

Abstract
Background
Red Ginseng has been used for many years to treat diseases. Ginsenoside Rg3 has documented therapeutic effects, including anticancer and anti-inflammatory activities. However, the anticancer effect of Rg3-enriched red ginseng extract (Rg3-RGE) and its underlying mechanisms have not been fully explored. We investigated whether Rg3-RGE plays an anti-tumor role in lung cancer cells.
Methods
To examine the effect of Rg3-RGE on lung cancer cells, we performed cell viability assays, flow cytometry, western blotting analysis, and immunofluorescence to monitor specific markers.
Results
Rg3-RGE significantly inhibited cell proliferation and induced mitochondria-dependent apoptosis. Furthermore, Rg3-RGE also increased expression of mitophagy-related proteins such as PINK1 and Parkin. In addition, treatment with Rg3-RGE and mitophagy inhibitors stimulated cell death by inducing mitochondria dysfunction.
Conclusions
Rg3-RGE could be used as a therapeutic agent against lung cancer.
Keywords: Rg3-RGE (Rg3-enriched red ginseng extract), apoptosis, mitophagy, PINK1-parkin signaling
Graphical abstract
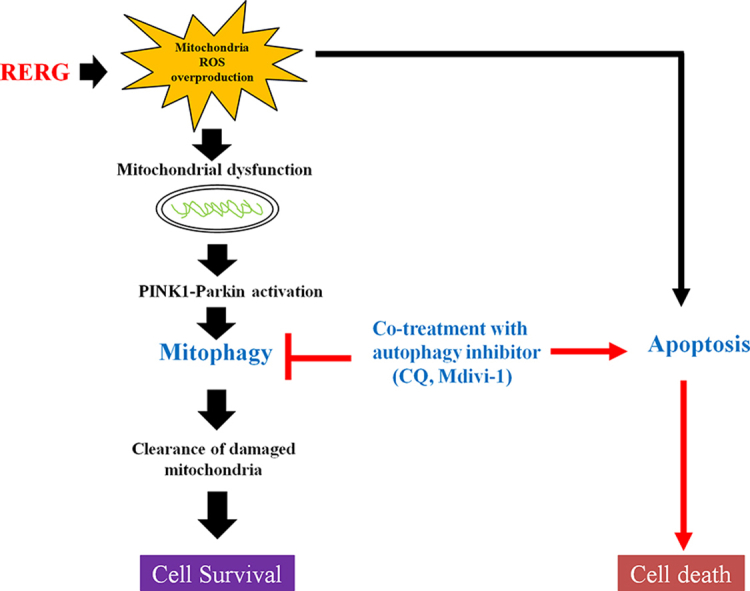
Abbreviations
- Rg3-RGE
Rg3-enriched red ginseng extract
- NSCLC
Non-small cell lung cancer
- PINK1
PTEN-induced putative kinase 1
- MMP
Mitochondrial membrane potential
- CQ
Chloroquine
- NAC
N-acetyl-cysteine
- LDH
lactate dehydrogenase
- Baf A1
Bafilomycin A1
1. Introduction
Lung cancer is the leading cause of cancer-related deaths, and non-small cell lung cancer (NSCLC) accounts for about 85% of all lung cancer cases [1]. Although chemotherapy drugs can improve the quality of life and increase initial survival rates, the therapeutic effects of such drugs are limited toxicity, side effects and drug resistance [2,3]. Furthermore, recent studies suggested that chemotherapy drugs induce tumor metastasis and invasion [4]. Accordingly, new therapeutic strategies for regulating non-small cell lung cancer are needed.
Mitochondria are important regulators of cell death that respond to many different stresses, including oxidative stress and DNA damage [5]. Damage to mitochondria often results in activation of both mitophagy (removal of damaged mitochondria through autophagy) or induce mitochondrial apoptosis [6,7]. Thus, mitophagy is critical for maintaining intracellular functions [8]. Mitophagy promotes tumorigenesis and cell survival in various types of cancers [9]. The toxicity of most chemotherapeutic agents can be attributed to the induction of mitochondrial dysfunction and oxidative stress [10]. In addition, clearance of damaged mitochondria by mitophagy mediates drug resistance in cancer cells [11]. However, mitochondrial clearance may induce cell death via mitophagy [12]. Therefore, mitophagy plays a dual role in cancer progression and suppression.
PTEN-induced putative kinase 1 (PINK1) and Parkin are key regulators of mitophagy [13]. When mitochondrial membrane potential (MMP) is impaired by ROS, irradiation, or chemotherapeutic agents, PINK1 is stabilized on the outer mitochondrial membrane, leading to recruitment of Parkin to damaged mitochondria [14,15]. However, the roles of mitophagy in various types of cancers, and their underlying mechanisms are complex and incompletely understood.
Korean Red Ginseng is a traditional medicinal plant with various pharmacological effects, including antioxidation and anti-tumor activities [16,17]. Ginsenosides, which are the main bioactive components in ginseng, are derived from the roots of diverse ginseng species. Ginsenoside Rg3 exert anti-inflammatory and anti-cancer effects in several disease and cancers [18]. However, the anticancer efficacy of Rg3-enriched red ginseng extract (Rg3-RGE) against lung cancer cells and the underlying mechanisms of action remain unexplored.
In this study, we focused on the role of mitophagy in cell death induced by Rg3-RGE. Our results provide the first demonstration that Rg3-RGE promotes apoptotic cell death by inhibiting mitophagy. We found that Rg3-RGE induced mitophagy via the PINK1-Parkin activation. In addition, treatment of lung cancer cells with Rg3-RGE and a mitophagy inhibitor significantly decreased cell viability and increased apoptosis through mitochondrial dysfunction. Taken together, these results imply that Rg3-RGE induces apoptotic cell death via suppression of PINK/Parkin-regulated mitophagy, suggesting that this substance could be used to develop an effective therapy for lung cancer.
2. Materials and methods
2.1. Materials
Rg3-RGE was obtained from the Korea Ginseng Corporation (Daejeon, Korea). Chloroquine (CQ), N-acetyl-cysteine (NAC) and Mdivi-1 were obtained from Sigma-Aldrich (St. Louis, MO, USA). JC-1, MitoTracker® Red CMXRos, MitoSOX Red, the BCA protein assay kit, and the LDH assay kit were obtained from Thermo Fisher Scientific (Waltham, MA, USA). Bafilomycin A1 (Baf A1) was purchased from Selleck (Houston, TX, USA). Antibodies against cleaved caspase-3, cleaved PARP, MFN2, OPA1, LC3A/B, PINK1, Parkin, phospho-Parkin (Ser 65), BNIP3, TFEB and ATG5 were obtained from Cell Signaling Technologies (Beverly, MA, USA). Antibodies against cytochrome c, PCNA, p62 and BECN1 antibodies were obtained from Santa Cruz Biotechnology (Delaware, CA, USA): and anti-β-actin was obtained from Sigma-Aldrich.
2.2. Cell culture
Human lung cancer cells (A549, H460) were cultured at 37°C in a humidified atmosphere of 5% CO2 in RPMI 1640 medium (Gibco) contained with 10% fetal bovine serum and 1% penicillin-streptomycin.
2.3. Cell viability assay
Lung cancer (5.0 × 103/well) were seeded in 96-well plates and cultured overnight in RPMI medium. After treatment with the indicated concentrations of Rg3-RGE (0, 50, 100, or 200 μg/ml) for 24 h, 10 μl WST-8 solution was added to each well and incubated at 37°C for an additional 2 h. After 2 h, cell viability was measured using the WST-8 assay kit (Roche Applied Science, Mannheim, Germany).
2.4. LDH assay
Human lung cancer cells (A549, H460) (5.0 × 103/well) were seeded in 96-well plates and cultured overnight. After treatment with the indicated concentrations of Rg3-RGE for 24h, cellular cytotoxicity was measured by detecting LDH release using the LDH assay kit.
2.5. Colony formation assay
Cells were seeded in 6-well plates (1.0 × 104/well) and cultured overnight. The cells were then exposed to the indicated concentrations of Rg3-RGE for 10days. Cells were fixed with 100% methanol and stained with a 0.1% crystal violet solution. Stained colonies were detected and counted.
2.6. Annexin V/PI flow cytometry assay
Lung cancer cells (A549 and H460) were incubated with Rg3-RGE (200 μg/ml) for 24 h and then incubated in 500 μl binding buffer, to which was added 10 μl Annexin V and PI solution from the Annexin V-FITC& PI Apoptosis Detection Kit (BD Biosciences, San Jose, CA, USA).
2.7. Western blot analysis
Cells were collected and lysed in RIPA lysis buffer. Cellular protein (25 g) was separated by SDS-PAGE gels and then transferred onto NC membranes. Primary antibodies were incubated for overnight at 4°C. After that, the membranes were incubated with a secondary antibody for 2 h at room temperature. Images were detected on a Bio-Rad ChemiDoc imaging system. Densities were measured using NIH ImageJ (NIH, Bethesda, MD, USA).
2.8. Immunofluorescence assay
A549 and H460 cells were seeded in two-chamber slides at a density of 1.0 × 104cells/well. Cells were treated with 200 μg/ml Rg3-RGE for 24 h, and then incubated with LC3A/B antibody for 24 h at 4°C. Cells were washed three times, incubated with FITC-conjugated secondary antibody for 1 h at room temperature, and stained with DAPI for 5min. Images were acquired using a Nikon confocal microscope.
2.9. siRNA transfection
Lung cancer cells were seed in 6-well plates and cultured overnight. siRNA (100 pmol) and 10 μl Trans IT-TKO solutions (Mirusbio, Madison, WI, USA) were incubated in OPTI-MEM for 20 min at room temperature. After transfection, the cells were treated with Rg3-RGE (200 μg/ml) for 24h, and expression levels of proteins were analyzed by western blotting. Sequences of siRNAs were as follows: PINK1: 5′-CGCUGUUCCUCGUUAUGAATT-3′; Parkin: 5′-GCCACGUGAUUUGCUUAGATT-3.
2.10. Cell staining assay
Cells were seeded in 4-well chamber slides and cultured overnight. After treatment with Rg3-RGE (200 μg/ml) for 24h, the cells were stained with MitoTracker Red CMXRos for 30 min at 37°C in the dark to detect mitochondria morphology. Also, cells were stained overnight in the dark with LC3A/B antibody to observe mitochondria-lysosome fusion. Images were acquired using a Nikon confocal microscope.
2.11. Detection of ROS generation
The cells were incubated with the indicated concentrations of Rg3-RGE for 24 h. 2′,7′-Dichlorofluorescein diacetate (Sigma-Aldrich) was added to the culture medium for 30 min. After washing with PBS, the fluorescence intensity of dichlorofluorescein was detected using a luminometer.
2.12. Measure of mitochondrial ROS
A549 and H460 cells were seed in 35mm dishes and treated with Rg3-RGE (200 μg/ml) for 24h. The cells were then exposed to MitoSOX Red (10 μM) for 30 min at 37°C in the dark. Images were captured on a Nikon confocal microscope. For quantitative analysis, A549 and H460 cells were seeded in black 96-well microplates (2 × 104 cells/well) and treated with MitoSOX Red. A microplate reader (Beckman Coulter, USA) was used to measure fluorescence intensity (Ex = 510 and Em = 580 nm for MitoSOX Red).
2.13. Mitochondrial membrane potential assay
Mitochondrial membrane potential (MMP) was detected by JC-1 staining assays. After treatment with Rg3-RGE (200 μg/ml) for 24 h, the cells were stained with JC-1 (5 M) for 30 min at 37°C in the dark. Mitochondria membrane potential (MMP) was detected using Nikon confocal microscope.
2.14. Statistical analyses
All statistical analyses and graphics were conducted using the GraphPad6 software. Students t-test was used to analyze statistical differences. All data represent means ± SE from at least three individual experiments. A p-value < 0.05 was considered statistically significant.
3. Results
3.1. Rg3-RGE inhibits cell proliferation
First, we used WST-8 assays to investigate whether Rg3-RGE could lead to lung cancer cell death. Treatment with Rg3-RGE for 24 h decreased the viability of two lung cancer cell lines in a dose-dependent manner (Fig. 1A). To further confirm the cytotoxic effects of Rg3-RGE on lung cancer cells, we performed lactate dehydrogenase (LDH) assays. Figure.1A shows that treatment with 200 μg/ml Rg3-RGE for 24h resulted in a 3-fold increase in lactate dehydrogenase concentration in A549 and H460 cells. Rg3-RGE also inhibited the proliferation of A549 and H460 cells (Fig. 1B), suggesting that Rg3-RGE might cause cell death in lung cancer cells. Activation of poly polymerase (PARP) serves as a marker for apoptotic cells [19]. We found that the Rg3-RGE dose-dependently increased the cleavage of PARP in both A549 and H460 cells. Caspase-3 was also increased cleavage significantly after Rg3-RGE treatment (Fig. 1C). In addition, we confirmed the effect of Rg3-RGE on apoptosis by performing annexin-PI staining. Flow cytometry revealed that the proportion of apoptotic cells were significantly increased by Rg3-RGE treatment (Fig. 1D). Caspase-3 activation was also markedly higher in Rg3-RGE treated cells than in non-treated cells (Fig. 1E), and cytochrome c was released from the mitochondria into the cytoplasm after Rg3-RGE treatment (Fig. 1F).
Fig. 1.

Rg3-RGE suppress proliferation of lung cancer cells. (A) A549 and H460 cells were treated with different concentrations of Rg3-RGE for 24h. Cell viability was measured by WST-8 and LDH assays. The experiment was repeated in triplicate. Results represent means ± SE. ∗,#: p < 0.05 or ∗∗,##: p < 0.01 ∗∗∗,###: p < 0.001 versus controls. (B) A549 and H460 cells were treated with the indicated concentrations of Rg3-RGE for 24h. Cell colonies were detected by crystal violet staining and counted. Data represent means ± SD of three independent experiments. ∗: p < 0.05 or ∗∗: p < 0.01 versus controls. (C) A549 and H460 cells were treated with the indicated concentrations of Rg3-RGE for 24 h. Protein expression levels of cleaved caspase3 and PARP were analyzed by western blotting. (D) Rates of apoptosis in A549 and H460 cells treated with Rg3-RGE (200 μg/ml) for 24 were detected by flow cytometry. (E) Caspase 3 activity was detected after treatment with Rg3-RGE (200 μg/ml) for 24 h. (F) A549 and H460 cells were treated with Rg3-RGE (200 μg/ml) for 24h, and cell lysates were divided into cytoplasmic and mitochondrial fraction. Protein expression levels of cytochrome c were analyzed by western blotting. β-actin and PCNA were used as loading controls.
3.2. Rg3-RGE induces autophagy
Given that Rg3 induces autophagy in cancer, we hypothesized that Rg3-RGE would have the same effect in lung cancer cells. As expected, after Rg3-RGE treatment for 24 h, LC3 accumulation (Fig. 2A) and autophagy vesicles were observed in both lung cancer cells (Fig. 2B). Moreover, Rg3-RGE also induced autophagosome formation, as determined by confocal microscopy analysis (Fig. 2C), providing further evidence that Rg3-RGE induced autophagy. To further confirm that Rg3-RGE induced autophagy, we co-treated Rg3-RGE treated lung cancer cells with chloroquine (CQ), an inhibitor of autophagy, and measured protein levels of LC3II and p62 by western blotting. LC3II levels were higher in CQ-treated cells than in non-treated (Fig. 2D). Because treatment with Rg3-RGE induced mitochondria-dependent apoptosis, we speculated that Rg3-RGE might affect mitophagy. As shown in Fig. 2E, the mitochondrial proteins Mfn2 and OPA1 were degraded in a dose-dependent manner in both lung cancer cell lines. To further confirm activation of mitophagy, we stained the cells with a mitochondria tracker and LC3II (autophagosome marker) antibody. As shown in Fig. 2F, the colocalization of mitochondria (red signal) with LC3II (green signal) confirmed that mitophagy was induced by Rg3-RGE treatment. These results indicated that Rg3-RGE induced autophagy.
Fig. 2.

Rg3-RGE induces mitophagy in lung cancer cells. (A) A549 and H460 cells were treated with the indicated concentrations of Rg3-RGE for 24 h, and levels of LC3-II and p62 were detected by western blotting. (B) A549 and H460 cells were incubated with the indicated concentrations of Rg3-RGE for 24 h. Cell morphology was observed under a light microscope. (C) After the indicated concentration of RG3-RGE for 24 h, A549 and H460 cells were immunostained with anti-LC3II antibody. (D) A549/H640 cells were co-treated with chloroquine (50 μM) for 24 h, and levels of p62 and LC3II were measured by western blotting. (E) A549 and H460 cells were treated with the indicated concentration of Rg3-RGE for 24h, and mitochondrial protein levels (MFN2, OPA1) were detected by western blotting. (F) A549 and H460 cells were treated with Rg3-RGE (200 μg/ml) for 24 h. Mitochondrial colocalization with LC3II was detected by a confocal microscope.
3.3. Parkin-dependent signaling involved in Rg3-RGE induced mitophagy
To determine whether Rg3-RGE regulates the expression levels of classical autophagy marker proteins, we performed western blots to measure the influence of Rg3-RGE on these proteins. Rg3-RGE does not affect the protein expression levels of classical autophagy marker such as Beclin1, Atg5, BNIP3, and TFEB (Fig. 3A). PINK1 and Parkin are two key factors regulating mitophagy and parkin is translocated to the mitochondria through upregulation of PINK1, which in turn initiates mitophagy [20]. Hence, we then investigated whether the PINK1-Parkin signaling was involved in Rg3-RGE induced mitophagy. The results showed that Rg3-RGE significantly increased the protein expression levels of PINK1 and phosphorylation of Parkin (Ser65) in both lung cancer cell lines (Fig. 3B). To further investigate the interaction of PINK1 with Parkin under Rg3-RGE treatment, we performed immunoprecipitation assays. Fig. 3C shows that the PINK1 and Parkin interaction was strengthened in Rg3-RGE -treated A549 cells. To reconfirm the role of PINK1 and Parkin in Rg3-RGE -induced mitophagy, we transfected A549 cells with siRNA targeting Parkin (siRNA-Parkin). As shown in Fig. 3D, Rg3-RGE treatment combined with siRNA Parkin decreased LC3II protein levels compared with Rg3-RGE only. Also, siRNA Parkin abolished colocalization of mitochondria with LC3II induced by 24 h treatment with Rg3-RGE (Fig. 3E). These results indicated that PINK1-Parkin are involved in Rg3-RGE induced mitophagy.
Fig. 3.

Rg3-RGE induces mitophagy through the PINK1/Parkin activation. (A) A549 and H460 cells were treated with the indicated concentrations of Rg3-RGE (200 μg/ml) for 24h, and the levels of autophagy-related proteins were detected. (B) A549 and H460 cells were treated with Rg3-RGE (200 μg/ml) for 24h, and protein levels of phospho-parkin (Ser65), total parkin and PINK1 were measured. (C) After treatment with Rg3-RGE for 24h, A549 cells were lysed with IP lysis buffer, and the interaction between PINK1 and Parkin was measured by IP assay. (D) A549 and H460 cells were co-treated with negative control (NC) siRNA, Parkin siRNA, and Rg3-RGE (200 μg/ml) for 24h. PINK1 and Parkin protein levels were determined by western blotting. (E) A549 and H460 cells were transfected with NC siRNA and Parkin siRNA for 24h. Colocalization of mitochondria with autophagosomes was detected by confocal microscopy.
3.4. Inhibition of mitophagy by Mdivi-1 enhanced Rg3-RGE induced cell death
To determine the role of PINK1-Parkin mediated mitophagy on Rg3-RGE induced cell death, we used siRNA to knock down the PINK1 in A549 cells. PINK1 knockdown significantly inhibited cell growth and increased LDH release in Rg3-RGE treated A549 cells (Fig. 4A and B). In addition, co-treatment with siRNA-PINK1 and Rg3-RGE significantly increased caspase3 and PARP cleavage. To further investigate the role of Rg3-RGE induced apoptosis, we used Mdivi-1, a mitophagy inhibitor. Co-treatment with Mdivi-1 and Rg3-RGE induced cell death and inhibited proliferation (Fig. 4D and E). Furthermore, pretreatment with Mdivi-1 inhibited the Rg3-RGE induced colocalization of mitochondria with LC3II (Fig. 4F). In addition, chloroquine (CQ) and Bafilomycin A1 (Baf1), which inhibits the fusion of autophagosomes with lysosomes, increased the rate of cell death in Rg3-RGE treated A549 cells (Fig. 4G and H). These findings support a critical protective role of PINK1-Parkin mediated mitophagy in preventing Rg3-RGE-induced cell death.
Fig. 4.

Suppression of mitophagy promotes cell death induced by Rg3-RGE treatment. (A) A549 and H460 cells were treated with Rg3-RGE and transfected with negative control (NC) siRNA and siRNA-PINK1 for 24h. Cell viability was measured by WST-8 assay. The experiment was repeated in triplicate. Data represent means ± SD. ∗: p < 0.05 or ∗∗: p < 0.01 versus controls. (B) A549 and H460 cells were incubated with Rg3-RGE and transfected with NC siRNA and siRNA-PINK1 for 24h. Cell cytotoxicity was measured by LDH assay. The experiment was repeated in triplicate. Data represent means ± SD. ∗: p < 0.05, ∗∗: p < 0.01, or ∗∗∗: p < 0.001 versus controls. (C) A549 and H460 cells co-treated with siRNA-NC, siRNA-PINK1, and Rg3-RGE for 24 h. Protein levels of cleaved PARP and cleaved caspase-3 were measured by western blotting. (D) A549 and H460 cells were pretreated with Mdivi-1 (50 μM) for 1h, followed by treatment with Rg3-RGE (200 μg/ml) for another 24 h. Cell viability was measured by WST-8 assay. Data represent means ± SD of three independent experiments. ∗: p < 0.05 or ∗∗: p < 0.01 versus controls. (E) A549 and H460 cells were pretreated with Mdivi-1(50 μM) for 1h, followed by treatment with Rg3-RGE (200 μg/ml) for another 24 h. Cell cytotoxicity was measured by LDH assay. Data represent means ± SD of three independent experiments. ∗: p < 0.05, ∗∗: p < 0.01, or ∗∗∗: p < 0.001 versus controls. (F) A549 and H460 cells were treated with Rg3-RGE (200 μg/ml) for 24 h. Colocalization of mitochondria with LC3II was detected by confocal microscopy. (G) A549 and H460 cells were pretreated with CQ for (50 μM) for 1 h, followed by treatment with Rg3-RGE (200 μg/ml) for another 24 h. Cell viability was measured by WST-8 assay. The experiment was repeated in triplicate. Data represent means ± SD. ∗: p < 0.05 or ∗∗: p < 0.01 versus controls. (H) A549 and H460 cells were pretreated with Baf1 (10nM) for 1 h, followed by treatment with Rg3-RGE (200 μg/ml) for another 24 h. Cell viability was measured by WST-8 assay. The experiment was repeated in triplicate. Data represent means ± SD. ∗: p < 0.05 or ∗∗: p < 0.01 versus controls.
3.5. ROS contributed mitophagy and cell death in Rg3-RGE treated lung cancer cells
Next, we attempted to determine the cause of Rg3-RGE induced mitophagy. Mitochondria regulate ROS production in cells, and excessive ROS levels lead to mitophagy [21]. Thus, we tested whether intracellular ROS and mitochondrial ROS following RG3-RGE treatment by ROS detection kit. As shown in Fig. 5A, total intracellular ROS were significantly increased in both lung cancer cells following Rg3-RGE treatment (Fig. 5A). Also, Rg3-RGE treated cells increased mitoSOX-based mitochondrial ROS (Fig. 5B). To investigate whether elevated ROS induced mitochondria damage by Rg3-RGE, we observed mitochondria fragmentation in Rg3-RGE treated cells (Fig. 5C). Moreover, the mitochondrial membrane potential of both lung cancer cells decreased after Rg3-RGE treatment (Fig. 5D). These data indicate that Rg3-RGE induced mitochondria damage. To confirm the role of ROS in mitophagy in Rg3-RGE treatment, we treated A549 and H460 cells with a ROS scavenger, N-acetly-Lcysteine (NAC). In Fig. 5F, NAC pretreatment decreased the co-localization of mitochondria and lysosomes, which indicated a lower occurrence of mitophagy (Fig. 5E). These results demonstrated that the induction of ROS contributes to the initiation of mitophagy following Rg3-RGE. Furthermore, ROS also play an important role in cell death. Pretreatment with NAC showed a considerable blocking effects on the Rg3-RGE induced decreased cell viability (Fig. 5F). Therefore, overproduction of ROS plays a role in the activation of mitophagy and induction of cell death in Rg3-RGE treated lung cancer cells.
Fig. 5.

Rg3-RGE induces apoptosis and mitophagy by upregulating ROS production. (A) A549 and H460 cells were treated with Rg3-RGE and NAC for 24 h, and intracellular ROS levels were measured using a microplate reader. (B) A549 and H460 cells were treated with Rg3-RGE for 24 h, and mitochondrial ROS levels were measured by confocal microscopy. (C) After treatment with Rg3-RGE for 24 h, A549 and H460 cells were stained with MitoTracker red (200 nM), and mitochondrial morphology was observed by confocal microscopy. (D) A549 and H460 cells were treated with Rg3-RGE for 24 h, and mitochondrial membrane potential was visualized by JC-1 staining and detected by confocal microscopy. (E) A549 and H460 cells were treated with Rg3-RGE and NAC for 24 h, stained with MitoTracker red, and immunostained with anti-LC3II antibody. Colocalization of mitochondria and autophagosomes was observed by a confocal microscope. (F) A549 and H460 cells were treated with Rg3-RGE for 24 h in the presence or absence of NAC (20 mM). Cell viability was measured by WST-8 assay. Data represent means ± SD of three independent experiments. ∗: p < 0.05, ∗∗: p < 0.01 or, ∗∗∗: p < 0.001 relative to control.
4. Discussion
In recent years, lung cancer has become the most common cancer and leading cause of cancer-related death. Consequently, an increasing number of novel diagnostic methods and treatments for cancer have been developed. Despite improvements in conventional therapies for non-small cell lung cancer, including (surgery, radiotherapy, and chemotherapy), survival remains poor, and new and effective treatments are required.
Ginsenoside Rg3 is a natural substance extracted from a Korean red ginseng. Several studies have suggested that ginsenoside Rg3 plays roles in tumor development, a process that includes cell proliferation, apoptosis, migration and angiogenesis [22]. However, no study of the anti-tumor effect of Rg3-RGE in cancer has been performed. Here, we studied the anticancer effect of Rg3-RGE on lung cancer cells. Treatment with Rg3-RGE induced mitochondria apoptosis and mitophagy. Furthermore, co-treatment Rg3-RGE with autophagy and mitophagy inhibitors (CQ and Mdivi-1) increased apoptosis of Rg3-RGE in lung cancer cells. Our results suggest that Rg3-RGE could be developed as a therapeutic agent for treating lung cancer.
Mitochondria are particularly vulnerable to damage because they are main source and targets of intracellular oxidative stress. Excessive production of ROS can lead to organelle dysfunction, disturbed redox homeostasis, and induction of apoptosis through a ROS-related mitochondria pathway [23,24]. Cytochrome c release from mitochondria, that triggers a caspase activation, appears to be largely mediated by direct ROS activation [25]. We found that Rg3-RGE promoted mitochondria-dependent apoptosis by releasing cytochrome c and activating caspase. Also, ROS have been reported to cause mitochondrial dysfunction and activate multiple signaling pathways to induce mitophagy [26]. We demonstrated that Rg3-RGE induced the significant accumulation of both cellular and mitochondria ROS. Pretreatment with NAC (N-acetly-Lcysteine) significantly inhibited the colocalization of mitochondria and lysosomes and cell death in Rg3-RGE treated cells. These results suggest the accumulation ROS induced by Rg3-RGE is an important intracellular factor that contributes to triggering apoptosis and mitophagy.
Mitophagy is a regulated catabolic process in which cells degrade their dysfunctional or damaged mitochondria to maintain a healthy mitochondrial population. When mitochondrial damage occurs, cells initiate mitophagy or mitochondria fission to clear damaged mitochondria [27]. We observed that Rg3-RGE could induce a decrease in mitochondria membrane potential and mitochondria fragmentation, which means the initiation of mitophagy. PINK1 and Parkin function in the first steps of a signaling pathway that activates mitochondrial control pathways in response to mitochondrial damage. Recent studies suggested that accumulation of PINK1 and Parkin on the outer mitochondrial membrane can trigger mitophagy [28]. In our study, a significant increase expression of PINK1 and Phosphorylation of Parkin, as well as the interaction Parkin with PINK1 showed that Rg3-RGE induced significant mitophagy in A549 cells, which was involved in the activation of the PINK1-Parkin signaling pathway.
Several studies showed that mitophagy plays dual functions in cancer, playing roles in both tumor prevention and survival [29]. Mitophagy is supposed to play a protective role in response to mitochondria injury. However, under certain circumstances, excessive mitophagy may induce over-degradation of mitochondria and subsequent cell death [30]. In our results, PINK1 knockdown decreased cell viability and increased cell apoptosis, suggesting the protective role of mitophagy in protecting cells by treating Rg3-RGE.
In our study demonstrated that excessive intracellular ROS produced by Rg3-RGE induce mitochondria dysfunction and lead to mitophagy. This study first provides that Rg3-RGE can lead to apoptosis and mitophagy through the activation of ROS and PINK1-Parkin signaling pathway. Our work also showed that inhibition of mitophagy by siRNA-PINK1 and the mitophagy inhibitor (Mdivi-1) stimulated apoptosis under Rg3-RGE treatment. Therefore, further studies are required to confirm the anticancer effect of Rg3-RGE on lung cancer in vivo model. Overall, our present results indicate that the combination of Rg3-RGE with Mdivi-1 may help overcome the limitations of treatment with Rg3-RGE alone for lung cancer therapy.
Declaration of competing interest
The authors declare that they have no competing interests.
Acknowledgement
This research was supported by the Korean Society of Ginseng (2020).
References
- 1.Molina J.R., Yang P., Cassivi S.D., Schild S.E., Adjei A.A. Non-small cell lung cancer: epidemiology, risk factors, treatment, and survivorship. Mayo Clin Proc. 2008;83(5):584–594. doi: 10.4065/83.5.584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Tang C., Qin S., Wu W., Wu Y., Zhang T. Efficacy and potential application of neoadjuvant chemotherapy in patients with IIIa stage non-small cell lung cancer. Zhongguo Fei Ai Za Zhi. 2017;20(2):100–106. doi: 10.3779/j.issn.1009-3419.2017.02.04. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Rossi A., La Salvia A., Di Maio M. Chemotherapy and intercalated gefitinib or erlotinib in the treatment of advanced non-small-cell lung cancer. Expert Rev Respir Med. 2017;11(3):171–180. doi: 10.1080/17476348.2017.1290526. [DOI] [PubMed] [Google Scholar]
- 4.Tas F., Yildiz I., Kilic L., Ciftci R., Keskin S., Sen F. Same chemotherapy regimen leads different myelotoxicity in different malignancies: a comparison of chemotherapy-associated myelotoxicity in patients with advanced ovarian and non-small-cell lung cancer. Am J Ther. 2016;23(3):e670–e679. doi: 10.1097/MJT.0b013e31828232b8. [DOI] [PubMed] [Google Scholar]
- 5.Bock F.J., Tait S.W.G. Mitochondria as multifaceted regulators of cell death. Nat Rev Mol Cell Biol. 2020;21(2):85–100. doi: 10.1038/s41580-019-0173-8. [DOI] [PubMed] [Google Scholar]
- 6.Green D.R., Reed J.C. Mitochondria and apoptosis. Science. 1998;281(5381):1309–1312. doi: 10.1126/science.281.5381.1309. [DOI] [PubMed] [Google Scholar]
- 7.Chen Z., Berquez M., Luciani A. Mitochondria, mitophagy, and metabolic disease: towards assembling the puzzle. Cell Stress. 2020;4(6):147–150. doi: 10.15698/cst2020.06.222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Zhang J. Autophagy and mitophagy in cellular damage control. Redox Biol. 2013;1(1):19–23. doi: 10.1016/j.redox.2012.11.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kulikov A.V., Luchkina E.A., Gogvadze V., Zhivotovsky B. Mitophagy: link to cancer development and therapy. Biochem Biophys Res Commun. 2017;482(3):432–439. doi: 10.1016/j.bbrc.2016.10.088. [DOI] [PubMed] [Google Scholar]
- 10.Fulda S., Galluzzi L., Kroemer G. Targeting mitochondria for cancer therapy. Nat Rev Drug Discov. 2010;9(6):447–464. doi: 10.1038/nrd3137. [DOI] [PubMed] [Google Scholar]
- 11.Smith A.G., Macleod K.F. Autophagy, cancer stem cells and drug resistance. J Pathol. 2019;247(5):708–718. doi: 10.1002/path.5222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ashrafi G., Schwarz T.L. The pathways of mitophagy for quality control and clearance of mitochondria. Cell Death Differ. 2013;20(1):31–42. doi: 10.1038/cdd.2012.81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Narendra D.P., Jin S.M., Tanaka A., Suen D.F., Gautier C.A., Shen J., et al. PINK1 is selectively stabilized on impaired mitochondria to activate Parkin. PLoS Biol. 2010;8(1) doi: 10.1371/journal.pbio.1000298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Xiao B., Goh J.Y., Xiao L., Xian H., Lim K.L., Liou Y.C. Reactive oxygen species trigger Parkin/PINK1 pathway-dependent mitophagy by inducing mitochondrial recruitment of Parkin. J Biol Chem. 2017;292(40):16697–16708. doi: 10.1074/jbc.M117.787739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Matsuda N., Sato S., Shiba K., Okatsu K., Saisho K., Gautier C.A., et al. PINK1 stabilized by mitochondrial depolarization recruits Parkin to damaged mitochondria and activates latent Parkin for mitophagy. J Cell Biol. 2010;189(2):211–221. doi: 10.1083/jcb.200910140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Lee J.W., Mo E.J., Choi J.E., Jo Y.H., Jang H., Jeong J.Y., et al. Effect of Korean Red Ginseng extraction conditions on antioxidant activity, extraction yield, and ginsenoside Rg1 and phenolic content: optimization using response surface methodology. J Ginseng Res. 2016;40(3):229–236. doi: 10.1016/j.jgr.2015.08.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Wang C.Z., Anderson S., Du W., He T.C., Yuan C.S. Red ginseng and cancer treatment. Chin J Nat Med. 2016;14(1):7–16. doi: 10.3724/SP.J.1009.2016.00007. [DOI] [PubMed] [Google Scholar]
- 18.Mohanan P., Subramaniyam S., Mathiyalagan R., Yang D.C. Molecular signaling of ginsenosides Rb1, Rg1, and Rg3 and their mode of actions. J Ginseng Res. 2018;42(2):123–132. doi: 10.1016/j.jgr.2017.01.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Herceg Z., Wang Z.Q. Failure of poly(ADP-ribose) polymerase cleavage by caspases leads to induction of necrosis and enhanced apoptosis. Mol Cell Biol. 1999;19(7):5124–5133. doi: 10.1128/mcb.19.7.5124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Eiyama A., Okamoto K. PINK1/Parkin-mediated mitophagy in mammalian cells. Curr Opin Cell Biol. 2015;33:95–101. doi: 10.1016/j.ceb.2015.01.002. [DOI] [PubMed] [Google Scholar]
- 21.Yang Y., Karakhanova S., Hartwig W., D'Haese J.G., Philippov P.P., Werner J., et al. Mitochondria and mitochondrial ROS in cancer: novel targets for anticancer therapy. J Cell Physiol. 2016;231(12):2570–2581. doi: 10.1002/jcp.25349. [DOI] [PubMed] [Google Scholar]
- 22.Chen T., Li B., Qiu Y., Qiu Z., Qu P. Functional mechanism of Ginsenosides on tumor growth and metastasis. Saudi J Biol Sci. 2018;25(5):917–922. doi: 10.1016/j.sjbs.2018.01.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Wang C.H., Wu S.B., Wu Y.T., Wei Y.H. Oxidative stress response elicited by mitochondrial dysfunction: implication in the pathophysiology of aging. Exp Biol Med (Maywood) 2013;238(5):450–460. doi: 10.1177/1535370213493069. [DOI] [PubMed] [Google Scholar]
- 24.Sinha K., Das J., Pal P.B., Sil P.C. Oxidative stress: the mitochondria-dependent and mitochondria-independent pathways of apoptosis. Arch Toxicol. 2013;87(7):1157–1180. doi: 10.1007/s00204-013-1034-4. [DOI] [PubMed] [Google Scholar]
- 25.Moungjaroen J., Nimmannit U., Callery P.S., Wang L., Azad N., Lipipun V., et al. Reactive oxygen species mediate caspase activation and apoptosis induced by lipoic acid in human lung epithelial cancer cells through Bcl-2 down-regulation. J Pharmacol Exp Ther. 2006;319(3):1062–1069. doi: 10.1124/jpet.106.110965. [DOI] [PubMed] [Google Scholar]
- 26.Scherz-Shouval R., Elazar Z. Regulation of autophagy by ROS: physiology and pathology. Trends Biochem Sci. 2011;36(1):30–38. doi: 10.1016/j.tibs.2010.07.007. [DOI] [PubMed] [Google Scholar]
- 27.Yoo S.M., Jung Y.K. A molecular approach to mitophagy and mitochondrial dynamics. Mol Cells. 2018;41(1):18–26. doi: 10.14348/molcells.2018.2277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Narendra D., Tanaka A., Suen D.F., Youle R.J. Parkin is recruited selectively to impaired mitochondria and promotes their autophagy. J Cell Biol. 2008;183(5):795–803. doi: 10.1083/jcb.200809125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Tilija Pun N., Jang W.J., Jeong C.H. Role of autophagy in regulation of cancer cell death/apoptosis during anti-cancer therapy: focus on autophagy flux blockade. Arch Pharm Res. 2020;43(5):475–488. doi: 10.1007/s12272-020-01239-w. [DOI] [PubMed] [Google Scholar]
- 30.Bialik S., Dasari S.K., Kimchi A. Autophagy-dependent cell death - where, how and why a cell eats itself to death. J Cell Sci. 2018;131(18) doi: 10.1242/jcs.215152. [DOI] [PubMed] [Google Scholar]