

Abstract
Serine/threonine kinase 3 (STK3) is an essential member of the highly conserved Hippo tumor suppressor pathway that regulates Yes-associated protein 1 (YAP1) and TAZ. STK3 and its paralog STK4 initiate a phosphorylation cascade that regulates YAP1/TAZ inhibition and degradation, which is important for regulated cell growth and organ size. Deregulation of this pathway leads to hyperactivation of YAP1 in various cancers. Counter to the canonical tumor suppression role of STK3, we report that in the context of prostate cancer (PC), STK3 has a pro-tumorigenic role. Our investigation started with the observation that STK3, but not STK4, is frequently amplified in PC. Additionally, high STK3 expression is associated with decreased overall survival and positively correlates with androgen receptor (AR) activity in metastatic castrate-resistant PC. XMU-MP-1, an STK3/4 inhibitor, slowed cell proliferation, spheroid growth, and Matrigel invasion in multiple models. Genetic depletion of STK3 decreased proliferation in several PC cell lines. In a syngeneic allograft model, STK3 loss slowed tumor growth kinetics in vivo, and biochemical analysis suggests a mitotic growth arrest phenotype. To further probe the role of STK3 in PC, we identified and validated a new set of selective STK3 inhibitors, with enhanced kinase selectivity relative to XMU-MP-1, that inhibited tumor spheroid growth and invasion. Consistent with the canonical role, inhibition of STK3 induced cardiomyocyte growth and had chemoprotective effects. Our results indicate that STK3 has a non-canonical role in PC progression and that inhibition of STK3 may have a therapeutic potential for PC that merits further investigation.
Keywords: prostate cancer, STK3, Hippo tumor suppressor, non-canonical, kinase inhibitor
Graphical abstract

STK3, also known as MST2, is a key member of the Hippo tumor suppressor signaling pathway. In various cancer types, STK3 is known to have a tumor suppressive role. Here, Schirmer et al. report that counter to STK3 tumor suppressor function, STK3 is frequently amplified in breast and prostate cancer. Moreover, they show using genetic and pharmacological tools that STK3 has a non-canonical tumor supportive role in prostate cancer.
Introduction
Prostate cancer (PC) remains the second leading cause of cancer-related deaths in men. Metastatic castration-resistant PC (mCRPC) occurs when PC progresses despite continuous androgen deprivation therapy. This is largely due to aberrations in the androgen receptor (AR), including AR gene amplification, expression of splice variants, or mutations. During the past few years, a number of AR-targeted therapies have been US Food and Drug Administration (FDA) approved, including enzalutamide and abiraterone acetate for mCRPC, and most recently darolutamide and apalutamide. However, in spite of these life-prolonging therapeutic advances, there is no cure for mCRPC, resulting in patients eventually succumbing to the disease. Thus, there is a need to identify other molecular targets for mCRPC beyond the AR.
We have focused our efforts on understudied kinases that are correlated with poor cancer patient outcomes in PC. Kinases are high-value targets due to their druggability and have emerged as the most successful drug targets of the 21st century, with more than 60 small-molecule kinase inhibitors approved by the FDA for oncology.1 This enzyme family contains more than 500 members, presenting a wealth of opportunities for the continued introduction of life-extending medicines. However, a large number of kinases are understudied and thus overlooked as potential therapeutic targets.2,3 Herein, we investigate the role of one such overlooked kinase, serine/threonine kinase 3 (STK3), also known as STE20-like kinase (MST2), a key member of the Hippo tumor suppressor signaling pathway.
The term “Hippo” is derived from the overgrowth phenotype in Drosophila that is associated with genetic deletion of the hpo gene, which encodes a Ste-20 family protein kinase that restricts cell proliferation and regulates apoptosis.4 The mammalian homolog STK3 gene and its paralog STK4 encode what are commonly referred to as MST2 and MST1 kinases, respectively. STK3 and STK4 are essential members of the highly conserved Hippo tumor suppressor pathway (hereafter, simply STK3 and STK4 kinases).4,5 Mammalian STK3/4 kinases bind to adaptor protein salvador homolog 1 (SAV1) and phosphorylate large tumor suppressor homolog 1/2 (LATS1/2) and Mob kinase activator 1 (MOB1). Phosphorylation of homologous transcriptional co-activators Yes-associated protein (YAP) and transcriptional co-activator with PDZ binding motif (TAZ) by LATS1/2 ensues, which leads to YAP/TAZ cytoplasmic retention or degradation.6 Inactivation or loss of Hippo kinase signaling leads to non-phosphorylated active YAP/TAZ, which promotes cell proliferation and inhibits apoptosis. YAP/TAZ transcriptional oncogenes are hyperactivated in many cancer types.6,7 Hyperactivation of YAP in PC has been observed in a number of studies.8 Thus, the consensus would be that loss of STK3/4 Hippo kinase signaling results in hyperactivation of YAP/TAZ. However, our analysis of genomic data shows that STK3, but not STK4, is amplified in human PCs and correlates with worse patient outcomes. Thus, we hypothesized that STK3 may have a non-canonical pro-tumorigenic role in PC.
Given the high value of STK3 as an actionable target and the urgent need for new molecular targets in PC, we sought to investigate the potential non-canonical pro-tumorigenic role of STK3. To complement STK3 molecular, genetic studies, we utilized the available STK3/4 inhibitor XMU-MP-1 and identified a novel complementary series of STK3 small-molecule inhibitors with different structures, narrower kinase inhibition profiles, and more potent cellular activity. These small molecules may also be useful tools for the research community to elucidate STK3 function in other disease contexts. Our cumulative data support that STK3 has a non-canonical pro-tumorigenic role in PC, which may be targeted by small-molecule inhibitors to reduce PC growth and progression.
Results
STK3 gene is amplified in PC
Analysis of genomic data from 158 non-redundant studies in the cBioPortal with a cutoff of ≥10% copy number alteration (CNA) frequency shows that STK3 is amplified in a number of cancer cohorts (Figure 1A).9 Of the 14 cohorts with ≥10% STK3 amplification, 4 were PC cohorts. In addition, The Cancer Genome Atlas (TCGA) prostate adenocarcinoma (PRAD) and Memorial Sloan Kettering Cancer Center (MSKCC) prostate adenocarcinoma cohort datasets contained >5% STK3 amplification, that is, 8.5% and 5.85%, respectively (data not shown). In contrast, no PC cohort showed a ≥10% amplification of STK4 (Figure 1B).
Figure 1.

Correlative analysis of STK3 and cancer patient outcomes
(A and B) Cancer cohorts with >10% STK3 (A) and STK4 copy number amplification (B). Asterisks denotes PC cohorts. (C) Progression-free survival analysis of STK3 diploid and STK3 amplified patients from TCGA/PRAD (p = 0.0239). (D) STK3 mRNA expresses Z scores in diploid and STK3-amplified PCs. ∗∗∗∗p < 0.0001. (E and F) Survival analysis of advanced mCRPC by lower and upper median STK3 expression (E) and low and high STK4 expression groups (F). (G and H) Survival analysis of BC patient overall survival in patients stratified by upper (n = 503) and lower (n = 503) median expression Z scores of (G) STK3 and (H) STK4. (I) Spearman’s rank correlation analysis of STK4 and STK3 and AR response gene signature. GS, Gleason score; mCRPC, metastatic castration-resistant PC. Rho and p values denoted on graphs.
STK3 correlates with worse outcomes in PC
Progression-free survival analysis of patients from TCGA PRAD cohort (n = 489) shows a significant difference in time to progression in STK3 amplified patients (n = 41) compared to STK3 diploid patients (n = 448) (hazard ratio [HR] = 1.94; 95% confidence interval [CI], 0.91–4.15; p = 0.024, log-rank test) (Figure 1C). TCGA PRADs with increased STK3 gene copy number were associated with increased STK3 mRNA levels (Figure 1D). When all samples (n = 489) from the PRAD TCGA cohort were stratified into upper and lower STK3 mRNA median expression groups no significant correlation was observed. Next, we queried 71 patients with available RNA sequencing (RNA-seq) gene expression and survival data from the metastatic Stand Up 2 Cancer/Prostate Cancer Foundation cohort (SU2C/PCF).10 Patients were stratified into lower median (STK3 low, n = 36) and upper median STK3 expression (STK3 high, n = 35) groups by mRNA expression Z scores for survival analysis. Compared to the STK3 low expression group, the STK3 upper median expression group had a significantly decreased survival rate, that is, median survival 30.7 versus 22.2 months, respectively (HR = 1.78; 95% CI, 0.961–3.218; p = 0.042, log-rank test) (Figure 1E). Importantly, when we stratified patients by STK4 median expression, there was no statistically significant difference in survival between groups (p = 0.263, log-rank test) (Figure 1F).
Given that STK3 is frequently amplified in breast cancer (BC), we also queried BC outcomes (Figures 1G and 1H). Patient samples from TCGA invasive BC cohort stratified by STK3 above the median upper expression levels compared to lower STK3 expression showed significantly decreased overall survival (HR = 1.84; 95% CI, 1.314–2.580; p = 0.0005, log-rank test). When BC patients were stratified by STK4 median expression, there was no significant difference in overall survival (p = 0.825, log-rank test).
STK3 gene expression is correlated with AR activity in mCRPC
To determine whether STK3 plays a role in AR-dependent or AR-independent PCs, we next ran correlative analysis of STK3 or STK4 gene expression and the AR response signature (HALLMARK_ANDROGEN_RESPONSE) across PC disease states. For STK4 and the AR response, there was a negative or no significant correlation observed (Figure 1I). However, STK3 gene expression and the AR response gene signature had a significantly positive correlation at every disease state with the highest correlation in the mCRPC setting (Spearman’s rho value = 0.33, p = 0.001). Altogether, analyses of STK3 in human PC specimens suggest that STK3 may play a role in lethal AR-driven mCRPC.
STK3 expression in PC cell lines
Across commonly used PC cell lines, there was a trend for an inverse relationship between STK3 and STK4 protein expression (Figure 2A). In addition, in AR-positive cell lines, STK3 tends to be higher as shown by a higher STK3/AR ratio as compared AR null cell lines with a higher STK4/AR ratio (Figure 2B). Exploring PC cell lines for syngeneic studies, we found that the Hi-Myc ventral prostate 2 (HMVP2) cell line derived from the commonly Hi-Myc transgenic PC mouse model has significantly elevated levels of STK3 (Figure 2C). Importantly, note that HMVP2 cells do not express AR.11
Figure 2.

Pharmacological inhibition of Hippo kinases in PC and BC cells
(A) Expression of STK3 and STK4 across PC cell lines. (B) STK3- and STK4-to-AR protein ratios (densitometry). (C) Expression of STK3 in HMVP2 mouse PC cell line. (D) Dose response of XMU-MP-1 (nonlinear regression curve fit) in 22RV1, PC-3, and LNCaP cells treated for 96 h. (E) Representative bright-field images of LAPC-4 spheroids at the denoted time points. (F) Graphical representation of average LAPC-4 spheroid growth kinetics (spheroid area) over time treated as denoted with XMU-MP-1 (n = 5). (G) Western blot analysis of denoted PC cell lines. (H) BC cell lysates treated with 10 μM XMU-MP-1 collected at indicated time points after treatment. Vtp, verteporfin. (I) Representative bright-field images of BC spheroids in three cell line models. (J) Average BC spheroid growth kinetics in response to XMU-MP-1 (n = 5/dose). Data were represented as mean ± SD.
STK3/4 inhibitor, XMU-MP-1, slows PC and BC cell growth and Matrigel invasion
We next wanted to determine whether STK3 is a druggable target in PC. To this end, we utilized the available STK3/4 small-molecule inhibitor XMU-MP-1 across a battery of cell lines.12 In LNCaP, 22RV1, and PC-3 cells, we observed a dose-dependent decrease in proliferation rates with XMU-MP-1 treatment, with half-maximal inhibitory concentration (IC50) values of 0.8, 1.97, and 3.86 μM, respectively (Figure 2D). Importantly, PC-3 cells have lower levels of STK3, which may be reflective of a slightly higher IC50 and steeper curve slope. Hill slope values for XMU-MP-1 in LNCaP, 22RV1, and PC-3 cells were −0.73, −1.54, and −2.28, respectively. Nonetheless, observed growth inhibition may also be in part due to inhibition of STK4 or other documented XMU-MP-1 off-targets. We next found that growth of LAPC-4 3D spheroids was significantly blunted with XMU-MP-1 in a dose-dependent manner (Figures 2E and 2F). Western blot analysis of LNCaP and LAPC-4 cell lysates treated with 10 μM XMU-MP-1 showed decreased levels of phosphorylated (p-)MOB1 and p-YAP at both Ser127 and Ser397, denoting activated YAP, yet PC cells were growth-inhibited counter to the canonical pathway phenotype (Figure 2G). Consistent with slowed proliferation rates, the cell cycle progression marker cyclin D1 was downregulated 18 h after XMU-MP-1 treatment.
Given that STK3 is also frequently amplified in BC (Figure 1A), we also tested the effects of XMU-MP-1 on BC cells. Similar to our observations in PC cell lines, western blot analysis of BC cell lines treated with XMU-MP-1 also displayed reduced phosphorylation of YAP and a reduction in the cell proliferation marker cyclin D1 (Figure 2H). Verteporfin, a known YAP inhibitor, was used as a treatment for 18 h as a positive control. Similar results were observed in various 3D BC spheroid models (Figures 2I and 2J). Taken together, data from PC and BC cells show that STK3/4 inhibition with XMU-MP-1 results in reduced cell proliferation despite YAP1 activation (reduced YAP phosphorylation).
Genetic depletion of STK3 slows PC cell proliferation
We utilized short hairpin RNAs (shRNAs) to test whether STK3 is essential for PC cell proliferation. Western blot validation of STK3 inducible knockdown in C4-2 cells coincides with reduced levels of STK3 phosphorylation target p-MOB1 Thr35 (Figure 3A). Importantly, STK4 levels are not induced to compensate for STK3 loss. In addition, we observed increased levels of cell cycle inhibitor p27. Modest increases in YAP1 Ser127 were observed, but they appear in non-targeting doxycycline-treated cells as well. This indicates that YAP is not activated in this model due to loss of STK3. In C4-2, LNCaP, and HMVP2 cell lines, STK3 knockdown with two shRNAs compared to the non-targeting shRNA control resulted in significantly reduced proliferation rates (Figure 3B). In LAPC-4 (Figure 3C) and C4-2 (Figure 3D) spheroid models, inducible STK3 gene knockdown also resulted in slowed tumor spheroid growth kinetics.
Figure 3.

Assessment of STK3 genetic depletion in PC in vitro and in vivo
(A) Western blot validation of STK3 shRNA C4-2 inducible knockdown (doxycycline at 0.5 μg/mL) and assessment of denoted proteins. (B) Cell proliferation assays of C4-2, LNCaP, and HMVP2 PC cells with shNT or shSTK3s (n = 4). (C) Representative images of LAPC-4 spheroids and graphical depiction of growth kinetics and LAPC-4 with inducible ZsGreen1/shRNAs and western blot verification of knockdown. (D) Representative images of C4-2 spheroids and graphical depiction of growth kinetics with inducible ZsGreen1/shRNAs. (E) PI-based FACS analysis for cell cycle phase. (F) Western blot time course for cell cycle markers in shNT or shSTK3 C4-2 cell lysates after doxycycline treatment at denoted times. (G) Western blot analysis of LNCaP cell lysates grown in varying serum conditions for 72 h and R1881 (1 nM). (H) Cell proliferation assays of LNCaP in FBS or CSS (n = 5). (I) Analysis of STK3 knockdown and denoted proteins in HMVP2 cells in vitro. (J) Longitudinal growth kinetics of HMPV2 allograft tumors with shNT (n = 8), shSTK3-1 (n = 8), and shSTK3-2 (n = 7) (interaction time versus shRNA arm, p < 0.001; pairwise comparison shSTK3-1 versus shNT, p < 0.001; shSTK3-2 versus shNT, p = 0.23. (K) Final tumor wet weight at the terminal endpoint. (L) Western blot analysis of pooled tumor lysates (n = 6/shRNA) loaded in technical triplicate. (M) Phosbind gel assessment of YAP phosphorylation species. Data were represented as mean ± SD. ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001, ∗∗∗∗p < 0.0001.
Genetic depletion of STK3 alters cell cycle progression
Fluorescence-activated cell sorting (FACS)-based cell cycle analysis on C4-2 cells 96 h after shRNA induction showed that shSTK3 cells had altered cell cycle marker progression profiles relative to shNT (non-targeting) cells (Figures 3E and S1A). Interestingly, the two shSTK3s induced accumulation in two distinct phases, shSTK3-1 in the G1 phase and shSTK3-2 in the G2/M phase. Statistical analysis shows a significant difference between all three groups in the G1 and G2/M phases (p < 0.0001), but no significant differences in the S phase (Figure S1B). Biochemical analysis of C4-2 protein lysates 24–96 h after shRNA induction with doxycycline shows altered expression of cell cycle markers in shSTK3 cells relative to shNT cell lysates (Figure 3F). Consistent with FACS analysis, shSTK3-1 shows accumulation of cyclin E, which is induced in the G1 phase, while shSTK3-2 shows sustained p-CDK1 Y15 levels and a reduction of the p-H3 mitotic marker, signifying G2/M arrest. Taken together, these data suggest that STK3 may act at multiple points of the cell cycle.
STK3 in castrate growth conditions
Given that STK3 is amplified in advanced mCRPC cohorts, we asked whether STK3 plays a role in hormone-independent PC cell growth. For these studies we utilized charcoal-stripped serum (CSS) to mimic androgen deprivation conditions, and as a control for Hippo pathway activation we used low-serum growth conditions. Interestingly, after 72 h of growth in CSS, STK3 protein levels and activity (p-MOB1) were increased (Figure 3G). Importantly, add-back of synthetic androgen R1881, as evident by induction of AR the gene target prostate-specific antigen (PSA), reduced p-MOB1 levels. In LNCaP cell lines grown in CSS compared to fetal bovine serum (FBS), STK3 loss resulted in added inhibition of proliferation compared to non-targeting control (Figure 3H). Taken together, these data indicate that STK3 may support PC cell proliferation in castrate-like conditions, counter to the role of STK3 as a putative tumor suppressor.
STK3 knockdown slows allograft tumor growth in vivo
To determine the role of STK3 on PC growth in vivo, we utilized an HMVP2 syngeneic allograft model. Depletion of STK3 in HMVP2 cells in vitro resulted in reduced p-MOB1 levels and induction of p27, but no significant changes in p-YAP levels (Figure 3I). Loss of STK3 in vivo showed an overall statistically significant interaction (p < 0.0001) between treatment arm and time, which tests whether tumor volume differs by treatment (i.e., shSTK3 versus shNT) over time (Figure 3J). On a pairwise comparison, shSTK3-1 (p < 0.001), but not shSTK3-2 (p = 0.23), was significantly different compared to the shNT control arm. At the experimental endpoint, similar results were observed with tumor weights (Figure 3K). Biochemical analysis of pooled tumor protein lysates (n = 6 tumors per group) confirmed reduced levels of STK3 in shSTK3 tumors compared to shNT control tumors (Figure 3L). Interestingly, we did not observe reduced levels of p-MOB1 in STK3-depleted tumors. In fact, contrary to in vitro STK3 knockdown (Figure 3I), we observed increased levels of p-MOB1 in shSTK3 tumors relative to shNT tumor lysates. Analysis of YAP phosphorylation species in tumor lysates using phosphorylation tag acrylamide gels also showed increased YAP phosphorylation (Figure 3M). Loss of STK3 in vivo resulted in reduced levels of p-CDK1 Tyr15. In addition, we observed decreased levels of cleaved caspase-3 and the pro-apoptotic protein PUMA in shSTK3 tumors compared to shNT tumors (Figure 3L). These data suggest that STK3 loss in vivo results in slowed tumor growth kinetics due to cell cycle/proliferative alterations and not cell death due to hyperactivation of YAP as described in other models.13
Identification and validation of novel STK3 small-molecule inhibitors
To identify new medicinal chemistry starting points for targeting STK3, our team screened diverse kinase inhibitors in a large assay panel and carefully scrutinized previously published kinome data.14 Through this literature mining, we identified two independent scaffolds (Figure 4A) disclosed as inhibitors of well-studied kinases that also possess what we perceived to be exploitable off-target STK3 inhibition.15,16 One lead, alias UNC-BE4-017, referred to as compound A1 hereafter, is a pyrrolopyrazine that was originally disclosed in a medicinal chemistry campaign targeted at the JAK kinases.16 The compound potently inhibits JAK1, JAK2, and JAK3, but only three kinases (with one being STK3) out of the panel of 48 are inhibited >90%, indicating good kinase selectivity for a starting point. The second lead, alias UNC-SOB-5-16, referred to as compound B1 hereafter, a pyrrolopyrimidine, came from a literature series designed to target the kinase LRRK2.15 It is also an inhibitor of STK3, with an IC50 of 22 nM, and in the DiscoveRx KINOMEscan panel of 451 kinase assays, only 10 kinases give a percent of control (PoC) <90% at 1 μM. Thus, compounds A1 and B1 are very selective across the kinome with only a handful of off-targets. We synthesized these lead compounds and some additional analogs from each series to test for STK3 engagement and inhibition (Supplemental materials and methods).
Figure 4.

Identification and assessment of STK3 small-molecule inhibitors
(A) Structure of lead compound A1 and B2. (B) Western blot analysis of STK3 target p-MOB1 in HMVP2 cells treated with denoted compound for 30 min. (C) STK3-NanoBRET intracellular 11-point IC50 dose-response curves with denoted compounds. (D) p-MOB1 and p-STAT3 western blots. (E) Plotted densitometry normalized to GAPDH plotted relative to DMSO control (n = 4). ∗p = 0.0473, ∗∗p = 0.0017, ∗∗∗p < 0.0007; ns, not significant. (F) Efficacy of lead STK3 compounds on HMVP2 spheroid growth at day 9 (n = 4). (G) Representative bright-field images of vehicle-, A1-, or B2 (10 μM)-treated HMVP2 spheroids at 9 day endpoint. (H) H9C2 cardiomyocyte proliferation assay treated with denoted compounds 72 h post-treatment (n = 4). (I) H9C2 proliferation assay at 72 h with non-targeting control or shSTK3 (n = 3/shRNA). (J) H9C2 proliferation and cell death monitored by cytotox green dye (GFP) over time treated with vehicle, doxorubicin (Doxo, 500 nM), or STK3 inhibitors (1 μΜ). (K) Kinome tree for A1 and B2 at 1 μM doses. Kinases with 0%–35% of control remaining activity depicted. STK3 is denoted in blue. Data were represented as mean ± SD. ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001, ∗∗∗∗p < 0.0001.
Validation of lead scaffolds and STK3 target engagement
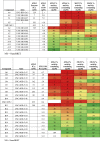
We utilized p-MOB1 Thr35, a widely validated STK3/4 phosphorylation substrate as a readout for STK3 inhibition in cells.17 As shown in Figure 4B, a 30-min treatment with compounds A1, A2, B1, B2, and XMU-MP-1 resulted in reduced STK3 activity at the 10 μM dose. However, A1 and B2 were more potent, blocking MOB1 phosphorylation at lower doses (Figure 4B). We next used an STK3-NanoBRET assay to quantitate the occupancy and affinity of our compounds to STK3 in live cells.18 In the STK3-NanoBRET assay, A1 and B2 were found to have the lowest in-cell IC50 values, that is, 7.6 and 2.7 μM, respectively (Figure 4C). XMU-MP-1 was found to have an IC50 of 8.4 μM, which coincided well with western blot results. Of note, B3 showed no activity in western blot assays and an IC50 out of range (>10 μM) in the STK3 NanoBRET assay, indicating strong congruency between these two assays. Compounds were further evaluated using radiometric enzyme kinases assays using human recombinant STK3, STK4, STK24 (aka MST3), and STK26 (aka MST4) kinase assays (Table 1). Enzymatic IC50 values for A1 and B2 were in the low nanomolar range, that is, 41 and 33 nM, respectively.
Table 1.
Pyrrolopyridazines (Series A) and Pyrrolopyrimidine (Series B) MST family selectivity and NB correlation with literature compound (XMU-MP-1) for comparison. STK3 IC50 and STK3, STK4, 2, 3 and 4% activity remaining determinations performed in an enzyme assay at Eurofins at the Km of ATP.
 |
Quantitative densitometry analysis of western blots of p-MOB1 in HMVP2 cells treated with 1 μM for 30 min showed an approximately 75% reduction of p-MOB1 in cell lysates treated with A1 and B2 (Figures 4D and 4E). Compounds in series A were derived from JAK inhibitors, and, accordingly, we observed a modest but significant reduction of p-STAT3 Tyr705 in A1-treated cells only. Compounds in series B were derived from LRRK2 inhibitors; however, phosphorylation of LRRK2 at autophosphorylation site S1292 was not detected in PC cells.19 We tested A1 and B2 against HMVP2 tumor spheroid growth, both of which have a high level of STK3 expression. As shown in Figures 4F and 4G, both compounds inhibited HVMP2 spheroid growth by more than 50% at 10 μM doses.
STK3 chemical tools have a protective effect in H9C2 cardiomyocytes
To determine whether our investigational compounds are specific to STK3 or have a general toxic effect, we utilized the rat H9C2 cardiomyocyte cell line, the cells of which are protected from reactive oxygen species (ROS)-induced apoptosis by activation of YAP, including with XMU-MP-1.20 As shown in Figure 4H, treatment with increasing doses of A1 and B2 for 72 h had modest growth stimulatory effects on H9C2; nonetheless, the effects were divergent from those observed in PC or BC cells. Consistently, shRNA-mediated knockdown of STK3 did not inhibit H9C2 cell proliferation (Figure 4I). To determine whether inhibition of STK3 has cardioprotective effects, we treated cells with a lethal dose of doxorubicin (500 nM) alone or in combination with pretreatment (1 h) of A1 and B2 (1 μM). Real-time quantitation of cell death showed a steady induction of H9C2 cell death with doxorubicin, which was partially, but significantly, blunted by co-treatment with STK3 inhibitors A1 and B2 (Figure 4J). Similar chemo-protective effects were observed with the taxane docetaxel (Figure 4J). These data are consistent with inhibition of canonical Hippo tumor suppressor function that leads to activation of YAP/TAZ and provides protective effects in cardiomyocytes.20
Kinome selectivity screening
A1 and B2 were screened for selectivity against a panel of 468 kinases at a 1 μM dose. Both compounds showed a high degree of selectivity (Figure 4K). A1 inhibited only 10 kinases other than STK3 by more than 90% with an S10 (1 μM) of 0.027. B2 inhibited only eight other kinases by >90% of control with an S10 (1 μM) of 0.022. Kinases with >90% inhibition for A1 and B2 are depicted in Table S1. In addition to STK3 overlap, compounds A1 and B2 S10 overlapping hits included LRRK2, LRRK2 (G2019S), and NUAK2. In this assay, the STK4 percent of control for A1 and B2 was 69% and 38%, respectively. However, in recombinant enzymatic assays, A1 and B2 inhibited STK4 by 98% and 94%, respectively (Table 1).
Pyrrolopyrimidine (series B) analogs have increased potency.
Given our encouraging previous results, we proceeded to synthesize and screen additional analogs from both BE4 and SOB-5 scaffolds. Compounds were tested using recombinant protein radiometric assays and in-cell STK3 NanoBRET target engagement assays (Table 1). From these two assays, compounds with a NanoBRET IC50 <2.5 μM and enzymatic IC50 <200 nM were screened for in vitro efficacy. Among these, we tested B1, B4, B5, B6, A3, and A4 on HMVP2 spheroids at a 5 μM dose (chemical structures in Supplemental materials and methods). During the course of a 9-day HMVP2 spheroid assay, B5 and B6 showed the highest efficacy (Figures 5A and 5B). Western blot analyses of HMVP2 lysates treated with B5 and B6 (1 μM) confirm inhibition of STK3 activity as measured by loss of p-MOB1 T35 to a level of approximately 75% inhibition for both compounds (Figures 5C and 5D). We also tested a number of these compounds against off-targets identified in our kinome selectivity screen using recombinant radiometric assays (Table S2). Consistent with recombinant JAK3 enzyme assays (Table S2), B6, but not B5, significantly reduced levels of JAK3 phosphorylation target p-STAT3 Tyr705, albeit with a modest reduction relative to STK3 inhibition (Figures 5C and 5D).
Figure 5.

Assessment of lead analogs
(A) Representative images of HMVP2 spheroids. (B) Percent growth bar graphs relative to vehicle control at endpoint day 9 treated with 5 μM of indicated STK3 inhibitors. (C and D) Western blot analysis of HMVP2 cells treated for 30 min with B5 and B6 at 1 μM (C) and densitometric analysis normalized to GAPDH (D). (E and F) Quantification of STK3 in indicated cell lines (E) plotted by a STK3/GAPDH ratio (F). (G) Representative images of HMVP2 spheroid area and graphed growth kinetics treated with denoted inhibitor. (H) Graphical representation of LAPC-4 spheroid growth kinetics. (I) Curve fit of HMVP2 and LAPC-4 spheroid growth assays with B5 and B6 for IC50 calculation. (J) Western blot analysis of B6 dose response assessed by p-MOB1 in STK3 overexpressing and empty vector C4-2 cells. (K) Proliferation assay C4-2 control or C4-2 pLX-STK3 cells treated with indicated inhibitor for 6 days. (L) Western blot assessment of B6 after 24 h in H9C2 and LAPC-4 cells with low, medium, and high exposures for p-MOB1. Data were represented as mean ± SD. ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001, ∗∗∗∗p < 0.0001.
As shown in Figure 5E, HMVP2 cells have inherently higher levels of STK3 relative to LAPC-4 cells. Thus, we next conducted dose-response curves using STK3 high-expressing HMVP2 (Figure 5G) and STK3 low-expressing LAPC-4 (Figure 5H) spheroid models. A non-linear curve fit was used to determine IC50 values of B5 and B6, which showed lower IC50 values in HMVP2 cells compared to LAPC-4 cells (Figure 5I). This finding suggests that STK3 high-expressing cells are more dependent on STK3 activity. In addition, we generated C4-2 cells stably overexpressing STK3 to test the specificity of our compounds (Figure 5J). However, despite increased STK3 levels, we did not see increased p-MOB1 basal levels. However, there was a modest level of sustained p-MOB1 in response to compound B6 in STK3 overexpressing cells. In addition, pLX-STK3 cells were partially resistant to compound B6 at the 5 μM dose, but not at 10 μM (Figure 5K). This suggests that STK3 inhibition is at least partially responsible for the response to our compounds, but other factors are likely necessary to activate STK3 kinase activity in overexpression models. For example, unlike C4-2 STK3 overexpressing cells, H9C2 cells, which have higher levels of STK3, did show increased basal levels of STK3 kinase activity relative to LAPC-4 cells (Figure 5L). This coincided with sustained activity in response to increasing concentrations of compound B6 in H9C2 cells compared to LAPC-4 cells.
STK3 inhibition and loss inhibit PC cell invasion
Next, we tested the effects of STK3 inhibition on 3D Matrigel invasion of HMVP2 cells. In this model, we first tested XMU-MP-1 and our two leads, A1 and B2. Compared to vehicle control, HMPV2 spheroids treated with each of the three compounds had statistically significant reduced overall spheroid size and invasion fronts (Figure S2). We next tested B6, which showed increased STK3 target engagement and efficacy in the HMVP2 3D invasion model (Figure 6A). B6 dramatically reduced overall 3D Matrigel invasion of the spheroid area in a dose-dependent manner (Figure 6B). In addition, the invading cell front (blue mask) was universally inhibited at all three doses tested (Figure 6C). Similarly, genetic depletion of STK3 in HMVP2 cells reduced invasion rates compared to the non-targeting control (Figures 6D and 6E). However, genetic depletion of STK3 was not as potent as inhibition with B6, likely due to modest STK3 shRNA knockdown in HMVP2 cells. Taken together, our chemical and genetic data show that STK3 plays a role in cell invasion counter to its role as a tumor suppressor in other contexts.
Figure 6.

Pharmacological inhibition and genetic depletion of STK3 slows PC cell invasion
(A) Representative images of 3D invasion of HMVP2 cells treated as denoted and imaged over time. (B and C) Graphical representation of real-time 3D HMVP2 total spheroid area kinetics (B) and cell invasion front (C; blue mask). (D–F) Representative images of 3D invasion of HMVP2 with denoted shRNAs (D), total spheroid area (E), and invasion front (F). Data were represented as mean ± SD. Two-way repeated measures ANOVA: ∗p = 0.01, ∗∗∗∗p < 0.0001.
Discussion
Our investigation originated from the simple observation that STK3, a known tumor suppressor, is amplified in a subset of cancer types including PC. For this reason, we sought to determine whether STK3 plays a non-canonical pro-tumorigenic role in PC. Although this notion contradicts the widely accepted Hippo tumor suppressor signaling dogma, the combined value of STK3 as a druggable target and the need for added molecular targets to combat mCRPC motivated us to investigate this likelihood. Our study provides genetic and pharmacological evidence that in PC, STK3 is essential for PC cell proliferation and in vivo tumor growth.
There is ample evidence that STK3 acts as a tumor suppressor in various tumor types other than PC; however, data from the literature does support the possibility that STK3 may have a pro-tumorigenic role in a cancer type-dependent manner. For example, Hippo tumor suppressor kinases LATS1 and LATS2, the immediate downstream phosphorylation targets of STK3/4, were also shown to be essential for tumorigenesis in a mouse colon cancer model.13 Likewise, a subset of acute myeloid leukemias were found to be dependent on STK3 signaling in vitro.21 Lastly, in a retrospective PC study, STK3 expression was positively correlated with higher Gleason grade and predicted biochemical recurrence in Taiwanese PC patients.22 Our study expands on the idea that STK3 has a non-canonical role in a cancer type-dependent manner.
Our pharmacological and genetic knockdown studies show that inhibiting STK3 slows PC cell proliferation (in vitro and in vivo) and Matrigel invasion. The small-molecule inhibitors had effects across multiple cell lines, with the greatest phenotypic consequences in cell lines with the highest expression of STK3, implicating STK3 as the driver of the response. The impact of the loss of STK3 on invasion is particularly noteworthy, as PC metastasis is the main cause of death in patients. A target that is specific to this phenotype in metastatic PC or BC, but does not inhibit growth or have cytotoxic effects in normal cells, could have pronounced translational impacts. Data from STK3-depleted PC cells in vitro and HMVP2 tumors suggest a cell cycle arrest phenotype as shown by altered expression patterns of cell cycle progression markers and loss of p-CDK1/accumulation of cyclin B1 in HMVP2 tumors. This is consistent with STK3-dependent leukemias that suggest STK3 regulates CDK1 to promote cell proliferation.21 In HMVP2 STK3-depleted tumors, we did not observe increased cell death as was observed by hyperactive YAP/TAZ due to loss of Hippo kinase (LATS1/2) signaling in a colon cancer model.13 This suggests that in PC cells, the STK3 pro-tumorigenic role differs from the non-canonical role of LATS1/2 in colon cancer. Importantly, note that in HMVP2 syngeneic tumors, STK3 knockdown tumors unexpectedly showed increased levels of p-MOB1 and p-YAP species (Figures 3K and 3L). Although STK3 knockdown in HMVP2 tumors was modest, this does not explain this observation. Given that this is a syngeneic model in immune intact mice, it is possible that tumor-infiltrating cells may account for increased phosphorylation of MOB1 and YAP.23,24 However, this remains to be tested and is out of the scope of this study.
Lastly, we found that STK3 gene expression correlates with the AR response gene signature in mCRPC. Androgen deprivation (CSS) in vitro induced STK3 expression and activity. Importantly, loss of STK3 in androgen-deprived conditions further slowed cell proliferation. Taken together, the data suggest that STK3 may promote PC progression from hormone sensitivity to castrate resistance. Overall, our data are consistent with correlative analyses showing that STK3 is frequently amplified in PCs and correlates with worse outcomes in advanced mCRPC. Nonetheless, we have not established a direct link between AR and STK3 or a role for STK3 in mCRPC, and these observations remain correlative.
Our study also presents a new set of small-molecule tools to dissect the role and functions of STK3/4. In a screening assay of more than 450 kinases, A1 and B2 proved to have a narrow spectrum of activity that is improved compared to XMU-MP-1. While XMU-MP-1 is a potent STK3 inhibitor, it also inhibits 23 other kinases by 90% or more, resulting in a S10 (1 μM) of 0.05.12 In comparison, A1 (S10 [1 μM] of 0.03) and B2 (S10 [1 μM] = 0.02) inhibited 11 and 8 other kinases, respectively, by 90% or more. Our medicinal chemistry campaign yielded compounds with improved efficacy (B5 and B6) in the low micromolar range on 3D tumor spheroids and in the sub-micromolar range on 3D Matrigel invasion. Further optimization and in vivo evaluation of our compounds is needed, yet at this point these compounds may serve as tools to further investigate STK3 function in different contexts.
To determine potential off-target or general toxicity effects of our STK3 kinase inhibitors, we tested H9C2 cardiomyocytes. Contrary to our observations in PC cells, STK3 inhibitors did not slow H9C2 cell proliferation. Since STK3 inhibition and activation of YAP have been shown to reduce the effect of apoptotic stimuli in H9C2 cells, we tested the effects of STK3 inhibitors in combination with the commonly used chemotherapeutics doxorubicin and docetaxel.20 Consistent with a canonical role for STK3, we found that chemical inhibition of STK3 with our investigational compounds blunted chemotherapy-induced cardiomyocyte cell death. While these are initial observations, it is understood that an STK3 kinase-targeted therapy with combined anti-tumor and cardiac chemoprotective effects would hold high clinical significance.
Overall, our study illustrates that STK3 does not always act as a tumor suppressor and that in fact, for a subset of PCs, STK3 provides a pro-tumorigenic growth advantage. Our studies show that inhibition or loss of STK3 slowed PC growth and limited invasion potential. We developed potent and specific inhibitors to expand the tools available for probing STK3 and the Hippo/YAP pathway. Importantly, these inhibitors did not slow proliferation in cardiomyocytes and instead provided protection from apoptosis, which illustrates the canonical function of STK3. While deeper mechanistic studies are necessary, our studies support a non-canonical and actionable role for STK3 in PC growth and progression.
Materials and methods
Cell lines and culture conditions
HMVP2 cells were a gift from Dr. John DiGiovanni.11 Cell lines were authenticated by short tandem repeat (STR) profiling and tested for mycoplasma at the Duke University cell culture facility by the MycoAlert Plus test (Lonza). H9C2, DU-145, and 293T cells were grown in Dulbecco’s modified Eagle’s medium (DMEM) + 10% FBS. PC3, 22RV1, HMVP2, C4-2, and LNCaP cells were cultured in RPMI 1640 + 10% FBS. LAPC-4 cells, a gift from Dr. William Aronson at UCLA, were cultured in Iscove’s modified Dulbecco’s medium (IMDM) + 10% FBS + 1 nM R1881. For androgen deprivation conditions, CSS (MilliporeSigma, St. Louis, MO, USA) was used.
Western blots
Cells were collected in radioimmunoprecipitation assay (RIPA) lysis buffer with protease cocktail inhibitor 1 and phosphatase cocktail inhibitors 2 and 3 (MilliporeSigma). For tumor biochemical analysis, tumoral tissues were ground in liquid nitrogen with a mortar and pestle and then extracted with RIPA buffer. Protein was quantified using a DC (detergent compatible) Bio-Rad protein assay (Bio-Rad, Hercules, CA, USA) and 30–60 μg of protein was run on SDS-PAGE gels and transferred onto 0.45- or 0.22-μm nitrocellulose. The following antibodies were used at a 1:1,000 dilution: p-MOB1-Thr35 (Cell Signaling Technology [CST], Danvers, MA, USA, #8699), MOB1 (CST #13730S), cyclin D1 (CST #2922S), p-YAP-Ser397 (CST #13619S), p-YAP-Ser127 (CST #13008S), YAP (CST #14074S), STK4 (CST #1496S), TAZ (CST #83669S), p-CDK1-Tyr15 (CST #4539T), p-STAT3-Tyr705 (CST #9145S), cyclin E1 (CST #20808), cyclin B1 (CST #12231), p-histone H3-Ser10 (CST #53348), Puma (CST #98672), p27 (CST #3686), cleaved caspase-3 (CST #9664), and thymidine kinase 1 (CST #8064). Antibodies for GAPDH (CST #5174S) and AR (CST #5153S) were used at 1:4,000 and 1:2,000 dilution, respectively. Antibody for STK3 (Thermo Fisher Scientific, Waltham, MA, USA, #703027) was used at 1:2,000 dilution. For phosphorylation binding (Phosbind) gels, samples were extracted in 50 mM Tris (pH 6.8), 2% SDS, 0.05% bromophenol blue, 10% glycerol, and 5% β-mercaptoethanol. Samples were boiled for 5 min at 95°C and then spun down at 14,800 rpm for 15 min. For the resolving gel, Phosbind acrylamide (PB-A) (APExBIO, Houston, TX, USA, #F4002) was used to pour 6% acrylamide concentration and 50 μM PB-A concentration gel in Mini-PROTEAN 12-well empty cassettes. Gels were run at 80 V for 20 min and then 150 V for 1 h. Gels were rinsed in transfer buffer containing 10 mM EDTA for 10 min, three times, then transfer buffer alone for 10 min, and transferred onto nitrocellulose membrane, then immunoblotted for total YAP. For western blot quantification, ImageJ 1.52a software was utilized to quantify band densitometry and actin or GAPDH loading controls for normalization. When shown as a percentage, treatment group ratios were divided by a control group ratio and multiplied by 100. For AR null cell lines, densitometry background levels were used for STK/AR ratios.
Cell proliferation assays
For standard 2D proliferation assays, cells were plated in a 96-well in 100 μL of medium and, after 24 h, cells were treated as specified for 120–192 h using DMSO as a vehicle or 2× drug in 100 μL of medium. For 3D spheroid assays, cells were plated in 96-well, U-bottom, BioPrime ultra-low attachment plates (Fujifilm Wako Chemicals, Richmond, VA, USA) in 100 μl of medium, and, after 24 h when spheres formed, cells were treated as denoted with 2× drug or DMSO vehicle control. Confluence and spheroid measures were conducted on an Incucyte S3 live-cell imaging system (Sartorious, Ann Arbor, MI, USA) on the 96 whole-well scan mode. Plates were imaged every 8–24 h and quantified with bundled image analysis software. For real-time cell death assays, Incucyte cytotox green reagent (Sartorius) was used to indicate dead cells.
Gene knockdown
For inducible knockdown of STK3, a shERWOOD-UltramiR lentiviral inducible shRNA system was used (Transomic Technologies, Huntsville, AL, USA). To deplete mouse STK3 gene expression in HMVP2 cells, TRC2 MISSION shRNAs in pLKO.1-puro vector backbone were used (MilliporeSigma). STK3 overexpression construct and empty vector control were purchased from VectorBuilder (Chicago, IL, USA). Lentivirus was generated with psPAX2 and pMD2.G packaging plasmids and used with the Polyplus-transfection jetPRIME transfection kit in 293T cells. Virus was collected at 48 h post-transfection, and a 1:1 ratio of viral medium to fresh culture medium with 8 μg/mL Polybrene infection reagent was incubated for 24 h on target cells. Cells were selected with 1–2 μg/mL puromycin.
FACS cell cycle analysis
Doxycycline was added to plates to induce shRNAs at final concentration of 0.25 μg/mL for 96 h, trypsinized, and fixed with 70% ethanol. Cells were resuspended in 800 μL of PBS-BSA solution, and 10 μL of 10 mg/mL RNase A (Thermo Fisher Scientific, #EN0531) was added and incubated at 37°C for 30 min. After incubation, propidium iodide (PI) (Thermo Fisher Scientific, #P3566) was added to each tube for a final concentration of 250 μg/mL. Cell solutions were transferred to 5-mL round-bottom polystyrene tubes and sorted based on PI intensity using the BD FACSCanto flow cytometry system and BD FACSDiva software (BD Biosciences, Franklin Lakes, NJ, USA). FCSalyzer 0.9.22-alpha free software (SourceForge, San Diego, CA, USA) was then used for analysis of FACS data.
Kinetic cell migration and Matrigel invasion
For 3D spheroid invasions, 500 cells were plated in PrimeSurface 3D culture, ultra-low attachment, 96-well, U-bottom, clear plates in 50 μL of medium for 24 h to allow spheres to form (S-BIO, Hudson, NH, USA). Then, Matrigel was gently mixed in for a final concentration of 3 μg/mL and incubated for 1 h at 37°C, and then overlaid with 100 μL of RPMI 1640 medium and the indicated treatment. Quantification for total spheroid growth of the invasion front was conducted using the Incucyte spheroid software module.
In vivo syngeneic allograft study
Mouse experiments were conducted as approved by the Duke University Institutional Animal Care and Use Committee (IACUC; board protocol no. IACUC006565). Briefly, 32 male FVB/N mice aged 6 weeks (Charles River Laboratories, Raleigh, NC, USA) were injected with HMVP2 spheroids grown in ultra-low attachment plates as previously described.11 Approximately 2 × 105 cells were plated in six-well ultra-low attachment plates and allowed to form spheroids for 24 h, after which spheroids were injected in a 1:1 RPMI 1640-to-Matrigel ratio per animal subcutaneously in the right flank. Caliper measurements were commenced 14 days after engraftment three times per week. and body weights measured twice a week. Tumor volumes were calculated using the formula length × (width)2/2. At the terminal endpoint, wet tumor weights were recorded; tumoral tissues were divided and fixed in formalin or flash-frozen in liquid nitrogen.
STK3 NanoBRET kinase assays
Intracellular NanoBRET assays were performed as previously described using NanoLuc-STK3 fusion vector (Promega, Madison, WI, USA, #NV4301).18 Briefly, NanoLuc-STK3 along with carrier DNA was transfected into HEK293 cells in a 96-well format. NanoBRET Tracer K10 was used at a concentration of 1 μM to test compounds at 11 concentrations. To measure the NanoBRET signal, Nano-Glo substrate (Promega) at 1:166 to Opti-MEM medium in combination with extracellular NanoLuc inhibitor (Promega) diluted 1:500 was combined to create a 3× stock solution. A total of 50 μL of the 3× substrate/extracellular NanoLuc inhibitor was added to each well. The plates were read within 10 min on a GloMax discover luminometer (Promega). Biological replicates were normalized and fit using a sigmoidal, four-parameter logistic regression binding curve in GraphPad Prism software (GraphPad, La Jolla, CA, USA). The IC50 and standard error (SE) were calculated in GraphPad Prism software.
Enzyme assays
Assays were performed at Eurofins discovery services (Celle-l’Evescault, France) as previously described.25 Investigational compounds were provided as 10 mM stock solutions and screened in a 10-point dose-response curve at the Km (Michaelis constant) of ATP using two-end recombinant STK3 protein and myelin basic protein as a substrate.
Compound kinase selectivity
A KINOMEscan scanMAX assay panel was performed at Eurofins DiscoverX (San Diego, C., USA) across 403 wild-type human kinases and 65 mutant human kinases at a compound concentration of 1 μM as previously described.26 S10 (1 μM) was used for compound selectivity metric, where the threshold is 10% of activity remaining relative to control for a given kinase. The S score is calculated by dividing the number of inhibited protein kinases at or above this threshold, by the total number of tested protein kinases.
Statistical analysis
GraphPad Prism 9.0.2 was used for statistical analysis of in vitro studies and survival analysis of patient datasets. For time-course live cell proliferation assays and migration/invasion assays, two-way repeated-measures ANOVA multiple comparison analysis was conducted with Dunnett’s multiple comparison test for an adjusted p value both with α = 0.05. Tumor growth kinetics in vivo were analyzed using a generalized estimating equation with an exchangeable correlation to test whether tumor volume growth varied over time between the three treatment arms. Time was treated as a categorical variable to not assume linear growth. p < 0.05 was considered statistically significant. CNA frequency was queried in 187 studies that included 48,341 no overlapping samples on cBioPortal. Transcriptome profiles and patient clinical information of patient data were downloaded from cBioPortal for TCGA PRAD (n = 489), SU2C/PCF metastatic PRAD (RNA-seq fragments per kilobase of transcript per million mapped reads [FPKM] relative to all samples), and TCGA invasive breast cancer (BRCA) cohorts (log RNA-seq V2 RSEM).10,27 Patient samples with available time to progression (PRAD) or overall survival data (SU2C/PCF and BRCA) were used to perform a log-rank Mantel-Cox test to estimate significance of survival difference and HR between stratified groups. TCGA PRAD samples were stratified by STK3 copy number, while SU2C/PCF metastatic PRAD and TCGA invasive BRCA were stratified by upper and lower median expression levels, FPKM capture, and RSEM, respectively. For STK3/4 gene and AR activity correlation studies, we used a web-based software named the Prostate Cancer Transcriptome Atlas (PCTA, http://www.thepcta.org).28 For AR activity, the Androgen_Response hallmark (MSigDB 5908) gene set activation score was computed by using the Z score method.29 Correlations were computed by Spearman’s rank correlation method.
Acknowledgments
This work was supported in part by institutional funds to E.M. from the Department of Pathology, Duke University School of Medicine. This work was funded in part by the NIH Illuminating the Druggable Genome program (grant no. U24DK116204-01). The SGC is a registered charity (no. 1097737) that receives funds from AbbVie, Bayer Pharma AG, Boehringer Ingelheim, the Canada Foundation for Innovation, the Eshelman Institute for Innovation, Genome Canada, the Innovative Medicines Initiative (EU/EFPIA) (ULTRA-DD grant no. 115766), Janssen, Merck KGaA Darmstadt Germany, MSD, Novartis Pharma AG, the Ontario Ministry of Economic Development and Innovation, Pfizer, Takeda, and Wellcome (106169/ZZ14/Z). We are grateful for liquid chromatography-mass spectrometry/high-resolution mass spectrometry (LC-MS/HRMS) support provided by Dr. Brandie Ehrmann and Diane E. Wallace in the Mass Spectrometry Core Laboratory at the University of North Carolina at Chapel Hill (National Science Foundation under grant no. CHE-1726291). In addition, we are grateful for the Duke Cancer Institute shared resources, Flow Cytometry Core and Cancer Center Isolation Facility, supported by the NCI CCSG award no. P30CA014236.
Author contributions
A.U.S., C.I.W., D.H.D., and E.M. wrote, revised, and edited the manuscript. A.U.S. and E.M. carried out most of the in vitro and in vivo cell-based experiments. L.M.D. assisted with immunoblots and Matrigel invasion studies. M.T.Z. assisted with immunohistochemistry. C.I.W. and J.E.P. performed in-cell NanoBRET testing. D.H.D. identified literature hits, designed compounds, and developed chemical synthesis routes. S.N.O., B.J.E., and X.Y. assisted with medicinal chemistry aspects of the project, including design, synthesis, and compound scale-up as needed. L.H. and S.Y. performed biostatistical and bioinformatics analysis, respectively. G.R.D., J.D., S.J.F., and J.-T.C. provided input on study design and manuscript preparation. D.H.D. and E.M. provided project administration.
Declaration of interests
The authors declare no competing interests.
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.ymthe.2021.08.029.
Contributor Information
David H. Drewry, Email: david.drewry@unc.edu.
Everardo Macias, Email: everardo.macias@duke.edu.
Supplemental information
References
- 1.Bhullar K.S., Lagarón N.O., McGowan E.M., Parmar I., Jha A., Hubbard B.P., Rupasinghe H.P.V. Kinase-targeted cancer therapies: Progress, challenges and future directions. Mol. Cancer. 2018;17:48. doi: 10.1186/s12943-018-0804-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Rodgers G., Austin C., Anderson J., Pawlyk A., Colvis C., Margolis R., Baker J. Glimmers in illuminating the druggable genome. Nat. Rev. Drug Discov. 2018;17:301–302. doi: 10.1038/nrd.2017.252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Nguyen D.T., Mathias S., Bologa C., Brunak S., Fernandez N., Gaulton A., Hersey A., Holmes J., Jensen L.J., Karlsson A., et al. Pharos: Collating protein information to shed light on the druggable genome. Nucleic Acids Res. 2017;45(D1):D995–D1002. doi: 10.1093/nar/gkw1072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Wu S., Huang J., Dong J., Pan D. hippo encodes a Ste-20 family protein kinase that restricts cell proliferation and promotes apoptosis in conjunction with salvador and warts. Cell. 2003;114:445–456. doi: 10.1016/s0092-8674(03)00549-x. [DOI] [PubMed] [Google Scholar]
- 5.Hansen C.G., Moroishi T., Guan K.L. YAP and TAZ: A nexus for Hippo signaling and beyond. Trends Cell Biol. 2015;25:499–513. doi: 10.1016/j.tcb.2015.05.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Zheng Y., Pan D. The Hippo signaling pathway in development and disease. Dev. Cell. 2019;50:264–282. doi: 10.1016/j.devcel.2019.06.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Johnson R., Halder G. The two faces of Hippo: Targeting the Hippo pathway for regenerative medicine and cancer treatment. Nat. Rev. Drug Discov. 2014;13:63–79. doi: 10.1038/nrd4161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Salem O., Hansen C.G. The Hippo pathway in prostate cancer. Cells. 2019;8:E370. doi: 10.3390/cells8040370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Gao J., Aksoy B.A., Dogrusoz U., Dresdner G., Gross B., Sumer S.O., Sun Y., Jacobsen A., Sinha R., Larsson E., et al. Integrative analysis of complex cancer genomics and clinical profiles using the cBioPortal. Sci. Signal. 2013;6:pl1. doi: 10.1126/scisignal.2004088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Abida W., Cyrta J., Heller G., Prandi D., Armenia J., Coleman I., Cieslik M., Benelli M., Robinson D., Van Allen E.M., et al. Genomic correlates of clinical outcome in advanced prostate cancer. Proc. Natl. Acad. Sci. USA. 2019;116:11428–11436. doi: 10.1073/pnas.1902651116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Saha A., Blando J., Fernandez I., Kiguchi K., DiGiovanni J. Linneg Sca-1high CD49fhigh prostate cancer cells derived from the Hi-Myc mouse model are tumor-initiating cells with basal-epithelial characteristics and differentiation potential in vitro and in vivo. Oncotarget. 2016;7:25194–25207. doi: 10.18632/oncotarget.7535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Fan F., He Z., Kong L.L., Chen Q., Yuan Q., Zhang S., Ye J., Liu H., Sun X., Geng J., et al. Pharmacological targeting of kinases MST1 and MST2 augments tissue repair and regeneration. Sci. Transl. Med. 2016;8:352ra108. doi: 10.1126/scitranslmed.aaf2304. [DOI] [PubMed] [Google Scholar]
- 13.Pan W.W., Moroishi T., Koo J.H., Guan K.L. Cell type-dependent function of LATS1/2 in cancer cell growth. Oncogene. 2019;38:2595–2610. doi: 10.1038/s41388-018-0610-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Gaulton A., Hersey A., Nowotka M., Bento A.P., Chambers J., Mendez D., Mutowo P., Atkinson F., Bellis L.J., Cibrián-Uhalte E., et al. The ChEMBL database in 2017. Nucleic Acids Res. 2017;45(D1):D945–D954. doi: 10.1093/nar/gkw1074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Henderson J.L., Kormos B.L., Hayward M.M., Coffman K.J., Jasti J., Kurumbail R.G., Wager T.T., Verhoest P.R., Noell G.S., Chen Y., et al. Discovery and preclinical profiling of 3-[4-(morpholin-4-yl)-7H-pyrrolo[2,3-d]pyrimidin-5-yl]benzonitrile (PF-06447475), a highly potent, selective, brain penetrant, and in vivo active LRRK2 kinase inhibitor. J. Med. Chem. 2015;58:419–432. doi: 10.1021/jm5014055. [DOI] [PubMed] [Google Scholar]
- 16.Soth M., Hermann J.C., Yee C., Alam M., Barnett J.W., Berry P., Browner M.F., Frank K., Frauchiger S., Harris S., et al. 3-Amido pyrrolopyrazine JAK kinase inhibitors: Development of a JAK3 vs JAK1 selective inhibitor and evaluation in cellular and in vivo models. J. Med. Chem. 2013;56:345–356. doi: 10.1021/jm301646k. [DOI] [PubMed] [Google Scholar]
- 17.Gundogdu R., Hergovich A. MOB (Mps one binder) proteins in the Hippo pathway and cancer. Cells. 2019;8:E569. doi: 10.3390/cells8060569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Vasta J.D., Corona C.R., Wilkinson J., Zimprich C.A., Hartnett J.R., Ingold M.R., Zimmerman K., Machleidt T., Kirkland T.A., Huwiler K.G., et al. Quantitative, wide-spectrum kinase profiling in live cells for assessing the effect of cellular ATP on target engagement. Cell Chem. Biol. 2018;25:206–214.e11. doi: 10.1016/j.chembiol.2017.10.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kluss J.H., Conti M.M., Kaganovich A., Beilina A., Melrose H.L., Cookson M.R., Mamais A. Detection of endogenous S1292 LRRK2 autophosphorylation in mouse tissue as a readout for kinase activity. NPJ Parkinsons Dis. 2018;4:13. doi: 10.1038/s41531-018-0049-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Tian Y., Song H., Qin W., Ding Z., Zhang Y., Shan W., Jin D. Mammalian STE20-like kinase 2 promotes lipopolysaccharides-mediated cardiomyocyte inflammation and apoptosis by enhancing mitochondrial fission. Front. Physiol. 2020;11:897. doi: 10.3389/fphys.2020.00897. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 21.Camgoz A., Paszkowski-Rogacz M., Satpathy S., Wermke M., Hamann M.V., von Bonin M., Choudhary C., Knapp S., Buchholz F. STK3 is a therapeutic target for a subset of acute myeloid leukemias. Oncotarget. 2018;9:25458–25473. doi: 10.18632/oncotarget.25238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Huang C.Y., Huang S.P., Lin V.C., Yu C.C., Chang T.Y., Juang S.H., Bao B.Y. Genetic variants in the Hippo pathway predict biochemical recurrence after radical prostatectomy for localized prostate cancer. Sci. Rep. 2015;5:8556. doi: 10.1038/srep08556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Du X., Wen J., Wang Y., Karmaus P.W.F., Khatamian A., Tan H., Li Y., Guy C., Nguyen T.M., Dhungana Y., et al. Hippo/Mst signalling couples metabolic state and immune function of CD8α+ dendritic cells. Nature. 2018;558:141–145. doi: 10.1038/s41586-018-0177-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Pan Z., Tian Y., Cao C., Niu G. The emerging role of YAP/TAZ in tumor immunity. Mol. Cancer Res. 2019;17:1777–1786. doi: 10.1158/1541-7786.MCR-19-0375. [DOI] [PubMed] [Google Scholar]
- 25.Gao Y., Davies S.P., Augustin M., Woodward A., Patel U.A., Kovelman R., Harvey K.J. A broad activity screen in support of a chemogenomic map for kinase signalling research and drug discovery. Biochem. J. 2013;451:313–328. doi: 10.1042/BJ20121418. [DOI] [PubMed] [Google Scholar]
- 26.Davis M.I., Hunt J.P., Herrgard S., Ciceri P., Wodicka L.M., Pallares G., Hocker M., Treiber D.K., Zarrinkar P.P. Comprehensive analysis of kinase inhibitor selectivity. Nat. Biotechnol. 2011;29:1046–1051. doi: 10.1038/nbt.1990. [DOI] [PubMed] [Google Scholar]
- 27.Liu J., Lichtenberg T., Hoadley K.A., Poisson L.M., Lazar A.J., Cherniack A.D., Kovatich A.J., Benz C.C., Levine D.A., Lee A.V., et al. Cancer Genome Atlas Research Network An integrated TCGA pan-cancer clinical data resource to drive high-quality survival outcome analytics. Cell. 2018;173:400–416.e11. doi: 10.1016/j.cell.2018.02.052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.You S., Knudsen B.S., Erho N., Alshalalfa M., Takhar M., Al-Deen Ashab H., Davicioni E., Karnes R.J., Klein E.A., Den R.B., et al. Integrated classification of prostate cancer reveals a novel luminal subtype with poor outcome. Cancer Res. 2016;76:4948–4958. doi: 10.1158/0008-5472.CAN-16-0902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Levine D.M., Haynor D.R., Castle J.C., Stepaniants S.B., Pellegrini M., Mao M., Johnson J.M. Pathway and gene-set activation measurement from mRNA expression data: The tissue distribution of human pathways. Genome Biol. 2006;7:R93. doi: 10.1186/gb-2006-7-10-r93. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.