

Abstract
The programmable nuclease technology CRISPR-Cas9 has revolutionized gene editing in the last decade. Due to the risk of off-target editing, accurate and sensitive methods for off-target characterization are crucial prior to applying CRISPR-Cas9 therapeutically. Here, we utilized a rhesus macaque model to compare the predictive values of CIRCLE-seq, an in vitro off-target prediction method, with in silico prediction (ISP) based solely on genomic sequence comparisons. We use AmpliSeq HD error-corrected sequencing to validate off-target sites predicted by CIRCLE-seq and ISP for a CD33 guide RNA (gRNA) with thousands of off-target sites predicted by ISP and CIRCLE-seq. We found poor correlation between the sites predicted by the two methods. When almost 500 sites predicted by each method were analyzed by error-corrected sequencing of hematopoietic cells following transplantation, 19 off-target sites revealed insertion or deletion mutations. Of these sites, 8 were predicted by both methods, 8 by CIRCLE-seq only, and 3 by ISP only. The levels of cells with these off-target edits exhibited no expansion or abnormal behavior in vivo in animals followed for up to 2 years. In addition, we utilized an unbiased method termed CAST-seq to search for translocations between the on-target site and off-target sites present in animals following transplantation, detecting one specific translocation that persisted in blood cells for at least 1 year following transplantation. In conclusion, neither CIRCLE-seq or ISP predicted all sites, and a combination of careful gRNA design, followed by screening for predicted off-target sites in target cells by multiple methods, may be required for optimizing safety of clinical development.
Keywords: CRISPR, Ca9, gene editing, off-target, gene therapy, Macaque, translocation, error-corrected sequencing
Graphical abstract
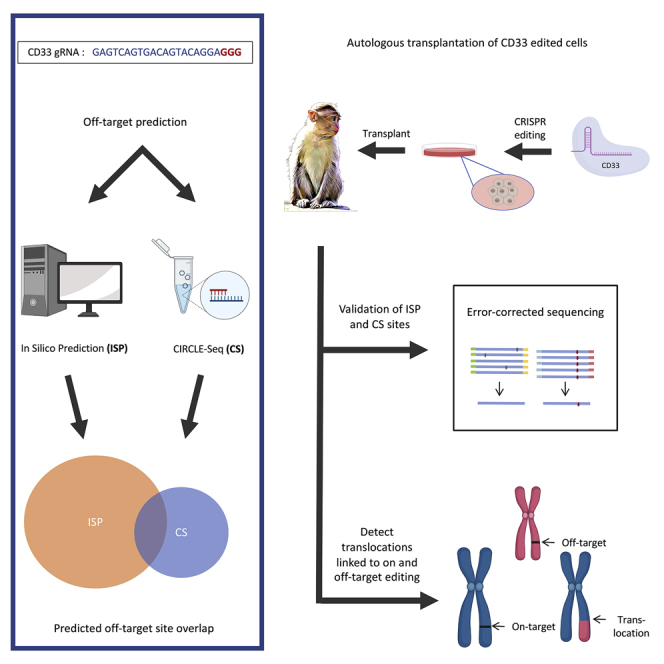
The authors compare in silico algorithms versus CIRCLE-seq for prediction of off-target CRISPR-Cas9 sites and then detected in rhesus macaques following transplantation with edited HSPCs. Both methods miss valid off-target sites; thus, a combination of approaches yields important preclinical information. Translocations resulting from off-target editing were present in vivo long term.
Introduction
Cas9 nucleases can be programmed by guide RNAs (gRNAs) to induce double-stranded DNA breaks (DSBs) at a genomic locus of interest.1, 2, 3 The ease with which this nuclease can be programmed and delivered to cells makes it an ideal system for gene therapies.4,5 However, Cas9 genome editors have been shown to result in unintended and potentially deleterious off-target editing.6, 7, 8, 9, 10 Accurate predictive methods for CRISPR-Cas9 off-target sites identification are crucial prior to applying such systems therapeutically.
In silico prediction (ISP) algorithms rely solely on sequence similarity between genomic loci and the gRNA. Sites similar to the guide are scored based on knowledge of gRNA and DNA binding dynamics.11,12 Even though ISP could be customized to patient genomes if genotyping is available, it is typically performed on reference genomes, is not specific to any individual patient or animal, and is based on assumptions regarding editing specificity. Cellular off-target prediction methodologies, such as genome-wide, unbiased identification of DSBs enabled by sequencing (GUIDE-seq), have been devised based on the principle of efficient integration of an end-protected DNA tag at a DSB, followed by tag-specific amplification and then sequencing to identify off-target sites.13 Although cellular assays are directly relevant to real off-target editing in living target cells, they are challenging to perform on certain primary cell types. Hematopoietic stem cells (HSCs), for example, are very sensitive to DSB. Even though HSCs can withstand the levels of editing resulting in sufficient modification of the on-target site, these cells exhibit cytotoxicity with high level or sustained DSBs. Thus, methodologies requiring high concentrations of nucleases and gRNAs for sensitive detection of off-target sites are challenging or impossible in these cell types. To overcome the sensitivity limitations of cellular assays and potential lack of biologic specificity of ISP, in vitro off-target prediction methods were developed.14,15 These methods rely on exposure of naked genomic DNA to high concentrations of Cas9 and gRNA ribonucleoprotein (RNP). In vitro methods bypass the cellular delivery requirement, allowing for the saturation of the reaction, which increases sensitivity and reproducibility.14
To date, only small numbers of potential ISP-predicted, off-target sites have been validated in cell lines or in target cells or tissues. Cell lines often have abnormal genomic composition and do not serve as representative models for human therapeutics; therefore, the field is trending toward large-scale, off-target validation in relevant cells or in vivo models.3,16 In order to better understand and prevent risks related to off-target editing prior to clinical applications of gene editing, the predictive power of these methods must be comparatively assessed in edited cells present in vivo in relevant preclinical animal models. Additionally, the impact of bona fide off-target editing should be tracked long term to ensure that there are no adverse effects, such as premalignant clonal expansion, linked to unintended off-target editing.
In the current study, we compare the predictive power of circularization for in vitro reporting of cleavage effects by sequencing, also known as CIRCLE-seq (CS),14,17 a widely used semiquantitative in vitro off-target prediction method, to a commonly utilized ISP algorithm (https://zlab.bio/guide-design-resources) adapted to the rhesus macaque (RM) genome.18,19 We focused on analysis of on- and off-target editing of RM hematopoietic stem and progenitor cells (HSPCs) followed by autologous transplantation, based on similarities between RM and human hematopoiesis and the strong predictive value of this large animal model for HSPC gene therapies.20,21 Following engraftment of edited cells, hematopoietic cells can be assessed over time for editing at on- and off-target sites as predicted by ISP or CS.
Results
Off-target sites predicted via in vitro CS or an in silico algorithm
For initial optimization of CS and comparison of predicted sites to ISP, we selected four gRNAs currently being utilized in our program to edit RM HSPCs, specifically targeting sites in the PPP1R12C (containing the AAVS1 “safe harbor” intronic sequence), DNMT3A, TET2, or CD33 genes.5,19 Loss-of-function mutations in TET2 and DNMT3A have been linked to clonal hematopoiesis of aging, and we have used editing to create a RM mode.22 We have reported that creation of CD33 loss-of-function mutations in HSPC can protect normal myelopoiesis from CAR-T cells targeting CD33 expressed by myeloid leukemias.5 The AAVS1 guide was initially reported in 2013.23 DNMT3 and TET2 gRNAs were designed using the Benchling webtool (https://www.benchling.com/crispr/). The CD33 gRNA was modified from a previously utilized human gRNA and truncated to 18 bp for improved specificity.5,24,25
We applied a widely utilized ISP algorithm (https://zlab.bio/guide-design-resources) to the RM genome (MMUL8.0) for each gRNA.18 It predicts sites with up to 4-bp mismatches. It was successful in predicting bona fide off-target sites in RM induced pluripotent stem cells.19 We performed CS14 for each gRNA on genomic DNA from RM blood cells collected and stored before these animals were transplanted with edited HSPCs to create a list of cleavage sites, ranked by the number of reads. The absolute number of predicted sites correlated well between CS and ISP across the 4 guide RNAs (Figure 1A). The TET2 gRNA had the lowest number of off-target sites predicted by both methods and the CD33 guide the highest. ISP consistently predicted higher numbers of off-target sites compared to CS.
Figure 1.

CIRCLE-seq reproducibility and comparison of retrieved sites to ISP
(A) Number of sites predicted by CIRCLE-seq versus ISP for 4 different gRNAs on a log scale. (B) Normalized read counts retrieved via TET2 gRNA CIRCLE-seq technical replicates performed on the same RM blood DNA sample are shown. (C) Normalized read counts retrieved via CD33 gRNA CIRCLE-seq technical replicates performed on the same RM blood DNA sample are shown. Pearson correlation R2 values are shown. (D–F) Visualization of differences between the sites predicted by CIRCLE-seq for the CD33 gRNA performed on blood DNA samples from animals ZJ52, ZL38, and ZL33, respectively, with an MA plot, where M is the log ratio scale and A is the mean average scale is shown (average read count for each predicted site calculated using all 3 sets of data). (G–I) Normalized read count plots (log scale) show the correlation between the reads for the CD33 gRNA off-target sites detected by CIRCLE-seq, 2 animals at a time, with pairwise Pearson correlations shown for each comparison. (J) Overlap between TET2 sites predicted with CIRCLE-seq on ZL26 DNA and by ISP is shown. (K) Overlap between CD33 sites predicted by CIRCLE-seq on DNA from ZL33, ZL38, and ZJ52 and by ISP is shown. (L and M) Plot of the ranks for off-target sites predicted by CIRCLE-seq versus ISP and Spearman correlations between the two rankings for the TET2 gRNA (L) and the CD33 gRNA (M) are shown.
The TET2 and CD33 gRNAs were selected for further investigation, as these two gRNAs had the lowest and highest predicted off-target editing, respectively. In addition, animals were available with excellent on-target editing following transplantation with HSPCs edited utilizing these gRNAs. To confirm reproducibility of CS, we performed technical replicates for these gRNAs, showing an R2 of ≥ 0.950 (Figures 1B, 1C, and S1). Sites not identified by both replicates had very low read counts.
Next, to define any impact of genetic variations (SNPs) between animals, we performed CS on DNA from 3 animals using the CD33 gRNA. CS off-target site read counts for each animal’s run were normalized to the on-target read counts and then compared. MA plots visualize differences in measurements taken between samples via comparison of M (log ratio for each site) versus A (mean for all samples; Figures 1D–1F). Despite differences in sequencing depth and resultant number of predicted sites, i.e., 2 replicates with a total of >16 million reads for animals ZL38 (2,227 sites) and ZJ52 (871 sites) and 1 replicate with slightly over 4 million reads for ZL33 (479 sites), and efficiency of CS between runs, pairwise comparisons showed reasonable correlations of at least R2 > 0.62 (Figures 1G–1I) when comparing sites and normalized read counts between animals. Correlations were less for the run performed on ZL33 DNA with the lower sequencing depth, likely related to sampling, but matched well for all but the lowest read count sites (Figures 1H and 1I).
The sites predicted by CS for all three animals were combined into a master list of 2,384 CS sites. The top 50 CS sites in the master list were predicted via CS on DNA from all three animals. In the top 200 sites, only 34 were not predicted for all three animals. 6/34 of these sites had unique SNPs between animals that could explain these differences. All but 25 of the sites ranked 201–500 were retrieved from both animals with high read depth (ZJ52 and ZL38). Of those 25 sites, 7 sites had SNPs that could explain these differences in prediction between ZJ52 and ZL38. We compared CS- versus ISP-predicted sites. Only 5 of 36 CS and 196 ISP TET2 gRNA sites were the same and only 407 of 2,384 CS and 2,587 ISP sites for the CD33 gRNA (Figures 1J and 1K). Spearman correlations for ranking of ISP versus CS sites were very low for both TET2 (R2 = 0.14) and CD33 (R2 = 0.02) gRNAs (Figures 1L and 1M).
Pilot validation of top CS and ISP CD33 gRNA off-target sites via targeted sequencing
An initial validation was performed on CD33-edited CD34+ HSPCs removed from the “infusion product” (IP) used for autologous transplantation of RM ZJ52 (Figure S2). Primers flanking the 15 top ranked sites from CS and the 15 top sites ranked by ISP were used on IP DNA for standard Illumina sequencing to a depth of >200,000 reads per site (Figure 2). The CD33 on-target site (ZJ52-CS1/ISP1) was 99.8% edited in the IP. Five of the predicted off-target sites had mutated reads of >1%, the limit of sensitivity for this methodology. Three of these sites were predicted by both CS and ISP and had typical insertions and deletions (indels) centered around the predicted cut site (Tables S1–S3). Two sites had reads with a single or a few deleted nucleotides, often not centered at a predicted cut site, and mutations in these sites were found as well in non-edited pre-transplant DNA and distributed evenly across sequencing reads, consistent with sequencing artifacts rather than bona fide off-target editing (Tables S4 and S5).
Figure 2.

Preliminary Illumina sequencing of ZJ52
Results from DAN Illumina sequencing of ZJ52 infusion product and post-transplantation granulocyte for the top 15 ZJ52 CIRCLE-seq sites (left) and top 15 in silico predicted sites (right) for the CD33 guide RNA. m, month post-transplantation.
The five sites identified as edited in the IP were next assessed in granulocytes collected from the blood of ZJ52 at 1 and 5 months following transplantation. Granulocytes turn over every several days and thus reflect ongoing production from HSPCs. Sites ZJ52-CS2/ISP8, ZJ52-CS4/ISP9, and ZJ52-CS26/ISP2 also had convincing indel patterns detected in granulocytes, with higher indel percentages at 1 month compared to 5 months, a similar pattern observed for the on-target site in this animal and others transplanted in our program, reflecting more efficient editing of short-term as compared to long-term engrafting HSPCs by CRISPR-Cas9 editing.5,26 Sites ZJ52-CS10/ISP1259 and ZJ52-CS14 again had mutations not typical for Cas9-mediated editing, further suggesting that these changes resulted instead from sequencing artifacts (Figure 2; Tables S4 and S5). This pilot analysis confirmed that both CS and ISP were able to predict off-target sites present in vivo in a clinically relevant transplantation model. However, sequencing artifacts occurred at some sites, and this approach could not reliably be used for detection of edits present at allele fractions of less than 1%.
Application of error-corrected sequencing for validation of off-target sites of TET2 and CD33
We applied error-corrected AmpliSeq-HD targeted sequencing to improve sensitivity and accuracy of mutation detection. Error-corrected sequencing adds a unique molecular index (UMI) to each amplicon, allowing sensitive discrimination between sequencing artifacts and actual mutations (Figure 3A). We created targeted panels for TET2 off-target sites identified via CS on DNA obtained from animal ZL26 prior to transplantation and via ISP, including all 36 sites predicted by CS and the top 40 (20%) by ISP. We validated quantitation at the on-target site in ZL26 granulocytes, comparing on-target indel quantitation from error-corrected versus standard Illumina sequencing (Figure 3B), revealing close matching between the two methodologies (R2 = 0.99) and clonal expansion of cells containing loss-of-function TET2 indels over the 2-year follow-up. No off-target editing was detected at any of the CS- or ISP-predicted sites using the error-corrected targeted panel in granulocytes or lymphocytes for up to 22 months post-transplantation, confirming the relatively low off-target editing risk for this gRNA predicted by both CS and ISP.
Figure 3.

AmpliSeq HD error-corrected sequencing panels
(A) AmpliSeq HD schematic. (1) Genomic DNA (blue) is extracted from cells, some with CRISPR mutations (red star). (2) The region of interest (on or off target) is amplified using the primer panel, adding UMIs to each end (different colors) and 5′ and 3′ universal adaptors (purple and green, respectively). (3) Each UMI-labeled molecule is amplified redundantly. (4) The molecules are sequenced and computationally sorted into molecular families based on the UMI. (5) A consensus sequence for each molecular family is computed. (B) Quantitation of on-target editing in TET2-edited RM ZL26 with Illumina targeted sequencing versus AmpliSeq HD is shown. (C) Read counts of each CD33 CIRCLE-seq site for the three animals individually are shown. (D) The source and overlap of the 500 CIRCLE-seq sites selected for AmpliSeq HD are shown. (E) Source (ISP and/or CIRCLE-seq) for the 1,000 sites selected for AmpliSeq HD is shown.
We then created a panel including the top 500 sites from the CS master list and the top 500 sites predicted by ISP for the CD33 gRNA. To verify whether selection of only the top 500 CS sites based on combined read rank from the master list was adequate, we plotted the read counts for each site from each of the three individual animal’s runs (Figure 3C). Within these top 500 sites, all CS sites with a normalized read count of ≥25 reads or more from any run on any of the three individual animal’s DNA were included. Venn diagrams confirmed the consistent overlap between these 500 sites for the three animals (Figure 3D). However, only 67 sites overlapped between the top 500 CS sites and top 500 ISP sites (Figure 3E). We were unable to design appropriate unique primers based on flanking sequence characteristics for 28 sites. Thus, the final CD33 panel consisted of a total of 906 sites (Figure S3).
Even with error-corrected sequencing, application of criteria for bona fide off-target editing from the raw sequencing output is required. The error rate for AmpliSeq HD is estimated to be 5- to 10-fold lower than for standard Illumina sequencing, but it is not zero. We applied the following rationale and literature-supported criteria to score a site as edited in a sequenced blood cell sample: (1) only indels were considered as edited, given that non-homologous end joining (NHEJ) very rarely results in SNPs and most sequencing errors mimic SNPs.3,27 (2) We removed indels that are completely non-overlapping with the 18-bp window of the gRNA. (3) Non-edited cells from the same animal obtained pre-transplant analyzed concurrently could not contain the edited site. (4) Indels at site >0.05%, based on sensitivity of AmpliSeq HD. (5) Meets at least 1 of the following criteria (a, has at least 2 UMIs; b, present in more than one animal or at more than one time point or in more than one lineage in an individual animal; c, multiple edit types at one site; d, non-repetitive indel >5 bp in length). These criteria were designed to avoid false positives. However, as this is the most sensitive sequencing method available, we have no means of confirming that all sites that meet these criteria were truly edited. Nonetheless, we assume that any false positives or negatives will not be biased toward CS or ISP.
We sequenced post-transplantation granulocytes from four RMs transplanted with CD33-edited HSPCs. Two of the animals (ZJ52 and ZM36) had significantly higher editing rates, likely due to using chemically modified gRNAs. Details have been previously published for animals ZL38 and ZL33, and Figure S2 and Table S6 summarize experimental parameters.5 By applying the above criteria, editing was detected at 17 predicted off-target sites. Of note, all 17 showed the same trend as the on-target site, with higher indels at earlier times following engraftment, dropping to stable lower levels several months post-transplantation, as more efficiently edited short-term HSPCs are replaced by more difficult to edit long-term HSCs (Figure 4).5
Figure 4.

Tracking of CD33 off-target editing in granulocytes over time
The left graph for each animal has a y axis fixed at 100%. The y axis shows fraction of edited alleles at each off-target site in relation to the on-target site ISP1,CS1 (blue dashed line). The right graph for each animal only shows the off-target sites with the y axis adjusted to allow visualization of the editing levels for each site. The sites are designated in the legend by their CS and/or ISP ranking.
In order to ensure that off-target editing perturbing lineage output were not being missed by focusing solely on granulocytes, 40 additional T, B, and natural killer (NK) cell samples were analyzed. Editing at two additional off-target sites was detected: CS103 at ≤0.15% in T cells of ZL33 and ZL38 and CS391 (0.23%) in NK cells of ZJ52, thus increasing the total number of off-target sites confirmed in vivo to 19 (Figure 5). Details of all edited sites detected from in vivo samples are summarized in Table S7 and Figure S4. Almost half of the sites (9/19) were detected in more than one lineage, with those off-target sites contributing at the highest levels in any lineage more likely to be found in multiple lineages, suggesting that sampling-based detection limits rather than lineage bias resulting from off-target editing accounted for lack of lineage concordance.
Figure 5.

Summary of the sequencing results of the 19 bona fide off-target sites
The shown mutation rates were obtained from sequencing granulocytes and T, B, and NK cells in all 4 CD33 animals. ZJ52 sites are in red, ZM36 in orange, ZL33 in blue, and ZL38 in green.
12/19 edited sites were intragenic, with two sites located in exons (Table S7). None of the perturbed genes or any genes within 200 kB of the sites are known cancer driver genes,28 which is consistent with lack of any expansion of cells containing these off-target edits over time (Table S7; Figures 4 and 5).
8/19 off-targets sites found in vivo following transplantation were ranked in the top 500 by both CS and ISP, despite the overall low overlap between CS- and ISP-predicted sites. Of the remaining sites, 8 were predicted in the top 500 by CS only, although 3 were predicted in the top 500 by ISP only. One of the ISP-only sites was predicted by CS but ranked 1,567th and therefore not scored as a CS top 500 site (Figure 6A). The Levenshtein distance, based on the number of nucleotides differing between the gRNA and off-target site, was analyzed regarding an explanation for why the 8 CS-only sites were not predicted by ISP. Notably, 2 sites were excluded by ISP due to >4-bp mismatches. Additionally, 3 sites were excluded by ISP due to indels (also referred to as gaps) between the DNA and gRNA, which the ISP algorithm utilized does not consider (Figures 6B and 6C). CS versus the ISP missed fewer valid sites detectable in vivo, although the difference did not reach statistical significance (p = 0.0625 with Fischer’s exact test).
Figure 6.

Off-target predictive power of CIRCLE-seq versus ISP
(A) In vivo detected bona fide sites predicted by ISP versus CS or both. (B) The Levenshtein distance between the bona fide off-target sites and the gRNA is shown. (C) Mapping of discrepancies between the gRNA and the bona fide off-target sites retrieved in vivo is shown.
To compare our results to more recent ISP algorithms, we ran CCtop, COSMID, CRISPOR, E-CRISP, and Cas-OFFinder.12,29, 30, 31, 32 For each of these algorithms, we allowed 6 mismatches (or the maximum allowed) and 2 indels (or the maximum allowed; Figure 7A; Tables S8 and S9). 3 valid sites predicted by CS were not identified by any algorithm (Figure 7B; Table S9). Cas-OFFinder predicted 14 out of the 19 sites, the highest of any algorithm (Figure 7C); however, this algorithm predicted a large number of sites and did not provide any ranking for the predicted sites, so we could not ask whether the valid sites were ranked high by this algorithm, an issue given the overall number of sites predicted. Collectively, these 5 additional algorithms predicted 18,581 unique sites. 2,673 were predicted by more than 1 algorithm (2,007 predicted by 2 algorithms, 558 by 3 algorithms, 79 by 4 algorithms, and 29 by all 5 algorithms).
Figure 7.

Investigating the predictive power of newer ISP algorithms
(A) Number of unique off-target sites predicted via 5 newer ISP algorithms. (B) The number of verified off-target sites predicted by the newer ISP algorithms is shown. (C) The number of times each verified off-target site was predicted by the 5 newer ISP algorithms is shown.
We looked at the average percent edit of each bona fide off-target site in all 40 sequenced samples from the 4 animals (Figures S5 and S6). Spearman correlation between the rank of the off-target sites by both prediction methods and the average percent edit in all 40 samples showed no correlation between the CS ranks and the average percent editing (R2 = 0.002) and only very low correlation with the ISP ranks (R2 = 0.231). Eight out of the 12 valid ISP off-target sites were ranked in the top 30 or higher. The remaining had rankings of 300 or higher (Figure S6).
Potential effects of chromatin accessibility on CD33 off-target editing
To explore the possible effect of chromatin accessibility on the presence or absence of predicted off-targets in edited hematopoietic cells, we performed a hypothesis-generating experiment and compared chromatin accessibility as assessed by available “assay for transposase-accessible chromatin” sequencing (ATAC-seq) data derived from human HSPCs for the 906 CD33 predicted off-target sites included on the AmpliSeq HD panel versus the 19 detected in hematopoietic cell samples from the macaques.33,34 Human CD34+ cell ATAC-seq data were utilized (GEO: GSE96772) due to lack of ATAC-seq data for RM HSPCs. CS is performed on naked DNA and would not be predicted to be sensitive to chromatin structure, and current ISP algorithms do not take into account chromatin features.
ATAC-seq peaks from human genomic coordinates (hg19) were mapped to macaque genomic coordinates (rheMac8), and predicted off-target sites were mapped onto the ATAC-seq features.35 We found that 47/906 sites were predicted to be in open chromatin of CD34+ HPSCs, and of those, 4 were bona fide off-target sites detected in RM blood cells (Table S10). The presence of open chromatin correlated with a higher likelihood of validated off-target editing (chi square statistic = 8.9913; p = 0.002—without Yates correction). Although a larger analysis of multiple gRNAs with additional statistical power will be required to strengthen claims for a relationship, these results support a connection between editing and chromatin accessibility, supporting our recent paper showing such a relationship editing on- and off-target sites.16
Detection of persistent CD33 translocations in vivo resulting from off-target editing
As large genomic rearrangements are a cause of concern for CRISPR-Cas9-mediated gene therapies,36, 37, 38 we searched for translocations resulting from fusion of on-target and any off-target DSBs via “chromosomal aberrations analysis by single targeted Ligation-mediated-PCR” sequencing (CAST-seq)39 on DNA from peripheral blood mononuclear cells (PBMCs) and granulocytes collected 2 weeks and 1 month following transplantation of ZJ52. This methodology detects on-target or off-target translocations down to a frequency of 1 in 10,000 cells and requires only knowledge of the on-target sequence and thus searches for translocation partners in an unbiased way, potentially encompassing both off-target sites already identified in our analysis, as well as any other off-target sites not previously identified. CAST-seq at both time points detected a translocation between the on-target site (Chr19:46493369-46504459) and the bona fide off-target site CS28/ISP23 (Chr3:10674353-10674859; Figure 8) but no translocations between the on-target site and any other off-target site, whether the other 18 off-target sites previously detected as edited in vivo or any other previously unvalidated or unpredicted off-target site.
Figure 8.

Detection of chromosomal translocation in engineered HSPCs after CRISPR-Cas9 gene editing
(A) Visualization of chromosomal rearrangements found by CAST-seq. Circos plot shows CD33 target region enlarged on the left. On-target site cluster is shown in green. Significant scores are accentuated by red dots (on-target-mediated translocations) or blue dots (homology-mediated translocation). Gray dots represent natural break sites. (B) Details of the sites involved in the CAST-seq-detected translocation are shown, with the mismatches between the off-target site and the gRNA highlighted in red text. (C) Sequence of the on-target and off-target translocation is shown. On-target sequences are shown in green. Off-target sequences are shown in red. The shared base pairs between the two sequences are highlighted in dark gray, and the surrounding sequences are in light gray. The gRNA is in bold, and the PAM sequence is underlined. The expected cut site is marked with a blue arrow.
To verify the CAST-seq results and to search for the translocation at later time points and in an additional animal, we performed targeted sequencing using primers spanning the putative translocation (Figure 8B; Table S11) on 1-month and 5-month granulocyte samples from ZJ52 and on 2-month and 12-month granulocyte samples from ZM36. This translocation was detected in all four samples. This confirms the presence of this translocation and its persistence for at least 1 year in animal ZM36.
Discussion
It has been challenging to come to a consensus on clinically relevant approaches for the prediction and detection of off-target mutations and large rearrangements following CRISPR-Cas gene editing. Following in vivo delivery of editing machinery to target muscle in animal models, off-target editing has not been detected; however, generally less than 10 sites were analyzed, despite far more sites predicted by the algorithms.40, 41, 42, 43, 44, 45, 46 Because gRNAs are designed using in silico algorithms, predicting off targets using the same or similar ISP off-target algorithms can be insensitive to sites not considered relevant by these approaches or ranked lower in the list of possible sites. Additionally, insensitive detection methods, such as T7E1 PCR or non-error corrected sequencing, were utilized and thus could not detect off-target editing accurately at levels of less than 2%–3%. Sites predicted by CS have been detected in vivo in mice, following editing of a locus in the liver; however, the predictive value of CS was not compared to in silico approaches in this prior report.3
Our studies confirm that gRNA design plays a major role in determining off-target effects, whether predicted by ISP or CS. A gRNA targeting TET2 designed using modern algorithms and predicted to have low off-target risk by both ISP and CS resulted in no detectable mutations at off-target sites in hematopoietic cells following transplantation, even when applying highly sensitive error-corrected sequencing. In contrast, a CD33 gRNA designed with over 10-fold more off-targets sites predicted by both ISP and CS resulted in multiple detected off-target mutations in hematopoietic cells in vivo persisting over time and in multiple lineages post-transplantation. Of note, even truncated guides, initially developed as more specific,47,48 also resulted in significant off-target editing, supporting our previous findings.13 We confirm that mismatches at the 5′ end of the guide were tolerated by Cas9, even when using a truncated guide (Figure 6C).49,50 In addition, we utilized RNP delivery, which is predicted to minimize off-target editing due to short intracellular editing machinery half-life.4,47,51, 52, 53 We were also surprised to see a potentially different spectrum of valid off-target sites when utilizing standard in vitro transcribed gRNAs in the animals (ZL33 and ZL38) versus chemically modified gRNAs (animals ZJ52 and ZM36; Figure 5). This difference could not be explained solely by the efficiency of editing. To our knowledge, similar findings have not been reported; however, a published study did find a difference in the off targets for guides transcribed with a U6 promoter as opposed to T7 promoter.12
Current ISP algorithms search the reference genome for sequence similarity to the gRNA. ISP is generally set to exclude sites with >4-bp mismatches; otherwise, thousands or tens of thousands of off-target sites would be predicted for every gRNA.48 However, Cas9 has been shown to tolerate more than 4-bp mismatches and bulges between off-target sites and the gRNA, both previously and in the current study.8,13,50 Individual-specific SNPs are not taken into account by ISP, even though SNPs have been shown to impact off-target editing.14,54 Additionally, although ISP uses the short gRNA as a query and searches for sites with mismatches to the sequence, in vitro approaches have the advantage of more specific alignment due to the larger query sequence (∼300 bp for CS).14 We speculate that the few bona fide sites predicted by ISP, but not by CS, were missed either because the sites are in regions that are difficult to amplify during library preparation or difficult to sequence on the Illumina platform.
Our exploratory study of chromatin accessibility via ATAC-seq in relation to off-target editing showed a slight preference of off-target editing to open chromatin, in support of prior data on the impact of chromatin structure on the efficiency of on-target editing.16 However, the majority of the sites detected in vivo, including the on-target site, did not fall in open chromatin. This is consistent with previous reports documenting that Cas9 is able to access heterochromatin.55 Therefore, chromatin inaccessibility does not absolutely prevent off-target editing and cannot be used to gauge the relevance of off-target sites predicted by in vitro or ISP methods.
Although error-corrected sequencing is the most sensitive sequencing method available, with a 10-fold increase in sensitivity, it is still limited by PCR error rates. PCR polymerases can make errors in the same position repeatedly, mimicking a mutation.56, 57, 58 This can result in false positives, as seen in our data for some putative sites not consistent with editing based on lack of a PAM sequence or by presence in control unedited DNA. Additionally, AmpliSeq HD was optimized for 20 ng of DNA, amounting to ∼3,000 cells (∼6,000 rhesus genome copies). This limits detection of low-frequency, off-target sites. ISP29 found in both ZL33 and ZL38 is an example of this limitation. A mutation in ISP29 is detectable in low frequencies at early time points and then disappears at some time points, only to appear again in later. Multiple replicate samples could improve sensitivity or the process could be optimized for larger amounts of DNA.
Predicting and detecting chromosomal translocations resulting from DSBs induced by gene editing is challenging, because they are difficult to predict and happen at low frequencies. However, as translocations can result in fusion genes or dysregulate gene expression resulting in neoplasia,59 the importance of searching for these translocations and other large genomic rearrangements has been realized, as well as taking advantage of this phenomenon to model neoplasia linked to chromosomal translocations. Previous reports have detected translocations when multiple gRNAs were used to target two different on-target sites that then fuse and translocate with each other.37,38,60 Such translocations have been shown to persist in vivo following infusion of CRISPR-Cas9 edited CAR-T cells.60 In the current study, we utilized the recently described CAST-seq sensitive and unbiased approach to detect a translocation between the on-target site and one off-target site that had been predicted via both CS and ISP. To our knowledge, this represents the first in vivo detection of a translocation between an on-target site and an off-target site. The translocation persisted for as long as 12 months post-transplantation. Searching for such translocations via knowledge of valid off-target sites will be important in preclinical models and human clinical trials. The lack of detection of translocations involving other off-target sites may be due to negative selection for cells containing translocations, as documented during prolonged in vitro culture of edited CD34+ HSPCs in the original CAST-seq study.
In conclusion, both CS and ISP predicted valid off-target sites; however, both methods missed valid sites detected in vivo. Although most bona fide CS sites could be predicted by using a more liberal ISP, such an ISP algorithm would expand the list of potential off-target sites to levels impractical to screen for validity, even with large targeted panels. Therefore, at this time, we believe there is added value in using both CS and ISP to predict off-target sites, ensuring that sites in cancer-linked genes are not predicted by either method and that overall numbers of sites are predicted to be low. Choice of gRNAs to move forward clinically could be based on analysis of results from both ISP and CS. Large panel-based, error-corrected sequencing can be used to screen for bona fide off targets in relevant primary cells prior to choice of specific gRNAs for clinical applications and in patients enrolled in early-stage clinical trials. Given that we found off-target site rankings generated by either method were not accurate at predicting which sites were found to be edited in vivo, a panel consisting of all predicted off-target sites should be utilized.
In the future, comparative studies performed on a larger number of gRNAs with validation of sites using in vivo preclinical models or samples from early-phase clinical trials could be utilized in a machine-learning approach to modify in silico algorithms to capture valid sites found by CS not currently predicted by available algorithms. GUIDE-seq has recently been optimized for human CD34+ cells, and a comparison of sites predicted by this methodology to in vivo off editing will be welcome.61 In addition, chromosomal translocations or other large genomic rearrangements should be looked for via sensitive methodologies, such as CAST-seq. These approaches are particularly important when performing clinical editing on cells, such as HSPCs, that are highly susceptible to genotoxicity due to lifelong self-renewal.
Materials and methods
gRNA design
The RM CD33 gRNA was designed based on homology to a previously reported human gRNA, along with truncation to 18 bp based on reports suggesting increased specificity from shorter gRNAs.5,24,25 The TET2 guide was designed using Benchling webtool (https://www.benchling.com/crispr/). All gRNA sequences are in Table S6.
In silico prediction
In silico off-target prediction was performed with an in-house Python script modifying the previously published and recently updated (https://zlab.bio/guide-design-resources) algorithm for the rhesus macaque (Macaca mulatta) genome, utilizing the unmasked reference RM genome from Ensembl (http://www.ensembl.org//useast.ensembl.org/?redirectsrc=//www.ensembl.org%2F, release 89).18,19 This algorithm calculates a score for each potential off-target site in the reference genome that has 4 or less bp mismatches to the gRNA.
CS
Genomic DNA was extracted with the Gentra Puregene Kit (QIAGEN). CS was performed as previously described.14,17 In short, RM genomic DNA from CD34− cells, which were collected prior to any animal editing, was sheared to an average of 300 bp (Covaris S2). The sheared molecules were end-repaired, A-tailed, and ligated to a looped adaptor containing a uracil using the KAPA HTP Library Preparation Kit PCR-Free (KAPA Biosystems). Lambda exonuclease (New England Biolabs) and E. coli exonuclease I (New England Biolabs) were used to digest all molecules with free ends. Adaptor-ligated, exonuclease-treated DNA molecules were then treated with USER-enzyme (New England Biolabs) and T4 polynucleotide kinase (New England Biolabs), followed by intramolecular ligation using T4 DNA ligase (New England Biolabs). Remaining linear DNA molecules were eliminated with Plasmid-Safe ATP-dependent DNase (Epicenter). In vitro cleavage was performed in a 50-μL reaction consisting of 125 ng of circularized DNA, 90 nM of SpCas9 protein (New England Biolabs), Cas9 nuclease buffer (New England Biolabs), and 90 nM of gRNA (in vitro transcribed using the GeneArt kit for TET2; Thermo Fisher Scientific). Chemically modified synthetic gRNAs for CD33 (Synthego) were used for all CD33 CS analysis, as the runs were more reproducible with synthetic guides (data not shown). The cleaved molecules were A tailed and ligated with a hair-pin adaptor (New England Biolabs), treated with USER enzyme, and then amplified using universal Next Multiplex Oligos for Illumina (New England Biolabs) and Kapa HiFi Polymerase (KAPA Biosystems). Libraries were sequenced on an Illumina MiSeq with 150-bp paired-end reads.
CS data analysis
Raw data were analyzed using open-source CS software (https://github.com/tsailabSJ/circleseq). For normalization between replicates, the “identified_matched.txt” CS output files for each technical replicate were utilized. The number of reads for the on-target site with least reads comparing all replicates was used as readRef#. The multiplication factor for each technical replicate was calculated as multiplication factor = readRef#/on target read count for that replicate. Each read count for each off target for the replicate was then normalized as read count × multiplication factor. A python script named replicateCombiner.py (https://github.com/aljanahiaa/off-targets) was used to combine the normalized replicates to create a combined technical replicate file. The same python script also calculated the Levenshtein distance between the guide and the predicted off target. If the site has a different distance and/or coordinates, depending on whether a gap was allowed or not, the python script will use the distance and coordinates that allow for a gap.
In order to normalize read counts from CS between individual animals, “identified_matched.txt” or combined technical replicate files for each animal were used, depending on whether technical replicates were performed for each animal or not. Similar to replicate normalization, a multiplication factor was calculated based on the smallest read number for an on-target site between animals and applied to all the read counts from that run of CS in order to normalize read counts between animals. Then, all normalized files were combined with the python masterlistCreator.py (https://github.com/aljanahiaa/off-targets), and the average read count per site was calculated as such: average read count = sum of reads for site/number of animals in which this site was predicted. Calculating the average this way insured that, if there are sites specific to one animal due to unique SNPs, then that site would not be penalized for not being predicted in other animals. The average read count was used to rank the sites in the master list of off-target sites for that gRNA.
Autologous transplantation of rhesus macaques with CRISPR-Cas-edited HSPCs
Autologous transplantation of rhesus macaques with CRISPR-Cas9-edited CD34+-enriched mobilized peripheral blood HSPCs was performed as previously described under protocols approved by the NHLBI Animal Care and Use Committee and as shown (Figure S2).5,62 On day −2, CD34+ cells were purified from the apheresis PBMC collection via immunoabsorption and cultured overnight at 37°C in X-VIVOTM 10 (Lonza) supplemented with 1% HSA (Baxter) and cytokines (SCF 100 ng/mL, FLT3L 100 ng/mL, and TPO 100 ng/mL; all from PeproTech).62 The next day (day −1), RNPs were prepared by mixing Cas9 protein 30–60 μg (PNA Bio) and 15–30 μg of gRNA per aliquot followed by incubation of 10 min at room temperature. Target CD34+ cells were removed from culture, washed with PBS, and then resuspended in aliquots of 3–5 × 106 CD34+ cells in a total volume of 750 μL of Opti-MEM (Thermo Fisher Scientific). An aliquot of RNPs was added to the cell suspension and electroporated using the BTX ECM 830 Square Wave Electroporation System (Harvard Apparatus) with a single pulse of 400 V for 5 ms. The electroporated cells were pooled and incubated at 32°C overnight in X-VIVOTM 10; 1% HSA; and SCF, FLT3L, and TPO. The autologous RM underwent total body irradiation with 400–500 cGy/day on days −1 and 0. Several hours following TBI on day 0, the edited CD34+ cells were infused intravenously. More information on the transplantation and editing parameters is given in Table S6.
Collection and purification of hematopoietic cells post-transplantation
Peripheral blood samples are layered onto Lymphocyte Separation Medium (MP Biomedicals) to separate mononuclear cells (MNCs) and granulocytes. Red blood cells in each fraction were lysed with ACK lysis buffer (Quality Biological). The MNCs were stained with lineage-specific antibodies (Table S12). T cells, B cells, and NK cells were purified via fluorescence-activated cell sorting on a BD FACSAria II instrument.
AmpliSeq HD
The custom primer panels for the AmpliSeq HD sites were designed by Thermo Fisher Scientific using the rheMac8 reference genome (https://www.ncbi.nlm.nih.gov/assembly/GCF_000772875.2/). Amplicons were 70–225 bp for TET2 and 59–161 bp for CD33. The libraries were prepared using the Ion AmpliSeq HD Library Kit (Thermo Fisher Scientific) using the custom primer panels for the first PCR and the Ion AmpliSeq HD Dual Barcode Kit (Thermo Fisher Scientific) for the second PCR. Each 3 samples for TET2 and 2 samples for CD33 were pooled together and templated on the IonChef instrument and then sequenced on an IonTorrent S5 instrument on Ion 530 and Ion 550 chips for TET2 and CD33, respectively. The S5 Torrent Server was used to analyze the samples. Torrent Variant Caller 5.12 plug in was used with custom parameters made available at https://github.com/aljanahiaa/off-targets as a JavaScript Object Notation file. The variant caller Excel output files were downloaded and inspected manually for edits that met criteria for valid indels.
Targeted Illumina sequencing
Targeted sequencing for specific off-target or on-target sites was performed via a standard 2-step PCR using gene-specific primers with adaptors in the first round of PCR amplification and NEBNext Multiplex Oligos for Illumina (Dual Index Primers Set 1; New England Biolabs) for the second round of PCR amplification. Gene-specific primers were designed using Primer3Plus (https://www.bioinformatics.nl/cgi-bin/primer3plus/primer3plusHelp.cgi). Amplicons were approximately 250 bp. Forward and reverse adaptors for the NEBNext Multiplex were added 5′ of the gene-specific primers. The adaptor sequences are provided in Table S13. For the first round, 12.5 μL of KAPA HiFi HotStart ReadyMix polymerase (KAPA Biosystems) and 0.75 μL of each 10 μM primer was added to 20 ng of cellular DNA. For the second round of PCR, a unique combination of 1.5 μL of i5 and i7 primers from the NEBNext kit were added, plus an additional 22 μL of Kapa polymerase was added. Cycling conditions were 3 min at 95°; 20 cycles of 98° for 20 s, 62° for 15 s, and 72° for 30 s; followed by 1 min at 72°. Sequencing was performed on an Illumina MiSeq with 150-bp paired-end reads at a depth generally >500,000 reads per site. To determine % reads with indels, CRISPResso was utilized (https://crispresso.pinellolab.partners.org/) with the following arguments: “CRISPResso -r1 read1.fastq -r2 read2.fastq -a amplicon -g gRNA/off-target site seq -w 40 -q 30–ignore_substitutions.”
Detection of translocations
CAST-seq (https://github.com/AG-Boerries/CAST-Seq) was performed as described.39 In brief, positive- and negative-strand linker oligos were annealed to create adaptors needed for PCR I (Table S14). DNA from edited cells was fragmented, end repaired, and A tailed using the NEBNext Ultra II FS DNA Library Prep Kit for Illumina (New England Biolabs). The annealed adaptor was ligated to the treated DNA using the NEBNext Ultra II Ligation Master Mix and Ligation Enhancer (New England Biolabs). PCR I was carried out with Q5 Hot Start HF DNA Polymerase (New England Biolabs), linker prey primer, CD33 bait primer, and CD33 decoy forward and reverse oligos (Table S14). After purifying the DNA, PCR II was performed with Q5 polymerase, nested linker prey primer, and the CD33 nested bait primer (Table S14). The DNA was purified, and PCR III was performed using NEBNext Ultra II Q5 Master Mix (New England Biolabs) and primers from NEBNext Multiplex Oligos for Illumina (New England Biolabs) to add the Illumina barcodes and sequencing adaptors. The DNA was purified and sequenced via Illumina MiSeq. Computational analysis of the sequences was performed as described.39 The CAST-seq bioinformatic pipeline allowed us to retrieve the translocation sites that were identified in two replicates and significant compared to the untreated samples (p < 0.05).
Targeted sequencing of the amplicon was performed using 2-step PCR as described above with some modifications to account for the likely rarity of the translocation event. The first PCR was performed with 150 ng of DNA using NEBNext Ultra II Q5 Master Mix (New England Biolabs) with 5 min at 96°; 50 cycles of 95° for 15 s, 68° for 40 s, and 72° for 40 s; followed by 2 min at 72°. The PCR product was run on a 2% agarose gel, and the expected size band was purified with QIAquick Gel Extraction Kit (QIAGEN). The second PCR was performed as described above for the 2-step targeted Illumina sequencing library prep.
Acknowledgments
We thank Alec R. Nickolls, Thomas Winkler, Sarah Davies, Nathan Edwards, Eric Glasgow, Karen Ross, Dean Rosenthal, and Diego Espinoza for their helpful advice; AbdulAziz AlJanahi for technical assistance; the NHLBI DNA Sequencing and Genomics Core for DNA sequencing; Keyvan Keyvanfar for cell sorting; and Eric Moon for providing AmpliSeq HD sequencing materials. We thank the animal care staff for their support of all animal care and procedures. This work was supported by the Intramural Program of the National Heart, Lung, and Blood Institute; St. Jude Children's Research Hospital and ALSAC; National Institutes of Health grant U01AI157189; St. Jude Children’s Research Hospital Collaborative Research Consortium on Novel Gene Therapies for Sickle Cell Disease (SCD); the Doris Duke Charitable Foundation (2017093); the German Ministry of Education and Research (IFB-01EO1303); and the Saudi Arabian Cultural Mission to the United States.
Author contributions
Conceptualization, A.A.A., S.Q.T., and C.E.D.; methodology, K.-R.Y., M.Y.K., D.J.Y., T.C., and S.Q.T.; software, A.A.A., S. Cordes, G.A., and I.T.; visualization, A.A.A., G.A., and L.L.T.; investigation, A.A.A., C.R.L., S. Chen, T.-H.S., X.F., I.J., Y.Z., B.-C.L., S.G.H., and J.K.; resources, Y.L. and B.T.; writing – original draft, A.A.A. and C.E.D.; writing – review & editing, A.A.A., C.R.L., T.-H.S., S.G.H., Y.Z., J.K., G.A., S.G., T.C., S.Q.T., and C.E.D.; funding acquisition, C.E.D. and A.A.A.; supervision, T.C., S.G., S.Q.T., and C.E.D.
Declaration of interests
The authors have no conflicts of interest.
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.ymthe.2021.06.016.
Supplemental information
References
- 1.Jinek M., Chylinski K., Fonfara I., Hauer M., Doudna J.A., Charpentier E. A programmable dual-RNA–guided DNA endonuclease in adaptive bacterial immunity. Science. 2012;337:816–821. doi: 10.1126/science.1225829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Ran F.A., Hsu P.D., Wright J., Agarwala V., Scott D.A., Zhang F. Genome engineering using the CRISPR-Cas9 system. Nat. Protoc. 2013;8:2281–2308. doi: 10.1038/nprot.2013.143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Akcakaya P., Bobbin M.L., Guo J.A., Malagon-Lopez J., Clement K., Garcia S.P., Fellows M.D., Porritt M.J., Firth M.A., Carreras A., et al. In vivo CRISPR editing with no detectable genome-wide off-target mutations. Nature. 2018;561:416–419. doi: 10.1038/s41586-018-0500-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Koo T., Kim J.-S. Therapeutic applications of CRISPR RNA-guided genome editing. Brief. Funct. Genomics. 2017;16:38–45. doi: 10.1093/bfgp/elw032. [DOI] [PubMed] [Google Scholar]
- 5.Kim M.Y., Yu K.-R., Kenderian S.S., Ruella M., Chen S., Shin T.-H., Aljanahi A.A., Schreeder D., Klichinsky M., Shestova O., et al. Genetic inactivation of CD33 in hematopoietic stem cells to enable CAR T cell immunotherapy for acute myeloid leukemia. Cell. 2018;173:1439–1453.e19. doi: 10.1016/j.cell.2018.05.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Anderson K.R., Haeussler M., Watanabe C., Janakiraman V., Lund J., Modrusan Z., Stinson J., Bei Q., Buechler A., Yu C., et al. CRISPR off-target analysis in genetically engineered rats and mice. Nat. Methods. 2018;15:512–514. doi: 10.1038/s41592-018-0011-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Li C., Zhou S., Li Y., Li G., Ding Y., Li L., Liu J., Qu L., Sonstegard T., Huang X., et al. Trio-based deep sequencing reveals a low incidence of off-target mutations in the offspring of genetically edited goats. Front. Genet. 2018;9:449. doi: 10.3389/fgene.2018.00449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Lin Y., Cradick T.J., Brown M.T., Deshmukh H., Ranjan P., Sarode N., Wile B.M., Vertino P.M., Stewart F.J., Bao G. CRISPR/Cas9 systems have off-target activity with insertions or deletions between target DNA and guide RNA sequences. Nucleic Acids Res. 2014;42:7473–7485. doi: 10.1093/nar/gku402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ayabe S., Nakashima K., Yoshiki A. Off- and on-target effects of genome editing in mouse embryos. J. Reprod. Dev. 2019;65:1–5. doi: 10.1262/jrd.2018-128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Yee J.K. Off-target effects of engineered nucleases. FEBS J. 2016;283:3239–3248. doi: 10.1111/febs.13760. [DOI] [PubMed] [Google Scholar]
- 11.Martin F., Sánchez-Hernández S., Gutiérrez-Guerrero A., Pinedo-Gomez J., Benabdellah K. Biased and unbiased methods for the detection of off-target cleavage by CRISPR/Cas9: an overview. Int. J. Mol. Sci. 2016;17:1507. doi: 10.3390/ijms17091507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Haeussler M., Schönig K., Eckert H., Eschstruth A., Mianné J., Renaud J.-B., Schneider-Maunoury S., Shkumatava A., Teboul L., Kent J., et al. Evaluation of off-target and on-target scoring algorithms and integration into the guide RNA selection tool CRISPOR. Genome Biol. 2016;17:148. doi: 10.1186/s13059-016-1012-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Tsai S.Q., Zheng Z., Nguyen N.T., Liebers M., Topkar V.V., Thapar V., Wyvekens N., Khayter C., Iafrate A.J., Le L.P., et al. GUIDE-seq enables genome-wide profiling of off-target cleavage by CRISPR-Cas nucleases. Nat. Biotechnol. 2015;33:187–197. doi: 10.1038/nbt.3117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Tsai S.Q., Nguyen N.T., Malagon-Lopez J., Topkar V.V., Aryee M.J., Joung J.K. CIRCLE-seq: a highly sensitive in vitro screen for genome-wide CRISPR-Cas9 nuclease off-targets. Nat. Methods. 2017;14:607–614. doi: 10.1038/nmeth.4278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kim D., Bae S., Park J., Kim E., Kim S., Yu H.R., Hwang J., Kim J.I., Kim J.S. Digenome-seq: genome-wide profiling of CRISPR-Cas9 off-target effects in human cells. Nat. Methods. 2015;12:237–243, 1, 243. doi: 10.1038/nmeth.3284. [DOI] [PubMed] [Google Scholar]
- 16.Lazzarotto C.R., Malinin N.L., Li Y., Zhang R., Yang Y., Lee G., Cowley E., He Y., Lan X., Jividen K., et al. CHANGE-seq reveals genetic and epigenetic effects on CRISPR-Cas9 genome-wide activity. Nat. Biotechnol. 2020;38:1317–1327. doi: 10.1038/s41587-020-0555-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Lazzarotto C.R., Nguyen N.T., Tang X., Malagon-Lopez J., Guo J.A., Aryee M.J., Joung J.K., Tsai S.Q. Defining CRISPR-Cas9 genome-wide nuclease activities with CIRCLE-seq. Nat. Protoc. 2018;13:2615–2642. doi: 10.1038/s41596-018-0055-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hsu P.D., Scott D.A., Weinstein J.A., Ran F.A., Konermann S., Agarwala V., Li Y., Fine E.J., Wu X., Shalem O., et al. DNA targeting specificity of RNA-guided Cas9 nucleases. Nat. Biotechnol. 2013;31:827–832. doi: 10.1038/nbt.2647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hong S.G., Yada R.C., Choi K., Carpentier A., Liang T.J., Merling R.K., Sweeney C.L., Malech H.L., Jung M., Corat M.A.F., et al. Rhesus iPSC safe harbor gene-editing platform for stable expression of transgenes in differentiated cells of all germ layers. Mol. Ther. 2017;25:44–53. doi: 10.1016/j.ymthe.2016.10.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Dunbar C.E., High K.A., Joung J.K., Kohn D.B., Ozawa K., Sadelain M. Gene therapy comes of age. Science. 2018;359:eaan4672. doi: 10.1126/science.aan4672. [DOI] [PubMed] [Google Scholar]
- 21.Larochelle A., Dunbar C.E. Hematopoietic stem cell gene therapy:assessing the relevance of preclinical models. Semin. Hematol. 2013;50:101–130. doi: 10.1053/j.seminhematol.2013.03.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Yu K.-R., Dunbar C.E., Corat M., Chen S., Aljanahi A., Baek E.J., Metais J.-Y., Natanson H., Winkler T., Donahue R.E. A non-human primate CRISPR/Cas9 model of clonal hematopoiesis of indeterminate potential demonstrates expansion of TET2-disrupted clones. Blood. 2017;130:117. [Google Scholar]
- 23.Mali P., Yang L., Esvelt K.M., Aach J., Guell M., DiCarlo J.E., Norville J.E., Church G.M. RNA-guided human genome engineering via Cas9. Science. 2013;339:823–826. doi: 10.1126/science.1232033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Doench J.G., Hartenian E., Graham D.B., Tothova Z., Hegde M., Smith I., Sullender M., Ebert B.L., Xavier R.J., Root D.E. Rational design of highly active sgRNAs for CRISPR-Cas9-mediated gene inactivation. Nat. Biotechnol. 2014;32:1262–1267. doi: 10.1038/nbt.3026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Fu Y., Sander J.D., Reyon D., Cascio V.M., Joung J.K. Improving CRISPR-Cas nuclease specificity using truncated guide RNAs. Nat. Biotechnol. 2014;32:279–284. doi: 10.1038/nbt.2808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Shin T.H., Baek E.J., Corat M.A.F., Chen S., Metais J.Y., AlJanahi A.A., Zhou Y., Donahue R.E., Yu K.R., Dunbar C.E. CRISPR/Cas9 PIG-A gene editing in nonhuman primate model demonstrates no intrinsic clonal expansion of PNH HSPCs. Blood. 2019;133:2542–2545. doi: 10.1182/blood.2019000800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ma X., Shao Y., Tian L., Flasch D.A., Mulder H.L., Edmonson M.N., Liu Y., Chen X., Newman S., Nakitandwe J., et al. Analysis of error profiles in deep next-generation sequencing data. Genome Biol. 2019;20:50. doi: 10.1186/s13059-019-1659-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Bailey M.H., Tokheim C., Porta-Pardo E., Sengupta S., Bertrand D., Weerasinghe A., Colaprico A., Wendl M.C., Kim J., Reardon B., et al. MC3 Working Group. Cancer Genome Atlas Research Network Comprehensive characterization of cancer driver genes and mutations. Cell. 2018;173:371–385.e18. doi: 10.1016/j.cell.2018.02.060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Stemmer M., Thumberger T., Del Sol Keyer M., Wittbrodt J., Mateo J.L. CCTop: An intuitive, flexible and reliable CRISPR/Cas9 target prediction tool. PLoS ONE. 2015;10:e0124633. doi: 10.1371/journal.pone.0124633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Cradick T.J., Qiu P., Lee C.M., Fine E.J., Bao G. COSMID: a web-based tool for identifying and validating CRISPR/Cas off-target sites. Mol. Ther. Nucleic Acids. 2014;3:e214. doi: 10.1038/mtna.2014.64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Heigwer F., Kerr G., Boutros M. E-CRISP: fast CRISPR target site identification. Nat. Methods. 2014;11:122–123. doi: 10.1038/nmeth.2812. [DOI] [PubMed] [Google Scholar]
- 32.Bae S., Park J., Kim J.S. Cas-OFFinder: a fast and versatile algorithm that searches for potential off-target sites of Cas9 RNA-guided endonucleases. Bioinformatics. 2014;30:1473–1475. doi: 10.1093/bioinformatics/btu048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Buenrostro J.D., Wu B., Chang H.Y., Greenleaf W.J. ATAC-seq: a method for assaying chromatin accessibility genome-wide. Curr. Protoc. Mol. Biol. 2015;109:21.29.1–21.29.9. doi: 10.1002/0471142727.mb2129s109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Buenrostro J.D., Corces M.R., Lareau C.A., Wu B., Schep A.N., Aryee M.J., Majeti R., Chang H.Y., Greenleaf W.J. Integrated single-cell analysis maps the continuous regulatory landscape of human hematopoietic differentiation. Cell. 2018;173:1535–1548.e16. doi: 10.1016/j.cell.2018.03.074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Espinoza D.A., Fan X., Yang D., Cordes S.F., Truitt L.L., Calvo K.R., Yabe I.M., Demirci S., Hope K.J., Hong S.G., et al. Aberrant clonal hematopoiesis following lentiviral vector transduction of HSPCs in a rhesus macaque. Mol. Ther. 2019;27:1074–1086. doi: 10.1016/j.ymthe.2019.04.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Brunet E., Jasin M. In: Advances in Experimental Medicine and Biology. Zhang Y., editor. Springer Singapore; 2018. Induction of chromosomal translocations with CRISPR-Cas9 and other nucleases: understanding the repair mechanisms that give rise to translocations; pp. 15–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Reimer J., Knöß S., Labuhn M., Charpentier E.M., Göhring G., Schlegelberger B., Klusmann J.H., Heckl D. CRISPR-Cas9-induced t(11;19)/MLL-ENL translocations initiate leukemia in human hematopoietic progenitor cells in vivo. Haematologica. 2017;102:1558–1566. doi: 10.3324/haematol.2017.164046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Jeong J., Jager A., Domizi P., Pavel-Dinu M., Gojenola L., Iwasaki M., Wei M.C., Pan F., Zehnder J.L., Porteus M.H., et al. High-efficiency CRISPR induction of t(9;11) chromosomal translocations and acute leukemias in human blood stem cells. Blood Adv. 2019;3:2825–2835. doi: 10.1182/bloodadvances.2019000450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Turchiano G., Andrieux G., Klermund J., Blattner G., Pennucci V., El Gaz M., Monaco G., Poddar S., Mussolino C., Cornu T.I., et al. Quantitative evaluation of chromosomal rearrangements in gene-edited human stem cells by CAST-seq. Cell Stem Cell. 2021;28:1136–1147.e5. doi: 10.1016/j.stem.2021.02.002. [DOI] [PubMed] [Google Scholar]
- 40.Ding Q., Strong A., Patel K.M., Ng S.-L., Gosis B.S., Regan S.N., Cowan C.A., Rader D.J., Musunuru K. Permanent alteration of PCSK9 with in vivo CRISPR-Cas9 genome editing. Circ. Res. 2014;115:488–492. doi: 10.1161/CIRCRESAHA.115.304351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Nelson C.E., Hakim C.H., Ousterout D.G., Thakore P.I., Moreb E.A., Castellanos Rivera R.M., Madhavan S., Pan X., Ran F.A., Yan W.X., et al. In vivo genome editing improves muscle function in a mouse model of Duchenne muscular dystrophy. Science. 2016;351:403–407. doi: 10.1126/science.aad5143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Long C., Amoasii L., Mireault A.A., McAnally J.R., Li H., Sanchez-Ortiz E., Bhattacharyya S., Shelton J.M., Bassel-Duby R., Olson E.N. Postnatal genome editing partially restores dystrophin expression in a mouse model of muscular dystrophy. Science. 2016;351:400–403. doi: 10.1126/science.aad5725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Wu Y., Liang D., Wang Y., Bai M., Tang W., Bao S., Yan Z., Li D., Li J. Correction of a genetic disease in mouse via use of CRISPR-Cas9. Cell Stem Cell. 2013;13:659–662. doi: 10.1016/j.stem.2013.10.016. [DOI] [PubMed] [Google Scholar]
- 44.Amoasii L., Hildyard J.C.W., Li H., Sanchez-Ortiz E., Mireault A., Caballero D., Harron R., Stathopoulou T.-R., Massey C., Shelton J.M., et al. Gene editing restores dystrophin expression in a canine model of Duchenne muscular dystrophy. Science. 2018;362:86–91. doi: 10.1126/science.aau1549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Chen Y., Zheng Y., Kang Y., Yang W., Niu Y., Guo X., Tu Z., Si C., Wang H., Xing R., et al. Functional disruption of the dystrophin gene in rhesus monkey using CRISPR/Cas9. Hum. Mol. Genet. 2015;24:3764–3774. doi: 10.1093/hmg/ddv120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Wang S., Ren S., Bai R., Xiao P., Zhou Q., Zhou Y., Zhou Z., Niu Y., Ji W., Chen Y. No off-target mutations in functional genome regions of a CRISPR/Cas9-generated monkey model of muscular dystrophy. J. Biol. Chem. 2018;293:11654–11658. doi: 10.1074/jbc.AC118.004404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Komor A.C., Badran A.H., Liu D.R. CRISPR-based technologies for the manipulation of eukaryotic genomes. Cell. 2017;169:559. doi: 10.1016/j.cell.2017.04.005. [DOI] [PubMed] [Google Scholar]
- 48.Tsai S.Q., Joung J.K. Defining and improving the genome-wide specificities of CRISPR-Cas9 nucleases. Nat. Rev. Genet. 2016;17:300–312. doi: 10.1038/nrg.2016.28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Kuscu C., Arslan S., Singh R., Thorpe J., Adli M. Genome-wide analysis reveals characteristics of off-target sites bound by the Cas9 endonuclease. Nat. Biotechnol. 2014;32:677–683. doi: 10.1038/nbt.2916. [DOI] [PubMed] [Google Scholar]
- 50.Fu Y., Foden J.A., Khayter C., Maeder M.L., Reyon D., Joung J.K., Sander J.D. High-frequency off-target mutagenesis induced by CRISPR-Cas nucleases in human cells. Nat. Biotechnol. 2013;31:822–826. doi: 10.1038/nbt.2623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Yin H., Song C.-Q., Dorkin J.R., Zhu L.J., Li Y., Wu Q., Park A., Yang J., Suresh S., Bizhanova A., et al. Therapeutic genome editing by combined viral and non-viral delivery of CRISPR system components in vivo. Nat. Biotechnol. 2016;34:328–333. doi: 10.1038/nbt.3471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Kim S., Kim D., Cho S.W., Kim J., Kim J.S. Highly efficient RNA-guided genome editing in human cells via delivery of purified Cas9 ribonucleoproteins. Genome Res. 2014;24:1012–1019. doi: 10.1101/gr.171322.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Gao X., Tao Y., Lamas V., Huang M., Yeh W.H., Pan B., Hu Y.J., Hu J.H., Thompson D.B., Shu Y., et al. Treatment of autosomal dominant hearing loss by in vivo delivery of genome editing agents. Nature. 2018;553:217–221. doi: 10.1038/nature25164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Yang L., Grishin D., Wang G., Aach J., Zhang C.-Z., Chari R., Homsy J., Cai X., Zhao Y., Fan J.-B., et al. Targeted and genome-wide sequencing reveal single nucleotide variations impacting specificity of Cas9 in human stem cells. Nat. Commun. 2014;5:5507. doi: 10.1038/ncomms6507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Barkal A.A., Srinivasan S., Hashimoto T., Gifford D.K., Sherwood R.I. Cas9 functionally opens chromatin. PLoS ONE. 2016;11:e0152683. doi: 10.1371/journal.pone.0152683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.McInerney P., Adams P., Hadi M.Z. Error rate comparison during polymerase chain reaction by DNA polymerase. Mol. Biol. Int. 2014;2014:287430. doi: 10.1155/2014/287430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Filges S., Yamada E., Ståhlberg A., Godfrey T.E. Impact of polymerase fidelity on background error rates in next-generation sequencing with unique molecular identifiers/barcodes. Sci. Rep. 2019;9:3503. doi: 10.1038/s41598-019-39762-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.de Paz A.M., Cybulski T.R., Marblestone A.H., Zamft B.M., Church G.M., Boyden E.S., Kording K.P., Tyo K.E.J. High-resolution mapping of DNA polymerase fidelity using nucleotide imbalances and next-generation sequencing. Nucleic Acids Res. 2018;46:e78. doi: 10.1093/nar/gky296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Mertens F., Johansson B., Fioretos T., Mitelman F. The emerging complexity of gene fusions in cancer. Nat. Rev. Cancer. 2015;15:371–381. doi: 10.1038/nrc3947. [DOI] [PubMed] [Google Scholar]
- 60.Stadtmauer E.A., Fraietta J.A., Davis M.M., Cohen A.D., Weber K.L., Lancaster E., Mangan P.A., Kulikovskaya I., Gupta M., Chen F., et al. CRISPR-engineered T cells in patients with refractory cancer. Science. 2020;367:976–977. doi: 10.1126/science.aba7365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Frangoul H., Altshuler D., Cappellini M.D., Chen Y.-S., Domm J., Eustace B.K., Foell J., de la Fuente J., Grupp S., Handgretinger R., et al. CRISPR-Cas9 gene editing for sickle cell disease and β-thalassemia. N. Engl. J. Med. 2021;384:252–260. doi: 10.1056/NEJMoa2031054. [DOI] [PubMed] [Google Scholar]
- 62.Donahue R.E., Kuramoto K., Dunbar C.E. Large animal models for stem and progenitor cell analysis. Curr. Protoc. Immunol. 2005;Chapter 22:1–29. doi: 10.1002/0471142735.im22a01s69. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.