

Abstract
Acinetobacter baumannii is an insidious emerging nosocomial pathogen that has developed resistance to all available antimicrobials, including the last resort antibiotic, colistin. Colistin resistance often occurs due to mutations in the PmrAB two-component regulatory system. To better understand the regulatory mechanisms contributing to colistin resistance, we have biochemically characterized the A. baumannii PmrA response regulator. Initial DNA-binding analysis shows that A. baumannii PmrA bound to the Klebsiella pneumoniae PmrA box motif. This prompted analysis of the putative A. baumannii PmrAB regulon that indicated that the A. baumannii PmrA consensus box is 5′-HTTAAD N5 HTTAAD. Additionally, we provide the first structural information for the A. baumannii PmrA N-terminal domain through X-ray crystallography and we present a full-length model using molecular modelling. From these studies, we were able to infer the effects of two critical PmrA mutations, PmrA::I13M and PmrA::P102R, both of which confer increased colistin resistance. Based on these data, we suggest structural and dynamic reasons for how these mutations can affect PmrA function and hence encourage resistive traits. Understanding these mechanisms will aid in the development of new targeted antimicrobial therapies.
Keywords: Acinetobacter baumannii, colistin resistance, PmrA, response regulator, two-component system
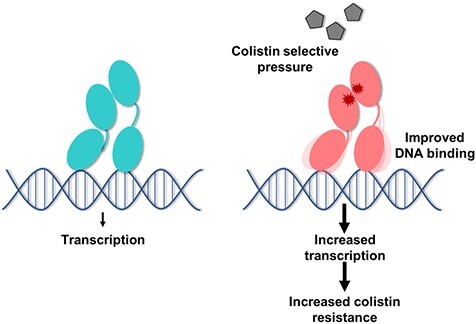
Graphical Abstract
Graphical Abstract.
Abbreviations
- Ara4N
4-amino-4-deoxy-L-arabinose
- BeF3−
Beryllium fluoride
- EMSA
electrophoretic mobility shift assay
- LPS
lipopolysaccharide
- MIC
minimum inhibitory concentration
- PDB
protein data bank
- pEtN
phosphoethanolamine
- SDS-PAGE
sodium dodecyl-sulphate polyacrylamide gel electrophoresis
- wt
wild type
Originally thought to be benign, the Gram-negative bacterium Acinetobacter baumannii is now a leading cause of hospital-acquired infections worldwide (1, 2). Acinetobacter baumannii predominantly causes ventilator-associated pneumonia in addition to urinary tract infections, skin and soft tissue infections, meningitis and bacteremia (3, 4). Outbreaks are often seen in intensive care units due to sustained colonization and resistance to environmental stresses. Acinetobacter baumannii can survive for months on hospital surfaces and medical equipment, allowing easy, indirect patient-to-patient transmission (5–8). In addition to such persistence, A. baumannii clinical isolates frequently display high levels of antibiotic resistance, with some studies estimating up to a 70% mortality rate due to multidrug-resistant infections (9). From the standpoint of economic impact, the Centers for Disease Control reports that $281 million in healthcare costs are attributable to carbapenem-resistance A. baumannii infections alone (10). Most alarmingly, there are reports of A. baumannii strains that are resistant to all available antibiotics (11–13). With all this in mind, we are in dire need of novel antimicrobial strategies to tackle A. baumannii.
Colistin (polymyxin E) is a cationic polypeptide and is used as a last resort antibiotic due to its severe side effects, which include nephrotoxicity and neurotoxicity (14). Colistin electrostatically interacts with the anionic lipid A component of lipopolysaccharide (LPS) in Gram-negative bacteria, causing permeabilization of the outer membrane. This disruption allows colistin to translocate across the outer membrane and destabilize the cytoplasmic membrane, resulting in cell death (15). Acinetobacter baumannii specifically has developed mechanisms to evade this last resort antibiotic; one study reports colistin resistance in 5.3% of Acinetobacter spp. clinical isolates found in the USA (16). In general, colistin resistance often occurs due to changes in the outer envelope. These modifications reduce the overall negative charge of LPS, lowering colistin’s affinity for the outer membrane (17). Gram-negative bacteria achieve this by either addition of 4-amino-4-deoxy-L-arabinose (Ara4N) to the lipid A component of LPS, addition of phosphoethanolamine (pEtN) to lipid A or by complete loss of LPS (17–19). Identification of the genes responsible for these mechanisms is vital for combating the rise in antimicrobial resistance.
Genetic mutations in the PmrAB two-component regulatory system are frequently identified in colistin-resistant A. baumannii strains (14, 18, 20–24). The PmrAB two-component system has also been implicated in colistin resistance in other Gram-negative bacteria (25), especially among the Enterobacteriaceae family members, including Klebsiella pneumoniae, Escherichia coli and Salmonella spp. (26–28). Most pmr mutations arise in patients receiving colistin treatment (20, 29). The pmrCAB operon encodes a pEtN transferase (PmrC), an OmpR/PhoB family response regulator (PmrA) and PmrA’s cognate sensor kinase (PmrB).
The prototypical two-component system phosphorylation cascade triggers when a sensor kinase, such as PmrB, detects a change in the environment and autophosphorylates at a conserved histidine residue. The subsequent phosphotransfer to a conserved aspartate residue on the matching response regulator protein, PmrA, causes dimerization and activation of the response regulator. This leads to DNA binding and a change in transcription. This response to the initial stimulus allows bacteria to rapidly adapt to changes in the environment (30, 31). Thus, studying two-component systems is highly important to understand bacterial stress response mechanisms, especially among multidrug-resistant bacteria.
Recently, two PmrA mutations that significantly increased colistin resistance in A. baumannii were identified. The PmrA::I13M mutant strain conferred a 16–32-fold increase in colistin minimum inhibitory concentration (MIC), and the PmrA::P102R mutant strain conferred a 32-fold increase in colistin MIC (24). Analysis showed that both strains had increased transcription of the pmrCAB operon compared to the parent strain. This led to the addition of pEtN to lipid A, resulting in increased colistin resistance. Since the PmrAB two-component system plays a vital role in A. baumannii, we wanted to explore the structural and biochemical impact of these PmrA mutations.
In this work, we first identified and characterized the previously unknown A. baumannii PmrA DNA-binding motif and began exploration of the PmrAB regulon. This then allowed us to study and compare the DNA-binding ability of wild-type (wt) PmrA and a colistin-resistant mutant. Until this work, structural information for A. baumannii PmrA did not exist, so we elucidated the high-resolution structure of the PmrA N-terminal domain (which contains residues I13 and P102) using X-ray crystallography. Since full-length structures of response regulators are notoriously difficult to solve due to their flexible nature, we generated a computational homology model of full-length PmrA. Finally, using our structure and the full-length model, we investigated the PmrA::I13M and PmrA::P102R mutations to provide insight into how associated changes in structure and dynamics could promote DNA-binding, thereby leading to observed colistin resistance among A. baumannii clinical isolates. This work provides the initial structural groundwork for understanding the ramifications of the critical A. baumannii PmrA::I13M and PmrA::P102R mutants.
Materials and Methods
Protein purification
Strains and plasmids used in this study are listed in Supplementary Table S1. Full-length PmrA and the PmrA amino-terminal domain (PmrAN) from A. baumannii strain ATCC 19606 were expressed with an amino-terminal His6 affinity tag using the expression vector pET28a (Novagen). pET28a-PmrA and pET28a-PmrAN were transformed into BL21 (DE3) E. coli cells. Overnight cultures were used to subculture into 2 L LB, that were then grown at 37°C, 160 rpm, to an optical density at 600 nm (OD600) of 0.7. Cultures were then induced with 1 mM IPTG and shifted to 25°C, 120 rpm overnight. Cells were harvested and pellets were resuspended in 30 mL lysis buffer containing 25 mM Tris pH 7.9 and 500 mM NaCl. Cells were lysed by sonication, and the resulting clarified supernatants were loaded onto a Ni-NTA agarose column (Qiagen), which had been equilibrated with lysis buffer. Next, the proteins were washed with 100 mL lysis buffer, followed by 100 mL 25 mM Tris pH 7.9 and 1 M NaCl. Proteins were eluted using an imidazole gradient (0–300 mM) in lysis buffer, and fractions were then dialyzed into 20 mM Tris pH 7.9, 400 mM NaCl and 5 mM EDTA. The His6 affinity tag was cleaved with the addition of 100 U thrombin for 2 h at room temperature and the reaction was quenched with 20 μM 4-(2-Aminoethyl)benzenesulfonyl fluoride hydrochloride (AEBSF) for 30 min at room temperature. Protein was concentrated using Millipore 10 K spin columns according to the manufacturer’s protocol.
pET28a-PmrA::I13M was expressed and purified using the above protocol, except dialysis buffer contained 25 mM Tris pH 7.9 and 100 mM NaCl. Following cleavage and quenching as above, PmrA::I13M was loaded onto a Q Sepharose resin column, equilibrated with Q Sepharose buffer, as per the manufacturer’s protocol (GE Healthcare). Protein was eluted using a NaCl gradient (0–500 mM) in 25 mM Tris pH 7.9, and concentrated using Millipore 10 K spin columns according to the manufacturer’s protocol.
Protein samples to be used for crystallography were further purified using a HiPrep™ 16/60 Sephacryl™ S-100 HR size exclusion column (Cytiva) equilibrated with 20 mM Tris pH 7.9 and 400 mM NaCl. Fractions containing purified protein (based on SDS-PAGE) were combined and concentrated using Millipore 10 K spin columns according to the manufacturer’s protocol. Protein was stored at 4°C for crystallography experiments.
Chemical activation of proteins with beryllium fluoride
Samples were activated using beryllium fluoride (BeF3−) as previously described (32–34). Briefly, 1 mg mL−1 purified protein was activated by addition of 7 mM MgCl2, 5 mM BeCl2 and 35 mM NaF. The solution was mixed and incubated at room temperature for at least 1 h.
X-ray crystallography
PmrAN was purified as described above. Crystals were grown at room temperature using hanging-drop vapour diffusion with a 1:1 ratio of protein to reservoir drop volumes. Hexagonal crystals were harvested from wells containing 0.2 M lithium sulphate monohydrate, 0.1 M Bis-Tris pH 5.5 and 25% w/v polyethylene glycol 3350 and were flash-frozen in liquid nitrogen. X-ray diffraction data was collected using the APS beamline 23-ID-D (GM/CA) at a wavelength of 1.0332 Å. Data was indexed, merged and scaled using HKL-2000 (35) and molecular replacement was achieved using PHENIX Phaser-MR (36) with Francisella tularensis subsp. novicida BfpR as a search model [Protein Data Bank (PDB) ID 6ONT] (37). The structure was refined in Coot (38) and using PHENIX.refine (36). Structure figures were produced using PyMOL (39).
Molecular modelling of full-length PmrA
A full-length model of PmrA was generated using MODELER v9.12 (40). The model was constructed using the A. baumannii PmrAN crystal structure (PDB ID 7M0S) and homologs PhoP from Mycobacterium tuberculosis (PDB ID 3R0J) (41), PmrA from K. pneumoniae (PDB IDs 4S04 and 4S05) (42), KdpE from E. coli (PDB IDs 4KFC and 4KNY) (43), BfmR from A. baumannii (PDB ID 5HM6) (32), QseB from F. novicida (PDB ID 5UIC) (44) and BfpR from F. novicida (PDB ID 6ONT) (37). A total of 500 models were generated from the sequence alignment of the above-mentioned protein structures with PmrA. Resulting models were scored based on the normalized DOPE (zDOPE) method (40). The structure with the lowest zDOPE score was subsequently run through MolSoft ICM Full Model Builder version 3.8-7c to generate a fully refined model (MolSoft LLC). Finally, the model was run through PROCHECK and PSVS to evaluate the quality of the model (45, 46). The ‘best’ model had the lowest zDOPE score, highest percentage of favoured and allowed Ramachandran regions and lowest MolProbity clash score. The model was further validated through comparison to the RoseTTAFold prediction of the PmrA C-terminal domain (47). The sequence of the DNA-binding domain was provided to the Robetta server (robetta.bakerlab.org). Output models were overlaid with the full-length model and RMSDs were calculated in PyMol (39).
Simulations of point mutants
The prediction of changes in protein stability due to point mutations was carried out in ICM-Pro (MolSoft LLC). The full-length PmrA model was subjected to 10 rounds of biassed probability Monte Carlo simulations using the Mutation–Protein Stability protocol outlined in the ICM User’s Guide. The simulation uses flexible side chains for the specific mutated residue and neighbouring residues while the rest of the structure remains rigid.
Electrophoretic mobility shift assays
DNA promoter fragments (Supplementary Table S2) were designed based on the K. pneumoniae PmrA box (PDB ID 4S04) (42), or using the A. baumannii ACICU genome (48). Equimolar concentrations of single-stranded DNA (Integrated DNA Technologies, Inc.) were annealed by heating at 95°C for 5 min in 20 mM Tris pH 7.9, 100 mM NaCl, 2 mM MgCl2 and 1 mM DTT, followed by slow cooling to room temperature. Binding reactions containing 1 μM DNA and indicated concentrations of protein were incubated for 5 min at room temperature before loading onto pre-chilled 8% TBE-acrylamide native gels. Samples were electrophoresed at 80 V in 1x TBE running buffer and stained with ethidium bromide for 5 min before visualizing bands. Following this, gels were stained with Coomassie blue and imaged.
Fluorescent anisotropy DNA-binding assay
A 25-bp oligonucleotide with a 5′-FAM (6-carboxyfluorescein) label was designed based on the K. pneumoniae PmrA box (49, 50). Equimolar concentrations of this KpPmrA_25mer_FAM-F single-stranded DNA fragment and the complimentary KpPmrA_25mer-R fragment (Supplementary Table S2) (Integrated DNA Technologies, Inc.) were mixed and annealed by incubating on ice for 1 h. Serial dilutions of the indicated protein concentrations were added to wells of a black 96-well plate (BrandTech) containing 5′-FAM labelled dsDNA in 20 mM Tris pH 7.9, 100 mM NaCl, 2 mM MgCl2, 0.1 mM DTT and 0.1 mg mL−1 BSA. The final reaction volume was 100 μl, and all assays were performed at room temperature. Fluorescent polarization was measured using Synergy H1 Hybrid Multi-Mode microplate reader (BioTek) and Gen5 software (BioTek) at an excitation wavelength of 482 nm and an emission wavelength of 528 nm. To determine Kd values, data were analysed using DynaFit software (BioKin Ltd.) to fit a standard equilibrium mechanism, a + b ab (51). Each experimental assay was performed at least three times for each protein.
Results and Discussion
A. baumannii PmrA binds to the K. pneumoniae PmrA box motif
The OmpR/PhoB family of response regulators controls transcription of numerous genes, including the pmrCAB operon, in response to a stimulus. It was originally shown that A. baumannii strains carrying PmrA::I13M and PmrA::P102R mutations had upregulated pmrCAB expression, which in turn led to increased colistin resistance (24). Our hypothesis was that the DNA-binding affinity of the mutants compared to wt would be altered, resulting in the observed changes in pmrCAB transcription. To further expand on this finding, we wanted to compare the DNA-binding ability of wt PmrA and the mutants. To do this, we first needed to identify the A. baumannii DNA-binding motif.
Although the A. baumannii PmrAB regulon has not been fully identified, PmrA-regulated genes have been well defined in other Gram-negative bacteria, such as Salmonella enterica and K. pneumoniae. The majority of these gene products function in LPS production and modification and include pEtN transferases (pmrC and cptA), Ara4N biosynthesis genes (pmrHFIJKLM and pmrE), genes that alter the LPS core (pmrG) and genes that determine the LPS O-antigen chain length (cld) (27, 28, 50). The PmrA DNA-binding motifs in these Gram-negative species appear to be homologous. The S. enterica PmrA recognition sequence consists of 5′-YTTAAK direct repeats (27) and the K. pneumoniae PmrA box consists of is 5′-CTTAAT and 5′-CCTAAG (50). Salmonella enterica PmrA and K. pneumoniae PmrA share 79.6% protein sequence identity, with 76.3% identity between their C-terminal DNA-binding domains (52). It is therefore unsurprising that the two organisms have homologous DNA-binding motifs.
Acinetobacter baumannii PmrA and K. pneumoniae PmrA share 39.6% sequence identity overall, with their C-terminal domains sharing 36.6% identity. However, the A. baumannii PmrA DNA-binding helix shares 54.5% sequence identity with the K. pneumoniae PmrA DNA-binding helix. Moreover, both helices contain four identical positively charged residues that potentially interact with DNA (A. baumannii PmrA residues H201, R204, K206 and K209) (53). Due to this conservation, and the fact that K. pneumoniae PmrA and S. enterica PmrA share homologous DNA-binding motifs, we decided to test A. baumannii PmrA’s ability to bind the K. pneumoniae PmrA box motif (KpPmrA_25mer; Fig. 1a). Electrophoretic mobility shift assays (EMSAs) showed that wt PmrA and PmrA::I13M bind to the KpPmrA_25mer (Fig. 1b). This suggests that the A. baumannii PmrA binding motif is similar to the K. pneumoniae PmrA box. We also generated a non-reversible activated construct of wt PmrA with the phosphomimic BeF3− (PmrA-BeF3−). This activation method has been shown to promote functional dimerization (32–34). Dimerization is an important activation step as it often facilitates response regulator binding to tandem DNA motifs (54). As expected, the EMSA showed that PmrA-BeF3− also binds to the KpPmrA_25mer (Fig. 1b). (All EMSA ethidium bromide-stained images and Coomassie Brilliant Blue-stained images have been overlaid in Supplementary Fig. S1). While we would have liked to similarly study PmrA::P102R, regrettably, this mutant expressed mostly in the insoluble pellet fraction, so no studies were possible. We are currently working to overcome this challenge. Unlike the full-length wt PmrA protein, the PmrA C-terminal domain alone was unable to bind to the KpPmrA_25mer (data not shown). This was somewhat expected since response regulators require communication between the N- and C-terminal domains via the flexible linker to coordinate structural and dynamic changes for DNA binding (55, 56). EMSA analysis of other response regulators, such as A. baumannii BfmR, also support this explanation (32).
Fig. 1.
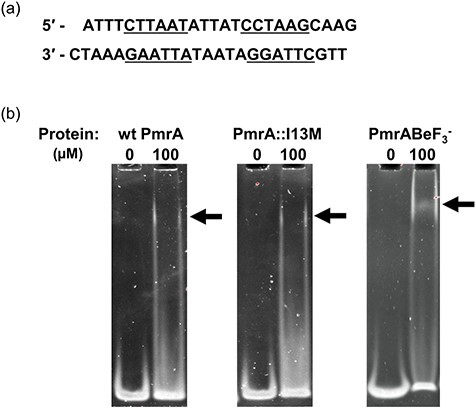
Defining the A. baumannii PmrA binding motif. (a) KpPmrA_25mer DNA sequence derived from the K. pneumoniae PmrA box (42). Inverted repeat sequences are underlined. (b) EMSAs showing binding of wt PmrA, PmrA::I13M and wt PmrA-BeF3− to 1 μM KpPmrA_25mer. Arrows indicate positions of mobility shifts.
We next performed fluorescent anisotropy to determine the DNA-binding affinity of both wt PmrA and PmrA::I13M to KpPmrA_25mer with a 5′-FAM (6-carboxyfluorescein) label (KpPmrA_25mer_FAM). Wt PmrA binds to this DNA fragment with a 2-fold higher affinity (Kd = 8.5 ± 1.7 μM; Fig. 2a) than the PmrA::I13M mutated protein (Kd = 16.6 ± 2.3 μM; Fig. 2b). Next we tested wt PmrA and PmrA::I13M, which had been activated using the phosphomimic BeF3− (Fig. 2c and d). As expected, the DNA-binding affinity of wt PmrA increased over 10-fold when activated with BeF3− (Kd = 0.78 ± 0.24 μM; Fig. 2c). Surprisingly, PmrA::I13M-BeF3− bound 55-fold tighter (Kd = 0.301 ± 0.088 μM; Fig. 2d) than inactive PmrA::I13M (Fig. 2b) and 2.5-fold higher than wt PmrA-BeF3− (Fig. 2c). These data suggest that the I13M mutation causes structural changes during activation that likely promote the dimerized conformation and therefore enhance DNA binding. These DNA-binding assays give further support for the upregulation of pmrCAB transcription reported previously in PmrA mutants (24) with the assumption that PmrA binds to its own promoter.
Fig. 2.
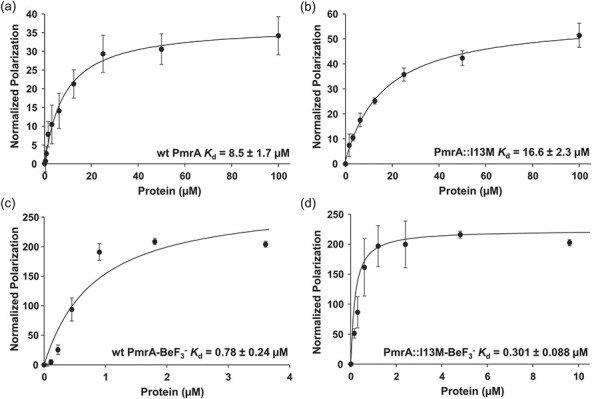
DNA-binding affinities of A. baumannii PmrA. DNA fluorescent anisotropy was used to determine the DNA-binding affinities of (a) wt PmrA, (b) PmrA::I13M, (c) wt PmrA-BeF3− and (d) PmrA::I13M-BeF3− binding to the KpPmrA_25mer_FAM dsDNA sequence.
The putative PmrAB regulon includes numerous genes required for LPS modifications
Several response regulators are capable of binding to their own promoters for autoactivation, including PmrA in Salmonella spp. (27, 57). Since PmrA mutations in A. baumannii are associated with increased pmrCAB transcription (24) we posited that A. baumannii PmrA also possesses autoregulatory activity. We analysed the pmrCAB promoter region for motifs similar to the KpPmrA_25mer and identified a similar motif, 5′-TTTAAG N5 TTTAAG, 48 bp upstream from the predicted translational start site of pmrC (Table 1). Figure 3 shows that both wt PmrA and wt PmrA-BeF3− bind to this DNA fragment. This observation is indicative that PmrA is indeed autoregulatory and further confirms the A. baumannii DNA-binding motif.
Table 1.
Putative A. baumannii PmrA Regulon
| ACICU locus taga | Predicted functionb | Motif identified (5′ → 3′) | Distance between motif and TSS (Bp) |
|---|---|---|---|
| ACICU_03004–ACICU_03002 | PmrC, PmrA, PmrB | TTTAAG N5 TTTAAG | 48 |
| ACICU_03001 | Putative lipid A 4′-phosphatase (LpxF-like) | ATTAAA N3 ATTAAA | 103 |
| ACICU_01072 | Putative lipid A pEtN transferase | TTTAAT N6 ATTAAT | 79 |
| ACICU_02907 | Diacylglycerol kinase | TTTAAT N5 TTTAAG | 44 |
| TTTAAA N5 TTTAAT | 55 | ||
| ACICU_02865–ACICU_02868 | Hypothetical glycosyl transferase family 2 | TTTAAG N5 ATTAAG | 85 |
| CTTAAG N5 TTTAAG | 96 | ||
| ACICU_00900–ACICU_00902 | PgaD, PgaC, PgaB | CTTAAG N0 TTTAAA | 41 |
| CTTAAG N5 AATAAA | 67 | ||
| TTTAAA N2 TTTAAG | 148 | ||
| ACICU_02895 | Putative polyketide synthase modules | TTTAAG N4 CTTAAT | 92 |
| TTTAAT N5 TTTAAG | 102 | ||
| ACICU_03041 | SbmA | TTTAAA N3 ATTAAA | 227 |
| ACICU_01552–ACICU_01551 | Hypothetical proteins | TTTAAG N1 TTTAAT | 51 |
| TTTAAG N5 TTTAAG | 58 | ||
| ACICU_01518 | Hypothetical protein | CTTAAA N5 ATTAAA | 126 |
| ACICU_00741 | Uncharacterized (COG3216) | TTTAAG N2 TTTAAT | 60 |
| TTTAAT N1 TTTAAG | 68 |
Fig. 3.
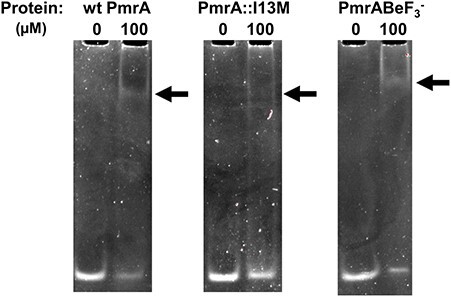
Acinetobacter baumannii PmrA binds to the pmrCAB promoter. EMSAs showing binding of wt PmrA, PmrA::I13M and wt PmrA-BeF3− binding to 2 μM AbPmrC_27mer. Arrows indicate positions of mobility shifts.
We next analysed promoter sequences from the putative A. baumannii PmrAB regulon. This regulon was originally suggested in a transcriptome study (22) that analysed differentially expressed genes in clinical A. baumannii isolates that had accumulated mutations over the course of an infection. Clinical isolates containing mutations in the pmr operon shared 21 dysregulated genes, some of which, expectedly, have also been identified as colistin-responsive genes (21, 22). From this gene set, we identified 11 promoter regions that each contains homologous sequences to the KpPmrA_25mer sequence. Table 1 shows the motifs identified along with the predicted downstream gene functions.
As anticipated, half of the promoter regions identified in Table 1 regulate genes predicted to function in LPS biosynthesis and modification mechanisms. pmrC, ACICU_03001 and ACICU_01072 encode enzymes that modify lipid A and have all been associated with colistin resistance (19). ACICU_02907 is also predicted to encode an enzyme involved in membrane phospholipid biosynthesis (58). ACICU_02865 potentially encodes a mannosyltransferase, and ACICU_02867 belongs to the glycosyltransferase family 2, both of which may have a role in cell wall biosynthesis (59). The remaining sets of promoters are associated with various other gene functions, including biofilm adhesion (pgaBCD) (60), or have not yet been characterized. Overall, these data clearly suggest that the PmrAB regulon is predominantly involved in production and maintenance of LPS. We propose that the A. baumannii PmrA consensus box is as follows: 5′-HTTAAD N5 HTTAAD. We are currently pursuing this further to identify how each of these genes is regulated by PmrA.
Crystal structure of the A. baumannii PmrA N-terminal receiver domain
Our initial goal was to better understand the effects of mutations in the N-terminal receiver domain of PmrA (PmrAN). Since there was no structural information available on PmrAN, we solved the high-resolution structure of this domain through X-ray crystallography (Fig. 4). The protein crystallized in the P1211 space group with unit cell parameters of a = 32.40 Å, b = 39.23 Å and c = 91.27 Å and α = γ = 90° and β = 95.66°. The structure was solved through molecular replacement using F. novicida BfpR (PDB ID 6ONT) (37) as a starting model and refined to 1.64 Å resolution with a crystallographic Rwork of 0.1741 and Rfree of 0.2136. Refinement corrected all Ramachandran outliers. Final statistics for data collection and refinement are listed in Supplementary Table S3.
Fig. 4.
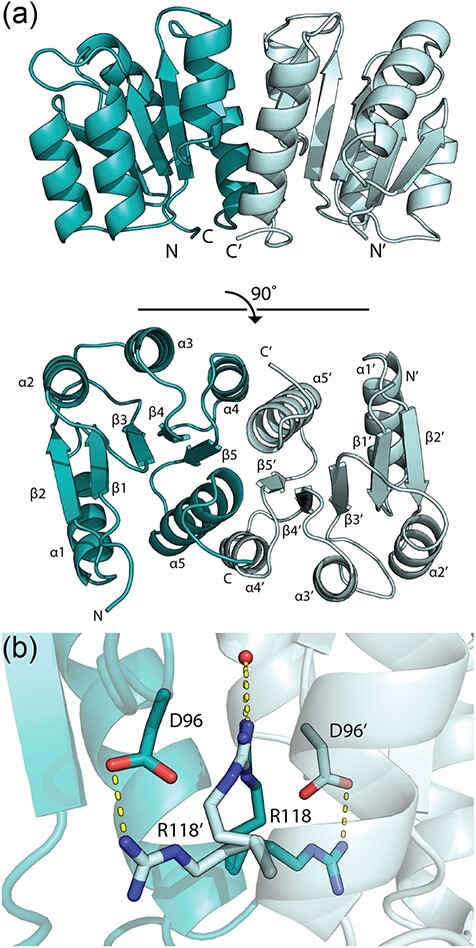
X-ray crystal structure of A. baumannii PmrAN. (a) The N-terminal domain of PmrA is a homodimer with each monomer coloured individually. The structure was solved and refined to 1.64 Å. Secondary structural elements are labelled. (b) PmrAN Arg118 shares density between the two monomer chains, allowing two alternative conformations for each. An interacting water molecule is shown in red. H-bonds are shown in yellow.
The PmrAN asymmetric unit contained two molecules representing a biologically relevant dimer in the active conformation (Fig. 4a). The dimeric state was confirmed through structural alignment with other known dimeric response regulator structures (Supplementary Fig. S2). The final model (Fig. 4a) contains residues 1–224 (100%) of monomer A and residues 1–223 (99.6%) of monomer B. As expected, each monomer follows the characteristic receiver domain α5/β5 fold, whereby five parallel β-sheets alternate with five amphiphilic α-helices, giving the final conserved β1-α1-β2-α2-β3-α3-β4-α4-β5-α5 conformation (44, 61–63). There were no structural discrepancies between the two monomers that interact via the α4-β5-α5 interface (Fig. 4). This specific interaction is highly conserved among OmpR/PhoB-type response regulators, providing a common dimerization mechanism (32, 37, 61, 64, 65).
Although PmrAN was not activated with a phosphomimic prior to crystallization, we expected it to crystallize in a dimeric conformation based on previous receiver domain structures. It is generally accepted that phosphorylation of response regulators by their cognate sensor kinases stimulates dimerization and activation. However, within a crystal lattice, active conformations of response regulators are often observed in the absence of phosphorylation, leading to the more plausible hypothesis that the inactive and active conformations are in constant equilibrium (55). A phosphorylation event simply shifts this balance to favour dimerization (66). The N-terminal domains of response regulators frequently crystalize in an active state, examples of which include A. baumannii BfmR (PDB ID H5M6), E. coli PhoP (PDB ID 2PKX) and Staphylococcus aureus ArlR (PDB ID 6IS1) (32, 64, 67).
A notable structural feature of the PmrAN crystal structure is that R118 is captured in two alternative confirmations, which share density between the two chains (Fig. 4b). R118 is located on the α5 helix and is conserved among the OmpR/PhoB family of response regulators. The typical orientation of R118, such as in PhoP (PDB ID 2PKX) (68) and ArlR (PDB ID 6IS1) (67), allows H-bonding to the conserved D96 residue (located on the α4–β5 loop), that connects the two monomers at the dimer interface (61). Alternatively, in our PmrAN structure, a second conformation of R118 H-bonds to a network of water molecules within the dimer interface.
To illustrate the structural conservation of response regulator receiver domains, we performed structural alignments of PmrAN against numerous other response regulators using PyMol (Fig. 5) (39). These alignments have an average RMSD of 0.912 Å, indicating strong structural similarity between PmrAN and other OmpR/PhoB receiver domains (Fig. 5b).
Fig. 5.
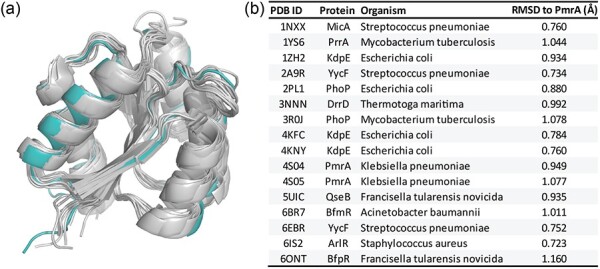
Structural alignment of A. baumannii PmrAN with other response regulators. (a) Structural alignment of our PmrAN structure to 16 other response regulator protein structures from the PDB. Acinetobacter baumannii PmrA is shown in teal and other response regulators are shown in grey. (b) Table showing RMSD values for alignment of each structure with A. baumannii PmrAN. A lower RMSD value indicates higher similarity between structures. Alignments were performed to chain A of each PDB structure. Values were calculated using PyMol.
Modelling of the A. baumannii full-length PmrA structure
The flexible nature of full-length response regulators that allows them to adopt multiple conformations often makes structural determination challenging. This was the case for full-length PmrA where, regrettably, all efforts to produce crystals that adequately diffracted were unsuccessful. To combat this, we have previously developed a hybrid method in which full-length structures are solved as separate N- and C-terminal domains. The individual domains are then spaced and oriented using distance restraints provided by chemical cross-linking and mass spectrometry (32). With that in mind, we attempted to express and crystallize the C-terminal DNA-binding domain of A. baumannii PmrA (PmrAC), but we have been unsuccessful to this point.
In the meantime, we have generated a full-length model for A. baumannii PmrA (Fig. 6a) using our solved PmrAN structure (Fig. 4) and other OmpR/PhoB response regulator structures as homologs for the C-terminal domain. Overall, PmrA is predicted to bear significant structural homology to other OmpR/PhoB response regulators. To validate our model, we utilized RoseTTAFold to predict the structure of PmrAC (47). RoseTTAFold is the latest advancement in protein structure prediction using deep learning to build computational models from protein sequences. Our full-length model is in close agreement with the RoseTTAFold results with an αC RMSD of 1.101 Å (Fig. 6b). This strongly suggests that we have built the best PmrA homolog possible with the currently available technology. The resulting model follows the archetypal OmpR/PhoB winged helix-turn-helix DNA-binding motif (Fig. 6a).
Fig. 6.
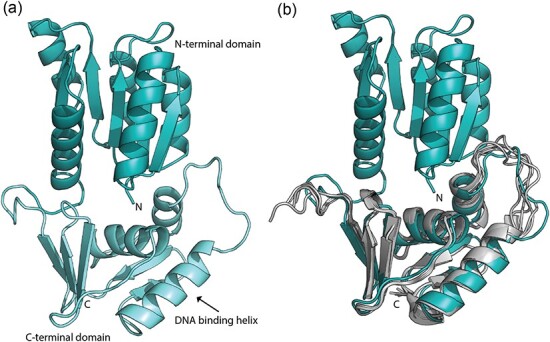
Model of full-length A. baumannii PmrA. (a) A full-length model of PmrA was generated based on the A. baumannii PmrAN crystal structure (Fig. 4) in addition to other homologous structures, as listed in the Materials and Methods. (b) Structural alignment of our A. baumannii PmrA model with a RoseTTAFold structural prediction of the PmrA C-terminal domain. RoseTTAFold output the top five models. The top model (model01) has a confidence score of 0.8391 (RMSD between model01 and PmrA is 1.101 Å).
The full-length PmrA monomer model (Fig. 6a) consists of the N- and C-terminal domains joined by a 12-amino acid linker. As noted, the OmpR/PhoB family uses this flexible linker region between the two domains to allow the protein to sample a range of conformations. The two most extreme conformations are described as ‘tucked’ (N- and C-terminal domains proximal) and ‘extended’ (N- and C-terminal domains distal) and play a role in correct binding of DNA (42, 43, 56). Both conformations have been captured in the K. pneumoniae PmrA crystal structures (PDB IDs 4S04 and 4S05) (42). In our A. baumannii model, PmrA is seen in the ‘tucked’ state (Fig. 6a).
As expected, our full-length PmrA model displays the distinctive structural characteristics of the OmpR/PhoB family. Since response regulators have highly conserved structural motifs, we were curious how specific mutations such as I13M and P102R could cause potential perturbations leading to altered protein activity, such as DNA-binding ability (as demonstrated in Fig. 2 for PmrA::I13M) (24). At this time, we have been unable to produce appropriate crystals of the mutants. With this in mind, we have generated homology models for comparative investigations.
The PmrA::I13M mutation likely alters the overall structure, plasticity and intra-protein communication networks of PmrA to promote DNA binding
Although Sun et al. (24) were the first to identify the novel PmrA::I13M mutation, other studies have identified other mutations located around this position that confer colistin resistance (PmrA::M12I (69, 70); PmrA::A14V (69)). Both PmrA::M12I and PmrA::A14V mutations arose in A. baumannii strains isolated from patients undergoing colistin therapy (69). PmrA::M12I was also identified as a spontaneous mutant on a colistin gradient plate (70), and PmrA::I13M was isolated in a transposon mutant library after exposure to colistin (24). These findings indicate that this region of PmrA is susceptible to advantageous mutations during exposure of A. baumannii to colistin.
The impact of the I13M mutation on the overall stability of PmrA was simulated using Molsoft ICM software (Molsoft LLC) (Fig. 7a). This program calculates the change in free energy of a mutant using the equation ΔΔG = ΔGmutant – ΔGwild type, whereby ΔΔG >0 kcal/mol represents destabilization. We modelled the PmrA::I13M mutation in the N-terminal structure as a single domain and also in the full length model. We then compared each mutant to their respective wts. The data show that the I13M mutant has a ΔΔG value of 0.51 kcal/mol when compared to the PmrAN structure and 0.31 kcal/mol when compared to the full-length PmrA model. Since both values are >0 kcal/mol, the I13M mutation is predicted to cause structural instability whereby the protein becomes more amenable to conformational changes.
Fig. 7.
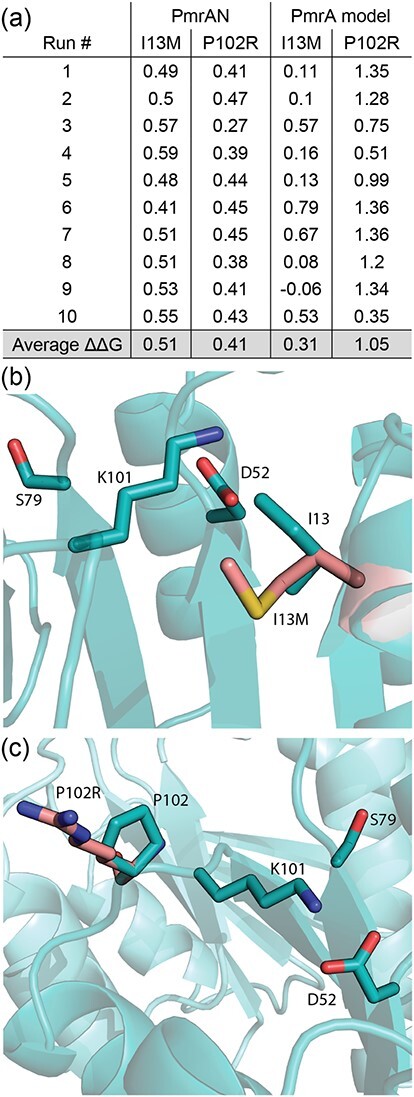
Simulations of biologically relevant PmrA point mutants. (a) The PmrA::I13M and PmrA::P102R mutated proteins were compared to the wt PmrAN structure and the PmrA full-length model to predict structural stability, whereby a ΔΔG value of >0 kcal/mol is considered destabilizing. (b) The PmrA::I13M mutation (pink) was modelled and overlaid onto the wt I13 residue (teal). Other relevant residues are also labelled. (c) The PmrA::P102R mutation (pink) was modelled and overlaid onto the wt P102 residue (teal). Other relevant residues are also labelled.
Both Ile and Met are non-polar, hydrophobic amino acids and are interchangeable via a single nucleotide mutation. However, unlike Ile, Met has unbranching side chains that, due to low rotational barrier energy, offer increased flexibility (71, 72). This is consistent with the predictions from Fig. 7a, which suggest that the PmrA::I13M mutant is less stable and more flexible than the wt. Structural plasticity is an important functional feature of response regulators since it enables continuous sampling of different conformations (73). Thus, this supports the hypothesis that the introduction of a Met residue causes structural perturbations throughout the N-terminal domain, which also propagate to the C-terminal DNA-binding domain, promoting an active conformation. This could explain the increase in pmrCAB expression in the PmrA::I13M mutant that lead to higher colistin MICs (24).
We also propose several other hypotheses for how the I13M structural changes impact PmrA’s function. Since structural analysis of the well-studied response regulator Spo0F (Bacillus subtilis, PDB ID 1PUX) has identified a number of residues that are important for response regulator structure–function relationships, our other hypotheses predicting the effects of the PmrA::I13M mutation are based on homology of PmrA to Spo0F. The important residues identified in Spo0F include the sensor kinase and phosphatase interaction residues (Spo0F G14, I15, I17, L18, E21 and V22) (74–76), the aspartyl pocket residues (Spo0F D10, D11 and D54) (77) and residues surrounding the aspartyl pocket (Spo0F K104, R16 and T82) (78). These correspond to PmrA residues M12, I13, E15, S16, T19 L20, D9, D10, D52, K101, A14 and S79, respectively (Supplementary Fig. S3) and are discussed below. The PmrA::I13M mutation was modelled and overlaid onto the wt I13 residue, and Fig. 7b demonstrates the orientations of some of these key residues in relation to I13.
It is predicted that residues M12, I13, S16, T19 and L20, all of which are located on the PmrA N-terminal α1 helix, likely interact with sensor kinases (74). Each of these residues is in proximity of the PmrA::I13M mutation. Local structural perturbations due to the mutation would dramatically impact this whole sensor kinase interaction domain, suggesting that PmrA::I13M has altered recognition of and interactions with its cognate sensor kinase, PmrB. Since pmrCAB transcription was elevated in the PmrA::I13M mutant (24), it is tempting to speculate that this mutation causes an altered interaction between PmrA and PmrB, which promotes the active conformation of PmrA, resulting in increased DNA-binding activity. Additionally, the I13M mutation could cause relaxed kinase specificity, allowing more sensor kinases (other than PmrB) to interact with and phosphorylate PmrA (74, 79). This too could explain increased DNA-binding activity of the PmrA::I13M mutant. Congruently, it is predicted that PmrA residues M12, I13, E15 and S16 are also involved in interactions with phosphatases (75,76). A mutation in this region could alter interactions between PmrA and phosphatases, leading to reduced dephosphorylation, and hence increased DNA-binding. This reiterates the importance of the α1 helix for recognition by both kinases and phosphatases. Thus, it is not surprising that a single mutation such as PmrA::I13M could have a huge effect on protein–protein interactions, leading to phosphorylation-induced changes in PmrA activity.
In addition to altering interactions with sensor kinases and phosphatases, the PmrA::I13M mutation could also affect local interactions between the aspartyl pocket itself (PmrA residues D9, D10 and D52; Fig. 7b) and the surrounding residues, thus affecting the phosphorylation state. The residues surrounding the aspartyl pocket form an H-bonding network between their amino acid side chains and the phosphate oxygen. Three major residues predicted to interact with the aspartyl-phosphate (PmrA D52) are PmrA K101, A14 and S79 (Fig. 7b) (78). PmrA K101 is an invariant residue at the end of the β5 sheet that is required for proper phosphorylation of response regulators and subsequent conformational changes (63, 78). PmrA I13 is in close proximity to K101 (Fig. 7b). Therefore, we surmise that an I13M mutation would alter this interaction and could bring K101 closer to the aspartyl pocket, stabilizing its interaction with the aspartyl phosphate. Similarly, since PmrA A14 is next to the I13M mutation (Fig. 7b), local perturbations would alter the positioning of A14, also leading to altered interactions with the aspartyl-phosphate. Although the data in Fig. 7a suggest that the PmrA::I13M mutant is more flexible overall, this is a global prediction of whole protein stability and small-scale, local stabilizations are also possible.
Phosphorylation of the aspartyl pocket also causes additional structural changes away from the site of phosphorylation via intra-protein communication networks (78). Response regulators have two highly conserved ‘switch’ residues, PmrA Y98 and S79, that are flipped during activation to allow reorientation and interaction with the aspartyl-phosphate (63, 66). Therefore, these conserved residues are required for phosphorylation-induced conformational changes (63). PmrA K101 is proximal to the ‘switch’ residue, S79 (Fig. 7b). We suggest that the altered interaction between I13M, K101 and S79 could also bring S79 closer into proximity of the aspartyl pocket, further promoting this active interaction. This agrees with our data showing that PmrA::I13M-BeF3− binds to DNA with a tighter affinity than wt PmrA-BeF3− (Fig. 2d and c, respectively).
Intra-protein communication networks in two-domain response regulators are also vital in relaying phospho-activation signals from the N-terminal receiver domain to the C-terminal DNA-binding domain, so that it can bind DNA (55, 78). By analogy to Spo0F, the PmrA residues M12, I13, A14 (α1 helix), S79, Y98 (switch residues) and K101 that surround the phosphorylation site (aspartyl pocket D9, D10, D52) propagate information concerning the phosphorylation state. Such conformational changes are transmitted throughout the protein via buried residues L51 and L64 to the C-terminal end of the α3 helix, particularly R86 (Fig. 8). This region appears to be an integral part of the interface between the receiver and DNA-binding domain, with ~70% of response regulators having a Lys or Arg in this position. It is suggested that this highly conserved residue may be involved in limiting the movement of the C-terminal domain when the protein is unphosphorylated (78). Interactions between R68 and the C-terminal domain can be perturbed upon activation such that the C-terminal domain is released, enabling increased conformation sampling to provide more opportunities for DNA-binding. Consequently, we hypothesize that the PmrA::I13M mutation can affect the concerted motions and conformations of the aforementioned residues in the intra-protein communication network, increasing the ability of PmrA to bind DNA, and therefore promoting colistin resistance.
Fig. 8.
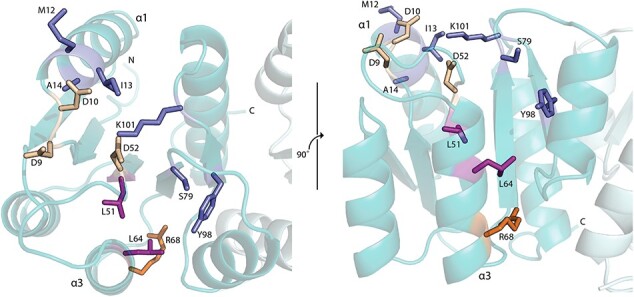
Intra-protein communication network. Top and side view of PmrA highlighting residues involved in intra-protein communication from the phosphorylation site to the DNA-binding domain. Aspartyl pocket residues (D9, D10 and D52) are shown in cream. Key residues surrounding the aspartyl pocket (M12, M13, A14 and K101) and switch residues (S79 and Y98) are shown in blue. Buried hydrophobic residues that transmit the signal through the protein (L51 and L64) are in purple. R68 is highlighted in orange.
To summarize, we predict that the PmrA::I13M mutation alters interactions with PmrB via the recognition face, enhances phosphorylation state and alters the overall structure and plasticity of PmrA to promote an active conformation and therefore DNA binding. This explains the increased pmrCAB transcription reported previously in the PmrA::I13M strain (24) and demonstrates how a single nucleotide mutation can dramatically alter protein structure and function, leading to antimicrobial resistance. An additional possibility is that the PmrA::I13M mutation affects interactions with RNA polymerase, and this too could contribute to altered transcription of the pmrCAB operon (80).
The PmrA::P102R mutation can affect multiple components of the PmrA N-terminal domain activation mechanism
Interestingly, multiple studies have additionally reported mutations in the A. baumannii PmrA residue P102 (P102R (24), P102H (18)) and also in other residues near this location (F105L (69)). The PmrA::P102R and PmrA::P102H mutations occurred after successive passaging of A. baumannii in increasing colistin concentrations (18, 24), whereas the strain containing the PmrA::F105L mutation was isolated from a patient receiving colistin treatment (69). This suggests that this particular region of PmrA, in addition to the region around residue I13, is also susceptible to mutagenesis that promotes pathogenicity, especially in response to colistin selective pressure.
PmrA residue P102 is in the cis conformation and is located in the PmrA N-terminal domain β5-α5 loop. Although this is a somewhat unusual structural feature, this cis-Pro has been observed among many response regulators (63). Pro residues contain a pyrrolidine ring, which covalently binds to the backbone and restricts the conformational freedom of this amino acid (81, 82). This restriction means that Pro residues are often found in tight turns and loops in protein structures, as is the case in PmrA (81).
We calculated the structural stability of PmrA::P102R, as we did for PmrA::I13M (Fig. 7a). The restrictive nature of the pyrrolidine ring in Pro residues decreases the conformational entropy for the unfolded state. Consequently, Pro residues such as P102 can help stabilize protein structures (83). Therefore, we hypothesized that the P102R mutation would decrease the stability of PmrA. Additionally, Arg residues benefit from conformational flexibility due to their long side chains (84) and studies show that Arg residues are less rigid than Pro residues (85). This further supports our hypothesis that the PmrA::P102R structure is more flexible than wt PmrA. Simulations of PmrA::P102R predict that this mutated protein has a ΔΔG value of 0.41 kcal/mol when compared to the PmrAN structure, and ΔΔG = 1.05 kcal/mol when compared to the PmrA full-length model (Fig. 7a). Since these values are >0 kcal/mol, these data confirm our hypothesis, indicating a reduction in protein stability and an increase in general flexibility in the PmrA::P102R mutant.
Limited conformations of P102 induced by the pyrrolidine ring also cause steric hindrance of preceding residues, such as K101 (Fig. 7c) (86). Hence, the PmrA::P102R mutation would remove the cis-peptide bond to K101, relieving the conformational restraints caused by the pyrrolidine ring. This dynamic freedom would alter interactions between K101 and the aspartyl-phosphate, D52, and it is conceivable that these structural changes would promote phosphorylation and activation. This could explain the increased pmrCAB transcription reported in the PmrA::P102R mutant strain (24).
Since the PmrA::P102R mutation perturbs the conserved K101 residue, this would also affect the S79 ‘switch’ residue, as described above for PmrA::I13M. Alterations to S79 (Fig. 7c) may shift the equilibrium to increase sampling of the activated ‘switched’ conformation, promoting DNA binding (66). Further supporting this, the Y98 ‘switch’ residue is located near the P102 residue. The introduction of an Arg residue would perturb the backbone through to residue Y98, and this relaxation could allow Y98 to move more freely into the active conformation. This provides another possible reason for increased colistin resistance in the PmrA::P102R strain.
As mentioned, P102R is located on the β5-α5 loop, which forms part of the conserved α4-β5-α5 dimer interface (61). Models of other response regulator dimer interaction domains allowed us to hypothesize that residue P102 is directly involved in forming the hydrophobic interface between two PmrA monomers (68, 87). The specific P102R mutation exchanges a hydrophobic residue for a hydrophilic one. Hydrophilic residues such as Arg are usually surface exposed and form hydrogen bonds with water molecules, whereas hydrophobic residues such as Pro are typically less exposed (84). Hence, altered interactions between the exposed positive Arg and a negative residue on the partnering monomer would contribute to the network of salt bridges at the dimer interface (61). This could alter the affinity between two monomers, promoting dimerization, and further confirming that the P102R mutation would enhance the DNA-binding activity of PmrA.
Furthermore, it is likely that the location of residue P102 is also accessible to sensor kinases prior to dimerization (88, 89). Similar to the PmrA::I13M mutation, this suggests that the PmrA::P102R mutation could affect the interaction between PmrA and PmrB. For instance, the affinity of the PmrA/PmrB interaction could increase, leading to enhanced phosphorylation. Additionally, the mutation could alter sensor kinase specificity, especially due to the flexible nature of Arg residues, leading to more interactions and phosphorylation events. Both of these occurrences would cause increased transcription of pmrCAB in the PmrA::P102R mutant.
Overall, we predict that PmrA::P102R impacts the N-terminal domain by altering the orientation of key residues (K101, S79, Y98) and by affecting interactions at both the dimer interface and the sensor kinase interface. Therefore, P102R affects multiple components of the activation mechanism that will have downstream structural perturbations throughout the N- and C-terminal domains. Flexibility is an essential feature of response regulators to allow sampling of multiple conformations for DNA binding (56), thus showing the potential pathogenic advantages of mutations such as P102R. Furthermore, this mutation could also alter binding to RNA polymerase, as mentioned for PmrA::I13M.
To summarize, we have predicted the A. baumannii PmrA consensus box as 5′-HTTAAD N5 HTTAAD. From this, we were able to identify numerous genes in the putative PmrA regulon, including its own pmrCAB promoter. We have solved the structure for the A. baumannii PmrA N-terminal domain using X-ray crystallography and have provided a model for the full-length PmrA structure from this multidrug-resistant pathogen. Lastly, we investigated two biologically relevant PmrA mutants through biochemical and computational approaches and have provided a detailed discussion on their potential structural perturbations. These structural changes could alter PmrA function, leading to colistin resistance among A. baumannii clinical isolates.
Author contributions
All authors conceived the study. S.P. and M.E.M. designed the experimental approaches. S.P. performed experiments. M.E.M. assisted S.P. with X-ray data collection and structure solving. M.E.M. performed computational work. All authors contributed to the writing of the manuscript.
Supplementary Material
Supplementary Data
Supplementary Data are available at JB Online.
Acknowledgements
Crystallographic data were collected at General Medical Sciences and Cancer Institute’s Structural Biology Facility (GM/CA) 23-ID at the Advanced Photon Source, Argonne National Laboratory. We would like to thank J. Hyatt and F. Jaimes for their assistance with protein purification. We would also like to thank A. J. Milton for reviewing the manuscript.
Contributor Information
Samantha Palethorpe, Department of Microbiology and Immunology Brody School of Medicine East Carolina University Greenville, NC 27834 United States.
Morgan E Milton, Department of Biochemistry and Molecular Biology Brody School of Medicine East Carolina University Greenville, NC 27834 United States.
Everett C Pesci, Department of Microbiology and Immunology Brody School of Medicine East Carolina University Greenville, NC 27834 United States.
John Cavanagh, Department of Biochemistry and Molecular Biology Brody School of Medicine East Carolina University Greenville, NC 27834 United States.
Funding
This work was supported by the National Institutes of Health [R01 AI136904].
Conflict of interest
None declared.
Accession numbers
The structure of PmrAN has been deposited to the Protein Data Bank with PDB entry ID 7M0S.
REFERENCES
- 1. Almasaudi, S.B. (2018) Acinetobacter spp. as nosocomial pathogens: epidemiology and resistance features. Saudi J. Biol. Sci. 25, 586–596 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Ayobami, O., Willrich, N., Harder, T., Okeke, I.N., Eckmanns, T., and Markwart, R. (2019) The incidence and prevalence of hospital-acquired (carbapenem-resistant) Acinetobacter baumannii in Europe, Eastern Mediterranean and Africa: a systematic review and meta-analysis. Emerg. Microbes Infect. 8, 1747–1759 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Howard, A., O’Donoghue, M., Feeney, A., and Sleator, R.D. (2012) Acinetobacter baumannii: an emerging opportunistic pathogen. Virulence 3, 243–250 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Morris, F.C., Dexter, C., Kostoulias, X., Uddin, M.I., and Peleg, A.Y. (2019) The mechanisms of disease caused by Acinetobacter baumannii. Front. Microbiol. 10, 1601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Sherertz, R.J., and Sullivan, M.L. (1985) An outbreak of infections with Acinetobacter calcoaceticus in burn patients: contamination of patients’ mattresses. J. Infect. Dis. 151, 252–258 [DOI] [PubMed] [Google Scholar]
- 6. Wang, S.H., Sheng, W.H., Chang, Y.Y., Wang, L.H., Lin, H.C., Chen, M.L., Pan, H.J., Ko, Y.J., Chang, S.C., and Lin, F.Y. (2003) Healthcare-associated outbreak due to pan-drug resistant Acinetobacter baumannii in a surgical intensive care unit. J. Hosp. Infect. 53, 97–102 [DOI] [PubMed] [Google Scholar]
- 7. Fournier, P.E., Richet, H., and Weinstein, R.A. (2006) The epidemiology and control of Acinetobacter baumannii in health care facilities. Clin. Infect. Dis. 42, 692–699 [DOI] [PubMed] [Google Scholar]
- 8. Farrow, J.M., III, Wells, G., and Pesci, E.C. (2018) Desiccation tolerance in Acinetobacter baumannii is mediated by the two-component response regulator BfmR. PLoS One 13, e0205638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Wong, D., Nielsen, T.B., Bonomo, R.A., Pantapalangkoor, P., Luna, B., and Spellberg, B. (2017) Clinical and pathophysiological overview of Acinetobacter infections: a century of challenges. Clin. Microbiol. Rev. 30, 409–447 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Centers for Disease Control and Prevention (U.S.), National Center for Emerging Zoonotic and Infectious Diseases (U.S.), Division of Healthcare Quality Promotion, and Antibiotic Resistance Coordination and Strategy Unit (2019) Antibiotic resistance threats in the United States, 2019. 10.15620/cdc:82532 [DOI]
- 11. Scott, P.T., Petersen, K., Fishbain, J., Craft, D.W., Ewell, A.J., Moran, K. et al. (2004) Morb. Mortal. Wkly. Rep. 2004 [Google Scholar]
- 12. Matthaiou, D.K., Michalopoulos, A., Rafailidis, P.I., Karageorgopoulos, D.E., Papaioannou, V., Ntani, G. et al. (2008) Risk factors associated with the isolation of colistin-resistant Gram-negative bacteria: a matched case-control study. Crit. Care Med. 36, 807–811 [DOI] [PubMed] [Google Scholar]
- 13. Taccone, F.S., Rodriguez-Villalobos, H., De Backer, D., De Moor, V., Deviere, J., Vincent, J.-L. et al. (2006) Successful treatment of septic shock due to pan-resistant Acinetobacter baumannii using combined antimicrobial therapy including tigecycline. Eur. J. Clin. Microbiol. Infect. Dis. Off Publ. Eur. Soc. Clin. Microbiol. 25, 257–260 [DOI] [PubMed] [Google Scholar]
- 14. Park, Y.K., Choi, J.Y., Shin, D., and Ko, K.S. (2011) Correlation between overexpression and amino acid substitution of the PmrAB locus and colistin resistance in Acinetobacter baumannii. Int. J. Antimicrob. Agents 37, 525–530 [DOI] [PubMed] [Google Scholar]
- 15. Wiese, A., Gutsmann, T., and Seydel, U. (2003) Towards antibacterial strategies: studies on the mechanisms of interaction between antibacterial peptides and model membranes. J. Endotoxin. Res. 9, 67–84 [DOI] [PubMed] [Google Scholar]
- 16. Queenan, A.M., Pillar, C.M., Deane, J., Sahm, D.F., Lynch, A.S., Flamm, R.K. et al. (2012) Multidrug resistance among Acinetobacter spp. in the USA and activity profile of key agents: results from CAPITAL Surveillance 2010. Diagn. Microbiol. Infect. Dis. 73, 267–270 [DOI] [PubMed] [Google Scholar]
- 17. El-Sayed Ahmed, M.A.E.-G., Zhong, L.-L., Shen, C., Yang, Y., Doi, Y., and Tian, G.-B. (2020) Colistin and its role in the era of antibiotic resistance: an extended review (2000–2019). Emerg. Microbes Infect. 9, 868–885 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Adams, M.D., Nickel, G.C., Bajaksouzian, S., Lavender, H., Murthy, A.R., Jacobs, M.R. et al. (2009) Resistance to colistin in Acinetobacter baumannii associated with mutations in the PmrAB two-component system. Antimicrob. Agents Chemother. 53, 3628–3634 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Moffatt, J.H., Harper, M., Harrison, P., Hale, J.D.F., Vinogradov, E., Seemann, T. et al. (2010) Colistin resistance in Acinetobacter baumannii is mediated by complete loss of lipopolysaccharide production. Antimicrob. Agents Chemother. 54, 4971–4977 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Lesho, E., Yoon, E.-J., McGann, P., Snesrud, E., Kwak, Y., Milillo, M. et al. (2013) Emergence of colistin-resistance in extremely drug-resistant Acinetobacter baumannii containing a novel pmrCAB operon during colistin therapy of wound infections. J. Infect. Dis. 208, 1142–1151 [DOI] [PubMed] [Google Scholar]
- 21. Cheah, S.-E., Johnson, M.D., Zhu, Y., Tsuji, B.T., Forrest, A., Bulitta, J.B. et al. (2016) Polymyxin resistance in Acinetobacter baumannii: genetic mutations and transcriptomic changes in response to clinically relevant dosage regimens. Sci. Rep. 6, 26233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Wright, M.S., Jacobs, M.R., Bonomo, R.A., and Adams, M.D. (2017) Transcriptome remodeling of Acinetobacter baumannii during infection and treatment. MBio 8, e02193–e02116 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Trebosc, V., Gartenmann, S., Tötzl, M., Lucchini, V., Schellhorn, B., Pieren, M. et al. (2019) Dissecting colistin resistance mechanisms in extensively drug-resistant Acinetobacter baumannii clinical isolates. MBio 10, e01083–19 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Sun, B., Liu, H., Jiang, Y., Shao, L., Yang, S., and Chen, D. (2020) New mutations involved in colistin resistance in Acinetobacter baumannii. mSphere 5, e00895-19 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Huang, J., Li, C., Song, J., Velkov, T., Wang, L., Zhu, Y. et al. (2020) Regulating polymyxin resistance in Gram-negative bacteria: roles of two-component systems PhoPQ and PmrAB. Future Microbiol. 15, 445–459 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Mmatli, M., Mbelle, N.M., Maningi, N.E., and Osei, S.J. (2020) Emerging transcriptional and genomic mechanisms mediating carbapenem and polymyxin resistance in Enterobacteriaceae: a systematic review of current reports. mSystems 5, e00783-20 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Gunn, J.S. (2008) The salmonella PmrAB regulon: lipopolysaccharide modifications, antimicrobial peptide resistance and more. Trends Microbiol. 16, 284–290 [DOI] [PubMed] [Google Scholar]
- 28. Wösten, M.M., and Groisman, E.A. (1999) Molecular characterization of the PmrA regulon. J. Biol. Chem. 274, 27185–27190 [DOI] [PubMed] [Google Scholar]
- 29. Wright, M.S., Iovleva, A., Jacobs, M.R., Bonomo, R.A., and Adams, M.D. (2016) Genome dynamics of multidrug-resistant Acinetobacter baumannii during infection and treatment. Genome Med. 8, 26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Bourret, R.B., and Silversmith, R.E. (2010) Two-component signal transduction. Curr. Opin. Microbiol. 13, 113–115 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Desai, S.K., and Kenney, L.J. (2017) To ∼P or not to ∼P? Non-canonical activation by two-component response regulators. Mol. Microbiol. 103, 203–213 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Draughn, G.L., Milton, M.E., Feldmann, E.A., Bobay, B.G., Roth, B.M., Olson, A.L. et al. (2018) The structure of the biofilm-controlling response regulator BfmR from Acinetobacter baumannii reveals details of its DNA-binding mechanism. J. Mol. Biol. 430, 806–821 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Bachhawat, P., Swapna, G.V.T., Montelione, G.T., and Stock, A.M. (2005) Mechanism of activation for transcription factor PhoB suggested by different modes of dimerization in the inactive and active states. Structure 13, 1353–1363 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Cho, H., Wang, W., Kim, R., Yokota, H., Damo, S., Kim, S.H. et al. (2001) BeF(3)(−) acts as a phosphate analog in proteins phosphorylated on aspartate: structure of a BeF(3)(−) complex with phosphoserine phosphatase. Proc. Natl. Acad. Sci. U S A 98, 8525–8530 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Otwinowski, Z., and Minor, W. (1997) [20] Processing of X-ray diffraction data collected in oscillation mode. Methods Enzymol. 1997, 307–326 [DOI] [PubMed] [Google Scholar]
- 36. Liebschner, D., Afonine, P.V., Baker, M.L., Bunkóczi, G., Chen, V.B., Croll, T.I. et al. (2019) Macromolecular structure determination using X-rays, neutrons and electrons: recent developments in Phenix. Acta Crystallogr. D Struct. Biol. 75, 861–877 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Dean, S.N., Milton, M.E., Cavanagh, J., and van Hoek, M.L. (2020) Francisella novicida two-component system response regulator BfpR modulates iglC gene expression, antimicrobial peptide resistance, and biofilm production. Front. Cell Infect. Microbiol. 10, 82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Emsley, P., Lohkamp, B., Scott, W.G., and Cowtan, K. (2010) Features and development of coot. Acta Crystallogr. D Biol. Crystallogr. 66, 486–501 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Schrodinger, L. (2010) The PyMOL molecular graphics system. Version 1.3r1.
- 40. Eswar, N., Webb, B., Marti-Renom, M.A., Madhusudhan, M.S., Eramian, D., Shen, M.-Y. et al. (2006) Comparative protein structure modeling using Modeller. Curr. Protoc. Bioinformatics 54, 5.6.1–5.6.37 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Menon, S., and Wang, S. (2011) Structure of the response regulator PhoP from mycobacterium tuberculosis reveals a dimer through the receiver domain. Biochemistry 50, 5948–5957 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Lou, Y.-C., Weng, T.-H., Li, Y.-C., Kao, Y.-F., Lin, W.-F., Peng, H.-L. et al. (2015) Structure and dynamics of polymyxin-resistance-associated response regulator PmrA in complex with promoter DNA. Nat. Commun. 6, 8838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Narayanan, A., Kumar, S., Evrard, A.N., Paul, L.N., and Yernool, D.A. (2014) An asymmetric heterodomain interface stabilizes a response regulator–DNA complex. Nat. Commun. 5, 3282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Milton, M.E., Allen, C.L., Feldmann, E.A., Bobay, B.G., Jung, D.K., Stephens, M.D. et al. (2017) Structure of the Francisella response regulator QseB receiver domain, and characterization of QseB inhibition by antibiofilm 2-aminoimidazole-based compounds. Mol. Microbiol. 106, 223–235 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Laskowski, R.A., MacArthur, M.W., Moss, D.S., and Thornton, J.M. (1993) PROCHECK - a program to check the stereochemical quality of protein structures. J. App. Cryst. 26, 283–291 [Google Scholar]
- 46. Bhattacharya, A., Tejero, R., and Montelione, G.T. (2007) Evaluating protein structures determined by structural genomics consortia. Proteins 66, 778–795 [DOI] [PubMed] [Google Scholar]
- 47. Baek, M., DiMaio, F., Anishchenko, I., Dauparas, J., Ovchinnikov, S., Lee, G.R. et al. (2021) Accurate prediction of protein structures and interactions using a 3-track network. Science 373, 871–876 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Hamidian, M., Wick, R.R., Hartstein, R.M., Judd, L.M., Holt, K.E., and Hall, R.M. (2019) Insights from the revised complete genome sequences of Acinetobacter baumannii strains AB307-0294 and ACICU belonging to global clones 1 and 2. Microb. Genomics 5, e000298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Cheng, H.-Y., Chen, Y.-F., and Peng, H.-L. (2010) Molecular characterization of the PhoPQ-PmrD-PmrAB mediated pathway regulating polymyxin B resistance in Klebsiella pneumoniae CG43. J. Biomed. Sci. 17, 60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Lou, Y.-C., Wang, I., Rajasekaran, M., Kao, Y.-F., Ho, M.-R., Hsu, S.-T.D. et al. (2014) Solution structure and tandem DNA recognition of the C-terminal effector domain of PmrA from Klebsiella pneumoniae. Nucleic Acids Res. 42, 4080–4093 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Kuzmic, P. (1996) Program DYNAFIT for the analysis of enzyme kinetic data: application to HIV proteinase. Anal. Biochem. 237, 260–273 [DOI] [PubMed] [Google Scholar]
- 52. Gasteiger, E., Gattiker, A., Hoogland, C., Ivanyi, I., Appel, R.D., and Bairoch, A. (2003) ExPASy: The proteomics server for in-depth protein knowledge and analysis. Nucleic Acids Res. 31, 3784–3788 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Khosa, S., Hoeppner, A., Gohlke, H., Schmitt, L., and Smits, S.H.J. (2016) Structure of the response regulator NsrR from Streptococcus agalactiae, which is involved in Lantibiotic resistance. PLoS One 11, e0149903 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Gao, R., and Stock, A.M. (2010) Molecular strategies for phosphorylation-mediated regulation of response regulator activity. Curr. Opin. Microbiol. 13, 160–167 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Feher, V.A., and Cavanagh, J. (1999) Millisecond-timescale motions contribute to the function of the bacterial response regulator protein Spo0F. Nature 400, 289–293 [DOI] [PubMed] [Google Scholar]
- 56. Milton, M.E., Minrovic, B.M., Harris, D.L., Kang, B., Jung, D., Lewis, C.P. et al. (2018) Re-sensitizing multidrug resistant bacteria to antibiotics by targeting bacterial response regulators: characterization and comparison of interactions between 2-Aminoimidazoles and the response regulators BfmR from Acinetobacter baumannii and QseB from Fr. Front. Mol. Biosci. 5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Goulian, M. (2010) Two-component signaling circuit structure and properties. Curr. Opin. Microbiol. 13, 184–189 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Raetz, C.R., and Newman, K.F. (1979) Diglyceride kinase mutants of Escherichia coli: inner membrane association of 1,2-diglyceride and its relation to synthesis of membrane-derived oligosaccharides. J. Bacteriol. 137, 860–868 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Cote, J.M., and Taylor, E.A. (2017) The glycosyltransferases of LPS core: a review of four heptosyltransferase enzymes in context. Int. J. Mol. Sci. 18, 2256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Wang, X., Preston, J.F., 3rd, and Romeo, T. (2004) The pgaABCD locus of Escherichia coli promotes the synthesis of a polysaccharide adhesin required for biofilm formation. J. Bacteriol. 186, 2724–2734 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Toro-Roman, A., Wu, T., and Stock, A.M. (2005) A common dimerization interface in bacterial response regulators KdpE and TorR. Protein Sci. 14, 3077–3088 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Volz, K. (1993) Structural conservation in the CheY superfamily. Biochemistry 32, 11741–11753 [DOI] [PubMed] [Google Scholar]
- 63. Bourret, R.B. (2010) Receiver domain structure and function in response regulator proteins. Curr. Opin. Microbiol. 13, 142–149 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Bachhawat, P., and Stock, A.M. (2007) Crystal structures of the receiver domain of the response regulator PhoP from Escherichia coli in the absence and presence of the phosphoryl analog Beryllofluoride. J. Bacteriol. 189, 5987LP–5995 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Toro-Roman, A., Mack, T.R., and Stock, A.M. (2005) Structural analysis and solution studies of the activated regulatory domain of the response regulator ArcA: a symmetric dimer mediated by the alpha4-beta5-alpha5 face. J. Mol. Biol. 349, 11–26 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Stock, A.M., and Guhaniyogi, J. (2006) A new perspective on response regulator activation. J. Bacteriol. 188, 7328–7330 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Ouyang, Z., Zheng, F., Chew, J.Y., Pei, Y., Zhou, J., Wen, K. et al. (2019) Deciphering the activation and recognition mechanisms of Staphylococcus aureus response regulator ArlR. Nucleic Acids Res. 47, 11418–11429 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Qing, X.-Y., Steenackers, H., Venken, T., De Maeyer, M., and Voet, A. (2017) Computational studies of the active and inactive regulatory domains of response regulator PhoP using molecular dynamics simulations. Mol. Inform. 36 [DOI] [PubMed] [Google Scholar]
- 69. Mustapha, M.M., Li, B., Pacey, M.P., Mettus, R.T., McElheny, C.L., Marshall, C.W. et al. (2018) Phylogenomics of colistin-susceptible and resistant XDR Acinetobacter baumannii. J. Antimicrob. Chemother. 73, 2952–2959 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Arroyo, L.A., Herrera, C.M., Fernandez, L., Hankins, J.V., Trent, M.S., and Hancock, R.E.W. (2011) The pmrCAB operon mediates polymyxin resistance in Acinetobacter baumannii ATCC 17978 and clinical isolates through phosphoethanolamine modification of lipid A. Antimicrob. Agents Chemother. 55, 3743–3751 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Gellman, S.H. (1991) On the role of methionine residues in the sequence-independent recognition of nonpolar protein surfaces. Biochemistry 30, 6633–6636 [DOI] [PubMed] [Google Scholar]
- 72. Aledo, J.C. (2019) Methionine in proteins: the Cinderella of the proteinogenic amino acids. Protein Sci. 28, 1785–1796 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. McEvoy, M.M., Hausrath, A.C., Randolph, G.B., Remington, S.J., and Dahlquist, F.W. (1998) Two binding modes reveal flexibility in kinase/response regulator interactions in the bacterial chemotaxis pathway. Proc. Natl. Acad. Sci. U S A. 95, 7333–7338 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Szurmant, H., Bobay, B.G., White, R.A., Sullivan, D.M., Thompson, R.J., Hwa, T. et al. (2008) Co-evolving motions at protein-protein interfaces of two-component signaling systems identified by covariance analysis. Biochemistry 47, 7782–7784 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Tzeng, Y.L., Feher, V.A., Cavanagh, J., Perego, M., and Hoch, J.A. (1998) Characterization of interactions between a two-component response regulator, Spo0F, and its phosphatase, RapB. Biochemistry 37, 16538–16545 [DOI] [PubMed] [Google Scholar]
- 76. McLaughlin, P.D., Bobay, B.G., Regel, E.J., Thompson, R.J., Hoch, J.A., and Cavanagh, J. (2007) Predominantly buried residues in the response regulator Spo0F influence specific sensor kinase recognition. FEBS Lett. 581, 1425–1429 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Tzeng, Y.-L., and Hoch, J.A. (1997) Molecular recognition in signal transduction: the interaction surfaces of the Spo0F response regulator with its cognate phosphorelay proteins revealed by alanine scanning mutagenesis. J. Mol. Biol. 272, 200–212 [DOI] [PubMed] [Google Scholar]
- 78. Feher, V.A., Tzeng, Y.L., Hoch, J.A., and Cavanagh, J. (1998) Identification of communication networks in Spo0F: a model for phosphorylation-induced conformational change and implications for activation of multiple domain bacterial response regulators. FEBS Lett. 425, 1–6 [DOI] [PubMed] [Google Scholar]
- 79. Jiang, M., Tzeng, Y.L., Feher, V.A., Perego, M., and Hoch, J.A. (1999) Alanine mutants of the Spo0F response regulator modifying specificity for sensor kinases in sporulation initiation. Mol. Microbiol. 33, 389–395 [DOI] [PubMed] [Google Scholar]
- 80. Blanco, A.G., Canals, A., Bernués, J., Solà, M., and Coll, M. (2011) The structure of a transcription activation subcomplex reveals how σ(70) is recruited to PhoB promoters. EMBO J. 30, 3776–3785 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. MacArthur, M.W., and Thornton, J.M. (1991) Influence of proline residues on protein conformation. J. Mol. Biol. 218, 397–412 [DOI] [PubMed] [Google Scholar]
- 82. Ho, B.K., and Brasseur, R. (2005) The Ramachandran plots of glycine and pre-proline. BMC Struct. Biol. 5, 14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Choi, E.J., and Mayo, S.L. (2006) Generation and analysis of proline mutants in protein G. Protein Eng. Des. Sel. 19, 285–289 [DOI] [PubMed] [Google Scholar]
- 84. Shalit, Y., and Tuvi-Arad, I. (2020) Side chain flexibility and the symmetry of protein homodimers. PLoS One 15, e0235863–e0235863 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Huang, F., and Nau, W.M. (2003) A conformational flexibility scale for amino acids in peptides. Angew. Chem. Int. Ed. Engl. 42, 2269–2272 [DOI] [PubMed] [Google Scholar]
- 86. Schimmel, P.R., and Flory, P.J. (1968) Conformational energies and configurational statistics of copolypeptides containing L-proline. J. Mol. Biol. 34, 105–120 [DOI] [PubMed] [Google Scholar]
- 87. Solà, M., Gomis-Rüth, F.X., Serrano, L., González, A., and Coll, M. (1999) Three-dimensional crystal structure of the transcription factor PhoB receiver domain. J. Mol. Biol. 285, 675–687 [DOI] [PubMed] [Google Scholar]
- 88. Xie, M., Wu, M., and Han, A. (2020) Structural insights into the signal transduction mechanism of the K+-sensing two-component system KdpDE. Sci. Signal. 13, eaaz2970 [DOI] [PubMed] [Google Scholar]
- 89. Zapf, J., Sen, U., Madhusudan, H.J.A., and Varughese, K.I. (2000) A transient interaction between two phosphorelay proteins trapped in a crystal lattice reveals the mechanism of molecular recognition and phosphotransfer in signal transduction. Structure 8, 851–862 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.