

Abstract
B cells have been engineered ex vivo to express an HIV-1 broadly neutralizing antibody (bNAb). B cell reprograming may be scientifically and therapeutically useful, but current approaches limit B cell repertoire diversity and disrupt the organization of the heavy-chain locus. A more diverse and physiologic B cell repertoire targeting a key HIV-1 epitope could facilitate evaluation of vaccines designed to elicit bNAbs, help identify more potent and bioavailable bNAb variants, or directly enhance viral control in vivo. Here we address the challenges of generating such a repertoire by replacing the heavy-chain CDR3 (HCDR3) regions of primary human B cells. To do so, we identified and utilized an uncharacterized Cas12a ortholog that recognizes PAM motifs present in human JH genes. We also optimized the design of 200 nucleotide homology-directed repair templates (HDRT) by minimizing the required 3′-5′ deletion of the HDRT-complementary strand. Using these techniques, we edited primary human B cells to express a hemagglutinin epitope tag and the HCDR3 regions of the bNAbs PG9 and PG16. Those edited with bNAb HCDR3 efficiently bound trimeric HIV-1 antigens, implying they could affinity mature in vivo in response to the same antigens. This approach generates diverse B cell repertoires recognizing a key HIV-1 neutralizing epitope.
Keywords: Cas12a, HDR, B cell receptor, HCDR3, HIV-1 V2-glycan antibody, Mb2Cas12a, PG9, PG16, SOSIP
Graphical abstract
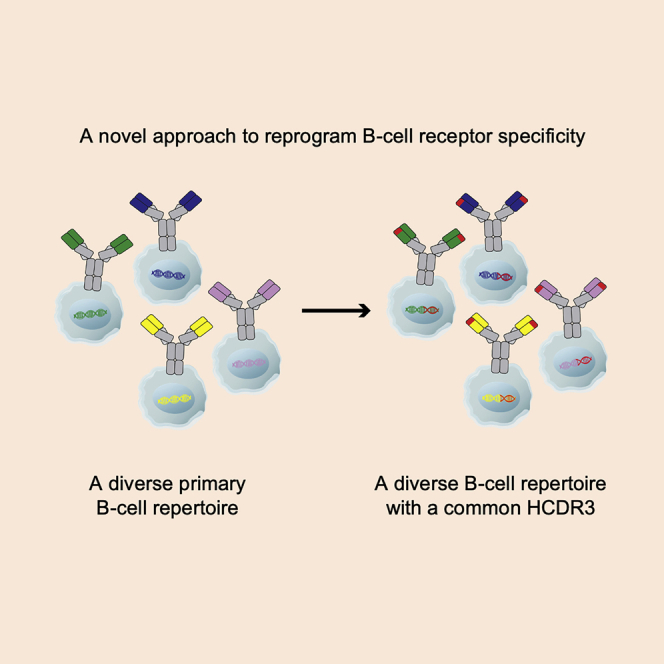
Ou and colleagues edit primary human B cells so that their diverse B cell receptors include a common human-derived heavy-chain CDR3. Many of these B cells produce antibodies that neutralize HIV-1. This approach creates a diverse set of B cells that could generate an individualized response to virus in an HIV-positive person.
Introduction
Traditional vaccination approaches do not elicit broadly neutralizing antibodies (bNAbs) that target conserved epitopes of HIV-1 envelope glycoprotein trimer (Env).1, 2, 3, 4 Human precursor B cell receptors (BCRs) that can develop into bNAbs are rare,5,6 and mature bNAbs have properties that are difficult to access through antibody maturation.1,7,8 A number of groups have begun to explore an alternative to conventional vaccines in which B cells themselves are reprogrammed.9, 10, 11, 12, 13 This approach employs CRISPR-mediated editing of the BCR loci so that the edited B cell expresses a mature HIV-1 bNAb. In addition to its long-term potential for reprograming human immune responses, BCR editing can be applied more immediately to generate animal models useful for assessing vaccination strategies, and for developing more potent and bioavailable bNAb variants.
The BCR includes a membrane-bound heavy chain (H) covalently associated with a light chain (L). Both chains are composed of a variable and a constant region. The heavy-chain variable domain is formed by a process of VDJ recombination of the immunoglobulin heavy-chain (IgH) gene. In humans, one of the 38 to 46 functional variable (VH) genes recombines with one of 23 diversity (DH) and one of six joining (JH) genes.14 The recombination process also introduces diversity at the junctions of VH, DH, and JH genes through removal and addition of nucleotides. The light-chain variable domain is formed similarly by VJ recombination of the IgL gene. The naive B cell repertoire thus reflects extensive combinatorial diversity.15, 16, 17, 18 This diversity is further amplified after antigen exposure. B cells undergo somatic hypermutation (SHM) as they compete for access to antigens in the lymph-node germinal centers, a process resulting in affinity maturation of the BCR.19
The combinatorial diversity of the B cell repertoire complicates efforts to reprogram BCR. To date, investigators have bypassed this challenge by targeting an unvarying intron between the recombined variable region and the IgM constant region (Cμ).9,11, 12, 13 This strategy introduces a single cassette encoding an exogenous promoter and bNAb heavy- and light-chain sequences into this heavy-chain intron. By design, these constructs halt expression of the native variable heavy chain. Expression of the native B cell light chain is usually also prevented through various mechanisms. While powerful and convenient, this approach eliminates combinatorial diversity and relies solely on SHM to broaden the HIV-1 neutralizing response. In addition, it introduces several less physiologic elements, including novel locations for both variable genes, use of exogenous promoter, and some architectural differences between the expressed bNAb-like construct and native antibodies. These limitations may be especially important if edited B cells need to adapt efficiently to a diverse HIV-1 reservoir,20,21 or when the edited repertoire is used to study B cell biology.22
Here we develop a complementary approach in which sequence encoding a bNAb HCDR3 is introduced into a diverse BCR repertoire at its native location. This approach is useful with antibodies that are highly dependent on the HCDR3 to bind antigen, including members of an exceptionally potent class of bNAbs that recognize the Env apex/V2-glycan epitope.23,24 However, retaining combinatorial diversity in a natural B cell setting poses several challenges. First, long homology arms of a homology-dependent repair (HDR) template (HDRT) can overwrite the V-encoded region of a heavy chain. Second, the region 5′ of the HCDR3-encoding region is necessarily diverse, and thus editing can be variably efficient due to mismatch of a homology arm with its chromosomal complement. Third, introducing exogenous HCDR3 requires deletion of chromosomal material of unknown length and content, rather than simply insertion of a sequence, or direct replacement of a known sequence. To address these challenges, we identified a previously uncharacterized Cas12a variant25 that efficiently recognizes a specific four-nucleotide protospacer adjacent motif (PAM) present in the 3′ region of the most commonly used JH genes in humans. We also optimized the use of 200-nucleotide (nt) single-stranded HDRT with short (∼50 nt) homology arms, demonstrating in the process that editing efficiency is primarily determined by the length of a 3′ mismatch tail rather than the relationship of the HDRT to transcription direction (sense or anti-sense) or to the target strand of the CRISPR guide RNA (gRNA). With these procedures, we altered the specificity of primary human B cells by editing their HCDR3 regions to bind HIV-1 Env, while retaining the original diversity of the VH and VL repertoire. These studies demonstrate the feasibility of an alternative approach to human B cell reprogramming.
Results
Targeting a conserved region of the IgH locus with a Cas12a ortholog
A major challenge of precisely replacing the HCDR3-encoding region of a diverse primary B cell population is the variability of the mature IgH locus. This variability arises from the random combinations of V, D, and J segments that are joined imprecisely and unpredictably.16 It complicates two processes necessary for CRISPR-mediated editing of the B cell locus, namely the selection of a gRNA that must complement a 20- to 24-nt genome sequence, and the design of HDRT whose 5′ and 3′ homology arms must complement even longer genomic regions.26, 27, 28
We began by designing a gRNA that recognizes a large proportion of BCR and targets genome cleavage to site of insertion, where it is most efficient. The HCDR3 is encoded by the 3′ end of a VH gene, a DH gene, and the 5′ end of a JH gene (Figure 1A). There are six human JH segments, and a JH4 alone participates in about 50% of productive human VDJ-recombination events.29,30 Due to junctional diversity, the 3′ JH region is conserved, but the 5′ is less predictable. However, the 3′ of JH4 did not contain any canonical Cas12a PAM31 sequences (TTTV), and the available Cas9 PAM (NGG)32 mediates cleavage too distal from the site of insertion. Instead, two potential non-canonical Cas12a PAM sites, GTTC and TTCC,33 were well positioned to facilitate gRNA recognition of conserved JH regions while cleaving where a new HCDR3 would be inserted. We therefore characterized a number of Cas12a orthologs for their ability to recognize these divergent PAM sites. To do so, we tested several uncharacterized Cas12a orthologs25 in a human B cell line Jeko-1. Jeko-1 cells were transfected with plasmids encoding BsCas12a, TsCas12a, Mb2Cas12a, or Mb3Cas12a along with plasmids encoding gRNAs adjacent to the GTTC and TTCC PAM regions. DNA cleavage within the HCDR3 frequently results in error-prone non-homologous end-joining (NHEJ) that eliminates expression of the Jeko-1 BCR.34 Thus, loss of IgM expression indicates a successful double-strand break. Among the Cas12a orthologs tested, Mb2Cas12a most efficiently cleaved the Jeko-1 JH4 region initiated with GTTC and TTCC, with the highest efficiency (∼18%) observed when the GTTC PAM was targeted (Figure 1B).
Figure 1.

Targeting the conserved region of JH4 gene requires a Cas12a ortholog recognizing non-canonical PAMs
(A) A representation of the coding region of an antibody heavy-chain variable region is presented. As indicated, the HCDR3 (green) is encoded by the 3′ of a recombined V gene, a D gene, and the 5′ of a J-chain. To insert a common HCDR3 into a diverse population of BCR, the gRNA of a CRISPR effector protein must complement a conserved HC region at the 3′ end of the recombined J-gene, while cleaving a more variable region near the site of HCDR3 insertion. Note that, unlike Cas9, Cas12a cleaves distally from its PAM and seed regions. The preferred PAM recognition sequence of commonly studied Cas12a orthologs is TTTV. However, as shown, JH4, the most frequently used JH gene in all species, contains optimally located GTTC and TTCC PAM sequences, located 3′ of the HCDR3-encoding sequence but oriented Cas12a cleavage within this sequence. This PAM, sequence of the gRNA, and the Cas12a cut sites are indicated. (B) To identify a Cas12a ortholog efficient at cleaving these non-canonical PAM motifs, the human B cell line Jeko-1 was co-transfected with two plasmids encoding the CRISPR protein (BsCas12a, TsCas12a, Mb2Cas12a, or Mb3Cas12a), and their corresponding gRNA. Control samples were transfected without gRNA plasmids. Targeting efficiency was measured by flow cytometry as loss of IgM expression. Among these Cas12a orthologs, Mb2Cas12 most efficiently cleaved the J-chain region initiated with GTTC and TTCC (orange). Error bars indicate standard error (SEM) of two independent experiments, and asterisks indicate statistical significance relative to controls. Statistical difference was determined by non-paired Student’s t-test (∗∗∗∗p < 0.0001). (C) Mb2Cas12a RNP was compared with commercial AsCas12a RNP for their ability cleave four distinct regions in the HCDR3-encoding region of Jeko-1 cells. Loss of IgM expression indicates successful introduction of a double-strand break and NHEJ. RNP with gRNA targeting an irrelevant site (EMX1) was used as control. (D) Results of three experiments similar to that shown in (B). Error bars indicate SEM of at least two independent experiments. Asterisks indicate significant differences from the canonical TTTG PAM (Mb2Cas12a or AsCas12a, respectively). Statistical difference was determined by non-paired Student’s t-test (∗∗∗∗p < 0.0001).
Ribonucleoprotein (RNP) forms of CRISPR effector proteins, electroporated into cells, are typically more efficient than plasmids expressing the same protein.28,35 We accordingly produced Mb2Cas12a RNP and compared its editing efficiency with a commercial AsCas12a RNP in Jeko-1 cells, again as determined through loss of IgM expression. These RNPs cleaved a canonical Cas12a TTTG with comparable efficiency but Mb2Cas12a cleaved three non-canonical PAM regions more efficiently (Figures 1C and 1D), demonstrating that Mb2Cas12a has a broad PAM specificity and efficiently edits the HCDR3 region of Jeko-1 cells. Notably, Mb2Cas12a RNP efficiently cleaved JH4 when initiated with a GTTC PAM, and this PAM region is conserved in the JH genes of both humans and rodents.
We also characterized the target specificity of Mb2Cas12a RNP in Jeko-1 cells using the iGUIDE assay.36 Perhaps due to its broader PAM specificity, Mb2Cas12a showed a somewhat higher off-target profile compared with that of AsCas12a (IDT) when a region with a TTTN PAM, used by both enzymes, was targeted (Figure S1, Table S1). Notably off-target NHEJ was less frequent when targeting a GTTN PAM-initiated JH4 region used in subsequent primary B cell editing studies.
Optimization of gene editing using single-stranded HDR templates
We also optimized the design of HDRT used to replace a native HCDR3 region. Again, the underlying diversity of the recombined heavy chains limited our options. Most importantly, the HDRT homology arms needed to remain short to maximize complementarity to the 3′ VH region. We therefore optimized a strategy based on short single-stranded DNA (ssDNA) HDRT with 50-nt homology arms by monitoring the efficiency with which a hemagglutinin (HA) tag could replace the Jeko-1 HCDR3 region. Specifically, we compared sense and anti-sense forms of two distinct HDRTs (Figure 2A), each with different length linkers bounding the HA tag, and cleavage at four distinct Mb2Cas12a sites and four proximal SpCas9 sites (Figure S2A). Note that some of these sites are unique to the Jeko-1 HCDR3 region, and are therefore not generalizable to primary B cells. Knock-in efficiency was determined by flow cytometry with fluorescently labeled anti-HA antibodies (Figure 2B). We observed that on average, with four different cut sites and two sets of distinct HDRT, Mb2Cas12a and SpCas9 edited with comparable efficiencies (Figure 2C). We analyzed these same data by comparing a number of parameters proposed to impact editing efficiencies in other systems.27,28,37,38 However, no significant differences were detected when sense or anti-sense-strand HDRTs were used (Figure 2D), and only a modest difference for Mb2Cas12a, but not for SpCas9, was observed when target (complementary to gRNA) or non-target strand HDRT was used (Figure 2E). At each site, SpCas9 and Mb2Cas12a always preferred the same strand HDRT regardless of it being sense/anti-sense or target/non-target (Figure S2B). These observations were distinct from those of previous studies,27,38 perhaps because both DNA deletion and insertion are required to replace an HCDR3.
Figure 2.

Optimization of ssDNA templates for Mb2Cas12a-mediated editing the HCDR3-encoding region of a human B cell line
(A) A diagram representing four HDRTs used in (B–E). Specifically, sense and anti-sense forms of HDRT-A, were used to replace a 9-nt region (gray) with a 39-nt insert (green), and both forms of HDRT-B were used to replace a 36-nt region with a 69-nt region; 50-nt homology arms of the sense and anti-sense forms are represented in red and blue, respectively. SpCas9 (cyan) and Mb2Cas12A (orange) cleavage sites of the target strand (complementary to gRNA) are indicated by arrows. Note that paired Cas9 and Cas12a cleavage sites are separated by at most 5 nts. (B) A representative example of an experiment used to generate (C–E) in which editing efficiency of Mb2Cas12A or SpCas9 RNP was monitored through recognition of an HA tag introduced into the HCDR3 of the Jeko-1 cell BCR by flow cytometry. Control cells were electroporated with Mb2Cas12a RNP without an HDRT. (C) A comparison of Mb2Cas12a (Mb2) and SpCas9 (Cas9) knock-in efficiencies, measured as described in (B), with the four different HDRTs and for all four sites shown in (A). Differences between Mb2 and Cas9, and among the four sites, are not significant (n.s.). The same data generated for (C) was replotted according to whether the sense or anti-sense HDRT was used (D), or whether the HDRT complemented the gRNA target or non-target strand. (E) Non-target strand is the PAM-containing strand, and the target strand is the strand annealed to gRNA. Again, as indicated, most differences were not significant. However, the HDRT-complementary to the Mb2Cas12a gRNA target strand were slightly more efficient than those complementary to the non-target strand (p = 0.027). Dots in (C–E) represent pooled data from two independent experiments. Statistical significance was calculated by one-way ANOVA with Tukey's multiple comparison test.
Figure 3A presents a model of a system in which deletion of chromosomal DNA and insertion of novel sequence are both necessary for successful editing. After CRISPR-mediated formation of a double-strand break, HDR is initiated by 5′ to 3′ resection of DNA, exposing two 3′ ends, one of which can anneal to a single-stranded HDRT homology arm. However, at least one 3′ end necessarily includes sequence that must be deleted 3′ to 5′, either on the HDRT-complementary strand, creating a 3′ mismatch tail, on the opposing strand, or on both. As shown in Figures 3B and 3C, the length of this 3′ mismatch tail, determined as shown in Table S2, strongly predicts editing efficiency in this system regardless of whether the double-stranded break was mediated by Mb2Cas12a or SpCas9. Specifically, editing is significantly more efficient when mismatch tails are shorter than 10 nt, presumably because longer tails prevent polymerase priming and templated extension of the 3′ arm. We presume that this principle can be extended to other systems in which a templated sequence must replace a chromosomal region by HDR.
Figure 3.

The length of the 3′ mismatch tail determines replacement efficiency with short single-stranded HDRT
(A) A model showing where a 3′ mismatch tail occurs. A cut site (yellow) is introduced into a region of the gene targeted for replacement (gray), asymmetrically dividing this region. Efficient 5′ to 3′ resection exposes two 3′ ends. An HDRT can complement a strand with a short (left figures) or long 3′-mismatch tail (right figures), which must be removed before the remaining 3′ end can be extended to complement the HDRT insert region and its distal homology arm. We propose that the removal of this 3′ mismatch tail is a rate-limiting step determining editing efficiency when genomic sequences are replaced. (B) The predicted length of the 3′-mismatch tail in experiments using HDRT-A and HDRT-B presented in Figure 2 are plotted against the efficiency with which an HA tag is introduced into the HCDR3 region, as determined by flow cytometry. Error bar indicates SEM from two independent experiments. (C) A comparison of editing efficiency between those with short (<10 nt) or long (>10 nt) 3′ mismatch tails. Editing by SpCas9 or Mb2Cas12a is significantly more efficient with short 3′ mismatch tails, as determined by one-way ANOVA with Tukey's multiple comparison test (p < 0.0001). Dots represent pooled data from two independent experiments.
Reprogramming B cell specificity toward HIV through HCDR3 replacement
Using the strategies described above, we then designed HDRT that is optimal for site 4 (Figure S2A) to replace the Jeko-1 HCDR3 with those of PG9 or PG16 (Figure 4A), two potent HIV bNAbs directed against a V2 apex of the HIV-1 Env trimer.39,40 The gRNA used in site 4 (Figure S2A) targets the conserved region of JH4 (Figure 1A), and is the only sequence tested that is also consistently present in primary B cells. To monitor the successful introduction of these HCDR3s in contexts in which the resulting BCR does not bind soluble native-like HIV-1 Env trimer (SOSIP),41,42 we employed an antibody, PSG2,43 that recognizes sulfated tyrosines present at the tips of the PG9 and PG16 HCDR3. In addition, we monitored HIV-1 Env binding with two reagents, a SOSIP protein derived from the HIV-1 isolate BG505, and the multivalent nanoparticle (E2p)44 based on the same BG505 HIV-1 isolate. Jeko-1 cells were edited with HDRT-PG9-CVR and HDRT-PG9-CAR to express two forms of the PG9 HCDR3, distinguished by an alanine (A) or valine (V) immediately adjacent to the HCDR3-initiating cysteine. The original PG9 antibody has the valine, while its germline VH3-33 has the alanine. Cells edited with either HDRT could be recognized by all three binding reagents, indicating that the resulting BCR could bind the BG505 Env (Figures 4B and 4C). As expected, none of these three antigens bound Jeko-1 cells in which a control HDRT, introducing an HA tag into the HCDR3, was employed. However, Jeko-1 cells edited to express the PG16 HCDR3 were recognized only by PSG2, indicating that editing was efficient, but the resulting BCR did not bind HIV-1 Env. This is plausibly due to the incompatibility of the PG16-HCDR3 with the Jeko-1 VH, a supposition supported by subsequent experiments in primary human B cells. Finally, we tested whether increasing one end of the homology arm could further enhance the editing efficiency. We observed that, at the same total length, symmetric homology arms facilitated more efficient editing than did asymmetric arms (Figure S2C).
Figure 4.

The BCR specificity of Jeko-1 cells can be reprogrammed with a novel HCDR3
(A) The amino acid sequence of the native Jeko-1 cell HCDR3 region and those of the HIV-1 neutralizing antibodies PG9 and PG16 are shown. The amino acid translations of the new sequences from the four HDRTs used in the subsequent panels are represented in green italics, in the context of the remaining Jeko-1 region. (B) Mb2Cas12a RNP targeting the GTTC PAM of site 4 in Jeko-1 cells shown in Figure 2B were co-electroporated with the indicated HDRT. Editing efficiency was monitored on the vertical axis by flow cytometry with fluorescently labeled PSG2, an antibody that recognizes sulfotyrosines within the PG9 and PG16 HCDR3 region, a similarly labeled HIV SOSIP or E2p. The horizontal axis indicates IgM expression, and its loss indicates imprecise NHEJ after Mb2Cas12a-mediated cleavage. Note that introduction of a PG16 HCDR3 was efficient, as indicated by PSG2 recognition, but unlike the PG9 HCDR3, it did not bind the Env trimer. Cells edited to express an HA tag did not bind any reagent. SOSIP proteins were derived from the BG505 HIV-1 isolate. (C) A summary of three independent experiments similar to that shown in (B) of flow cytometric studies used to generate (B). Error bars indicate SD. (D) Jeko-1 edited with PG9-CAR HDRT were enriched by FACS with the anti-sulfotyrosine antibody PSG2. (E) Cells enriched in (D) were analyzed 2 weeks later by flow cytometry for their ability to bind PSG2, a BG505-based nanoparticle (BG505-E2p), SOSIP trimers derived from the indicated HIV-1 isolate, or an V2 apex negative mutant (dBG505-SOSIP). Gray control indicates wild-type Jeko-1 cells. (F) Unedited Jeko-1 cells and those edited with PG9-CAR HDRT without sorting, or sorted with PSG2 or with E2p, were analyzed by NGS of the VDJ region. Sequences were divided into four categories, depending on whether the edited sequence exactly matched the HDRT (Perfect HDR), whether the HDRT sequence was visible but modified (Imperfect HDR), whether the original Jeko-1 HCDR3 region was intact (Original), or whether this region was modified by NHEJ as indicated by the presence of insertions or deletions (Indel). Representative examples of each category are shown below the charts.
We further characterized Jeko-1 cells edited with HDRT-PG9-CAR by enriching edited cells by fluorescence-activated cell sorting (FACS) with the PSG2 antibody (Figure 4D). Sorted cells were then analyzed by flow cytometry for their ability to interact with PSG2, SOSIP variants derived from three HIV-1 isolates and from a negative mutant, and BG505-E2p (Figure 4E). Each of these reagents bound PG9-HCDR3-edited cells efficiently, with CRF250 SOSIP proteins binding most efficiently and therefore used in subsequent experiments. In parallel, next-generation sequencing (NGS) analysis was performed on the HCDR3 region of unedited Jeko-1 cells, cells edited with HDRT-PG9-CAR before they were sorted, and the same cells sorted with either PSG2 or the E2p nanoparticle presenting multiple BG505 proteins. HCDR3 sequences were divided based on whether HDR was successful and whether the introduced sequence exactly matched that in the HDRT (Figure 4F). We observed that before sorting, the original Jeko-1 HCDR3 bearing indels reflecting NHEJ predominated. After sorting, HCDR3 that matched the HDRT predominated. Collectively, the data shown in Figure 4 indicate that the HCDR3 of Jeko-1 cells can be replaced by that of PG9 to generate a BCR that efficiently binds multiple HIV-1 SOSIP proteins. This approach can be used to insert the HCDR3 of other apex bNAbs, such as CAP256 (Figure S2D).
Using consensus sequences of multiple VH families to edit primary human B cells
The preceding studies showed that Mb2Cas12a could cleave a conserved region of the JH4 gene useful for introducing an exogenous HCDR3 sequence, that editing with sense-strand HDRT in this setting is optimal because it minimizes the length the 3′ mismatch tail, and that the PG9 HCDR3 could function with the divergent Jeko-1 heavy and light chain to bind multiple HIV-1 Env trimers. However, primary human B cells pose an additional challenge: in contrast to Jeko-1 cells, the VH-gene sequences of primary cells are variable and unpredictable. This difficulty complicates the design of the 5′ homology arm, which must complement the 3′ region of the VH gene. Alignment of the 3′ regions of the most commonly used VH gene families, namely VH1, VH3, and VH4 (Figure S3A), revealed a good deal of interfamily diversity, but showed that intrafamily diversity was limited among the 3′ nts. We accordingly evaluated HDRT with 5′ homology arms based on consensus sequences for each of these VH families in primary human B cells. These cells were isolated from peripheral blood and activated by an anti-CD180 antibody11 for 48 h before electroporation with Mb2Cas12a RNP along with different HDRTs. Editing efficiency was measured 48 h post electroporation by flow cytometry using the anti-sulfotyrosine antibody PSG2 (Figure 5A). Two different fluorophores were used to label PSG2 to eliminate non-specific binding from either fluorophore. As negative controls, primary human primary B cells were activated in the same way, but they were not electroporated (null), or electroporated with Mb2Cas12a RNP and an HDRT that was not homologous to any sequence in the human genome. The HDRT homologous to VH1 had relatively lower efficiency than to the other two families, largely due to the lower VH1 usage frequency in mature human B cells (Figure 5B). An equal mixture of three HDRTs (those of VH1, VH3, and VH4) edited more cells than any individual HDRT, suggesting a diverse pool of B cells could be targeted simultaneously. NGS performed on two sets of B cells from different donors edited with the mixed HDRT, and the frequency of successful in-frame editing reflected the efficiencies of the individual HDRT (Figure 5C). These data show that, using consensus HDRT homology arms, approximately 1% of primary human B cells can be edited to express the PG9 HCDR3. Note that all primary human cells we tested, from multiple donors, expressed the JH4∗02 allele. A modified gRNA should be used for the other JH4∗01 and JH4∗03 for optimal results. We also compared the editing efficiency of Mb2Cas12a and SpCas9 for this particular JH4 site in human primary B cells. While the knock-out efficiency was comparable (Figure S3B), the Mb2Cas12a more efficiently than SpCas9 inserted an HA tag using the optimized HDRT (Figure S3C).
Figure 5.

Editing primary human B cells with HDRT recognizing consensus sequences of multiple VH families
(A) A panel of PG9-CAR HDRT with homology arms complementary to JH4 and to consensus VH1-, VH3-, and VH4-family sequences were evaluated for their ability to edit primary human B cells. Cells electroporated with Mb2Cas12a RNP and PG9-CAR HDRT were analyzed by flow cytometry with the anti-sulfotyrosine antibody PSG2 modified with two distinct fluorophores to eliminate non-specific binding from either fluorophore. (B) A summary of results from experiments similar to that shown in (A), using primary B cells from three independent donors. Note that a mixture of three HDRTs edited more cells than any individual HDRT. Null indicates that cells were not electroporated and control indicates cells electroporated with Mb2Cas12a RNP and an HDRT that is not homologous to any sequence in the human genome. Mix indicates cells electroporated with RNP and an equimolar mixture of HDRT with VH1-, VH3-, and VH4-specific homology arms. Error bars indicate range of three independent experiments, and asterisks indicate statistical significance calculated by one-way ANOVA with Tukey's multiple comparison test (∗p < 0.5; ∗∗p < 0.01; ∗∗∗∗p < 0.0001). (C) NGS analysis of primary B cells from two human donors. %HDR was quantified as described in Figure 4F, including both perfect and imperfect. The portion of VH-family of edited cells in HDR-positive sequences was also counted.
The diversity and specificity of HCDR3-edited primary human B cell repertoires
To determine if HCDR3-edited BCR acquired their reprogrammed specificity and retained their VH diversity, edited B cells were expanded for a week and then sorted with an appropriate antigen. To evaluate their specificity and diversity, we tested HDRT encoding the PG9 HCDR3, PG16 HCDR3, or an HA tag (Figure 6). As in Figure 5, Mb2Cas12a RNPs were used to cleave the JH4 region, and mixtures of three HDRTs, recognizing consensus 3′ VH1, VH3, or VH4 sequences, directed the insertion of the novel HCDR3. Edited cells were sorted with an anti-HA antibody (Figure 6A) or the CRF250 SOSIP trimer derived from the CRF_AG_250 isolate (Figures 6B and 6C). Heavy-chain sequences were analyzed by NGS before (blue) and after (red) sorting. As anticipated, sorting changed the frequency of successfully edited B cells. Critically, in each case, multiple VH1, VH3, and VH4 genes continued to be represented after sorting, suggesting that the combinatorial diversity of the repertoire could be preserved after introducing the PG9 HCDR3 into primary B cells. Light-chain sequences from sorted PG9-HCDR3 inserted population were also analyzed by NGS (Figure 6D). The distribution of light chains was largely preserved after sorting, but a notable decrease in kappa chains was observed. Thus the combinatorial diversity of the repertoire of HCDR3-edited B cells can be maintained.
Figure 6.

Reprogrammed primary human B cells retain VH-gene and light-chain diversity
Primary cells were electroporated with Mb2Cas12a RNP and HDRT encoding an HA tag (A) or the HCDR3 regions of the HIV-1 neutralizing antibodies PG9 (B) and PG16 (C), with the same mixture of homology arms as those used in Figure 5. Cells were sorted with an anti-HA antibody (HA tag [A]) or an SOSIP trimer derived from the CRF_AG_250 isolate (B and C). Approximately 2% to 3% of cells were sorted by the SOSIP trimer. Edited cells were analyzed by NGS before and after sorting, the percentages of PG9 and PG16-HCDR3 inserts were enriched from ∼1% to 30%. The frequency of each VH1-, VH3-, and VH4-family genes from BCR sequences bearing the desired inserts was measured. Flow cytometry histograms display one of two experiments with similar results, and bar graphs indicate the mean of those two experiments. (D) The frequency of each light chain genes of PG9-HCR3 inserted primary cells pre (blue) and post (red) sorting was analyzed by NGS. Bar graphs indicate the mean of two independent experiments.
To confirm that this HCDR3 could function when expressed with multiple variable genes, we expressed in HEK293T cells antibody variants with the PG9 HCDR3 and light chain, but encoded with several distinct VH genes. We focused on VH1-, VH3-, and VH4-family genes that generated a high signal after sorting, and included VH3-33, the original PG9 variable gene, for comparison. All five VH genes generated antibodies that could detectably express and bind a SOSIP trimer, with BCR expressed from the VH3-30 sequence binding most efficiently (Figure 7). Similarly, soluble forms of these antibodies could neutralize at least one HIV-1 isolate (Figure S4). Notably, antibodies generated from the VH3-30 gene bound SOSIP trimers and neutralized HIV-1 more efficiently than those based on VH3-33, indicating that VH3-30 and perhaps several other VH genes could serve as alternative starting points for PG9-like antibodies. The same test was performed for PG16-HCDR3 with three VH genes identified from sorting. Each bound the HIV SOSIP trimer (Figure 7C). In contrast, PG16 HCDR3 on the VH from Jeko-1 cells (VH2-70) failed to bind HIV Env. Similarly, Jeko-1 cells edited with the CH01 HCDR3 failed to bind SOSIP trimers (Figure S2D), but primary human B cells edited with CH01 HCDR3 detectably bound these trimers (Figure S4A). We also further confirmed binding of antibodies composed of the PG9 germline light chain (Vl2-14), the most enriched kappa chain (Vk2-28), and each of two enriched lambda (Vl2-8, Vl1-51) light chains combined with the most efficient germline VH for PG9 (Figure 7). As a control, a light chain whose frequency was diminished after sorting, Vl3-21, impaired association with HIV Env.
Figure 7.

Binding of PG9 HCDR3 and PG16 HCDR3 pairing with germline VH and VL genes
293T cells were used to express PG9 HCDR3 and PG16 HCDR3 paring with multiple germline VH and VL genes. (A) Constructs composed of the heavy chains expressed from the indicated VH genes enriched in Figure 6 or that of PG9, the PG9 HCDR3, a transmembrane domain, and the native PG9 light chain were expressed on the surface of 293T cells and analyzed by flow cytometry. The no-VH construct expresses the Fc region (truncated at the hinge) with the transmembrane domain. (B) Mean fluorescence intensity of SOSIP staining from the successfully transfected cells (IgG-positive population) was normalized to the IgG expression. The mean of two experiments shown in (A) is presented. (C) Binding experiments similar in (A) with PG16. In addition to the VH genes enriched in Figure 6, the VH gene of Jeko-1 (VH2-70) paring with the PG16 HCDR3 was also tested. (D) Binding experiments of the best VH gene (VH3-30) for PG9 paring with the original (mature) and the germline light chains of the PG9, and the enriched light chains from Figure 6. One representative of two independent experiments is shown for (A, C, and D).
Collectively, Figures 6 and 7 show that diverse human B cells can be edited to express the PG9 and PG16 HCDR3, enabling these cells to bind an SOSIP trimer and express receptors primed to adapt to specific isolates in an HIV-positive person. However, these engineered repertoires have not been subjected to early immune-tolerance checkpoints, a potential risk for their human use.
To address this concern, we examined the autoreactivity of the PG9 HCDR3 engrafted on the primary human BCR repertoire. We performed an HEp-2 cell staining45 with the antibodies bearing the PG9 HCDR3 on six combinations of VH and VL chains. We observed than all six were markedly less autoreactive than the autoreactive HIV-1 neutralizing antibody 2F5 (Figure S5), suggesting that autoreactivity would not be a frequent result of this approach.
Discussion
Transgenic mice engineered to express human variable-chain sequences of bNAbs and inferred germline forms of these bNAbs have been used extensively to study bNAb maturation in response to HIV-1 antigens.46, 47, 48 These mice were developed primarily to study vaccination strategies, but they could potentially be used to improve the breadth and potency of bNAbs as well. The advent of CRISPR technologies enables ex vivo editing of mature B cells, and adoptive transfer edited B cells into a new murine host.9,11, 12, 13 CRISPR-mediated editing of B cells is more rapid and versatile than developing a transgenic mouse, but a more limited subset of B cells express the bNAb of interest. These cells can nonetheless be amplified through vaccination, and they undergo class switching and SHM. As this technology advances, reprogramming of mature naive B cells could replace transgenic mice as a means of evaluating and optimizing vaccine protocols. This approach could also be developed as an in vivo alternative to phage- and yeast-display technologies to improve the breadth, potency, or bioavailability of existing antibody or even another biologic. Finally, this technology may form the basis of future vaccines and cell therapies, following a path established by chimeric antigen-receptor (CAR) T cells. By retaining the diversity of the response, such engineered B cells could be stimulated to respond to a wider range of viruses than any monoclonal antibody, and/or adapt to specific viral variants reactivated in an infected person after interruption of anti-viral therapy.
Nearly every reported effort to date to reprogram primary B cells uses a conserved intron downstream of the VDJ-recombined variable region.9,11, 12, 13 A typical insertion cassette initiates with a poly(A) tail to terminate transcription of the native variable region, followed by an exogenous promoter, a human bNAb light-chain variable-region sequence, a P2A peptide or linker, and a heavy-chain variable-region sequence with a splice donor that promotes splicing to native constant genes. This approach is efficient because every mature B cell could theoretically be modified, because HDRT with long homology arms can be used, and because a single editing event introduces both variable chains at once. This efficiency makes this approach attractive to most investigators, but other approaches that preserve the organization of the heavy-chain locus have been attempted. For example, Voss et al.10 have explored an alternative in which the entire native variable-region was replaced by the heavy-chain variable of the bNAb PG9.
A common property of all previous investigations is that an entire heavy chain or heavy-chain/light-chain pair are introduced. Thus, initially B cells express a monoclonal antibody that then can diversify through SHM. As a consequence, one major contributor to antibody diversity, namely combinatorial diversity, is bypassed. Such an approach is necessary for many HIV-1 bNAbs because antigen recognition is distributed across multiple CDR loops; however, most known V2-glycan/apex bNAbs have long, acidic HCDR3 regions that make an unusually large contribution to Env recognition.23,24 Moreover, neutralization with these antibodies is especially potent, typically 10-fold higher than with other classes of bNAbs.49 Finally, antibodies of this class, uniquely recognizing a quaternary epitope, are especially sensitive to the quality of HIV-1 antigens.40 We therefore undertook to reprograming human B cells to express the HCDR3 of a potentV2-glycan/apex antibody while largely preserving the combinatorial diversity of the repertoire. Ultimately, we anticipate this diversity will enhance and personalize the adaptive humoral response to the diversity of HIV-1 isolates in reservoirs of infected humans.
However, before this concept can be tested, we had to address several challenges unique to introducing an HCDR3 into primary human B cells. These challenges arose from two sources. First, for optimal editing, a double-strand break should be introduced near the insert region, in this case at the 5′ of a commonly used J-gene such as JH4. However, due to junctional diversity, this region is highly variable in a diverse repertoire. Second, HDRT with long homology arms are typically more efficient, but long arms would complement only a narrow BCR subset or overwrite the native VH gene.
To address the first challenge, we initiated studies with the CRISPR effector protein Cas12a. We began with Cas12a because, unlike the more commonly employed Cas9, this CRISPR effector protein cleaves distally from its PAM and seed regions. Thus, a more variable region can be cleaved from a more predictable gRNA target sequence. However, most commonly studied Cas12a orthologs, including LbCas12a and AsCas12a, use restrictive PAM recognition sequences31 absent in human JH4 sequences. We therefore characterized a number of less studied Cas12a variants, and identified one, Mb2Cas12a, that efficiently recognized a GTTC PAM present at an optimal location in both human and rodent JH4 genes. Thus, electroporated Mb2Cas12a RNP efficiently introduced double-strand breaks near the 5′ of the JH4-encoded region in Jeko-1 cells and in primary human B cells.
Our second challenge arose from the unpredictability of the VH-encoded region in the diverse repertoire of primary B cells, precluding the use of long HDRT. We accordingly designed HDRT that recognized short consensus sequences at the 3′ of three VH-gene families. We also optimized the efficiency of editing using these shorter HDRTs. To do so, we first evaluated the impact of two parameters that have been proposed to alter editing efficiency. First, we asked whether the HDRT should complement the coding or non-coding sequence, and we also investigated whether it should complement the gRNA target strand or its opposite. Both parameters have be reported to contribute to editing efficiencies of other systems,27,38 but neither of these variables had a dramatic impact on editing efficiency in our study. Further analysis identified a distinct, decisive factor in editing efficiency, namely the length of the 3′ mismatch tail. While 5′-3′ resection is among the first events in HDR, removal of the 3′ end is rate limiting. Our data suggest that the pace of removal of the 3′ end of the HDRT-complementary strand is especially critical, and if the necessary deletion is greater than 10 nts, editing is significantly impaired. These data are consistent with previous observations that Pol δ, the polymerase responsible for 3′ extension of the HDRT-associated strand, has modest 3′ exonuclease activity,50,51 and conforms to a previously proposed model, the synthesis-dependent strand annealing (SDSA) model.52 Regardless of the underlying mechanism, this optimization enabled editing efficiencies with Mb2Cas12a and short single-stranded HDRT comparable to those reported with Cas9 and much longer HDRTs.
With these tools in hand, we showed that the HCDR3 regions of primary human B cells could be reprogrammed to encode three novel sequences, two of which derived from HIV-1 bNAbs. In each case, edited cells could be enriched by FACS with an appropriate antigen while still largely retaining the diversity of the edited repertoire. Interestingly, in the case of cells edited to express the PG9 HCDR, this approach more efficiently enriched BCR encoded by the V3-30 heavy chain than for V3-33, the heavy-chain gene from which the bNAb PG9 originally derived. We confirmed this observation by showing that a PG9 variant constructed from this germline form of this VH gene neutralizes more efficiently than one constructed from the germline V3-33 gene. As importantly, a number of VH genes enriched in this manner bound SOSIP trimers and neutralized HIV-1. Thus, at least in the case of PG9, a range of modified BCR could respond to an SOSIP antigen or to HIV-1 emerging from a reactivated reservoir.
Despite its several advantages, this approach has some limitations. First, editing efficiency is of necessity lower than when the target region is well defined. Further optimization using combinations of HDRT recognize a broader range of VH families may help increase the proportion of successfully edited B cells. In addition, antigen stimulation in vivo can amplify an initially small fraction of edited and engrafted B cells, compensating in part for this limitation. Second, this approach requires some initial affinity between the HCDR3 region and a target antigen. Of course, as we show with the HA tag, many kinds of peptides and small folded domains can be introduced and perhaps affinity matured to an appropriate antigen or binding protein. Finally, ex vivo editing of B cells is less efficient and more expensive than in vivo editing. Interestingly, the small HDRT used here could be combined with Mb2Cas12a and a gRNA in a single adeno-associated virus vector to facilitate direct in vivo editing, although the efficiency and utility of such editing remains to be demonstrated.
In short, we have overcome several challenges associated with introducing an exogenous HCDR3 sequence into a diverse repertoire of human BCR. In the process, we have identified and characterized a Cas12a ortholog especially useful in introducing double-strand breaks near the DJ junction of a recombined heavy chain, and described an optimized approach for replacing genomic regions with short HDRT. Finally, we showed that this approach could create a diverse repertoire of B cells capable of recognizing a critical epitope of HIV-1 Env. These studies establish foundations for proof-of-concept studies in primate models of HIV-1 infection.
Material and methods
Plasmids
Wild-type Mb2Cas12a (pcDNA3.1-hMb2Cpf1), Mb3Cas12a (pcDNA3.1-hMb3Cpf1), TsCas12a (pcDNA3.1-hTsCpf1), and BsCas12a (pcDNA3.1-hBsCpf1) plasmids were gifts from Dr. Feng Zhang (Addgene: 92,292, 92,293, 92,267, 92,300). pMAL-his-LbCpf1-EC was a gift from Dr. Jin-Soo Kim (Addgene: 79,008) and was used to express Mb2Cas12a in E. coli for protein production. For Cas12a protein production, each Cas12a gene was codon optimized for E. coli, synthesized by IDT, and cloned into pMAL-his-LbCpf1-EC vector.
Mb2Cas12a protein production and purification
Expression cassette of maltose binding protein (MBP)-Mb2Cas12a-His in pMal vector was transformed to Rosetta 2 (DE3) (Novagen) competent cells. A single colony was first grown in 5 mL, then scaled up to 10 L for production, in LB broth with 4 μm/mL chloramphenicol and 100 μg/mL carbenicillin. Cell cultures were grown to OD ∼0.5 before placing on ice for 15 min, and added with 0.5 mM IPTG at 16°C to induce expression. After 18 h of incubation, cells were resuspended in the buffer with 50 mM NaH2PO4, 500 mM NaCl, 15 mM imidazole, 10% glycerol, and 10 mM Tris at pH 8.0, then sonicated on ice for 20 min at 18 W output before clarified by centrifugation for 25 min at 50,000 × g. Clarified supernatant was loaded to the HisTrap FF column (GE Healthcare) and eluted with linear imidazole gradient from 10 mM to 300 mM using ÄKTA explorer (GE Healthcare). To remove the N-terminal MBP tag, the protein elution fractions were pooled and concentrated with a 50-kDa molecular weight cutoff ultrafiltration unit (Millipore), then 1 mg of TEV protease was used per 50 mg protein for cleavage during dialyzing to the buffer with 250 mM NaCl, 0.5 mM EDTA, 1 mM DTT, and 20 mM HEPES with pH 7.4 for 48 h at 4°C. For cation exchange chromatography, the protein was diluted with 2-fold volume of 20 mM HEPES with pH 7.0 and loaded on HiTrap SP HP column (GE Healthcare) equilibrated with 100 mM NaCl, 20 mM HEPES at pH 7.0. Proteins were eluted with a linear NaCl gradient from 100 mM to 2 M, then further purified by size exclusion chromatography with Superdex 200 26/60 column (GE Healthcare) with the protein storage buffer (500 mM NaCl, 0.1 mM EDTA, 1 mM DTT, 10% glycerol, and 20 mM HEPES at pH 7.5). Pure protein fractions were pooled and concentrated followed with endotoxin removal with columns from Pierce.
RNP formation and electroporation
Mb2Cas12a, AsCas12a, and SpCas9 gRNAs were ordered from IDT. RNAs were resuspended in RNase-free water and refolded by incubation at 95°C for 5 min and cooling down at room temperature for 1 h. For each electroporation sample, RNP complexes were formed by mixing 200 pmol of Mb2Cas12a, AsCas12a (Alt-R AsCas12a [Cpf1] V3 from IDT) or SpCas9 (Alt-R SpCas9 Nuclease V3 from IDT) with 300 pmol of crispr RNA and PBS. The RNP mixture was incubated at room temperature for 15 to 30 min, then added with 600 pmol of ssDNA HDRT. For the mixture of HDRT (VH1, VH3, and VH4) to target primary cells, 200 pmol of each was used. HDRT and gRNA sequences used in this study are listed in Table S3. HDRT contains PS modification as advised by IDT. Jeko-1 cells (2 million/sample) and human primary B cells (4 to 5 million/sample) were harvested and rinsed with PBS before resuspension in electroporation solution. Cells were electroporated using Lonza 4D modules according to Lonza's protocols. After electroporation, cells were incubated in the cuvette for 15 min at room temperature before transferring to the antibiotic-free media. Culture media was refreshed in 24 h.
iGUIDE assay
Off-target identification assay was performed and analyzed as described previously.36 Briefly, 1 million of Jeko-1 cells were electroporated with RNP complexes and 200 pmol of iGUIDE dsODN using Lonza 4D. Cells were harvested 48 h post electroporation to extract genomic DNA, following by genome shearing, ligation of adaptors, and introduction of UMI tags. Amplicons were sequenced by Illumina Miseq using the 2 × 150 platform.
HIV protein and antibodies
BG505 SOSIP v5.2 ds (E64K A316W A73C-A561C I201C-A433C)42 and BG505 E2p44 were constructed as previously described. Amino acid sequences were codon optimized and synthesized by IDT and cloned into the CMV/R expression plasmid following a human IgH signal peptide. The apex negative mutant (dBG505) was constructed by altering the V2 basic patch of the BG505 apex from RDKKQK to IDNVQQ to abolish PGT145 and PG9 binding. All proteins were produced in transiently transfected Expi293F (Invitrogen) cells. Protein constructs were co-transfected with plasmids encoding furin, FGE (formylglycine generating enzyme), and PDI (protein disulfide isomerase), respectively (at 4:1:1:1 ratio) using FectoPRO (Polyplus) according to the manufacturer's protocol. Supernatants were harvested 5 days after transfection, filtered, and purified with CH01 or PGT145 affinity column. Proteins were eluted with gentle Ag/Ab elution buffer (21027; Thermo). The elution was exchanged to buffer (358 mM HEPES, 75 mM NaCl pH 8.0). For antibody production, heavy- and light-chain plasmids were co-transfected (1:1.25 ratio) in Expi293F cells. The supernatants were harvested 5 days later, and the IgG was purified using protein A Sepharose (GE Healthcare) and eluted with gentle Ag/Ab elution buffer (21027; Thermo), following buffer-exchanged into PBS.
Human cell culture
Human blood samples were obtained through OneBlood (Florida), and were conducted in accordance with IRB protocols approved by the Institutional Review Board at the Scripps Research Institute (TSRI). Peripheral blood mononuclear cells were isolated by density gradient centrifugation with Ficoll (GE Healthcare), stored in liquid nitrogen, then thawed in a 37°C water bath and resuspended in human B cell medium composed of RPMI-1640 with GlutaMAX, supplemented with 10% FBS, 10 mM HEPES, 1 mM sodium pyruvate, and 53 μM 2-mercaptoethanol (all from Gibco). B cells were isolated by magnetic sorting using the Human B Cell Isolation Kit II (130-091-151) from Miltenyi according to the manufacturer's instructions and cultured in the above medium supplemented with 2 μg/mL anti-human RP105 antibody clone (312907; BioLegend).
Flow cytometry and cell sorting
IgM expression was detected by fluorescein isothiocyanate (FITC) anti-human IgM antibody (clone MHM-88; BioLegend). HA insertion was detected by APC anti-HA antibody (clone 16B12; BioLegend). Reprogrammed specificity of B cells were validated by binding with PSG2, SOSIP, E2p proteins conjugated with different fluorophores using Lightning-Link Antibody Labeling Kits according to manufacturer's instructions. Cultured cells were harvested by centrifugation and rinsed by FACS buffer (PBS, 0.5% BSA, 1 mM EDTA) and resuspend to 10 million/mL in 100 μL. Cells were stained for 20 min on ice with fluorescently labeled antibodies (1 μg/mL) or proteins (3 μg/mL), then washed twice with FACS buffer before measuring fluorescence by BD Accuri C6 flow cytometer or sorting by BD FACSAria Fusion sorter. Flow data were analyzed by FlowJo software.
Sequencing and analysis of the edited B cell IgH and IgL repertoire
Human B cells harvested post gene editing or cell sorting were lysed for RNA extraction by the RNeasy Micro Kit (74004; Qiagen). Primers used for reverse transcription and library amplification were modified from previous29 (see Table S4). IgH and IgL mRNA were reverse transcribed using different methods. For the IgH mRNA, first-strand cDNA synthesis was carried out by SuperScript III (Thermo Fisher) using primers specific for IgM and IgG. For the IgL mRNA, template-switching oligo was appended to the cDNA by SMARTScribe transcriptase (Takara Bio) during reverse transcription. Residual primers and dNTPs were degraded enzymatically (ExoSAP-IT; Thermo Fisher) according to the manufacturer's protocol. Second-strand synthesis reaction was carried out using HotStarTaq Plus (Qiagen). Residual primers and dNTPs were again degraded enzymatically (ExoSAP-IT) and dsDNA was purified using SPRI beads (SPRIselect, Beckman Coulter Genomics). Eluted dsDNA was amplified (HotStarTaq Plus), and the PCR products were using SPRI beads (SPRIselect). Of the eluted PCR product, 10 μL was used in a final indexing using NEBNext Multiplex Oligos for Illumina (E7710S, NEB) following the manufacturer's instructions. PCR products were purified with 0.7 volumes of SPRI beads (SPRIselect). SPRI-purified libraries were sequenced on an Illumina MiSeq using 2 × 250 base pairs. Sequencing reads were processed and analyzed. Briefly, paired reads were merged with PANDAseq53 using the default merging algorithm then trimmed and collapsed by UMI through Migec using the “checkout” algorithm. Processed reads were mapped by MiGMAP based on IgBlast.54,55
Autoreactivity assay
Reactivity of antibodies bearing the PG9 HCDR3 paring with various VH and VL genes to human epithelial HEp-2 cells was determined by indirect immunofluorescence on HEp-2 slides using FITC-conjugated goat anti-human IgG (Zeus Scientific). The anti-HIV-1 antibody 2F5 is used as a positive control. Slides were incubated with 150 μg/mL of antibodies for 30 min following two PBS rinses. Slides were photographed on an Olympus fluorescent microscope.
Neutralization assay
Pseudoviruses were produced by co-transfection of different HIV envelope plasmids acquired through the NIH AIDS Reagents Program along with NL4-3-ΔEnv in HEK293T cells using PEIpro (Polyplus). Supernatant was harvested 48 h post transfection, clarified by centrifugation and 0.45 μm filter, and aliquoted for storage at −80°C. TZM-bl neutralization assays were performed as previously described.56 Briefly, titrated antibodies in 96-well plates were incubated with pseudotyped viruses at 37°C for 1 h. TZM-bl cells were then added to the wells with 50,000 cells/well. Cells were then incubated for 48 h at 37°C. At 48 h post infection, cells were lysed in wells and subjected to Firefly luciferase assays. Viral entry was determined using Britelite Plus (PerkinElmer), and luciferase expression was measured using a Victor X3 plate reader (PerkinElmer).
Statistical analysis
Data were expressed as mean values ±SD or SEM, and all statistical analyses were performed in GraphPad Prism 7.0 software. IC50 of antibody neutralization was analyzed using default settings for log(inhibitor) versus normalized response method. Statistical difference was determined using non-paired Student's t-test or one-way ANOVA with Tukey's test. Differences were considered significant at p < 0.05.
Data availability
The datasets generated in this study are available upon reasonable request.
Acknowledgments
This work is supported by NIH R37 AI091476 and DP1 DA043912 (PI: M.F.). The authors thank Xiaohua Wu, PhD for scientific advice on DNA repair mechanisms, and Hyeryun Choe, PhD for reviewing the manuscript.
Author contributions
T.O., W.H., G.Z., and M.F. conceived of this study. G.Z. and M.F. provided guidance for experimental design. T.O., W.H., B.Q., Y.G., P.K., H.P., M.D.G., M.H.T., Y.Y., X.Z., and H.W. performed all experiments. T.O., W.H., and M.F. wrote the manuscript.
Declaration of interests
The authors declare no competing interests.
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.ymthe.2021.10.027.
Supplemental information
Full length sequence of Jeko-1 HCDR3 target sites
References
- 1.West A.P., Jr., Scharf L., Scheid J.F., Klein F., Bjorkman P.J., Nussenzweig M.C. Structural insights on the role of antibodies in HIV-1 vaccine and therapy. Cell. 2014;156:633–648. doi: 10.1016/j.cell.2014.01.052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Mascola J.R., Haynes B.F. HIV-1 neutralizing antibodies: understanding nature's pathways. Immunol. Rev. 2013;254:225–244. doi: 10.1111/imr.12075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Burton D.R., Hangartner L. Broadly neutralizing antibodies to HIV and their role in vaccine design. Annu. Rev. Immunol. 2016;34:635–659. doi: 10.1146/annurev-immunol-041015-055515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Escolano A., Dosenovic P., Nussenzweig M.C. Progress toward active or passive HIV-1 vaccination. J. Exp. Med. 2017;214:3–16. doi: 10.1084/jem.20161765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Jardine J.G., Kulp D.W., Havenar-Daughton C., Sarkar A., Briney B., Sok D., Sesterhenn F., Ereño-Orbea J., Kalyuzhniy O., Deresa I. HIV-1 broadly neutralizing antibody precursor B cells revealed by germline-targeting immunogen. Science. 2016;351:1458–1463. doi: 10.1126/science.aad9195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Steichen J.M., Lin Y.-C., Havenar-Daughton C., Pecetta S., Ozorowski G., Willis J.R., Toy L., Sok D., Liguori A., Kratochvil S. A generalized HIV vaccine design strategy for priming of broadly neutralizing antibody responses. Science. 2019;366:eaax4380. doi: 10.1126/science.aax4380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Klein F., Diskin R., Scheid J.F., Gaebler C., Mouquet H., Georgiev I.S., Pancera M., Zhou T., Incesu R.-B., Fu B.Z. Somatic mutations of the immunoglobulin framework are generally required for broad and potent HIV-1 neutralization. Cell. 2013;153:126–138. doi: 10.1016/j.cell.2013.03.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Walker L.M., Huber M., Doores K.J., Falkowska E., Pejchal R., Julien J.-P., Wang S.-K., Ramos A., Chan-Hui P.-Y., Moyle M. Broad neutralization coverage of HIV by multiple highly potent antibodies. Nature. 2011;477:466–470. doi: 10.1038/nature10373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Huang D., Tran J.T., Olson A., Vollbrecht T., Tenuta M., Guryleva M.V., Fuller R.P., Schiffner T., Abadejos J.R., Couvrette L. Vaccine elicitation of HIV broadly neutralizing antibodies from engineered B cells. Nat. Commun. 2020;11:1–10. doi: 10.1038/s41467-020-19650-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Voss J.E., Gonzalez-Martin A., Andrabi R., Fuller R.P., Murrell B., McCoy L.E., Porter K., Huang D., Li W., Sok D. Reprogramming the antigen specificity of B cells using genome-editing technologies. Elife. 2019;8:e42995. doi: 10.7554/eLife.42995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hartweger H., McGuire A.T., Horning M., Taylor J.J., Dosenovic P., Yost D., Gazumyan A., Seaman M.S., Stamatatos L., Jankovic M. HIV-specific humoral immune responses by CRISPR/Cas9-edited B cells. J. Exp. Med. 2019;216:1301–1310. doi: 10.1084/jem.20190287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Moffett H.F., Harms C.K., Fitzpatrick K.S., Tooley M.R., Boonyaratanakornkit J., Taylor J.J. B cells engineered to express pathogen-specific antibodies protect against infection. Sci. Immunol. 2019;4,:eaax0644. doi: 10.1126/sciimmunol.aax0644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Nahmad A.D., Raviv Y., Horovitz-Fried M., Sofer I., Akriv T., Nataf D., Dotan I., Carmi Y., Burstein D., Wine Y. Engineered B cells expressing an anti-HIV antibody enable memory retention, isotype switching and clonal expansion. Nat. Commun. 2020;11:5851. doi: 10.1038/s41467-020-19649-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Giudicelli V., Duroux P., Ginestoux C., Folch G., Jabado-Michaloud J., Chaume D., Lefranc M.-P. IMGT/LIGM-DB, the IMGT® comprehensive database of immunoglobulin and T cell receptor nucleotide sequences. Nucleic Acids Res. 2006;34:D781–D784. doi: 10.1093/nar/gkj088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Rolink A., Melchers F. Molecular and cellular origins of B lymphocyte diversity. Cell. 1991;66:1081–1094. doi: 10.1016/0092-8674(91)90032-t. [DOI] [PubMed] [Google Scholar]
- 16.Elhanati Y., Sethna Z., Marcou Q., Callan C.G., Jr., Mora T., Walczak A.M. Inferring processes underlying B-cell repertoire diversity. Philos. Trans. R. Soc. B Biol. Sci. 2015;370:20140243. doi: 10.1098/rstb.2014.0243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Schatz D.G., Ji Y. Recombination centres and the orchestration of V (D) J recombination. Nat. Rev. Immunol. 2011;11:251–263. doi: 10.1038/nri2941. [DOI] [PubMed] [Google Scholar]
- 18.Bassing C.H., Swat W., Alt F.W. The mechanism and regulation of chromosomal V (D) J recombination. Cell. 2002;109:S45–S55. doi: 10.1016/s0092-8674(02)00675-x. [DOI] [PubMed] [Google Scholar]
- 19.Mesin L., Ersching J., Victora G.D. Germinal center B cell dynamics. Immunity. 2016;45:471–482. doi: 10.1016/j.immuni.2016.09.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Leslie A., Pfafferott K., Chetty P., Draenert R., Addo M., Feeney M., Tang Y., Holmes E., Allen T., Prado J. HIV evolution: CTL escape mutation and reversion after transmission. Nat. Med. 2004;10:282–289. doi: 10.1038/nm992. [DOI] [PubMed] [Google Scholar]
- 21.Abbott R.K., Lee J.H., Menis S., Skog P., Rossi M., Ota T., Kulp D.W., Bhullar D., Kalyuzhniy O., Havenar-Daughton C. Precursor frequency and affinity determine B cell competitive fitness in germinal centers, tested with germline-targeting HIV vaccine immunogens. Immunity. 2018;48:133–146.e6. doi: 10.1016/j.immuni.2017.11.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Dosenovic P., Kara E.E., Pettersson A.-K., McGuire A.T., Gray M., Hartweger H., Thientosapol E.S., Stamatatos L., Nussenzweig M.C. Anti–HIV-1 B cell responses are dependent on B cell precursor frequency and antigen-binding affinity. Proc. Natl. Acad. Sci. U S A. 2018;115:4743–4748. doi: 10.1073/pnas.1803457115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Andrabi R., Voss J.E., Liang C.-H., Briney B., McCoy L.E., Wu C.-Y., Wong C.-H., Poignard P., Burton D.R. Identification of common features in prototype broadly neutralizing antibodies to HIV envelope V2 apex to facilitate vaccine design. Immunity. 2015;43:959–973. doi: 10.1016/j.immuni.2015.10.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Lee J.H., Andrabi R., Su C.-Y., Yasmeen A., Julien J.-P., Kong L., Wu N.C., McBride R., Sok D., Pauthner M. A broadly neutralizing antibody targets the dynamic HIV envelope trimer apex via a long, rigidified, and anionic β-hairpin structure. Immunity. 2017;46:690–702. doi: 10.1016/j.immuni.2017.03.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Zetsche B., Strecker J., Abudayyeh O.O., Gootenberg J.S., Scott D.A., Zhang F. A survey of genome editing activity for 16 Cpf1 orthologs. bioRxiv. 2017:134015. doi: 10.1101/134015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Doudna J.A., Charpentier E. The new frontier of genome engineering with CRISPR-Cas9. Science. 2014;346,:1258096. doi: 10.1126/science.1258096. [DOI] [PubMed] [Google Scholar]
- 27.Wang Y., Liu K.I., Sutrisnoh N.-A.B., Srinivasan H., Zhang J., Li J., Zhang F., Lalith C.R.J., Xing H., Shanmugam R. Systematic evaluation of CRISPR-Cas systems reveals design principles for genome editing in human cells. Genome Biol. 2018;19:62. doi: 10.1186/s13059-018-1445-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Liang X., Potter J., Kumar S., Ravinder N., Chesnut J.D. Enhanced CRISPR/Cas9-mediated precise genome editing by improved design and delivery of gRNA, Cas9 nuclease, and donor DNA. J. Biotechnol. 2017;241:136–146. doi: 10.1016/j.jbiotec.2016.11.011. [DOI] [PubMed] [Google Scholar]
- 29.Briney B., Inderbitzin A., Joyce C., Burton D.R. Commonality despite exceptional diversity in the baseline human antibody repertoire. Nature. 2019;566:393–397. doi: 10.1038/s41586-019-0879-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Volpe J.M., Kepler T.B. Large-scale analysis of human heavy chain V (D) J recombination patterns. Immunome Res. 2008;4:3. doi: 10.1186/1745-7580-4-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Zetsche B., Gootenberg J.S., Abudayyeh O.O., Slaymaker I.M., Makarova K.S., Essletzbichler P., Volz S.E., Joung J., Van Der Oost J., Regev A. Cpf1 is a single RNA-guided endonuclease of a class 2 CRISPR-Cas system. Cell. 2015;163:759–771. doi: 10.1016/j.cell.2015.09.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Jinek M., Chylinski K., Fonfara I., Hauer M., Doudna J.A., Charpentier E. A programmable dual-RNA–guided DNA endonuclease in adaptive bacterial immunity. Science. 2012;337:816–821. doi: 10.1126/science.1225829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Yamano T., Zetsche B., Ishitani R., Zhang F., Nishimasu H., Nureki O. Structural basis for the canonical and non-canonical PAM recognition by CRISPR-Cpf1. Mol. Cell. 2017;67:633–645.e3. doi: 10.1016/j.molcel.2017.06.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Shalem O., Sanjana N.E., Hartenian E., Shi X., Scott D.A., Mikkelsen T.S., Heckl D., Ebert B.L., Root D.E., Doench J.G. Genome-scale CRISPR-Cas9 knockout screening in human cells. Science. 2014;343:84–87. doi: 10.1126/science.1247005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Hur J.K., Kim K., Been K.W., Baek G., Ye S., Hur J.W., Ryu S.-M., Lee Y.S., Kim J.-S. Targeted mutagenesis in mice by electroporation of Cpf1 ribonucleoproteins. Nat. Biotechnol. 2016;34:807–808. doi: 10.1038/nbt.3596. [DOI] [PubMed] [Google Scholar]
- 36.Nobles C.L., Reddy S., Salas-McKee J., Liu X., June C.H., Melenhorst J.J., Davis M.M., Zhao Y., Bushman F.D. iGUIDE: an improved pipeline for analyzing CRISPR cleavage specificity. Genome Biol. 2019;20:14. doi: 10.1186/s13059-019-1625-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Paix A., Folkmann A., Goldman D.H., Kulaga H., Grzelak M.J., Rasoloson D., Paidemarry S., Green R., Reed R.R., Seydoux G. Precision genome editing using synthesis-dependent repair of Cas9-induced DNA breaks. Proc. Natl. Acad. Sci. U S A. 2017;114:E10745–E10754. doi: 10.1073/pnas.1711979114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Richardson C.D., Ray G.J., DeWitt M.A., Curie G.L., Corn J.E. Enhancing homology-directed genome editing by catalytically active and inactive CRISPR-Cas9 using asymmetric donor DNA. Nat. Biotechnol. 2016;34:339–344. doi: 10.1038/nbt.3481. [DOI] [PubMed] [Google Scholar]
- 39.Walker L.M., Phogat S.K., Chan-Hui P.-Y., Wagner D., Phung P., Goss J.L., Wrin T., Simek M.D., Fling S., Mitcham J.L. Broad and potent neutralizing antibodies from an African donor reveal a new HIV-1 vaccine target. Science. 2009;326:285–289. doi: 10.1126/science.1178746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Sok D., van Gils M.J., Pauthner M., Julien J.-P., Saye-Francisco K.L., Hsueh J., Briney B., Lee J.H., Le K.M., Lee P.S. Recombinant HIV envelope trimer selects for quaternary-dependent antibodies targeting the trimer apex. Proc. Natl. Acad. Sci. U S A. 2014;111:17624–17629. doi: 10.1073/pnas.1415789111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Sanders R.W., Derking R., Cupo A., Julien J.-P., Yasmeen A., de Val N., Kim H.J., Blattner C., de la Peña A.T., Korzun J. A next-generation cleaved, soluble HIV-1 Env trimer, BG505 SOSIP. 664 gp140, expresses multiple epitopes for broadly neutralizing but not non-neutralizing antibodies. PLoS Pathog. 2013;9:e1003618. doi: 10.1371/journal.ppat.1003618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.de la Peña A.T., Julien J.-P., de Taeye S.W., Garces F., Guttman M., Ozorowski G., Pritchard L.K., Behrens A.-J., Go E.P., Burger J.A. Improving the immunogenicity of native-like HIV-1 envelope trimers by hyperstabilization. Cell Rep. 2017;20:1805–1817. doi: 10.1016/j.celrep.2017.07.077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Hoffhines A.J., Damoc E., Bridges K.G., Leary J.A., Moore K.L. Detection and purification of tyrosine-sulfated proteins using a novel anti-sulfotyrosine monoclonal antibody. J. Biol. Chem. 2006;281:37877–37887. doi: 10.1074/jbc.M609398200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.He L., De Val N., Morris C.D., Vora N., Thinnes T.C., Kong L., Azadnia P., Sok D., Zhou B., Burton D.R. Presenting native-like trimeric HIV-1 antigens with self-assembling nanoparticles. Nat. Commun. 2016;7:12041. doi: 10.1038/ncomms12041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Haynes B.F., Fleming J., Clair E.W.S., Katinger H., Stiegler G., Kunert R., Robinson J., Scearce R.M., Plonk K., Staats H.F. Cardiolipin polyspecific autoreactivity in two broadly neutralizing HIV-1 antibodies. Science. 2005;308:1906–1908. doi: 10.1126/science.1111781. [DOI] [PubMed] [Google Scholar]
- 46.Briney B., Sok D., Jardine J.G., Kulp D.W., Skog P., Menis S., Jacak R., Kalyuzhniy O., De Val N., Sesterhenn F. Tailored immunogens direct affinity maturation toward HIV neutralizing antibodies. Cell. 2016;166:1459–1470.e11. doi: 10.1016/j.cell.2016.08.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Escolano A., Steichen J.M., Dosenovic P., Kulp D.W., Golijanin J., Sok D., Freund N.T., Gitlin A.D., Oliveira T., Araki T. Sequential immunization elicits broadly neutralizing anti-HIV-1 antibodies in Ig knockin mice. Cell. 2016;166:1445–1458.e12. doi: 10.1016/j.cell.2016.07.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Tian M., Cheng C., Chen X., Duan H., Cheng H.-L., Dao M., Sheng Z., Kimble M., Wang L., Lin S. Induction of HIV neutralizing antibody lineages in mice with diverse precursor repertoires. Cell. 2016;166:1471–1484.e18. doi: 10.1016/j.cell.2016.07.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Doria-Rose N.A., Bhiman J.N., Roark R.S., Schramm C.A., Gorman J., Chuang G.-Y., Pancera M., Cale E.M., Ernandes M.J., Louder M.K. New member of the V1V2-directed CAP256-VRC26 lineage that shows increased breadth and exceptional potency. J. Virol. 2016;90:76–91. doi: 10.1128/JVI.01791-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Pâques F., Haber J.E. Two pathways for removal of nonhomologous DNA ends during double-strand break repair in Saccharomyces cerevisiae. Mol. Cell Biol. 1997;17:6765–6771. doi: 10.1128/mcb.17.11.6765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Anand R., Beach A., Li K., Haber J. Rad51-mediated double-strand break repair and mismatch correction of divergent substrates. Nature. 2017;544:377–380. doi: 10.1038/nature22046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Kan Y., Ruis B., Takasugi T., Hendrickson E.A. Mechanisms of precise genome editing using oligonucleotide donors. Genome Res. 2017;27:1099–1111. doi: 10.1101/gr.214775.116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Masella A.P., Bartram A.K., Truszkowski J.M., Brown D.G., Neufeld J.D. PANDAseq: paired-end assembler for illumina sequences. BMC Bioinformatics. 2012;13:31. doi: 10.1186/1471-2105-13-31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Shugay M., Britanova O.V., Merzlyak E.M., Turchaninova M.A., Mamedov I.Z., Tuganbaev T.R., Bolotin D.A., Staroverov D.B., Putintseva E.V., Plevova K. Towards error-free profiling of immune repertoires. Nat. Methods. 2014;11:653–655. doi: 10.1038/nmeth.2960. [DOI] [PubMed] [Google Scholar]
- 55.Shugay M., Bagaev D.V., Turchaninova M.A., Bolotin D.A., Britanova O.V., Putintseva E.V., Pogorelyy M.V., Nazarov V.I., Zvyagin I.V., Kirgizova V.I. VDJtools: unifying post-analysis of T cell receptor repertoires. PLoS Comput. Biol. 2015;11:e1004503. doi: 10.1371/journal.pcbi.1004503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Montefiori D.C. HIV Protocols. Springer; 2009. Measuring HIV neutralization in a luciferase reporter gene assay; pp. 395–405. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Full length sequence of Jeko-1 HCDR3 target sites
Data Availability Statement
The datasets generated in this study are available upon reasonable request.