

Abstract
Emerging data indicate that G-protein coupled receptor (GPCR) signaling is determined by not only the agonist and a given receptor but also a variety of cell-type-specific factors that can influence a receptor’s response. For example, the metabotropic glutamate receptor, mGlu5, which is implicated in a number of neuropsychiatric disorders such as depression, anxiety, and autism, also signals from inside the cell which leads to sustained Ca2+ mobilization versus rapid transient responses. Because mGlu5 is an important drug target, many negative allosteric modulators (NAMs) have been generated to modulate its activity. Here we show that NAMs such as AFQ056, AZD2066, and RG7090 elicit very different end points when tested in postnatal neuronal cultures expressing endogenous mGlu5 receptors. For example, AFQ056 fails to block intracellular mGlu5-mediated Ca2+ increases whereas RG7090 is very effective. These differences are not due to differential receptor levels, since about the same number of mGlu5 receptors are present on neurons from the cortex, hippocampus, and striatum based on pharmacological, biochemical, and molecular data. Moreover, biotinylation studies reveal that more than 90% of the receptor is intracellular in these neurons. Taken together, these data indicate that the tested NAMs exhibit both location-dependent and cell type specific bias for mGlu5-mediated Ca2+ mobilization which may affect clinical outcomes.
Keywords: GPCR, mGlu5, NAM, intracellular, calcium, neuron
Graphical Abstract
INTRODUCTION
G-protein coupled receptors (GPCRs) are known for coupling a diverse range of signals to a broad variety of amplified responses. Because they sit at the top of many different signaling pathways, GPCRs are considered a central target for drug development.1,2 Toward this end, substantial efforts have gone into developing compounds that either affect the natural binding pocket (orthosteric site) or a different site on the protein (allosteric site) that can increase (positive allosteric modulator, PAM) or decrease (negative allosteric modulator, NAM) receptor responses to the natural ligand. As allosteric modulators, PAMs or NAMs work by modifying the affinity or efficacy of the natural ligand or by stabilizing the receptor in a given conformation.3–6 Allosteric modulators have opened the door to enhanced selectivity, specificity, and kinetics, rendering “undruggable” receptors amenable to manipulation.7 Collectively, these compounds offer a promising new approach to increased drug efficacy with reduced side effects.
Despite these advances, allosteric modulators also present new challenges since a given GPCR can couple to multiple signaling pathways and the context of receptor modulation may dramatically affect ligand efficacy. This is relevant since emerging studies have shown that many GPCRs are also found on intracellular membranes such as the endoplasmic reticulum (ER), Golgi apparatus, nucleus, and mitochondria where they may activate distinct signaling pathways with unique biological outputs.8–12 One such intracellular receptor is the metabotropic glutamate receptor, mGlu5. Our studies show mGlu5 receptors signal from the cell surface as well as the ER and nuclear membranes in the striatum, hippocampus, cortex, and spinal cord dorsal horn (SCDH) where they couple to Gq/11/PLC/IP3 to release intracellular Ca2+.13–16 Intracellular mGlu5 is activated by glutamate after transport into the cell via various transporters or exchangers such as the excitatory amino acid transporter 3 (EAAT3) or cystine/glutamate exchanger (xCT) located at the cell surface and on intracellular membranes.13,15 To date, we have used pharmacological isolation to determine intracellular responses. This is achieved via specific impermeable, nontransported drugs, including the antagonist LY39305317 and the agonist DHPG, as well as transported or permeable drugs, such as the agonists, glutamate and quisqualate (Quis), and the NAMs, MPEP18,19 and Fenobam.20 These tools show that cell surface and intracellular mGlu5 receptors exhibit unique Ca2+ patterns and downstream signaling cascades.14 For example, in striatal and spinal cord dorsal horn neurons, intracellular mGlu5 activation triggers a sustained Ca2+ response whereas activation of cell surface mGlu5 triggers a rapid, transient Ca2+ response.14,16 In contrast, both intracellular and cell surface mGlu5’s are responsible for oscillatory Ca2+ responses in the hippocampus, whereas sustained Ca2+ responses are only associated with intracellular dendritic mGlu5.15 These data suggest cell-type-specific differences in mGlu5 signaling cascades or inherent differences in the cell types tested.13–16
Because mGlu5 plays a critical role in multiple central nervous system (CNS) disorders such as anxiety, depression, autism spectrum disorders (ASD,21,22), and pain,23 many mGlu5 NAMs have been developed, several of which are in clinical trials albeit with varying degrees of success.24–28 Although most drugs targeted for clinical use are designed to be potent, bioavailable, and metabolically stable, each drug scaffold has unique chemical properties. As different cells also have unique membrane constituents and lipophilic properties, drug parameters established in heterologous cell types might not reflect endogenous constraints. Thus, the cellular context of mGlu5 activation may dramatically modify its efficacy and/or the ability of a given ligand to reach an intracellular receptor.8–12 Moreover, at least initially, most NAMs are tested for efficacy and potency in heterologous cell lines (e.g., HeLa, HEK); thus, drug effectiveness in the cell types being targeted might be missed. To test the hypothesis that different mGlu5 NAMs will exhibit location-specific and cell-type-specific signaling patterns, we characterized several representative mGlu5 NAMs including mavoglurant (AFQ05624), AZD2066,25 and basimglurant (RG709026) as well as MPEP18,19 and Fenobam20 as controls. Here we show that, despite similar log P values, the available NAMs exhibit unique location-dependent and cell-type-specific profiles that are not due to variable levels of cell surface and/or intracellular mGlu5 receptors. Moreover, we show that, in dissociated neuronal cultures from the cortex, hippocampus, and striatum, >90% of mGlu5 is intracellular.
RESULTS AND DISCUSSION
Location-Dependent Bias of Various mGlu5 NAMs.
Recent data show that numerous GPCRs are not just on the plasma membrane but also on intracellular organelles including the nucleus, ER, mitochondria, Golgi apparatus, and vesicles.8–12 These new locales contribute to the complexity of receptor efficacy since a given ligand may not have the same intracellular access and/or might be more or less influenced by the composition of a particular membrane.8,9 For example, mGlu5 is largely intracellular where it can be activated by agonist uptake (glutamate, Quis) via facilitated transport (glutamate transporters and/or exchangers13,15). The mGlu5 agonist, DHPG, however, is neither transported into the cell nor is it freely permeable (consensus log Po/w of −1.07); hence, DHPG is unable to activate intracellular receptors29. Glutamate and Quis are not freely permeable either since both have consensus log Po/w values of −2.67 and −2.29, respectively. (A consensus log Po/w is computed using the arithmetic mean of five different predictive models based on the partition coefficient between n-octanol and water: a log P value > 2 is considered readily membrane permeant). SwissADME (http://www.swissadme.ch30) consensus log Po/w values for mGlu5-specific NAMs also indicate that they are membrane permeant (Table 1). Moreover, we have functionally shown that MPEP and Fenobam inhibit intracellular mGlu5 receptors.13,15,16 Based on their log Po/w values, additional mGlu5 NAMs used in this study (AZD2066, RG7090, and AFQ056) similarly have positive log Po/w values (Table 1) and thus would be predicted to functionally block intracellular mGlu5 responses.
Table 1.
Summary of Chemical Structures, Molecular Weight (MW), Values of Consensus Log Po/w, TPSA, Log S, Log BB, Log PS, H-Bond Donor, H-Bond Acceptor and Free Rotatable Bonds of the Tested mGlu5 NAMs
| Name | Structure | M.W.a | Cons. LogPo/wa | TPSAa | LogSa (Log mol/L) | LogBBb | LogPSb | H-bond donorc | H-bond acceptorc | Free rotatable bondc |
|---|---|---|---|---|---|---|---|---|---|---|
| MPEP |
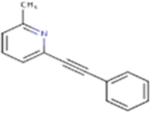
|
193.24 | 3.15 | 12.89 Å2 | −3.008 | 0.449 | −1.415 | 0 | 1 | 0 |
| Fenobam |
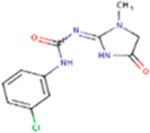
|
266.68 | 1.25 | 73.80 Å2 | −2.753 | −0.587 | −2.433 | 2 | 6 | 4 |
| FQ056 |
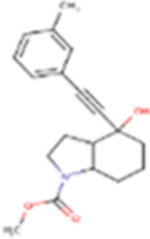
|
313.39 | 2.85 | 49.77 Å2 | −3.731 | 0.711 | −1.894 | 1 | 3 | 3 |
| G7090 |
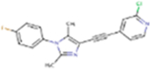
|
325.77 | 4.16 | 30.71 Å2 | −3.035 | 0.04 | −1.48 | 0 | 3 | 3 |
| ZD2066 |
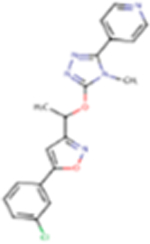
|
381.82 | 3.28 | 78.86 Å2 | −2.89 | −1.189 | −2.961 | 1 | 6 | 5 |
Predicted values from SwissADME (http://www.swissadme.ch30) including log Po/w (partition coefficient between n-octanol and water), an indicator of lipophilicity. The consensus (cons.) log Po/w is the average value of the five available predictive models in SwissADME. The topological polar surface area (TPSA), defined as the surface sum over all polar atoms or molecules, is a measure of permeability in that molecules > 140 Å2 are unlikely to permeate the blood brain barrier.
Predicted values from http://biosig.unimelb.edu.au/pkcsm/prediction.38 The water solubility of a compound (log S) reflects the solubility of the molecule in water at 25 °C. For a given compound, a log BB > 0.3 can readily cross the blood-brain barrier (BB), while molecules with log BB < −1 are poorly distributed in the brain. Compounds with a log PS > −2 are predicted to penetrate the CNS, while those with log PS < −3 are unlikely to cross the BBB or CNS membranes.39
The numbers of H-bond donor, H-bond acceptor, and free rotatable bonds of each NAM were obtained from PubChem (https://pubchem.ncbi.nlm.nih.gov/) and The IUPHAR/BPS Guide to PHARMACOLOGY (https://www.guidetopharmacology.org/).
To test the hypothesis that the selected NAMs may exhibit significant cell type-specific and location-dependent effects, we used single cell Ca2+ imaging to determine whether AZD2066, RG7090, and AFQ056 also block intracellular mGlu5 Gq/11-mediated activity. In these studies, we used mGlu5/HEK cells31 as well as primary neuronal cultures expressing endogenous mGlu5 derived from the striatum and hippocampus.14,15 Cultures grown on glass coverslips were loaded with the Ca2+ indicator, Oregon Green BAPTA-AM, and subsequently treated with agonists and/or NAMs with variable intracellular access. Agonist concentrations were chosen based on previous dose dependency responses taking into account cell surface versus intracellular mGlu5 receptor pools.32,33 For example, in striatal neurons, only a single transient Ca2+ peak is observed at Quis doses < 250 nM, whereas both transient and sustained peaks are seen with higher Quis concentrations (>5 μM; Supplemental Figure S1C,D in ref 32).
Figure 1 highlights agonist-mediated single cell Ca2+ responses due to either pre- or post-treatment with AFQ056. This particular NAM exhibited the most disparate location and cell-specific effects. Bath application of nonpermeant, nontransported DHPG (100 μM) in mGlu5/HEK cells led to an oscillatory Ca2+ response which was subsequently inhibited by AFQ056 (Figure 1A, left panel) and all mGlu5 NAMs at 1, 5, and 10 μM (not shown). The nonpermeant, transported agonist Quis (10 μM) also produced oscillatory Ca2+ responses in mGlu5/HEK cells and was subsequently inhibited by AFQ056 (Figure 1A, middle panel) as well as the other tested NAMs (1, 5, and 10 μM; not shown). Finally, pretreatment with AFQ056 (Figure 1A, right panel) and all of the other NAMs (1, 5, and 10 μM; not shown) blocked either DHPG or Quis effects in mGlu5/HEK cells. Quantitation of all pretreatment NAM results in mGlu5/HEK cells at all doses tested is shown in Figure 2A. Similarly, neuronal cultures prepared from the hippocampus exhibited oscillatory Ca2+ responses following DHPG (1, 10 μM) or Quis (2, 10 μM) and were similarly inhibited by pre- or post-treatment with all NAMs at 1, 5, and 10 μM (Figure 1B; Figure 2B; and not shown). Interestingly, although pre- or post-treatment of striatal cultures with 1 μM AFQ056 blocked the cell-surface only agonist, DHPG (Figure 1C left and right panels), neither 1, 5, nor 10 μM AFQ056 blocked Quis-mediated Ca2+ responses (Figure 1C, middle panel; Figure 2C). Fenobam and AZD2066 were also less effective in striatal neurons at the tested doses (Figure 2C). RG7090 was the most effective drug at blocking single cell Quis effects in the striatum (Figure 2C). As expected, neither 5MPEP,34 a neutral allosteric modulator, nor LY393053 affected Quis-induced Ca2+ rises (refs 14 and 15, and data not shown). Taken together, these data show that all tested NAMs inhibit mGlu5 cell surface receptors in either heterologous cells or striatal or hippocampal neurons. For the most part, the intracellular pool of receptors accessed by Quis are also inhibited by the selected NAMs, although at least one NAM that was designed to be brain penetrant was unable to block the striatal intracellular receptor versus the cell surface receptor at a concentration 100-fold its published IC50 (AFQ056 IC50 = 30 nM24) whereas it was able to do so in other cell types. Fenobam and AZD2066 did not block single cell intracellular responses at 10-fold their IC50 values.20,25 Conceivably, single cell Ca2+ responses differ between striatal neurons and hippocampal tissues because the latter contain many different types of mGlu5-expressing neurons including excitatory pyramidal neurons and inhibitory interneurons, whereas all of the mGlu5-expressing medium spiny neurons from the striatum are GABAergic.13 Nonetheless, drugs that are potent and effective in heterologous cell types and some types of neurons may exhibit significant cell type-specific and location-dependent effects in other neuronal cell types.
Figure 1.
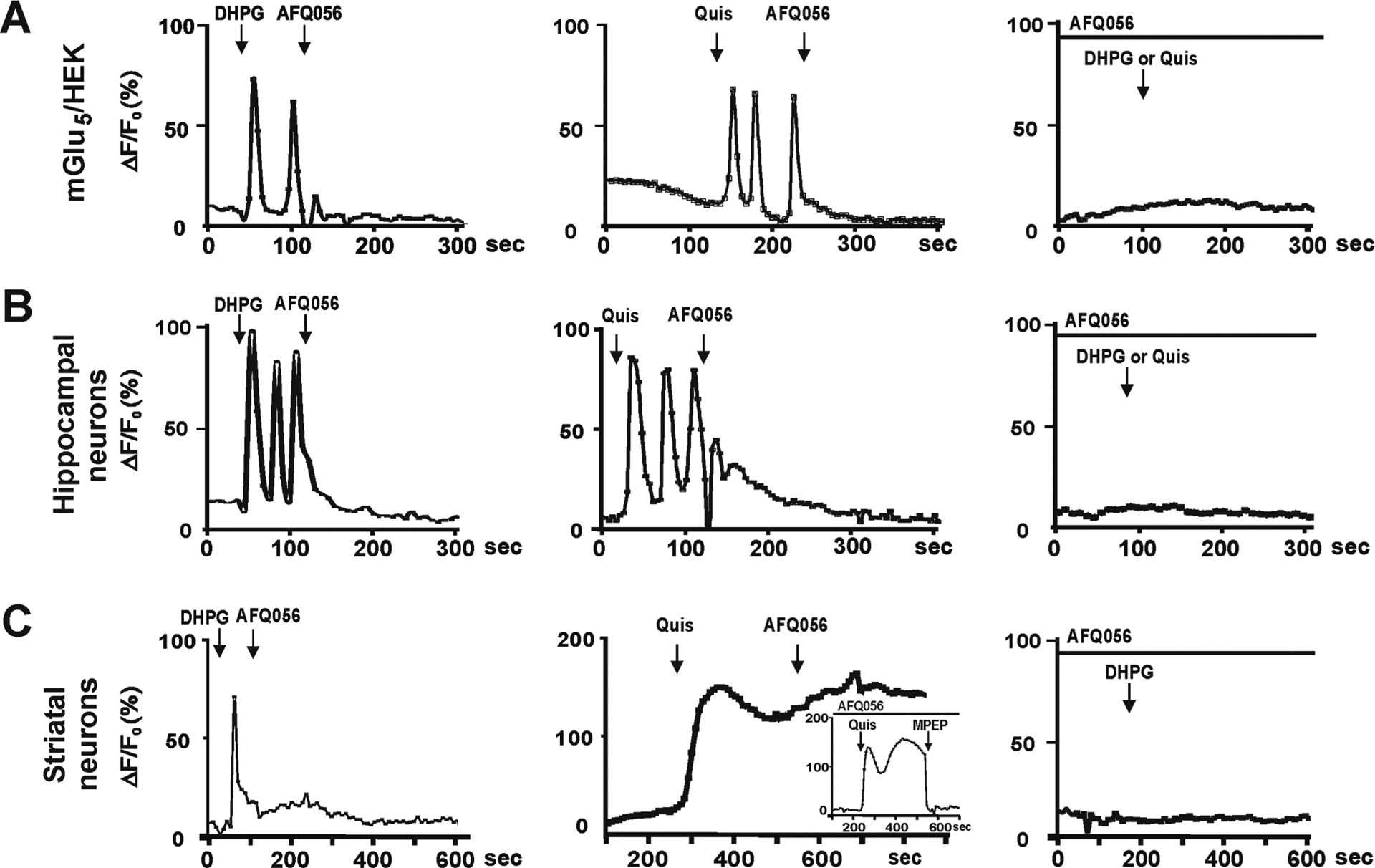
Single cell imaging of intracellular Ca2+ showing location-dependent bias of various mGlu5 NAMs. (A–C) Traces derived from the mGlu5/HEK cell line, hippocampal, or striatal cultures loaded with Ca2+ fluorophore and then treated as indicated. Traces represent the fractional change in fluorescence relative to the basal level.13–15 (A, Left) Cell surface mGlu5 agonist DHPG generated Ca2+ oscillations in mGlu5/HEK cells which could be blocked by 10 μM AFQ056 (shown) or MPEP, Fenobam, RG7090, or AZD2066 (not shown). (A, Middle) Quis-induced Ca2+ oscillations could also be blocked with AFQ056 as well as the other tested NAMs. (A, Right) Pretreatment of mGlu5/HEK cells with AFQ056 (shown) or MPEP, Fenobam, RG7090, or AZD2066 completely blocked DHPG or Quis-mediated Ca2+ responses. DHPG (B, left) and Quis (B, middle) also induced Ca2+ oscillations in hippocampal neurons which could be blocked by all NAMs. NAM pretreatment (B, right) completely prevented subsequent DHPG or Quis responses. Pre- (C, right) or post- (C, left) treatment with AFQ056 blocked DHPG effects in striatal neurons. (C, Middle) In contrast, AFQ056 did not block Quis-mediated Ca2+ changes in striatal neurons and pretreatment did not prevent a Quis-induced Ca2+ response (inset) Although only single traces are shown here, quantitation (Figure 2) was from 80 to 100 neurons for each condition and each experiment was performed three times. In these experiments, DHPG was applied at 100 μM, Quis at 10 μM, and all NAMs at 1, 5, and 10 μM. In the traces shown, AFQ056 is 10 μM.
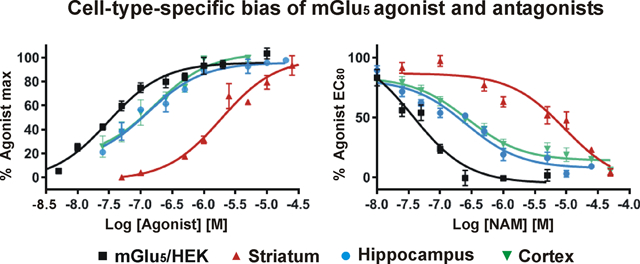
Figure 2.
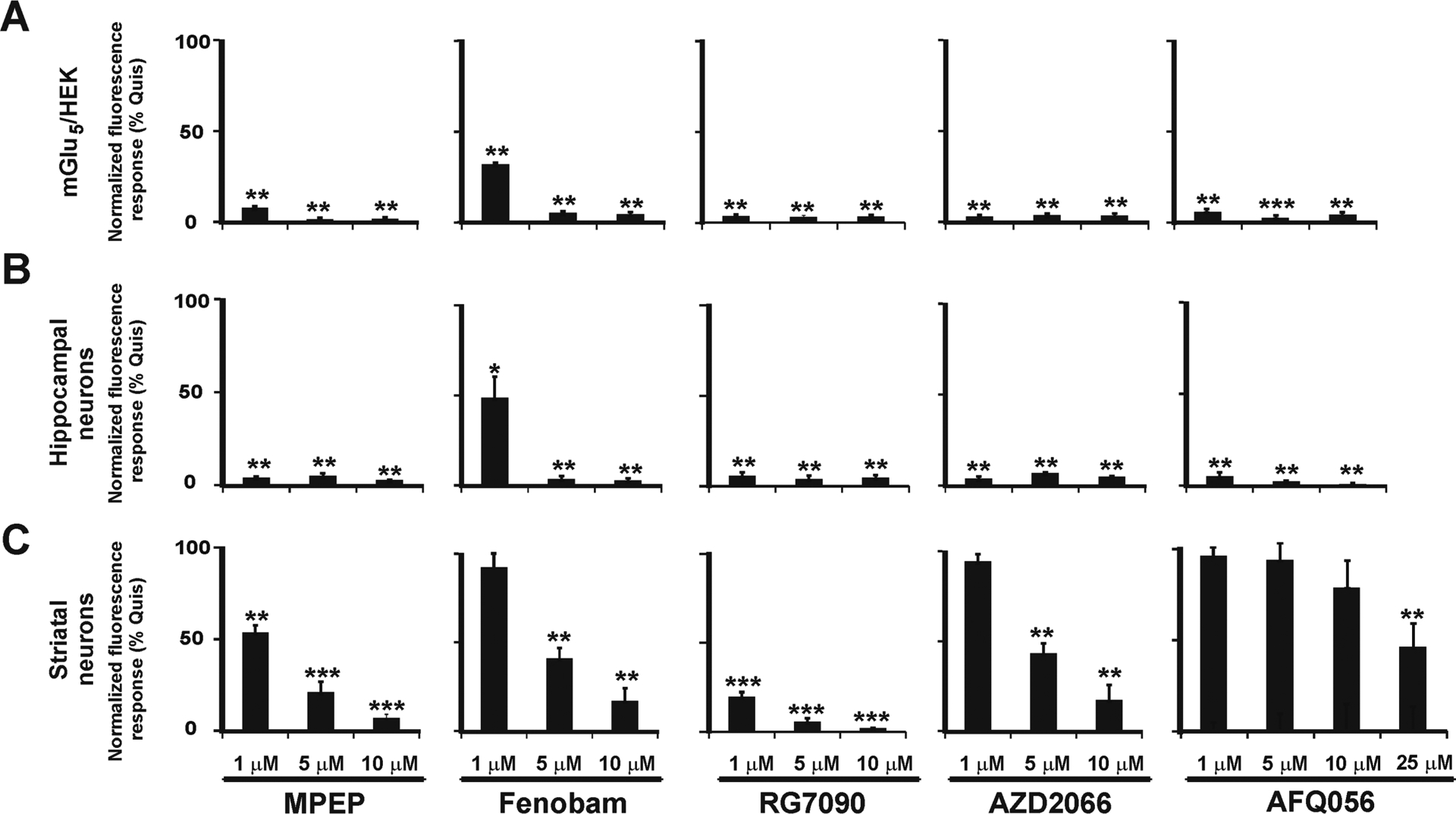
Quantitation of single cell Ca2+ imaging showing cell-type and location-dependent bias of tested mGlu5 NAMs. Compiled data from Quis-induced Ca2+ responses (10 μM) as normalized fluorescence response (% Quis) following pretreatment with MPEP, Fenobam, RG7090, AZD2066, or AFQ056, at the indicated concentrations in mGlu5/HEK cells and hippocampal and striatal neurons. At 5 and 10 μM, all NAMs tested blocked Quis-induced Ca2+ responses in HEK cells or hippocampal neurons whereas AFQ056 did not completely block the Quis-induced Ca2+ rise in striatal neurons at any dose. At 1 μM, Fenobam was less effective in mGlu5/HEK, hippocampal and striatal cells as well. Bars represent the mean of three independent experiments ± SEM from 80 to 100 neurons/condition. **Denotes statistical significance compared with control (Quis only) with Student’s t test (**p < 0.01, ***p < 0.001).
Inasmuch as mGlu5-mediated Ca2+ induction triggers a variety of signaling pathways in neurons and glia which affect neuronal survival, differentiation, and synaptic plasticity,35 it would be of great interest to examine additional pathways to determine whether there are differential responses to the various NAMs. For example, several pathways frequently associated with mGlu5 include Ca2+/calmodulin-dependent protein kinase (CaMK) and the mitogen-activated protein kinase (MAPK) pathways via extracellular signal-regulated kinase 1/2 (ERK1/2), p38 MAPK, or stress-activated protein kinase/c-Jun N-terminal kinase (JNK).10 Previously, we showed that Quis treatment of striatal mGlu5 led to the activation of all of these pathways whereas DHPG only activated a subset.14 We subsequently used an unbiased bioinformatics approach to confirm and extend these findings showing that activation of both cell-surface and intracellular mGlu5 led to the upregulation of many early response genes involved in growth and differentiation, but that only activation of intracellular mGlu5 led to the upregulation of genes involved in synaptic plasticity (e.g., Arc, Atf3, FOS, JUNB, EGR1, NR4A1).32 In these examples, MPEP blocked all responses whereas LY393053 only blocked those activated by DHPG. Based on these results, AFQ056 might not be able to block the Quis induction of ERK1/2 in striatal neurons whereas RG7090 should. New biosensors to measure ERK1/2 activation in situ may allow us to achieve the requisite sensitivity and selectivity necessary to explore this question in future studies. Differential activation of multiple signaling pathways downstream of mGlu5 activation broadens the repertoire of receptor-mediated sequelae and potentially enhances integrated cellular responses and possible feedback regulation.
Quis Exhibits Cell Type-Specific Bias.
Given the variability, labor, and time-intensive nature of single cell data collection and in order to minimize system and measurement bias, we established an assay system using a fluorescence-based Ca2+ flux plate reader by scaling up our dissociated neuronal cultures such that dose–response curves from multiple tissue sources could be assayed in the same time frame using the same reagents. Specifically, ~50 000 neurons prepared from postnatal day (P) 1–3 neonates were plated per well and allowed to mature until DIV 14 (days in vitro 14). The baseline 340/380 nm excitation ratio for Fura-2 was collected for 5 s before injecting various concentrations of Quis. We used Quis as a more specific agonist to test for location-dependent or cell-type-specific bias because establishing a glutamate dose–response curve is affected by the complexity and heterogeneity of cell types in the brain as well as the variance and density of ionotropic and metabotropic glutamate receptors, transporters, and exchangers. All of these factors contribute to high levels of nonspecific binding. Because Quis would also activate α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid receptor (AMPAR) as well as mGlu1 receptors, it was bath-applied in the presence of 25 μM SYM2206, an AMPAR antagonist, and CPCCOEt, an mGlu1 antagonist (20 μM). Data were collected for an additional 30 s and then analyzed. Exemplar traces for equi-effective concentrations of Quis in each of the cell populations are shown in Figure 3A. Although glial responses35 might confound these types of population assays, not only did our standard tissue preparation minimize the presence of glia (no feeder layer, Neurobasal media, ARA-C treatment) but also glial responses were not observed based on the morphological appearance of responding cells during single cell Ca2+ imaging or via astrocytic Ca2+ oscillations in population traces (Figure 3A).
Figure 3.
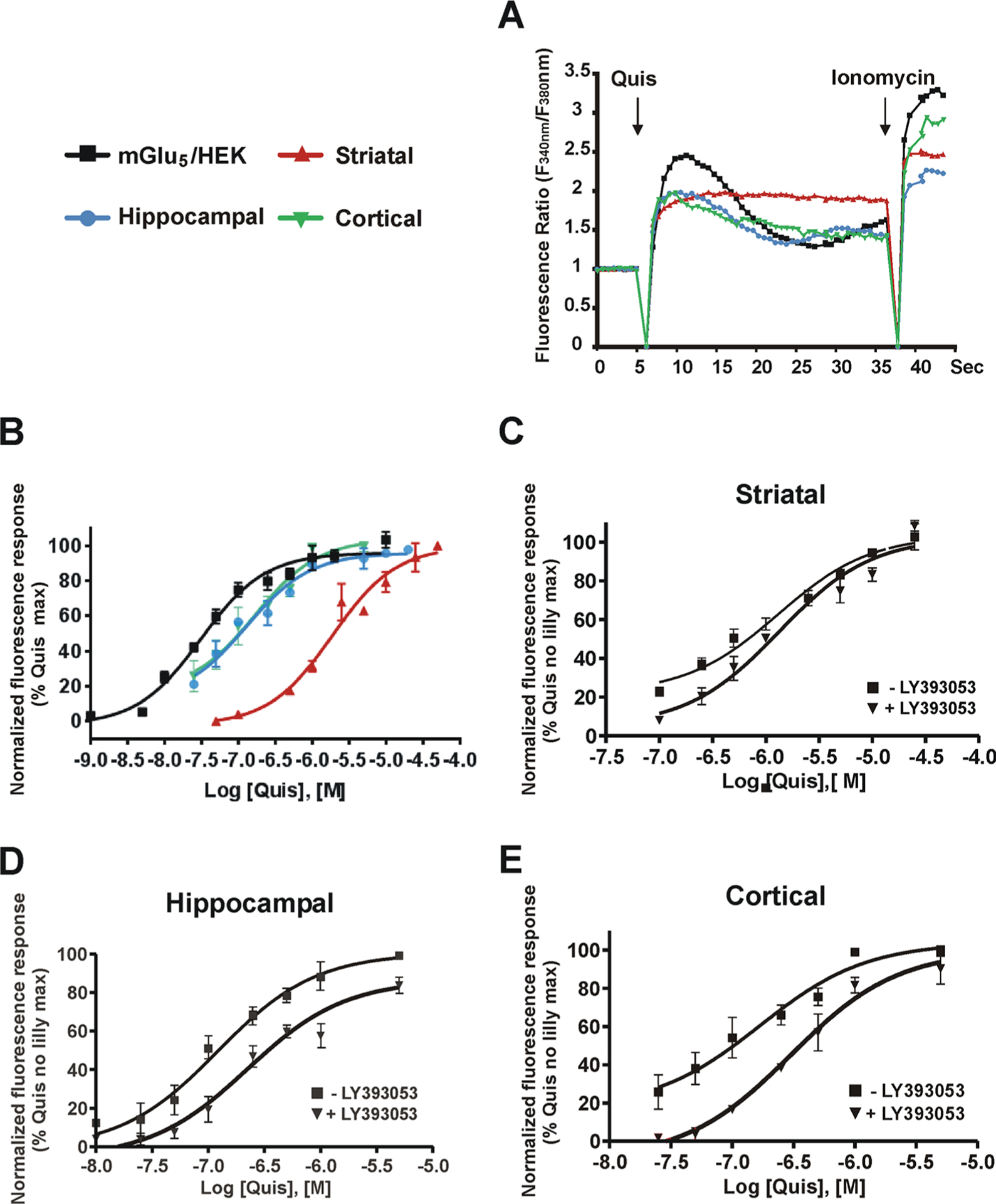
Quis exhibits cell type-specific Ca2+ responses. Cells were plated in 96-well plates, loaded with the Ca2+ sensitive fluorescent dye Fura-2 AM, and placed in a BioTek Synergy H4 Hybrid microplate reader. A range of concentrations of Quis was added after 5 s of baseline determination. (A) Ca2+ traces from equi-effective concentrations of Quis determined in mGlu5/HEK (black solid square), striatal (red solid up triangle), hippocampal (blue solid circle), or cortical (green solid down triangle) cultures. The Quis-induced response was defined as the average rise of normalized fluorescence ratio between 3 and 5 s after the agonist addition. Ionomycin 2 μM was added at the end to verify Fura-2 AM loading. (B) Dose–response curves of Quis-induced Ca2+ responses. Similar experiments were performed with (black solid down triangle) and without (black solid square) pretreatment with LY393053 followed by various concentrations of Quis in striatal (C), hippocampal (D), or cortical cultures (E). The maximum response induced by the highest Quis concentration applied was set as 100%. Data were normalized to a Quis control maximum. Dose–response curves were generated from mean data sets of at least three independent experiments performed in triplicate. Error bars represent SEM. EC50 values of Quis in all cell types were determined using GraphPad Prism, and the values are summarized in Table 2.
Population Ca2+ dose–response curves are shown in Figure 3B, and calculated values are shown in Table 2. Quis exhibits the highest efficacy in HEK cells stably expressing mGlu5 versus the endogenous receptors in neuronal populations derived from the cortex, hippocampus, and striatum. Interestingly, there is a 42-fold difference in the EC50 for Quis in HEK cells versus endogenous mGlu5-expressing striatal neurons and an 8–10-fold difference in EC50 between the cortical and hippocampal neurons and the striatum, respectively (Figure 3B, Table 2). These data imply that the physiological context of the different cell types expressing mGlu5 must be different in some regard.
Table 2.
Summary of EC50 or EC80 Values of Quis and Quis + LY393053 in mGlu5/HEK Cells and Striatal, Hippocampal, and Cortical Culturesa
| mGlu5 HEK | striatal | hippocampal | cortical | |
|---|---|---|---|---|
| Quis EC50 | 31.0 ± 8.3 nM | 1.31 ± 0.36 μM | 122 ± 12.1 nM | 168 ± 55.8 nM |
| Quis with LY393053 EC50 | 1.33 ± 0.15 μM | 225 ± 35.7 nM* | 301 ± 59.2 nM** | |
| Quis EC80 | 125 ± 32.3 nM | 5.25 ± 1.43 μM | 515 ± 51.4 nM | 670 ± 223 nM |
| max fluorescence response (Emax); (Quis with LY393053)/Quis (%) | 97.1 ± 0.565 | 84.5 ± 1.75* | 89.1 ± 2.29 |
Besides establishing a dose–response curve for Quis in tissues expressing endogenous mGlu5, LY393053 (20 μM) was used to block cell surface receptors so as to establish the presence and efficacy of intracellular mGlu5 receptors. In all three tissues, a robust intracellular mGlu5 response was observed (Figure 3C–E; Table 2), confirming and extending our previous observations with single cell Ca2+ imaging in the striatum,14 hippocampus,15 and spinal cord.16 LY393053 induced a significant rightward shift of the Quis concentration–response curves in hippocampal and cortical neurons but not in striatal cultures (Figure 3C–E; Table 2). There is about a 1.8-fold difference in potency with the cell surface receptor in the hippocampus and the cortex, whereas there was no difference in EC50 between cell surface and intracellular mGlu5 in the striatum. By analogy with receptor desensitization and internalization studies of ligand binding affinity (e.g., dopamine D2 receptors36), we used percent change in Emax as a rough estimation of internal mGlu5 receptor density and, by extrapolation, cell surface mGlu5 receptor density. Neither striatal nor cortical Emax values in the presence of LY393053 exhibited significantly different Emax values with or without LY393053 when examined in sibling culture preparations treated at the same time. Thus, we estimate that <10% mGlu5 is on the cell surface of these neurons. There was however a significant 15% difference in the hippocampal Emax values (p = 0.03), suggesting either more mGlu5 was on hippocampal plasma membranes or that perhaps the cell surface pool of mGlu5 was internalizing at a faster rate than it did in striatal or cortical neurons. These estimations of cell surface mGlu5 were largely in agreement with biotinylation experiments shown below (Figure 5)
Figure 5.

Most of mGlu5 is intracellular in striatal, hippocampal, and cortical cultures. (A) mGlu5 receptors are expressed on dendrites, within cell bodies, and on nuclear membranes in neuronal cultures. DIV 14 striatal, hippocampal, or cortical cultures showing colocalization of mGlu5 receptor immunoreactivity (red) together with the inner nuclear membrane marker Lamin B2 (green). (B) Western blots of DIV 14 striatal lysates prepared from knockout (−/−) and wildtype (+/+) littermates generated by mating mGlu5 (+/−) heterozygote mice. (C) Western blots of neuronal culture lysates probed with the antibody against mGlu5. (D) Comparison of mGlu5 levels in hippocampus or cortex to striatal levels following normalization to β-actin. Data shown represent the mean of three experiments (hippocampal/striatal = 112.96 ± 9.17%; cortical/striatal = 101.06 ± 9.09%). (E) Cell surface proteins from DIV 14 striatal, hippocampal, and cortical cultures were labeled with cell-membrane-impermeable EZ-Link Sulfo-NHS-LC-Biotin for 30 min prior to quenching. Cells were lysed, and an aliquot representing 25% of the total protein was removed. The remaining 75% was incubated with NeutrAvidin agarose, washed with RIPA buffer, and bound proteins were resuspended in SDS sample buffer and heated at 60 °C. An amount of 10 μg of total protein (tot) was run on a gel next to the biotinylated proteins (bio) derived from 100 μg of the equivalent neuronal lysate. The Na+-K+ ATPase was used as a plasma membrane positive control, whereas β-actin served as a loading control. (F) Glycosylation analysis of mGlu5 in cortical cultures. Biotinylated cortical cell lysate was either treated or not with PNGaseF and then run on SDS-PAGE along with 10 μg of total cell lysate followed by Western blotting with anti-mGlu5. The biotinylated surface mGlu5 was sensitive to the PNGase F treatment. (G) Quantitative analysis of Western blotting results in (E) showing that only a small percentage of total mGlu5 is on the cell surface in striatal, hippocampal, and cortical cultures: 1.81 ± 0.84% for striatal, 5.58 ± 1.52% for hippocampal, and 6.37 ± 1.38% for cortical cultures. These values were derived following densitometric analyses using the ChemiDoc MP system (Bio-Rad) together with associated Image Lab software. The percentage of the cell surface receptor = (the background-subtracted density of the biotinylated band from 100 μg of total lysate/10 × the background-subtracted density of 10 μg of total lysate) × 100. Bars represent the mean of three independent experiments ± SE. *Denotes statistical significance compared with control with Student’s t test (p < 0.05).
mGlu5 NAMs Exhibit Cell-Type-Specific Bias.
In order to determine whether the NAMs tested for location-dependent bias also exhibited cell type-specific bias, we measured receptor-mediated mobilization of intracellular Ca2+ after pretreating with different concentrations of MPEP, Fenobam, RG7090, AZD2066, and AFQ056. Quis was added at its previously determined EC80 (Figure 3 and Table 2) for each cell type. Inhibition curves of the chosen NAMs for cultures from each brain region are shown in Figure 4, and IC50 data are summarized in Table 3. All NAMs tested exhibited the greatest response in the mGlu5/HEK cells (Figure 4). They were less potent in hippocampal and cortical neurons which generally tracked more closely together (~14-fold difference versus HEK cells) except RG7090 with a 45-fold difference in cortical neurons (Table 3). All tested NAMs were the least potent in striatal neurons exhibiting a 16- (Fenobam) to a 250-fold shift (AFQ056) between mGlu5/HEK cells and striatal cultures. These data emphasize that not only does an agonist exhibit cell type-dependent bias but NAMs do as well. We also tried to test varying concentrations of the various NAMs in the presence of EC80 DHPG to obtain their IC50 values in this context. Given the low level of mGlu5 receptors on the cell surface (Figure 5, below), the maximum DHPG-induced Ca2+ increase was only 15–20% of the Quis-induced response. We could not achieve consistent EC50 values in repetitive experiments with such low responses, and we could not determine the IC50 of each NAM under the current experimental configuration.
Figure 4.
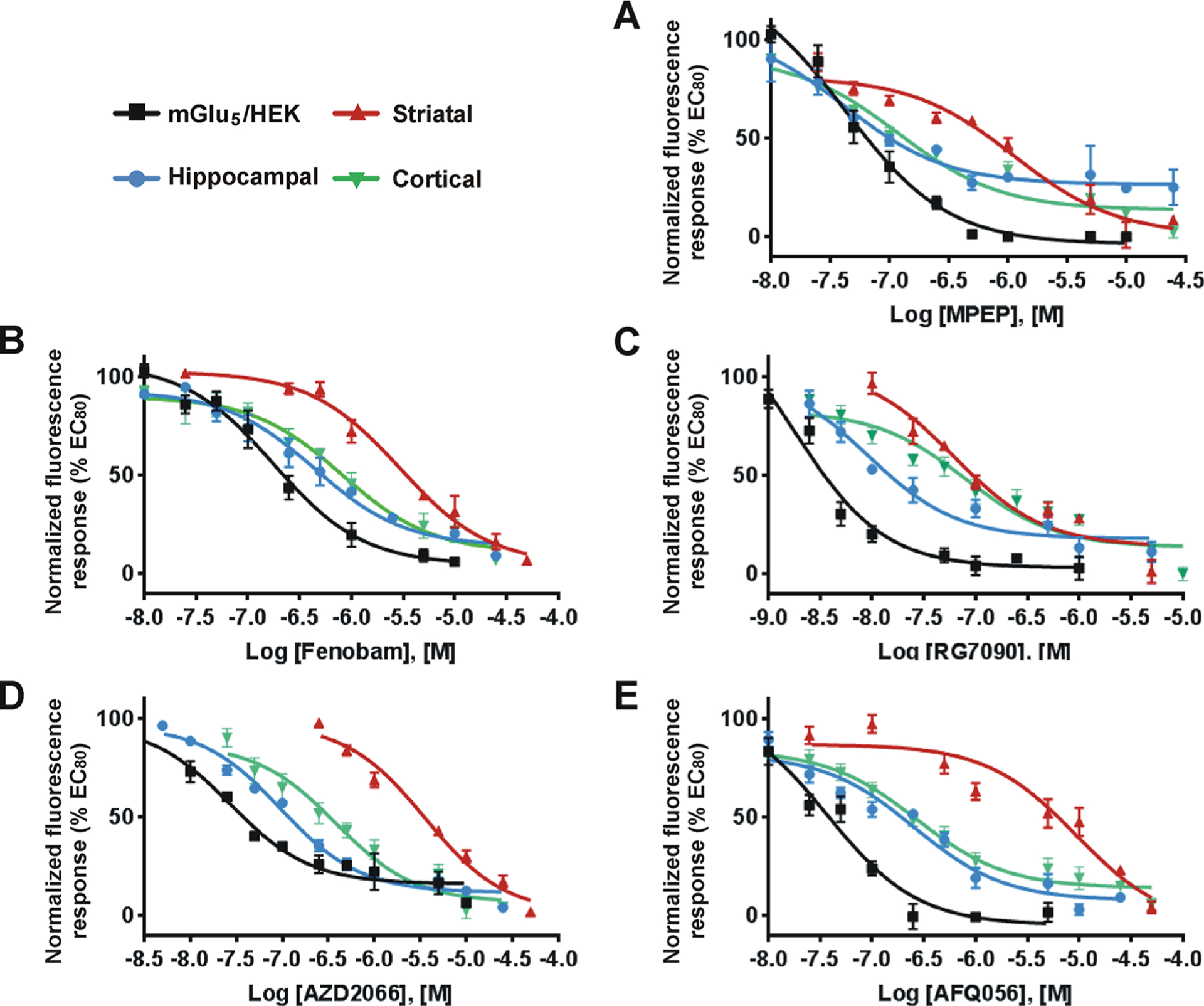
mGlu5 NAMs exhibit cell type-specific bias. HEK cells expressing mGlu5, striatal, hippocampal, and cortical cultures were prepared as described in Methods. A range of concentrations of mGlu5 NAMs (MPEP, Fenobam, RG7090, AZD2066, and AFQ056) were added to the Fura-2 AM loaded cells in 96-well plates and incubated for 20 min. Quis was added at the cell specific EC80 value determined in Figure 3, and Ca2+ responses were measured as described in Methods. (A–E) Inhibition curves for each NAM are as indicated. Inhibition curves were generated from the mean data of three independent experiments performed in triplicate. Error bars represent SEM. Data are plotted as a percentage of the EC80 response to Quis. IC50 values of each mGlu5 NAM in all cell types were determined using Prism curve fitting software from GraphPad (Table 3).
Table 3.
Summary of IC50 Values of Various mGlu5 NAMs in mGlu5/HEK Cells and Striatal, Hippocampal, and Cortical Culturesa
| IC50 | ||||
|---|---|---|---|---|
| NAM | mGlu5 HEK | striatal | hippocampal | cortical |
| MPEP | 42.4 ± 7.5 nM | 1.13 ± 0.36 μM | 41.7 ± 4.7 nM | 119 ± 34.1 nM |
| Fenobam | 178 ± 40 nM | 2.93 ± 0.48 μM | 442 ± 150 nM | 807 ± 53.5 nM |
| AFQ056 | 37.6 ± 8.9 nM | 9.44 ± 3.78 μM | 257 ± 27.4 nM | 270 ± 73.0 nM |
| RG7090 | 1.76 ± 0.88 nM | 62.0 ± 17.5 nM | 8.97 ± 3.40 nM | 80.2 ± 16.8 nM |
| AZD2066 | 27.2 ± 9.1 nM | 3.56 ± 0.52 μM | 96.2 ± 17.8 nM | 380 ± 78.0 nM |
The IC50 values of various mGlu5 NAMs were determined from the inhibition curves in Figure 4 using GraphPad Prism.
Since all of the tested NAMs have the topological polar surface area (TPSA) < 90 Å2 and molecular weight < 45037 and all of the tested NAMs except for Fenobam have consensus log Po/w values within optimal limits of about 3 (Table 129,38,39), our initial hypothesis was that the chosen compounds would exhibit fairly equivalent effects in the tissues tested. Thus, it was surprising that AFQ056 in particular was so disparate. In examining other parameters related to solubility (log S), blood-brain barrier (BBB) permeability (log BB), and especially CNS permeability (log PS; Table 138,39), AZD2066 seemed to have the poorest ability to cross the BBB and access the CNS but it was AFQ056 that was the least effective in blocking Quis-mediated striatal mGlu5 activity. Although these hallmarks of permeability have been successful in predicting a compound’s ability to cross biological membranes, other physicochemical properties that influence membrane permeability include hydrogen-bond accepting groups as well as hydrogen-bond donating groups. In general, increased numbers of either type of hydrogen bond are associated with poorer compound permeability.40 Freely rotatable bonds are another factor in that the greater the number of rotatable bonds, the poorer is the diffusion.41 All of these values are included in Table 1 for the tested NAMs. There are no obvious differences between these compounds when accounting for the empirical data (Table 1).
To further assess the differences between AFQ056 and the other NAMs, we subsequently explored ligand efficiency metrics which incorporate aspects of biological activity with the inherent structural features of the ligand in binding to its target as well as lipophilic ligand efficiency (LLE) which combines in vitro potency and lipophilicity.42–45 Defined as the difference of log Po/w and the negative logarithm of a potency measure such as IC50, the LLE is frequently used as a rule of thumb to optimize promising compound leads. LLE was proposed as a measure of specificity by Leeson and Springthorpe46 who suggested that an ideal LLE value is ~5–7 units or greater. Applying this concept to our data (Table 4), in every tissue but striatum, the tested compounds exhibited LLE values of ~3 or greater. In contrast, LLE values of all NAMs in the striatum were lower than those in other tissues with AFQ056 being the worst (1.88). Treating the data in this way reveals that factors other than ligand size, permeability, and selectivity can affect potency and importantly that the milieu, i.e., the biological membrane and membrane constituents, that the receptor is in must also play a role. LLE might be a useful parameter to explore during compound optimization especially for drugs targeting the CNS.
Table 4.
Summary of Lipophilic Ligand Efficiency (LLE) Values of Tested mGlu5 NAMs in mGlu5/HEK Cells and Striatal, Hippocampal, and Cortical Culturesa
| LLE | ||||
|---|---|---|---|---|
| mGlu5 HEK | striatal | hippocampal | cortical | |
| MPEP | 4.22 | 2.80 | 4.23 | 3.77 |
| Fenobam | 5.50 | 2.38 | 3.20 | 2.94 |
| AFQ056 | 4.57 | 1.88 | 3.44 | 3.42 |
| RG7090 | 4.59 | 4.06 | 4.90 | 3.95 |
| AZD2066 | 4.29 | 2.30 | 3.87 | 3.27 |
It is also worth noting that recent structure-based optimization analyses of Fenobam, M-MPEP (a methoxy-substituted derivative of MPEP),47 and AFQ056 complexed with mGlu5 affirm the difficulty of the optimization process.48 The X-ray crystal structures of docked Fenobam and M-MPEP show that these ligands bind in the proposed allosteric pocket as does AFQ056 but the headgroup of the latter is rotated 180°. These data emphasize that each ligand, but especially AFQ056, induces a different binding site conformation and that binding sites are also modified by the water network within the allosteric pocket. Even the X-ray structure of Fenobam which more closely overlays that of M-MPEP is significantly different largely due to water network interactions. Taken together, these new structural data48 together with empirical data in biological membranes further emphasize the challenges of drug design and the absolute need to incorporate the milieu the drug is being targeted for.
What other physiological differences might contribute to the cell type specificity observed in Quis- or NAM-treated cultures? Besides differences in receptor number described below, another possibility is that the membrane environment and/or its specific lipid composition might also modulate receptor function.49 For example, lipids such as cholesterol and phospholipids serve as both structural elements and important signaling molecules which contribute to receptor and channel regulation. Moreover, cholesterol significantly modulates the stability and ligand-binding properties of many GPCRs including mGlus50–52 by enhancing53–57 or inhibiting ligand binding.58–61 Inasmuch as cholesterol is enriched in the plasma membrane (20–25% of lipid molecules62) but is only 1% of ER membrane lipids,62 it is conceivable that membrane-specific cholesterol levels might differentially affect GPCR function at the cell surface versus ER or nuclear membranes. Thus, a drug that can readily access the brain parenchyma and easily modulate a cell surface GPCR may not have the same effect on an intracellular receptor due to membrane cholesterol composition. Other lipids may also play a role since recent lipidomic studies point to hundreds of lipid differences between different brain regions, many of which can change dramatically depending upon the condition to which they are exposed.63 Whether cholesterol or other lipids affect mGlu5 function in these neurons remains to be established.
More than 90% of Endogenous mGlu5 Is Intracellular in Cortical, Hippocampal, and Striatal Dissociated Cultures.
Given the differences in Quis and NAM efficacies across the cell line and tissues examined, we tested whether there were significant differences in receptor numbers either on the cell surface or inside the cell and whether the transporters and/or exchanger necessary for intracellular mGlu5 activation were also present. Thus we used anti-mGlu5 immunohistochemistry of dissociated striatal, hippocampal, and cortical cultures as well as western blotting of cellular lysates and biotinylation to determine not only the overall pattern of expression but also the cell surface levels of mGlu5. Figure 5A confirms and extends our previous observations showing large amounts of mGlu5 within neurons from these areas. Moreover, in all brain regions, there appeared to be significant yet varying amounts of mGlu5 on nuclear membranes as demarcated by Lamin B2 staining (Figure 5A) and as shown previously.13,15 Western blotting (Figure 5B–D) also revealed similar levels of mGlu5 receptor in cellular lysates from these brain regions. The specificity of the antibody is indicated by its lack of staining in mGlu5 KO versus mGlu5 wildtype striatal cultures (Figure 5B). One approach to determine cell surface levels of mGlu5 is to use antibodies directed against its N-terminal extracellular domain in nonpermeabilized cultures. However, no specific mGlu5 staining was observed in wildtype littermate striatal, hippocampal or cortical cultures versus mGlu5KO cultures when we tested several commercially available N-terminal antibodies (not shown). Therefore we used biotinylation of cell surface proteins to quantify cell surface mGlu5. Using this approach, only 2% of total mGlu5 receptors were on striatal neuronal surfaces and only ~5–7% of mGlu5 receptors were found on the plasma membrane of hippocampal or cortical cells (Figure, 5E–G). Because the biotinylated fraction ran at a higher molecular weight than the input sample, we also tested whether it was sensitive to PNGase F treatment. As shown in Figure 5F, deglycosylated, biotinylated mGlu5 exhibited a mobility shift akin to the input fraction. Thus, endogenous mGlu5 receptors present on neuronal plasma membranes are glycosylated whereas most of the intracellular receptor pool appears to be nonglycosylated (Figure 5F). Finally, we used western blotting to determine the approximate levels of the neuronal excitatory amino acid transporter, EAAT3, and the cystine/glutamate exchanger, xCT, which together, provide facilitated transport of glutamate or Quis passage across cellular membranes.13,15 Both EAAT3 and xCT were abundantly and evenly expressed in cortical, hippocampal and striatal cultures (Figure 6).
Figure 6.
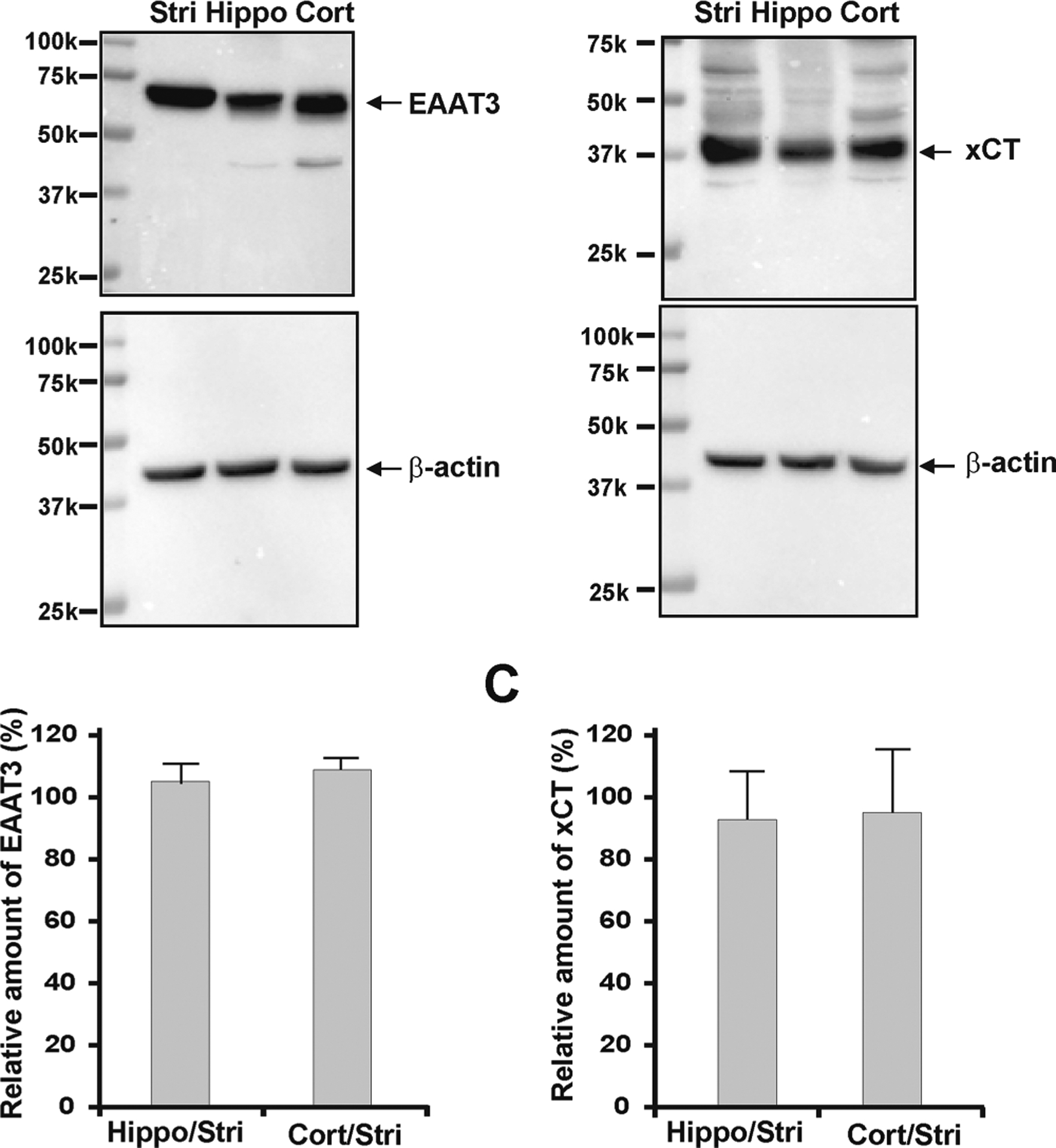
EAAT3 and xCT are evenly expressed in striatal, hippocampal, and cortical cultures. (A) Western blots of DIV 14 neuronal lysates probed with antibodies against EAAT3 or xCT. EAAT3 or xCT expression was normalized to β-actin, to control for equal protein loading. (B) Quantitation of EAAT3: hippocampal/striatal = 105.08 ± 6.01%, cortical/striatal = 108.95 ± 4.82%. (C) Quantitation of xCT: hippocampal/striatal = 92.75 ± 15.09%, cortical/striatal = 94.97 ± 20.41%. Data shown represent the mean of three experiments.
Previous electron microscopy results indicated that between 60 and 90% of mGlu5 is intracellular although levels varied with age and brain region examined.64,65 Our own studies indicated about 91% of immunogold labeling was intracellular in rat visual cortex66 and >85% in rat spinal cord dorsal horn.16 The cell surface biotinylation paradigm used here revealed even fewer cell surface mGlu5 receptors than were measured using electron microscopy. These lower numbers might be due to the neonatal nature of the culture system and the absence of a normal neuronal circuit context. For example, studies have demonstrated that cell surface expression of mGlu5 is highly dependent upon protein–protein interactions, many of which are activity-dependent. Thus lower numbers of cell surface receptors measured using biotinylation might be due to circuit properties present in vivo which are missing in the dissociated paradigm. Nonetheless, the biotinylation paradigm suggested >90% of mGlu5 was inside the cell versus on the cell surface as did the maximum fluorescence response (Emax) values in the presence and absence of the cell surface mGlu5 inhibitor LY393053, (Figure 3, Table 2). Taken together these data are entirely consistent with the bulk of the receptor (>90%) being present and signaling from inside the cell vs on the cell surface at this developmental time point.
In summary, emerging data indicate that GPCR signaling is determined not only by the agonist and a given receptor but also by a variety of cell-type-specific factors that can influence a given response. These might include the lipid composition of the bilayer, the scaffolding proteins available, the levels and composition of signaling pathways in proximity, and where the receptor normally resides or is trafficked to. Thus the clinical efficacy of a given ligand might be dramatically affected by cell-type- and location-specific bias. By evaluating ligand responses of native mGlu5 receptors in primary neuronal cultures with unique membrane constituents and lipophilic properties, these studies demonstrate that receptor location and membrane environment play key roles in drug potency and efficacy. Given that the drugs tested have all been in clinical trials, our studies highlight the importance for drug candidates to be screened under conditions mimicking the environment targeted in vivo. Although drug discovery programs are largely focused on identifying ligands for cell surface receptors using heterologous cell types, there is a growing recognition that compartmentalized and cell type-specific GPCR signaling can play critical roles in physiological and/or pathophysiological paradigms. These and other studies emphasize the importance of intracellular receptors as potential therapeutic targets. Challenges exist, however, in the development of effective pharmacological agents that target a chosen receptor that signals through a desired pathway in the preferred subcellular compartment of the most relevant cell type. In spite of these challenges, recent progress especially in the pain field shows that at least targeting endosomal signaling can provide new opportunities for therapeutic benefits.67
METHODS
Materials.
Glutamate, Quisqualate (Quis), (S)-3,5-dihydroxyphenylglycine (DHPG), 2-methyl-6-(phenylethynyl)pyridine (MPEP), 7-(hydroxyimino)-cyclopropan[b]chromen-1a-carboxylate ethyl ester (CPCCOEt), (±)-4-(4-aminophenyl)-1,2-dihydro-1-methyl-2-propyl-carbamoyl-6,7-methylenedioxyphthalazine (SYM2206), D-2-amino-5-phosphonopentanoic acid (D-AP5), 6-cyano-7-nitroquinoxaline-2,3-dione (CNQX), and N-(3-chlorophenyl)-N′-(4,5-dihydro-1-methyl-4-oxo-1H-imidazol-2-yl)urea (Fenobam) were purchased from Tocris (Minneapolis, MN). 2-Amino-2-(3-cis/trans-carboxycyclobutyl)-3-(9H-thioxanthen-9-yl) propionic acid (LY393053), 5-methyl-6-(phenylethynyl)-pyridine (5MPEP), methyl (3aR,4S,7aR)-4-hydroxy-4-[(3-methylphenyl)ethynyl]octahydro-1H-indole-1-carboxylate (mavoglurant, AFQ056), 2-chloro-4-((1-(4-fluorophenyl)-2,5-dimethyl-1H-imidazol-4-yl)ethynyl)pyridine (basimglurant, RG-7090, RO-4917523), and 4-[5-[(1R)-1-[5-(3-chlorophenyl)-3-isoxazolyl]ethoxy]-4-methyl-4H-1,2,4-triazol-3-yl]pyridine (AZD2066) were originally obtained from Lilly Research Laboratories, Eli Lilly and Company (Indianapolis, IN). Subsequently, they were purchased from MedChemExpress (Monmouth Junction, NJ) or Tocris.
Cell Cultures.
The mGlu5/HEK stable cell line was generated and maintained as described.31,68 Primary striatal, cortical, and hippocampal neuronal cultures using neonatal 1 day old rat pups were prepared and maintained as previously described.13,15
Biotinylation.
Biochemical measurements of surface expressed receptors. Days in vitro (DIV) 14 cultured primary neurons plated in 12-well plates (1 million/well, minimum 4 wells for each cell type) were washed three times with ice-cold PBS (pH 8.0). Cultures were then incubated with PBS (pH 8.0) containing 2 mM EZ-Link Sulfo-NHS-LC-Biotin (Thermo Fisher Scientific, Waltham, MA) at room temperature for 30 min. Cultures were rinsed in PBS (pH 8.0) containing 100 mM glycine to quench the biotin reaction. Cultures were lysed in 75 μL/well modified RIPA buffer (50 mM Tris, pH 7.5, 150 mM NaCl, 1% NP-40, 0.1% SDS, 0.5% sodium deoxycholate, 1 mM phenylmethylsulfonyl fluoride (PMSF), complete protease and phosphatase inhibitor cocktail; MilliporeSigma). The homogenates were centrifuged at 14 000g for 15 min at 4 °C. Twenty-five percent of the supernatant was removed to measure total mGlu5; 75% of the remaining supernatant was incubated with NeutrAvidin agarose (Thermo Fisher Scientific) for 3 h at 4 °C and washed three times with RIPA buffer, and then bound proteins were resuspended in SDS sample buffer and heated at 60 °C for 10 min. Quantitative Western blots were performed on both total and biotinylated (surface) proteins using anti-mGlu5.
Deglycosylation.
Deglycosylation of biotinylated DIV 14 cortical cultures with PNGase F (New England Biolabs, Ipswich, MA) was performed as described.69
Immunocytochemistry and Western Blotting.
Primary striatal, hippocampal, or cortical cultures were fixed at DIV 14 and stained as described previously.14 Primary antibodies included rabbit polyclonal anti-mGlu5 (1:200; Abcam Inc., Cambridge, MA) and monoclonal anti-lamin B2 (1:200; Zymed Laboratories, San Francisco, CA). Secondary antibodies included goat anti-rabbit Cy3 (1:300; Jackson Immunoresearch, West Grove, PA) and goat anti-mouse Alexa 488 (1:300; Molecular Probes, Eugene, OR). Western blotting was performed using whole cell extracts or biotinylated fractions from DIV 14 striatal, hippocampal, or cortical cultures. Protein concentrations of whole cell lysates were determined using the Bradford assay (Bio-Rad, Hercules, CA). Proteins were separated by SDS-PAGE, blotted, and probed with rabbit monoclonal anti-mGlu5 (1:1500; Abcam Inc.), mouse monoclonal β-actin (1:2500; Millipore Sigma, St. Louis, MO), or mouse monoclonal anti-Na+-K+ ATPase (1:500; Developmental Studies Hybridoma Bank, University of Iowa, Iowa City, IA). Rabbit polyclonal anti-EAAT3 and xCT antibodies were gifts from Dr. J. Rothstein (The Johns Hopkins University) and used at dilutions of 1:200. A horseradish peroxidase conjugated with goat anti-rabbit IgG (1:2000; Cell Signaling Technology, Inc., Danvers, MA) or anti-mouse IgG (1:2500; MilliporeSigma) was used in conjunction with enhanced chemiluminescence (Bio-Rad) to detect the signal. Densitometric analyses were performed using the ChemiDoc MP system (Bio-Rad) together with associated Image Lab software.
Confocal Microscopy and Data Analysis: Fluorescent Measurements of Intracellular Ca2+.
The mGlu5/HEK cell line and primary striatal and hippocampal cultures were loaded with Ca2+ fluorophore, imaged, and quantitated as described previously.13 Briefly, cells were grown on 35 mm dishes with glass grids, loaded with 5 μM Oregon Green 488 BAPTA-AM (Molecular Probes, Eugene, OR), and imaged using a laser confocal microscope (Olympus BX 50WI) with an Olympus LUMPlanFl/lR 40×/0.80w or 60×/0.90w objective. Cultures were preincubated with 20 μM mGlu1 antagonist CPCCOEt and 25 μM AMPAR antagonist SYM220 to measure mGlu5 specific activation by Quis. NAMs were added at 10 μM. The real time images were captured by using an Olympus Fluoview FVX confocal laser scanning system using Fluoview 2.0 acquisition software. Images were converted to TIF files for processing with MetaMorph (version 7.7) Professional Image Analysis software, produced by Universal Imaging Corporation (Downingtown, PA).
Fluorescence-Based Ca2+ Flux Assay with a Microplate Reader.
The mGlu5/HEK cells were plated at 40 000 cells per well in black-walled, clear-bottomed, poly(D-lysine)-coated 96-well plates (Corning Incorporated, Corning, NY). Cells were grown overnight at 37 °C in the presence of 5% CO2. The next day, the cells were loaded with 1 μM Fura-2 AM for 30 min at 37 °C and washed with Hanks’ balanced salt solution (HBSS). Primary rat striatal, hippocampal, SCDH, or cortical cultures were plated at 50 000 cells per well in black-walled, clear-bottomed, poly(D-lysine)-coated 96-well plates. After 14 days, the primary cultures were loaded with 1 μM Fura-2 AM for 30 min at 37 °C and then washed with HBSS. To determine the EC50 for Quis in each cell type, mGlu5/HEK cells or primary neuronal cultures were then preincubated with HBSS buffer containing the mGlu1 antagonist (20 μM CPCCOEt) and the AMPA receptor antagonist (25 μM SYM2206) to measure mGlu5 specific activation by Quis. A range of concentrations of Quis was added after 5 s of baseline determination; readings were collected per 0.7 s for an additional 30 s. To determine mGlu5 NAM potency, mGlu5/HEK cells or primary neuronal cultures were preincubated with various concentrations of compound for 20 min at 37 °C in assay buffer (HBSS containing 20 μM CPCCOEt and 25 μM SYM2206) prior to Ca2+ flux measurement. Fura-2 fluorescence was measured using a BioTek Synergy H4 Hybrid Microplate Reader. Baseline recordings were acquired for 5 s before injecting Quis at a final concentration equal to the EC80 previously determined for each cell type. Thirty additional seconds of data were collected, and then the 340/380 nm excitation ratio for Fura-2 was analyzed with Gen5 analysis software. Normalized fold change of the 340/380 ratio from NAM-treated wells was compared to those of control wells, and the percentage of inhibition was calculated. The IC50 value was determined using the curve fitting software developed by GraphPad Prism. Specifically, an XY data table was created where X equals the logarithmic concentration of each NAM and Y represents the percentage of the response. In all cases, “Y” included three sets of data, each in triplicate. The data table was analyzed with nonlinear regression and “dose–response curves – inhibition” with the equation “log(inhibitor) vs response”: .
ACKNOWLEDGMENTS
We thank Dr. David L. McKinzie (Lilly USA) for reagents and advice. We also thank Dr. Ron Dolle (Washington University in St. Louis) for helpful discussions and critical reading of the manuscript. Microscopes and software were provided in part through the use of Washington University Center for Cellular Imaging (WUCCI) supported by Washington University School of Medicine, The Children’s Discovery Institute of Washington University and St. Louis Children’s Hospital (CDI-CORE-2015-505), and the NINDS, National Institutes of Health (NS086741).
Funding
This work was supported, in whole or in part, by National Institutes of Health Grants RO1 MH119197, MH109019 and NS102783, IDDRC Grant #U54 HD087011, and the Lilly Research Award Program.
ABBREVIATIONS
- AMPAR
amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid receptor
- ASD
autism spectrum disorder
- CNS
central nervous system
- EAAT3
excitatory amino acid transporter 3
- ER
endoplasmic reticulum
- ERK1/2
extracellular signal-regulated kinase 1/2
- GPCRs
G-protein coupled receptors
- LLE
lipophilic ligand efficiency
- mGlu5
metabotropic glutamate receptor 5
- NAM
negative allosteric modulators
- PAM
positive allosteric modulator
- Quis
quisqualate
- SCDH
spinal cord dorsal horn
- xCT
cystine/glutamate exchanger
- TPSA
topological polar surface area
Footnotes
The authors declare no competing financial interest.
REFERENCES
- (1).Sriram K, and Insel PA (2018) G Protein-Coupled Receptors as Targets for Approved Drugs: How Many Targets and How Many Drugs? Mol. Pharmacol 93, 251–258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (2).Hauser AS, Attwood MM, Rask-Andersen M, Schiöth HB, and Gloriam DE (2017) Trends in GPCR drug discovery: new agents, targets and indications. Nat. Rev. Drug Discovery 16, 829–842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (3).Conn PJ, Lindsley CW, Meiler J, and Niswender CM (2014) Opportunities and challenges in the discovery of allosteric modulators of GPCRs for treating CNS disorders. Nat. Rev. Drug Discovery 13, 692–708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (4).Sengmany K, and Gregory KJ (2016) Metabotropic glutamate receptor subtype 5: molecular pharmacology, allosteric modulation and stimulus bias. Br. J. Pharmacol 173, 3001–3017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (5).Lane JR, May LT, Parton RG, Sexton PM, and Christopoulos A (2017) A kinetic view of GPCR allostery and biased agonism. Nat. Chem. Biol 13, 929–937. [DOI] [PubMed] [Google Scholar]
- (6).Changeux JP, and Christopoulos A (2016) Allosteric modulation as a unifying mechanism for receptor function and regulation. Cell 166, 1084–1102. [DOI] [PubMed] [Google Scholar]
- (7).Stansley BJ, and Conn PJ (2019) Neuropharmacological Insight from Allosteric Modulation of mGlu Receptors. Trends Pharmacol. Sci 40, 240–252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (8).Jong YI, Harmon SK, and O’Malley KL (2018) Intracellular GPCRs Play Key Roles in Synaptic Plasticity. ACS Chem. Neurosci 9, 2162–2172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (9).Jong YI, Harmon SK, and O’Malley KL (2018) GPCR signalling from within the cell. Br. J. Pharmacol 175, 4026–4035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (10).Weinberg ZY, Crilly SE, and Puthenveedu MA (2019) Spatial encoding of GPCR signaling in the nervous system. Curr. Opin. Cell Biol 57, 83–89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (11).Calebiro D, and Koszegi Z (2019) The subcellular dynamics of GPCR signaling. Mol. Cell. Endocrinol 483, 24–30. [DOI] [PubMed] [Google Scholar]
- (12).Lobingier BT, and von Zastrow M (2019) When trafficking and signaling mix: How subcellular location shapes G protein-coupled receptor activation of heterotrimeric G proteins. Traffic 20, 130–136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (13).Jong YJ, Kumar V, Kingston AE, Romano C, and O’Malley KL (2005) Functional metabotropic glutamate receptors on nuclei from brain and primary cultured striatal neurons. Role of transporters in delivering ligand. J. Biol. Chem 280, 30469–30480. [DOI] [PubMed] [Google Scholar]
- (14).Jong YJ, Kumar V, and O’Malley KL (2009) Intracellular metabotropic glutamate receptor 5 (mGluR5) activates signaling cascades distinct from cell surface counterparts. J. Biol. Chem 284, 35827–35838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (15).Purgert CA, Izumi Y, Jong YJ, Kumar V, Zorumski CF, and O’Malley KL (2014) Intracellular mGlu5 can mediate synaptic plasticity in the hippocampus. J. Neurosci 34, 4589–4598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (16).Vincent K, Cornea VM, Jong YJ, Laferrière A, Kumar N, Mickeviciute A, Fung JS, Bandegi P, Ribeiro-Da-Silva A, O’Malley KL, and Coderre TJ (2016) Intracellular mGluR5 plays a critical role in neuropathic pain. Nat. Commun 7, 10604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (17).Chen Y, Bacon G, Sher E, Clark BP, Kallman MJ, Wright RA, Johnson BG, Schoepp DD, and Kingston AE (1999) Evaluation of the activity of a novel metabotropic glutamate receptor antagonist (±)-2-amino-2-(3-cis and trans-carboxycyclobutyl-3-(9-thioxanthyl)propionic acid) in the in vitro neonatal spinal cord and in an in vivo pain model. Neuroscience 95, 787–793. [DOI] [PubMed] [Google Scholar]
- (18).Gasparini F, Lingenhöhl K, Stoehr N, Flor PJ, Heinrich M, Vranesic I, Biollaz M, Allgeier H, Heckendorn R, Urwyler S, Varney MA, Johnson EC, Hess SD, Rao SP, Sacaan AI, Santori EM, Velicelebi G, and Kuhn R (1999) 2-Methyl-6-(phenylethynyl)-pyridine (MPEP), a potent, selective and systemically active mGlu5 receptor antagonist. Neuropharmacology 38, 1493–1503. [DOI] [PubMed] [Google Scholar]
- (19).Pagano A, Ruegg D, Litschig S, Stoehr N, Stierlin C, Heinrich M, Floersheim P, Prezeau L, Carroll F, Pin JP, Cambria A, Vranesic I, Flor PJ, Gasparini F, and Kuhn R (2000) The non-competitive antagonists 2-methyl-6-(phenylethynyl)-pyridine and 7-hydroxyiminocyclopropan[b]chromen-1a-carboxylic acid ethyl ester interact with overlapping binding pockets in the transmembrane region of group I metabotropic glutamate receptors. J. Biol. Chem 275, 33750–33758. [DOI] [PubMed] [Google Scholar]
- (20).Porter RH, Jaeschke G, Spooren W, Ballard TM, Büttelmann B, Kolczewski S, Peters JU, Prinssen E, Wichmann J, Vieira E, Mühlemann A, Gatti S, Mutel V, and Malherbe P (2005) Fenobam: a clinically validated nonbenzodiazepine anxiolytic is a potent, selective, and noncompetitive mGlu5 receptor antagonist with inverse agonist activity. J. Pharmacol. Exp. Ther 315, 711–721. [DOI] [PubMed] [Google Scholar]
- (21).Gregory KJ, Noetzel MJ, and Niswender CM (2013) Pharmacology of metabotropic glutamate receptor allosteric modulators: structural basis and therapeutic potential for CNS disorders. Prog. Mol. Biol. Transl 115, 61–121. [DOI] [PubMed] [Google Scholar]
- (22).Crupi R, Impellizzeri D, and Cuzzocrea S (2019) Role of Metabotropic Glutamate Receptors in Neurological Disorders. Front. Mol. Neurosci 12, 20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (23).Pereira V, and Goudet C (2019) Emerging Trends in Pain Modulation by Metabotropic Glutamate Receptors. Front. Mol. Neurosci 11, 464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (24).Vranesic I, Ofner S, Flor PJ, Bilbe G, Bouhelal R, Enz A, Desrayaud S, McAllister K, Kuhn R, and Gasparini F (2014) AFQ056/mavoglurant, a novel clinically effective mGluR5 antagonist: identification, SAR and pharmacological characterization. Bioorg. Med. Chem 22, 5790–5803. [DOI] [PubMed] [Google Scholar]
- (25).Swedberg MD, and Raboisson P (2014) AZD9272 and AZD2066: selective and highly central nervous system penetrant mGluR5 antagonists characterized by their discriminative effects. J. Pharmacol. Exp. Ther 350, 212–222. [DOI] [PubMed] [Google Scholar]
- (26).Lindemann L, Porter RH, Scharf SH, Kuennecke B, Bruns A, von Kienlin M, Harrison AC, Paehler A, Funk C, Gloge A, Schneider M, Parrott NJ, Polonchuk L, Niederhauser U, Morairty SR, Kilduff TS, Vieira E, Kolczewski S, Wichmann J, Hartung T, Honer M, Borroni E, Moreau JL, Prinssen E, Spooren W, Wettstein JG, and Jaeschke G (2015) Pharmacology of basimglurant (RO4917523, RG7090), a unique metabotropic glutamate receptor 5 negative allosteric modulator in clinical development for depression. J. Pharmacol. Exp. Ther 353, 213–233. [DOI] [PubMed] [Google Scholar]
- (27).Berry-Kravis EM, Lindemann L, Jønch AE, Apostol G, Bear MF, Carpenter RL, Crawley JN, Curie A, Des Portes V, Hossain F, Gasparini F, Gomez-Mancilla B, Hessl D, Loth E, Scharf SH, Wang PP, Von Raison F, Hagerman R, Spooren W, and Jacquemont S (2018) Drug development for neurodevelopmental disorders: lessons learned from fragile X syndro me. Nat. Rev. Drug Discovery 17, 280–299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (28).Yamasue H, Aran A, and Berry-Kravis E (2019) Emerging pharmacological therapies in fragile X syndrome and autism. Curr. Opin. Neurol 32, 635–640. [DOI] [PubMed] [Google Scholar]
- (29).Lester HA, Miwa JM, and Srinivasan R (2012) Psychiatric drugs bind to classical targets within early exocytotic pathways; therapeutic effects. Biol. Psychiatry 72, 907–915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (30).Daina A, Michielin O, and Zoete V (2017) SwissADME: a free web tool to evaluate pharmacokinetics, drug-likeness and medicinal chemistry friendliness of small molecules. Sci. Rep 7, 42717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (31).Romano C, Yang WL, and O’Malley KL (1996) Metabotropic glutamate receptor 5 is a disulfide-linked dimer. J. Biol. Chem 271, 28612–28616. [DOI] [PubMed] [Google Scholar]
- (32).Kumar V, Fahey PG, Jong YJ, Ramanan N, and O’Malley KL (2012) Activation of intracellular metabotropic glutamate receptor 5 in striatal neurons leads to up-regulation of genes associated with sustained synaptic transmission including Arc/Arg3.1 protein. J. Biol. Chem 287, 5412–5425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (33).Jong YI, and O’Malley KL (2017) Mechanisms Associated with Activation of Intracellular Metabotropic Glutamate Receptor, mGluR5. Neurochem. Res 42, 166–172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (34).Rodriguez AL, Nong Y, Sekaran NK, Alagille D, Tamagnan GD, and Conn PJ (2005) A close structural analog of 2-methyl-6-(phenylethynyl)-pyridine acts as a neutral allosteric site ligand on metabotropic glutamate receptor subtype 5 and blocks the effects of multiple allosteric modulators. Mol. Pharmacol 68, 1793–1802. [DOI] [PubMed] [Google Scholar]
- (35).Spampinato SF, Copani A, Nicoletti F, Sortino MA, and Caraci F (2018) Metabotropic Glutamate Receptors in Glial Cells: A New Potential Target for Neuroprotection? Front. Mol. Neurosci 11, 414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (36).Guo N, Guo W, Kralikova M, Jiang M, Schieren I, Narendran R, Slifstein M, Abi-Dargham A, Laruelle M, Javitch JA, and Rayport S (2010) Impact of D2 receptor internalization on binding affinity of neuroimaging radiotracers. Neuropsychopharmacology 35, 806–817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (37).Pajouhesh H, and Lenz GR (2005) Medicinal chemical properties of successful central nervous system drugs. NeuroRx 2, 541–553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (38).Pires DE, Blundell L, and Ascher DB (2015) pkCSM: Predicting Small-Molecule Pharmacokinetic and Toxicity Properties Using Graph-Based Signatures. J. Med. Chem 58, 4066–4072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (39).Vilar S, Chakrabarti M, and Costanzi S (2010) Prediction of passive blood-brain partitioning: straightforward and effective classification models based on in silico derived physicochemical descriptors. J. Mol. Graphics Modell 28, 899–903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (40).Goodwin JT, Conradi RA, Ho NF, and Burton PS (2001) Physicochemical determinants of passive membrane permeability: role of solute hydrogen-bonding potential and volume. J. Med. Chem 44, 3721–3729. [DOI] [PubMed] [Google Scholar]
- (41).Veber DF, Johnson SR, Cheng HY, Smith BR, Ward KW, and Kopple KD (2002) Molecular properties that influence the oral bioavailability of drug candidates. J. Med. Chem 45, 2615–2623. [DOI] [PubMed] [Google Scholar]
- (42).Hopkins AL, Keserü GM, Leeson PD, Rees DC, and Reynolds CH (2014) The role of ligand efficiency metrics in drug discovery. Nat. Rev. Drug Discovery 13, 105–121. [DOI] [PubMed] [Google Scholar]
- (43).Barret R (2018) Importance and Evaluation of Lipophilicity. Therapeutical Chemistry, 53–78. [Google Scholar]
- (44).Scott JS, and Waring MJ (2018) Practical application of ligand efficiency metrics in lead optimization. Bioorg. Med. Chem 26, 3006–3015. [DOI] [PubMed] [Google Scholar]
- (45).Shultz MD (2019) Two Decades under the Influence of the Rule of Five and the Changing Properties of Approved Oral Drugs. J. Med. Chem 62, 1701–1714. [DOI] [PubMed] [Google Scholar]
- (46).Leeson PD, and Springthorpe B (2007) The influence of drug-like concepts on decision-making in medicinal chemistry. Nat. Rev. Drug Discovery 6, 881–890. [DOI] [PubMed] [Google Scholar]
- (47).Gasparini F, Andres H, Flor PJ, Heinrich M, Inderbitzin W, Lingenhöhl K, Müller H, Munk VC, Omilusik K, Stierlin C, Stoehr N, Vranesic I, and Kuhn R (2002) [(3)H]-M-MPEP, a potent, subtype-selective radioligand for the metabotropic glutamate receptor subtype 5. Bioorg. Med. Chem. Lett 12, 407–409. [DOI] [PubMed] [Google Scholar]
- (48).Christopher JA, Orgován Z, Congreve M, Doré AS, Errey JC, Marshall FH, Mason JS, Okrasa K, Rucktooa P, Serrano-Vega MJ, Ferenczy GG, and Keserü GM (2019) Structure-Based Optimization Strategies for G Protein-Coupled Receptor (GPCR) Allosteric Modulators: A Case Study from Analyses of New Metabotropic Glutamate Receptor 5 (mGlu5) X-ray Structures. J. Med. Chem 62, 207–222. [DOI] [PubMed] [Google Scholar]
- (49).Dawaliby R, Trubbia C, Delporte C, Masureel M, Van Antwerpen P, Kobilka BK, and Govaerts C (2016) Allosteric regulation of G protein-coupled receptor activity by phospholipids. Nat. Chem. Biol 12, 35–39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (50).Sengupta D, and Chattopadhyay A (2015) Molecular dynamics simulations of GPCR-cholesterol interaction: An emerging paradigm. Biochim. Biophys. Acta, Biomembr 1848, 1775–1782. [DOI] [PubMed] [Google Scholar]
- (51).Gimpl G (2016) Interaction of G protein coupled receptors and cholesterol. Chem. Phys. Lipids 199, 61–73. [DOI] [PubMed] [Google Scholar]
- (52).Guixà-González R, Albasanz J, Rodriguez-Espigares IL, Pastor M, Sanz F, MartíSolano M, Manna M, Martinez-Seara H, Hildebrand PW, Martín M, and Selent J (2017) Membrane cholesterol access into a G-protein-coupled receptor. Nat. Commun 8, 14505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (53).Gimpl G, Burger K, and Fahrenholz F (1997) Cholesterol as modulator of receptor function. Biochemistry 36, 10959–10974. [DOI] [PubMed] [Google Scholar]
- (54).Nguyen DH, and Taub D (2002) CXCR4 function requires membrane cholesterol: implications for HIV infection. J. Immunol 168, 4121–4126. [DOI] [PubMed] [Google Scholar]
- (55).Nguyen DH, and Taub DD (2003) Inhibition of chemokine receptor function by membrane cholesterol oxidation. Exp. Cell Res 291, 36–45. [DOI] [PubMed] [Google Scholar]
- (56).Harikumar KG, Puri V, Singh RD, Hanada K, Pagano RE, and Miller LJ (2005) Differential effects of modification of membrane cholesterol and sphingolipids on the conformation, function, and trafficking of the G protein-coupled cholecystokinin receptor. J. Biol. Chem 280, 2176–2185. [DOI] [PubMed] [Google Scholar]
- (57).Huang P, Xu W, Yoon SI, Chen C, Chong PL, and Liu-Chen LY (2007) Cholesterol reduction by methyl-β-cyclodextrin attenuates the delta opioid receptor-mediated signaling in neuronal cells but enhances it in non-neuronal cells. Biochem. Pharmacol 73, 534–549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (58).Bari M, Paradisi A, Pasquariello N, and Maccarrone M (2005) Cholesterol-dependent modulation of type 1 cannabinoid receptors in nerve cells. J. Neurosci. Res 81, 275–283. [DOI] [PubMed] [Google Scholar]
- (59).Colozo AT, Park PSH, Sum CS, Pisterzi LF, and Wells JW (2007) Cholesterol as a determinant of cooperativity in the M2 muscarinic cholinergic receptor. Biochem. Pharmacol 74, 236–255. [DOI] [PubMed] [Google Scholar]
- (60).Michal P, Rudajev V, El-Fakahany EE, and Dolezal V (2009) Membrane cholesterol content influences binding properties of muscarinic M2 receptors and differentially impacts activation of second messenger pathways. Eur. J. Pharmacol 606, 50–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (61).Xu W, Yoon SI, Huang P, Wang Y, Chen C, Chong PL, and Liu-Chen LY (2006) Localization of the kappa opioid receptor in lipid rafts. J. Pharmacol. Exp. Ther 317, 1295–1306. [DOI] [PubMed] [Google Scholar]
- (62).Ikonen E (2008) Cellular cholesterol trafficking and compartmentalization. Nat. Rev. Mol. Cell Biol 9, 125–138. [DOI] [PubMed] [Google Scholar]
- (63).Lin Y, Gu H, Jiang L, Xu W, Liu C, Li Y, Qian X, Li D, Li Z, Hu J, Zhang H, Guo W, Zhao Y, and Cen X (2017) Cocaine modifies brain lipidome in mice. Mol. Cell. Neurosci 85, 29–44. [DOI] [PubMed] [Google Scholar]
- (64).Hubert GW, Paquet M, and Smith Y (2001) Differential subcellular localization of mGluR1a and mGluR5 in the rat and monkey substantia nigra. J. Neurosci 21, 1838–1847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (65).Kuwajima M, Hall RA, Aiba A, and Smith Y (2004) Subcellular and subsynaptic localization of group I metabotropic glutamate receptors in the monkey subthalamic nucleus. J. Comp. Neurol 474, 589–602. [DOI] [PubMed] [Google Scholar]
- (66).O’Malley KL, Jong YJ, Gonchar Y, Burkhalter A, and Romano C (2003) Activation of metabotropic glutamate receptor mGlu5 on nuclear membranes mediates intranuclear Ca2+ changes in heterologous cell types and neurons. J. Biol. Chem 278, 28210–28219. [DOI] [PubMed] [Google Scholar]
- (67).Thomsen ARB, Jensen DD, Hicks GA, and Bunnett NW (2018) Therapeutic Targeting of Endosomal G-Protein-Coupled Receptors. Trends Pharmacol. Sci 39, 879–891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (68).Romano C, Miller JK, Hyrc K, Dikranian S, Mennerick S, Takeuchi Y, Goldberg MP, and O’Malley KL (2001) Covalent and noncovalent interactions mediate metabotropic glutamate receptor mGlu5 dimerization. Mol. Pharmacol 59, 46–53. [PubMed] [Google Scholar]
- (69).Sergin I, Jong YI, Harmon SK, Kumar V, and O’Malley KL (2017) Sequences within the C Terminus of the Metabotropic Glutamate Receptor 5 (mGluR5) Are Responsible for Inner Nuclear Membrane Localization. J. Biol. Chem 292, 3637–3655. [DOI] [PMC free article] [PubMed] [Google Scholar]