

Abstract
Sickle cell disease (SCD) is caused by a mutation in the β-globin gene leading to polymerization of the sickle hemoglobin (HbS) and deformation of red blood cells. Autologous transplantation of hematopoietic stem/progenitor cells (HSPCs) genetically modified using lentiviral vectors (LVs) to express an anti-sickling β-globin leads to some clinical benefit in SCD patients, but it requires high-level transgene expression (i.e., high vector copy number [VCN]) to counteract HbS polymerization.
Here, we developed therapeutic approaches combining LV-based gene addition and CRISPR-Cas9 strategies aimed to either knock down the sickle β-globin and increase the incorporation of an anti-sickling globin (AS3) in hemoglobin tetramers, or to induce the expression of anti-sickling fetal γ-globins. HSPCs from SCD patients were transduced with LVs expressing AS3 and a guide RNA either targeting the endogenous β-globin gene or regions involved in fetal hemoglobin silencing. Transfection of transduced cells with Cas9 protein resulted in high editing efficiency, elevated levels of anti-sickling hemoglobins, and rescue of the SCD phenotype at a significantly lower VCN compared to the conventional LV-based approach.
This versatile platform can improve the efficacy of current gene addition approaches by combining different therapeutic strategies, thus reducing the vector amount required to achieve a therapeutic VCN and the associated genotoxicity risk.
Keywords: lentiviral vectors, genome editing, CRISPR-Cas9 nuclease, sickle cell disease
Graphical abstract
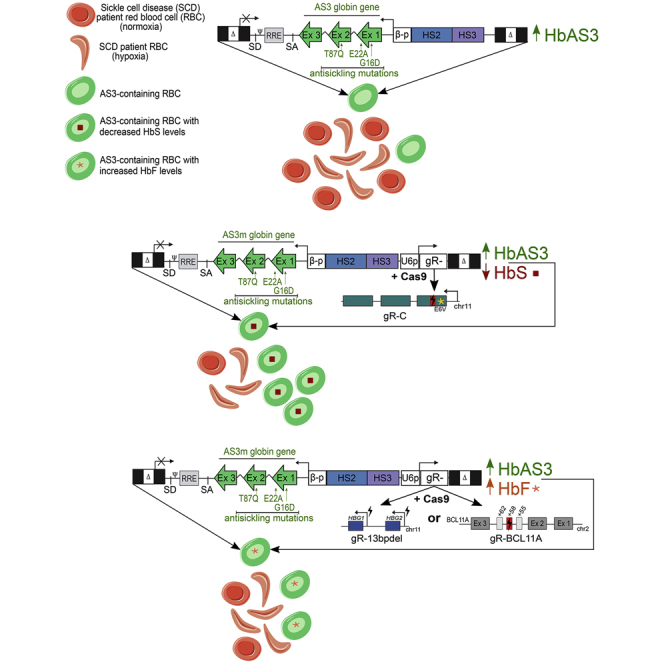
Miccio, Meneghini, and colleagues developed effective therapeutic approaches for sickle cell disease combining gene addition of a therapeutic transgene and CRISPR-Cas9 to either knock down the sickle β-globin or induce anti-sickling fetal γ-globin. This novel approach can be exploited to simultaneously induce the expression of therapeutic proteins and downregulate disease-causing genes.
Introduction
β-Hemoglobinopathies are severe anemias affecting ∼350,000 newborns each year.1 Sickle cell disease (SCD) is caused by a point mutation in the sixth codon of the β-globin gene (HBB), which leads to the E6V substitution. Hemoglobin tetramers containing the defective sickle βS-globin (HbS) polymerize under hypoxia, and red blood cells (RBCs) assume a sickle shape and become inflexible. Sickle RBCs have a short half-life and obstruct microvessels, causing a chronic multi-organ disease associated with poor quality of life and short life expectancy.2 β-Thalassemia is caused by mutations that reduce or abrogate β-globin production. The uncoupled α-globin chains cause apoptosis of erythroid precursors and hemolytic anemia.
Current treatments of SCD and β-thalassemia include regular RBC transfusions, which are associated with significant side effects, such as iron overload and organ damage. The only definitive cure for β-hemoglobinopathies is the allogeneic hematopoietic stem cell (HSC) transplantation from human leukocyte antigen (HLA)-matched sibling donors, which is available only to a fraction of the patients, requires an immunosuppressive regiment, and can be associated with chronic graft-versus-host disease.3,4 Transplantation of autologous HSCs transduced with lentiviral vectors (LVs) carrying an anti-sickling β-globin transgene (i.e., encoding a β-globin chain inhibiting Hb polymerization) is a promising therapeutic option for patients lacking a compatible donor.5,6 In particular, early data demonstrated a clinical benefit in a SCD patient transplanted with HSCs genetically corrected with a LV expressing a β-globin chain containing a single anti-sickling amino acid (Q at position 87, derived from the natural anti-sickling fetal γ-globin).7 However, the analysis of larger cohorts of patients showed that this treatment is only partially effective in the case of poor transgene transfer in HSCs,8 which results in therapeutic β-globin levels insufficient to compete with βS-globin for the incorporation into the Hb tetramers. Therefore, the achievement of clinically relevant transgene expression in SCD patients required a high number of integrated LV copies per cell, which could increase the potential genotoxicity risks associated to LV semi-random integration in the genome.9,10 We have recently optimized a high-titer β-globin-expressing LV (currently used in a clinical trial for β-thalassemia11) for the treatment of SCD by introducing three anti-sickling amino acids substitutions (G16D, E22A, T87Q) in the β-globin chain (AS3),12 which prevent the formation of axial and lateral contacts necessary for the generation of Hb polymers and increase the affinity for α-globin compared to the βS-globin.6,13 However, despite these improvements, the RBC sickling phenotype was only partially corrected even in the presence of a high vector copy number (VCN).6
Genome editing approaches have been recently developed for the treatment of β-hemoglobinopathies. Strategies based on the high-fidelity homology-directed repair (HDR) pathway have been exploited to correct the SCD mutation by providing a DNA donor template containing the wild-type (WT) β-globin sequence.14, 15, 16 However, the efficiency of HDR-mediated gene correction is limited in HSCs, and gene disruption by non-homologous end joining (NHEJ), a more active DNA repair pathway in HSCs,17 may be more frequent, generating a β-thalassemic phenotype instead of correcting the SCD mutation.18
The clinical severity of SCD and β-thalassemia is alleviated by the co-inheritance of mutations causing fetal γ-globin expression in adult RBCs (a condition termed hereditary persistence of fetal hemoglobin [HPFH]19). The NHEJ pathway has been exploited to induce fetal γ-globin re-activation by disrupting cis-regulatory silencer regions in the γ-globin (HBG) promoters20, 21, 22, 23 (e.g., binding sites for BCL11A and LRF transcriptional repressors) and in the β-globin locus,24 or to downregulate the BCL11A HbF repressor by targeting its erythroid-specific enhancer.25, 26, 27 The extent and impact of HbF reactivation on the recovery of SCD and β-thalassemic phenotypes is currently under investigation in clinical studies.28
Overall, these pre-clinical and clinical studies show that gene therapy approaches for β-hemoglobinopathies require highly efficient genetic modification of HSCs and robust expression of therapeutic globins to achieve clinical benefit.
In this study, we developed a novel LV platform based on the concomitant expression of the anti-sickling AS3 transgene and a single guide RNA (sgRNA) targeting the HBB gene to induce the downregulation of the endogenous βS globin in SCD erythroid cells and favor the incorporation of the therapeutic AS3 β-globin chain into Hb tetramers. The versatility of this platform was exploited by combining the AS3 expression with γ-globin re-activation induced by CRISPR-Cas9-mediated disruption of the BCL11A binding site in the HBG promoters or BCL11A downregulation, thus potentially extending its application to the treatment of β-thalassemia.
Our results demonstrate that editing the HBB gene or the HBG promoters is safe and enhances the therapeutic effect of the AS3 gene addition strategy, correcting the SCD pathological phenotype with a limited number of vector copies per cell.
Results
Selection of an efficient sgRNA downregulating HBB expression
To select a sgRNA able to efficiently and safely knock down the mutant sickle HBB gene, we tested sgRNAs recognizing sequences within HBB exon 1 (gR-A, gR-B, gR-C, gR-D29,30) (Figure 1A). Plasmids expressing the different sgRNAs under the control of the human U6 promoter were individually delivered together with a plasmid carrying SpCas9 nuclease fused with the green fluorescent protein (Cas9-GFP) in fetal K562 and adult HUDEP-2 erythroid cell lines.31 The frequency of insertion and deletions (indels) ranged from ∼40% to ∼80% (Figure 1B), as measured by Sanger sequencing and tracking of indels by decomposition (TIDE) analysis.32 The frequency of out-of-frame mutations (likely causing a decreased HBB expression) was substantially higher for gR-C and gR-D (70%–95%) than for gR-A and gR-B (25%–60%) in both K562 and HUDEP-2 cell lines (Figure 1C).
Figure 1.

Test and validation of an effective sgRNA to knock down the HBB gene
(A) sgRNAs are aligned to their complementary on-target loci on exon 1 of the HBB gene. gR-B targets a region containing the SCD mutation, and therefore its sequence was modified to target the WT HBB gene during the validation experiments in K562 and HUDEP-2 erythroid cell lines, and in HD HSPCs. The codon containing the SCD mutation is highlighted in red. (B) Editing efficiency by TIDE analysis after PCR amplification of the target region and Sanger sequencing in Cas9-GFP+ K562 (n = 3 for gR-A, 4 for gR-B, 6 for gR-C, and 5 for gR-D) and HUDEP-2 (n = 5 for gR-C; n = 4 for gR-A, gR-B, and gR-D) erythroid cell lines. Transfection efficiency was 55%–70% for K562 and 30%–60% for HUDEP-2. Data are expressed as mean ± SEM. (C) Out-of-frame mutations identified using TIDE after PCR amplification of the target region and Sanger sequencing in K562 (n = 3 for gR-A, 4 for gR-B, 6 for gR-C, and 5 for gR-D) and HUDEP-2 (n = 5 for gR-C; n = 4 for gR-A, gR-B, and gR-D) erythroid cell lines. Data are expressed as mean ± SEM. The frequency of out-of-frame mutations for gR-C was significantly higher than for gR-A (p < 0.0001 in K562 and p < 0.05 in HUDEP-2) and gR-B (p < 0.005 in K562). In K562, the frequency of out-of-frame mutations for gR-D was significantly higher than for gR-A and gR-B (p < 0.0005). In both K562 and HUDEP-2, no differences were observed between gR-C and gR-D (one-way ANOVA test: Tukey’s multiple comparisons test). (D) Relative expression of HBB, HBD, and HBG mRNAs normalized to α-globin, detected by qRT-PCR in HUDEP-2 cells. Data are expressed as mean ± SEM (n = 3). ∗∗p < 0.01 between gR-A or gR-B versus control (Ctr)-Cas9; ∗∗∗p < 0.001 between gR-C or gR-D versus Ctr-Cas9 (two-way ANOVA test: Dunnett’s multiple comparisons test). Ctr-Cas9 samples are HUDEP-2 cells transfected only with the Cas9-GFP plasmid. (E) Relative expression of the β-globin chain normalized to the α-globin chain, as measured by western blot analysis in differentiated HUDEP-2 cells. Data are expressed as mean ± SEM (n = 4 for gR-C; n = 3 for gR-A, gR-B, and gR-D). ∗p < 0.05 and ∗∗p < 0.01 versus Ctr-Cas9 (two-way ANOVA test: Sidak’s multiple comparisons test). A representative western blot image is shown. Ctr-Cas9 samples are HUDEP-2 cells transfected only with the Cas9-GFP plasmid. (F) Editing efficiency (indels) and frequency of out-of-frame mutations measured by TIDE analysis after PCR amplification of the target region and Sanger sequencing in G-CSF-mobilized peripheral blood (mPB) HD HSPCs edited with gR-C (n = 1 for HD1, n = 2 for HD2, and n = 2 for HD3). Data are expressed as mean ± SEM. (G) Relative expression of HBB, HBD, and HBG mRNAs normalized to α-globin, measured by qRT-PCR in erythroid cells derived from G-CSF-mPB HD HSPCs transfected with Cas9-GFP plasmid only (Ctr-Cas9) or with both Cas9-GFP and gR-C plasmids (gR-C). HBB decrease upon gR-C treatment is reported as percentage above the histogram bars (n = 1 for HD1, n = 2 for HD2, and n = 2 for HD3). Data are expressed as mean ± SEM. (H) RP-HPLC quantification of globin chains in erythroid cells derived from G-CSF-mPB HD HSPCs transfected with Cas9-GFP plasmid only (Ctr-Cas9) or with both Cas9-GFP and gR-C plasmids (gR-C). β-Like globin chains are normalized over α-globin chains (n = 1 for HD1, n = 2 for HD2, and n = 2 for HD3; n = 5 biological replicates). The percentage of β-globin decreases upon gR-C treatment and the α-globin/non-α-globin ratio is reported above the histogram bars. Data are expressed as mean ± SEM. A representative chromatogram is reported in the left panel. (I) CE-HPLC analysis of Hb tetramers in β-thalassemic cells (thal) and in erythroid cells derived from G-CSF-mPB HD HSPCs treated or not with gR-C. gR-C-treated samples have a hemoglobin expression pattern similar to the profile observed in in vitro-differentiated erythroid cells from a β-thalassemia patient. We calculated the percentage of Hb types over the total Hb tetramers (n = 2 donors). CE-HPLC chromatograms are reported on the left panel. αp, α-precipitates. Data are expressed as mean ± SEM.
The ability of the different sgRNAs to downregulate HBB expression was evaluated in HUDEP-2 cells (expressing mainly adult β-globin) differentiated in mature erythroid precursors. qRT-PCR and western blot analysis showed a strong downregulation of β-globin expression at both mRNA (70%–85%) and protein (70%–90%) levels in comparison to HUDEP-2 cells transfected only with the Cas9-GFP plasmid (Figures 1D and 1E). Of note, all of the gRNAs generated premature stop codons (data not shown) that likely cause mRNA degradation through nonsense-mediated decay. As expected from editing data, gR-C and gR-D caused a more robust β-globin downregulation compared to gR-A and gR-B (Figures 1D and 1E). Of note, sgRNAs targeting the HBB gene did not alter mRNA expression of the other β-like globin genes (i.e., adult HBD and fetal HBG genes; Figure 1D), suggesting a specific targeting of the HBB coding sequence. Reversed-phase high-pressure liquid chromatography (RP-HPLC) and cation-exchange HPLC (CE-HPLC) analyses of differentiated HUDEP-2 cells treated with gR-C showed a decrease in β-globin production, resulting in an imbalance between α-globin and non-α-globin chains and accumulation of α-globin precipitates, a typical hallmark of β-thalassemia (Figures S1A and S1B). Notably, the presence of δ- and γ-globin chains was not sufficient to compensate for the lack of β-globin (Figure S1A).
The off-target activity of the best performing sgRNAs (gR-C and gR-D) was initially evaluated in the HBD gene, the HBB paralog with the highest sequence homology, which is often reported as the major off-target locus when targeting the HBB gene.33 gR-C and gR-D target sequences have four and two mismatches with the potential off-target loci in HBD exon 1 (Figure S1C). TIDE analysis showed an absence of indels in the HBD loci in both K562 and HUDEP-2 cells treated with gR-C or gR-D (Figure S1C). However, plasmid delivery of the CRISPR-Cas9 system in primary healthy donor (HD) hematopoietic stem/progenitor cells (HSPCs) resulted in ∼3% of edited HBD alleles in gR-D-treated cells, while no off-target events in the HBD locus were detected in samples transfected with gR-C (Figure S1C). These findings are in line with the lower number of mismatches between the gR-D target sequence and the HBD off-target locus as compared to gR-C (Figure S1C). Based on these findings, we further characterized the off-target activity of gR-C in plasmid-transfected HUDEP-2 cells and primary HD HSPCs by analyzing the top-15 off-targets predicted by COSMID34 (Table S2). TIDE analysis revealed no off-target activity in 14 out of 15 loci, including HBD and HBG genes. Indels were detected only at the off-target locus 4 (OT4) in HUDEP-2 cells (∼2% indels) and at relatively low levels in primary HSPCs (∼0.6% indels) (Figure S1D). However, this off-target maps to an intergenic region, which does not contain known hematopoietic cis-regulatory regions (Figure S1E).
The gR-C was selected for further analyses aimed at evaluating β-globin downregulation in HSPC-derived erythroid cells, because of its high on-target activity and low off-target editing. We transfected adult HD HPSCs with plasmids encoding gR-C and Cas9-GFP, and fluorescence-activated cell sorting (FACS)-sorted Cas9-GFP+ cells were differentiated toward the erythroid lineage. Genome editing efficiency ranged from 32% to 54% of indels with 30%–49% of out-of-frame mutations in HSPC-derived erythroid cells (Figures 1F; Figure S1F). We observed β-globin downregulation at both mRNA and protein levels, which was well correlated with the frequency of frameshift mutations (Figures 1F–1H). In HSPC-derived erythroid cells, RP-HPLC analysis revealed that β-globin downregulation induced an imbalance between the α- and β-like globin chains, which was not compensated by the presence of γ (Aγ+Gγ)-globin and δ-globin chains (Figure 1H). Importantly, CE-HPLC analysis demonstrated that gR-C led to a substantial reduction of HbA tetramers and accumulation of α-globin precipitates (Figure 1I).
Overall, these data showed that gR-C is efficient in generating frameshift mutations in HBB, leading to a robust downregulation of β-globin synthesis in both erythroid cell lines and primary cells.
A bifunctional LV to concomitantly express the AS3 anti-sickling globin and downregulate the endogenous β-globin
Transduction of HSPCs isolated from SCD patients with a LV carrying the anti-sickling AS3-globin transgene (LV AS3) led to a partial correction of the SCD phenotype of HSPC-derived RBCs.6 To enhance the therapeutic efficacy of gene addition strategies, we introduced a gR-C expression cassette in LV AS3 to concomitantly induce the expression of the AS3 transgene and the knockdown of the endogenous β-globin gene upon transient Cas9 delivery, thus favoring the incorporation of the AS3-globin chain into Hb tetramers. Importantly, the combination of gene addition and gene silencing strategies allows the knockdown of the mutant sickle β-globin gene only in cells expressing the therapeutic β-globin chain, which will compensate for the HbS reduction, thus avoiding the potential generation of a β-thalassemic phenotype.
The sgRNA expression cassette was inserted downstream of the AS3-expressing cassette in reverse orientation to the AS3 transgene transcription (Figure 2A). To further increase gR-C editing efficiency, we used an optimized sgRNA scaffold carrying a mutation in the RNA polymerase (Pol) II transcription pause sequence and an extended sgRNA duplex to increase U6-driven sgRNA transcription and sgRNA-Cas9 interactions, respectively.35 Plasmid transfection of both K562 cells and adult HD HSPCs showed that the use of the optimized scaffold increased indel frequency, while maintaining a similar percentage of out-of-frame mutations (∼90%) compared to the original scaffold (Figures S1G and S1H). To avoid the downregulation of transgene expression, we introduced six silent mutations in the AS3 sequence complementary to gR-C (AS3m transgene) using synonymous codons commonly found in β-like globin genes (Figure 2A). These silent mutations are expected to impair AS3m targeting and editing by gR-C, which will then recognize only the endogenous β-globin gene. The insertion of the sgRNA expression cassette did not compromise LV titer (∼109 transduction unit [TU]/mL) and infectivity (∼7 × 104 TU/ng p24) (LV AS3m.C vector; Figure S2A).
Figure 2.

Combination of lentiviral and genome editing technologies is efficient in HUDEP-2 cells
(A) Schematic representation of the LVs carrying either the AS3 globin gene (LV AS3, strategy #1) or the AS3m globin gene and a sgRNA-expressing cassette (LV AS3m.gR). Both AS3- and AS3m-globin genes are under the control of a short β-globin promoter (β-p) and a mini-βLCR containing HS2 and HS3. The anti-sickling amino acid substitutions are reported in red. The modified nucleotides in LV AS3m, avoiding its targeting by the gR-C, are reported in green. We indicated the amino acids below the nucleotide sequence. sgRNA expression is under the control of the human U6 promoter (U6p). gR-C targets a region in exon 1 (Ex1) of HBB (strategy #2), gR-BCL11A targets the +58-kb erythroid-specific enhancer of BCL11A (strategy #3), and gR-13bpdel targets the BCL11A binding site within the HBG promoters (strategy #4). (B) Schematic representation of the LV transduction and plasmid transfection protocol used in HUDEP-2 cells. Cells were transduced with LV AS3m carrying gR-C, BCL11A, and 13bpdel sgRNAs and transfected after 10 days with a Cas9-GFP plasmid. Transfection efficiency was 30%–60%. FACS-sorted Cas9-GFP+ cells were differentiated into mature erythroblasts. (C) Relative expression of HBB and AS3m mRNAs normalized to α-globin, as detected by qRT-PCR. VCN values are reported in blue and indels in black below the graph. Ctr UTs are untreated HUDEP-2 cells and Ctr TEs are HUDEP-2 cells transfected only with TE buffer. Transduced samples that were mock transfected are indicated with “–.” The percentage of HBB decrease upon LV AS3m.C treatment is reported above the histogram bars. Data are expressed as mean ± SEM. (D) RP-HPLC quantification of globin chains in Cas9-treated and control (mock-transfected) LV AS3m.C-transduced cells. β-Like globin chains were normalized to α-globin chains. VCN values are reported in blue and indels in black below the graph. Ctr UTs are untreated HUDEP-2 cells and Ctr TEs are HUDEP-2 cells transfected only with TE buffer. Transduced samples that were mock transfected are indicated with “–” and harbored no indels. The percentage of β-globin decrease upon LV AS3m.C treatment and the α-globin/non-α-globin ratio are reported above the histogram bars. (E) CE-HPLC chromatograms (left panel) and quantification (right panel) of Hb tetramers in Cas9-treated and control mock-transfected LV AS3m.C-transduced cells. We calculated the percentage of each Hb type over the total Hb tetramers. VCN values are reported in blue and indels in black below the graph. Ctr UTs are untreated HUDEP-2 cells and Ctr TEs are HUDEP-2 cells transfected only with TE buffer. The decrease in HbA expression upon LV AS3m.C treatment is reported as percentage above the histogram bars. (F) Editing efficiency (indel frequency) in LV AS3m.C-treated HUDEP-2 cells measured by TIDE analysis after PCR amplification and Sanger sequencing of the target region in HBB exon 1 and the potential off-target region in the transgene (AS3m) in Cas9-GFP+ HUDEP-2 erythroid cells. (G) Relative HBG expression normalized to α-globin, as detected by qRT-PCR in Cas9-treated and mock-transfected cells transduced with LV AS3m.BCL11A or LV AS3m.13bpdel. VCN values are reported in blue and indels in black below the graph. Ctr UTs are untreated HUDEP-2 cells and Ctr TEs are HUDEP-2 cells transfected only with TE buffer. Data are expressed as mean ± SEM. (H) RP-HPLC quantification of globin chains in Cas9-treated and mock-transfected cells transduced with LV AS3m.BCL11A or LV AS3m.13bpdel. β-Like globin chains are normalized over α-globin. VCN values are reported in blue and indels in black below the graph. α-Globin/non-α-globin ratios are reported above the histogram bars. Ctr UTs are untreated HUDEP-2 cells and Ctr TEs are HUDEP-2 cells transfected only with TE buffer. (I) CE-HPLC chromatograms (left panel) and quantification (right panel) of Hb tetramers in Cas9-treated and mock-transfected LV AS3m.BCL11A- and LV AS3m.13bpdel-transduced cells. We plotted the percentage of each Hb type over the total Hb tetramers. VCN values are reported in blue and indels in black below the graph. The percentage of HbA decrease is reported above the histogram bars. Ctr UTs are untreated HUDEP-2 cells and Ctr TEs are HUDEP-2 cells transfected only with TE buffer.
To evaluate whether the silent mutations impair the production of AS3 chains, we compared LVs harboring the original (LV AS3) or the modified (LV AS3m) AS3 transgene in a β-thalassemic HUDEP-2 cell line36 (HBB knockout [KO]). In differentiated HBB KO HUDEP-2 cells, β-globin expression is abolished, resulting in the imbalance between α and non-α chains and accumulation of α-globin precipitates (Figures S2B and S2C). We transduced HBB KO HUDEP-2 cells with LV AS3 and LV AS3m at increasing multiplicities of infection (MOIs) obtaining a VCN ranging from 0.5 to ∼9 (Figures S2B and S2C). HPLC analyses showed that AS3 expression was comparable in LV AS3- and LV AS3m-transduced erythroid precursors harboring a similar VCN (Figures S2B and S2C). Both LVs partially restored the β-thalassemic phenotype in cells harboring a low VCN (0.5–0.9), while a higher gene marking (VCN > 4) fully corrected the α-globin/non-α-globin imbalance, as evaluated by RP-HPLC (Figure S2B). These results demonstrated that the silent mutations in the modified transgene do not affect AS3 expression and the production of functional HbAS3 tetramers.
To test the capability of LV AS3m.C to downregulate the expression of the endogenous HBB gene, WT HUDEP-2 cells were transduced with increasing doses of LV AS3m.C and then either transfected with the Cas9-GFP-expressing plasmid or mock transfected. FACS-sorted Cas9-GFP+ cells were then differentiated into mature erythroblasts (Figure 2B). The indel frequency at the on-target HBB gene was positively correlated with the VCN (Figure 2C). HBB editing resulted in the knockdown of the endogenous β-globin at both mRNA and protein levels, with higher indel frequency resulting in more robust downregulation of HBB expression (Figures 2C–2E). Of note, no indels were detected in the AS3m transgene even in samples displaying a high on-target activity at the endogenous HBB gene, demonstrating that silent mutations in AS3m avoid its targeting by gR-C, which specifically recognizes the endogenous β-globin gene (Figure 2F). Concordantly, AS3m mRNA levels were comparable between the control and edited samples (Figure 2C). Importantly, the downregulation of the endogenous β-globin expression favored incorporation of the AS3 globin chain into the Hb tetramers (Figure 2E). In cells harboring a relatively low gene marking (VCN of 3 associated with an editing efficiency of 40%), this resulted in a balance between HbA and HbAS3 tetramers, which could not be reached in unedited cells even at high VCN (11.8; Figure 2E). Notably, the reduction of the β-globin chain content was compensated by the AS3-globin production and incorporation into the Hb tetramers, thus avoiding the imbalance between the α and non-α chains and the formation of α-precipitates (Figures 2D and 2E). Accordingly, the erythroid differentiation of HUDEP-2 cells was not impacted by editing of the HBB gene, as the cell composition was similar in edited and control HUDEP-2 cells, while HBB KO HUDEP-2 cells showed a delay in differentiation typical of β-thalassemia37 (Figure S3).
We achieved similar results using a clinically relevant, plasmid-free transfection protocol in LV-transduced WT HUDEP-2 cells. In particular, we compared Cas9-GFP plasmid or protein delivery methods in LV AS3m.C-transduced WT HUDEP-2 cells. Transfection with Cas9-GFP protein was less cytotoxic (68% ± 4% of alive cells in untransfected samples, 58% ± 4% and 37% ± 6% in samples transfected with 15 μg of Cas9-GFP protein and Cas9-GFP-expressing plasmid, respectively), more efficient (∼70% and 32.5% of GFP+ cells upon protein and plasmid transfection, respectively), and led to a higher editing efficiency at the on-target HBB locus in unsorted bulk populations (Figure S4A). LV AS3m.C-transduced WT HUDEP-2 cells transfected with 15 μg of Cas9-GFP protein were terminally differentiated in mature erythroid precursors. The decrease of HbA levels and the concomitant increase of HbAS3 content were significantly correlated with the VCN (Figure S4B) and the genome editing efficiency at the HBB locus (Figure S4C). Importantly, similar amounts of HbA and HbAS3 were detected in cells harboring a VCN of ∼2.5% and ∼46% of edited alleles (Figures S4B and S4C). On the contrary, in mock-transfected LV AS3m.C-transduced cells, HbA content exceeded the levels of HbAS3 even in cells harboring a high VCN (Figure S4B).
Overall, these data showed that the treatment with LV AS3m.C can safely and efficiently downregulate the expression of the endogenous β-globin and increase the production of Hb tetramers containing the therapeutic AS3-globin compared to the classical gene addition approach.
A bifunctional LV to induce the expression of the anti-sickling AS3-globin and γ-globin chains
Considering the therapeutic benefit of high fetal γ-globin expression in SCD and β-thalassemia patients,38, 39, 40 we exploited the flexibility of this novel LV platform to simultaneously express the AS3m transgene and reactivate HbF expression. The combined expression of AS3 and γ chains can elevate the total Hb content in β-thalassemia patients and, as both of these globins display anti-sickling properties, it can also benefit SCD patients.
We generated LVs expressing the AS3m transgene and a sgRNA that either disrupts a sequence in the BCL11A erythroid-specific enhancer critical for its expression (AS3m.BCL11A)26 or generates 13-bp deletions encompassing the BCL11A binding site in the fetal γ-globin promoters (AS3m.13bpdel)20 (Figure 2A). As observed for LV AS3m.C, the insertion of the sgRNA cassette did not impair LV titer and infectivity (Figure S2A).
WT HUDEP-2 cells were transduced with LV AS3m.BCL11A or LV AS3m.13bpdel and then transfected with Cas9-GFP plasmid or mock transfected. FACS-sorted Cas9-GFP+ cells and control samples were differentiated in mature erythroid precursors (Figure 2B). Editing of the BCL11A enhancer or the γ-globin promoters did not impact the differentiation of HUDEP-2 cells (Figure S3). LV AS3m.BCL11A-transduced cells showed up to 63% of indel frequency at the BCL11A enhancer, which caused a strong decrease in the levels of BCL11A-XL, the isoform mainly involved in the inhibition of HbF expression41,42 (Figure S5A). In LV AS3m.13bpdel-transduced cells, around one third (31.7% ± 0.5%) of the total editing events generated by gR-13bpdel were MMEJ-mediated 13-bp deletions removing the entire BCL11A binding site (Figure S5B). All other events also led to the disruption of the BCL11A binding site in the HBG promoters (Figure S5B). Editing of the BCL11A enhancer or the γ-globin promoters resulted in HBG gene induction, as well as increased γ-globin chain and HbF production (Figures 2G–2I). In particular, the concomitant expression of AS3 and γ-globin resulted in up to 37% and 55% of total anti-sickling hemoglobins (HbF+HbAS3) in LV AS3m.BCL11A- and LV AS3m.13bpdel-transduced cells, respectively (Figure 2I). The combined expression of HbF and HbAS3 decreased adult HbA production in samples with the higher genome editing frequencies (Figure 2I). Flow cytometry analysis confirmed the increased HbF production with up to 61% and 74% of F-cells in samples treated with LV AS3m.BCL11A and LV AS3m.13bpdel, respectively (Figure S5C).
These data show that CRISPR-Cas9-mediated induction of HbF expression can increase the total Hb content in cells expressing a β-globin therapeutic transgene, thus representing a promising strategy to correct both SCD and β-thalassemia phenotypes.
The combination of gene addition and genome editing approaches increased the content of anti-sickling hemoglobins in SCD RBCs
The efficacy and safety of our approach was evaluated in adult plerixafor-mobilized peripheral blood (mPB) HSPCs from SCD patients. HSPCs were transduced with increasing doses of LV AS3m.C and transfected 48 h later with Cas9-GFP protein using previously optimized protocols6,43 (Figure 3A). Control and edited SCD HSPCs were plated in clonogenic cultures (colony forming cell [CFC] assay) allowing the growth of erythroid (burst-forming unit-erythroid cells [BFU-Es]) and granulomonocytic (colony-forming unit granulocyte-macrophages [CFU-GMs]) progenitors. The electroporation of transduced cells modestly reduced the number of progenitors, although this decrease was not statistically significant (Figure S6A). Importantly, no significant difference was observed between LV AS3m.C-transduced, mock-transfected, and edited samples (Figure S6A). Upon differentiation in mature erythroblasts, we observed a significant correlation between editing efficiency at the HBB locus and VCN, with an indel frequency ranging from 21% to 67% (Figure 3B). A positive correlation between editing frequency and VCN was also observed in pools of BFU-Es derived from SCD HSPCs treated with LV AS3m.C and Cas9-GFP protein (Figure S6B). As previously observed in HUDEP-2 cells, no indels were detected in the sequence of the AS3m transgene in any of the treated samples (Figure 3C). Deep sequencing analysis in edited erythroblasts revealed a low off-target activity only in one out of the four off-target sites detected by GUIDE-seq (genome-wide unbiased identification of double-stranded breaks enabled by sequencing) analysis (Figure 3D). This locus corresponds to the OT4 previously identified in primary HSPCs by Sanger sequencing (Figures S1D and S1E).
Figure 3.

Testing of the bifunctional LV AS3m.C in SCD HSPCs
(A) Schematic representation of the protocol used to transduce and transfect SCD HSPCs. 24 h after thawing, HSPCs were transduced with LV AS3m.C and after 48 h cells were transfected with the Cas9-GFP protein and differentiated into mature RBCs following a 21-day differentiation protocol. (B) Correlation between VCN and indel frequency in cells treated with LV AS3m.C (two mobilized SCD donors). R2 and line-of-best-fit equation are indicated. (C) Indel frequency in LV AS3m.C-treated SCD HSPCs measured by TIDE analysis after PCR amplification and Sanger sequencing of the target region in HBB exon 1 and the potential off-target region in the transgene (AS3m). (D) Evaluation of off-target activity. GUIDE-seq analysis of gR-C in 293T cells (left panel). The protospacer targeted by gR-C and the protospacer adjacent motifs (PAMs) are shown in the first line. Off-targets and their mismatches with the on-target (highlighted in color), sequencing read counts, and chromosomal position are reported. Deep-sequencing analysis of off-target editing events in mature erythroblasts derived from SCD HSPCs treated with LV AS3m.C (right panel). Transduced and mock-transfected SCD cells (Ctr) are indicated in red and edited samples (AS3m.C) in black. (E) Correlation between VCN (left panel) or indel frequency (right panel) and percentage of HbS and HbAS3 (determined by CE-HPLC) in SCD RBCs derived from mock-transfected (orange and light blue) and Cas9-transfected (red and dark blue) SCD HSPCs transduced with LV AS3m.C (two mobilized SCD donors). R2 and line-of-best-fit equation are indicated. Dashed lines indicate the VCN required to achieve equal amounts of HbS and HbAS3. (F) Relative expression of HBB mRNA normalized to α-globin in untreated SCD cells (Ctr) (n = 6) and in cells treated with LV AS3m.C (n = 8, 2 mobilized SCD donors). ∗∗∗∗p < 0.0001 (unpaired t test). Horizontal lines indicate median and first and third quartiles. (G) Representative CE-HPLC chromatograms showing the Hb profile of in vitro-differentiated mature erythroblasts. From top to bottom: β-thalassemic cells (thal), healthy donor cells (HD), HD cells treated with gR-C plasmid (HD+gR-C), SCD cells (SCD), SCD cells transduced with LV AS3m.C and mock transfected, and SCD cells transduced with LV AS3m.C and transfected with Cas9-GFP protein.
Erythroblasts were further differentiated in enucleated RBCs using a three-phase protocol44 (Figure 3A). We observed a positive correlation between VCN and the HbAS3 content in RBCs derived from mock-transfected, LV AS3m.C-transduced HSPCs achieving equivalent HbS and HbAS3 levels at a VCN of ∼5.5 (Figure 3E). Cas9 treatment led to a decreased expression of the sickle HBB gene at both mRNA and protein levels, thus favoring the incorporation of the AS3 chain in Hb tetramers (Figures 3E and 3F; Figure S6C). Importantly, in RBCs derived from edited SCD HSPCs, similar amounts of HbS and HbAS3 were observed already at a VCN of ∼2.2 (indel frequency of ∼41%), reproducing the Hb profile of asymptomatic SCD carriers (Figure 3E). HbS downregulation and increased incorporation of the AS3 chain in Hb tetramers were still evident at low VCN (Figures 3E and 3F; Figure S6C). Notably, CE-HPLC showed an absence of α-globin precipitates in RBCs obtained from edited HSPCs, indicating that the expression of the AS3 chain was able to efficiently compensate for the lack of βS-globin expression and avoid the generation of a β-thalassemic phenotype (Figure 3G).
In parallel, SCD mPB HSPCs were transduced with increasing doses of LV AS3m.13bpdel transfected with Cas9-GFP protein and then differentiated toward the erythroid lineage (Figure 4A). No significant differences were observed in the number of BFU-Es and CFU-GMs between control and edited samples (Figure S6A). We observed a direct correlation between VCN and indel frequency in BFU-Es and mature erythroblasts (Figure 4B; Figure S6B). LV AS3m.13bpdel-transduced erythroblasts showed ∼30% of edited loci at a VCN of 3–4, and up to 50% at higher VCNs (Figure 4B; Figure S6B). On the contrary, SCD mPB HSPCs transduced with LV AS3m.BCL11A were poorly edited, reaching a maximum of ∼20% of indel efficiency (Figure 4B). Deep-sequencing analyses of the only gR-13bpdel off-target previously detected by GUIDE-seq23 revealed no off-target activity in LV AS3m.13bpdel-transduced erythroblasts (Figure 4C). GUIDE-seq analysis in 293T cells showed a high number of off-target sites for the sgRNA targeting the BCL11A enhancer (Figure 4D).
Figure 4.

Testing of LV AS3m.13bpdel and LV AS3m.BCL11A in SCD HSPCs
(A) Schematic representation of the protocol used to transduce and transfect SCD HSPCs. 24 h after thawing, HSPCs were transduced with LV AS3m.13bpdel and LV AS3m.BCL11A, and after 48 h cells were transfected with the Cas9-GFP protein and differentiated into mature RBCs following a 21-day differentiation protocol. (B) Correlation between VCN and indel frequency in cells treated with LVs AS3m.BCL11A (one mobilized SCD donor) and AS3m.13bpdel (two mobilized SCD donors). R2 and line-of-best-fit equation are indicated. (C) Deep-sequencing analyses of the 13bpdel off-target in mature erythroblasts derived from adult SCD HSPCs treated with LV AS3m.13bpdel. Transduced and mock-transfected SCD cells (Ctr) are indicated in red and edited samples (AS3m.13bpdel) in black. (D) GUIDE-seq analysis of gR-BCL11A in 293T cells. The protospacer targeted by gR-BCL11A and the PAMs are shown in the first line. Off-targets and their mismatches with the on-target (highlighted in color), sequencing read counts, and chromosomal position are reported. (E) Relative expression of HBG mRNA normalized to α-globin in untreated SCD cells (Ctr) (n = 5) and cells treated with LV AS3m.13bpdel (n = 14, 2 SCD donors). ∗∗∗p < 0.001 (unpaired t test). Horizontal lines indicate median and first and third quartiles. (F) Representative flow cytometry analysis of F cells in RBCs derived from untreated (Ctr UT; light blue), and LV AS3m.13bpdel-treated SCD HSPCs mock transfected (medium blue) or transfected with Cas9-GFP (dark blue) (two mobilized SCD donors). (G) Correlation between VCN and percentage of HbS and HbAS3 (determined by CE-HPLC) in cells derived from mock-transfected (orange and light blue) and Cas9-transfected (red and dark blue) SCD HSPCs transduced with LV AS3m.13bpdel (two mobilized SCD donors). We plotted the percentage of HbS or HbAS3+HbF over the total Hbs. R2 and line-of-best-fit equation are indicated. Dashed lines indicate the VCN required to achieve equal amounts of HbS and HbAS3. (H) Correlation between indel frequency and percentage of HbS and HbAS3 (determined by CE-HPLC) in cells derived from Cas9-transfected SCD HSPCs transduced with LV AS3m.13bpdel (two mobilized SCD donors). R2 and line-of-best-fit equation are indicated.
Given the low efficiency of LV AS3m.BCL11A and the high number of off-target sites, we further differentiated into enucleated RBCs only the cells derived from edited, LV AS3m.13bpdel-transduced HSPCs to evaluate the content of anti-sickling globins. The treatment of LV AS3m.13bpdel-transduced SCD HSPCs with Cas9-GFP protein led to an increased HBG mRNA expression (Figure 4E), reaching a percentage of F cells ranging from 40% to 85% (Figure 4F). CE-HPLC analysis showed that the total amount of anti-sickling hemoglobins (HbF+HbAS3) was positively correlated with the VCN and the editing frequency (Figures 4G and 4H). The concomitant AS3-globin chain expression and γ-globin reactivation resulted in reduced HbS levels (Figures 4G and 4H). Importantly, similar amounts of HbS and anti-sickling hemoglobins were obtained at a lower VCN (VCN of ∼3.7; 34% of indels) in edited cells compared to the mock-transfected samples (VCN of ∼5.4) (Figures 4G and 4H). Importantly, γ-globin reactivation was still observed in edited samples at low VCN (Figures 4G and 4H; Figure S6D).
The increased content of anti-sickling hemoglobins rescues the SCD cell phenotype
To assess the effect of the increased production of anti-sickling Hb tetramers on RBC sickling, we performed a deoxygenation assay to measure the proportion of sickle-shaped RBCs. In vitro-generated RBCs were exposed to an oxygen-deprived atmosphere, which induces HbS polymerization and RBC sickling. The percentage of sickled cells was decreased in RBC populations derived from mock-transfected, LV AS3m.C- and AS3m.13bpdel-transduced HSPCs compared to untreated SCD controls as a consequence of the expression of the AS3-globin chain, with 40%–60% of normal, doughnut-shaped RBCs at a VCN of ∼2–3 (Figure 5). Interestingly, in RBCs derived from edited LV AS3m.C-transduced HSPCs, HbS downregulation combined with the high HbAS3 expression resulted in a substantial increase in the proportion of corrected RBCs (Figure 5). This effect was still evident by comparing RBCs generated from edited and mock-transfected LV AS3m.C-transduced HSPCs with a higher VCN (Figure 5). Finally, the increased HbF amount induced by the editing of the HBG promoters led to a modest but still significant reduction in the percentage of sickle-shaped RBCs compared to samples obtained from unedited, LV AS3m.13bpdel-transduced HSPCs showing a similar VCN (Figure 5).
Figure 5.

The bifunctional LVs AS3m.C and AS3m.13bpdel reduce RBC sickling
In vitro sickling assay measuring the proportion of sickled RBCs under hypoxic conditions (0% O2). We reported the percentage of sickling RBCs (left panel) and representative photomicrographs of RBCs derived from control (Ctr), LVs AS3m.C and AS3m.13bpdel samples that were either mock transfected or transfected with Cas9-GFP (right panel; 2 SCD donors). Arrows indicate sickling cells. Data are expressed as mean ± SEM. Scale bar, 20 μm (upper left).
Erythroid cell differentiation and RBC properties remain largely unchanged upon editing of SCD HSPCs
To exclude any impairment in erythroid differentiation of edited HSPCs, we monitored over time the erythroid liquid cultures using flow cytometry and quantitative phase imaging. In particular, although the co-expression of the AS3-globin and the sgRNA targeting HBB should avoid the excessive reduction of β-like globin chains, a potential issue of this strategy is the possibility to generate a β-thalassemic phenotype in the absence of a sufficient amount of transgene-derived AS3-globin to compensate βS-downregulation.
SCD HSPCs were transduced with the LV AS3m.C, transfected with Cas9-GFP protein, and then in vitro differentiated into mature RBCs (Figure 6A). Erythroid cultures obtained from SCD or HD HSPCs transfected with Cas9 ribonucleoprotein (RNP) complexes containing gR-C were used as β-thalassemia-like control cells (Figure 6A). Mature RBCs derived from SCD or HD HSPCs treated with gR-C-RNP displayed a strong reduction in the HbS and HbA content, respectively, and an increased α chain/non-α chain ratio in comparison to untreated controls (Figure 6B). On the contrary, in RBCs derived from HBB-edited, LV AS3m.C-transduced SCD HSPCs, α-chain/non-α chain ratios remained largely unchanged (1.08 ± 0.04; n = 14) compared to untreated controls and mock-transfected, LV AS3m.C-treated samples (1.05 ± 0.03; n = 15)
Figure 6.

The bifunctional LV downregulating HbS does not generate a β-thalassemic phenotype
(A) Schematic representation of the protocol used to evaluate erythroid differentiation and RBC parameters. We transduced SCD/HD HSPCs with LV AS3m.C and transfected them with Cas9 protein. As controls, we transfected SCD/HD HSPCs with RNPs containing gR-C. Images of in vitro-cultured RBCs were taken using the Phasics SID4-HR GE camera. The BIO-Data interface software was used to analyze RBC images. (B) CE-HPLC quantification of Hb tetramers in untreated HD and SCD cells (Ctr UT), gR-C-treated HD and SCD cells (gR-C), and LV AS3m.C-treated SCD HSPCs that were mock transfected (“–”) or transfected with Cas9-GFP protein. We plotted the percentage of each Hb type over the total Hb tetramers. VCN values are reported in blue and indels in black below the graph. The α-globin/non-α-globin ratio (determined by RP-HPLC) is reported on top of the histograms.
The erythroid differentiation of edited HSPCs cells treated with LV AS3m.C was not impaired. Indeed, the expression of surface markers identifying the different erythroid populations (CD36, CD71, CD235A, CD49d, and Band3) and the frequency of enucleated cells were similar in control and treated samples all along the differentiation (Figures 7A–7E; Figure S7A). On the contrary, in β-thalassemia-like cells erythroid differentiation was delayed as well as RBC enucleation (Figures 7A–7E).
Figure 7.

Erythroid differentiation and RBC parameters were not impaired in cells derived from LV AS3m.C-transduced, edited SCD HSPCs
(A–E) Flow cytometry analysis of the enucleation rate and of the early (CD71, CD36, and CD49d) and late (CD235A and Band3) erythroid markers at day 13, 16, and 19 or 20 of erythroid differentiation of untreated HD (n = 1) and SCD (n = 3) cells (Ctr UT), gR-C-treated HD (n = 1) and SCD (n = 1) cells (gR-C), and LV AS3m.C-treated SCD HSPCs that were mock transfected or transfected with Cas9-GFP protein (n = 3). VCN and indel values are reported below the graph as mean ± SEM. (A–D) Data are expressed as mean ± SEM. (A) Enucleation rate measured using DRAQ5 nuclear staining. (B–D) Proportion of CD71+, CD36+, and CD235A+ cells during erythroid differentiation. (E) Expression of CD49d and Band3 during the erythroid differentiation. During terminal erythroid differentiation, cells lose CD49d expression. (F–I) RBC parameters extracted using the BIO-Data software. RBCs were obtained after 19 days of differentiation from SCD HSPCs transduced with LV AS3m.C and either mock or Cas9 transfected. As controls, we used RBCs obtained from SCD/HD HSPCs transfected with RNPs containing gR-C For each population; data were normalized to the total number of RBCs and are reported as overlaid histograms. Darker colors represent controls and lighter colors edited samples. From top to bottom: control HD RBCs compared with HD RBCs derived from HSPCs treated with gR-C; control SCD RBCs compared with SCD RBCs derived from HSPCs treated with gR-C; control SCD RBCs derived from HSPCs transduced with LV AS3m.C and mock transfected compared to RBCs derived from HSPCs transduced with LV AS3m.C and transfected with Cas9 protein. (F) Dry mass (pg). (G) Surface (μm2). (H) Perimeter (μm). (I) Ellipticity.
Furthermore, we acquired images from in vitro-differentiated RBCs derived from SCD HSPCs treated with LV AS3m.C to evaluate several morphological and physical parameters in a quantitative manner (Figure 6A). β-Thalassemia-like RBCs were characterized by a reduced dry mass compared to mock-treated control samples, due to the lower Hb content (Figure 7F; Figure S7B). Surface, perimeter, and ellipticity parameters were altered in β-thalassemia-like RBCs (Figures 7G–7I; Figures S7C–S7E), reflecting the anisocytosis and poikilocytosis characterizing β-thalassemic RBCs.45,46 In RBCs derived from LV AS3m.C-treated SCD HSPCs harboring ∼42% of edited HBB alleles, only a small percentage of cells showed reduced dry mass, surface, and perimeter (6.2% ± 2.3%, 5.4% ± 3.6% and 6.5% ± 3.7%; n = 2, respectively) in comparison to RBCs obtained from mock-transfected LV AS3m.C HSPCs (Figures 7F–7H; Figures S7B and S7D), while ellipticity was overall unaffected (Figure 7I; Figure S7E). These frequencies were modestly increased in RBCs derived from SCD HSPCs harboring 63% of edited alleles (10.1% for dry mass, 13.4% for surface, and 10.2% for perimeter).
Finally, editing of HBG promoters did not impact the differentiation and enucleation of LV AS3m.13bpdel-transduced SCD HSPCs (Figures S8B–S8E). α-Chain/non-α chain ratios, surface, dry mass, perimeter, and ellipticity were similar in RBCs derived from edited LV AS3m.13bpdel-transduced HSPCs and control samples (Figures S8A and S8F–S8I).
In conclusion, these analyses showed that erythroid differentiation of SCD HSPCs edited using bifunctional LVs and Cas9 transfection is largely unaltered.
Discussion
LVs expressing a β-globin transgene to compensate β-globin deficiency in β-thalassemia, or to inhibit Hb polymerization in SCD, have shown promising clinical outcomes7,11,47. However, to be effective, gene addition strategies require the sustained engraftment of highly transduced HSCs.8 This could increase the genotoxic risk particularly in SCD patients who have an increased probability of developing hematological malignancies compared to the general population.48,49 While promising approaches aimed at improving LV-derived transgene expression have been investigated,50 reaching therapeutic globin expression with a low VCN is still challenging, as LVs cannot accommodate the entire β-globin locus control region (βLCR) responsible for high-level expression of the β-like globin genes.
Interestingly, clinical observations in a compound β0/βS heterozygous SCD patient indicate that β-globin deficiency was associated with amelioration of the disease phenotype despite the relatively low gene marking in engrafted HSCs.8 Furthermore, a mild disease phenotype is observed in SCD and β-thalassemia patients harboring HPFH mutations.19,51,52 These clinical outcomes support the rationale of the design of gene therapy approaches that combine the expression of an anti-sickling β-globin transgene with editing strategies aimed to either reduce the βS-globin expression or to increase the γ-globin content to counteract HbS polymerization in SCD or to increase Hb content in β-thalassemia.
We designed LVs simultaneously driving the expression of a β-globin chain carrying three anti-sickling amino acids,13 and a sgRNA either to introduce frameshift mutations in the HBB gene (LV AS3m.C) or to reactivate HbF expression by disrupting the BCL11A binding site in the γ-globin promoters (LV AS3m.13bpdel)5 or by downregulating BCL11A expression (LV AS3m.BCL11A).5
The editing efficiency at the on-target loci was positively correlated with the number of integrated LV copies in HUDEP-2 cells and SCD primary HSPCs, demonstrating the efficient LV-derived sgRNA expression and the formation of the functional CRISPR-Cas9 RNP complexes in transfected cells. These data are in line with combinatorial systems based on the stable LV-mediated delivery of sgRNAs coupled with transient transfection of Cas9 mRNA in human HD HSPCs to downregulate the expression of cell surface proteins.53 Importantly, we achieved high editing efficiencies using Cas9 protein delivery that we previously demonstrated to be less toxic and more efficient than mRNA delivery in primary HSPCs.43 A recent report showed that precomplexing of Cas9 protein with a non-targeting sgRNA was required to achieve high genome editing frequency using LV-delivered sgRNAs.54 Interestingly, our system was already efficient by transfecting Cas9 protein alone, and the editing efficiency was not further increased by complexing the Cas9 with a nontargeting sgRNA (data not shown). Importantly, we applied this technology to develop a gene therapy approach for β-hemoglobinopathies by combining the efficient LV-based delivery of sgRNAs with the expression of a therapeutic β-globin transgene.
The delivery of Cas9 protein in clinically relevant SCD HSPCs transduced with LV AS3m.C resulted in robust downregulation of HBB expression at both the mRNA and protein levels. Of note, introduction of silent mutations in the AS3 sequence prevented undesired editing and inactivation of the transgene. The increased availability of α-globin chains caused by β-globin downregulation favored the formation of AS3-containing Hb tetramers, thus increasing the total anti-sickling Hb content in RBCs derived from edited HSPCs, as compared to RBCs obtained from control HSPCs harboring a similar number of LV copies. In a parallel approach, the editing of the γ-globin promoters in SCD HSPCs transduced with LV AS3m.13bpdel increased the total content of anti-sickling Hb tetramers by inducing the expression of both Gγ- and Aγ-globins. As previously reported,20,23,55,56 editing of the −115 region of the HBG promoters disrupts the BCL11A binding site and evicts BCL11A, allowing the recruitment of the NF-Y transcriptional activator at the −86 region and γ-globin reactivation. The sgRNAs targeting the HBB gene or the HBG promoters showed minimal off-target activity as detected by GUIDE-seq and targeted NGS sequencing of the potential off-targets. However, although GUIDE-seq was identified as the best performing assay for ex vivo therapeutics,57, 58, 59 it has a limited sensitivity (0.1%) and therefore could fail to detect off-target events that occur at a low frequency but can still potentially lead to deleterious outcomes (e.g., clonal expansion and malignant neoplasms). More sensitive assays to assess the potential CRISPR-Cas9-associated genotoxic risk should be developed in particular for clinical applications aimed to modify hundreds of millions of cells.
Surprisingly, LV AS3m.BCL11A showed poor transduction and editing efficiencies in the BCL11A erythroid-specific enhancer in SCD HSPCs. We have not clarified the mechanisms of poor transduction by LV AS3m.BCL11A. In cell lines, we have not observed any significant difference in titer and infectivity between LV AS3m.BCL11A and the other vectors, although the LV harboring the sgRNA targeting the BCL11A enhancer tends to have a lower infectivity that could impact the efficiency of transduction of primary HSPCs. In our previous study, we also compared LVs with similar titer and infectivity (as determined in cell lines) and observed, for some of these vectors, a low transduction of HSPCs, likely related to specific sequences that are detrimental to the vector performance in primary cells.43 Furthermore, bi-allelic disruption of this enhancer is required to achieve HbF reactivation, and the selected sgRNA targeting BCL11A26 showed numerous potential off-target sites. For these reasons, we decided not to further pursue this approach. The selection of a more efficient and specific sgRNA disrupting critical regions in the BCL11A enhancer will allow us to ameliorate this therapeutic approach in future experiments.
Interestingly, the high content of anti-sickling hemoglobins observed with both LV AS3m.C and LV AS3m.13bpdel resulted in reduced HbS levels, Hb polymerization, and frequency of sickled RBCs compared to the conventional gene addition approach. Importantly, compared to a LV expressing only the anti-sickling transgene, our combined approach requires a lower VCN to achieve a therapeutic effect, thus reducing the potential genotoxicity risk associated with LV integration. In particular, we achieved similar amounts of HbS and anti-sickling hemoglobins (resembling the Hb profile of an asymptomatic heterozygous SCD carrier) with a VCN of ∼5.5 using the conventional gene addition strategy, and a VCN of ∼2.2 and ∼3.7 (associated with 30%–35% of edited HBB or HBG genes) using LV AS3m.C and LV AS3m.13bpdel, respectively. As a higher VCN was required to reach a similar editing frequency for LV AS3m.13bpdel compared to LV AS3m.C, we hypothesize that the difference in the extent of the correction of the disease phenotype between these two approaches is due to the higher editing efficiency of the gR-C compared to the sgRNA targeting the HBG promoters. Alternative sgRNAs targeting HbF inhibitory sequences23,60 could be tested in future studies to further enhance γ-globin reactivation with a lower VCN.
Importantly, we used flow cytometry and quantitative phase imaging to evaluate erythroid differentiation of edited HSPCs and quantify at a single-cell level morphological and physical parameters of in vitro-differentiated RBCs. This study is fundamental to demonstrate the safety of HSC-based treatments for β-hemoglobinopathies. β-Thalassemic erythroid cells showed delayed differentiation and enucleation, imbalance in the α-chain/non-α chain ratio, α-precipitates, anisocytosis, and poikilocytosis. Editing of the HBB gene or the γ-globin promoters in HSPCs did not negatively impact their differentiation and enucleation rate, allowing the production of mature RBCs with Hb content and morphological parameters largely similar to untreated controls. In particular, in erythroid cells derived from HBB-edited HSPCs, AS3-globin chain expression was sufficient to compensate for the lack of βS-globin expression in the vast majority of RBCs. In fact, only a small percentage of RBCs (∼5%–10%) displayed a β-thalassemic-like phenotype. We can speculate that a small fraction of poorly transduced HSPCs harboring bi-allelic disruption of the HBB gene give rise to this RBC subpopulation producing AS3 levels, which are insufficient to compensate βS downregulation. Importantly, preclinical and clinical data indicate that in allotransplanted or gene therapy-treated β-thalassemia subjects, β-thalassemic cells are counterselected in vivo because the erythroid precursors undergo apoptosis, and mature RBCs have a shorter lifespan compared to HD or corrected cells.5,11,61 Furthermore, we do not expect that such a low frequency of β-thalassemic RBCs will impact the correction of the SCD phenotype in vivo.
Other therapeutic strategies for SCD aim at reverting the SCD mutation by cleaving the sickle HBB gene using the CRISPR-Cas9 system and inserting through the HDR pathway a donor template containing the WT HBB sequence that is either provided as a single-strand oligonucleotide or delivered by a viral vector.14,62,63 More recently, we proposed a combined treatment for β-thalassemia based on the insertion of the AS3 transgene in the α-globin (HBA) locus to simultaneously express the therapeutic β-globin while reducing HBA expression levels36 and α-globin precipitates. However, although more promising than the standard gene addition strategy, these approaches currently suffer from poor HDR efficiency in HSCs. On the contrary, our combined approach is likely more prone to produce the desired genome modification, as it relies on the NHEJ pathway, known to be preferentially active and more efficient in HSCs compared to HDR.64, 65, 66, 67 Moreover, in SCD, failed HDR-mediated gene correction likely results in the NHEJ-mediated HBB gene disruption, which could likely not be compensated by the residual endogenous β-like globin chains, thus potentially generating a β-thalassemic phenotype.68 The generation of a β-thalassemia phenotype was observed only in a small fraction of RBCs using our combined strategy, as the same LV is co-expressing the sgRNA targeting HBB and the therapeutic AS3 chain that compensates for the knockdown of the endogenous HBB.
Ongoing trials for β-hemoglobinopathies using a genome editing strategy efficiently targeting the BCL11A enhancer showed early promising results.28 However, HbF reactivation and therapeutic benefit were modest when editing occurred at low efficiency.40 Furthermore, as reported in trials based on LV gene addition approaches, some patients did not attain normal levels of total Hb, and in SCD subjects HbS levels were not always decreased to the values observed in an asymptomatic heterozygous SCD carrier, suggesting the persistence of a subset of non-corrected RBCs.69
Our strategies combine two gene therapy platforms currently under evaluation in experimental clinical trials for β-hemoglobinopathies, potentially improving the efficacy of gene therapy to treat SCD and even β-thalassemia when AS3 expression is combined with HbF re-activation. Notably, in our strategies bi-allelic editing is likely not required when targeting either the sickle β-globin gene or the HBG promoters. However, there are some concerns related to the risks of combining LV and CRISPR-Cas9 approaches, which can both lead to potential deleterious DNA mutagenesis and DNA damage response. Although our study suggests that the treatment of patient HSPCs with both LV and Cas9 did not affect progenitor growth and differentiation, predictive assays, such as xenotransplantation in immunodeficient mouse models, are required to demonstrate the safety of the proposed approaches in repopulating stem cells and their progeny.
Finally, bifunctional LVs can have a wider range of potential applications in this field. In fact, by simply modifying the sgRNA sequence, the bifunctional LVs could be exploited to develop different therapeutic approaches for β-hemoglobinopathies (e.g., sickle β-globin or BCL11A downregulation;26 inactivation of HBG inhibitory sequences;20,23 insertion of HBG activating sequences70,71) by using a variety of editing tools (e.g., Cas9 nucleases, base editors, epigenome editors, and prime editors72, 73, 74).
More generally, the combination of gene addition and genome editing strategies has the potential to simultaneously induce the expression of therapeutic proteins and downregulate disease-causing genes. By way of example, we envision that this versatile technology can allow the development of therapeutic strategies for the treatment of autosomal dominant disorders, which requires the downregulation of the dominant allele. As it is difficult to achieve targeted downregulation of the mutant gene without impairing the expression of the WT allele, our strategy could allow the disruption of the endogenous genes, and simultaneously the LV-derived expression of a non-targeted WT allele (i.e., harboring silent mutations, which impair sgRNA binding and transgene disruption).
Materials and methods
LV production and titration
For LV production, the expression cassette consisting of DNase I hypersensitive sites HS2 (genomic coordinates [hg38]: chr11:5280255–5281665) and HS3 (genomic coordinates [hg38]: chr11:5284251–5285452) of the βLCR,75 and a HBB mini-gene extending from −265 bp upstream of the transcriptional start site to +300 bp downstream of the poly(A) addition site (genomic coordinates [hg38]: chr11:5225174–5227336) with a short version of intron 2 (genomic coordinates [hg38]: chr11:5203703–5204295) was cloned into a pCCL LV backbone to generate the pCCL.β-globin plasmid. The mutations determining three anti-sickling amino acid substitutions (G16D, E22A, and T87Q)13 were introduced in the pCCL.β-globin plasmid by in vitro site-directed mutagenesis to obtain the pCCL.AS3 plasmid. Silent mutations were inserted in the 19-bp AS3 transgene sequence recognized by gR-C to generate the pCCL.AS3m plasmid. To insert silent mutations, we used synonymous codons from HBB, HBD, HBG1, and HBG2 genes.
Synthetic oligonucleotides (Integrated DNA Technologies) containing the sgRNA expression cassette (with the optimized sgRNA scaffold35) were inserted in pCCL.AS3m, generating the transfer vectors (pCCL.AS3m.gRs) for the LV production.
Third-generation LVs were produced by calcium phosphate transient transfection of HEK293T cells with the transfer vector (pCCL.AS3m or pCCL.AS3m.gRs), the packaging plasmid pKLg/p.RRE, the Rev-encoding plasmid pK.REV, and the vesicular stomatitis virus glycoprotein G (VSV-G) envelope-encoding plasmid pK.G. The physical titer of vector preparations was measured using the HIV-1 Gag p24 antigen immunocapture assay kit (PerkinElmer, Waltham, MA, USA) and expressed as p24 ng/mL. The viral infectious titer was calculated by transducing HCT116 cells with serial vector dilutions, as previously described.76 VCN per diploid genome was calculated to determine the viral infectious titer, expressed as TU/mL. Viral infectivity was calculated as the ratio between infectious and physical titers (TU/ng p24).
HSPC transduction and transfection
We isolated cord blood CD34+ HSPCs and adult granulocyte colony-stimulating factor (G-CSF)-mobilized HSPCs from healthy donors. Human adult CD34+ HSPCs were isolated from the blood of SCD patients either after plerixafor mobilization (ClinicalTrials.gov: NCT02212535, Necker Hospital, Paris, France) or after erythrocytapheresis. Written informed consent was obtained from all of the subjects. All experiments were performed in accordance with the Declaration of Helsinki. The study was approved by the Regional Investigational Review Board (reference: DC 2014-2272, CPP Ile-de-France II “Hôpital Necker-Enfants malades”). HSPCs were purified by immunomagnetic selection with autoMACS (Miltenyi Biotec) after immunostaining with a CD34 MicroBead kit (Miltenyi Biotec). CD34+ cells were cultured (106 cells/mL) 24 h before transduction in X-VIVO 20 supplemented with penicillin/streptomycin (Gibco, Carlsbad, CA, USA), StemRegenin1 (SR1) at 250 nM (STEMCELL Technologies), and the following recombinant human cytokines (PeproTech): 300 ng/mL stem cell factor (SCF), 300 ng/mL Flt-3L, 100 ng/mL thyroperoxidase (TPO), and 60 ng/mL interleukin (IL)-3 (“expansion medium”).
Two hours before LV transduction, CD34+ cells were plated in RetroNectin-coated plates (10 μg/cm2, Takara Bio, Kusatsu, Japan) at 3 × 106 cells/mL in the expansion medium supplemented with 16,16-dimethyl-prostaglandin E2 (PGE2) (10 μM PGE2; Cayman Chemical).77,78 Cells were then transduced for 24 h in the expansion medium supplemented with protamine sulfate (4 μg/mL, Sigma-Aldrich, St. Louis, MO, USA or APP Pharmaceuticals, Schaumburg, IL, USA), PGE2, and LentiBOOST (Sirion Biotech69). After expansion of CD34+ cells for 48 h, 100,000 cells were transfected with 15 μg of Cas9-GFP protein (provided by Dr. De Cian) by using the P3 Primary Cell 4D-Nucleofector X kit S (Lonza) and the AMAXA 4D CA137 program (Lonza). After transfection, cells were plated at a concentration of 50,000 cells/mL in the erythroid differentiation medium.
Erythroid differentiation of HSPCs
After transfection, HSPCs were differentiated into mature RBCs using a three-phase protocol.44 From day 0 to day 6, cells were grown in a basal erythroid medium6 supplemented with 10−6 M hydrocortisone (Sigma), 100 ng/mL SCF (PeproTech), 5 ng/mL IL-3 (PeproTech), and 3 IU/mL erythropoietin (EPO) (Eprex, Janssen-Cilag). From day 6 to day 9, cells were cultured onto a layer of murine stromal MS-5 cells in the basal erythroid medium supplemented only with 3 IU/mL EPO. Finally, from day 9 to day 20, cells were cultured on a layer of MS-5 cells in the basal erythroid medium without any cytokines. Erythroid differentiation and enucleation rate were monitored by flow cytometry analysis.
RP- and CE-HPLC analysis
For HPLC analyses, HUDEP-2 cells were collected at day 9 of erythroid differentiation, whereas in vitro-differentiated RBCs were collected at day 20 of erythroid differentiation.
RP-HPLC analyses were performed using a Nexera X2 SIL-30AC chromatograph (Shimadzu) and the Lc Solution software. Globin chains were separated by HPLC using a 250 × 4.6 mm, 3.6-μm Aeris Widepore column (Phenomenex). Samples were eluted with a gradient mixture of solution A (water/acetonitrile/trifluoroacetic acid, 95:5:0.1) and solution B (water/acetonitrile/trifluoroacetic acid, 5:95:0.1). The absorbance was measured at 220 nm.
CA-HPLC analyses were performed using a Nexera X2 SIL-30AC chromatograph (Shimadzu) and the Lc Solution software. Hemoglobin tetramers were separated by HPLC using a two cation-exchange column (PolyCAT A, PolyLC, Columbia, MD, USA). Samples were eluted with a gradient mixture of solution A (20 mM bis-Tris, 2 mM KCN [pH 6.5]) and solution B (20 mM bis-Tris, 2 mM KCN, 250 mM NaCl [pH 6.8]). The absorbance was measured at 415 nm.
Sickling assay
In vitro-differentiated RBCs (day 19–20 of differentiation) derived from treated and control SCD HSPCs were exposed to an oxygen-deprived atmosphere (0% O2) and the time course of sickling was monitored in real time by video microscopy for 1 h, capturing images every 20 min using the Axio Observer Z1 microscope (Zeiss) and a ×40 objective. At 0% O2, around 400 cells per condition were counted and processed with ImageJ to determine the percentage of sickled RBCs in the total cell population.
Quantitative phase image microscopy of RBCs
50,000 to 100,00 in vitro-differentiated RBCs were centrifuged at 1,500 rpm for 5 min, resuspended in 50 or 700 μL of PBS 0.5% BSA, and placed in a μ-Slide with 15 or 8 wells (ibidi), respectively. We used the SID4 HR GE camera (Phasics, Saint-Aubin, France) with an inverted microscope (Eclispe Ti-E, Nikon) to obtain quantitative phase images of label-free RBCs.79 Images were taken using a ×40/0.60 objective. The BIO-Data R&D software (version 2.7.1.46) was used to perform a segmentation procedure to isolate each RBC from an image. We analyzed only enucleated RBCs, discarding cells with a surface density (dry mass/surface) higher than 0.55 pg/μm2. For each RBC, we evaluated dry mass (pg), surface (μm2), perimeter (μm), and ellipticity.80
Statistical analysis
Data were analyzed with GraphPad Prism software (version 7.0) and expressed as mean ± standard error of the mean (SEM), unless stated otherwise. Parametric tests (Student’s t test, unpaired t tests, two- or pne-way ANOVA test, and Sidak’s, Dunnett’s, and Tukey’s multiple comparisons test) were used according to datasets. The threshold for statistical significance was set to p < 0.05. Linear or non-linear regression analyses were performed to assess the correlation between frequency of genome editing and VCN or between frequency of genome editing or VCN and Hb expression.
Acknowledgments
This work was supported by State funding from the Agence Nationale de la Recherche under “Investissements d’avenir” program (ANR-10-IAHU-01), the Paris Ile de France Region under “DIM Thérapie génique” initiative, the Société d’Accélération du Transfert de Technologies-SATT IDF Innov, the Fondation Maladies Rare, and the Sanofi Innovation Award. We thank Sylvie Fabrega (Plateforme Vecteurs Viraux et Transfert de Gènes, SFR Necker, US 24, UMS 3633, 75014 Paris, France) for the LV production
Author contributions
S.R. designed and performed experiments, analyzed data, and wrote the paper; A.C., T.F., G.F., M.B., A. Casini, and G.M. performed experiments and analyzed data; N.O. and S.A. analyzed quantitative phase microscopy, C.M. analyzed off-target NGS data; A.D.C. provided reagents; J.-P.C., M.C., A. Cereseto, W.E.N., M.A., and B.W. contributed to the design of the experimental strategy; V.M. conceived the study, designed and performed experiments, analyzed data, and wrote the paper; A.M. conceived the study, designed experiments, analyzed data, and wrote the paper.
Declaration of interests
V.M. and A.M. are the inventors of two patents describing this gene addition/genome editing platform (WO/2018/220211, “Viral vectors combining gene therapy and genome editing approaches for gene therapy of genetic disorders”; WO/2018/220210, “Recombinant lentiviral vector for stem cell-based gene therapy of sickle cell disorder). The remaining authors declare no competing interests.
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.ymthe.2021.08.019.
Contributor Information
Vasco Meneghini, Email: meneghini.vasco@hsr.it.
Annarita Miccio, Email: annarita.miccio@institutimagine.org.
Supplemental information
References
- 1.Cavazzana M., Antoniani C., Miccio A. Gene therapy for β-hemoglobinopathies. Mol. Ther. 2017;25:1142–1154. doi: 10.1016/j.ymthe.2017.03.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Ngo S., Bartolucci P., Lobo D., Mekontso-Dessap A., Gellen-Dautremer J., Noizat-Pirenne F., Audard V., Godeau B., Galacteros F., Habibi A., ( Causes of death in sickle cell disease adult patients: Old and new trends. Blood. 2014;124:2715. [Google Scholar]
- 3.Sadelain M., Boulad F., Galanello R., Giardina P., Locatelli F., Maggio A., Rivella S., Riviere I., Tisdale J. Therapeutic options for patients with severe β-thalassemia: The need for globin gene therapy. Hum. Gene Ther. 2007;18:1–9. doi: 10.1089/hum.2006.151. [DOI] [PubMed] [Google Scholar]
- 4.Besse K., Maiers M., Confer D., Albrecht M. On modeling human leukocyte antigen-identical sibling match probability for allogeneic hematopoietic cell transplantation: Estimating the need for an unrelated donor source. Biol. Blood Marrow Transplant. 2016;22:410–417. doi: 10.1016/j.bbmt.2015.09.012. [DOI] [PubMed] [Google Scholar]
- 5.Miccio A., Cesari R., Lotti F., Rossi C., Sanvito F., Ponzoni M., Routledge S.J., Chow C.M., Antoniou M.N., Ferrari G. In vivo selection of genetically modified erythroblastic progenitors leads to long-term correction of β-thalassemia. Proc. Natl. Acad. Sci. USA. 2008;105:10547–10552. doi: 10.1073/pnas.0711666105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Weber L., Poletti V., Magrin E., Antoniani C., Martin S., Bayard C., Sadek H., Felix T., Meneghini V., Antoniou M.N., et al. An optimized lentiviral vector efficiently corrects the human sickle cell disease phenotype. Mol. Ther. Methods Clin. Dev. 2018;10:268–280. doi: 10.1016/j.omtm.2018.07.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ribeil J.-A., Hacein-Bey-Abina S., Payen E., Magnani A., Semeraro M., Magrin E., Caccavelli L., Neven B., Bourget P., El Nemer W., et al. Gene therapy in a patient with sickle cell disease. N. Engl. J. Med. 2017;376:848–855. doi: 10.1056/NEJMoa1609677. [DOI] [PubMed] [Google Scholar]
- 8.Magrin E., Miccio A., Cavazzana M. Lentiviral and genome-editing strategies for the treatment of β-hemoglobinopathies. Blood. 2019;134:1203–1213. doi: 10.1182/blood.2019000949. [DOI] [PubMed] [Google Scholar]
- 9.Cavazzana-Calvo M., Payen E., Negre O., Wang G., Hehir K., Fusil F., Down J., Denaro M., Brady T., Westerman K., et al. Transfusion independence and HMGA2 activation after gene therapy of human β-thalassaemia. Nature. 2010;467:318–322. doi: 10.1038/nature09328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kaiser J. 2021. Gene therapy trials for sickle cell disease halted after two patients develop cancer.https://www.sciencemag.org/news/2021/02/gene-therapy-trials-sickle-cell-disease-halted-after-two-patients-develop-cancer [Google Scholar]
- 11.Marktel S., Scaramuzza S., Cicalese M.P., Giglio F., Galimberti S., Lidonnici M.R., Calbi V., Assanelli A., Bernardo M.E., Rossi C., et al. Intrabone hematopoietic stem cell gene therapy for adult and pediatric patients affected by transfusion-dependent β-thalassemia. Nat. Med. 2019;25:234–241. doi: 10.1038/s41591-018-0301-6. [DOI] [PubMed] [Google Scholar]
- 12.Miccio A., Poletti V., Tiboni F., Rossi C., Antonelli A., Mavilio F., Ferrari G. The GATA1-HS2 enhancer allows persistent and position-independent expression of a β-globin transgene. PLoS ONE. 2011;6:e27955. doi: 10.1371/journal.pone.0027955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Levasseur D.N., Ryan T.M., Reilly M.P., McCune S.L., Asakura T., Townes T.M. A recombinant human hemoglobin with anti-sickling properties greater than fetal hemoglobin. J. Biol. Chem. 2004;279:27518–27524. doi: 10.1074/jbc.M402578200. [DOI] [PubMed] [Google Scholar]
- 14.Dever D.P., Bak R.O., Reinisch A., Camarena J., Washington G., Nicolas C.E., Pavel-Dinu M., Saxena N., Wilkens A.B., Mantri S., et al. CRISPR/Cas9 β-globin gene targeting in human haematopoietic stem cells. Nature. 2016;539:384–389. doi: 10.1038/nature20134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Wilkinson A.C., Dever D.P., Baik R., Camarena J., Hsu I., Charlesworth C.T., Morita C., Nakauchi H., Porteus M.H. Cas9-AAV6 gene correction of beta-globin in autologous HSCs improves sickle cell disease erythropoiesis in mice. Nat. Commun. 2021;12:686. doi: 10.1038/s41467-021-20909-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Park S.H., Lee C.M., Dever D.P., Davis T.H., Camarena J., Srifa W., Zhang Y., Paikari A., Chang A.K., Porteus M.H., et al. Highly efficient editing of the β-globin gene in patient-derived hematopoietic stem and progenitor cells to treat sickle cell disease. Nucleic Acids Res. 2019;47:7955–7972. doi: 10.1093/nar/gkz475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Genovese P., Schiroli G., Escobar G., Tomaso T.D., Firrito C., Calabria A., Moi D., Mazzieri R., Bonini C., Holmes M.C., et al. Targeted genome editing in human repopulating haematopoietic stem cells. Nature. 2014;510:235–240. doi: 10.1038/nature13420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Magis W., DeWitt M.A., Wyman S.K., et al. High-level correction of the sickle mutation amplified in vivo during erythroid differentiation. bioRxiv. 2019:432716. doi: 10.1016/j.isci.2022.104374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Forget B.G. Molecular basis of hereditary persistence of fetal hemoglobin. Ann. NY Acad. Sci. 1998;850:38–44. doi: 10.1111/j.1749-6632.1998.tb10460.x. [DOI] [PubMed] [Google Scholar]
- 20.Traxler E.A., Yao Y., Wang Y.-D., Woodard K.J., Kurita R., Nakamura Y., Hughes J.R., Hardison R.C., Blobel G.A., Li C., Weiss M.J. A genome-editing strategy to treat β-hemoglobinopathies that recapitulates a mutation associated with a benign genetic condition. Nat. Med. 2016;22:987–990. doi: 10.1038/nm.4170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Li C., Psatha N., Sova P., Gil S., Wang H., Kim J., Kulkarni C., Valensisi C., Hawkins R.D., Stamatoyannopoulos G., Lieber A. Reactivation of γ-globin in adult β-YAC mice after ex vivo and in vivo hematopoietic stem cell genome editing. Blood. 2018;131:2915–2928. doi: 10.1182/blood-2018-03-838540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Lux C.T., Pattabhi S., Berger M., Nourigat C., Flowers D.A., Negre O., Humbert O., Yang J.G., Lee C., Jacoby K., et al. TALEN-mediated gene editing of HBG in human hematopoietic stem cells leads to therapeutic fetal hemoglobin induction. Mol. Ther. Methods Clin. Dev. 2018;12:175–183. doi: 10.1016/j.omtm.2018.12.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Weber L., Frati G., Felix T., Hardouin G., Casini A., Wollenschlaeger C., Meneghini V., Masson C., De Cian A., Chalumeau A., et al. Editing a γ-globin repressor binding site restores fetal hemoglobin synthesis and corrects the sickle cell disease phenotype. Sci. Adv. 2020;6:eaay9392. doi: 10.1126/sciadv.aay9392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Antoniani C., Meneghini V., Lattanzi A., Felix T., Romano O., Magrin E., Weber L., Pavani G., El Hoss S., Kurita R., et al. Induction of fetal hemoglobin synthesis by CRISPR/Cas9-mediated editing of the human β-globin locus. Blood. 2018;131:1960–1973. doi: 10.1182/blood-2017-10-811505. [DOI] [PubMed] [Google Scholar]
- 25.Brendel C., Guda S., Renella R., Bauer D.E., Canver M.C., Kim Y.J., Heeney M.M., Klatt D., Fogel J., Milsom M.D., et al. Lineage-specific BCL11A knockdown circumvents toxicities and reverses sickle phenotype. J. Clin. Invest. 2016;126:3868–3878. doi: 10.1172/JCI87885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Canver M.C., Smith E.C., Sher F., Pinello L., Sanjana N.E., Shalem O., Chen D.D., Schupp P.G., Vinjamur D.S., Garcia S.P., et al. BCL11A enhancer dissection by Cas9-mediated in situ saturating mutagenesis. Nature. 2015;527:192–197. doi: 10.1038/nature15521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Chang K.-H., Smith S.E., Sullivan T., Chen K., Zhou Q., West J.A., Liu M., Liu Y., Vieira B.F., Sun C., et al. Long-term engraftment and fetal globin induction upon BCL11A gene editing in bone-marrow-derived CD34+ hematopoietic stem and progenitor cells. Mol. Ther. Methods Clin. Dev. 2017;4:137–148. doi: 10.1016/j.omtm.2016.12.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Frangoul H., Altshuler D., Cappellini M.D., Chen Y.S., Domm J., Eustace B.K., Foell J., de la Fuente J., Grupp S., Handgretinger R., et al. CRISPR-Cas9 gene editing for sickle cell disease and β-thalassemia. N. Engl. J. Med. 2021;384:252–260. doi: 10.1056/NEJMoa2031054. [DOI] [PubMed] [Google Scholar]
- 29.Liang P., Xu Y., Zhang X., Ding C., Huang R., Zhang Z., Lv J., Xie X., Chen Y., Li Y., et al. CRISPR/Cas9-mediated gene editing in human tripronuclear zygotes. Protein Cell. 2015;6:363–372. doi: 10.1007/s13238-015-0153-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Cradick T.J., Fine E.J., Antico C.J., Bao G. CRISPR/Cas9 systems targeting β-globin and CCR5 genes have substantial off-target activity. Nucleic Acids Res. 2013;41:9584–9592. doi: 10.1093/nar/gkt714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kurita R., Suda N., Sudo K., Miharada K., Hiroyama T., Miyoshi H., Tani K., Nakamura Y. Establishment of immortalized human erythroid progenitor cell lines able to produce enucleated red blood cells. PLoS ONE. 2013;8:e59890. doi: 10.1371/journal.pone.0059890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Brinkman E.K., Chen T., Amendola M., van Steensel B. Easy quantitative assessment of genome editing by sequence trace decomposition. Nucleic Acids Res. 2014;42:e168. doi: 10.1093/nar/gku936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Luo Y., Zhu D., Zhang Z., Chen Y., Sun X. Integrative analysis of CRISPR/Cas9 target sites in the human HBB gene. Biomed. Res. Int. 2015;2015:514709. doi: 10.1155/2015/514709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Cradick T.J., Qiu P., Lee C.M., Fine E.J., Bao G. COSMID: A web-based tool for identifying and validating CRISPR/Cas off-target sites. Mol. Ther. Nucleic Acids. 2014;3:e214. doi: 10.1038/mtna.2014.64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Dang Y., Jia G., Choi J., Ma H., Anaya E., Ye C., Shankar P., Wu H. Optimizing sgRNA structure to improve CRISPR-Cas9 knockout efficiency. Genome Biol. 2015;16:280. doi: 10.1186/s13059-015-0846-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Pavani G., Fabiano A., Laurent M., Amor F., Cantelli E., Chalumeau A., Maule G., Tachtsidi A., Concordet J.P., Cereseto A., et al. Correction of β-thalassemia by CRISPR/Cas9 editing of the α-globin locus in human hematopoietic stem cells. Blood Adv. 2021;5:1137–1153. doi: 10.1182/bloodadvances.2020001996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Roselli E.A., Mezzadra R., Frittoli M.C., Maruggi G., Biral E., Mavilio F., Mastropietro F., Amato A., Tonon G., Refaldi C., et al. Correction of β-thalassemia major by gene transfer in haematopoietic progenitors of pediatric patients. EMBO Mol. Med. 2010;2:315–328. doi: 10.1002/emmm.201000083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Esrick E.B., Lehmann L.E., Biffi A., Achebe M., Brendel C., Ciuculescu M.F., Daley H., MacKinnon B., Morris E., Federico A., et al. Post-transcriptional genetic silencing of BCL11A to treat sickle cell disease. N. Engl. J. Med. 2021;384:205–215. doi: 10.1056/NEJMoa2029392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Ahle S. 2020. Gene editing therapy shows early benefit for patients with SCD and beta thalassemia.https://www.ashclinicalnews.org/news/latest-and-greatest/crispr-shows-early-benefit-patients-sickle-cell-disease-beta-thalassemia/ [Google Scholar]
- 40.Smith A.R., Schiller G.J., Vercellotti G.M., et al. Preliminary results of a phase 1/2 clinical study of zinc finger nuclease-mediated editing of BCL11A in autologous hematopoietic stem cells for transfusion-dependent beta thalassemia. Blood. 2019;134(Suppl 1):3544. [Google Scholar]
- 41.Sankaran V.G., Menne T.F., Xu J., Akie T.E., Lettre G., Van Handel B., Mikkola H.K., Hirschhorn J.N., Cantor A.B., Orkin S.H. Human fetal hemoglobin expression is regulated by the developmental stage-specific repressor BCL11A. Science. 2008;322:1839–1842. doi: 10.1126/science.1165409. [DOI] [PubMed] [Google Scholar]
- 42.Yin J., Xie X., Ye Y., Wang L., Che F. BCL11A: A potential diagnostic biomarker and therapeutic target in human diseases. Biosci. Rep. 2019;39 doi: 10.1042/BSR20190604. BSR20190604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Lattanzi A., Meneghini V., Pavani G., Amor F., Ramadier S., Felix T., Antoniani C., Masson C., Alibeu O., Lee C., et al. Optimization of CRISPR/Cas9 delivery to human hematopoietic stem and progenitor cells for therapeutic genomic rearrangements. Mol. Ther. 2019;27:137–150. doi: 10.1016/j.ymthe.2018.10.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Giarratana M.-C., Kobari L., Lapillonne H., Chalmers D., Kiger L., Cynober T., Marden M.C., Wajcman H., Douay L. Ex vivo generation of fully mature human red blood cells from hematopoietic stem cells. Nat. Biotechnol. 2005;23:69–74. doi: 10.1038/nbt1047. [DOI] [PubMed] [Google Scholar]
- 45.Galanello R., Origa R. Beta-thalassemia. Orphanet J. Rare Dis. 2010;5:11. doi: 10.1186/1750-1172-5-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Needs T., Gonzalez-Mosquera L.F., Lynch D.T. StatPearls [Internet] StatPearls Publishing; 2021. Beta thalassemia.https://www.ncbi.nlm.nih.gov/books/NBK531481/ [Google Scholar]
- 47.Thompson A.A., Walters M.C., Kwiatkowski J., Rasko J.E.J., Ribeil J.A., Hongeng S., Magrin E., Schiller G.J., Payen E., Semeraro M., et al. Gene therapy in patients with transfusion-dependent β-thalassemia. N. Engl. J. Med. 2018;378:1479–1493. doi: 10.1056/NEJMoa1705342. [DOI] [PubMed] [Google Scholar]
- 48.Seminog O.O., Ogunlaja O.I., Yeates D., Goldacre M.J. Risk of individual malignant neoplasms in patients with sickle cell disease: English national record linkage study. J. R. Soc. Med. 2016;109:303–309. doi: 10.1177/0141076816651037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Brunson A., Keegan T.H.M., Bang H., Mahajan A., Paulukonis S., Wun T. Increased risk of leukemia among sickle cell disease patients in California. Blood. 2017;130:1597–1599. doi: 10.1182/blood-2017-05-783233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Breda L., Ghiaccio V., Tanaka N., Jarocha D., Ikawa Y., Abdulmalik O., Dong A., Casu C., Raabe T.D., Shan X., et al. Lentiviral vector ALS20 yields high hemoglobin levels with low genomic integrations for treatment of beta-globinopathies. Mol. Ther. 2021;29:1625–1638. doi: 10.1016/j.ymthe.2020.12.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Akinsheye I., Alsultan A., Solovieff N., Ngo D., Baldwin C.T., Sebastiani P., Chui D.H., Steinberg M.H. Fetal hemoglobin in sickle cell anemia. Blood. 2011;118:19–27. doi: 10.1182/blood-2011-03-325258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Steinberg M.H., Chui D.H.K., Dover G.J., Sebastiani P., Alsultan A. Fetal hemoglobin in sickle cell anemia: A glass half full? Blood. 2014;123:481–485. doi: 10.1182/blood-2013-09-528067. [DOI] [PubMed] [Google Scholar]
- 53.Yudovich D., Bäckström A., Schmiderer L., Žemaitis K., Subramaniam A., Larsson J. Combined lentiviral- and RNA-mediated CRISPR/Cas9 delivery for efficient and traceable gene editing in human hematopoietic stem and progenitor cells. Sci. Rep. 2020;10:22393. doi: 10.1038/s41598-020-79724-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Ting P.Y., Parker A.E., Lee J.S., Trussell C., Sharif O., Luna F., Federe G., Barnes S.W., Walker J.R., Vance J., et al. Guide Swap enables genome-scale pooled CRISPR-Cas9 screening in human primary cells. Nat. Methods. 2018;15:941–946. doi: 10.1038/s41592-018-0149-1. [DOI] [PubMed] [Google Scholar]
- 55.Martyn G.E., Wienert B., Yang L., Shah M., Norton L.J., Burdach J., Kurita R., Nakamura Y., Pearson R.C.M., Funnell A.P.W., et al. Natural regulatory mutations elevate the fetal globin gene via disruption of BCL11A or ZBTB7A binding. Nat. Genet. 2018;50:498–503. doi: 10.1038/s41588-018-0085-0. [DOI] [PubMed] [Google Scholar]
- 56.Liu N., Xu S., Yao Q., Zhu Q., Kai Y., Hsu J.Y., Sakon P., Pinello L., Yuan G.C., Bauer D.E., Orkin S.H. Transcription factor competition at the γ-globin promoters controls hemoglobin switching. Nat. Genet. 2021;53:511–520. doi: 10.1038/s41588-021-00798-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Chaudhari H.G., Penterman J., Whitton H.J., Spencer S.J., Flanagan N., Lei Zhang M.C., Huang E., Khedkar A.S., Toomey J.M., Shearer C.A., et al. Evaluation of homology-independent CRISPR-Cas9 off-target assessment methods. CRISPR J. 2020;3:440–453. doi: 10.1089/crispr.2020.0053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Shapiro J., Iancu O., Jacobi A.M., McNeill M.S., Turk R., Rettig G.R., Amit I., Tovin-Recht A., Yakhini Z., Behlke M.A., Hendel A. Increasing CRISPR efficiency and measuring its specificity in HSPCs using a clinically relevant system. Mol. Ther. Methods Clin. Dev. 2020;17:1097–1107. doi: 10.1016/j.omtm.2020.04.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Tsai S.Q., Joung J.K. Defining and improving the genome-wide specificities of CRISPR-Cas9 nucleases. Nat. Rev. Genet. 2016;17:300–312. doi: 10.1038/nrg.2016.28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Wu Y., Zeng J., Roscoe B.P., Liu P., Yao Q., Lazzarotto C.R., Clement K., Cole M.A., Luk K., Baricordi C., et al. Highly efficient therapeutic gene editing of human hematopoietic stem cells. Nat. Med. 2019;25:776–783. doi: 10.1038/s41591-019-0401-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Gaziev J., Lucarelli G. Stem cell transplantation for hemoglobinopathies. Curr. Opin. Pediatr. 2003;15:24–31. doi: 10.1097/00008480-200302000-00005. [DOI] [PubMed] [Google Scholar]
- 62.Cromer M.K., Vaidyanathan S., Ryan D.E., Curry B., Lucas A.B., Camarena J., Kaushik M., Hay S.R., Martin R.M., Steinfeld I., et al. Global transcriptional response to CRISPR/Cas9-AAV6-based genome editing in CD34+ hematopoietic stem and progenitor cells. Mol. Ther. 2018;26:2431–2442. doi: 10.1016/j.ymthe.2018.06.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Uchida N., Drysdale C.M., Nassehi T., Gamer J., Yapundich M., DiNicola J., Shibata Y., Hinds M., Gudmundsdottir B., Haro-Mora J.J., et al. Cas9 protein delivery non-integrating lentiviral vectors for gene correction in sickle cell disease. Mol. Ther. Methods Clin. Dev. 2021;21:121–132. doi: 10.1016/j.omtm.2021.02.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Chang H.H.Y., Pannunzio N.R., Adachi N., Lieber M.R. Non-homologous DNA end joining and alternative pathways to double-strand break repair. Nat. Rev. Mol. Cell Biol. 2017;18:495–506. doi: 10.1038/nrm.2017.48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Hustedt N., Durocher D. The control of DNA repair by the cell cycle. Nat. Cell Biol. 2016;19:1–9. doi: 10.1038/ncb3452. [DOI] [PubMed] [Google Scholar]
- 66.Nakamura K., Saredi G., Becker J.R., Foster B.M., Nguyen N.V., Beyer T.E., Cesa L.C., Faull P.A., Lukauskas S., Frimurer T., et al. H4K20me0 recognition by BRCA1-BARD1 directs homologous recombination to sister chromatids. Nat. Cell Biol. 2019;21:311–318. doi: 10.1038/s41556-019-0282-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Vítor A.C., Huertas P., Legube G., de Almeida S.F. Studying DNA double-strand break repair: An ever-growing toolbox. Front. Mol. Biosci. 2020;7:24. doi: 10.3389/fmolb.2020.00024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Magis W., DeWitt M.A., Wyman S.K., Vu J.T., Heo S.-J., Shao S.J., Hennig F., Romero Z.G., Campo-Fernandez B., McNeill M., et al. High-level correction of the sickle mutation amplified in vivo during erythroid differentiation. bioRxiv. 2018 doi: 10.1101/432716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Frangoul H., Bobruff Y., Cappellini M.D., Corbacioglu S., Fernandez C.M., de la Fuente J., Grupp S.A., Handgretinger R., Ho T.W., Imren S., et al. Safety and efficacy of CTX001 in patients with transfusion-dependent β-thalassemia and sickle cell disease: Early results from the Climb THAL-111 and Climb SCD-121 studies of autologous CRISPR-CAS9-Modified CD34+ hematopoietic stem and progenitor cells. Blood. 2020;136(Suppl 1):3–4. [Google Scholar]
- 70.Gaudelli N.M., Lam D.K., Rees H.A., Solá-Esteves N.M., Barrera L.A., Born D.A., Edwards A., Gehrke J.M., Lee S.J., Liquori A.J., et al. Directed evolution of adenine base editors with increased activity and therapeutic application. Nat. Biotechnol. 2020;38:892–900. doi: 10.1038/s41587-020-0491-6. [DOI] [PubMed] [Google Scholar]
- 71.Richter M.F., Zhao K.T., Eton E., Lapinaite A., Newby G.A., Thuronyi B.W., Wilson C., Koblan L.W., Zeng J., Bauer D.E., et al. Phage-assisted evolution of an adenine base editor with improved Cas domain compatibility and activity. Nat. Biotechnol. 2020;38:883–891. doi: 10.1038/s41587-020-0453-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Rees H.A., Liu D.R. Base editing: Precision chemistry on the genome and transcriptome of living cells. Nat. Rev. Genet. 2018;19:770–788. doi: 10.1038/s41576-018-0059-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Nakamura M., Gao Y., Dominguez A.A., Qi L.S. CRISPR technologies for precise epigenome editing. Nat. Cell Biol. 2021;23:11–22. doi: 10.1038/s41556-020-00620-7. [DOI] [PubMed] [Google Scholar]
- 74.Anzalone A.V., Randolph P.B., Davis J.R., Sousa A.A., Koblan L.W., Levy J.M., Chen P.J., Wilson C., Newby G.A., Raguram A., Liu D.R. Search-and-replace genome editing without double-strand breaks or donor DNA. Nature. 2019;576:149–157. doi: 10.1038/s41586-019-1711-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Hardison R., Slightom J.L., Gumucio D.L., Goodman M., Stojanovic N., Miller W. Locus control regions of mammalian beta-globin gene clusters: Combining phylogenetic analyses and experimental results to gain functional insights. Gene. 1997;205:73–94. doi: 10.1016/s0378-1119(97)00474-5. [DOI] [PubMed] [Google Scholar]
- 76.Lattanzi A., Duguez S., Moiani A., Izmiryan A., Barbon E., Martin S., Mamchaoui K., Mouly V., Bernardi F., Mavilio F., Bovolenta M. Correction of the exon 2 duplication in DMD myoblasts by a single CRISPR/Cas9 system. Mol. Ther. Nucleic Acids. 2017;7:11–19. doi: 10.1016/j.omtn.2017.02.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Zonari E., Desantis G., Petrillo C., Boccalatte F.E., Lidonnici M.R., Kajaste-Rudnitski A., Aiuti A., Ferrari G., Naldini L., Gentner B. Efficient ex vivo engineering and expansion of highly purified human hematopoietic stem and progenitor cell populations for gene therapy. Stem Cell Reports. 2017;8:977–990. doi: 10.1016/j.stemcr.2017.02.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Delville M., Soheili T., Bellier F., Durand A., Denis A., Lagresle-Peyrou C., Cavazzana M., Andre-Schmutz I., Six E. A nontoxic transduction enhancer enables highly efficient lentiviral transduction of primary murine T cells and hematopoietic stem cells. Mol. Ther. Methods Clin. Dev. 2018;10:341–347. doi: 10.1016/j.omtm.2018.08.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Bon P., Maucort G., Wattellier B., Monneret S. Quadriwave lateral shearing interferometry for quantitative phase microscopy of living cells. Opt. Express. 2009;17:13080–13094. doi: 10.1364/oe.17.013080. [DOI] [PubMed] [Google Scholar]
- 80.Aknoun S., Savatier J., Bon P., Galland F., Abdeladim L., Wattellier B., Monneret S. Living cell dry mass measurement using quantitative phase imaging with quadriwave lateral shearing interferometry: An accuracy and sensitivity discussion. J. Biomed. Opt. 2015;20:126009. doi: 10.1117/1.JBO.20.12.126009. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.