

Abstract
Fibroblasts can be reprogrammed into cardiovascular progenitor cells (CPCs) using transgenic approaches, although the underlying mechanism remains unclear. We determined whether activation of endogenous genes such as Gata4, Nkx2.5, and Tbx5 can rapidly establish autoregulatory loops and initiate CPC generation in adult extracardiac fibroblasts using a CRISPR activation system. The induced fibroblasts (>80%) showed phenotypic changes as indicated by an Nkx2.5 cardiac enhancer reporter. The progenitor characteristics were confirmed by colony formation and expression of cardiovascular genes. Cardiac sphere induction segregated the early and late reprogrammed cells that can generate functional cardiomyocytes and vascular cells in vitro. Therefore, they were termed CRISPR-induced CPCs (ciCPCs). Transcriptomic analysis showed that cell cycle and heart development pathways were important to accelerate CPC formation during the early reprogramming stage. The CRISPR system opened the silenced chromatin locus, thereby allowing transcriptional factors to access their own promoters and eventually forming a positive feedback loop. The regenerative potential of ciCPCs was assessed after implantation in mouse myocardial infarction models. The engrafted ciCPCs differentiated into cardiovascular cells in vivo but also significantly improved contractile function and scar formation. In conclusion, multiplex gene activation was sufficient to drive CPC reprogramming, providing a new cell source for regenerative therapeutics.
Keywords: cell reprogramming, cardiovascular progenitor cells, CRISPR, heart regeneration, myocardial infarction
Graphical abstract
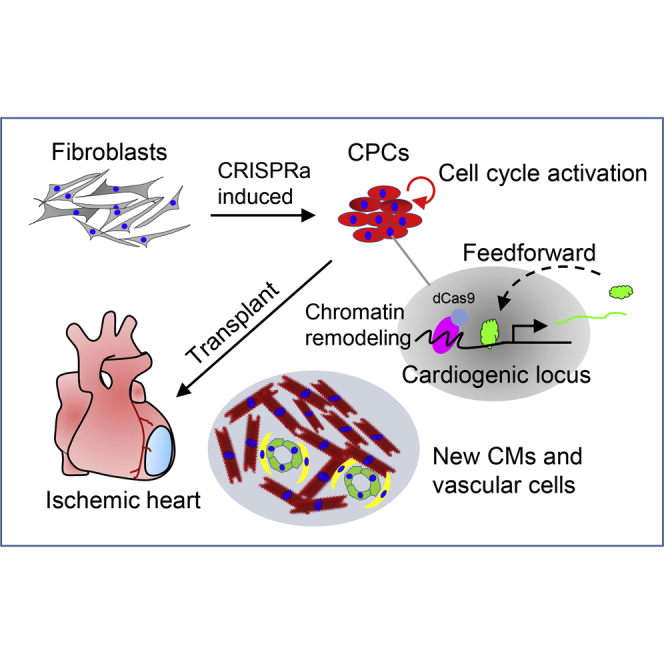
This study from Jiang et al. identifies essential loci controlling CPC formation and helps understanding of the mechanisms underlying cardiac gene autoregulation during cell lineage reprogramming. It also provides a new epigenetic reprogramming system of CPC generation using CRISPR activation approaches for myocardial infarction therapy.
Introduction
The search for an abundant source of cardiovascular progenitor cells (CPCs) has taken on great importance within the field of cardiac regenerative medicine. CPCs derived from pluripotent stem cells (PSCs) emerge as a promising cell source to treat ischemic heart disease such as myocardial infarction (MI) through simultaneous neovascularization and cardiomyogenesis.1,2 Recent studies have shown that various fibroblasts can be directly reprogrammed into induced CPCs (iCPCs) by the forced expression of cardiac transcription factors (TFs).3, 4, 5 However, achieving robust and well-defined reprogrammed cell populations remains a critical challenge for heart regeneration. Further investigation of the mechanisms underlying heart development may help to discover new reprogramming targets for iCPC generation.
Although Gata4, Nkx2.5, and other core TFs have been well studied in heart development and differentiation,6,7 the transcriptional mechanism is still unclear during iCPC reprogramming with respect to chromatin landscape or DNA accessibility. In a conventional transgenic reprogramming approach, TF expression from exogenous vectors may rely on stochastic processes to reactivate the corresponding endogenous genes that are occluded by repressive chromatin marks in somatic cells.8, 9, 10 Interestingly, pluripotent factors can bind together to the promoters of their own genes and form regulatory circuits consisting of autoregulatory and feedforward loops for controlling cell identity.11,12 Furthermore, TFs such as Gata4 and Nkx2.5 reinforce their own expression by directly binding their own promoter or enhancer elements in cardiac cells,7,13 thereby creating feedforward loops. This autostimulatory network likely enables self-perpetuation and stability of cardiac progenitor states,14,15 although its role in initiating CPC induction is largely unknown. Therefore, identifying the essential loci targeted by reprogramming factors helps to elucidate the epigenetic mechanism and further develop more efficient approaches.
Among the programmable DNA-binding proteins, CRISPR/Cas9 has been shown to facilitate gene editing easily and reproducibly in cells,16,17 due to its simplicity and effectiveness of engineering. Recently, a deactivated Cas9 (dCas9) has been engineered with various transcriptional activators or repressors to regulate gene expression by directly binding the DNA sequence near the transcription start site (TSS) or promoter region.16,18 This technology allows for precise targeting of an individual locus using customized single guide RNAs (sgRNAs).19,20 Therefore, the dCas9-based system provides a beneficial tool that can precisely remodel endogenous chromatin loci for various models of cellular reprogramming.21,22 The CRISPR-derived systems also provide a powerful platform to perform genome-scale screens in a wide range of cell types, dissect developmental differentiation pathways, and model disease.18,23 Recently, the CRISPRa-SAM (synergistic activation mediator) system has been used to reprogram human fibroblasts into induced cardiac progenitor cells.24 However, the cell phenotype was not clearly characterized, and it remains unknown whether the CRISPR-induced cells can be used for heart regenerative therapeutics.
The reactivation of endogenous genes has been considered an important criterion of high-quality fully reprogrammed cells.25,26 In this study, we tested a hypothesis that direct activation of endogenous loci using the dCas9-based system can rapidly establish a positive feedback loop of the cardiac TF network, fulfilling iCPC reprogramming. To this end, the CRISPRa-SAM system was employed to induce fibroblast reprogramming through targeting promoters of critical TFs. Multiple benchmarks were utilized in parallel to demonstrate cell lineage conversion. Moreover, transcriptomic profiling of induced cells was revealed by next-generation sequencing before and after cardiac sphere formation. Progenitor cell properties including self-renewal and differentiation potential were further characterized. Finally, the regenerative abilities of reprogrammed cells were assessed in mouse MI models.
Results
Reprogramming of extracardiac fibroblasts into CPCs by CRISPR-based activation of cardiac mesodermal genes
Cardiogenic genes such as Gata4 and Nkx2.5 were moderately expressed in cardiac fibroblasts and may have interrupted the interpretation of CPC generation, but they were scarcely expressed in extracardiac fibroblasts such as tail-tip fibroblasts (TTFs) that were used as starting cells in the present study (Figure S1A). Transgenic mice bearing the Nkx2.5 cardiac enhancer-driven eGFP reporter (Nkx2.5eGFP)27 were used to indicate cell conversion toward CPC lineage, and the isolated TTFs did not express eGFP (Figure S1B). We began cell reprogramming with CRISPRa using a simplified reprogramming scheme targeting promoters of cardiac mesodermal genes. An sgRNA pool was designed to target 11 critical genes that have been demonstrated to regulate mammalian heart development (listed in Table S1).7,28 The CRISPRa-SAM system was transduced in TTFs using lentivirus (TTFCRISPRa, TTF with a scrambled sequence [TTFScr] as a negative control) to screen essential genes capable of inducing CPC reprogramming. At least three sgRNAs per gene were designed and the sgRNA with the highest gene expression was selected for our experiments, as confirmed by qPCR and immunostaining (Figures S1C–S1E). Nkx2.5eGFP was activated in TTFCRISPRa early at day 4 of transduction, and the percentage was increased to ∼60% for 2 weeks (Figures 1A and 1B). The eGFP+ cells lost their parental fibroblast morphology and formed cell clusters (Figure 1A). Approximately nine eGFP+ cell clusters were generated from 105 starting cells (Figure S1F).
Figure 1.

Activation of endogenous genes for reprogramming fibroblasts into CPCs
(A) Cell phenotype change of TTFs after transduction of CRISPRa targeting 11 genes. Scale bars, 100 μm. (B) FACS analysis of eGFP+ cells in TTFs after transduction of CRISPRa targeting 11 genes. (C) Morphological imaging of TTFs transduced with various CRISPRa targets for 14 days. Scale bars, 500 μm. (D) Quantitation of eGFP+ cluster formation. Data measures are presented as mean ± standard error. Versus TTFBGIMNT: #p < 0.05; ns, no significance. (E and F) FACS showing the expression of Flk1, Ssea1, and Cxcr4 in TTFs after transduction of various CRISPRa targets for 14 days. (G) Western blotting of Flk1 expression in TTFs transduced with various CRISPRa targets for 14 days. Uncropped blots refer to Figure S9. (H and I) FACS showing the eGFP+ cell percentage in TTFs after transduction of various CRISPRa targets for 14 days. Data are presented as mean ± standard error. Versus scramble control: ∗p < 0.05. (J) Progenitor cell sphere phenotype of TTFGNT during suspension culture. Scale bar, 1.0 mm. (K) Representative images of eGFP expression in colonies after reattachment on gelatin-coated dishes. Scale bars, 100 μm. (L) Immunostaining of cardiovascular markers in differentiated cells after suspension culture and reattachment. Scale bars, 100 μm. See also Figures S1 and S2.
Additionally, we assessed whether fibroblasts from different tissues of origin can be reprogrammed into CPCs by activation of endogenous genes. Fibroblasts were isolated from lung (LuFib) and liver (LiFib) explants of Nkx2.5eGFP mice and then transduced with the CRISPRa system. Under the same induced conditions as used for TTFs, Nkx2.5eGFP+ colonies were formed in LuFib and LiFib, and the cells were expanded for differentiation after suspension culture and replating (Figure S2A). When cultured in the differentiation medium for 14 days, Gata4+ LuFib- and LiFib-derived reprogrammed cells can differentiate into cardiomyocyte (CM)-like cells expressing with cardiac troponin T (cTnT) (Figure S2B). Pluripotency markers (such as Oct4) were not expressed during the reprogramming process (Figure S2C). Therefore, the CRISPRa-modified cells that expressed CPC genes and could differentiate into the cardiovascular cells were termed CRISPRa-induced CPCs (ciCPCs).
Activation of Gata4, Nkx2.5, or Tbx5 promoter is indispensable for reprogramming fibroblasts into a cardiac progenitor phenotype
Identification of essential genes can facilitate simplification of the reprogramming approach. The efficiency of eGFP+ cluster formation was analyzed by testing different sgRNA combinations to identify the essential loci required for Nkx2.5 activation. The removal of sgRNAs targeting Gata6, Irx4, Hand1, Hand2, or Srf had no significant impact on eGFP+ cluster formation (Figure S1F). Therefore, the CRISPRa system targeting six genes including Baf60c, Gata4, Isl1, Mesp1, Nkx2.5, and Tbx5 (BGIMNT) was employed to induce eGFP+ cell formation in TTFs (TTFBGIMNT) for generating ciCPCs (Figures 1C and 1D), which was consistent with the previous finding of iCPC formation by the introduction of exogenous TFs.4 Additionally, expressions of putative CPC markers including Flk1,29 Ssea1,30 and Cxcr431 were assessed. Flk1 and Ssea1 were expressed in 20% of TTFBGIMNT, while Cxcr4 was expressed in more than 70% of TTFBGIMNT (Figures 1E and 1F). Notably, the removal of individual sgRNAs targeting Baf60c, Gata4, Isl1, Nkx2.5, or Tbx5 led to decreasing eGFP+ cluster numbers and decreasing Flk1+ and Ssea1+ cell numbers in the TTFBGIMNT, but they were not significantly affected by the removal of Mesp1 sgRNA (Figures 1C, 1D, and S1G). Immunoblotting also showed that Flk1 expression was decreased after removal of sgRNAs targeting Isl1, Gata4, Tbx5, or Nkx2.5 in the TTFBGIMNT (Figure 1G), while Cxcr4 expression was markedly decreased by the removal of Gata4 sgRNA (Figure 1F). The reduction in CPC markers was more pronounced after the removal of Gata4, Nkx2.5, or Tbx5 sgRNAs that were further focused on in the present study. The eGFP reporter was expressed in ∼30% of TTFs transduced with CRISPRa targeting Nkx2.5 but was moderately activated in less than 5% of TTFs with Gata4 and Tbx5 sgRNAs (Figures 1H and 1I). Remarkably, Nkx2.5eGFP was expressed in over 80% of TTFs transduced with multiplex CRISPRa targeting Gata4, Nkx2.5, and Tbx5 (TTFGNT) (Figures 1H and 1I). Nkx2.5eGFP+ cell colonies were significantly formed in TTFGNT but not in TTFs with other CRISPRa combinations (Figures S1H and S1I). These data suggested that activation of Nkx2.5 was essential for cell reprogramming and that other genes including Gata4 and Tbx5 served as important facilitators.
Because dCas9/sgRNA complexes may directly activate Nkx2.5eGFP transgenic reporter through undefined promoter-enhancer interactions,32 an additional benchmark is needed to demonstrate cell lineage conversion. Nkx2.5eGFP+ cell colonies were chosen for further characterization to define whether the CRISPRa-induced phenotypes were functional cardiac precursors. These cells were dissociated for subculture and aggregated into spheroids during suspension culture (Figure 1J), reminiscent of the phenotype of embryonic CPCs.30 When the cells were replated on gelatin-coated dishes, there was outgrowth of monolayer cells with minimal expression of Nkx2.5eGFP (Figure 1K). The change of mechanobiological environment from suspension culture to reattachment may render cells prone to differentiation,33 as indicated by decreasing Nkx2.5eGFP activity. Furthermore, cardiovascular differentiation markers including cTnT (∼25%), α-smooth muscle actin (αSMA) (∼40%), and CD31 (∼15%) were expressed in TTFGNT after fetal bovine serum (FBS) induction for 14 days (Figure 1L). Although spontaneous contraction was not found in the ciCPCs during suspension culture or reattachment, a beating cluster was formed in our long-term coculture and differentiation system for 6 weeks (Video S1).
ciCPCs sustain long-term self-renewal under chemically defined conditions
Self-renewal and proliferation are a hallmark of CPC phenotype. Developmental cues such as Wnt/β-catenin signaling, fibroblast growth factor (FGF) activation, and transforming growth factor β (TGF-β) inhibition are important for self-renewal and expansion of CPCs.34,35 To develop an in vitro culture system for large-scale production, we tested the effect of supplements (Figure S3A) on the proliferative activity of induced cells. A stepwise iteration approach was designed to identify the most effective cocktail of factors. Proliferation assays indicated that the cell number of TTFGNT was increased by the combination of leukemia inhibitory factor (LIF), basic FGF (bFGF), SB431542, and Chir99021 (Figure S3B). Unexpectedly, insulin-like growth factor 1 (IGF1, which has been reported to enhance human CPC proliferation)36 did not affect the proliferative activity of TTFGNT. Furthermore, the number of Ki67+ cells and formation of Nkx2.5eGFP+ colonies were increased with the addition of the chemical cocktail (CC) containing these molecules (Figures S3C and S3D). The CC treatment was also favorable for the clonal expansion of single ciCPCs (Figure S3E). Therefore, CC supplements were employed in the expansion medium of reprogrammed cells. TTFGNT cells were able to stably propagate in the medium for more than 20 passages (∼1014-fold expansion) and yielded a large number of ciCPCs (Figure S3F). The activation of Nkx2.5eGFP was retained for up to 20 cell passages of ciCPCs (Figure S3G).
The features of proliferative progenitors were analyzed in long-term expanded ciCPCs in comparison with the early-induced fibroblasts. Proliferative markers (Ki67 and proliferating cell nuclear antigen [Pnca]) were stably expressed in the eGFP+ ciCPCs after serial passages (P10 to P20) as shown by immunostaining (Figures 2A–2D). The proliferative markers were coexpressed with Nkx2.5eGFP in ciCPCs, which was consistent with embryonic stem cell (ESC)-derived CPCs.30 Furthermore, Ssea1+ or Cxcr4+ cell populations were enriched in the ciCPCs after serial passages (P10 to P20) (Figures 2E–2H). Our three-dimensional cell expansion approach allowed for the generation of ciCPCs with a percentage of over 90% as indicated by fluorescence-activated cell sorting (FACS) of Cxcr4 (Figures 2F and 2H). Interestingly, the markers of cell cycle arrest, p53 and p16 (which are responsible for inhibiting cell proliferation in the nucleus),37 were not activated in the nuclei of ciCPCs, while the nuclear localization of p53 and p16 was significantly increased in wild-type TTFs under the same culture conditions (Figures 2I–2L). Therefore, cell cycle arrest can be prevented in ciCPCs after undergoing a limited number of divisions.
Figure 2.

Self-renewal capacity of CRISPR-induced progenitors
(A–D) Imaging and quantitation of eGFP, Ki67, and Pnca expression in ciCPCs after serial passages. Scale bars, 50 μm. Versus P0: ∗p < 0.05. (E–H) FACS and quantitation of cells expressing Ssea1 and Cxcr4 after serial passages. Versus P0: ∗p < 0.05, ∗∗p < 0.01. (I–L) Immunostaining of p16 and p53 in long-term expanded ciCPCs and TTFs as a phenotype control. Scale bars, 50 μm. Quantitation of p16 and p53 expression enriched in nuclei. Versus TTF: ∗∗p < 0.01. P0, TTFs transduced with the CRISPRa system for 4 days before cell dissociation for expansion; P10, cell passage 10; P20, cell passage 20. Data measures are presented as mean ± standard error. See also Figure S3.
In vitro differentiation of ciCPCs into functional cardiovascular cells
Heterogeneity of endothelial progenitors38,39 motivated us to further determine the cardiogenic potential of ciCPCs in terms of early and late phases. A strategy for cell aggregate (spheroid formation) via suspension culture was devised to facilitate ciCPC maturation toward a stable phenotype. Therefore, ciCPCs that were initially derived from TTFs and expanded during early passages (<10) were referred to as early ciCPCs, while ciCPCs that grew out from the suspended spheroids replated on a dish for 3 days before inducing differentiation were referred to as late ciCPCs (Figures 3A and S4A). Beating clusters were formed in the late ciCPCs after inducing cardiac differentiation for 14–20 days (Video S2). Action potentials were detected by intracellular electrical recordings from the single beating ciCPCs (Figure S4B). Compared with TTFs, CPC genes (such as Gata4, Nkx2.5, and Tbx5) and embryonic genes encoding cardiac contractile proteins (such as cTnT and αMHC) were upregulated in both early and late ciCPCs (Figure 3B). Vascular markers (such as αSMA, CD31, and Cdh5) were not significantly activated in early ciCPCs but were robustly upregulated in the late ciCPCs and the differentiated cells (DCs). Therefore, both cardiac and vascular cells were formed in the DCs after spontaneous differentiation.
Figure 3.

Differentiation potential of ciCPCs under in vitro conditions
(A) Schematic of cell expansion and differentiation procedures. (B) Heatmap of cardiovascular genes detected by qPCR in TTFs, early ciCPCs, late ciCPCs, and differentiated cells (DCs). (C) Immunostaining of sarcomeric α-Actinin and Cx43 in ciCPCs treated by CM differentiation medium. Scale bars, 50 μm. (D) Representative Ca2+ waves detected by Fluo-4 dye in ciCPC-CMs at Ca2+ maximum and minimum using live-cell imaging. Fluorescent images correspond to the Video S3. Scale bar, 20 μm. (E) Representative traces of action potentials in ciCPC-CMs (18 cell beating clusters) analyzed by the intracellular electrical recording. (F) Immunostaining of αSMA and SM-MHC in ciCPCs treated by differentiation medium. Scale bars, 100 μm. (G) Corresponding I-V plot of ciCPC-SMCs in response to the vehicle (n = 5), nifedipine (n = 5), or FPL641176 (n = 7) as detected by the whole-cell patch-clamp. Current is given in pA/pF and membrane potential in mV. Data measures are presented as mean ± standard error. (H) Immunostaining of CD31 and vWF in ciCPCs treated by differentiation medium. Scale bars, 100 μm. (I) In vitro tube formation potential of ciCPC-ECs analyzed on Matrigel. Scale bar, 500 μm. See also Figure S4.
The tri-lineage potential of ciCPCs was further assessed by using the molecular cues that drive cardiovascular differentiation. ciCPCs were converted into CM-like cells in CM differentiation medium after 21 days, as evidenced by immunostaining of sarcomeric α-actinin and the gap-junction protein Cx43 (Figure 3C). Furthermore, intracellular calcium flux was detected in a subset of ciCPC-derived CMs (ciCPC-CMs), showing spontaneous calcium oscillations (Figure 3D and Video S3). Ventricular-like action potentials were mainly observed in the beating clusters of ciCPC-CMs (Figure 3E), suggesting that ciCPCs were composed of ventricular precursors. With the smooth muscle cell (SMC) differentiation medium, ciCPCs were converted into SMC-like cells as evidenced by immunostaining of αSMA and SM-MHC (Figure 3F). Contraction of SMCs can be elicited through the L-type calcium channels.40 The ciCPC-SMCs exhibited voltage-dependent increases of inward current that were enhanced by FPL64176 (L-type channel agonist) but were not affected by nifedipine (L-type channel antagonist) (Figure 3G). With endothelial cell (EC) differentiation medium, ciCPCs were converted into EC-like cells as evidenced by immunostaining of CD31 and von Willebrand factor (vWF) (Figure 3H). The newly formed ciCPC-ECs were verified using an in vitro tube formation assay (Figure 3I). To determine the differentiation efficiency in vitro, cTnT+, CD31+, or αSMA+ cells were analyzed by FACS in ciCPCs under CM, EC, SMC, or tri-lineage (mixed) differentiation conditions (Figure S4C). More than 90% of ciCPCs can directly differentiate into the cardiovascular tri-lineage cells (Figure S4D).
differentiation
ciCPCs possess transcriptional profiles similar to those of cardiac precursors
RNA sequencing (RNA-seq) was performed to characterize the transcriptome of reprogrammed cells (early ciCPCs and late ciCPCs) and the uninfected TTFs (Figure S5A). To determine the biological reproducibility of the sample replicates in a heatmap, the 500 most variable genes were analyzed by hierarchical clustering, yielding distinct gene clusters (Figure 4A): 361 variable genes (samples listed in clusters 1–3) upregulated in late ciCPCs were related to the gene ontology (GO) terms of heart development, while 139 genes (samples listed in cluster 4) downregulated in early ciCPCs were involved in cell adhesion or other processes. Clusters 1 and 2 represented a transcriptomic difference between early and late ciCPCs. Differential expression analysis was performed to elucidate the biological processes that occurred in ciCPC generation. There were 1,931 genes (such as Gata4, Nkx2.5, and Isl1) upregulated in early ciCPCs compared with TTFs (Figure S5B), and the GO term analysis showed that the gene subsets were associated with cell cycle, cell division, and other cellular responses involved in the early induction stage (Figure 4B). Furthermore, there were 3,318 genes including CPC markers and cardiac muscle markers upregulated in late ciCPCs compared with TTFs, while fibroblast genes such as Fsp1, Thy1, and Col1a1 were downregulated (Figure S5C). The upregulated gene-related GO terms in late ciCPCs included categories such as sarcomere organization, cardiac muscle contraction, and heart development (Figure 4C), suggesting a conversion toward cardiac lineage. GO terms of downregulated genes belong to categories such as cell adhesion, collagen catabolic process, and immune system process (Figure S5D). Non-cardiac cell fate conversions did not occur at both early and late ciCPCs, while robust expression of cardiac lineage genes was restricted in late ciCPCs (Figure S5E), suggesting that late ciCPCs were distinct from early ciCPCs in terms of cardiac differentiation stage.
Figure 4.

Transcriptional signatures of ciCPCs
(A) Heatmap of 500 most variable genes expressed in TTFs, early ciCPCs, and late ciCPCs. The gene examples are shown with their functional annotations. (B and C) GO analyses of upregulated genes in early or late ciCPCs compared with TTFs. (D) Principal component (PC) analysis of the global gene profiling in our tested cell types and the previously reported CPCs. Directions of cell lineage conversion are depicted by the colored, dotted arrows in different studies. (E) Heatmap of gene expression related to the GO terms of interest across different studies. CF, cardiac fibroblast; iCPC, induced cardiovascular progenitor cell; MEF, mouse embryonic fibroblast; CD, cardiac derivative; ESC, embryonic stem cell. See also Figure S5.
Additionally, bioinformatics analysis was used to assess the differentiation stages of ciCPCs by comparing the RNA-seq dataset of mouse ESC-CPCs and previously reported iCPCs.4,41,42 RUVSeq was employed to remove unwanted variations from the RNA-seq data of different studies and methods,43 and the systematic shifts in normalized datasets were improved among the samples as shown in Figure S5F. Principal component analysis of the normalized datasets revealed that early ciCPCs had a transcription profile similar to that of cardiac progenitors, while late ciCPCs were more similar to differentiated CMs (Figure 4D). Pearson's correlation analysis also revealed that transcription profiles of early ciCPCs had a higher correlation with the reported ESC-CPCs and iCPCs compared with late ciCPCs (Figure S5G). We further evaluated a panel of well-studied genes involved in the cell cycle, heart development, and cardiac muscle contraction as identified by the GO term analysis in ciCPCs (Figure 4E). Cell cycle genes (such as Cdkn1b) were upregulated in early ciCPCs and other CPC samples but downregulated in late ciCPCs and differentiated CM samples. Heart development genes (such as Isl1) were moderately upregulated in various CPC samples or differentiated CM samples, but were highly expressed in early ciCPCs. Moreover, the genes of cardiac muscle contraction (such as Tnnt2) were specifically expressed in late ciCPCs and differentiated CM samples. Although heterogeneity was found in different datasets of gene expression, these results demonstrate that TTFs were reprogrammed to an intermediate proliferative progenitor stage (early ciCPCs) and further initiated cardiac lineage commitment (late ciCPCs), recapitulating heart development processes.
CRISPRa develops an autoregulatory loop targeting Gata4, Nkx2.5, and Tbx5
Next, we determined how GNT genes were regulated by CRISPRa during cell reprogramming. To assess whether activation of CPC genes was dependent upon the transgenic approach, a doxycycline (Dox)-inducible system was employed to produce on-demand gene expression. Activation of endogenous GNT was manipulated by the Dox-inducible CRISPRa system through targeting the gene promoters in TTFs (TTFiGNT) (Figure S6A). The potential off-target effect of GNT sgRNA sequences on gene expression was excluded (Figures S6B and S6C). The transgenic expression controlled by Dox was indicated by mCherry (Figure S6D). TTFiGNT were treated with Dox for 21 days and the Nkx2.5eGFP was gradually activated (Figure 5A). The percentage of Nkx2.5eGFP+ cells induced in TTFiGNT was retained for up to 35 days even after Dox withdrawal (Figures 5A and 5B). Furthermore, endogenous CPC genes were analyzed by qPCR. Nkx2.5, Gata4, and Tbx5 were upregulated in the TTFiGNT activated by Dox treatments compared with day 0, while other CPC genes such as Mesp1, Irx4, and MyoCD were upregulated after 14 days of induction (Figures 5C and S6E). These genes were activated at different levels.
Figure 5.

Formation of an autoregulatory feedback loop in ciCPCs
(A and B) Schematic of the period of Dox treatment. FACS and quantitation of eGFP+ cells in TTFs transduced with a Dox-inducible CRISPRa targeting GNT genes. (C) Expressions of endogenous GNT genes detected by qPCR after Dox treatment. (D) Schematic of DNA sites of GNT targeted by CRISPRa and other DNA binding assays. (E) Chromatin accessibility in the promoter region of GNT as analyzed by qPCR. (F) H3K4me3 modification near the promoters of GNT as analyzed by ChIP-qPCR. (G) ChIP-qPCR analysis of Nkx2.5 binding Nkx2.5 promoter. (H) ChIP-qPCR analysis of Gata4 binding Gata4 promoter. (I) ChIP-qPCR analysis of Tbx5 binding Tbx5 promoter. Data measures are presented as mean ± standard error. Versus day-0 control: ∗p < 0.05, ∗∗p < 0.01. See also Figure S6.
The dCas9VP64 protein was reported to open chromatin at inaccessible genomic loci;44 therefore, we assessed whether this epigenetic mechanism was involved in iCPC generation. The opening of the targeted locus (0.1–1 kbp distant from the sgRNA site) was analyzed by chromatin accessibility assay (Figure 5D). The accessibility of the GNT promoters in the TTFiGNT was transiently increased after 3-day Dox induction compared with the starting cells at day 0 (Figure 5E). Although there was a decreasing trend in DNA accessibility after Dox withdrawal, it was significantly higher in the day-35 cells than in the day-0 cells. There was likely a time window for dCas9VP64 influencing the accessibility of targeted chromatin, thereby allowing for settling and binding of epigenetic modifiers to effectively facilitate gene transcription. To test this possibility, an active chromatin mark (such as H3K4me3) related to cardiac reprogramming45 was analyzed by chromatin immunoprecipitation (ChIP)-qPCR (Figure 5F). H3K4me3 on the promoter regions of GNT was not changed within early induction, while it was significantly increased in TTFiGNT after Dox withdrawal compared with the starting cells (day 0). Stable, active epigenetic modifications are important for the accessibility of TFs to the promoter or enhancer regions.46 Although there was no difference within early induction, Gata4, Nkx2.5, and Tbx2.5 could bind to their own promoter regions in TTFiGNT after Dox withdrawal (Figures 5G–5I). These data indicated that an autoregulatory loop was established during the late reprogramming stage after depletion of initial induction factors.
Engrafted ciCPCs give rise to cardiovascular cells in the infarcted heart
Late ciCPCs undergoing tri-lineage commitment were injected into the infarcted heart of syngeneic mice to evaluate the regenerative efficacy, and TTFs were used as a control treatment of cell therapy. The lineage of ciCPCs was traced to determine whether engrafted cells contributed to heart regeneration after MI. Nkx2.5Cre/RosaRFP(tdTomato) transgenic mice were used to permanently label ciCPCs that were expressed with Nkx2.5 (Figure 6A); therefore RFP+ ciCPC colonies were generated from TTFs for cell transplantation. Additionally, the collected cells were expanded with the maintenance medium and characterized with cardiogenic gene expression before transplantation to minimize the potential batch effect of ciCPCs. Mice that constitutively expressed GFP were used as transplant recipients to assess the extent to which cell fusion occurred between the ciCPCs and the host cells. Heart sections from 2 to 28 days post MI showed that cell fusion events were rare and that there were no RFP+ ciCPCs that significantly coexpressed GFP (Figures 6B and 6C).
Figure 6.

Tracking the ciCPCs implanted in infarcted hearts
(A) Schematic representation of using Nkx2.5Cre/RosaRFP(tdTomato) TTF-derived ciCPCs to determine cell fusion in GFP reporter mice or identify cell differentiation in wild-type mice. IRES, internal ribosome entry site. Scale bars, 200 μm. (B and C): Representative imaging of RFP+ cell retention in GFP myocardium after injection. Scale bars, 100 μm. (D–F) Representative imaging of cTnT+ CMs, αSMA+ SMCs, or CD31+ ECs derived from RFP+ ciCPCs at 4 weeks post MI. White arrows indicate the integration of ciCPCs with the host's microvessels. Scale bars, 100 μm. (G) Overall differentiation efficiency of ciCPCs in infarcted hearts. See also Figure S7.
Furthermore, the differentiation potential of ciCPCs was assessed by immunostaining of individual cardiac markers in wild-type mice. Cells coexpressing the striated CM marker (cTnT) and RFP were identified in the border zone of infarcted myocardium that possessed viable CMs (Figure 6D). Quantification of immunostaining indicated that ∼36% of engrafted RFP+ ciCPCs differentiated into cTnT+ CMs, while TTFs failed to initiate transdifferentiation (Figures S7A and S7B). The vasculogenic potential of ciCPCs was also assessed by the regeneration of vascular ECs and SMCs. Most of ciCPC-derived αSMA+ SMCs and CD31+ ECs were integrated with microvessels in the infarcted area of MI mice (Figures 6E and 6F). The quantification of immunostaining indicated that ∼24% and ∼39% of engrafted RFP+ ciCPCs differentiated into SMCs and ECs, respectively (Figure 6G). Although control TTFs can convert into αSMA+ myofibroblasts after implantation, they were scattered in the infarcted area and did not incorporate into the host's vascular vessels (Figures S7A and B). We attempted to perform in vivo imaging to analyze the survival of the implanted cells labeled with RFP, but the fluorescence intensity was low or did not pass the detection threshold (<104 RFP+ cells) (Figures S7C and S7D). Therefore, it was estimated that few cells (<1%) were retained in hearts after injection for 1 week. These findings indicated that ciCPCs can give rise to new cardiovascular cells under in vivo environmental conditions despite low cell retention.
Delivery of ciCPCs enhances the functional restoration of the infarcted heart
To determine the therapeutic efficacy of ciCPCs, the heart function of MI mice was assessed by echocardiography after surgery. Adverse remodeling such as left ventricular dilation was reduced in mice that received ciCPCs compared with phosphate-buffered saline (PBS) or TTF treatment at 4 weeks post MI (Figure 7A). Progressive deterioration of heart function in MI mice was prevented by injection of ciCPCs after 2 weeks (Figure S8A). Heart functional improvement by ciCPC treatment was indicated by decreasing left ventricular end-diastolic or end-systolic diameters and increasing ejection fraction (EF) and fractional shortening (FS) in comparison with PBS or TTF treatment at 4 weeks post MI (Figure 7B). Furthermore, interstitial collagen staining showed that scar formation in MI mice was improved by ciCPC treatment compared with PBS or TTF treatment (Figures 7C and 7D). Microscopic analysis by Masson's trichrome staining showed that the infarct size of MI mice injected with ciCPCs was smaller than that of the PBS or TTF group (Figures 7E and 7F). There was no tumor growth in the athymic nude mouse hearts injected with ciCPCs during a 12-week observation (Figure 7G). Unlike ESCs, injection of ciCPCs did not induce teratoma formation in the skin of nude mice (Figure 7H). Moreover, no arrhythmia was found in the mice transplanted with ciCPCs that were electrically coupled with native myocardium, similar to the sham group (Figure S8B).
Figure 7.

Functional assays of infarcted heart in response to cell therapy
(A) Representative imaging of M-mode echocardiography at 4 weeks after surgery. (B) Quantification of left ventricular end-systolic dimension (LVDs), left ventricular end-diastolic dimension (LVDd), EF, and fractional shortening (FS) at 4 weeks post MI (n = 8 per group). (C and D) Collagen staining of heart tissues at 4 weeks post MI and quantification of fibrotic size (n = 5 per group). Scale bars, 1 mm. (E and F) Masson's trichrome staining of heart tissues and quantification of infarct size at 4 weeks post MI (n = 5 per group). Scale bars, 1 mm. (G) Exposure of a nude mouse heart injected with ciCPCs for 12 weeks. (H) Teratoma-forming ability of ESCs or iCPCs for 12 weeks after subcutaneous injection in nude mice. Data measures are presented as mean ± standard error. ns, no significance; ∗p < 0.05, ∗∗p < 0.01. See also Figure S8.
Discussion
Reprogramming of non-myocytes or fibroblasts into developmental CPCs with self-renewal and cardiac tri-potency offers a promising paradigm for heart regeneration. In this study, we identified promoter regions for initiating cell conversion toward cardiogenic lineage using CRISPRa approaches. The multiplex targeting promoters of GNT genes enabled reprogramming of fibroblasts into a precursor stage that underwent active cell cycle and heart development pathways. The induced cells in the late stage of reprogramming can be expanded to a large scale and differentiate into functional cardiovascular cells. Importantly, ciCPCs integrated and generated new cardiovascular cells after transplantation into an infarcted heart, leading to cardiac functional improvement.
The CRISPRa system can serve as an alternative approach for cellular reprogramming. Most fibroblasts were robustly expressed with the Nkx2.5eGFP by targeting the promoter regions of cardiac TFs, suggesting that multiplex gene activation was sufficient to initiate CPC formation. Our simplified scheme of sgRNA screening indicated that the promoter region close to TSS of the Nkx2.5 gene was a pivotal locus for inducing CPC formation of fibroblasts. Approximately 4–10 cell spheres were formed per 1 × 105 starting cells in TTFs induced by the CRISPRa system, which was comparable with the reprogramming efficiency (4–6 colonies per 50,000 starting cells) of adult cardiac fibroblasts that were reported using a transgenic approach.4 Our study and others indicated that a single gene activation cannot sufficiently induce cell reprogramming or CPC colony formation. Therefore, the cooperation of multiple genes plays a critical role in the cell reprogramming process, and further investigation is needed. The progenitor phenotype also was determined by the formation of various cell populations such as Flk1-, Ssea1-, and Cxcr4-positive subtypes. This finding indicated heterogeneity in the reprogrammed cells, and further characterization at single-cell resolution is necessary to determine whether they resemble native CPC populations such as the first and second heart fields.
A hallmark of progenitor cells is their proliferative capacity for self-renewal before differentiation. Similar to the reported PSC-derived CPC populations,47 progenitor markers such as Nkx2.5eGFP of ciCPCs waned during subculture, concomitant with increasing differentiation. The two key properties including self-renewal and differentiation of CPCs were coordinated by developmental signaling such as BMP/JNK.48 Therefore, the in vitro culture system of CPCs for long-term expansion requires further refinement by mimicking the developmental environments, such as using feeder cells, high-level serum, growth factors, small molecules, and genetic manipulation.30,34,36,49 Chemically defined conditions are ultimately more attractive for the establishment of a GMP-grade cell culture system for further clinical applications. We found that the addition of CC containing LIF, bFGF, SB431542, and Chir99021 allowed the expansion of cycling ciCPCs with stable characteristics to a large scale. However, the self-renewal proliferation of ciCPCs was not pronounced by the IGF1 supplement. As shown by the transcriptomic analysis, the CRISPRa system (targeting GNT) upregulated Isl1, which is known to activate insulin gene promoter and likely formed negative feedback in response to an excessive IGF1 stimulus,50 although this possibility remains to be determined. Importantly, the issue of low production efficiency can be addressed by the expansion of reprogrammed iCPCs with a robust proliferative capability. Avoiding activation of cell senescence signals also may contribute to the reprogramming efficiency of fibroblasts, which can be influenced by age-associated inflammation through inflammatory cytokines.51 Interestingly, antioxidant supplements that are extensively used to prevent cellular oxidative stress may maintain an undifferentiated state by repressing the differentiation of cardiac stem cells.52,53 The molecular mechanisms involved in inflammation and redox signaling are still poorly understood and merit further investigation to fine-tune existing CPC culture methods.
The tri-lineage differentiation potential of CPCs is critical for complete heart regeneration. Unlike adult cardiac stem cells that become ECs rather than CMs,54 fetal CPCs can spontaneously differentiate into the major heart cell types including CMs, SMCs, and ECs. Cardiac developmental genes such as αMHC and cTnT were expressed in early and late ciCPCs, suggesting an onset of cardiogenic potential during the early reprogramming stage. However, it remains inconclusive whether this potential was a leak from poorly maintained progenitor cells because promoter regions of these cardiac genes can be transiently targeted by the activation of TFs such as Gata4 and Tbx5 that were used in direct cardiac reprogramming.55 Although the route for iCPC reprogramming is distinct from that of direct cardiac reprogramming that does not pass through a progenitor fate,55 the conversion processes may share a common development pathway that needs further investigation. Notably, vascular genes were not expressed in early ciCPCs, which may have contributed to their lack of regulatory elements directly targeted by cardiac TFs. However, they were further activated in late ciCPCs, which may result from the procedure of cardiac sphere induction that mimics the developmental heart's vasculature and microenvironment.56 Indeed, sphere formation has been reported to enhance the differentiation potentials of cardiac progenitors through an autoparacrine loop.57
Regeneration of functional cardiac cells is a hallmark of CPCs. The ciCPC-derived CMs displayed the characteristic striations and exhibited contractions in a long-term coculture system. In contrast, iCPCs generated by transiently expressing pluripotency factors were shown to rapidly generate spontaneously contracting cells at day 3 of differentiation.41 In the present study, coupling with pre-existing CMs was required to trigger the contractility of ciCPC-CMs, consistent with the reported iCPC-CMs.4 Functional discrepancies among reprogrammed cells may result from the use of different reprogramming factors. For instance, over 90% of ciCPCs were Cxcr4 positive with a dim expression of Flk1 (which was consistent with the iCPCs generated by overexpression of cardiac TFs),4,58 while >80% of iCPCs generated by transiently expressing pluripotency factors were Flk1 positive.41 Therefore, a genome-wide screen such as using CRISPR tools is necessary to identify essential genes that control the cell fate of fibroblasts during the reprogramming process.59,60 Other experimental variations such as starting cell sources and culture conditions could contribute to the functional variability of CPCs induced by different reprogramming approaches. Embryonic or neonatal cardiac fibroblasts are considered to be more easily reprogrammed into cardiac lineage as compared with adult or aging cells.61 We found that extracardiac fibroblasts such as TTFs, LiFib, and LuFib can be reprogrammed into ciCPCs with similar efficiencies. Cardiogenic genes are expressed in a population of cardiac fibroblasts and may prime them to transdifferentiate with higher efficiency.62 Thus, reprogramming approaches remain to be refined using various cell models or sources before translational studies can be attempted.
The biological processes involved in the route for iCPC reprogramming were characterized by transcriptional profiling. Genes related to heart development were upregulated in both early and late ciCPCs, while cardiac differentiation genes were activated in the late ciCPCs. The early ciCPCs possessed high similarity with the reported iCPCs and ESC-CPCs, but the late ciCPCs were more similar to differentiated cardiac cells. These results suggested that ciCPCs were progressively derived via cardiac transdifferentiation of fibroblasts reactivating the gene pattern of heart development. The cycling genes such as Cdkn1b were highly expressed at the early ciCPCs but downregulated at the late ciCPCs when differentiation began. It is known that cardiac differentiation is controlled by the cell cycle pathways that maintain CPC self-renewal.63,64 Also, cell cycle acceleration promoted somatic cell fate transition into pluripotency through epigenetic remodeling.65 A study indicated that cell cycle exit was a prerequisite for successful fate conversion of fibroblasts toward CM lineage.66 Therefore, the cell cycle dynamics likely control the cell fate specification in iCPC reprogramming, although further investigation is needed to elucidate the underlying mechanism.
The establishment of positive feedback networks activating endogenous genes is essential to generate a new cellular identity after reprogramming. We found that activation of endogenous loci (such as Gata4, Nkx2.5, and Tbx5) by CRISPRa targeting of the promoter regions was sufficient to drive iCPC reprogramming in fibroblasts. Results of epigenetic studies suggested that the CRISPRa system can serve as a locus-specific activator to open the silenced chromatin locus that tightly represses cardiac gene expression in fibroblasts. Subsequently, the subsequent increasing level of transcripts and proteins can bind their own promoters and eventually create a positive feedback loop. Moreover, the interactions between various TFs, promoters, and enhancer elements can form larger regulatory circuits that would result in an explosive increase in gene activity.67 Therefore, CRISPRa can be harnessed to reveal the TF networks and provide significant insights to interpret the mechanism of cell lineage conversion. The importance of activating endogenous genes is also highlighted by other studies of CRISPRa. For instance, CRISPRa induced the precise epigenetic remodeling of endogenous loci including Sox2 and Oct4 for induced PSC (iPSC) generation, which was followed by the induction of other pluripotent genes and the formation of the pluripotency network.68 The CRISPRa system has also been shown to rapidly remodel the epigenetic state of the target loci, and induced sustained endogenous gene expression during neuronal cell reprogramming.69 In future studies, genome-wide assays (e.g., ChIP-seq, ATAC-seq, and chromosome conformation capture technologies)70 will be helpful to investigate epigenomics in the context of reprogramming cardiac genes.
Delivery of the late ciCPCs provides a cell reservoir to regenerate the damaged CMs and vascular cells while simultaneously creating an environment more conducive to proper healing of the infarcted heart. Speculation based on the in vitro differentiation studies would be that ciCPCs gradually differentiated into functional CMs after exposure to a heart microenvironment. The results of cardiac regeneration may be mixed by cell fusion.71 Although the engrafted ciCPCs integrated with host tissue, rare cell fusion was found between them after implantation. Indeed, new CMs were derived from ciCPCs in the infarct border zone as demonstrated by the Nkx2.5 tracing and identified by the formation of sarcomeric striations that are known to be a fundamental unit of CM contractility.72 The engrafted ciCPCs became vascular cells composed of SMCs and ECs that contribute to neovascularization and increasing blood supply and play an essential role in CM regeneration and cardiac repair.73 Interestingly, ciCPCs generated three cardiovascular lineages with roughly similar efficiencies, while the vascular SMCs and ECs accounted for a large proportion of ciCPC derivatives in vivo. It is known that stem cell fate is controlled by a combination of intrinsic mechanisms (such as cell-autonomous gene expression) and extrinsic environmental cues (non-cell-autonomous).74 Our transcriptomic analysis showed that ciCPCs expressed various genes related to cardiac muscle contraction or angiogenesis, demonstrating the involved intrinsic mechanisms. However, the hypoxic microenvironment enables the heart to produce numerous growth factors (such as vascular endothelial growth factor A [VEGFA] and angiopoietin 1)75 that make progenitors more prone to differentiate toward ECs or SMCs. Moreover, cell fate determination of embryonic progenitors into CMs, SMCs, or ECs is dependent on a relative proportion of Nkx2.5 and Isl1 expression,76 recapitulating a potential mechanism of how ciCPCs undergo a fate decision after implantation in a damaged heart. The complex interplay between donor cells and the host's microenvironment remains to be investigated.
Various intercellular interactions between the donor cells and host tissues also contribute to the mechanism of cell patterning in vivo.77 In addition, other cell therapy effects such as stimulating cardiac cell proliferation, endogenous angiogenesis, and antiapoptosis through paracrine mechanisms may be involved in the cellular mechanisms of action.78 Therefore, the incorporation of ciCPC-derived cardiovascular cells can salvage ischemic myocardium at the early stage of MI and minimize the adverse scar growth, ultimately improving heart function. Notably, the heart function deficits of MI mice were not completely resolved by cell injection, which was related to low cell retention and engraftment. Low cell survival remains a major challenge hindering the application of cell-based therapies for MI. Current advances in tissue engineering technologies potentially provide a more effective delivery approach that addresses this limitation with a new regeneration strategy employing CPCs.79,80
In summary, our results have demonstrated that endogenous gene activation by the CRISPRa complexes can induce reprogramming of mouse fibroblasts into CPCs. This approach bypasses pluripotency induction, thereby avoiding the risk of teratoma formation from immature iPSC/ESC derivatives. A positive feedback loop of autoregulatory cardiac TFs can be created by targeting the promoter accessibility in ciCPCs. Furthermore, ciCPCs are self-renewing and expandable, have cardiac tri-lineage potential, and show transcriptional profiling similar to that of embryonic CPCs. Therefore, the use of CRISPR tools serves to reveal the mechanism of cardiac TF loop formation and bring a new reprogramming strategy for cardiac regenerative therapy through precise targeting of endogenous genes.
Materials and methods
All research protocols conformed to the Guidelines for the Care and Use of Laboratory Animals published by the National Institutes of Health (National Academies Press, eighth edition, 2011). All animal use protocols and experiments conducted in this study were approved and oversighted by the University of Cincinnati Animal Care and Use Committee.
Mouse lines
Nkx2.5eGFP reporter mice were purchased from The Jackson Laboratory (stock no. 029489). Nkx2.5Cre/RosaRFP(tdTomato) mice were obtained by crossing Nkx2.5Cre mice (from The Jackson Laboratory [stock no. 024637]) and td-Tomato mice (from The Jackson Laboratory [stock no. 007914]). GFP mice for whole-body imaging were purchased from The Jackson Laboratory (stock no. 003291). Inbred C57BL/6 mice were used as the wild type. Athymic nude mice were purchased from The Jackson Laboratory (stock no. 002019). The transgenic mice were identified by standard PCR according to the manufacturer's protocols. Their health status was routinely checked to maintain consistency.
Isolation of mouse fibroblasts
Six-week old postnatal mice (Nkx2.5eGFP or Nkx2.5Cre/RosaRFP(tdTomato)) were euthanized and the heart, liver, lung, and tail-tip tissues were harvested for fibroblast isolation. The tissues were washed using PBS and minced into pieces nearly 1 mm3. Subsequently, the tissues were digested with 0.05% trypsin- EDTA (Thermo Fisher, cat. #25300062) for 1 h in a rotated incubator (12 rpm) at 37°C. Digestion medium was removed after brief centrifugation (200 rpm, 3 min), after which settled-down tissues were plated on 0.2% gelatin (Sigma-Aldrich, cat. #1.04078)-coated dishes with fibroblast growth medium consisting of high-glucose Dulbecco’s modified Eagle’s medium (DMEM; Thermo Fisher, cat. #10566016) supplemented with 15% FBS (Thermo Fisher, cat. #10082147), 1× non-essential acids (NEAA; Thermo Fisher, cat. #11140050), 1× GlutaMAX (Thermo Fisher; cat. #35050061), 1× β-mercaptoethanol (Thermo Fisher, cat. #21985023), 25 ng/mL bFGF (Sigma-Aldrich, cat. #F0291-25UG), 2 ng/mL EGF (Sigma-Aldrich, cat. #E9644-.5MG), and 1× penicillin (pen)/streptomycin (strep) (Thermo Fisher, cat. #15140122). The medium was changed every 2 days, and the tissues were cultured for 7–12 days until fibroblasts robustly migrated from the explants. Finally, fibroblasts were digested by 0.25% trypsin and harvested for cell passages by filtering with a 40-μm cell strainer (Corning Falcon, cat. #352098) to avoid tissue fragment contamination.
Vector construction
The sgRNA sequences were designed by using the GPP sgRNA Designer available at Broad Institute and Zhang Lab. Three sgRNAs were designed and synthesized for each gene, and the optimal sgRNA was selected after being confirmed by qPCR analysis. The sgRNA sequences used in the present study are listed in Table S2. Off-target was assessed by Cas-OFFinder81 and CCtop82 algorithms. Plasmids encoding sgRNAs, dCas9VP64, and MS2-P65-HSF1 were a gift from Feng Zhang (Addgene, cat. #73797, 61425, and 61426). To establish the Dox-inducible CRISPRa system, the dCas9VP64 was subcloned into the downstream of tetracycline (Tet) response element in a lentiviral vector (Takara, cat. #631844). Multiplexed sgRNAs were subcloned into one single vector (Addgene, cat. #85745) through the Golden Gate Assembly as previously described.83
Lentiviral transduction
HEK293T cells were obtained from the American Tissue Collection Center (ATCC; cat. #CRL-3216) and were maintained in high-glucose DMEM supplemented with 10% FBS and 1% pen/strep at 37°C with 5% CO2. One day before transfection, HEK293T cells were seeded on a 10-cm culture dish (Corning Falcon, cat. #353003) at 50% confluency. For each dish, 8 μg of psPAX2 plasmid (Addgene, cat. #12260), 3.5 μg of pMD2.G plasmid (Addgene, cat. #12259), and 8 μg of the plasmid containing the vector of interest were cotransfected into HEK293T cells using 30 μL of Lipofectamine 2000 (Thermo Fisher, cat. #11668019), according to manufacturer's instructions. Cells were cultured with the transfection medium overnight and replaced with 10 mL of fibroblast growth medium on the following day. After 48 h of transfection, the lentiviral supernatant was collected, filtered using 0.2-μm polyvinylidene fluoride syringe filters (Fisher Scientific, cat. #13-100-104), and concentrated using a polyethylene glycol virus precipitation kit (BioVision, cat. #K904), kept frozen at −80°C. Fibroblasts (2–5 passages) were resuspended at the density of 1 × 105 cells per well with 400 μL of fibroblast growth medium on a 24-well plate (Corning Falcon, cat. #353047) and supplemented with 100 μL of concentrated lentiviral supernatant (plus 8 μg/mL Polybrene [Sigma-Aldrich, cat. #TR-1003]). Cells were transduced with lentivirus via spinfection (centrifugation at 800 × g/min at 37°C for 2 h) and then incubated at 37°C with 5% CO2 for 48–72 h. Subsequently, the transduced cells were maintained with the relevant antibiotics based on the antibiotic resistance genes. The cells transduced with CRISPRa were stably subcultured with 10 μg/mL blasticidin (InvivoGen), 200 μg/mL hygromycin (Thermo Fisher, cat. #10687010), and 5 μg/mL puromycin (Thermo Fisher, cat. #A1113803) for 4 days.
Reprogramming of fibroblasts into CPCs
After gene transduction for 4 days, the fibroblasts (1 × 105 cells per well) were split and seeded on fibronectin (Sigma-Aldrich, cat. #11051407001)-coated 6-well plates with reprogramming medium (high-glucose DMEM supplemented with 10% FBS, 1% NEAA, 1% L-glutamine, 1% pen/strep, 10 ng/mL bFGF, and 103 units/mL LIF [Millipore, cat. #ESG1107]) at 37°C with 5% CO2. The culture medium was refreshed every 2 days until eGFP+ cell colonies were formed.
CPC expansion
On day 7 (after gene transduction), the cells were incubated with CPC expansion medium (high-glucose DMEM supplemented with 10% FBS, 1% NEAA, 1% L-glutamine, 1% pen/strep, 10 ng/mL bFGF, 3 μM CHIR99021 [Hello-bio, cat. #HB1261], 3 μM SB431542 [Sigma-Aldrich, cat. #616461], and 103 units/mL LIF). The culture medium was refreshed every 2 days. Cell colonies were split when ∼90% confluency occurred. The cells could be maintained for at least 20 passages.
In vitro differentiation of CRISPR-induced CPCs
A strategy for cell aggregate (spheroid formation) via suspension culture was devised to eliminate unreprogrammed fibroblasts and mimic the developmental heart's microenvironment. iCPC colonies were picked, dissociated, and suspended in low-attachment plates (Corning Costar, cat. #CLS3471) with the CPC expansion medium, allowing formation and growth of cell spheroids for 2–3 days. To enhance the differentiation, the cell spheroids were collected and seeded on 0.2% gelatin-coated dishes with various differentiation media. For CM differentiation, the attached cell spheroids were incubated with CM differentiation medium (high-glucose DMEM containing 1% NEAA, 1% L-glutamine, 1×β-mercaptoethanol, 1× N2 supplement [ThermoFisher, cat. #17502048], 1× B27 supplement [ThermoFisher, cat. #17504044], 0.1% BSA [Sigma-Aldrich, cat. #A9418], 12.5 ng/mL bFGF, 40 ng/mL BMP4 [R&D Systems, cat. #314-BP-010/CF], 10 ng/mL Activin A [R&D Systems, cat. #338-AC-010/CF], 15 ng/mL Wnt-3a [R&D Systems, cat. #5036-WN-010/CF], 5 μM IWP4 [REPROCELL Stemolecule, cat. #04-0036], 5 ng/mL VEGFA [R&D Systems, cat. #293-VE-010/CF], and 1% pen/strep). For SMC differentiation, the attached cell spheroids were incubated with SMC differentiation medium containing StemPro-34 medium, 20 ng/mL TGF-β1 (R&D Systems, cat. #240-B-002/CF), and 10 ng/mL PDGF-BB (R&D Systems, cat. #220-BB-010). For EC differentiation, the attached cell spheroids were incubated with EC differentiation medium containing StemPro-34 medium (Thermo Fisher, cat. #10639011) and 30 ng/mL VEGFA. For tri-lineage commitment, the attached cell spheroids were incubated with Tri-differentiation medium (StemPro-34 medium, 12.5 ng/mL bFGF, 5 ng/mL VEGFA, 20 ng/mL BMP4, 5 μM IWP4, and 10 ng/mL TGF-β1).
To generate more CMs with mature functions, ciCPCs were further induced with electrical coupling (IonOptix, C-Pace EP) through a coculture system with beating human induced PSC (hiPSC)-CMs as we previously described.84 The CytoSelect cell coculture system (Cell Biolabs, cat. #CBA-160) was devised by a 24-well plate containing a proprietary treated plastic insert in each well. CiCPCs were cultured with CM differentiation medium until they formed a monolayer around the insert, creating a defined 8-mm-diameter cell-free zone. Once the insert was removed, hiPSC-derived CMs (as previously reported)84 were seeded into the exposed zone. After coculture for 7 days, 200 μg/mL hygromycin to which ciCPCs were insensitive due to gene transduction was added into the medium for 7 days to eliminate the hiPSC-CMs. Finally, the beating cell clusters were analyzed by live-cell imaging and intracellular electrical recording.
Quantitative PCR
After CRISPRa transduction at different time points, total RNA was isolated using the Pure-link RNeasy Mini kit (Thermo Fisher, cat. #12183020), and reverse transcription was prepared using the Omniscript-RT kit (Qiagen, cat. #205113). Real-time PCR was performed on the Bio-Rad CFX96 system using the QuantiTect SYBR Green PCR kit (Qiagen, cat. #204145). Expression levels of the transcript were normalized to the averaged expression of the housekeeping gene (GAPDH) and expressed as fold changes. All PCR primer sequences are shown in Table S3.
Flow cytometry and FACS
Cells were harvested with 0.25% trypsin-EDTA digestion. The single-cell suspension was then fixed on ice with 4% paraformaldehyde (PFA; Thermo Fisher, cat. #AAJ19943K2) for 15 min, permeabilized, and blocked in block buffer consisting of 0.1% Triton X-100 and 5% BSA for 15 min on ice. Staining was done in PBS containing 1 mM EDTA (Thermo Fisher, cat. #AM9912) and 0.2% BSA; the antibodies and dilutions used are shown in Table S4 and followed the manufacturer's recommendation. GFP reporter activation was measured with the fluorescein isothiocyanate (FITC) channel in a blue laser (488 nm). Data collection was performed using a flow cytometer (Canto and LSRII, BD Bioscience) and analyzed via FlowJo v10 software. Living cell sorting was performed on a FACSAria III instrument (BD Bioscience).
Immunocytochemistry
Immunofluorescence staining was performed on cells seeded onto 0.2% gelatin-coated slices in an 8-well chamber (Thermo Fisher) or 35-mm tissue culture dish (Falcon). Cells were fixed with 4% PFA for 10 min and permeabilized in 0.2% Triton X-100 for 10 min. After treatment with 2% BSA blocking buffer for 30 min, samples were incubated with specific primary antibodies followed by their related secondary antibody. All antibodies used are listed in Table S4 and were diluted in PBS with 0.1% BSA. Following staining, samples were covered with VECTASHIELD Antifade Mounting Medium with DAPI (Vector Labs, cat. #H-1200-10), observed under a fluorescent microscope (Olympus).
Chromatin accessibility assay
TTFs were transduced with the Dox-inducible CRISPRa system targeting the GNT genes. Mouse-specific chromatin accessibility was analyzed by nuclease-dependent chromatin degradation via qPCR using EpiQuik chromatin accessibility assay kit (Epigentek, cat. #P-1047-48). Chromatin DNA was isolated from 1 × 106 cells and treated with or without a nuclease mix (Nse) following the manufacturer's instructions. DNA was purified with RNase I (Thermo Fisher Scientific, cat. #EN0601) and amplified using real-time PCR targeting specific promoter regions. The fold enrichment (FE) was calculated using a ratio of amplification efficiency of Nse-treated DNA sample over that of no-Nse control sample: FE = 2(Ct(Nse) − Ct(no-Nse)) × 100%. The primers used for chromatin accessibility sequences are listed in Table S3.
Chromatin immunoprecipitation-qPCR
TTFs were transduced with the Dox-inducible CRISPRa system targeting GNT genes. For each experimental condition, cells were expanded to 80% of confluency in a 15-cm culture dish. ChIP was performed using the CHIP-IT Express Enzymatic Kit (Active Motif, cat. #53009) precisely according to the manufacturer's instructions. After Dox (Sigma-Aldrich, cat. #1225984) treatment at different time points, the cells were fixed with 1% formaldehyde for 10 min and then treated with glycine stop-fix solution. Chromatin was subjected to enzymatic digestion for 10 min at 37°C and then stopped with EDTA. A total of 1 μg of anti-H3K4me3, Gata4, Nkx2.5, or Tbx5 antibodies were incubated with 3 μg of chromatin per reaction at 4°C overnight. The pull-down chromatin was isolated by using magnetic beads. One percent of starting chromatin is used as input. Furthermore, DNA fragments were recovered by using the ChIP DNA Purification Kit (Active Motif, cat. #58002). The column-purified DNA was subjected to qPCR using the protocol recommended for the SYBR Green from Bio-Rad (CFX96 system). The primers designed for ChIP-qPCR are listed in Table S3. The ChIP signals were measured by using the percent input method: %input = 1% × 2(Ct(input) − Ct(sample)).
RNA sequencing
To analyze the gene expression profile changes across the reprogramming process, we focused on the early-stage ciCPCs (day 7 after CRISPRa transduction), late-stage ciCPCs (day 22 after CRISPRa transduction and cell spheroid formation), and the original TTFs without CRISPRa. Using two biological replicate samples for each cell type, total RNA was extracted by using a Pure-link RNeasy Mini kit. Only the RNA samples that passed the quality control step with an RNA integrity number score ≥7.0 were used. Approximately 200 ng of total RNA for each sample was prepared for conventional sequencing libraries using the TruSeq RNA Library Prep Kit (Illumina, cat. #RS-122-2001) as per manufacturer's instructions, with mRNA enriched via poly-A-selection using oligoDT beads. RNA was then thermally fragmented with random priming, converted to cDNA, end repair, dA-tailing, adenylated for adapter ligation, and PCR amplified. The libraries were sequenced using the Illumina HiSeq 2500 platform with 100 nucleotide paired-end reads as per the manufacturer's instructions. The raw sequencing data can be accessible from the Gene Expression Omnibus (Database: GSE158084).
Bioinformatics analysis
Sequence reads were trimmed to remove possible adapter sequences and nucleotides with poor quality using Trimmomatic v.0.36. The trimmed reads were mapped to the Mus musculus GRCm38 reference genome available on ENSEMBL using the STAR aligner v.2.5.2b. The STAR aligner is a spliced aligner that detects splice junctions and incorporates them to help align the entire read sequences. Unique gene hit counts were calculated by using featureCounts from the Subread package v.1.5.2. The hit counts were summarized and reported using the gene_id feature in the annotation file. Unique reads that fell within exon regions were counted. After extraction of gene hit counts, the gene hit counts table was used for downstream differential expression analysis. Using DESeq2,85 a comparison of gene expression between the defined groups of samples was performed. The Wald test was used to generate p values and log2 fold changes. Genes with a p value of <0.05 and absolute log2 fold change of >1 were called differentially expressed genes for each comparison. The DAVID Functional Annotation Tool86 was used for GO analyses of gene set enrichment among different samples.
To compare our ciCPCs with the reported CPCs, gene expression data from Wamstad et al.,42 Zhang et al.,41 and Lalit et al.4 were obtained from the GEO dataset. All FASTQ files were input, and mapping of reads to reference transcripts was performed by using the Salmon package.87 Next, the tximport package was used to assemble estimated count and offset matrices for use with Bioconductor differential gene expression packages such as DESeq2.88 Furthermore, all samples were normalized using the RUVseq43 Bioconductor package in R. Genes that had no counts or only a single count across all samples were removed. From the remaining set of genes, coefficients of variation were computed for each gene across all samples. Genes that had an adjusted p value of >0.1 between samples were then chosen as control genes. As justified by the relative gene expression plots, five factors (k = 5) were supplied to the RUVg function to remove the unwanted variation associated with the lab of origin for each sample. The gene expression was shown using pheatmap package in R. The principal component analysis of all samples was performed using the plotPCA function in R, and the Pearson's correlation analysis was performed by using the corrr package in R.
Immunoblotting
Samples from various treatment groups were lysed with ice-cold lysis buffer with 1:100 protease inhibitor cocktail (Sigma-Aldrich, cat. #P8340) for 20 min on ice. The protein samples were centrifuged at 12,000 rpm at 4°C for 10 min, then resolved in 6× SDS-PAGE sample buffer and boiled for 10 min before loading on 10% polyacrylamide gels (Bio-Rad, cat. #4561033EDU) via electroblotting. Proteins were transferred to nitrocellulose membranes (Bio-Rad, cat. #1620112) that were blocked with 5% milk in Tris-buffered saline for 60 min. Membranes were probed with antibodies as described in Table S4. Membranes were visualized using an enhanced chemiluminescence system (Thermo Fisher Scientific, cat. #32106), exposed to X-ray film, and quantified by a laser scanner.
Ca2+ fluorescence imaging
ciCPC-CMs were seeded on 35-mm Petri dishes with a No. 1.5 cm coverglass window (MatTek) with CM growth medium for 14–28 days. Intracellular Ca+ was measured by loading with 1× Ca2+ sensitive probe Fluo-4 Direct reagent (Thermo Fisher Scientific, cat. #F10471) for 45 min at 37°C in a 5% CO2 incubator. The cell medium was then changed with a prewarmed CM differentiation medium plus 10% FBS. Finally, a Zeiss 780 confocal microscope was used for recording living cell imaging of the FITC-labeled calcium flux.
Intracellular electrical recording
Intracellular recording techniques were used to measure the spontaneous action potential of ciCPC-CMs at room temperature, as we previously described.89 The electrodes were pulled from borosilicate glass with filament (Sutter Instrument) using a P-97 Flaming/Brown Micropipette Puller (Sutter Instrument) and filled with prefiltered 2 M KCl. Pipette resistance ranged from 30 to 100 MΩ for impaling into the cell successfully. CM differentiation medium plus 10% FBS was used as the bath solution for action potential (AP) measurements. Ten percent FBS was added at least 30 min before measurement and the cells were incubated at 5% CO2 and 37°C until just before measurement. Signals were acquired at 10 kHz and filtered at 2 kHz with the Axopatch 200B amplifier via Axon Digidata 1550B digitizer hardware and pClamp11 software (Molecular Devices).
L-type Ca2+ current recording
The L-type Ca2+ current was recorded at room temperature using the whole-cell patch-clamp technique with the Axopatch 200B (Axon Instruments), as we previously described.90 Extracellular solution contained 140 mM NaCl, 2.7 mM KCl, 10 mM HEPES, and 10 mM EGTA (pH7.4). Glass pipettes were filled with solution containing 100 mM CsCl, 30 mM CsF, 4 mM ATP-Mg, 2 mM MgCl2, 10 mM HEPES, and 10 mM EGTA (pH 7.2 adjusted with CsOH). After the membrane was ruptured, cells were voltage-clamped at a holding potential of −70 mV, and inward currents were evoked by a 240-ms test pulse to +40 mV in 10-mV increments. After a further 30-min interval, the effects of 300 nM FPL64176 and 3 μM nifedipine (all from Sigma-Aldrich) were tested. Data collection and analysis were performed using pClamp 9.0 software (Axon Instruments).
Cell proliferation assay
The CellTiter 96 AQueous One Solution Cell Proliferation Assay (Promega, cat. #G3582) was used to determine the number of proliferative ciCPCs as per the manufacturer's instructions. Fifty microliters of the cell suspension (5,000 cells) was dispensed into all wells of a 96-well plate with the CPC expansion medium. After 3 days, assays were performed by adding a small amount of the CellTiter 96 AQueous One Solution Reagent (MTS) directly to culture wells, incubating for 4 h, and recording the absorbance at 490 nm with a 96-well plate reader. The quantity of formazan product as measured by the amount of 490-nm absorbance is directly proportional to the number of living cells in culture.
Tube formation assay
The In Vitro Angiogenesis Matrigel assay kit (Millipore, cat. #ECM625) was used to evaluate in vitro angiogenesis activity by observing tube formation of iCPC-EC following the manufacturer's protocol. Cells (1 × 105) suspended in 100 μL of endothelial basal medium (Lonza, cat. #CC-3121) were seeded on the solidified Matrigel at 37°C with 5% CO2. After further incubation for 10 h the tube-like structure could be observed, and bright-field images were captured under the microscope (Olympus) at 200× magnification.
Mouse MI model and cell transplantation
ciCPCs were generated by reprogramming the TTFs from Nkx2.5Cre/RosaRFP(tdTomato) mice. To generate RFP+ TTFs as control cells, TTFs were isolated from the td-Tomato mice and transduced with a Cre recombinase vector to activate RFP expression (Addgene #30205). MI models were induced by permanent ligation of the left anterior descending coronary (LAD) in 10-week-old female C57BL/6J mice or GFP mice, and the cell injection was performed according to our previous publication.91 In brief, mice were anesthetized by intraperitoneal administration of 0.1% ketamine and 0.02% xylene. The heart was exposed by left-sided minimal thoracotomy and the LAD ligated using 6.0 silk. After LAD ligation for 10 min, 30 μL of ciCPCs (1.5 × 106 cells), control TTFs (1.5 × 106 cells), or PBS were injected at three different areas along the boundary between the infarct and border zones.
In vivo fluorescence imaging
Fluorescence imaging was acquired by an IVIS Spectrum (PerkinElmer Health Sciences, USA). To determine the correlation between cell number and RFP signal, approximately 106, 105, 104, or 0 (blank) ciCPCs (derived from the TTFs of Nkx2.5Cre/RosaRFP(tdTomato) mice) per well were seeded on a 6-well plate for 12 h of culture. The ex vivo fluorescence signal was captured by epi-illumination. The fluorescence intensity was measured within a region of interest by the built-in Living Image 4.5.4 software according to the manufacturer's manual. Linear regression between cell number and RFP signal was analyzed. To monitor the in vivo survival of TTFs and ciCPCs, 1.5 × 106 cells were injected into the infarcted hearts of C57BL/6 mice for 1 week. Because the RNA signal was undetectable from a living animal's heart, the hearts (kidneys as negative control) were from various groups for ex vivo imaging after euthanasia. Images were captured by epi-illumination of the IVIS Spectrum.
Echocardiography
Cardiac function was analyzed with transthoracic echocardiography (VisualSonics Vevo 2100 Imaging System, with 15-MHz probe) after MI according to our previous publication.92 Mice were placed supine on an electrical heating pad at 37°C under a low dose of isoflurane anesthesia (usual maintenance level: 1.5% of isoflurane/98.5% of oxygen) for echo examination. Two-dimensional targeted M-mode traces were obtained in the position of perpendicular left ventricular (LV) anterior and posterior walls. LV internal diameter during diastole (LVDd) and LV internal diameter during systole (LVDs) were measured from M-mode recording. EF and FS values were calculated with the formula EF = (LVDd3 − LVDs3)/LVDd3 × 100% and FS = (LVDd − LVDs)/LVDd × 100%. The electrical signal was monitored by an electrocardiogram. All measurements were performed according to the American Society for Echocardiography leading-edge technique standards and measured in at least three consecutive cardiac cycles.
Immunohistochemistry
For cryosection, heart tissues were harvested, fixed in 4% PFA overnight at 4°C, and perfused with 30% sucrose for 1 day at 4°C. The fixed tissue was embedded in an OCT compound and then frozen by liquid nitrogen. The frozen tissue was sectioned at 8-μm thickness using a cryostat (−20°C) and collected by Polysine slides. The tissue sections were treated with 0.1% protein kinase A solution for 5 min and then with 0.1% Triton X-100 with serum blocking buffer for 10 min. After washing with PBS, the antibody solutions (such as cTnT, αSMA, and CD31) were added to the slides and incubated for 2 h at room temperature. After removing the primary antibody, the fluorescein-labeled secondary antibody was added and incubated for 1 h at room temperature. The autofluorescence of RFP and GFP in the frozen section was displayed under the microscope. Nuclei were stained with DAPI. For paraffin sections, heart tissues were harvested, fixed in 10% formalin, embedded in paraffin, and sectioned at 8-μm thickness according to our previous publication.92 After deparaffinization and rehydration, the sections were treated with citric acid/microwave antigen retrieval for 10 min. The tissue sections were treated with the serum blocking buffer for 10–30 min, after which the slides were incubated with antibodies (such as collagen I) for 2 h at room temperature. After removing the primary antibody, the fluorescein-labeled secondary antibody was added and incubated for 1 h at room temperature. Nuclei were stained with DAPI. The immunostaining was observed under the microscope and the positive vessel numbers per high-power field counted. All of the antibody information in this study is presented in Table S4. Additionally, Masson's trichrome staining was performed following the manufacturer's instructions before observation under the light microscope.
Teratoma formation
Mouse ESCs (ATCC, cat. #SCRC-1039) and ciCPCs were harvested at lower passage numbers (<10) for injection and transplanted into 12-week-old female nude mice at the gastrocnemius muscles and heart tissue, both of which are well suited for teratoma growth and surgical access. In brief, mice were anesthetized, and a total of 1 × 107 cells were prepared in a 50-μL serum-free DMEM medium and slowly injected into gastrocnemius intramuscularly. Likewise, mice were supported on a ventilator and their chests were opened aseptically to expose the heart, then 1.5 × 106 cells were suspended in 30 μL of serum-free DMEM medium and injected into the anterior wall of the left ventricle. The mice were then monitored for 12 weeks to evaluate whether teratoma was growing at the injection site (n = 5).
Statistical analysis
Statistical analysis was performed using GraphPad Prism 10 (GraphPad Software). Differences between two mean values were evaluated by an unpaired Student's t test, while the data of multiple groups were tested for statistical significance using one-way ANOVA followed by Bonferroni’s post hoc analysis. All graphic data were presented as mean ± standard error. p values of <0.05 were considered statistically significant.
Acknowledgments
This work was supported by the American Heart Association 18CDA34110223 (J.L.), 20CDA35310176 (W.H.), and NIH grants including R01HL136025, R01HL143490, and R01HL157456 (Y.W.). We would like to acknowledge the assistance of the Research Flow Cytometry Core in the Division of Rheumatology at Cincinnati Children's Hospital Medical Center, and thank Xiangning Wang (University of Cincinnati) for in vivo fluorescence imaging assistance.
Author contributions
L.J. and J.L.: conception and design, performed experiments, data collection and assembly, analysis and interpretation, and manuscript writing. W.H. and Z.W.: performed animal surgery, cell injection, and echocardiographic data collection. J.M., K.H.P., P.C., and W.C.: performed whole-cell patch-clamp, calcium imaging, and flow cytometer data collection. C.P.: confocal microscopy data collection and manuscript editing. H.Z., J.-J.M., L.N., G.-C.F., H.-S.W., O.K., and M.X.: comments, discussion, and manuscript editing. Y.W.: conception, experiment design, and manuscript writing.
Declaration of interests
The authors declare no competing interests.
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.ymthe.2021.10.015.
Contributor Information
Jialiang Liang, Email: liangjl@ucmail.uc.edu.
Yigang Wang, Email: yi-gang.wang@uc.edu.
Supplemental information
References
- 1.Menasche P., Vanneaux V., Hagege A., Bel A., Cholley B., Cacciapuoti I., Parouchev A., Benhamouda N., Tachdjian G., Tosca L., et al. Human embryonic stem cell-derived cardiac progenitors for severe heart failure treatment: first clinical case report. Eur. Heart J. 2015;36:2011–2017. doi: 10.1093/eurheartj/ehv189. [DOI] [PubMed] [Google Scholar]
- 2.Menasche P., Vanneaux V., Fabreguettes J.R., Bel A., Tosca L., Garcia S., Bellamy V., Farouz Y., Pouly J., Damour O., et al. Towards a clinical use of human embryonic stem cell-derived cardiac progenitors: a translational experience. Eur. Heart J. 2015;36:743–750. doi: 10.1093/eurheartj/ehu192. [DOI] [PubMed] [Google Scholar]
- 3.Islas J.F., Liu Y., Weng K.C., Robertson M.J., Zhang S., Prejusa A., Harger J., Tikhomirova D., Chopra M., Iyer D., et al. Transcription factors ETS2 and MESP1 transdifferentiate human dermal fibroblasts into cardiac progenitors. Proc. Natl. Acad. Sci. U S A. 2012;109:13016–13021. doi: 10.1073/pnas.1120299109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Lalit P.A., Salick M.R., Nelson D.O., Squirrell J.M., Shafer C.M., Patel N.G., Saeed I., Schmuck E.G., Markandeya Y.S., Wong R., et al. Lineage reprogramming of fibroblasts into proliferative induced cardiac progenitor cells by defined factors. Cell Stem Cell. 2016;18:354–367. doi: 10.1016/j.stem.2015.12.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Li X.H., Li Q., Jiang L., Deng C., Liu Z., Fu Y., Zhang M., Tan H., Feng Y., Shan Z., et al. Generation of functional human cardiac progenitor cells by high-efficiency protein transduction. Stem Cells Transl. Med. 2015;4:1415–1424. doi: 10.5966/sctm.2015-0136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Paige S.L., Plonowska K., Xu A., Wu S.M. Molecular regulation of cardiomyocyte differentiation. Circ. Res. 2015;116:341–353. doi: 10.1161/CIRCRESAHA.116.302752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Olson E.N. Gene regulatory networks in the evolution and development of the heart. Science. 2006;313:1922–1927. doi: 10.1126/science.1132292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Hanna J., Saha K., Pando B., van Zon J., Lengner C.J., Creyghton M.P., van Oudenaarden A., Jaenisch R. Direct cell reprogramming is a stochastic process amenable to acceleration. Nature. 2009;462:595–601. doi: 10.1038/nature08592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Vierbuchen T., Wernig M. Molecular roadblocks for cellular reprogramming. Mol. Cell. 2012;47:827–838. doi: 10.1016/j.molcel.2012.09.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Warlich E., Kuehle J., Cantz T., Brugman M.H., Maetzig T., Galla M., Filipczyk A.A., Halle S., Klump H., Scholer H.R., et al. Lentiviral vector design and imaging approaches to visualize the early stages of cellular reprogramming. Mol. Ther. 2011;19:782–789. doi: 10.1038/mt.2010.314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Boyer L.A., Lee T.I., Cole M.F., Johnstone S.E., Levine S.S., Zucker J.P., Guenther M.G., Kumar R.M., Murray H.L., Jenner R.G., et al. Core transcriptional regulatory circuitry in human embryonic stem cells. Cell. 2005;122:947–956. doi: 10.1016/j.cell.2005.08.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Zhang H., Jiao W., Sun L., Fan J., Chen M., Wang H., Xu X., Shen A., Li T., Niu B., et al. Intrachromosomal looping is required for activation of endogenous pluripotency genes during reprogramming. Cell Stem Cell. 2013;13:30–35. doi: 10.1016/j.stem.2013.05.012. [DOI] [PubMed] [Google Scholar]
- 13.He A., Kong S.W., Ma Q., Pu W.T. Co-occupancy by multiple cardiac transcription factors identifies transcriptional enhancers active in heart. Proc. Natl. Acad. Sci. U S A. 2011;108:5632–5637. doi: 10.1073/pnas.1016959108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Prall O.W., Menon M.K., Solloway M.J., Watanabe Y., Zaffran S., Bajolle F., Biben C., McBride J.J., Robertson B.R., Chaulet H., et al. An Nkx2-5/Bmp2/Smad1 negative feedback loop controls heart progenitor specification and proliferation. Cell. 2007;128:947–959. doi: 10.1016/j.cell.2007.01.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Zhou Y., Kim J., Yuan X., Braun T. Epigenetic modifications of stem cells: a paradigm for the control of cardiac progenitor cells. Circ. Res. 2011;109:1067–1081. doi: 10.1161/CIRCRESAHA.111.243709. [DOI] [PubMed] [Google Scholar]
- 16.Konermann S., Brigham M.D., Trevino A.E., Joung J., Abudayyeh O.O., Barcena C., Hsu P.D., Habib N., Gootenberg J.S., Nishimasu H., et al. Genome-scale transcriptional activation by an engineered CRISPR-Cas9 complex. Nature. 2015;517:583–588. doi: 10.1038/nature14136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Dominguez A.A., Lim W.A., Qi L.S. Beyond editing: repurposing CRISPR-Cas9 for precision genome regulation and interrogation. Nat. Rev. Mol. Cell Biol. 2016;17:5–15. doi: 10.1038/nrm.2015.2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Gilbert L.A., Horlbeck M.A., Adamson B., Villalta J.E., Chen Y., Whitehead E.H., Guimaraes C., Panning B., Ploegh H.L., Bassik M.C., et al. Genome-scale CRISPR-mediated control of gene repression and activation. Cell. 2014;159:647–661. doi: 10.1016/j.cell.2014.09.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hsu P.D., Scott D.A., Weinstein J.A., Ran F.A., Konermann S., Agarwala V., Li Y., Fine E.J., Wu X., Shalem O., et al. DNA targeting specificity of RNA-guided Cas9 nucleases. Nat. Biotechnol. 2013;31:827–832. doi: 10.1038/nbt.2647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Sander J.D., Joung J.K. CRISPR-Cas systems for editing, regulating and targeting genomes. Nat. Biotechnol. 2014;32:347–355. doi: 10.1038/nbt.2842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Balboa D., Weltner J., Eurola S., Trokovic R., Wartiovaara K., Otonkoski T. Conditionally stabilized dCas9 activator for controlling gene expression in human cell reprogramming and differentiation. Stem Cell Reports. 2015;5:448–459. doi: 10.1016/j.stemcr.2015.08.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Gilbert L.A., Larson M.H., Morsut L., Liu Z., Brar G.A., Torres S.E., Stern-Ginossar N., Brandman O., Whitehead E.H., Doudna J.A., et al. CRISPR-mediated modular RNA-guided regulation of transcription in eukaryotes. Cell. 2013;154:442–451. doi: 10.1016/j.cell.2013.06.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Mandegar M.A., Huebsch N., Frolov E.B., Shin E., Truong A., Olvera M.P., Chan A.H., Miyaoka Y., Holmes K., Spencer C.I., et al. CRISPR interference efficiently induces specific and reversible gene silencing in human iPSCs. Cell Stem Cell. 2016;18:541–553. doi: 10.1016/j.stem.2016.01.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Wang J., Jiang X., Zhao L., Zuo S., Chen X., Zhang L., Lin Z., Zhao X., Qin Y., Zhou X., et al. Lineage reprogramming of fibroblasts into induced cardiac progenitor cells by CRISPR/Cas9-based transcriptional activators. Acta Pharm. Sin. B. 2020;10:313–326. doi: 10.1016/j.apsb.2019.09.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Smith Z.D., Sindhu C., Meissner A. Molecular features of cellular reprogramming and development. Nat. Rev. Mol. Cell Biol. 2016;17:139–154. doi: 10.1038/nrm.2016.6. [DOI] [PubMed] [Google Scholar]
- 26.Buganim Y., Faddah D.A., Jaenisch R. Mechanisms and models of somatic cell reprogramming. Nat. Rev. Genet. 2013;14:427–439. doi: 10.1038/nrg3473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Wu S.M., Fujiwara Y., Cibulsky S.M., Clapham D.E., Lien C.L., Schultheiss T.M., Orkin S.H. Developmental origin of a bipotential myocardial and smooth muscle cell precursor in the mammalian heart. Cell. 2006;127:1137–1150. doi: 10.1016/j.cell.2006.10.028. [DOI] [PubMed] [Google Scholar]
- 28.Takeuchi J.K., Bruneau B.G. Directed transdifferentiation of mouse mesoderm to heart tissue by defined factors. Nature. 2009;459:708–711. doi: 10.1038/nature08039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kattman S.J., Huber T.L., Keller G.M. Multipotent flk-1+ cardiovascular progenitor cells give rise to the cardiomyocyte, endothelial, and vascular smooth muscle lineages. Dev. Cell. 2006;11:723–732. doi: 10.1016/j.devcel.2006.10.002. [DOI] [PubMed] [Google Scholar]
- 30.Christoforou N., Miller R.A., Hill C.M., Jie C.C., McCallion A.S., Gearhart J.D. Mouse ES cell-derived cardiac precursor cells are multipotent and facilitate identification of novel cardiac genes. J. Clin. Invest. 2008;118:894–903. doi: 10.1172/JCI33942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Andersen P., Tampakakis E., Jimenez D.V., Kannan S., Miyamoto M., Shin H.K., Saberi A., Murphy S., Sulistio E., Chelko S.P., et al. Precardiac organoids form two heart fields via Bmp/Wnt signaling. Nat. Commun. 2018;9:3140. doi: 10.1038/s41467-018-05604-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Schoenfelder S., Fraser P. Long-range enhancer-promoter contacts in gene expression control. Nat. Rev. Genet. 2019;20:437–455. doi: 10.1038/s41576-019-0128-0. [DOI] [PubMed] [Google Scholar]
- 33.Engler A.J., Sen S., Sweeney H.L., Discher D.E. Matrix elasticity directs stem cell lineage specification. Cell. 2006;126:677–689. doi: 10.1016/j.cell.2006.06.044. [DOI] [PubMed] [Google Scholar]
- 34.Cohen E.D., Wang Z., Lepore J.J., Lu M.M., Taketo M.M., Epstein D.J., Morrisey E.E. Wnt/beta-catenin signaling promotes expansion of Isl-1-positive cardiac progenitor cells through regulation of FGF signaling. J. Clin. Invest. 2007;117:1794–1804. doi: 10.1172/JCI31731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Bao X., Lian X., Hacker T.A., Schmuck E.G., Qian T., Bhute V.J., Han T., Shi M., Drowley L., Plowright A., et al. Long-term self-renewing human epicardial cells generated from pluripotent stem cells under defined xeno-free conditions. Nat. Biomed. Eng. 2016;1:0003. doi: 10.1038/s41551-016-0003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Birket M.J., Ribeiro M.C., Verkerk A.O., Ward D., Leitoguinho A.R., den Hartogh S.C., Orlova V.V., Devalla H.D., Schwach V., Bellin M., et al. Expansion and patterning of cardiovascular progenitors derived from human pluripotent stem cells. Nat. Biotechnol. 2015;33:970–979. doi: 10.1038/nbt.3271. [DOI] [PubMed] [Google Scholar]
- 37.Beausejour C.M., Krtolica A., Galimi F., Narita M., Lowe S.W., Yaswen P., Campisi J. Reversal of human cellular senescence: roles of the p53 and p16 pathways. EMBO J. 2003;22:4212–4222. doi: 10.1093/emboj/cdg417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Lee S., Park C., Han J.W., Kim J.Y., Cho K., Kim E.J., Kim S., Lee S.J., Oh S.Y., Tanaka Y., et al. Direct reprogramming of human dermal fibroblasts into endothelial cells using ER71/ETV2. Circ. Res. 2017;120:848–861. doi: 10.1161/CIRCRESAHA.116.309833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Yoon C.H., Hur J., Park K.W., Kim J.H., Lee C.S., Oh I.Y., Kim T.Y., Cho H.J., Kang H.J., Chae I.H., et al. Synergistic neovascularization by mixed transplantation of early endothelial progenitor cells and late outgrowth endothelial cells: the role of angiogenic cytokines and matrix metalloproteinases. Circulation. 2005;112:1618–1627. doi: 10.1161/CIRCULATIONAHA.104.503433. [DOI] [PubMed] [Google Scholar]
- 40.Ross J.J., Hong Z., Willenbring B., Zeng L., Isenberg B., Lee E.H., Reyes M., Keirstead S.A., Weir E.K., Tranquillo R.T., et al. Cytokine-induced differentiation of multipotent adult progenitor cells into functional smooth muscle cells. J. Clin. Invest. 2006;116:3139–3149. doi: 10.1172/JCI28184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Zhang Y., Cao N., Huang Y., Spencer C.I., Fu J.D., Yu C., Liu K., Nie B., Xu T., Li K., et al. Expandable cardiovascular progenitor cells reprogrammed from fibroblasts. Cell Stem Cell. 2016;18:368–381. doi: 10.1016/j.stem.2016.02.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Wamstad J.A., Alexander J.M., Truty R.M., Shrikumar A., Li F., Eilertson K.E., Ding H., Wylie J.N., Pico A.R., Capra J.A., et al. Dynamic and coordinated epigenetic regulation of developmental transitions in the cardiac lineage. Cell. 2012;151:206–220. doi: 10.1016/j.cell.2012.07.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Risso D., Ngai J., Speed T.P., Dudoit S. Normalization of RNA-seq data using factor analysis of control genes or samples. Nat. Biotechnol. 2014;32:896–902. doi: 10.1038/nbt.2931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Barkal A.A., Srinivasan S., Hashimoto T., Gifford D.K., Sherwood R.I. Cas9 functionally opens chromatin. PLoS One. 2016;11:e0152683. doi: 10.1371/journal.pone.0152683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Liu Z., Chen O., Zheng M., Wang L., Zhou Y., Yin C., Liu J., Qian L. Re-patterning of H3K27me3, H3K4me3 and DNA methylation during fibroblast conversion into induced cardiomyocytes. Stem Cell Res. 2016;16:507–518. doi: 10.1016/j.scr.2016.02.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Liu L., Jin G., Zhou X. Modeling the relationship of epigenetic modifications to transcription factor binding. Nucleic Acids Res. 2015;43:3873–3885. doi: 10.1093/nar/gkv255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Domian I.J., Chiravuri M., van der Meer P., Feinberg A.W., Shi X., Shao Y., Wu S.M., Parker K.K., Chien K.R. Generation of functional ventricular heart muscle from mouse ventricular progenitor cells. Science. 2009;326:426–429. doi: 10.1126/science.1177350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Bylund J.B., Trinh L.T., Awgulewitsch C.P., Paik D.T., Jetter C., Jha R., Zhang J., Nolan K., Xu C., Thompson T.B., et al. Coordinated proliferation and differentiation of human-induced pluripotent stem cell-derived cardiac progenitor cells depend on bone morphogenetic protein signaling regulation by GREMLIN 2. Stem Cells Dev. 2017;26:678–693. doi: 10.1089/scd.2016.0226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Blin G., Nury D., Stefanovic S., Neri T., Guillevic O., Brinon B., Bellamy V., Rucker-Martin C., Barbry P., Bel A., et al. A purified population of multipotent cardiovascular progenitors derived from primate pluripotent stem cells engrafts in postmyocardial infarcted nonhuman primates. J. Clin. Invest. 2010;120:1125–1139. doi: 10.1172/JCI40120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Zhang H., Wang W.P., Guo T., Yang J.C., Chen P., Ma K.T., Guan Y.F., Zhou C.Y. The LIM-homeodomain protein ISL1 activates insulin gene promoter directly through synergy with BETA2. J. Mol. Biol. 2009;392:566–577. doi: 10.1016/j.jmb.2009.07.036. [DOI] [PubMed] [Google Scholar]
- 51.Mahmoudi S., Mancini E., Xu L., Moore A., Jahanbani F., Hebestreit K., Srinivasan R., Li X., Devarajan K., Prélot L., et al. Heterogeneity in old fibroblasts is linked to variability in reprogramming and wound healing. Nature. 2019;574:553–558. doi: 10.1038/s41586-019-1658-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Ji J., Sharma V., Qi S., Guarch M.E., Zhao P., Luo Z., Fan W., Wang Y., Mbabaali F., Neculai D., et al. Antioxidant supplementation reduces genomic aberrations in human induced pluripotent stem cells. Stem Cell Rep. 2014;2:44–51. doi: 10.1016/j.stemcr.2013.11.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Tu C., Allen A., Deng W., Conroy O., Nambiar M., Zoldan J. Commonly used thiol-containing antioxidants reduce cardiac differentiation and alter gene expression ratios of sarcomeric isoforms. Exp. Cell Res. 2018;370:150–159. doi: 10.1016/j.yexcr.2018.06.017. [DOI] [PubMed] [Google Scholar]
- 54.Lee R.T. Adult cardiac stem cell concept and the process of science. Circulation. 2018;138:2940–2942. doi: 10.1161/CIRCULATIONAHA.118.036407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Ieda M., Fu J.D., Delgado-Olguin P., Vedantham V., Hayashi Y., Bruneau B.G., Srivastava D. Direct reprogramming of fibroblasts into functional cardiomyocytes by defined factors. Cell. 2010;142:375–386. doi: 10.1016/j.cell.2010.07.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Li T.S., Cheng K., Lee S.T., Matsushita S., Davis D., Malliaras K., Zhang Y., Matsushita N., Smith R.R., Marban E. Cardiospheres recapitulate a niche-like microenvironment rich in stemness and cell-matrix interactions, rationalizing their enhanced functional potency for myocardial repair. Stem Cells. 2010;28:2088–2098. doi: 10.1002/stem.532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Cho H.J., Lee H.J., Youn S.W., Koh S.J., Won J.Y., Chung Y.J., Cho H.J., Yoon C.H., Lee S.W., Lee E.J., et al. Secondary sphere formation enhances the functionality of cardiac progenitor cells. Mol. Ther. 2012;20:1750–1766. doi: 10.1038/mt.2012.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Alexanian R.A., Mahapatra K., Lang D., Vaidyanathan R., Markandeya Y.S., Gill R.K., Zhai A.J., Dhillon A., Lea M.R., Abozeid S., et al. Induced cardiac progenitor cells repopulate decellularized mouse heart scaffolds and differentiate to generate cardiac tissue. Biochim. Biophys. Acta Mol. Cell Res. 2020;1867:118559. doi: 10.1016/j.bbamcr.2019.118559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Yu J.S.L., Palano G., Lim C., Moggio A., Drowley L., Plowright A.T., Bohlooly Y.M., Rosen B.S., Hansson E.M., Wang Q.D., et al. CRISPR-knockout screen identifies Dmap1 as a regulator of chemically induced reprogramming and differentiation of cardiac progenitors. Stem Cells. 2019;37:958–972. doi: 10.1002/stem.3012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Chong Z.S., Wright G.J., Sharma S. Investigating cellular recognition using CRISPR/Cas9 genetic screening. Trends Cell Biol. 2020;30:619–627. doi: 10.1016/j.tcb.2020.05.005. [DOI] [PubMed] [Google Scholar]
- 61.Srivastava D., Ieda M. Critical factors for cardiac reprogramming. Circ. Res. 2012;111:5–8. doi: 10.1161/CIRCRESAHA.112.271452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Furtado M.B., Costa M.W., Pranoto E.A., Salimova E., Pinto A.R., Lam N.T., Park A., Snider P., Chandran A., Harvey R.P., et al. Cardiogenic genes expressed in cardiac fibroblasts contribute to heart development and repair. Circ. Res. 2014;114:1422–1434. doi: 10.1161/CIRCRESAHA.114.302530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.He S., Nakada D., Morrison S.J. Mechanisms of stem cell self-renewal. Annu. Rev. Cell Dev. Biol. 2009;25:377–406. doi: 10.1146/annurev.cellbio.042308.113248. [DOI] [PubMed] [Google Scholar]
- 64.Ahuja P., Sdek P., MacLellan W.R. Cardiac myocyte cell cycle control in development, disease, and regeneration. Physiol. Rev. 2007;87:521–544. doi: 10.1152/physrev.00032.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Guo S., Zi X., Schulz V.P., Cheng J., Zhong M., Koochaki S.H., Megyola C.M., Pan X., Heydari K., Weissman S.M., et al. Nonstochastic reprogramming from a privileged somatic cell state. Cell. 2014;156:649–662. doi: 10.1016/j.cell.2014.01.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Zhou Y., Liu Z., Welch J.D., Gao X., Wang L., Garbutt T., Keepers B., Ma H., Prins J.F., Shen W., et al. Single-cell transcriptomic analyses of cell fate transitions during human cardiac reprogramming. Cell Stem Cell. 2019;25:149–164.e9. doi: 10.1016/j.stem.2019.05.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Kathiriya I.S., Nora E.P., Bruneau B.G. Investigating the transcriptional control of cardiovascular development. Circ. Res. 2015;116:700–714. doi: 10.1161/CIRCRESAHA.116.302832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Liu P., Chen M., Liu Y., Qi L.S., Ding S. CRISPR-based chromatin remodeling of the endogenous Oct4 or Sox2 locus enables reprogramming to pluripotency. Cell Stem Cell. 2018;22:252–261.e4. doi: 10.1016/j.stem.2017.12.001. [DOI] [PubMed] [Google Scholar]
- 69.Black J.B., Adler A.F., Wang H.G., D'Ippolito A.M., Hutchinson H.A., Reddy T.E., Pitt G.S., Leong K.W., Gersbach C.A. Targeted epigenetic remodeling of endogenous loci by CRISPR/Cas9-Based transcriptional activators directly converts fibroblasts to neuronal cells. Cell Stem Cell. 2016;19:406–414. doi: 10.1016/j.stem.2016.07.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Kumasaka N., Knights A.J., Gaffney D.J. High-resolution genetic mapping of putative causal interactions between regions of open chromatin. Nat. Genet. 2019;51:128–137. doi: 10.1038/s41588-018-0278-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Nygren J.M., Jovinge S., Breitbach M., Sawen P., Roll W., Hescheler J., Taneera J., Fleischmann B.K., Jacobsen S.E. Bone marrow-derived hematopoietic cells generate cardiomyocytes at a low frequency through cell fusion, but not transdifferentiation. Nat. Med. 2004;10:494–501. doi: 10.1038/nm1040. [DOI] [PubMed] [Google Scholar]
- 72.Llewellyn M.E., Barretto R.P.J., Delp S.L., Schnitzer M.J. Minimally invasive high-speed imaging of sarcomere contractile dynamics in mice and humans. Nature. 2008;454:784–788. doi: 10.1038/nature07104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Marin-Juez R., El-Sammak H., Helker C.S.M., Kamezaki A., Mullapuli S.T., Bibli S.I., Foglia M.J., Fleming I., Poss K.D., Stainier D.Y.R. Coronary revascularization during heart regeneration is regulated by epicardial and endocardial cues and forms a scaffold for cardiomyocyte repopulation. Dev. Cell. 2019;51:503–515.e4. doi: 10.1016/j.devcel.2019.10.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Watt F.M., Huck W.T. Role of the extracellular matrix in regulating stem cell fate. Nat. Rev. Mol. Cell Biol. 2013;14:467–473. doi: 10.1038/nrm3620. [DOI] [PubMed] [Google Scholar]
- 75.Lupu I.E., De Val S., Smart N. Coronary vessel formation in development and disease: mechanisms and insights for therapy. Nat. Rev. Cardiol. 2020;17:790–806. doi: 10.1038/s41569-020-0400-1. [DOI] [PubMed] [Google Scholar]
- 76.Jia G., Preussner J., Chen X., Guenther S., Yuan X., Yekelchyk M., Kuenne C., Looso M., Zhou Y., Teichmann S., et al. Single cell RNA-seq and ATAC-seq analysis of cardiac progenitor cell transition states and lineage settlement. Nat. Commun. 2018;9:4877. doi: 10.1038/s41467-018-07307-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Honig B., Shapiro L. Adhesion protein structure, molecular affinities, and principles of cell-cell recognition. Cell. 2020;181:520–535. doi: 10.1016/j.cell.2020.04.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Hodgkinson C.P., Bareja A., Gomez J.A., Dzau V.J. Emerging concepts in paracrine mechanisms in regenerative cardiovascular medicine and biology. Circ. Res. 2016;118:95–107. doi: 10.1161/CIRCRESAHA.115.305373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Yanamandala M., Zhu W., Garry D.J., Kamp T.J., Hare J.M., Jun H.W., Yoon Y.S., Bursac N., Prabhu S.D., Dorn G.W., 2nd, et al. Overcoming the roadblocks to cardiac cell therapy using tissue engineering. J. Am. Coll. Cardiol. 2017;70:766–775. doi: 10.1016/j.jacc.2017.06.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Gaetani R., Doevendans P.A., Metz C.H., Alblas J., Messina E., Giacomello A., Sluijter J.P. Cardiac tissue engineering using tissue printing technology and human cardiac progenitor cells. Biomaterials. 2012;33:1782–1790. doi: 10.1016/j.biomaterials.2011.11.003. [DOI] [PubMed] [Google Scholar]
- 81.Bae S., Park J., Kim J.-S. Cas-OFFinder: a fast and versatile algorithm that searches for potential off-target sites of Cas9 RNA-guided endonucleases. Bioinformatics. 2014;30:1473–1475. doi: 10.1093/bioinformatics/btu048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Stemmer M., Thumberger T., Del Sol Keyer M., Wittbrodt J., Mateo J.L. CCTop: an intuitive, flexible and reliable CRISPR/Cas9 target prediction tool. PLoS One. 2015;10:e0124633. doi: 10.1371/journal.pone.0124633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Cao J., Wu L., Zhang S.M., Lu M., Cheung W.K., Cai W., Gale M., Xu Q., Yan Q. An easy and efficient inducible CRISPR/Cas9 platform with improved specificity for multiple gene targeting. Nucleic Acids Res. 2016;44:e149. doi: 10.1093/nar/gkw660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Ma R., Liang J., Huang W., Guo L., Cai W., Wang L., Paul C., Yang H.T., Kim H.W., Wang Y. Electrical stimulation enhances cardiac differentiation of human induced pluripotent stem cells for myocardial infarction therapy. Antioxid. Redox Signal. 2018;28:371–384. doi: 10.1089/ars.2016.6766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Love M.I., Huber W., Anders S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014;15:550. doi: 10.1186/s13059-014-0550-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Huang da W., Sherman B.T., Lempicki R.A. Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat. Protoc. 2009;4:44–57. doi: 10.1038/nprot.2008.211. [DOI] [PubMed] [Google Scholar]
- 87.Patro R., Duggal G., Love M.I., Irizarry R.A., Kingsford C. Salmon provides fast and bias-aware quantification of transcript expression. Nat. Methods. 2017;14:417–419. doi: 10.1038/nmeth.4197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Soneson C., Love M.I., Robinson M.D. Differential analyses for RNA-seq: transcript-level estimates improve gene-level inferences. F1000Res. 2015;4:1521. doi: 10.12688/f1000research.7563.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Park K.H., Brotto L., Lehoang O., Brotto M., Ma J., Zhao X. Ex vivo assessment of contractility, fatigability and alternans in isolated skeletal muscles. J. Vis. Exp. 2012:e4198. doi: 10.3791/4198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Huang W., Liang J., Feng Y., Jia Z., Jiang L., Cai W., Paul C., Gu J.G., Stambrook P.J., Millard R.W., et al. Heterogeneity of adult masseter muscle satellite cells with cardiomyocyte differentiation potential. Exp. Cell Res. 2018;371:20–30. doi: 10.1016/j.yexcr.2018.05.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Liang J., Huang W., Cai W., Wang L., Guo L., Paul C., Yu X.Y., Wang Y. Inhibition of microRNA-495 enhances therapeutic angiogenesis of human induced pluripotent stem cells. Stem Cells. 2017;35:337–350. doi: 10.1002/stem.2477. [DOI] [PubMed] [Google Scholar]
- 92.Huang W., Feng Y., Liang J., Yu H., Wang C., Wang B., Wang M., Jiang L., Meng W., Cai W., et al. Loss of microRNA-128 promotes cardiomyocyte proliferation and heart regeneration. Nat. Commun. 2018;9:700. doi: 10.1038/s41467-018-03019-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
differentiation