

Abstract
Cold tumor microenvironment (TME) marked with low effector T cell infiltration leads to weak response to immune checkpoint inhibitor (ICI) treatment. Thus, switching cold to hot TME is critical to improve potent ICI therapy. Previously, we reported extracellular vesicle (EV)-like ginseng-derived nanoparticles (GDNPs) that were isolated from Panax ginseng C.A. Mey and can alter M2 polarization to delay the hot tumor B16F10 progression. However, the cold tumor is more common and challenging in the real world. Here, we explored a combinatorial strategy with both GDNPs and PD-1 (programmed cell death protein-1) monoclonal antibody (mAb), which exhibited the ability to alter cold TME and subsequently induce a durable systemic anti-tumor immunity in multiple murine tumor models. GDNPs enhanced PD-1 mAb anti-tumor efficacy in activating tumor-infiltrated T lymphocytes. Our results demonstrated that GDNPs could reprogram tumor-associated macrophages (TAMs) to increase CCL5 and CXCL9 secretion for recruiting CD8+ T cells into the tumor bed, which have the synergism to PD-1 mAb therapy with no detected systemic toxicity. In situ activation of TAMs by GDNPs may broadly serve as a facile platform to modulate the suppressive cold TME and optimize the PD-1 mAb immunotherapy in future clinical application.
Keywords: ginseng-derived nanoparticles, GDNPs, macrophages, cold tumor, immune checkpoint inhibitor, PD-1 mAb, chemokines
Graphical abstract
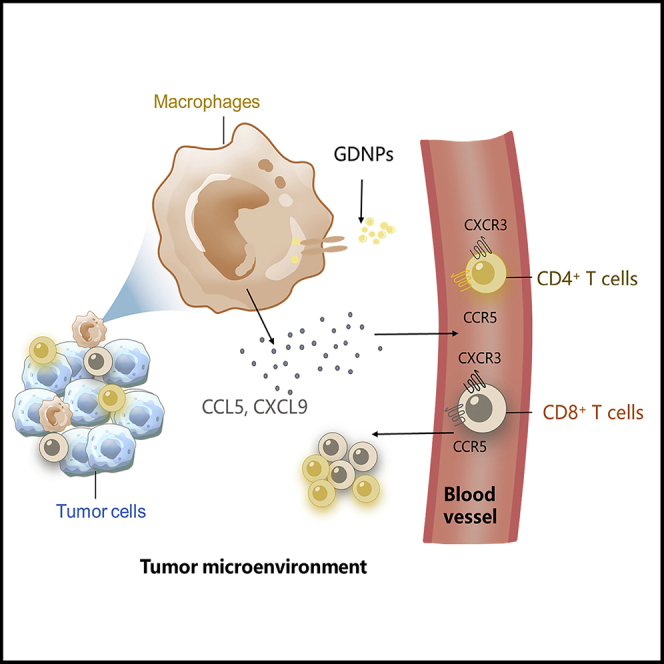
In this paper, Cao and colleagues found that GDNPs are a more efficient PD-1 mAb therapy by simulating TAMs secreting CCL5 and CXCL9 to recruit CD8+ T cells into tumors. GDNPs and PD-1 mAb combinatorial therapy effectively convert cold tumors into hot tumors in mice and increase the ICI treatment effectiveness.
Introduction
By inactivating inhibitory immune receptors (immune checkpoints), immune checkpoint inhibitor (ICI) therapies have greatly expanded the landscape of carcinoma treatment.1,2 Among ICI therapies, monoclonal antibodies (mAbs) targeting programmed cell death protein-1 (PD-1) or its ligand PD-L1 have become popular inhibitors that can activate tumor-infiltrated T lymphocytes and induce an anti-tumor immune response. Currently, many clinical trials on PD-1/PD-L1 therapies have shown promising therapeutic outcomes in multiple types of cancer.3 However, there has been a relatively low overall response rate in a large proportion of patients who undergo ICI treatments.1 T cell infiltration into tumors may be the primary factor for effective ICI treatments, and tumors with high T cell infiltration are defined as “hot” or “T cell-inflamed” tumors. When tumors per se lack T cell infiltration, or there is a paucity, they are characterized as “cold” or “non-inflamed” tumors.4, 5, 6 Cold tumors typically do not respond well to ICI therapy, as in the case of colon, breast, and pancreatic cancers.6,7 Therefore, boosting T cell infiltration into tumors is critical for improving the effects of ICI treatments.8
Tumor-associated macrophages (TAMs) play critical roles in supporting tumor progression, metastasis, and therapy resistance.9,10 In the early stages of tumor formation, tumoricidal macrophages (M1-like, CD206loCD86hi) are the main phenotype of TAMs. As the tumor microenvironment (TME) is modified to support tumor progression, tumor-supportive macrophages (M2-like, CD206hiCD86lo) represent the majority of TAMs in advanced tumor stage.11 Thus, TAMs with plasticity can be further therapeutically exploited to reactivate anti-tumor properties.12 Recent studies indicated that TAMs mediate the resistance of ICI or adoptive T cell immunotherapies.13,14 So they have emerged as a target of immunotherapy. Notably, the expression of pro-tumor M2-like phenotype on TAMs is strongly correlated with poor prognosis.15,16 Furthermore, TAM-targeting treatments in clinical trials and pre-clinical studies, such as colony-stimulating factor-1 receptor blockade,17 interferon (IFN)-γ,18 Toll-like receptor 7/8 (TLR7/8) agonists imiquimod,15,19 and iron oxide nanoparticles,20 indicated that reprogramming M2-like macrophage to M1 like or decreasing TAM tumor infiltration percentage is promising to effectively delay tumor progression. However, whether macrophage polarization by reprogramming macrophage could inflame the cold tumor immunophenotype to hot has not been reported.
Our previous study demonstrated that ginseng-derived nanoparticles (GDNPs), isolated from fresh Panax ginseng C.A. Mey, induce macrophage polarization to the tumoricidal phenotype through the TLR4/myeloid differentiation antigen 88 (Myd88) signaling pathway.21 We found that GDNPs monotherapy significantly suppressed B16-F10 tumor growth. Moreover, GDNPs increased CD8+ T/regulatory T cells (Tregs) ratio in TME in vivo and promoted CD8+ T lymphocyte proliferation in vitro. As GDNPs showed potential to reverse cold TME to hot, we hypothesized that GDNPs could be an auxiliary factor for ICI therapy.
In this study, we explored whether GDNPs combined with PD-1 blockade could alter the pro-tumor environment to an anti-tumor environment, initiated by polarizing M2-like to M1-like TAMs. The results demonstrated that, compared with T cells in the Vehicle group (immunoglobulin G [IgG] isotype), more T cells were recruited by TAM-derived chemokines to the TME after Combo (combinatorial) treatment. This Combo strategy aided conversion of the TME from cold to hot and further enhanced the anti-tumor efficacy of PD-1 mAb.
Results
Immune checkpoint blockade response positively correlated with CD206loCD86hiCD8hi status
The phenotype of TAMs is significantly correlated with patients' survival.9 The high density of tumor-supportive (M2-like) macrophages in the TME may indicate poor prognosis.22, 23, 24 To better explore the correlation between tumor-supportive (CD206hiCD86lo)/tumoricidal (CD206loCD86hi) TAMs and tumor cytotoxic T lymphocytes (CTLs) in colon adenocarcinoma (COAD) and triple-negative breast cancers (TNBCs), gene transcript analyses using Gene Expression Omnibus (GEO) was performed. The results showed that CD8A gene expression is lower in patients with CD206hiCD86lo than that of CD206lo CD86hi patients in both COAD and TNBC (Figure 1A, p = 0.00028, gene sets GEO: GSE17356 + GEO: GSE17357; Figure 1B, p = 0.0047, gene set GEO: GSE58812). Moreover, we found that patients with higher CD8A expression may indicate longer overall survival in both COAD and TNBC (Figure 1A, log rank test p = 0.0035, n = 355, best cut-off; Figure 1B, log rank test p = 2.8793e-05, n = 252, best cut-off). As CD206 expression could be treated as a surrogate for M2-like TAMs in tumors,25 52 clinical colorectal cancer samples were stained for CD206 and CD8A by immunohistochemistry. The results showed negative correlations between CD206 and CD8A in COAD tumor samples (Figure 1C; n = 52, R = −0.4047, p = 0.0029), which indicated that lower tumor-supportive macrophage infiltration in tumors is accompanied with higher CD8+ T lymphocyte infiltration.
Figure 1.

CD8A and tumor-associated macrophages (TAMs) marker expression in clinical specimens from the Gene Expression Omnibus (GEO) cancer dataset and multiple tumor models
Samples across (A) colon adenocarcinoma (COAD) or (B) triple-negative breast cancer (TNBC) were divided into CD206hiCD86lo and CD206loCD86hi using the median of all specimens (analyzed by t test, p = 0.00028 for COAD and p = 0.0047 for TNBC). Kaplan-Meier analyses of patients stratified by CD8hi versus CD8lo in (A) COAD or (B) TNBC (p = 0.0035 for COAD and p = 2.8793e-05 for TNBC). (C) Immunohistochemical staining of CD206 and CD8 in COAD samples (scale bars, 200 μm). Correlation analyses CD8 expression and CD206 expression in n = 52, p = 0.0029.
To mimic the ICI treatment efficiency in human cancer, we applied anti-PD-1 mAbs to multiple murine tumor models. As expected, PD-1 mAb alone significantly inhibited the growth of hot tumors such as B16-F10 murine melanoma1,26 (Figures S1A and S1B), but mice bearing CT26 murine colon tumor and 4T1 murine breast tumor were resistant to PD-1 mAb treatment27,28 (Figure S1B). Subsequently, we analyzed the TME in three murine models on day 21 following tumor inoculation by flow cytometry, and the results showed that a ratio of F4/80+/CD45+ is more than 10% in the 4T1 murine breast tumor, CT26 murine colon tumor, or B16-F10 murine melanoma model (Figure S1C). Moreover, to understand the association between ICI therapy resistance and TAMs characterization, correlations between a ratio of CD8+/CD45+ and tumor-supportive macrophages/TAMs (CD206+/F4/80+) were analyzed, and the results showed negative correlation in a CT26 murine colorectal cancer model, which were consistent with the results of gene expression trends in COAD (Figure S1D).
Therefore, we hypothesized that polarizing TAMs to the tumoricidal phenotype could effectively increase infiltration of CD8+ T cells into the TME, which may inflame cold tumors to improve a response to ICI therapy.
Combo therapy of GDNPs and PD-1 mAb reverses TAMs polarization and effectively inhibits growth of multiple tumors
Our previous data demonstrated that GDNPs isolated from fresh ginseng root can effectively inhibit melanoma (hot tumor) progression by polarizing TAMs to M1-like macrophages.21 Moreover, we found that the percentage of T cells in TME increased markedly. Based on these findings, we speculated that a GDNPs-induced increase in M1-like macrophages may improve immunosuppressive status in TME. Furthermore, GDNPs treatment can induce the activation of T cells to inflame cold tumors to hot tumors and enhance the efficacy of ICI. To clarify the efficacy of GDNPs in different tumor immunophenotypes, we prepared GDNPs as previously reported21 and confirmed their characterization. The Nanosight tracking analysis showed that mean size GDNPs is around 200.4 nm (Figure S2A). Transmission electron microscopy (TEM) examination indicated that GDNPs are spherical in shape (Figure S2B). By developing a sensitive liquid chromatography-mass spectrometry (LC-MS) method, Ginsenoside Re contents of GDNPs in each batch were detected without significant changes (Figure S2C). Besides, the GDNPs macrophage targeting properties were reclarified as well (Figure S2D). Interestingly, a remarkable decrease in tumor growth in CT26 murine colon tumor and 4T1 murine breast tumor models with GDNPs treatment (Figure S1E) was observed. However, 5-time GDNPs treatment did not maintain the long-time survival of CT26 murine tumor-bearing mice (clinical end point, tumor volume ≥ 2,000 mm3; Figure S1F).
Thus, we evaluated whether the combination of PD-1 mAb with GDNPs could improve the anti-tumor response in cold tumors (Figure 2A). In the CT26 murine colon tumor model, the tumor progression in mice with Combo treatment was significantly delayed compared with that in the PD-1 mAb group and Vehicle group. Meanwhile, the GDNPs treatment was more efficient than Vehicle or PD-1 mAb treatments (Figure 2B). Consistently, the Combo group showed a much lower tumor weight (Figures 2C; Figure S3A) than Vehicle, PD-1 mAb, and GDNPs groups. Similar results were found in 4T1 murine breast tumor and MC38 murine colon tumor models (Figures S4A−S4D). Thus, we concluded that Combo treatment of PD-1 mAb and GDNPs enhanced anti-tumor efficiency in cold tumor models. In addition, monitoring mice weight (Figure S3B) and organ tissue histological staining (Figure S5) showed Combo treatment was tolerated without significant toxicity.
Figure 2.

Combinatorial (Combo) therapy using GDNPs and PD-1 mAb elicits rejections of the CT26 murine colon tumor by polarizing M2-like macrophages to M1-like phenotype
(A) Time schedule for tumor implantation and drug treatment. (B) Tumor volume and (C) tumor weight for different treatment types, such as Vehicle, PD-1 mAb, GDNPs, or Combo treatment in CT26 murine colon tumor model (n = 6 for each group, one-way ANOVA or two-way ANOVA, ∗∗p < 0.01, ∗∗∗∗p < 0.0001). (D) Ratio of CD45+ in tumor microenvironment (TME), CD11b+CD45+ in immune cells, F4/80+CD11b+ in immune cells in the CT26 murine colon TME (n = 5 for each group, one-way ANOVA, ∗p < 0.05). (E) Representative fluorescence-activated cell sorting (FACS) plots and quantification of M2/M1. Percentage of PD-L1+/F4/80+ in TAMs. Representative flow cytometry picture for M2-TAM and M1-TAM (n = 5 for each group, one-way ANOVA, ∗p < 0.05).
Further analyses of Tumor infiltrated lymphocytes (TILs) in TME were carried out on day 21 after tumor inoculation by flow cytometry. The results indicated that the percentage of CD45+ immune cells increased 2- to 3-fold in tumors treated with GDNPs or Combo treatment than in those treated with PD-1 mAb in CT26-bearing mice (Figure 2D). In TILs, the percentage of myeloid cells in TME immune cells significantly decreased in the GDNPs group compared with that in the Vehicle or PD-1 mAb group (Figure 2D). The ratio of TAMs in all TILs (CD11b+ F4/80+/CD45+) did not differ in each group; however, GDNPs and Combo therapy decreased the M2/M1 ratio in tumors (Figures 2D and 2E). We also tested PD-L1+ expression on TAMs and found no significant changes among different treatment groups (Figure 2E). Thus, these results suggested that re-polarizing of TAMs by GDNPs can enrich immune cells in the TME and improve the efficacy of ICIs in cold tumors.
Combination of GDNPs and PD-1 blockade promotes T lymphocyte infiltration and anti-tumor immune response
CTLs play a central role in killing tumor cells. The ratio of infiltrated CD8+ T cells in tumors is the major difference that distinguishes cold tumors from hot ones.24 As PD-1 mAb mainly targets CTLs, we validated the percentage of T cell infiltration and the protein level of cytotoxic cytokines in T lymphocytes. Analyses of the graft TME by flow cytometry showed an increase in T cell (CD3+/CD45+) infiltration in the CT26 murine colon cancer model after GDNPs or Combo treatment (Figure 3A). The ratio of CD8+/CD3+ increased in GDNP- and Combo-treated groups compared with that in the PD-1 mAb or Vehicle group (Figure 3B). We also analyzed CD8 protein expression in the CT26 murine colon tumor by immunohistochemistry (IHC), which showed the same trend as that in the flow cytometry analyses (Figure 3C).
Figure 3.

Combo treatment can activate tumor-infiltrated T lymphocytes
Ratio of different T cell phenotypes and T cell-related functional analyses in CT26 murine colon TME by FACS. (A) Ratio of CD3+ in CD45+ cells (n = 5 for each group, one-way ANOVA, ∗p < 0.05). (B) Ratio of CD8+ in CD3+ T cells (n = 5 for each group, ∗p < 0.05. ∗∗p < 0.01, ∗∗∗∗p < 0.0001). (C) Representative pictures of CD8 IHC staining in TME and the quantification of the H-score of IHC staining for CD8 in CT26 murine colon tumor samples (scale bar, 50 μm; n = 3 for each group, ∗p < 0.05). (D) Quantification of IFN-γ+ CD8+ T cells and representative FACS histograms (n = 5 for each group, ∗p < 0.05, ∗∗p < 0.01). (E) Quantification of TNF-α+ CD4+ T cells, granzyme B+ CD8+ T cells, and Ki67+ CD4+ T cells (n = 5 for each group, ∗p < 0.05, ∗∗p < 0.01). (F) Quantification of IFN-γ+ CD4+ T cells and representative FACS histograms (n = 5 for each group, ∗p < 0.05, ∗∗p < 0.01). (G) Ratio of TNF-α+ CD4+ T cell, granzyme B+ CD4+ T cell, Ki67+ CD4+ T cell, and Th1/Treg (n = 5 for each group, ∗p < 0.05, ∗∗p < 0.01). Data are presented as mean ± SEM and analyzed by one-way ANOVA.
CTLs are characterized by the secretion of IFN-γ, granzyme B, and tumor necrosis factor (TNF)-α, which kill tumor cells directly. In this study, the upregulation of IFN-γ and TNF-α was detected in tumor-infiltrated CD8+ T cells from the CT26 murine graft colon tumor in the Combo group (Figures 3D and 3E). Meanwhile, CD8+ T lymphocytes showed higher expression of Ki67+ in the Combo-treated group compared with that in the PD-1 mAb/Vehicle group, which showed higher proliferation ability of CTLs in the Combo group (Figure 3E). We next focused on the expression of co-inhibitory molecules on T cells. For CD8+ T cells, the lower expression of T cell Ig mucin domain-3 (TIM3; Figure S6A), inducible co-stimulator (ICOS; Figure S6B), and PD-1 (Figure S6C) was observed in the CT26 murine colon tumor with Combo treatment. These results indicated that combining GDNPs with PD-1 mAb therapy could induce CD8+ T cell activation in the TME.
In addition, we found a higher T helper 1 cells (Th1)/Treg ratio (Figure 3G) and upregulated expression of granzyme B+, IFN-γ+, and TNF-α+ CD4+ T lymphocytes in tumors in the Combo group as compared to that of Vehicle-treated mice. Besides, the improved percentages of IFN-γ+ and TNF-α+ CD4+ T lymphocytes were detected in the Combo group rather than those in the PD-1 mAb group. These results suggested that more CD4+ T cells were differentiated into Th1 cells in the Combo-treated TME (Figures 3F and 3G). Besides, the Milliplex Luminex assay was performed to clarify the cytokine changes in the plasma in Combo treatment. Results showed that the concentrations of interleukin (IL)-2 and IL-12 p40 strongly increased in plasma under Combo treatment. Although the IL-12 p70 concentration showed no significant changes, the data are in the same trend as IL-2 (Figure S3C).
Finally, to validate the importance of CD8+ and CD4+ T lymphocytes in activating anti-tumor effects in Combo treatment, CD8+ and CD4+ T lymphocytes were independently ablated in CT26 murine colon tumor-bearing mice (Figure 4A). The depletion efficiency was confirmed in peripheral blood before Combo or Vehicle treatment by flow cytometry (Figures S7A and S7C). The results indicated that CD8+ or CD4+ T lymphocyte depletion reduced the Combo tumoricidal efficiency (Figures 4B and 4C; Figures S7B and S7D). Thus, these results are consistent with our hypothesis that CD4+ and CD8+ T lymphocytes play crucial roles in achieving efficacy with the Combo treatment. Taken together, these findings suggest that anti-tumor efficacy of Combo therapy is T lymphocyte dependent.
Figure 4.

CD4+ T and CD8 + T lymphocytes play important roles in Combo treatment
(A) Paradigm of tumor implantation, CD4+ or CD8+ depletion, and drug treatment time schedule in CT26 murine colon cancer model. (B) Tumor volume and weight of Vehicle + IgG, Combo + IgG, Vehicle + anti-CD8, and Combo + anti-CD8 four groups (n = 6 for each group, ∗∗∗p < 0.001, ∗∗∗∗p < 0.0001). (C) Tumor volume and weight of Vehicle + IgG, Combo + IgG, Vehicle + anti-CD4, and Combo + anti-CD4 four groups (n = 5 for each group, ∗p < 0.05, ∗∗∗p < 0.001, ∗∗∗∗p < 0.0001). Data are presented as mean ± SEM and analyzed using one-way ANOVA or two-way ANOVA.
Combo therapy enhances long-term antigen-specific anti-tumor memory and inhibits tumor metastasis
Of note, 40% of CT26-bearing mice in the Combo group were ethically survived for over 50 days with treatment administered only 14 days after tumor inoculation (Figure 5A). As the surviving mice in the Combo group were accompanied with complete regression of CT26 murine colon tumors after the first inoculation, it raised the question whether the mice can survive the relapse of the tumors. A rechallenge assay revealed that approximately 50% of mice in the pre-Combo-treated group overcame CT26 murine colon tumor recurrence (Figures 5B and 5C). However, tumor rejection was not observed in the 4T1 murine breast tumor in the same mice. The results further verified the specificity of anti-tumor immune response induced by Combo treatment.
Figure 5.

GDNPs combined with PD-1 mAb therapy enhance long-term antigen-specific anti-tumor memory
(A) Survival curves for Vehicle, PD-1 mAb, GDNPs, and Combo groups in the CT26 murine colon tumor model (n = 10 for each group, p = 0.0132). (B) Paradigm of tumor rechallenge assay and time schedule. (C) Quantification of tumor volumes for tumor rechallenge assay. Representative pictures for tumor-bearing mice (n = 6 for each group, ∗∗∗∗p < 0.0001). (D) Quantification of CD44+ CD62L+ and CD44+ CD62L− for CD4+ or CD8+ T cells in the spleen of wild-type (WT) or Combo pre-treated mice (n = 5 for each group, ∗p < 0.05, ∗∗p < 0.01, ∗∗∗∗p < 0.0001). Data are presented as mean ± SEM and analyzed using two-way ANOVA, Student’s t test, or log rank (Mantel-Cox) test.
To further investigate the latent changes in the long-term antigen-specific anti-tumor memory behind the encouraging results, we evaluated memory T cells in the spleen by flow cytometry. In the spleen, central memory T lymphocytes (CD44+ CD62L+) and effector memory T lymphocytes (CD44+ CD62L−) in both CD4+ and CD8+ populations significantly increased in pre-Combo-treated survivors compared with that in tumor-bearing mice of the same age (Figure 5D). These results indicate that Combo treatment may elicit a durable anti-tumor immune memory.
As tumor metastasis highly affects the mobility and mortality of patients, we established a 4T1-Luc murine breast cancer lung metastasis model (Figure S8A). A significant reduction in tumor nodules in the lungs of the mice was observed with Combo treatment compared with that for treatment with PD-1 mAb alone (Figure S8B). Moreover, bioluminescent signals were also analyzed to assess lung metastasis. We found that Combo treatment decreased the luminescence in lungs of tumor-bearing mice (Figure S8C). These results indicate GDNPs combined with PD-1 mAb may activate the immune system to better depress tumor metastasis.
GDNPs elicited M2-like macrophage-secreting chemokines for T cell chemotaxis in vitro
Mounting evidence showed that GDNPs combined with PD-1 mAb optimized the TME by increasing TIL infiltration, which successfully converted cold tumors to hot. This led us to explore how GDNPs-treated macrophages enhance T cell infiltration in the TME. To further clarify whether CTLs were recruited under GDNPs-treated bone marrow-derived macrophages (BMDMs) or not, 1,1’-dioctadecyl-3,3,3’,3’-tetramethylindocarbocyanine perchlorate (DiI)-stained CD8+ T cells (CD8+ T microbeads isolated from spleen) were intravenously (i.v.) injected into tumor-bearing mice. The immunofluorescence staining data showed that GDNPs treatment recruited more CD8+ T cells in the tumors than the phosphate-buffered saline (PBS) treatment group (Figure 6A).
Figure 6.

GDNPs may elicit macrophages to secrete chemokines for T cell chemotaxis
(A) CD8+ T trafficking and biodistribution of DiI-labeled CD8+ T lymphocytes in CT26 murine colon tumor with BMDMs + PBS or BMDMs + GDNPs treatment administered intravenously (scale bar, 50 μm; n = 5 for each group, ∗∗p < 0.01). (B) Bubble plots showing correlations between Cd8a and chemotactic gene-related transcripts in COAD and TNBC from TCGA cancer datasets. (C) Volcano plot showing upregulated chemotactic gene expression from results of RNA sequencing for M2-BMDMs + GDNPs or M2-BMDMs + PBS (p < 0.05, fold change (FC) > 1.2, three samples each group). (D) Relative gene expressions of Ccl3, Ccl5, Cxcl9, and Cxcl10 in M2 like-BMDMs or GDNPs-stimulated M2-BMDMs for 12 h/24 h by real-time PCR (n = 3−4 for each group, ∗∗∗∗p < 0.0001). (E) CCL5 and CXCL9 concentration in the culture media of M2-BMDMs, GDNPs + M2-BMDMs for 12 h, and GDNPs + M2-BMDMs for 24 h by ELISA (n = 4 for each group, ∗p < 0.05, ∗∗∗p < 0.001). (F) Schematic diagram and quantification of chemotactic assay. CFSE-stained CD8+ T cells migrated toward the supernatant from TAM culture media of the Combo group in the presence of anti-CCL5 and anti-CXCL9 neutralization by flow cytometry (n = 3 for each group, ∗∗p < 0.01). For all panels, data are presented as mean ± SEM and analyzed using one-way ANOVA or Student’s t test.
The CD8+ T cell infiltration always has been measured by Cd8a transcript expression level in tumor tissue, and chemokines have been clarified to regulate immune cell trafficking and tumor development. Therefore, we selected Cd8a as a gene marker for quantifying chemokine-associated genes. We found that Cd8a expression significantly correlated to Ccl5, Cxcl9, Cxcl10, and Cxcl13 high expression in COAD and TNBC across The Cancer Genome Atlas (TCGA) data (Figure 6B). Thus, we performed whole transcriptome RNA sequencing (RNA-seq) of M2-BMDMs with or without GDNPs in vitro, and the gene differential expression analyses showed significantly higher pro-inflammatory cytokines and chemokines, such as Ccl2, Ccl3, Ccl5, Cxcl9, and Cxcl10 genes in the GDNP-treated group (Figure 6C). Based on the RNA-seq results and bioinformatic analyses, real-time polymerase chain reaction (PCR) for chemokines was performed. Our results confirmed the significantly increased expression of Ccl5 and Cxcl9 transcriptomes in M2-like macrophages with GDNPs treatment (Figure 6D). Based on the chemokine sequencing results,21 we further analyzed the secretion of CCL5 and CXCL9 in culture media of M2-BMDMs and M2-BMDMs with GDNPs. Our findings confirmed that GDNPs treatment markedly increased M2-BMDMs secretion of CCL5 and CXCL9 (Figure 6E). To further examine the importance of GDNPs-treated TAMs in CD8+ T lymphocyte chemotaxis, CCL5 and CXCL9 were inhibited using neutralizing antibodies in the culture media of TAMs under Combo treatment. The results showed a marked decrease of migrating T lymphocytes with the CCL5 and CXCL9 neutralization antibodies (Figure 6F). These findings indicated that TAMs under GDNPs treatment may promote more CCL5 and CXCL9 secretion to recruit T lymphocytes.
GDNPs facilitated TAM-derived CCL5 and CXCL9 secretion to recruit T lymphocytes in vivo
We analyzed the changes in chemokines secreted by TAMs in different treatment groups in CT26 murine colon cancer model. 2 weeks after CT26 colon tumor cell subcutaneous inoculation, we found that the expression of CCL5 (Figure 7A) and CXCL9 (Figure 7B) markedly increased in TAMs in the Combo group, and CXCL9+/F4/80+ significantly increased in the GDNPs group.
Figure 7.

GDNPs combined with PD-1 mAb elicit TAM-secreting chemokines to change the TME
(A) Ratio of CCL5+/F4/80+ and (B) CXCL9+/F4/80+ in CT26 colon tumor model TME under Vehicle, PD-1 mAb, GDNPs, or Combo treatment (n = 5 for each group, ∗∗p < 0.01, ∗∗∗∗p < 0.0001). (C) Ratio of CCR5+/CD8+, CXCR3+/CD8+, CCR5+/CD4+, and CXCR3+/CD4+ in tumors with Vehicle, PD-1 mAb, GDNPs, or Combo treatment in CT26 murine colon tumor models (n = 5 for each group, ∗∗p < 0.01, ∗∗∗p < 0.001, ∗∗∗∗p < 0.0001). Kaplan-Meier analyses for (D) COAD or TNBC datasets from GEO and TCGA were divided into two groups independently, stratified by CCL9 or CXCL5 median of all specimens (p = 0.04 for CCL5 in COAD, p = 0.015 for CXCL9 in COAD, p = 0.00018 for CCL5 in TNBC, p = 0.0023 for CXCL9 in TNBC). Data are presented as mean ± SEM and analyzed using one-way ANOVA or Kaplan-Meier analyses.
As the gene transcript of CXCL9 and CXCR3, CCL5 and CCR5 showed positive correlations in both COAD and BRCA, two tumor types (Figure S9),29 we investigated CCL5 and CXCL9 receptors, CCR5 and CXCR3 expression on T lymphocytes in tumors by flow cytometry. Flow cytometry analyses indicated that CCR5+/CD8+ and CCR5+/CD4+ were found to significantly increase with Combo treatment in tumor samples (Figure 7C). These results indicated that increased chemokines from TAMs and chemokine receptors on T cells could be the causes of increased T lymphocyte infiltration.
Moreover, we found that higher CCL5 and CXCL9 expression in COAD and TNBC patients indicated longer overall survival, using TCGA and GEO datasets (Figure 7D). Taken together, GDNPs increased TAM-derived CCL5 and CXCL9 secretion for further T lymphocytes recruitment in vivo and were more efficient with PD-1 mAb treatment in cold tumors. In addition, we deleted the macrophage in vivo in the Combo-treated group using clodronate liposome (CL) (Figure S10A). Macrophage depletion efficiency by CL was confirmed by flow cytometry in peripheral blood (Figure S7E). The results showed a reverse of tumor decrease after in vivo macrophage deletion in Combo treatment (Figure S7F), and there was a reduction in CCL5 or CXCL9 in the tumor suspension in the Combo + CL group (Figure S10A), which verified the important roles of CCL5 and CXCL9 in a Combo-induced tumor decrease. Then, CD8 infiltration in tumors were verified by immunofluorescence simultaneously. The statistical results showed that CD8 infiltration significantly decreased in the Combo + CL group compared with the Combo group (Figure S10B). Thus, we confirmed that GDNPs activated macrophages to initiate the CD8+ T cell infiltration in TME. Besides, we neutralized the CCL5 and CXCL9 in the CT26 murine colorectal cancer model in vivo; the results showed that both αCCL5 and αCXCL9 significantly weakened the Combo-induced tumor depression (Figure S11A).
Discussion
Among cancer immunotherapies, ICI treatment has yielded remarkable clinical benefits to many cancer patients. However, a large proportion of patients are refractory to PD-1/PD-L1 blockade treatment due to the paucity of tumor T cell infiltration that characterizes cold tumors. Thus, boosting T cell infiltration in tumors is critical for improving ICI treatment. Studies have reported many optional therapies for cold tumors, for example, radiotherapy, chemotherapy, targeted therapies, DNA repair-based therapy, adoptive cell therapy, oncolytic therapy, vaccine-based therapy, and T cell immunomodulators.8,30,31 However, those therapies have not been clearly verified to effectively change cold tumors to hot. Thus, employing ICI-related Combo treatment to improve therapy efficacy raises great interest in translational medicine of related research.7,32
For most tumor types, especially cold tumors, TAMs are the major immune cells in the TME, and a high percentage of M2-like TAMs infiltration indicates poor clinical prognosis. Thus, targeting of TAMs using pan-therapy strategies, including targeting polarization signaling pathway and TAMs phagocytosis, has gained great interest.9,33 Many studies have published various strategies for TAMs-M1 polarization in tumor therapy including extracellular vesicles (EVs).20,34 Some reports have demonstrated that different EVs from others mammalian cells with high stability and high bioavailability are a promising candidate for drug delivery systems. Compared with mammalian cell-derived EVs35 and artificial EVs, plant-derived EVs are more advantageous with respect to scale-up and non-toxicity.36 Many studies have identified that multiple plant-derived EVs have biological properties of tissue and organ specificity and low immunogenicity and are easily mass produced.37, 38, 39 GDNP, a plant-derived nanoparticle drug, is a discovery in the cutting-edge area of EV drug delivery, with many advantages similar to most of plant-derived nanoparticles. However, to data, GDNPs are the only reported fresh plant-derived EVs to cause TAMs polarization to the tumoricidal phenotype, and their high biosafety demonstrates that GDNPs could be safely used in combination with ICI.
By analyzing the transcript expression between CD8 and CD206hiCD86lo in multiple human cold tumors, the results indicated that M1-like macrophage polarization may improve the cold TME by increasing CD8 infiltration and reversing cold tumors to hot. In this study, GDNPs combined with PD-1 mAb therapy inhibited tumor progression more efficiently in three different murine tumor models compared with PD-1 mAb treatment alone. The Combo treatment successfully altered macrophage polarization and overcame low CD8+ T cell infiltration to improve the effects of anti-PD-1.
An activated immune system accompanied with immunological memory is necessary for resisting tumor relapse.26,27 Besides, tumor metastasis plays an important role in patient mortality, as lung metastasis mostly occurs in advanced breast cancer progress.40 The CT26 murine colon cancer rechallenge assay and 4T1-Luc murine breast cancer lung metastasis assay were performed, and the results indicated that GDNPs combined with PD-1 mAb effectively activated the immune system, which helps to reduce tumor relapse and depress tumor lung metastasis foci formation.
To better illustrate the mechanism through which GDNPs-reprogrammed macrophages increased the ratio of T cells in the TME, multiple assays were performed. The results all supported that chemokines secreted by GDNP-polarized macrophages are the main cause of T cell tumor infiltration. Chemokines regulate the migration of leukocytes, and inducible chemokines increase at the inflamed site, which recruits activated leukocytes.41,42 Indeed, chemokines such as CCL5, CXCL9, CXCL13, CXCL10, and CXCL16 have been reported to recruit effector CD8+ T cells in the tumors, resulting in an anti-carcinoma TME.31,37,42, 43, 44 Our data indicated that CCL5 and CXCL9 induced by GDNPs polarized M1-like macrophages, contributing to the hot tumor immunophenotype establishing, which is consistent with previous reports.45
Whereas T lymphocyte recruitment-related chemokines increased in the TME, some chemokine receptors, such as CCR5 and CXCR3, are highly expressed in T lymphocytes as well, which are important for T lymphocyte infiltration in tumor beds. In our study, the increased T lymphocyte receptors CXCR3 and CCR5 enhanced CTL activation under Combo treatment. Although PD-1 mAb treatment alone led to a marked increase in CCR5 expression on CD4+ T cells, it was not sufficient to change the cold tumor immunophenotype. The key role of CXCL9 that binds CXCR3 on T cells enhances recruitment of CD4+ T cells and promotes the differentiation of inflammatory Th1 CD4+ T cells as well. CCR5 and CXCR3 are known markers of activated T lymphocytes, especially Th1 cells, and their anti-tumor properties are well studied.46, 47, 48 These results indicate that the Combo treatment can effectively transform cold tumors to hot by activating peripheral T lymphocytes.
Based on most of the anti-tumor immune indicators in this research, the results showed that the treatment efficacy of Combo was superior to GDNPs or PD-1 mAb alone. We speculated that it is because GDNPs reverse TME to make the PD-1 mAb treatment more effective, and PD-1 mAb blocked PD-1 function in TAMs in the Combo group, which is important to emancipate reprogrammed TAM anti-tumor efficacy.49 Besides, our previous study indicated that ceramides, an important lipid component of GDNPs, polarized macrophages to the pro-inflamed phenotype by activating the TLR4-dependent signaling pathway.50 Following TLR4 activation, Th1 proinflammatory cytokines such as TNF-α and IL-12p70 are highly secreted by macrophages.51 In fact, Th1 activation is necessary for subsequent CTLs activation, which is important for T cell chemotaxis initiation.
Although the macrophage depletion effectively indicates the importance of CCL5 and CXCL9 in vivo, the results of the mean tumor volume in Combo + CL are significantly lower than the Vehicle group. Our previous research presented that granulocytes and dendric cells could phagocytose a small amount of GDNPs, which could be activated as well.21 As the CL could only deplete 83% macrophages in this research (Figures S7E and S7F), and monocytes could differentiate into macrophages continuously,10,52 there are still some macrophages that survived the CL treatment. In the future, we will continue to figure out the bioactivation for GDNPs on other immune cells. Besides, many studies have published that tumors with low tumor mutation burden (TMB) are accompanied with a low ICI treatment response rate.53 As CT26 and MC38 murine tumor cells have been reported with a high TMB, and 4T1 has been recognized with a low TMB,54,55 we hypothesized that GDNPs treatment could improve the PD-1 mAb efficacy in low TMB, and more research will be performed to verify the hypothesis in future.
Compared with other reported ICI-related Combo therapies, our GDNPs and ICI Combo therapy are the discovery of fresh plant-derived EVs, which are efficient in tumoricidal agents and lack obvious side effects. As the pharmaceutical content of GDNPs may have unintended reactions in tumors and other diseases, concrete steps are necessary to be taken. Moreover, the universality of GDNPs, combined with different ICI treatments in tumoricidal roles, and related clinical research are intensive further steps of this study.
Materials and methods
The ethics statement, patients’ samples, study design, GDNPs preparation, RNA-seq analysis, and real-time PCR are presented in online Supplemental materials and methods.
Isolation of tumor-infiltrating cells and splenocytes
Mouse tumor tissues were sliced into 5 mm pieces and minced before using the Tumor Dissociation Kit (130096730, MACS) per the manufacturer’s instructions.
The spleens were sliced into 5 mm pieces and passed through a 40-μm filter. After being mixed with red blood cell lysis buffer (C3702; Beyotime), the cells were resuspended in RPMI-1640 medium for future analyses.
After mice were anesthetized by isoflurane, 100 μL peripheral blood was collected from the mouse tail, and 5 mL red blood cell lysis buffer was added. After centrifugation at 300 × g for 5 min at 4°C, cells from peripheral blood were resuspended in RPMI-1640 medium for further analyses.
Flow cytometry of immune cells in the TME, splenocytes, and peripheral blood
Immune cells were isolated using Percoll (17-0891-09; GE Healthcare) from tumor cell suspension. These cells were incubated with CD16/32 (clone 93; BioLegend, San Diego, CA, USA) for 15 min on ice and then stained with various combinations of the following fluorochrome-conjugated antibodies at the appropriate dilutions for 30 min on ice: CD3-allophycocyanin (APC)/Cy7 (clone 145-2C11; BioLegend), CD8a-phycoerythrin (PE; clone 53-6.7; BioLegend), CD45-fluorescein isothiocyanate (FITC; clone 30-F11; BioLegend), CD11b-APC/Cy7 (clone M1/70; BioLegend), F4/80-PE/Cy7 (clone BM8; BioLegend), F4/80-BV421 (clone BM8; BioLegend), CD8a-APC (clone 53-6.7; BioLegend), CD4-APC (clone GK1.5; BioLegend), CD86-PE (clone PO3; BioLegend), CD206-APC (clone C068C2; BioLegend), CD25-APC (clone PC61.5; Invitrogen), CD4-PE/Cy7 (clone GK1.5; BioLegend), FVD450 (eflour 450; Invitrogen), FVD506 (eflour 506; Invitrogen), CD45-BV510 (clone 30-F11; BioLegend), CD62L-APC (clone MEL-14; BioLegend), CD44-PE (clone IM7; BioLegend), CCR5-APC (clone HIM-CCR5; Invitrogen), CXCR3-PE (clone CXCR3-173; BioLegend), TIM3-PE (clone 5D12/TIM-3; BD Pharmingen), ICOS-PE/Cy7 (clone 7E.17G9; Invitrogen), and PD-1-APC (clone 29F-1A12; BioLegend).
After cells were stained with surface markers, a Fixation/Permeabilization Kit (00-5123-43, 00-5223-56; Invitrogen) was used according to the manufacturer’s instructions. Then, CCL5-PE (clone 2E9/CCL5; BioLegend), CXCL9-PE (clone MIG-2F5.5; BioLegend), Forkhead box protein P3 (FOXP3)-PE (clone FJK-16 s; Invitrogen), or Ki67-Peridinin Chlorophyll-Protein Complex (Percp)-Cy5.5 (clone B56; BD Biosciences) was diluted in the permeabilization buffer 10× (1:20) and incubated for 60 min on ice.
For T cell-derived anti-tumor cytokines, 2 × 106 splenocytes or TILs were incubated in RPMI-1640 medium with Cell Stimulation Cocktail Plus Protein Transport Inhibitors (500×) for 6 h. Then, the cells were stained with surface markers, fixed, and permeabilized using a Fixation/Permeabilization Kit, IFN-γ-PE/Cy7 (clone XMG1.2; BioLegend), granzyme B-FITC (clone NGZB; BioLegend), and TNF-α-BB700 (clone MP6-XT22; BD Horizon), which were diluent in the permeabilization buffer 10× (1:20) for 45 min.
Stained cells were analyzed on a FACSAria II Flow Cytometer; BD Biosciences) using BD FACSDiva software (BD Biosciences), and data were processed using FlowJo version 10 (BD Biosciences). Gating strategies were showed in Figure S12.
Preparation and polarization of mouse BMDMs
Mouse bone marrow was collected by flushing tibias and femurs of 9-week-old male BALB/c mice with cold PBS. After collection, red blood cells were lysed with the red blood cell lysis buffer (Beyotime), and the remaining cells were washed twice with PBS. To induce macrophage differentiation, the cells were cultured in DMEM with 10% fetal bovine serum and 20 ng/mL mouse macrophage colony-stimulating factor (M-CSF) (Bioworld Biotech, USA) for 6 days. On day 7, 20 ng/mL IL-4 (R&D Systems [R&D], USA) and 20 ng/mL IL-13 (R&D, USA) were added to polarize macrophages to the M2 phenotype macrophage for 48 h. The cells were prepared for real-time PCR.
Chemokine analyses
The supernatants of the M2 + GDNPs/PBS culture medium and the Combo-derived TAM culture medium were collected. Enzyme-linked immunosorbent assay (ELISA) was performed to examine the concentration of CCL5 and CXCL9 (ELISA kits were all from Mlbio, China), according to the manufacturer’s instructions.
Chemotaxis assay
For CD8+ T chemotaxis assay in vivo, CT26 murine colon cancer cell-bearing mice were i.v. administrated with 5 × 105 BMDMs under GDNPs or Vehicle pre-treated on day 7, followed by DiI-labeled 4 × 104 CD8+ T cells i.v. on day 8. The mice were sacrificed, and the tumors were collected 5 days after CD8+ T cell injection. Immunofluorescent assay was performed to analyze the tumor-infiltrated DiI-CD8+ T lymphocytes.
For lymphocyte migration assay in vitro, the CD8+ T MicroBeads Mouse Cell Isolation Kit (Miltenyi Biotec, USA) was used to obtain CD8+ T lymphocytes from splenocytes. CD8+ T lymphocytes were labeled for 3 min at 37°C in 5 mΜ carboxyfluorescein diacetate, succinimidyl ester (CFSE, Invitrogen, USA). Conditioned media (150 μL/well) of TAMs from Combo in vitro were loaded in the bottom as part of the Transwell migration chamber containing anti-CXCL9 1 μg/mL and anti-CCL5 1 μg/mL. CFSE-labeled CD8+ T cells (3 × 104) were added to the top chamber in the DMEM culture media. Migration was evaluated by enumerating the number of migrated cells in the bottom chamber using fluorescence-activated cell sorting analyses after 24 h.
Bioinformatic analyses
Computations were performed with the R software system for statistics and figure generation.
For COAD and TNBC tumors, the multi-tumor gene expression microarray dataset from the Expression Project for Oncology (expO; http://www.intgen.org/; GEO: GSE17356 + GEO: GSE17357 COAD and GEO: GSE58812 TNBC) and survival data were analyzed using “survminer” R package.56 TCGA (https://www.cancer.gov/about-nci/organization/ccg/research/structural-genomics/tcga) was used for multi-tumor chemokine gene expression data analyzed with the RTCGAToolbox R package.
For chemotaxis-related RNA transcription variation analyses between M2-BMDMs + PBS and M2-BMDMs + GDNPs, “ggplot2” R package was used; p < 0.05 and fold change = 1.2 were set as the criteria.
Statistical analyses
The results are expressed as the mean ± standard error of mean (SEM). All data were analyzed using GraphPad Prism 7.0 (GraphPad Software, USA) by unpaired Student’s t test, one-way or two-way analyses of variance (ANOVA), and log rank (Mantel-Cox) test. p < 0.05 was considered statistically significant (∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001, ∗∗∗∗p < 0.0001).
Acknowledgments
We thank Dr. Lixin Wang (Medical School, Southeast University) and Dr. Liwei Lu (The University of Hong Kong) for helpful discussion and critical reading of this manuscript. This work was supported by grants from the National Natural Science Foundation of China (no. 82125037, no. 81673945), Open Project of Chinese Materia Medica First-Class Discipline of Nanjing University of Chinese Medicine (no. 2020YLXK001), WINFAST Charity Foundation (RHKY20201215), Priority Academic Program Development of Jiangsu Higher Education Institutions (Integration of Chinese and Western Medicine), and Technology Development Program of Traditional Chinese Medicine in Jiangsu Province (YB2020019).
Author contributions
The study was conceived, designed, and co-written by X.H., M.C., and P.C. The acquisition of data was performed by X.H., Q.W. (Qin Wei), Y.L., L.W., H.H., Y.M., M.L., and D.H. All authors contributed to the analyses of the data, discussed the results, edited the manuscript, and approved the final manuscript. Funding was obtained, and all studies were supervised by M.C. and P.C.
Declaration of interests
The authors declare no competing interests.
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.ymthe.2021.08.028.
Contributor Information
Meng Cao, Email: mcao1979@yahoo.com.
Peng Cao, Email: cao_peng@njucm.edu.cn.
Supplemental information
References
- 1.Gong J., Chehrazi-Raffle A., Reddi S., Salgia R. Development of PD-1 and PD-L1 inhibitors as a form of cancer immunotherapy: a comprehensive review of registration trials and future considerations. J. Immunother. Cancer. 2018;6:8. doi: 10.1186/s40425-018-0316-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Zhu X., Lang J. Soluble PD-1 and PD-L1: predictive and prognostic significance in cancer. Oncotarget. 2017;8:97671–97682. doi: 10.18632/oncotarget.18311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Balar A.V., Weber J.S. PD-1 and PD-L1 antibodies in cancer: current status and future directions. Cancer Immunol. Immunother. 2017;66:551–564. doi: 10.1007/s00262-017-1954-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Duan X., Chan C., Han W., Guo N., Weichselbaum R.R., Lin W. Immunostimulatory nanomedicines synergize with checkpoint blockade immunotherapy to eradicate colorectal tumors. Nat. Commun. 2019;10:1899. doi: 10.1038/s41467-019-09221-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Oliveira A.F., Bretes L., Furtado I. Review of PD-1/PD-L1 Inhibitors in Metastatic dMMR/MSI-H Colorectal Cancer. Front. Oncol. 2019;9:396. doi: 10.3389/fonc.2019.00396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Shin D.S., Zaretsky J.M., Escuin-Ordinas H., Garcia-Diaz A., Hu-Lieskovan S., Kalbasi A., Grasso C.S., Hugo W., Sandoval S., Torrejon D.Y., et al. Primary Resistance to PD-1 Blockade Mediated by JAK1/2 Mutations. Cancer Discov. 2017;7:188–201. doi: 10.1158/2159-8290.CD-16-1223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Galon J., Bruni D. Approaches to treat immune hot, altered and cold tumours with combination immunotherapies. Nat. Rev. Drug Discov. 2019;18:197–218. doi: 10.1038/s41573-018-0007-y. [DOI] [PubMed] [Google Scholar]
- 8.Zappasodi R., Merghoub T., Wolchok J.D. Emerging Concepts for Immune Checkpoint Blockade-Based Combination Therapies. Cancer Cell. 2018;33:581–598. doi: 10.1016/j.ccell.2018.03.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Cassetta L., Fragkogianni S., Sims A.H., Swierczak A., Forrester L.M., Zhang H., Soong D.Y.H., Cotechini T., Anur P., Lin E.Y., et al. Human Tumor-Associated Macrophage and Monocyte Transcriptional Landscapes Reveal Cancer-Specific Reprogramming, Biomarkers, and Therapeutic Targets. Cancer Cell. 2019;35:588–602.e10. doi: 10.1016/j.ccell.2019.02.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ma R.Y., Zhang H., Li X.F., Zhang C.B., Selli C., Tagliavini G., Lam A.D., Prost S., Sims A.H., Hu H.Y., et al. Monocyte-derived macrophages promote breast cancer bone metastasis outgrowth. J. Exp. Med. 2020;217:e20191820. doi: 10.1084/jem.20191820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Guerriero J.L. Macrophages: The Road Less Traveled, Changing Anticancer Therapy. Trends Mol. Med. 2018;24:472–489. doi: 10.1016/j.molmed.2018.03.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Cassetta L., Pollard J.W. Targeting macrophages: therapeutic approaches in cancer. Nat. Rev. Drug Discov. 2018;17:887–904. doi: 10.1038/nrd.2018.169. [DOI] [PubMed] [Google Scholar]
- 13.Veldman J., Visser L., Berg A.V.D., Diepstra A. Primary and acquired resistance mechanisms to immune checkpoint inhibition in Hodgkin lymphoma. Cancer Treat. Rev. 2020;82:101931. doi: 10.1016/j.ctrv.2019.101931. [DOI] [PubMed] [Google Scholar]
- 14.Peranzoni E., Lemoine J., Vimeux L., Feuillet V., Barrin S., Kantari-Mimoun C., Bercovici N., Guérin M., Biton J., Ouakrim H., et al. Macrophages impede CD8 T cells from reaching tumor cells and limit the efficacy of anti-PD-1 treatment. Proc. Natl. Acad. Sci. USA. 2018;115:E4041–E4050. doi: 10.1073/pnas.1720948115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Petty A.J., Yang Y. Tumor-associated macrophages: implications in cancer immunotherapy. Immunotherapy. 2017;9:289–302. doi: 10.2217/imt-2016-0135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Yang M., McKay D., Pollard J.W., Lewis C.E. Diverse Functions of Macrophages in Different Tumor Microenvironments. Cancer Res. 2018;78:5492–5503. doi: 10.1158/0008-5472.CAN-18-1367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Shi G., Yang Q., Zhang Y., Jiang Q., Lin Y., Yang S., Wang H., Cheng L., Zhang X., Li Y., et al. Modulating the Tumor Microenvironment via Oncolytic Viruses and CSF-1R Inhibition Synergistically Enhances Anti-PD-1 Immunotherapy. Mol. Ther. 2019;27:244–260. doi: 10.1016/j.ymthe.2018.11.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Gao Y., Yang J., Cai Y., Fu S., Zhang N., Fu X., Li L. IFN-γ-mediated inhibition of lung cancer correlates with PD-L1 expression and is regulated by PI3K-AKT signaling. Int. J. Cancer. 2018;143:931–943. doi: 10.1002/ijc.31357. [DOI] [PubMed] [Google Scholar]
- 19.Mullins S.R., Vasilakos J.P., Deschler K., Grigsby I., Gillis P., John J., Elder M.J., Swales J., Timosenko E., Cooper Z., et al. Intratumoral immunotherapy with TLR7/8 agonist MEDI9197 modulates the tumor microenvironment leading to enhanced activity when combined with other immunotherapies. J. Immunother. Cancer. 2019;7:244. doi: 10.1186/s40425-019-0724-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Zanganeh S., Hutter G., Spitler R., Lenkov O., Mahmoudi M., Shaw A., Pajarinen J.S., Nejadnik H., Goodman S., Moseley M., et al. Iron oxide nanoparticles inhibit tumour growth by inducing pro-inflammatory macrophage polarization in tumour tissues. Nat. Nanotechnol. 2016;11:986–994. doi: 10.1038/nnano.2016.168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Cao M., Yan H., Han X., Weng L., Wei Q., Sun X., Lu W., Wei Q., Ye J., Cai X., et al. Ginseng-derived nanoparticles alter macrophage polarization to inhibit melanoma growth. J. Immunother. Cancer. 2019;7:326. doi: 10.1186/s40425-019-0817-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Choi J., Gyamfi J., Jang H., Koo J.S. The role of tumor-associated macrophage in breast cancer biology. Histol. Histopathol. 2018;33:133–145. doi: 10.14670/HH-11-916. [DOI] [PubMed] [Google Scholar]
- 23.La Fleur L., Boura V.F., Alexeyenko A., Berglund A., Pontén V., Mattsson J.S.M., Djureinovic D., Persson J., Brunnström H., Isaksson J., et al. Expression of scavenger receptor MARCO defines a targetable tumor-associated macrophage subset in non-small cell lung cancer. Int. J. Cancer. 2018;143:1741–1752. doi: 10.1002/ijc.31545. [DOI] [PubMed] [Google Scholar]
- 24.Muhitch J.B., Hoffend N.C., Azabdaftari G., Miller A., Bshara W., Morrison C.D., Schwaab T., Abrams S.I. Tumor-associated macrophage expression of interferon regulatory Factor-8 (IRF8) is a predictor of progression and patient survival in renal cell carcinoma. J. Immunother. Cancer. 2019;7:155. doi: 10.1186/s40425-019-0630-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Jaynes J.M., Sable R., Ronzetti M., Bautista W., Knotts Z., Abisoye-Ogunniyan A., Li D., Calvo R., Dashnyam M., Singh A., et al. Mannose receptor (CD206) activation in tumor-associated macrophages enhances adaptive and innate antitumor immune responses. Sci. Transl. Med. 2020;12:eaax6337. doi: 10.1126/scitranslmed.aax6337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.De Henau O., Rausch M., Winkler D., Campesato L.F., Liu C., Cymerman D.H., Budhu S., Ghosh A., Pink M., Tchaicha J., et al. Overcoming resistance to checkpoint blockade therapy by targeting PI3Kγ in myeloid cells. Nature. 2016;539:443–447. doi: 10.1038/nature20554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ho W.S., Wang H., Maggio D., Kovach J.S., Zhang Q., Song Q., Marincola F.M., Heiss J.D., Gilbert M.R., Lu R., Zhuang Z. Pharmacologic inhibition of protein phosphatase-2A achieves durable immune-mediated antitumor activity when combined with PD-1 blockade. Nat. Commun. 2018;9:2126. doi: 10.1038/s41467-018-04425-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ma H.S., Poudel B., Torres E.R., Sidhom J.W., Robinson T.M., Christmas B., Scott B., Cruz K., Woolman S., Wall V.Z., et al. A CD40 Agonist and PD-1 Antagonist Antibody Reprogram the Microenvironment of Nonimmunogenic Tumors to Allow T-cell-Mediated Anticancer Activity. Cancer Immunol. Res. 2019;7:428–442. doi: 10.1158/2326-6066.CIR-18-0061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Li T., Fu J., Zeng Z., Cohen D., Li J., Chen Q., Li B., Liu X.S. TIMER2.0 for analysis of tumor-infiltrating immune cells. Nucleic Acids Res. 2020;48(W1):W509–W514. doi: 10.1093/nar/gkaa407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Trujillo J.A., Sweis R.F., Bao R., Luke J.J. T Cell-Inflamed versus Non-T Cell-Inflamed Tumors: A Conceptual Framework for Cancer Immunotherapy Drug Development and Combination Therapy Selection. Cancer Immunol. Res. 2018;6:990–1000. doi: 10.1158/2326-6066.CIR-18-0277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Araujo J.M., Gomez A.C., Aguilar A., Salgado R., Balko J.M., Bravo L., Doimi F., Bretel D., Morante Z., Flores C., et al. Effect of CCL5 expression in the recruitment of immune cells in triple negative breast cancer. Sci. Rep. 2018;8:4899. doi: 10.1038/s41598-018-23099-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Irvine D.J., Dane E.L. Enhancing cancer immunotherapy with nanomedicine. Nat. Rev. Immunol. 2020;20:321–334. doi: 10.1038/s41577-019-0269-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Cassetta L., Pollard J.W. Tumor-associated macrophages. Curr. Biol. 2020;30:R246–R248. doi: 10.1016/j.cub.2020.01.031. [DOI] [PubMed] [Google Scholar]
- 34.Rao L., Zhao S.-K., Wen C., Tian R., Lin L., Cai B., Sun Y., Kang F., Yang Z., He L., et al. Activating Macrophage-Mediated Cancer Immunotherapy by Genetically Edited Nanoparticles. Adv. Mater. 2020;32:e2004853. doi: 10.1002/adma.202004853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Yang Y., Guo Z., Chen W., Wang X., Cao M., Han X., Zhang, K, Teng, B, Cao, J, Wu W., et al. M2 macrophage-derived exosomes promote angiogenesis and growth of pancreatic ductal adenocarcinoma by targeting E2F2. Mol. Ther. 2021;29:1226–1238. doi: 10.1016/j.ymthe.2020.11.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Woith E., Fuhrmann G., Melzig M.F. Extracellular Vesicles-Connecting Kingdoms. Int. J. Mol. Sci. 2019;20:5695. doi: 10.3390/ijms20225695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Wang Q., Zhuang X., Mu J., Deng Z.B., Jiang H., Zhang L., Xiang X., Wang B., Yan J., Miller D., Zhang H.G. Delivery of therapeutic agents by nanoparticles made of grapefruit-derived lipids. Nat. Commun. 2013;4:1867. doi: 10.1038/ncomms2886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Li Z., Wang H., Yin H., Bennett C., Zhang H.G., Guo P. Arrowtail RNA for Ligand Display on Ginger Exosome-like Nanovesicles to Systemic Deliver siRNA for Cancer Suppression. Sci. Rep. 2018;8:14644. doi: 10.1038/s41598-018-32953-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Yang C., Zhang M., Merlin D. Advances in Plant-derived Edible Nanoparticle-based lipid Nano-drug Delivery Systems as Therapeutic Nanomedicines. J. Mater. Chem. B Mater. Biol. Med. 2018;6:1312–1321. doi: 10.1039/C7TB03207B. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Kitamura T., Qian B.Z., Soong D., Cassetta L., Noy R., Sugano G., Kato Y., Li J., Pollard J.W. CCL2-induced chemokine cascade promotes breast cancer metastasis by enhancing retention of metastasis-associated macrophages. J. Exp. Med. 2015;212:1043–1059. doi: 10.1084/jem.20141836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Griffith J.W., Sokol C.L., Luster A.D. Chemokines and chemokine receptors: positioning cells for host defense and immunity. Annu. Rev. Immunol. 2014;32:659–702. doi: 10.1146/annurev-immunol-032713-120145. [DOI] [PubMed] [Google Scholar]
- 42.Dangaj D., Bruand M., Grimm A.J., Ronet C., Barras D., Duttagupta P.A., Lanitis E., Duraiswamy J., Tanyi J.L., Benencia F., et al. Cooperation between Constitutive and Inducible Chemokines Enables T Cell Engraftment and Immune Attack in Solid Tumors. Cancer Cell. 2019;35:885–900.e10. doi: 10.1016/j.ccell.2019.05.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Zumwalt T.J., Arnold M., Goel A., Boland C.R. Active secretion of CXCL10 and CCL5 from colorectal cancer microenvironments associates with GranzymeB+ CD8+ T-cell infiltration. Oncotarget. 2015;6:2981–2991. doi: 10.18632/oncotarget.3205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Litchfield K., Reading J.L., Puttick C., Thakkar K., Abbosh C., Bentham R., Watkins T.B.K., Rosenthal R., Biswas D., Rowan A., et al. Meta-analysis of tumor- and T cell-intrinsic mechanisms of sensitization to checkpoint inhibition. Cell. 2021;184:596–614.e14. doi: 10.1016/j.cell.2021.01.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Garrido-Martin E.M., Mellows T.W.P., Clarke J., Ganesan A.P., Wood O., Cazaly A., Seumois G., Chee S.J., Alzetani A., King E.V., et al. M1hot tumor-associated macrophages boost tissue-resident memory T cells infiltration and survival in human lung cancer. J. Immunother. Cancer. 2020;8:e000778. doi: 10.1136/jitc-2020-000778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.González-Martín A., Gómez L., Lustgarten J., Mira E., Mañes S. Maximal T cell-mediated antitumor responses rely upon CCR5 expression in both CD4(+) and CD8(+) T cells. Cancer Res. 2011;71:5455–5466. doi: 10.1158/0008-5472.CAN-11-1687. [DOI] [PubMed] [Google Scholar]
- 47.Mikucki M.E., Fisher D.T., Matsuzaki J., Skitzki J.J., Gaulin N.B., Muhitch J.B., Ku A.W., Frelinger J.G., Odunsi K., Gajewski T.F., et al. Non-redundant requirement for CXCR3 signalling during tumoricidal T-cell trafficking across tumour vascular checkpoints. Nat. Commun. 2015;6:7458. doi: 10.1038/ncomms8458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Fu H., Ward E.J., Marelli-Berg F.M. Mechanisms of T cell organotropism. Cell. Mol. Life Sci. 2016;73:3009–3033. doi: 10.1007/s00018-016-2211-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Gordon S.R., Maute R.L., Dulken B.W., Hutter G., George B.M., McCracken M.N., Gupta R., Tsai J.M., Sinha R., Corey D., et al. PD-1 expression by tumour-associated macrophages inhibits phagocytosis and tumour immunity. Nature. 2017;545:495–499. doi: 10.1038/nature22396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Fujiwara N., Porcelli S.A., Naka T., Yano I., Maeda S., Kuwata H., Akira S., Uematsu S., Takii T., Ogura H., Kobayashi K. Bacterial sphingophospholipids containing non-hydroxy fatty acid activate murine macrophages via Toll-like receptor 4 and stimulate bacterial clearance. Biochim. Biophys. Acta. 2013;1831:1177–1184. doi: 10.1016/j.bbalip.2013.03.008. [DOI] [PubMed] [Google Scholar]
- 51.Li L., Jay S.M., Wang Y., Wu S.W., Xiao Z. IL-12 stimulates CTLs to secrete exosomes capable of activating bystander CD8+ T cells. Sci. Rep. 2017;7:13365. doi: 10.1038/s41598-017-14000-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Qian B.Z., Li J., Zhang H., Kitamura T., Zhang J., Campion L.R., Kaiser E.A., Snyder L.A., Pollard J.W. CCL2 recruits inflammatory monocytes to facilitate breast-tumour metastasis. Nature. 2011;475:222–225. doi: 10.1038/nature10138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Maleki Vareki S. High and low mutational burden tumors versus immunologically hot and cold tumors and response to immune checkpoint inhibitors. J. Immunother. Cancer. 2018;6:157. doi: 10.1186/s40425-018-0479-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Fabian K.P., Padget M.R., Fujii R., Schlom J., Hodge J.W. Differential combination immunotherapy requirements for inflamed (warm) tumors versus T cell excluded (cool) tumors: engage, expand, enable, and evolve. J. Immunother. Cancer. 2021;9:e001691. doi: 10.1136/jitc-2020-001691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Hos B.J., Camps M.G.M., van den Bulk J., Tondini E., van den Ende T.C., Ruano D., Franken K., Janssen G.M.C., Ru A., Filippov D.V., et al. Identification of a neo-epitope dominating endogenous CD8 T cell responses to MC-38 colorectal cancer. OncoImmunology. 2019;9:1673125. doi: 10.1080/2162402X.2019.1673125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Robinson M.D., McCarthy D.J., Smyth G.K. edgeR: a Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics. 2010;26:139–140. doi: 10.1093/bioinformatics/btp616. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.