked by white arrowheads) co-localized with ribonuclear foci in the cardiac cells derived from non-corrected iPSCs (FL8 clone 35, FL5 clone 28, L81clone 08, L23 clone 06) (Figure 1F; Figure S10). Alternatively, in the corrected cells (FL8 clone 07, FL5 clone 12, L81 clone 10, L23 clone 03), where there is complete absence of ribonuclear foci, we observed a concomitant delocalization of the MBNL1 protein in the nucleus (Figure 1F; Figure S10). We also analyzed the localization of DMPK protein in the DM1-iPSC-derived cardiomyocyte-like cells (Figure S13A) and DM1-iPSCs (Figure S13B) with IF-FISH co-staining (for the DMPK mutated transcript) and IF, respectively. Analysis of the stained cells showed no significant difference in DMPK localization in both DM1-iPSC-derived cardiomyocyte-like cells and DM1-iPSCs after correction.
We subsequently verified the transcript levels of selected progenitor and differentiated cardiomyogenic markers in the iPSC-derived differentiated cell population obtained after the fourth stage of the iPSC cardiomyogenic differentiation protocol (Figure 1A). Cardiomyogenic differentiation was consistent with increased expression of characteristic cardiac progenitor genes, including MEF-2C, GATA4, and MLC2A in CRISPR-Cas9-corrected and non-corrected DM1 and WT cardiomyocyte-like cells, relative to the housekeeping gene GAPDH. In contrast, there was no relative increase in expression of these cardiac progenitor genes in non-differentiated CRISPR-Cas9-corrected and non-corrected DM1-iPSCs (Figure 2A). In the fourth stage of the iPSC cardiomyogenic differentiation protocol (Figure 1A), where we obtain cardiomyogenic like cells from cardiac mesodermal cells, the differentiated cells showed increased expression of differentiated cardiac marker genes, including MYH7 and VCAM, relative to GAPDH control, in CRISPR-Cas9-corrected and non-corrected DM1 and WT cardiomyocyte-like cells. In contrast, these differentiated cardiac markers were not induced relative to GAPDH in the corresponding undifferentiated iPSCs used as a control (Figure 2B; Figure S4B). Furthermore, immunofluorescence analysis of CRISPR-Cas9-corrected and non-corrected DM1 and wild-type cardiomyocyte-like cells expressed the cardiac markers NKX2.5, cTNT (Figure 2C; Figures S2C, S3A, and S4A), and SCN5A (sodium channel protein type 5-subunit α) (Figure 2D; Figure S3B), whereas their undifferentiated iPSC counterparts failed to express these cardiac markers. In addition to molecular marker analysis, functional assays were conducted on the differentiated CRISPR-Cas9-corrected iPSC-derived contracting cardiomyocytes by assessing calcium cycling (Figure S5A). Spontaneous dynamic changes in intracellular calcium concentration were observed. These spontaneous rhythmic fluctuations of the intracellular calcium concentration, highlighted with the increase (peak level) and decrease (basal level) in fluorescence activity, i.e., the calcium transients, was consistent with the functional excitation-contraction coupling of the DM1 patient-derived cardiomyocyte-like cells (Figure S5A; Videos S1, S2, S3, S4, S5, and S6). After characterizing the differentiated cells for cardiac marker expression and calcium transport, we quantified the contractility of cardiomyocyte-like cells. To do so, we used an automated contractility analysis tool for the measurement of contracting cardiomyocytes to determine parameters such as beat rate, contraction/ relaxation time, and contraction velocity (Figure S12A).18,19 The analysis of the contraction parameters from differentiated cells derived from four corrected DM1-iPSC lines (FL8-C07, FL5-C12, L81-C10, and L23-C03), four non-corrected DM1-iPSC lines (FL8-C35, FL5-C28, L81-C08, and L23-C06) and two WT iPSC lines (WT-C1 and WT-C7) showed significant increase in contraction velocity in the corrected group (mean contraction velocity: 27.8 ± 5.56 pixels/s) similar to that of wild-type controls (mean contraction velocity: 23.12 ± 6.56 pixels/s), when compared to their non-corrected counterparts (mean contraction velocity: 14.08 ± 2.93 pixels/s) (Figure S12A). This was consistent with the outcomes in other studies where DM1 was compared to healthy controls.20,21 However, there was no significant difference in beat rate from isogenic lines. We observed variable beat rates, with mean beat rate ranging from 12 ± 2.0 beats/min to 63.2 ± 10.3 beats/min (Figure S12D). Additionally, based on our PULSE analysis of the contraction and relaxation time of the beating clusters, the isogenic lines were comparable with mean contraction time ranging between 0.35 ± 0.04 s and 0.61 ± 0.17 s and mean relaxation time ranging between 0.65 ± 0.07 s and 0.85 ± 0.12 s (Figures S12B and S12C). The PULSE analysis was based on beating clusters rather than comparing individual beating cells. Therefore, a more detailed evaluation and analysis of individual beating cells from WT/corrected and non-corrected cell lines might provide additional in-depth characteristics of electrophysiological abnormalities due to DM1.
Figure 2.

Characterization of DM1-iPSC-derived cardiomyogenic cells (iPSC-Cardio)
(A) Quantitative progenitor cardiac markers MEF-2C, GATA4, and MLC2A gene expression relative to iPSCs in all of the cardiomyogenic cells obtained after differentiation. (B) Quantitative differentiated cardiac markers MYH7 and VCAM gene expression relative to iPSCs in all of the cardiomyogenic cells obtained after differentiation. (C) Immunostaining for cardiomyogenic markers such as cTNT and NKX2.5 in CRISPR-Cas9-corrected and non-corrected DM1-iPSC- and WT iPSC-derived cardiomyogenic cells. Scale bars, 100 μm. Nuclei were counterstained with DAPI. (D) Immunostaining for cardiomyogenic marker SCN5A in CRISPR-Cas9-corrected and non-corrected DM1-iPSC- and WT iPSC-derived cardiomyogenic cells. Nuclei were counterstained with DAPI. Scale bars, 20 μm.
Video depicting the cyclic change in intensity is due to influx and efflux of permeable Fluo-4 dye, indicating functional beating cardiomyocytes
Video depicting the cyclic change in intensity is due to influx and efflux of permeable Fluo-4 dye, indicating functional beating cardiomyocytes
Video depicting the cyclic change in intensity is due to influx and efflux of permeable Fluo-4 dye, indicating functional beating cardiomyocytes
Video of contracting cardiomyogenic-like cells
Video of contracting cardiomyogenic-like cells
Video of contracting cardiomyogenic-like cells
Collectively, the above results demonstrate that CRISPR-Cas9-corrected and non-corrected DM1 and WT iPSCs were able to undergo relatively robust cardiomyogenic differentiation. This is consistent with the generation of functional contracting cardiomyocyte-like cells that expressed cardiac marker genes and were able to exhibit calcium transients in the contracting cells. Variable expression levels of cardiac marker genes were apparent between different cell lines derived from different DM1 patients (i.e., FL8, FL5, L81, and L23) and WT donor lines. This likely reflects the well-established intrinsic variability among different iPSC lines, which may in turn have impacted the kinetics and/or robustness of the cardiomyogenic differentiation.
When comparing isogenic cell lines with the same genetic background, two CRISPR-Cas9-corrected DM1-iPSC-derived cardiomyocyte-like cells showed a relatively robust increase in relative expression of cardiac differentiation markers (i.e., GATA4, MLC2A, and MYH7) compared to their non-corrected counterparts (i.e., FL5: C28/C12 and L81: C08/C10) (Figures 2A and 2B). This suggests that the CTGexp repeat may have adversely impacted the differentiation efficiency and/or kinetics in those lines that could potentially be reverted by CRISPR-Cas9-mediated excision of the CTGexp repeat. However, the effect was only partial, since for some marker genes, there appeared to be no substantial difference in expression (e.g., MEF2C and VCAM) in corrected versus non-corrected isogenic lines (Figures 2A and 2B; Figure S4B). Nevertheless, in some isogenic cell lines there was no difference in cardiac marker gene expressions between non-corrected and CRISPR-Cas9-corrected DM1-iPSC-derived cardiomyocyte-like cells (i.e., FL8 [C35/C07] and L23 [C06/C03]) (Figures 2A and 2B; Figure S4B). The exact reason for this difference is not fully understood but may be due to intrinsic clonal variation between different iPSC lines. Therefore, we additionally assessed the transcriptional status of the exogenous reprogramming factors (transgene-encoded exogenous hOCT4, hSOX2, hcMYC, and hKLF4) in the starting DM1-iPSC clones and also analyzed the hOCT4, hSOX2, hcMYC, and hKLF4 transgene integration sites (ISs) in these iPSCs. This was essential to better characterize the consequences of the retroviral reprogramming of the DM1- iPSCs on its subsequent cardiomyogenic differentiation. For analysis of the expression pattern, the overall expression of reprogramming factors (denoted as total hOCT4/hSOX2/hcMYC/hKLF4) was compared to the specific expression of the transgene-encoded reprogramming factors sensu stricto (denoted as transgene hOCT4/hSOX2/hcMYC/hKLF4 [Figures S11A–S11D]; using different primers (Table S2).22 Expression of the transgene-encoded reprogramming factors was repressed in the different iPSC lines, as expected.23,24 Consequently, hOCT4, hSOX2, hcMYC, and hKLF4 expression in iPSCs was mainly derived from the endogenous loci (Figures S11A–S11D). Although hOCT4 and hSOX2 pluripotency genes were expressed at relatively high levels in the iPSC clones, no expression was detected in the differentiated cardiomyocyte-like cells (Figures S11E and S11F), consistent with their more differentiated phenotype. In contrast, only basal hcMYC and hKLF4 expression was apparent in both differentiated cells and iPSCs (Figures S11G and S11H).25, 26, 27, 28, 29
Since expression of the exogenous reprogramming factors by the retroviral vectors is repressed or absent across all iPSC clones and their respective differentiated cardiomyocyte-like cells, it is unlikely that this would have contributed to some of the inter-clonal differences in the extent of cardiomyogenic differentiation.
The objective of the IS analysis (ISA) was to determine whether there were any known classical genes related to cardiac differentiation and functioning that were disrupted following integration of the retroviral vector encoding the reprogramming factors. Analysis of ISA data showed that from a total of 4.4 million sequencing reads, a total of 26 different integration loci could be mapped in the four iPSC lines (Table S3; human chromosome number and sequence orientation are indicated; the hg38 database was used as reference). Further analysis of the genes and integration loci (Table S3) revealed that none of the retroviral ISs was present in genes that are directly involved in cardiac differentiation or in normal cardiac functioning. Moreover, none of the integrations occurred in exons. Notably, 1 of the 26 different retroviral ISs (Ch16; −66927012) was located upstream of the RRAD (Ras-related glycolysis inhibitor and calcium channel regulator) gene that plays a role in electrical and cytoskeletal function and Ca2+ handling in cell cardiomyocytes.30,31 However, it is reassuring that the retroviral integration was not in the RRAD open reading frame itself and consequently the exonic regions of the RRAD gene remained unaffected.
Based on the analysis of the expression pattern of exogenous reprogramming factors and the absence of any disruptions of genes involved in cardiac differentiation and function (as revealed by comprehensive genome-wide ISA), it is unlikely that retroviral-mediated iPSC generation impacted the cardiac differentiation potential of the iPSCs lines used in the current study.
Transcriptome-wide assessment of differential expression pattern in corrected versus non-corrected DM1 cardiomyocyte-like cells
Subsequently, a comprehensive transcriptome-wide assessment was conducted on the global impact of CRISPR-Cas9-mediated excision of the CTGexp repeat on the transcriptome in DM1 patient-derived cardiomyocyte-like cells. In particular, differentially expressed genes (DEGs) and differentially expressed transcripts (DETs) were monitored by RNA sequencing (RNA-seq) in differentiated cardiomyocyte-like cells obtained from isogenic CRISPR-Cas9-corrected versus non-corrected DM1-iPSC lines. DEG analysis allows for assessment of differential gene expression based on all of the transcripts encoded by a given gene indiscriminately and irrespective of differential splicing. In contrast, DET analysis permits identification of each individual differentially expressed transcript encoded by a given gene. The availability of isogenic pairs presented an unprecedented opportunity and advantage to conduct these types of comprehensive comparative DEG and DET analyses, as opposed to merely relying on non-isogenic healthy controls as benchmarks.
DEG analysis of corrected versus non-corrected DM1 cardiomyocyte-like cells for each of the four lines (i.e., FL5, FL8, L23, and L81) revealed that there was a significant difference in expression in less than 1% of all the genes analyzed. Of these DEGs, 54 genes (FL5), 16 genes (FL8), 39 genes (L23), and 42 genes (L81) were upregulated with log2 fold change (FC) > 2 (Figure 3A; Tables S5–S8). Similarly, 45 genes (FL5), 83 genes (FL8), 60 genes (L23), and 57 genes (L81) were downregulated with log2 fold change < –2 (Figure 3A; Tables S5–S8). DEG analysis based on the Gene Ontology (GO) pathways database in cardiomyocyte-like cells derived from CRISPR-Cas-corrected versus non-corrected DM1-iPSCs revealed that the genes that are upregulated or downregulated are potentially linked to biological processes for cardiac and muscle function and their development (Table S9). In particular, the DEGs were associated with cytoskeletal structures, sarcomere, z-disc, nucleic acid binding, calcium ion binding, and transcription factor activity (Table S9).
Figure 3.

Differentially expressed genes (DEGs) and transcripts in the CRISPR-Cas9-corrected DM1 cardiomyogenic cells
(A) Number of DEGs between CRISPR-Cas9-corrected and non-corrected DM1 clones for each of the cell lines (FL5, FL8, L81, and L23). (B) Number of differentially expressed transcripts (DETs) between the four CRISPR-Cas9-corrected and non-corrected DM1 clonal lines. (C) Volcano plots of DETs between the four CRISPR-Cas9-corrected and non-corrected DM1 clonal lines together. The x axis depicts log2 fold change, and the y axis depicts –log10 p value for each of the DETs. (D) Heatmap of DETs between the four CRISPR-Cas9-corrected and non-corrected DM1 clonal lines plotted separately based on the Z score.
After analysis of the RNA sequencing data, we obtained a large dataset corresponding to DETs wherein 733 and 866 specific transcripts were either differentially upregulated or downregulated, respectively, upon CRISPR-Cas9-mediated excision of CTGexp (Figure 3B). In order to identify common DETs between CRISPR-Cas9-corrected and non-corrected lines, the four non-corrected cell lines and their four CRISPR-Cas9-corrected isogenic counterparts were grouped in two categories (i.e., group 1: non-corrected DM1 FL5-C28, FL8-C35, L23-C06, and L81-C08 versus group 2: CRISPR-Cas9-corrected DM1 FL5-C12, FL8-C07, L23-C03, and L81-C10). Out of the above-identified set of DETs, 99 most significant upregulated/downregulated transcripts based on the log2 fold change and p values were selected for downstream analysis (Figures 3C and 3D; Tables S10 and S11). From the 99 transcript candidates, 44 transcripts were upregulated in the control DM1 lines (i.e., log2 fold change > 6.06) relative to the isogenic CRISPR-Cas9-corrected DM1 lines. Furthermore, 55 transcripts were downregulated in the control DM1 lines (i.e., log2 fold change < −8.78), relative to the isogenic CRISPR-Cas9-corrected DM1 line controls (Figures 3C and 3D; Tables S10 and S11). Unbiased analysis of these selected upregulated and downregulated DETs based on the GO pathways database and literature data revealed that seven of the DETs (i.e., TPM4, CYP2J2, DMD, MBNL3, CACNA1H, ROCK2, and ACTB) were associated with cardiac development, maturation, and function, including expression of gated ion channels32, 33, 34, 35, 36, 37 (Figure 3C; Table S11). In addition, another three DET transcripts were involved in alternative splicing as splicing regulators or a component of the spliceosomal complex (i.e., SMN2, GCFC2, MBNL3)35,38 (Figure 3C; Table S11). The other candidate genes whose transcripts were differentially expressed were associated in neuronal development and function, glucose metabolism, regulation of transcription, and nucleic acid binding, among others (Table S11).
Hence, these newly identified DETs (Tables S10 and S11; Figures 3C and 3D) may play a role in DM1 pathology, but their exact contribution requires further investigation beyond the scope of the current study. Most importantly, the changes in gene expression patterns in isogenic CRISPR-Cas9-corrected versus non-corrected DM1 cardiomyocyte-like cells and the corresponding outcomes from the GO enrichment analysis are consistent with the notion that CTGexp repeats impact cardiac function and interfere with the splicing machinery.
Correcting the abnormal splicing landscape in DM1 cardiomyogenic cells using CRISPR-Cas9
A detailed analysis of the alternative splicing landscape in CRISPR-Cas-corrected versus non-corrected DM1 patient-derived cardiomyocyte-like cells was subsequently conducted. The RNA sequencing output data were therefore subjected to a computational analysis using the rMATS bioinformatics algorithm (replicate multivariate analysis of alternative splicing, version 4.0).39 Differentially spliced genes were categorized under five different spliced groups, namely (1) skipped exons (SEs), (2) alternative 5′ splice site (A5SS), (3) alternative 3′ splice site (A3SS), (4) mutually exclusive exons (MXEs), and (5) retained introns (RIs) (Figure 4A).
Figure 4.

Alternative splicing comparison analysis of the DM1 versus CRISPR-Cas9-corrected cardiomyogenic cells
(A) Schematic representation of five different categories of alternative splicing patterns. (B) Number of significant differentially alternate spliced genes (DASGs) detected under each splice category. (C) Venn chart depicting intersection of genes between different splicing categories. Selection threshold for the genes were as follows: SE, p ≤ 0.001, false discovery rate (FDR) ≤ 0.05; RI, p ≤ 0.05, FDR ≤ 0.69; MXE, p ≤ 0.05, FDR ≤ 0.58; A5SS, p ≤ 0.05, FDR ≤ 0.53; A3SS, p ≤ 0.05, FDR ≤ 0.52. (D) List depicting functional enriched candidates from human phenotype (HP) database obtained with set of DASGs from the DM1 versus corrected group. (D) Heatmap of selected DASGs between four DM1 clonal lines and four corrected clonal lines plotted separately based on the Z score calculated from percent splice in (PSI) values.
In order to identify DASGs common between all of the cardiomyogenic lines, the four non-corrected cell lines and their CRISPR-Cas9-corrected isogenic counterparts were grouped together in two categories (i.e., group 1: non-corrected DM1 FL5-C28, FL8-C35, L23-C06, and L81-C08 versus group 2: CRISPR-Cas9-corrected DM1 FL5-C12, FL8-C07, L23-C03, and L81-C10). The percent spliced in (or inclusion [PSI], or Ψ) was determined for all genes between the non-corrected versus CRISPR-Cas-corrected cohorts, on which basis delta Ψ (ΔΨ = Ψgroup1 − Ψgroup2) was determined for each gene.39 Genes with a highly significant p value and false discovery rate (FDR) were selected for downstream analysis. The number of DASGs with a high threshold were sorted according to the different alternative splicing categories as shown in Figure 4B. Most of the gene candidates encoding transcripts that had undergone a splicing event were mutually exclusive, belonging to one of the five distinct splicing categories, whereas a relatively smaller number of genes encoding alternatively spliced transcripts belonged to different splicing categories (Figure 4C; Table S12).
DASGs (Table S12) with high threshold values were subsequently analyzed using the human phenotype (HP) database.40 A prominent pattern consistent with the DM1 disease phenotype was apparent (Figure 4D; Table S13). In particular, functionally enriched candidate genes include genes affecting muscle tone and abnormal muscle physiology, movement, and myotonia, but also certain central nervous system abnormalities (Figure 4D; Table S13).41, 42, 43 Hence, the rMATS analysis in conjunction with the HP database revealed that the alternatively spliced genes detected in DM1 patient-derived cardiomyocyte-like cells reflect well-known DM1 patient characteristics and indicate a shift of DM1 spliceopathy toward a healthy state after CRISPR-Cas9 correction.
Following the identification of the candidate genes corresponding to the different splicing categories (i.e., SE, A5SS, A3SS, MXE, and RI) with significant ΔΨ, the high confidence target genes were plotted as heatmaps based on their Z scores (Figure 4E; Figure S8; Tables S14, S15, S16, and S17). Heatmaps showed a distinct set of DASGs, which were either upregulated or downregulated after CRISPR-Cas9-mediated excision of the CTGexp in the cardiomyocyte-like cells. For this list of DASGs, a subset of genes was selected, which are known to be abnormally alternatively spliced in DM1.44, 45, 46, 47, 48, 49 Ψ analysis in CRISPR-Cas9-corrected versus non-corrected cardiomyocyte-like cell lines revealed a difference in the splicing pattern for a number of candidate genes (Figure 5; Figures S6 and S7; Table S18), in particular genes that are associated with cardiac function (i.e., TNNT, TNNT2, TTN, TPM1, SYNE1, CACNA1A, MTMR1, NEBL, and TPM1), cellular signaling (i.e., NCOR2, CLIP1, LRRFIP2, CLASP1, and CAMK2G), and DM1 disease markers (i.e., NUMA1, MBNL2, and LDB3) in addition to the disease-causing DMPK gene itself.
Figure 5.

Graphs of selected candidate genes with PSI values
The PSI values (denoted as Ψ on the y axis) for selected genes reported to be associated with cardiomyogenic cells were plotted. Each data point represents each of the lines: CRISPR-Cas9-corrected (DM1 patient 1: FL5-C12 and FL8-C07; DM1 patient 2: L81-C10 and L23-C03) and non-corrected DM1-iPSC-derived (DM1 patient 1: FL5-C28 and FL8-C35; DM1 patient 2: L81-C08 and L23-C06) cardiomyogenic cells.
Validation of the CRISPR-Cas9-mediated correction of spliceopathy correction by reverse transcriptase PCR (RT-PCR)
To further validate the reversal of aberrant splicing by CRISPR-Cas9 in DM1 cardiomyocyte-like cells, RT-PCR analysis was conducted. In order to eliminate any non-differentiated and partially differentiated cells from the bulk cardiomyocyte-like cell population, fully differentiated cardiomyocyte-like cells with a single and double VCAM+/SIRPA+ surface marker were further enriched from the bulk population using magnetic-activated cell sorting (MACS) (Figure 1A). These MACS-enriched differentiated cells showed increased expression of differentiated cardiac marker genes including MYH7 and VCAM, compared to GAPDH control, in CRISPR-Cas9-corrected and non-corrected DM1 and WT cardiomyocyte-like cells (Figure S4B), consistent with the results obtained in the unsorted population (Figures 2A and 2B). Having obtained a purified cell population, we then analyzed the impact of the CRISPR-Cas9-mediated excision of the CTGexp repeat on the spliceopathy in DM1 cardiomyocyte-like cells. The splicing pattern was analyzed for five selected biomarkers, that is, MBNL1, MBNL2, insulin receptor (INSR), adducin 3 (ADD3) and CREB-regulated transcription co-activator 2 (CRTC2), known to be associated with DM1 pathogenesis in cardiac cells of DM1 affected patients.1,48,50 For MBNL1 and MBNL2, the isoform that excluded exon 7 (exon 7−) was increased in the CRISPR-Cas9-corrected DM1 cardiomyocyte-like cells (i.e., FL8, FL5, L81, and L23) as compared to the non-corrected DM1 cardiomyocyte-like cells for each of the respective isogenic cell lines (Figures 6A–6C). Similarly, in the case of MBNL2, there was a significant increase in the ratio of exon 7 exclusion in the CRISPR-Cas9-corrected DM1 cardiomyocyte-like cells (i.e., FL8, L81, and L23) as compared to the non-corrected DM1 cardiomyocyte-like cells for each of the respective isogenic cell lines. In the case of FL5 we did observe a similar trend, although the difference was not significant. For INSR, semiquantitative analysis of the exon 11 isoforms (Figure 6B) revealed in most clonal lines (i.e., FL8, FL5, and L23, with the exception of L81) a significant decrease in the ratio of exon 11 exclusion in the CRISPR-Cas9-corrected DM1 cardiomyocyte-like cells, as compared to the non-corrected DM1 cardiomyocyte-like cells (Figures 6B and 6D). With respect to the ADD3 alternative splicing pattern, the ratio of exon 16 inclusion was analyzed. For the CRISPR-Cas9-corrected FL8, FL5, and L23 cardiomyogenic lines, we observed a significant decrease in the exon 16 inclusion ratio compared to their non-corrected DM1 counterparts (Figures 6B and 6E). Finally, when analyzing the splicing pattern of CRTC2, we observed a significant increase in the exon 13 inclusion ratio for the CRISPR-Cas9-corrected FL8 and L81 lines compared to their non-corrected DM1 controls (Figure 6F). Taken together, these results demonstrate that there is an overall reversal of the DM1 alternate splicing pattern in the CRISPR-Cas9-corrected DM1-iPSC-derived differentiated cardiomyocyte-like cells when compared to their non-corrected isogenic controls. In particular, the reversal of the abnormal splicing pattern was observed for MBNL1, MBNL2, INSR, ADD3, and CRTC2 in the large majority of independent DM1-iPSC-derived cardiomyocyte-like cell cultures. However, in some of the DM1-iPSC-derived cardiomyogenic cell cultures there was a reversal of abnormal splicing for most but not all of the markers after CRISPR-Cas9-editing (e.g., L81, INSR and ADD3; FL5 and L23, CRTC2; Figure 6). This may be due to factors such as variability between patient samples and innate differences in progression of the cardiac differentiation process.
Figure 6.

Analysis of alternate splicing pattern in differentiated cardiomyocytes
(A–F) Reversal of defective alternative splicing pattern in CRISPR-Cas9-corrected (DM1 patient 1: FL5-C12 and FL8-C07; DM1 patient 2: L81-C10 and L23-C03) and non-corrected DM1-iPSC-derived (DM1 patient 1: FL5-C28 and FL8-C35; DM1 patient 2: L81-C08 and L23-C06) cardiomyogenic cells. Cardiomyogenic cells obtained from Cas9- and 5′ and 3′ CTGrepeat-sgRNA-treated DM1-iPSCs were analyzed for alternative splicing with primers specific to candidate splicing genes MBNL1, MBNL2, INSR, ADD3, and CRTC2. The excluded and included exons for each candidate gene are denoted on the right of the gel image (see Figure 3B). The ratios of the band intensity of the alternate isoforms are enumerated based on band intensity and depicted in the form of a graph. GAPDH was used as loading control. ∗∗∗p < 0.001, unpaired two-tailed t test (±SEM).
Discussion
DM1 patients mainly suffer from severe muscle and cardiac dysfunction. Although cardiomyopathy is less prevalent in these patients compared to muscle dysfunction,43 it is associated with worse outcomes and mortality.51,52 DM1 patients with heart ailments predominantly suffer from conduction defects and atrial or ventricular tachyarrhythmia. With increased age and disease severity, the cardiac conditions worsen with second- and third-degree heart block and left ventricular hypertrophy.1,2,43,53 Histopathological analysis of affected hearts from DM1 patients have reported fibrosis and multifocal myofibrillar loss.1,2,53,54 Although DM1 cardiac abnormalities stem from aberrant alternate cellular splicing due to CTGexp repeat mutation in the 3′ UTR region of the DMPK gene, the global impact of the CTGexp repeat on the cardiac spliceopathy in DM1 is not fully understood, since comprehensive transcriptome-wide studies are lacking.3,16
To our knowledge, this is the first comprehensive transcriptome-wide study assessing the global impact of excising the CTGexp repeat on differential expression and splicing in DM1 cardiomyocyte-like cells. The results demonstrate that excision of the CTGexp repeat using CRISPR-Cas9 corrects the underlying spliceopathy in DM1-specific cardiomyocyte-like cells. This is consistent with previous reports showing that CRISPR-Cas9-mediated excision can restore some of the splicing abnormalities in DM1 patient-specific iPSC-derived myotubes,16 transformed muscle cell lines,17 and iPSC-derived neuronal and cardiac cells55 and DM1 patient cardiac samples.48 Nevertheless, these studies were non-comprehensive and based only on a limited set of genes. Moreover, comparing cardiac cells derived from DM1 patients versus healthy controls is not ideal given the significant inter-individual variation. Furthermore, primary cardiomyocytes from DM1 cannot readily be expanded in culture, making it particularly challenging to conduct unbiased transcriptome-wide comprehensive analyses. To overcome these limitations and minimize any potential confounding effects of this inter-individual variation, the current study relied on isogenic CRISPR-Cas9-corrected versus non-corrected DM1 patient-specific iPSC-derived cardiomyocyte-like cells. These isogenic pairs of DM1 patient-specific iPSC-derived cardiomyocyte-like cells were specifically designed to differ with respect to the presence or absence of CTGexp repeats in the DMPK 3′ UTR. The CTGexp repeats were excised by non-viral transfection using a CRISPR-Cas9 ribonuclear particle complex, which represents a “hit and go” strategy that reduces long-term exposure to the CRISPR-Cas9 components, minimizing the potential risk of off-target genome editing or untoward immune reactions directed against the edited cells in vivo, as opposed to other CRISPR-Cas9-based strategies that rely on plasmid transfection55 or stable lentiviral transduction.16 To our knowledge, this is the first study addressing the impact of CRISPR-Cas9-mediated excision of the CTGexp repeats in DM1 patient iPSC-derived cardiac cells by transcriptome-wide analysis. In contrast, previous studies were based on targeted integration of a transcriptional termination site (i.e., polyadenylation site) in the DMPK locus using either TALENs47 or CRISPR-Cas9.55 Although targeted integration of the polyadenylation site resulted in suppression of the pathogenic CUGexp RNA expression, the overall efficacy was relatively modest compared to the present strategy, since it relied on homology-directed recombination (HDR) and selective clonal enrichment. In contrast, the current strategy is HDR-independent and relies on non-homologous end joining (NHEJ), which is far more efficient, obviating the need for selective enrichment.
The RNA sequencing revealed that less than 1% of all the genes showed significant differential expression in the DM1 patient-specific iPSC-derived cardiomyocyte-like cells after CRISPR-Cas9-mediated CTGexp excision. This is consistent with the notion that DM1 is not associated with gross developmental cardiac abnormalities and is primarily a spliceopathy that is expected to result in more subtle changes in expression and/or splicing patterns of transcripts rather than drastic and widespread differences in gene expression levels.56 The use of isogenic lines may also have reduced the intrinsic variation and narrowed down the most significant DEGs. GO pathways analysis of the DEGs revealed that the fraction of genes that are significantly upregulated or downregulated are linked to biological processes that are associated with cardiac and muscle function and development. Additionally, our analysis of DETs revealed a larger set of individual transcripts that were significantly differentially expressed between the CRISPR-Cas9-corrected and non-corrected samples. On further analysis of the DETs, certain transcripts were of particular interest since the corresponding genes are thought to play a role in cardiac development, maturation, and function (i.e., TPM4, CYP2J2, DMD, MBNL3, CACNA1H, ROCK2, and ACTB).32, 33, 34, 35, 36, 37 The DETs were also associated with alternate splicing or as components of the spliceosomal complex (SMN2, GCFC2, and MBNL3).35,38 In particular, the SMN complex helps to assemble the cellular machinery needed to process pre-mRNA. The SMN complex is also important for the development of dendrites and axons. GCFC2 is involved in pre-mRNA splicing through regulating spliceosome C complex formation and may play a role during late-stage splicing events and turnover of excised introns. Interestingly, diseases associated with GCFC2 include dyslexia and reading disorder. MBNL3 mediates pre-mRNA alternative splicing regulation such as on TNNT2 and INSR pre-mRNA (see below). Some of these DETs are potentially new candidate genes that may contribute to DM1 pathology and represent novel targets for therapeutic intervention. This warrants further validation that is beyond the scope of the current study. By focusing specifically on the DASGs using the rMATS algorithm, DASGs affecting muscle tone and abnormal muscle physiology, movement, and myotonia, but also certain central nervous system abnormalities were identified. When comparing CRISPR-Cas-corrected versus non-corrected cardiomyocyte-like cell lines, a significant difference in the splicing pattern for a number of candidate genes was apparent pertaining to genes that are associated with cardiac function (i.e., TNNT, TNNT2, TTN, TPM1, SYNE1, CACNA1A, MTMR1, NEBL, and TPM1), cellular signaling (i.e., NCOR2, CLIP1, LRRFIP2, CLASP1, and CAMK2G), and DM1 disease markers (i.e., NUMA1, LDB3, and MBNL2) in addition to the disease-causing DMPK gene itself. In conclusion, the transcriptome-wide comprehensive analyses based on DET and DASG analysis in conjunction with specific bioinformatics platforms revealed that CRISPR-Cas9-mediated excision of the CTGexp repeat in DM1 reverses the abnormal expression and splicing patterns in cardiomyocyte-like cells. This further corroborates the relevance of the in vitro DM1 patient-derived cardiomyocytes as an in vitro disease model for DM1 and its usefulness to test and validate CRISPR-Cas9-based gene editing modalities.
Our study confirmed the spliceopathy observed in clinical samples consistent with abnormal alternate splicing of the INSR, MBNL1, MBNL2, ADD3, and CRTC2 genes in the DM1 patient-specific iPSC-derived cardiomyocyte-like cells. Moreover, both the DM1-iPSCs and their cardiomyocyte-like progeny exhibited ribonuclear foci, one of the characteristic features of DM1. The abnormal splicing in these specific genes is associated with some of the clinical manifestations of DM1, including insulin resistance in muscle and heart cells,57, 58, 59, 60 abnormal transition from the fetal to adult splicing pattern in DM1 patients,11 and defective neuronal architecture and cardiac conduction defects.48,61, 62, 63, 64, 65
MBNL1 and MBNL2 play an important role in DM1 disease pathogenesis by forming a toxic ribonuclear complex with CUGexp RNA, thus severely disrupting normal cellular splicing of numerous other genes as well as their own expression. This results in the exclusion of exon 7 in the MBNL1 and MBNL2 transcripts in DM1 cells, as was demonstrated in the non-corrected DM1 cardiomyocyte-like cells. This aberrant MBNL1 and MBNL2 splicing pattern was restored in most CRISPR-Cas9-corrected DM1 patient-specific iPSC-derived cardiomyocyte-like cells, consistent with the disappearance of the ribonuclear foci and the restoration of the normal intracellular localization of MBNL1.16 This is also in line with previous studies in Mbnl1 knockout (KO) mouse models that result in a characteristic DM1 cardiac phenotype.1 However, depending on the genetic background of the mouse strain, different outcomes have been reported in transgenic mouse studies. In particular, in another Mbnl1−/− KO mice model based on a different genetic background, no such cardiac phenotype was apparent,66 suggesting that unknown genetic factors may account for differences in cardiac phenotypes.1 Hence, the use of DM1 patient-specific iPSC-derived cellular models is complementary to in vivo mouse studies. Nevertheless, epigenetic differences between iPSC-derived cardiomyocyte-like cells as compared to bona fide cardiomyocytes from DM1 patients can come with their own set of limitations.
In addition, other genes such as INSR, CRTC2, and ADD3 all play vital roles in the DM1 phenotype. For instance, INSR is an important gene that is aberrantly spliced in DM1 patients, consistent with abnormal splicing in the DM300 DM1 mouse model.67 The aberrant alternative splicing of INSR causes a reduced responsiveness of muscle cells to the metabolic effects of insulin, leading to insulin resistance in some DM1 patients.57, 58, 59, 60 Similarly, CRTC2 has a role in mitochondrial biogenesis and is abnormally spliced in DM1 disease models with cardiac conduction defects.61,62 ADD3 is also misspliced in DM1 heart muscle cells48,63 and plays an important role in actin capping, modulating actin filament growth, endothelial junctions, and cardiac conduction defects.48,64,65 Therefore, correction of the abnormal splicing pattern of MBNL1, MBNL2, INSR, CRTC2, and ADD3 that are associated with some of the main clinical manifestations of DM1 may have contributed to reversing DM1 to a more normal phenotype.
Nevertheless, in some of the DM1-iPSC-derived cardiomyogenic cell cultures there was a reversal of abnormal splicing after CRISPR-Cas9-mediated excision of the CTGexp repeat for most but not all of the abnormally spliced genes. The exact reason for this difference is not fully understood. However, it is well established that there is inter-clonal variation among individual iPSC clones. Hence, unknown genetic or epigenetic differences among the different iPSC-derived clones and/or their cardiomyogenic progeny may have accounted for the clone-dependent differences in splicing outcomes. Based on the analysis of the expression pattern of exogenous reprogramming factors and the absence of any disruption of genes involved in cardiac differentiation and function (as revealed by comprehensive genome-wide ISA), it is unlikely that retroviral-mediated iPSC generation impacted the cardiac differentiation potential of the iPSC lines used in the current study.
The functionality of our DM1 iPSC-derived cardiomyocyte-like cells was confirmed by analyzing their contractility parameters and expression of cardiac differentiation markers Nevertheless, there are certain aspects of the differentiation process that require further improvement. Cardiomyogenic differentiation cultures produce a mixture of different cardiac subtypes, e.g., ventricular, atrial, nodal, and Purkinje cardiomyocytes.68, 69, 70, 71, 72 This mixture of cardiac subtypes poses a significant challenge for in vivo applications and in vitro studies requiring specific cardiac subtype cells. Moreover, the kinetics and overall efficiency73,74 of cardiomyogenic differentiation may vary depending on the iPSC clone, reflecting epigenetic and/or genetic differences. Another aspect regarding in vitro differentiation of hiPSCs into cardiac cells in 2D and 3D cultures is impacted by incomplete maturation compared to bona fide human adult cardiomyocytes, despite expression of mature cardiac markers, the manifestation of synchronous coordinated contractions, and other functional characteristics.75,76 The ultimate goal of obtaining fully mature cardiac tissues derived from iPSCs that mimic all of the features of adult heart tissue has not yet been realized due to the block in the fetal-to-adult differentiation. This aspect of incomplete cardiomyocyte maturation is particularly relevant in the case of DM1 where the transition from fetal to adult splicing patterns is blocked due to MBNL1/2 sequestration.4 Despite these intrinsic limitations of in vitro DM1 disease modeling, one of the strengths of the current study consists of the use of isogenic CRISPR-Cas9-corrected versus non-corrected clonal lines. Consequently, this cellular platform facilitated the comprehensive analysis of DEGs and transcripts, providing further molecular insights into the spliceopathy associated with DM1. In the current study for some iPSC clones, it would seem that CRISPR-Cas9-mediated excision of the CTGexp repeat might have enhanced cardiomyogenic differentiation, in accordance with the increased expression of cardiac differentiation markers and contraction velocity. This would be consistent with previous reports suggesting that CTGexp repeat excision enhances myogenic differentiation by overcoming the fetal splicing pattern arrest.4,17 However, in some iPSC clones the CRISPR-Cas9-mediated excision of the CTGexp repeat did not seem to have any effect, which might be due to the limitations of cardiomyogenic differentiation as discussed above, which could interfere with the ability to assess the impact of CRISPR-Cas9 correction on their cardiomyogenic differentiation potential. Hence, there is a need to identify newer methods and molecules to reliably guide the differentiation so as to obtain a highly purified population of cardiac subtypes.73,74 Further optimizations are warranted such that the phenotype of the iPSC-derived differentiated cardiomyocyte-like cells matches the properties of bona fide human cardiomyocytes as much as possible, with respect to cell size and structure, mechanical properties, electrophysiology, metabolism, and calcium handling. The use of 3D scaffolds mimicking the native microenvironment might provide more optimized conditions to further facilitate cardiac differentiation.
Our current study sets the stage for subsequent preclinical studies in DM1 animal models to further validate the efficacy and safety of both cardiac and muscle repair in anticipation of future clinical trials in patients suffering from DM1. This will require viral vector (i.e., AAV)- or non-viral vector (i.e., nanoparticle)-mediated delivery of the CRISPR-Cas components and characterization of the cardiac and muscle phenotypes based on the current study and our previous study,16 respectively. The current study also revealed new potential disease marker genes that are differentially expressed and differentially alternatively spliced after CTGexp repeat excision, paving the way to new insights in the molecular pathophysiology of DM1.
Materials and methods
Culturing of hiPSCs
hiPSC clones from healthy donors and CRISPR-Cas9-corrected and non-corrected iPSC clones from DM1 patients were generated as described.16 Briefly, the iPSCs were obtained following transduction with retroviral vectors expressing the human reprogramming factors hOCT4, hSOX2, hKLF4, and hcMYC. The CTGexp repeat was excised based on a “hit-and-go” correction strategy using non-viral Cas9-sgRNA RNP complexes.16 The selected iPSC clonal lines were cultured on a feeder-free Geltrex (Thermo Scientific, USA) matrix with Essential 8 (Thermo Scientific, USA) medium. The iPSC lines were subcultured using 50 mM EDTA (ethylenediaminetetraacetate; Thermo Scientific, USA) in Dulbecco’s phosphate-buffered saline (DPBS; Thermo Scientific, USA) at 70%–80% confluency. The cultures were supplemented with fresh media daily and passaged at a split ratio of 1:4 to 1:6.
Cardiomyogenic differentiation of iPSCs
Cardiomyogenic differentiation of iPSC clonal lines was carried out using the PSC cardiomyocyte differentiation kit (Thermo Scientific, USA, catalog #A29212-01). During initiation of the differentiation protocol, confluent iPSCs with 70%–80% confluency were treated with TrypLE (Thermo Scientific, USA) and plated on Geltrex-coated six-well plates with 1.5 × 105 cells/well using Essential 8 medium supplemented with 10 μM Y-27632 rock inhibitor (RI, STEMCELL Technologies, Canada). Fresh Essential 8 medium without RI was replenished the next day, followed by media change every 24 h with Essential 8 until the cultures attained 60%–70% confluency. After attaining the desired confluency, the iPSC lines were exposed to PSC cardiomyocyte medium. The PSC cardiomyocyte medium kit involves a three-step differentiation protocol comprised of three media components for primary mesodermal commitment followed by a cardiac mesodermal induction and finally a cardiomyocyte maturation step. Focal areas with beating cells were the indication of cardiomyocyte maturation, which was observed beyond day 14 of the cardiomyocyte differentiation protocol. Cells for downstream experiments were harvested between day 16 and day 20.
MACS sorting of single and double VCAM+/ SIRPA+ cardiomyocytes
In order to obtain a homogenously differentiated cell population after cardiomyogenic differentiation, we performed MACS-based positive selection with phycoerythrin (PE)-conjugated anti-CD172a (SIRPA, Miltenyi Biotec, Germany, catalog #130099781) and anti-CD106 (VCAM, Miltenyi Biotec, Germany, catalog #130104125) antibodies. To prepare the cells for antibody staining, the formed cardiac aggregates were dissociated with 0.25% trypsin-EDTA (Thermo Scientific, USA) for 4 min at 37°C. After cellular dissociation, the required cells were stained with PE-conjugated anti-CD172a and anti-CD106 antibodies, according to the manufacturer’s protocol. The labeled cells were then resuspended in MACS buffer containing anti-PE microbeads (Miltenyi Biotec, Germany, catalog #130105639) and incubated for 15 min at 4°C. Microbead-conjugated cells were subsequently washed with MACS buffer to remove unbound microbeads. Thereafter, the conjugated cells were resuspended in MACS buffer and applied onto the primed MACS-LS columns (Miltenyi Biotec, Germany) for positive selection with subsequent washes, as per the manufacturer’s protocol. After MACS sorting the resultant CD172a+ and CD106+ cells were collected for further downstream gene expression and alternate splicing analysis.
Flow cytometry
For cardiac troponin T (cTNT) staining, cells were harvested as mentioned above and fixed with 4% paraformaldehyde (PFA). The fixed cells were washed and then permeabilized with 0.1% Triton X-100 for 10 min followed by a brief centrifugation at 1,800 rpm for 5 min. Thereafter, the cells were incubated with fluorescein isothiocyanate (FITC)-conjugated anti-cTNT (Miltenyi Biotec, Germany, catalog #130-119-674) in fluorescence-activated cell sorting (FACS) buffer (1% bovine serum albumin [BSA] with 0.1% sodium azide in PBS) for 10 min at room temperature (RT). For CD106 (VCAM) staining, the harvested cells were incubated with PE-conjugated anti-CD106 (Miltenyi Biotec, Germany # 130-104-163) in FACS buffer for 20 min at 4°C. Next, the labeled cells were washed and subjected to flow cytometry analysis. Flow analysis was performed on at least 10,000 events per sample using a LSRFortessa III (BD Biosciences, USA) flow cytometer. Data acquisition was performed using FACSDiva (BD Biosciences, USA) and analyzed using FCSalyzer 9.15.
Immunocytochemistry
The differentiated cells were plated onto chambered glass slides (BD Falcon, USA) and cultured for 3 days prior to immunostaining. For iPSCs, staining was performed 2 days after plating of cells. For immunostaining, cells were washed with PBS (Sigma-Aldrich, USA) and fixed with 4% PFA (Sigma-Aldrich, USA) for 10 min at RT. Fixed cells were permeabilized with 0.2% Triton X-100 (Sigma-Aldrich, USA) and 1% BSA (Sigma-Aldrich, USA) in PBS for 15 min at RT. After blocking with donkey (5%) or goat serum (5%) (Sigma-Aldrich, USA) for 30 min at RT, primary antibodies specific for TNT (cTNT, Abcam, UK, 1:200 dilution), NK2 homeobox 5 (Nkx2.5, Abcam, UK, 1:200 dilution), SCN5A (Abcam, UK, 1:200 dilution), and DMPK (catalog #ab183860, Abcam, UK, 1:100 dilution) were added to the respective chambers and incubated overnight at 4°C. Cells were washed and the respective secondary anti-mouse, anti-rabbit antibodies conjugated with Alexa Fluor 488 (Life Technologies, USA) and anti-rabbit, 546-nm fluorochrome (Molecular Probes, USA) were used at a 1:500 dilution for 1 h at RT followed by 4′,6-diamidino-2-phenylindole (DAPI) nuclear staining for 5 min (1:1,000 dilution). After three washes with PBS, the stained slides were mounted using a mounting medium (ProLong diamond antifade mountant; Molecular Probes) and observed under a fluorescent microscope (Olympus IX-81) and a confocal microscope (Zeiss, Germany). All images were analyzed using ImageJ (NIH, https://imagej.nih.gov/ij/) and Zen image analysis software (Zeiss).
RNA isolation and quantitative real-time PCR
To assess the expression of reprogramming factors and cardiac markers post-differentiation, total RNA was isolated from iPSCs and cardiomyocyte-like cells using a QIAGEN RNeasy mini kit (QIAGEN, Germany) according to the manufacturer’s protocol. One microgram of the total RNA per sample was used to synthesize complementary DNA (cDNA) using SuperScript III reverse transcriptase (Thermo Scientific, USA) according to the manufacturer’s instructions. Real-time quantitative (q)PCR was performed using SYBR Green (Thermo Scientific, USA) and specific primers. Primer sequences are included in Table S2. Human GAPDH-specific primer was used as an internal control for normalization.
DNA isolation and ISA
Genomic DNA from the iPSCs was extracted using a QIAGEN DNeasy blood and tissue kit (QIAGEN, Germany) according to the manufacturer’s protocol. The DNA concentrations were determined and adjusted using Qubit fluorometric quantitation. Thereafter, 500 ng of DNA per reaction was taken for downstream processing. The genomic DNA was sheared using Covaris M220, followed by purification and subsequent primer extension using long terminal repeat (LTR)-specific biotinylated primers as described previously.77 The extension product obtained was further purified, followed by the magnetic capture of biotinylated DNA. The captured biotinylated DNA was then processed to ligate the linker cassettes containing the unique barcode sequences. These barcoded DNA fragments were then PCR amplified using biotinylated vector and adaptor-specific primers.77 The resultant products obtained from the first round of PCR were further purified and used as a template for the second round of PCR with primers for downstream deep sequencing analysis by the MiSeq platform (Illumina, USA). Sample preparation for deep sequencing was performed as previously described.78,79 After Illumina sequencing, the data were analyzed to estimate the number of vector-genome junctions (ISs). The relative sequence count of all detected ISs was calculated in relationship to all sequences, which was mapped to a definite position in the genome. Based on that, we identified and analyzed the 10 most prominent ISs based on their sequence counts. ISs were recovered using the S-extension primer tag selection (EPTS)/ligation-mediated (LM)-PCR protocol77 and analyzed using the genome IS analysis pipeline (GENE-IS) analysis platform.80
Off-target analysis
For off-target analysis in iPSCs, candidate genomic loci for the 5′ CTGrepeat-gRNA and 3′ CTGrepeat-gRNA were predicted using the validated algorithm from the Zhang lab (https://zlab.bio/guide-design-resources). From the list of predicted off-target sites identified by this algorithm, we selected the four most probable off-target sites for the 5′ CTGrepeat-gRNA and 3′ CTGrepeat-gRNA that shared significant homology with the actual on-target sites (Figures S9A and S9B) as described previously.16 An additional off-target site (with four mismatched; one bulge) for 5′ CTGrepeat-gRNA located in the hSF3B2 gene on chromosome 11 designated as off-target site 3 was selected for analysis due to role of the hSF3B2 gene in alternate splicing in cells. Genomic DNA (gDNA) from the iPSCs was extracted using QIAGEN DNeasy blood and tissue kit (QIAGEN, Germany) according to the manufacturer’s protocol. Isolated gDNA was used to perform target region PCR amplification of the chosen off-target sites, followed by Sanger sequencing of the PCR amplicons in TOPO cloning vectors. Briefly, PCR amplification was performed by an initial denaturation for 2 min at 98°C, followed by 35 cycles consisting of 10 s at 98°C, 30 s at the 58°C, 30 s at 72°C, and concluded by a final extension of 8 min at 72°C using Phusion Hot Start II high-fidelity DNA polymerase (F549S; Life Technologies) and the respective primers (Table S4) to generate PCR products of sizes 226–387 bp. After PCR and agarose gel electrophoresis, the PCR products were purified using a QIAquick gel extraction kit (QIAGEN). PCR products were cloned into Zero Blunt TOPO vector (K2875-J10, Life Technologies) and transformed into bacteria. Plasmids containing PCR amplicon were extracted using the plasmid mini-prep system (Promega) and sequenced using a T3 sequencing primer (5′-ATTAACCCTCACTAAAGGGA-3′) by Sanger sequencing (GATC, Germany). The sequences were analyzed using Ape software (https://biologylabs.utah.edu). A total number of 240 TOPO cloning PCR amplicons were sequenced.
Intracellular calcium imaging and analysis of cardiac contraction
Intracellular calcium release from the beating cardiac aggregates was observed after incubating the cells with 10 μmol/L of the calcium indicator dye Fluo-4 acetoxymethyl ester (Fluo-4 AM; Life Technologies, USA). Briefly, the beating aggregates were incubated with the fluorescent dye for 15 min at 37°C followed by a 15-min incubation at RT in the dark. The cells were washed with the cardiomyocyte maintenance medium followed by microscopic observation of the calcium efflux under a fluorescence microscope (Olympus IX-81). Changes in Fluo-4 AM fluorescence were recorded while the cells were beating spontaneously, and the images were acquired and analyzed using the software cellSens (Olympus) and ImageJ, respectively.
For analysis of cardiac contraction, spontaneously beating cardiac aggregate recordings (Videos S1, S2, S3, S4, S5, and S6; four to six videos per iPSC line) from corrected (FL5-C12, FL8-C07, L81-C10, and L23-C03), non-corrected (FL5-C28, FL8-C35, L81-C08, and L23-C06), and healthy WT (WT-C1 and WT-C7) groups were recorded using an Olympus IX81 microscope with an XM10 camera (Olympus, Japan) after day 14 of cardiac differentiation. These contraction recordings were analyzed using automated contractility software PULSE (Dana Solutions, Palo Alto, CA, USA)18,19 to determine the contractility parameters (contraction/relaxation time, contraction velocity, and beat rate).
FISH
FISH staining for ribonuclear foci on corrected and non-corrected iPSC-derived cardiomyocyte-like cells was performed as previously described.16 Cells obtained from iPSCs and differentiated cardiac aggregates were pretreated for 1 h with 10 μM Y-27632 RI (STEMCELL Technologies, Canada) prior dissociating them with TrypLE Express (Thermo Scientific USA). Harvested cells were plated on Geltrex-coated chamber slides (Labtek, USA) in Essential 8 medium supplemented with 10 μM Y-27632. The next day, cells were washed three times with PBS and fixed using 4% PFA (Sigma-Aldrich, USA) at RT for 15 min. This was followed by two washes with 70% ethanol for 5 min each and subsequently two rehydration washes with 0.5 mM MgCl2 in PBS for 10 min. Rehydrated cells were then hybridized in a hybridization chamber with a custom peptide nucleic acid (PNA) probe (CY3-5′-CAGCAGCAGCAGCAG-3′) (Eurogentec, Belgium) at a 1:500 dilution in hybridization buffer (0.1% BSA [Sigma-Aldrich, USA] in water, 50% vol of formamide [Thermo Scientific, USA], and 25% vol of saline sodium citrate [SSC] buffer [Life Technologies, USA]) for 90 min at 37°C. Thereafter, the cells were washed with 0.1% Tween 20 (Sigma-Aldrich, USA) in PBS for 5 min at RT followed by an incubation in Tween 20 buffer for 30 min at 45°C. The cells were then washed three times with PBS and stained for DAPI at a 1:500 dilution for 7 min before mounting.
Combined FISH and immunofluorescence specific for MBNL1 was subsequently performed as described previously.81 A polyclonal antibody specific for MBNL1 (catalog #ab45899, Abcam) and DMPK (catalog #ab183860, Abcam) was used as primary antibody at a concentration of 1 μg/mL and 1:100 dilution, respectively, followed by Alexa Fluor 488-conjugated donkey anti-rabbit (Life Technologies) secondary antibody. Before immunofluorescence, FISH staining was performed as described above. Microscopy images were captured using an Olympus IX81 microscope, and z stack acquisition was performed with an Olympus IX81 microscope. Analysis of microscopy images was performed by ImageJ software (NIH, https://imagej.nih.gov/ij/).
RNA sequencing and bioinformatics analysis
Total RNA for the sequencing was isolated using a QIAGEN RNeasy mini kit (QIAGEN, Germany) according to the manufacturer’s protocol. The integrity of the RNA was then analyzed with a 2100 Bioanalyzer (Agilent Technologies, USA). Thereafter, RNA sequencing libraries were prepared with a TruSeq stranded mRNA kit (Illumina, USA) by using the manufacturer’s sample preparation protocol. After library preparation, quality control analysis and quantification were performed using an Agilent Technologies 2100 Bioanalyzer high-sensitivity DNA chip, followed by RNA sequencing on a NovaSeq 6000 platform (Illumina, USA) by LC Sciences (USA).
For data processing, we used Perl scripts and Cutadapt82 to clean the raw data followed by analysis of sequence quality using Fast QC. The clean reads obtained were then mapped to the hG38 genome assembly using HISAT v2.083 and assembled with StringTie v1.3.84 Thereafter, we used Perl scripts and gffcompare to reconstruct a comprehensive transcriptome, and subsequently used StringTie v1.3 and edgeR85 to estimate the expression levels. To compare different transcripts expression levels, FPKM (fragments per kilobase of transcript per million mapped reads) values were used, with the selection criteria log2 fold change > 1 or log2 fold change < −1; p < 0.05, by edge R. rMATS v4.039 in conjunction with AS profile v1.086 was used for the assessment and analysis of alternative splicing. The enrichment analyses of the genes were performed using online tool g:Profiler.40 The heatmaps were generated through the online tool Morpheus (https://software.broadinstitute.org/morpheus) and Venn diagrams through jvenn viewer (http://jvenn.toulouse.inra.fr/app/example.html).
RT-PCR analysis
To investigate the alternative splicing in the sorted and bulk population of the iPSC-derived cardiomyocyte-like cells isolated from the WT, corrected, and non-corrected DM1-iPSC clonal lines, RNA isolation and cDNA synthesis were performed as described above. RT-PCRs were performed with cDNA using the Phusion Hot Start II DNA polymerase kit (Thermo Scientific USA), as published previously,16 to analyze the splicing patterns of MBNL1 and MBNL2 each spanning exon 7, INSR spanning exon 11, and ADD3 spanning exon 16. PCR amplifications were performed under the following conditions: an initial denaturation for 2 min at 98°C, followed by 25–35 cycles at 98°C for 10 s, annealing at 60°C for 30 s and at 72°C for 30 s, followed by a final extension of 2 min at 72°C. All PCR reactions were performed on a S1000 thermal cycler (Bio-Rad, USA). The products were analyzed by electrophoresis on 3% agarose gels containing ethidium bromide and visualized using an AlphaImager HP (Cell Biosciences, USA). Densitometric measurements were performed by quantifying band intensities using the ImageJ analysis tool (NIH, https://imagej.nih.gov/ij/) followed by calculating the percentage of exon inclusion/exclusion of each gene for all cell lines from three independent differentiation experiments. Sequences of primers and PCR conditions used for validation are mentioned in Table S2.
Study design and statistical analysis
The datasets were based on three independent cardiomyogenic differentiation experiments for all 10 iPSC lines. The data are based on a total of four DM1-iPSC lines from two DM1 patients and their respective CRISPR-Cas9-corrected isogenic counterpart. Statistical methods such as an unpaired Student’s t test was performed as indicated with statistical significance level (α) set at 0.05: ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001. Ribonuclear foci were quantified by a z stack acquisition method, as specified accordingly.
Data availability statement
The data that support the findings of this study are available from the corresponding author upon reasonable request.
Acknowledgments
We would like to thank Prof. J. Keith Joung, Dr. Yanfang Fu, and Dr. Deepak Reyon for valuable suggestions and support. This work was supported by research grants to M.C. and T.V. from the Fonds voor Wetenschappelijk Onderzoek (FWO), EU Horizon 2020 (UPGRADE - Unlocking Precision Gene Therapy) - grant agreement N°825825, Koning Boudewijn Stichting - Walter Pyleman Fund grant, Association Belge contre les Maladies Neuromusculaires - ABMM, VUB/UZ Wetenschappelijk Fonds Willy Gepts, VUB Strategic Research Program “Groeier”, VUB Industrieel Onderzoeksfonds (Groups of Expertise in Applied Research -GEAR) and Association Française contre les Myopathies (AFM).
Author contributions
Conception and design, T.V., M.K.C., and S.D.; financial support, T.V. and M.K.C.; administrative support, T.V. and M.K.C.; collection and/or assembly of data, T.V., M.K.C., S.D., D.M., J.T., and K.S.; data analysis and interpretation, T.V., M.K.C., S.D., and D.M.; manuscript writing, T.V., M.K.C., S.D., and D.M.; final approval of manuscript, T.V. and M.K.C.
Declaration of interests
The authors declare no competing interests.
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.ymthe.2021.08.004.
Contributor Information
Marinee K. Chuah, Email: marinee.chuah@vub.be.
Thierry VandenDriessche, Email: thierry.vandendriessche@vub.be.
Supplemental information
References
- 1.Dixon D.M., Choi J., El-Ghazali A., Park S.Y., Roos K.P., Jordan M.C., Fishbein M.C., Comai L., Reddy S. Loss of muscleblind-like 1 results in cardiac pathology and persistence of embryonic splice isoforms. Sci. Rep. 2015;5:9042. doi: 10.1038/srep09042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.van Mil A., Balk G.M., Neef K., Buikema J.W., Asselbergs F.W., Wu S.M., Doevendans P.A., Sluijter J.P.G. Modelling inherited cardiac disease using human induced pluripotent stem cell-derived cardiomyocytes: Progress, pitfalls, and potential. Cardiovasc. Res. 2018;114:1828–1842. doi: 10.1093/cvr/cvy208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Klein A.F., Dastidar S., Furling D., Chuah M.K. Therapeutic approaches for dominant muscle diseases: Highlight on myotonic dystrophy. Curr. Gene Ther. 2015;15:329–337. doi: 10.2174/1566523215666150630120537. [DOI] [PubMed] [Google Scholar]
- 4.Lin X., Miller J.W., Mankodi A., Kanadia R.N., Yuan Y., Moxley R.T., Swanson M.S., Thornton C.A. Failure of MBNL1-dependent post-natal splicing transitions in myotonic dystrophy. Hum. Mol. Genet. 2006;15:2087–2097. doi: 10.1093/hmg/ddl132. [DOI] [PubMed] [Google Scholar]
- 5.Pettersson O.J., Aagaard L., Jensen T.G., Damgaard C.K. Molecular mechanisms in DM1—A focus on foci. Nucleic Acids Res. 2015;43:2433–2441. doi: 10.1093/nar/gkv029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Cho D.H., Tapscott S.J. Myotonic dystrophy: Emerging mechanisms for DM1 and DM2. Biochim. Biophys. Acta. 2007;1772:195–204. doi: 10.1016/j.bbadis.2006.05.013. [DOI] [PubMed] [Google Scholar]
- 7.Udd B., Krahe R. The myotonic dystrophies: Molecular, clinical, and therapeutic challenges. Lancet Neurol. 2012;11:891–905. doi: 10.1016/S1474-4422(12)70204-1. [DOI] [PubMed] [Google Scholar]
- 8.Furling D., Lemieux D., Taneja K., Puymirat J. Decreased levels of myotonic dystrophy protein kinase (DMPK) and delayed differentiation in human myotonic dystrophy myoblasts. Neuromuscul. Disord. 2001;11:728–735. doi: 10.1016/s0960-8966(01)00226-7. [DOI] [PubMed] [Google Scholar]
- 9.Lee J.E., Cooper T.A. Pathogenic mechanisms of myotonic dystrophy. Biochem. Soc. Trans. 2009;37:1281–1286. doi: 10.1042/BST0371281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Jiang H., Mankodi A., Swanson M.S., Moxley R.T., Thornton C.A. Myotonic dystrophy type 1 is associated with nuclear foci of mutant RNA, sequestration of muscleblind proteins and deregulated alternative splicing in neurons. Hum. Mol. Genet. 2004;13:3079–3088. doi: 10.1093/hmg/ddh327. [DOI] [PubMed] [Google Scholar]
- 11.Kalsotra A., Xiao X., Ward A.J., Castle J.C., Johnson J.M., Burge C.B., Cooper T.A. A postnatal switch of CELF and MBNL proteins reprograms alternative splicing in the developing heart. Proc. Natl. Acad. Sci. USA. 2008;105:20333–20338. doi: 10.1073/pnas.0809045105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Mooers B.H., Logue J.S., Berglund J.A. The structural basis of myotonic dystrophy from the crystal structure of CUG repeats. Proc. Natl. Acad. Sci. USA. 2005;102:16626–16631. doi: 10.1073/pnas.0505873102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Klein A.F., Gasnier E., Furling D. Gain of RNA function in pathological cases: Focus on myotonic dystrophy. Biochimie. 2011;93:2006–2012. doi: 10.1016/j.biochi.2011.06.028. [DOI] [PubMed] [Google Scholar]
- 14.Charlet-B N., Savkur R.S., Singh G., Philips A.V., Grice E.A., Cooper T.A. Loss of the muscle-specific chloride channel in type 1 myotonic dystrophy due to misregulated alternative splicing. Mol. Cell. 2002;10:45–53. doi: 10.1016/s1097-2765(02)00572-5. [DOI] [PubMed] [Google Scholar]
- 15.Mankodi A., Takahashi M.P., Jiang H., Beck C.L., Bowers W.J., Moxley R.T., Cannon S.C., Thornton C.A. Expanded CUG repeats trigger aberrant splicing of ClC-1 chloride channel pre-mRNA and hyperexcitability of skeletal muscle in myotonic dystrophy. Mol. Cell. 2002;10:35–44. doi: 10.1016/s1097-2765(02)00563-4. [DOI] [PubMed] [Google Scholar]
- 16.Dastidar S., Ardui S., Singh K., Majumdar D., Nair N., Fu Y., Reyon D., Samara E., Gerli M.F.M., Klein A.F., et al. Efficient CRISPR/Cas9-mediated editing of trinucleotide repeat expansion in myotonic dystrophy patient-derived iPS and myogenic cells. Nucleic Acids Res. 2018;46:8275–8298. doi: 10.1093/nar/gky548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.van Agtmaal E.L., André L.M., Willemse M., Cumming S.A., van Kessel I.D.G., van den Broek W.J.A.A., Gourdon G., Furling D., Mouly V., Monckton D.G., et al. CRISPR/Cas9-induced (CTG⋅CAG)n repeat instability in the myotonic dystrophy type 1 locus: Implications for therapeutic genome editing. Mol. Ther. 2017;25:24–43. doi: 10.1016/j.ymthe.2016.10.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Maddah M., Heidmann J.D., Mandegar M.A., Walker C.D., Bolouki S., Conklin B.R., Loewke K.E. A non-invasive platform for functional characterization of stem-cell-derived cardiomyocytes with applications in cardiotoxicity testing. Stem Cell Reports. 2015;4:621–631. doi: 10.1016/j.stemcr.2015.02.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Maddah M., Shoukat-Mumtaz U., Nassirpour S., Loewke K. A system for automated, noninvasive, morphology-based evaluation of induced pluripotent stem cell cultures. J. Lab. Autom. 2014;19:454–460. doi: 10.1177/2211068214537258. [DOI] [PubMed] [Google Scholar]
- 20.Kim M.S., Fleres B., Lovett J., Anfinson M., Samudrala S.S.K., Kelly L.J., Teigen L.E., Cavanaugh M., Marquez M., Geurts A.M., et al. Contractility of induced pluripotent stem cell-cardiomyocytes with an MYH6 head domain variant associated with hypoplastic left heart syndrome. Front. Cell Dev. Biol. 2020;8:440. doi: 10.3389/fcell.2020.00440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Spitalieri P., Talarico R.V., Caioli S., Murdocca M., Serafino A., Girasole M., Dinarelli S., Longo G., Pucci S., Botta A., et al. Modelling the pathogenesis of myotonic dystrophy type 1 cardiac phenotype through human iPSC-derived cardiomyocytes. J. Mol. Cell. Cardiol. 2018;118:95–109. doi: 10.1016/j.yjmcc.2018.03.012. [DOI] [PubMed] [Google Scholar]
- 22.Kim Y.M., Kang Y.G., Park S.H., Han M.K., Kim J.H., Shin J.W., Shin J.W. Effects of mechanical stimulation on the reprogramming of somatic cells into human-induced pluripotent stem cells. Stem Cell Res. Ther. 2017;8:139. doi: 10.1186/s13287-017-0594-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Takahashi K., Tanabe K., Ohnuki M., Narita M., Ichisaka T., Tomoda K., Yamanaka S. Induction of pluripotent stem cells from adult human fibroblasts by defined factors. Cell. 2007;131:861–872. doi: 10.1016/j.cell.2007.11.019. [DOI] [PubMed] [Google Scholar]
- 24.Lee J., Xia Y., Son M.Y., Jin G., Seol B., Kim M.J., Son M.J., Do M., Lee M., Kim D., et al. A novel small molecule facilitates the reprogramming of human somatic cells into a pluripotent state and supports the maintenance of an undifferentiated state of human pluripotent stem cells. Angew. Chem. Int. Ed. Engl. 2012;51:12509–12513. doi: 10.1002/anie.201206691. [DOI] [PubMed] [Google Scholar]
- 25.Henriksson M., Lüscher B. Proteins of the Myc network: Essential regulators of cell growth and differentiation. Adv. Cancer Res. 1996;68:109–182. doi: 10.1016/s0065-230x(08)60353-x. [DOI] [PubMed] [Google Scholar]
- 26.Kelly K., Cochran B.H., Stiles C.D., Leder P. Cell-specific regulation of the c-myc gene by lymphocyte mitogens and platelet-derived growth factor. Cell. 1983;35:603–610. doi: 10.1016/0092-8674(83)90092-2. [DOI] [PubMed] [Google Scholar]
- 27.McConnell B.B., Yang V.W. Mammalian Krüppel-like factors in health and diseases. Physiol. Rev. 2010;90:1337–1381. doi: 10.1152/physrev.00058.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Polo J.M., Anderssen E., Walsh R.M., Schwarz B.A., Nefzger C.M., Lim S.M., Borkent M., Apostolou E., Alaei S., Cloutier J., et al. A molecular roadmap of reprogramming somatic cells into iPS cells. Cell. 2012;151:1617–1632. doi: 10.1016/j.cell.2012.11.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Waters C.M., Littlewood T.D., Hancock D.C., Moore J.P., Evan G.I. c-myc protein expression in untransformed fibroblasts. Oncogene. 1991;6:797–805. [PubMed] [Google Scholar]
- 30.Belbachir N., Portero V., Al Sayed Z.R., Gourraud J.B., Dilasser F., Jesel L., Guo H., Wu H., Gaborit N., Guilluy C., et al. RRAD mutation causes electrical and cytoskeletal defects in cardiomyocytes derived from a familial case of Brugada syndrome. Eur. Heart J. 2019;40:3081–3094. doi: 10.1093/eurheartj/ehz308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Li Y., Chang Y., Li X., Li X., Gao J., Zhou Y., Wu F., Bai R., Dong T., Ma S., et al. RAD-deficient human cardiomyocytes develop hypertrophic cardiomyopathy phenotypes due to calcium dysregulation. Front. Cell Dev. Biol. 2020;8:585879. doi: 10.3389/fcell.2020.585879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Rajan S., Jagatheesan G., Karam C.N., Alves M.L., Bodi I., Schwartz A., Bulcao C.F., D’Souza K.M., Akhter S.A., Boivin G.P., et al. Molecular and functional characterization of a novel cardiac-specific human tropomyosin isoform. Circulation. 2010;121:410–418. doi: 10.1161/CIRCULATIONAHA.109.889725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Evangelista E.A., Aliwarga T., Sotoodehnia N., Jensen P.N., McKnight B., Lemaitre R.N., Totah R.A., Gharib S.A. CYP2J2 modulates diverse transcriptional programs in adult human cardiomyocytes. Sci. Rep. 2020;10:5329. doi: 10.1038/s41598-020-62174-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Kamdar F., Garry D.J. Dystrophin-deficient cardiomyopathy. J. Am. Coll. Cardiol. 2016;67:2533–2546. doi: 10.1016/j.jacc.2016.02.081. [DOI] [PubMed] [Google Scholar]
- 35.Lee K.S., Cao Y., Witwicka H.E., Tom S., Tapscott S.J., Wang E.H. RNA-binding protein muscleblind-like 3 (MBNL3) disrupts myocyte enhancer factor 2 (Mef2) beta-exon splicing. J. Biol. Chem. 2010;285:33779–33787. doi: 10.1074/jbc.M110.124255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Scholl U.I., Stölting G., Nelson-Williams C., Vichot A.A., Choi M., Loring E., Prasad M.L., Goh G., Carling T., Juhlin C.C., et al. Recurrent gain of function mutation in calcium channel CACNA1H causes early-onset hypertension with primary aldosteronism. eLife. 2015;4:e06315. doi: 10.7554/eLife.06315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Hartmann S., Ridley A.J., Lutz S. The function of Rho-associated kinases ROCK1 and ROCK2 in the pathogenesis of cardiovascular disease. Front. Pharmacol. 2015;6:276. doi: 10.3389/fphar.2015.00276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Singh R.N., Singh N.N. Mechanism of splicing regulation of spinal muscular atrophy genes. Adv. Neurobiol. 2018;20:31–61. doi: 10.1007/978-3-319-89689-2_2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Shen S., Park J.W., Lu Z.X., Lin L., Henry M.D., Wu Y.N., Zhou Q., Xing Y. rMATS: Robust and flexible detection of differential alternative splicing from replicate RNA-seq data. Proc. Natl. Acad. Sci. USA. 2014;111:E5593–E5601. doi: 10.1073/pnas.1419161111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Raudvere U., Kolberg L., Kuzmin I., Arak T., Adler P., Peterson H., Vilo J. g:Profiler: A web server for functional enrichment analysis and conversions of gene lists (2019 update) Nucleic Acids Res. 2019;47(W1):W191–W198. doi: 10.1093/nar/gkz369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Turner C., Hilton-Jones D. The myotonic dystrophies: Diagnosis and management. J. Neurol. Neurosurg. Psychiatry. 2010;81:358–367. doi: 10.1136/jnnp.2008.158261. [DOI] [PubMed] [Google Scholar]
- 42.Thornton C.A. Myotonic dystrophy. Neurol. Clin. 2014;32:705–719, viii. doi: 10.1016/j.ncl.2014.04.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Petri H., Vissing J., Witting N., Bundgaard H., Køber L. Cardiac manifestations of myotonic dystrophy type 1. Int. J. Cardiol. 2012;160:82–88. doi: 10.1016/j.ijcard.2011.08.037. [DOI] [PubMed] [Google Scholar]
- 44.Misra C., Bangru S., Lin F., Lam K., Koenig S.N., Lubbers E.R., Hedhli J., Murphy N.P., Parker D.J., Dobrucki L.W., et al. Aberrant expression of a non-muscle RBFOX2 isoform triggers cardiac conduction defects in myotonic dystrophy. Dev. Cell. 2020;52:748–763.e6. doi: 10.1016/j.devcel.2020.01.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Reddy K., Jenquin J.R., McConnell O.L., Cleary J.D., Richardson J.I., Pinto B.S., Haerle M.C., Delgado E., Planco L., Nakamori M., et al. A CTG repeat-selective chemical screen identifies microtubule inhibitors as selective modulators of toxic CUG RNA levels. Proc. Natl. Acad. Sci. USA. 2019;116:20991–21000. doi: 10.1073/pnas.1901893116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Wang E.T., Treacy D., Eichinger K., Struck A., Estabrook J., Olafson H., Wang T.T., Bhatt K., Westbrook T., Sedehizadeh S., et al. Transcriptome alterations in myotonic dystrophy skeletal muscle and heart. Hum. Mol. Genet. 2019;28:1312–1321. doi: 10.1093/hmg/ddy432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Gao Y., Guo X., Santostefano K., Wang Y., Reid T., Zeng D., Terada N., Ashizawa T., Xia G. Genome therapy of myotonic dystrophy type 1 iPS cells for development of autologous stem cell therapy. Mol. Ther. 2016;24:1378–1387. doi: 10.1038/mt.2016.97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Freyermuth F., Rau F., Kokunai Y., Linke T., Sellier C., Nakamori M., Kino Y., Arandel L., Jollet A., Thibault C., et al. Splicing misregulation of SCN5A contributes to cardiac-conduction delay and heart arrhythmia in myotonic dystrophy. Nat. Commun. 2016;7:11067. doi: 10.1038/ncomms11067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Kuyumcu-Martinez N.M., Cooper T.A. Misregulation of alternative splicing causes pathogenesis in myotonic dystrophy. Prog. Mol. Subcell. Biol. 2006;44:133–159. doi: 10.1007/978-3-540-34449-0_7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Ueki J., Nakamori M., Nakamura M., Nishikawa M., Yoshida Y., Tanaka A., Morizane A., Kamon M., Araki T., Takahashi M.P., et al. Myotonic dystrophy type 1 patient-derived iPSCs for the investigation of CTG repeat instability. Sci. Rep. 2017;7:42522. doi: 10.1038/srep42522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Bhakta D., Groh M.R., Shen C., Pascuzzi R.M., Groh W.J. Increased mortality with left ventricular systolic dysfunction and heart failure in adults with myotonic dystrophy type 1. Am. Heart J. 2010;160:1137–1141, 1141.e1. doi: 10.1016/j.ahj.2010.07.032. [DOI] [PubMed] [Google Scholar]
- 52.Choudhary P., Nandakumar R., Greig H., Broadhurst P., Dean J., Puranik R., Celermajer D.S., Hillis G.S. Structural and electrical cardiac abnormalities are prevalent in asymptomatic adults with myotonic dystrophy. Heart. 2016;102:1472–1478. doi: 10.1136/heartjnl-2015-308517. [DOI] [PubMed] [Google Scholar]
- 53.Sovari A.A., Bodine C.K., Farokhi F. Cardiovascular manifestations of myotonic dystrophy-1. Cardiol. Rev. 2007;15:191–194. doi: 10.1097/CRD.0b013e318070d1a7. [DOI] [PubMed] [Google Scholar]
- 54.Meola G., Cardani R. Myotonic dystrophies: An update on clinical aspects, genetic, pathology, and molecular pathomechanisms. Biochim. Biophys. Acta. 2015;1852:594–606. doi: 10.1016/j.bbadis.2014.05.019. [DOI] [PubMed] [Google Scholar]
- 55.Wang Y., Hao L., Wang H., Santostefano K., Thapa A., Cleary J., Li H., Guo X., Terada N., Ashizawa T., Xia G. Therapeutic genome editing for myotonic dystrophy type 1 using CRISPR/Cas9. Mol. Ther. 2018;26:2617–2630. doi: 10.1016/j.ymthe.2018.09.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Day J.W., Ranum L.P. RNA pathogenesis of the myotonic dystrophies. Neuromuscul. Disord. 2005;15:5–16. doi: 10.1016/j.nmd.2004.09.012. [DOI] [PubMed] [Google Scholar]
- 57.Moller D.E., Yokota A., Caro J.F., Flier J.S. Tissue-specific expression of two alternatively spliced insulin receptor mRNAs in man. Mol. Endocrinol. 1989;3:1263–1269. doi: 10.1210/mend-3-8-1263. [DOI] [PubMed] [Google Scholar]
- 58.Savkur R.S., Philips A.V., Cooper T.A. Aberrant regulation of insulin receptor alternative splicing is associated with insulin resistance in myotonic dystrophy. Nat. Genet. 2001;29:40–47. doi: 10.1038/ng704. [DOI] [PubMed] [Google Scholar]
- 59.Morrone A., Pegoraro E., Angelini C., Zammarchi E., Marconi G., Hoffman E.P. RNA metabolism in myotonic dystrophy: Patient muscle shows decreased insulin receptor RNA and protein consistent with abnormal insulin resistance. J. Clin. Invest. 1997;99:1691–1698. doi: 10.1172/JCI119332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Knudsen L., De Meyts P., Kiselyov V.V. Insight into the molecular basis for the kinetic differences between the two insulin receptor isoforms. Biochem. J. 2011;440:397–403. doi: 10.1042/BJ20110550. [DOI] [PubMed] [Google Scholar]
- 61.Wu Z., Huang X., Feng Y., Handschin C., Feng Y., Gullicksen P.S., Bare O., Labow M., Spiegelman B., Stevenson S.C. Transducer of regulated CREB-binding proteins (TORCs) induce PGC-1α transcription and mitochondrial biogenesis in muscle cells. Proc. Natl. Acad. Sci. USA. 2006;103:14379–14384. doi: 10.1073/pnas.0606714103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Katoh Y., Takemori H., Lin X.Z., Tamura M., Muraoka M., Satoh T., Tsuchiya Y., Min L., Doi J., Miyauchi A., et al. Silencing the constitutive active transcription factor CREB by the LKB1-SIK signaling cascade. FEBS J. 2006;273:2730–2748. doi: 10.1111/j.1742-4658.2006.05291.x. [DOI] [PubMed] [Google Scholar]
- 63.Arandel L., Polay Espinoza M., Matloka M., Bazinet A., De Dea Diniz D., Naouar N., Rau F., Jollet A., Edom-Vovard F., Mamchaoui K., et al. Immortalized human myotonic dystrophy muscle cell lines to assess therapeutic compounds. Dis. Model. Mech. 2017;10:487–497. doi: 10.1242/dmm.027367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Kruer M.C., Jepperson T., Dutta S., Steiner R.D., Cottenie E., Sanford L., Merkens M., Russman B.S., Blasco P.A., Fan G., et al. Mutations in γ adducin are associated with inherited cerebral palsy. Ann. Neurol. 2013;74:805–814. doi: 10.1002/ana.23971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Nakano M., Okumura N., Nakagawa H., Koizumi N., Ikeda Y., Ueno M., Yoshii K., Adachi H., Aleff R.A., Butz M.L., et al. Trinucleotide repeat expansion in the TCF4 gene in Fuchs’ endothelial corneal dystrophy in Japanese. Invest. Ophthalmol. Vis. Sci. 2015;56:4865–4869. doi: 10.1167/iovs.15-17082. [DOI] [PubMed] [Google Scholar]
- 66.Lee K.Y., Li M., Manchanda M., Batra R., Charizanis K., Mohan A., Warren S.A., Chamberlain C.M., Finn D., Hong H., et al. Compound loss of muscleblind-like function in myotonic dystrophy. EMBO Mol. Med. 2013;5:1887–1900. doi: 10.1002/emmm.201303275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Guiraud-Dogan C., Huguet A., Gomes-Pereira M., Brisson E., Bassez G., Junien C., Gourdon G. DM1 CTG expansions affect insulin receptor isoforms expression in various tissues of transgenic mice. Biochim. Biophys. Acta. 2007;1772:1183–1191. doi: 10.1016/j.bbadis.2007.08.004. [DOI] [PubMed] [Google Scholar]
- 68.Karakikes I., Senyei G.D., Hansen J., Kong C.W., Azeloglu E.U., Stillitano F., Lieu D.K., Wang J., Ren L., Hulot J.S., et al. Small molecule-mediated directed differentiation of human embryonic stem cells toward ventricular cardiomyocytes. Stem Cells Transl. Med. 2014;3:18–31. doi: 10.5966/sctm.2013-0110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Zhang J., Klos M., Wilson G.F., Herman A.M., Lian X., Raval K.K., Barron M.R., Hou L., Soerens A.G., Yu J., et al. Extracellular matrix promotes highly efficient cardiac differentiation of human pluripotent stem cells: The matrix sandwich method. Circ. Res. 2012;111:1125–1136. doi: 10.1161/CIRCRESAHA.112.273144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Zhu W.Z., Xie Y., Moyes K.W., Gold J.D., Askari B., Laflamme M.A. Neuregulin/ErbB signaling regulates cardiac subtype specification in differentiating human embryonic stem cells. Circ. Res. 2010;107:776–786. doi: 10.1161/CIRCRESAHA.110.223917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Moore J.C., Fu J., Chan Y.C., Lin D., Tran H., Tse H.F., Li R.A. Distinct cardiogenic preferences of two human embryonic stem cell (hESC) lines are imprinted in their proteomes in the pluripotent state. Biochem. Biophys. Res. Commun. 2008;372:553–558. doi: 10.1016/j.bbrc.2008.05.076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.He J.Q., Ma Y., Lee Y., Thomson J.A., Kamp T.J. Human embryonic stem cells develop into multiple types of cardiac myocytes: Action potential characterization. Circ. Res. 2003;93:32–39. doi: 10.1161/01.RES.0000080317.92718.99. [DOI] [PubMed] [Google Scholar]
- 73.Ionta V., Liang W., Kim E.H., Rafie R., Giacomello A., Marbán E., Cho H.C. SHOX2 overexpression favors differentiation of embryonic stem cells into cardiac pacemaker cells, improving biological pacing ability. Stem Cell Reports. 2015;4:129–142. doi: 10.1016/j.stemcr.2014.11.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Dorn T., Goedel A., Lam J.T., Haas J., Tian Q., Herrmann F., Bundschu K., Dobreva G., Schiemann M., Dirschinger R., et al. Direct Nkx2-5 transcriptional repression of isl1 controls cardiomyocyte subtype identity. Stem Cells. 2015;33:1113–1129. doi: 10.1002/stem.1923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Ahmed R.E., Anzai T., Chanthra N., Uosaki H. A brief review of current maturation methods for human induced pluripotent stem cells-derived cardiomyocytes. Front. Cell Dev. Biol. 2020;8:178. doi: 10.3389/fcell.2020.00178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Sacchetto C., Vitiello L., de Windt L.J., Rampazzo A., Calore M. Modeling cardiovascular diseases with hiPSC-derived cardiomyocytes in 2D and 3D cultures. Int. J. Mol. Sci. 2020;21:21. doi: 10.3390/ijms21093404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Schmidt M., Hoffmann G., Wissler M., Lemke N., Müssig A., Glimm H., Williams D.A., Ragg S., Hesemann C.U., von Kalle C. Detection and direct genomic sequencing of multiple rare unknown flanking DNA in highly complex samples. Hum. Gene Ther. 2001;12:743–749. doi: 10.1089/104303401750148649. [DOI] [PubMed] [Google Scholar]
- 78.Paruzynski A., Arens A., Gabriel R., Bartholomae C.C., Scholz S., Wang W., Wolf S., Glimm H., Schmidt M., von Kalle C. Genome-wide high-throughput integrome analyses by nrLAM-PCR and next-generation sequencing. Nat. Protoc. 2010;5:1379–1395. doi: 10.1038/nprot.2010.87. [DOI] [PubMed] [Google Scholar]
- 79.Gabriel R., Eckenberg R., Paruzynski A., Bartholomae C.C., Nowrouzi A., Arens A., Howe S.J., Recchia A., Cattoglio C., Wang W., et al. Comprehensive genomic access to vector integration in clinical gene therapy. Nat. Med. 2009;15:1431–1436. doi: 10.1038/nm.2057. [DOI] [PubMed] [Google Scholar]
- 80.Afzal S., Wilkening S., von Kalle C., Schmidt M., Fronza R. GENE-IS: Time-efficient and accurate analysis of viral integration events in large-scale gene therapy data. Mol. Ther. Nucleic Acids. 2017;6:133–139. doi: 10.1016/j.omtn.2016.12.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Holt I., Mittal S., Furling D., Butler-Browne G.S., Brook J.D., Morris G.E. Defective mRNA in myotonic dystrophy accumulates at the periphery of nuclear splicing speckles. Genes Cells. 2007;12:1035–1048. doi: 10.1111/j.1365-2443.2007.01112.x. [DOI] [PubMed] [Google Scholar]
- 82.Martin M. Cutadapt removes adapter sequences from high-throughput sequencing reads. EMBnet. J. 2011;17:10–12. [Google Scholar]
- 83.Kim D., Langmead B., Salzberg S.L. HISAT: A fast spliced aligner with low memory requirements. Nat. Methods. 2015;12:357–360. doi: 10.1038/nmeth.3317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Pertea M., Pertea G.M., Antonescu C.M., Chang T.C., Mendell J.T., Salzberg S.L. StringTie enables improved reconstruction of a transcriptome from RNA-seq reads. Nat. Biotechnol. 2015;33:290–295. doi: 10.1038/nbt.3122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Robinson M.D., McCarthy D.J., Smyth G.K. edgeR: A Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics. 2010;26:139–140. doi: 10.1093/bioinformatics/btp616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Florea L., Song L., Salzberg S.L. Thousands of exon skipping events differentiate among splicing patterns in sixteen human tissues. F1000Res. 2013;2:188. doi: 10.12688/f1000research.2-188.v1. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Video depicting the cyclic change in intensity is due to influx and efflux of permeable Fluo-4 dye, indicating functional beating cardiomyocytes
Video depicting the cyclic change in intensity is due to influx and efflux of permeable Fluo-4 dye, indicating functional beating cardiomyocytes
Video depicting the cyclic change in intensity is due to influx and efflux of permeable Fluo-4 dye, indicating functional beating cardiomyocytes
Video of contracting cardiomyogenic-like cells
Video of contracting cardiomyogenic-like cells
Video of contracting cardiomyogenic-like cells
Data Availability Statement
The data that support the findings of this study are available from the corresponding author upon reasonable request.