

Abstract
Objective
The heterogeneity of systemic lupus erythematosus (SLE) constitutes clinical and therapeutical challenges. We therefore studied whether unrecognized disease subgroups can be identified by using autoantibody profiling together with HLA‐DRB1 alleles and immunological and clinical data.
Methods
An unsupervised cluster analysis was performed based on detection of 13 SLE‐associated autoantibodies (double‐stranded DNA, nucleosomes, ribosomal P, ribonucleoprotein [RNP] 68, RNPA, Smith [Sm], Sm/RNP, Sjögren's syndrome antigen A [SSA]/Ro52, SSA/Ro60, Sjögren's syndrome antigen B [SSB]/La, cardiolipin [CL]‐Immunoglobulin G [IgG], CL–Immunoglobulin M [IgM], and β2 glycoprotein I [β2GPI]–IgG) in 911 patients with SLE from two cohorts. We evaluated whether each SLE subgroup is associated with HLA‐DRB1 alleles, clinical manifestations (n = 743), and cytokine levels in circulation (n = 446).
Results
Our analysis identified four subgroups among the patients with SLE. Subgroup 1 (29.3%) was dominated by anti‐SSA/Ro60/Ro52/SSB autoantibodies and was strongly associated with HLA‐DRB1*03 (odds ratio [OR] = 4.73; 95% confidence interval [CI] = 4.52‐4.94). Discoid lesions were more common for this disease subgroup (OR = 1.71, 95% CI = 1.18‐2.47). Subgroup 2 (28.7%) was dominated by anti‐nucleosome/SmRNP/DNA/RNPA autoantibodies and associated with HLA‐DRB1*15 (OR = 1.62, 95% CI = 1.41‐1.84). Nephritis was most common in this subgroup (OR = 1.61, 95% CI = 1.14‐2.26). Subgroup 3 (23.8%) was characterized by anti‐ß2GPI‐IgG/anti‐CL–IgG/IgM autoantibodies and a higher frequency of HLA‐DRB1*04 compared with the other patients with SLE. Vascular events were more common in Subgroup 3 (OR = 1.74, 95% CI = 1.2‐2.5). Subgroup 4 (18.2%) was negative for the investigated autoantibodies, and this subgroup was not associated with HLA‐DRB1. Additionally, the levels of eight cytokines significantly differed among the disease subgroups.
Conclusion
Our findings suggest that four fairly distinct subgroups can be identified on the basis of the autoantibody profile in SLE. These four SLE subgroups differ regarding associations with HLA‐DRB1 alleles and immunological and clinical features, suggesting dissimilar disease pathways.
INTRODUCTION
The diagnostic entities in autoimmune diseases are delineated by sets of consensus criteria. Consequently, there is often an extensive heterogeneity and overlap within and between these conditions. This overlap is based on the genetic pleiotropy of autoimmune diseases (1), as well as similar clinical symptoms, inflammatory biomarkers, patterns of autoantibodies, immune cell reactions, and long‐term outcomes (2). Better characterization of subgroups and the use of new biomarkers could improve our understanding of underlying pathogenesis, diagnostics, and disease prognosis and guide therapeutic interventions for individual patients. In a broader perspective, we may need to re‐evaluate the diagnostic framework for autoimmune diseases, among which, systemic lupus erythematosus (SLE) is one of the most heterogeneous.
The American College of Rheumatology (ACR) criteria for the classification of SLE from the year 1982 are based on expert clinical knowledge (3). Extended criteria were proposed by the Systemic Lupus International Collaborating Clinics group (4), and a joint ACR–European League Against Rheumatism effort published new SLE criteria (5). However, these later criteria do not question the unity of SLE as a single entity. On the basis of clinical experience and the need to identify more homogeneous disease subgroups, we set out to comprehensively evaluate whether the SLE heterogeneity can be dissected using autoantibody status. We thereafter studied the distribution of HLA‐DRB1 alleles, clinical manifestations, circulating cytokines levels, disease activity, and organ damage.
The diversity of SLE is apparent with regard to clinical manifestations, type of autoantibodies, inflammatory biomarkers, genetic associations, and prognosis (2). The strongest genetic associations to SLE map to the HLA locus on chromosome 6 (6, 7), a region with known associations to the occurrence of autoantibodies and subphenotypes in several autoimmune diseases (eg, rheumatoid arthritis [8], systemic sclerosis [9], and primary Sjögren syndrome [10, 11]). For instance, we previously demonstrated that antiphospholipid antibodies (aPL) are associated with the HLA‐DRB1*04/*13 genotypes within SLE (12).
In this study, we implemented an unsupervised cluster analysis for 13 SLE‐associated autoantibodies, measured in patients with European white ancestry. We identified four subgroups of patients with SLE, which were differentially associated with HLA‐DRB1 alleles, clinical manifestations, circulating cytokine levels, measures of disease activity, and organ damage. From a clinical perspective, our results suggest that the present SLE diagnosis can be subdivided into fairly distinct autoantibody‐defined phenotypes.
METHODS
Study population
A total of 911 patients with SLE from three Swedish and two United States centers were included in our study (Tables 1 and 2). At inclusion, the patients were adults and self‐reported to be of European white origin. All individuals fulfilled at least four of the ACR classification criteria for SLE (3). When individuals were related, only a single case from each family was included. The adult patients from Columbus, Ohio, were included at the Nationwide Children’s Hospital Rheumatology Clinic and the University Hospital Clinic of Ohio State University (13) (n = 188); in Sweden, patients were included at the Karolinska University Hospital (n = 452), Lund University Hospital (n = 155), and Uppsala University Hospital (n = 116). Blood samples were consecutively collected and stored at −70°C. All analyses were performed on samples taken at inclusion. Clinical manifestations were only available for the Swedish patients (Table 3). Disease activity was measured at inclusion by the SLE Disease Activity Score (SLEDAI‐2K) (14) and the Systemic Lupus Activity Measure (SLAM) (15), and organ damage was measured by the Systemic Lupus International Collaborating Clinics/ACR Damage Index (SDI) (16).
Table 1.
Cohorts include in the current study
| Population | Group | n [%] or Median [range in years] |
|---|---|---|
| Sweden | Controls | 3186 |
| Females | 2387 [74.92] | |
| Age | 55 [15‐84] | |
| SLE patients | 723 | |
| Females | 628 [86.86] | |
| Age | 52 [17‐88] | |
| Ohio, USA | Controls | 468 |
| Females | 423 [90.38] | |
| Age | 37 [16‐73] | |
| SLE patients | 188 | |
| Females | 177 [94.14] | |
| Age | 45 [22‐74] |
Table 2.
Distribution of the positive status for the 13 studied autoantibodies
| Autoantibodies | SLE patients from Sweden n=723 [%] | SLE patients from Ohio, USA n=188 [%] |
|---|---|---|
| Anti‐DNA | 218 [30.15] | 41 [21.81] |
| Anti‐nucleosome | 273 [37.76] | 48 [25.53] |
| Anti‐ribosomal P | 40 [5.56] | 12 [6.38] |
| Anti‐RNP68 | 59 [8.19] | 9 [4.79] |
| Anti‐RNPA | 162 [22.50] | 27 [14.36] |
| Anti‐Sm | 105 [14.58] | 20 [10.64] |
| Anti‐SmRNP | 159 [22.08] | 29 [15.43] |
| Anti‐SSA/Ro52 | 204 [28.33] | 37 [19.68] |
| Anti‐SSA/Ro60 | 293 [40.69] | 48 [25.53] |
| Anti‐SSB/La | 165 [22.92] | 21 [11.17] |
| Anti‐CL IgG | 146 [20.28] | 20 [10.64] |
| Anti‐CL IgM | 150 [20.83] | 27 [14.36] |
| Anti‐β2GP1‐IgG | 151 [20.97] | 31 [16.49] |
Abbreviations: β2GPI, β2 glycoprotein I; CL, cardiolipin; dsDNA, double stranded deoxyribonucleic acid; Ig, immunoglobulin; RNP, ribonucleoprotein; Sm, Smith; SSA/B, Sjögren’s syndrome antigen A/B.
Table 3.
Relation between clinical manifestations and autoantibody subgroups in Swedish patients (n = 720)
| Clinical Manifestations | Subgroup 1 (Anti‐Ro/La; n = 226 [31.4%]) | Subgroup 2 (Antinucleosome/sm/DNA/RNP; n = 219 [30.4%]) | Subgroup 3 (Anti‐β2GP1/CL IgG/CL–IgM; n = 180 [25%]) | Subgroup 4 (Negative for 13 Autoantibodies; n = 95 [13.2%]) | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Percentage | P Value a | FDRp b | OR [95% CI] c | Percentage | P value a | FDRp b | OR [95% CI] c | Percentage | P value a | FDRp b | OR [95% CI] c | Percentage | P value a | FDRp b | OR [95% CI] c | |
| SLE clinical criteria | ||||||||||||||||
| ANA positivity | 98.2 | 0.72 | 0.75 | 0.84 [0.25‐3.28] | 99.5 | 0.22 | 0.72 | 3.72 [0.68‐69.34] | 98.3 | 0.93 | 0.95 | 0.94 [0.27‐4.36] | 96.8 | 0.26 | 0.61 | 0.46 [0.13‐2.15] |
| Butterfly erythema | 59.3 | 0.11 | 0.19 | 1.29 [0.89‐2.46] | 55.7 | 0.85 | 0.89 | 1.03 [0.74‐1.44] | 52.2 | 0.39 | 0.56 | 1.59 [0.96‐2.63] | 48.4 | 0.17 | 0.37 | 0.72 [0.47‐1.12] |
| Discoid skin lesions | 29.6 | 4.40 × 10−3 | 2.90 × 10−2 | 1.71 [1.18‐2.47] | 22.8 | 0.21 | 0.80 | 1.29 [0.86‐1.91] | 16.1 | 9.80 × 10−3 | 4.90 × 10−2 | 0.55 [0.35‐0.86] | 15.8 | 0.09 | 0.16 | 0.52 [0.28‐0.92] |
| Photosensitivity | 77 | 6.20 × 10−3 | 3.10 × 10−2 | 1.68 [1.16‐2.44] | 64.8 | 0.15 | 0.80 | 0.77 [0.54‐1.1] | 64.4 | 0.08 | 0.17 | 0.71 [0.49‐1.03] | 73.7 | 0.59 | 0.91 | 1.15 [0.76‐1.92] |
| Oral ulcers | 31.4 | 0.32 | 0.37 | 1.19 [0.84‐1.68] | 27.9 | 0.4 | 0.81 | 0.86 [0.59‐1.23] | 28.9 | 0.95 | 0.95 | 1.01 [0.69‐1.47] | 27.4 | 0.73 | 0.91 | 0.92 [0.55‐1.48] |
| Arthritis | 74.8 | 0.044 | 0.09 | 0.68 [0.46‐0.99] | 84 | 0.08 | 0.80 | 1.47 [0.96‐2.28] | 76.7 | 0.36 | 0.48 | 0.83 [0.55‐1.25] | 85.3 | 0.12 | 0.30 | 1.65 [0.93‐3.14] |
| Serositis | 43.8 | 0.86 | 0.86 | 0.97 [0.71‐1.34] | 45.2 | 0.43 | 0.81 | 1.14 [0.82‐1.59] | 38.3 | 0.05 | 0.14 | 0.70 [0.49‐0.99] | 52.6 | 0.08 | 0.28 | 1.47 [0.95‐2.29] |
| Nephritis | 23.4 | 2.34 × 10−5 | 4.60 × 10−4 | 0.46 [0.31‐0.65] | 46.1 | 6.00 × 10−3 | 0.12 | 1.61 [1.14‐2.26] | 38.9 | 0.15 | 0.22 | 1.31 [0.91‐1.87] | 33.7 | 0.77 | 0.91 | 1.1 [0.66‐1.70] |
| Seizures | 7.1 | 0.16 | 0.24 | 0.66 [0.36‐1.16] | 9.6 | 0.83 | 0.89 | 1.1 [0.59‐1.83] | 12.2 | 0.13 | 0.21 | 1.52 [0.87‐2.59] | 8.4 | 0.75 | 0.91 | 0.88 [0.36‐1.81] |
| Psychosis | 1.8 | 0.69 | 0.73 | 0.79 [0.22‐2.35] | 1.4 | 0.45 | 0.81 | 0.6 [0.13‐1.97] | 3.3 | 0.22 | 0.31 | 1.93 [0.64‐5.46] | 2.1 | 0.96 | 0.99 | 0.96 [0.15‐3.64] |
| Neurological criterium | 7.1 | 0.04 | 0.09 | 0.55 [0.3‐0.95] | 10.5 | 0.98 | 0.98 | 1 [0.58‐1.69] | 15 | 0.03 | 0.12 | 1.76 [1.05‐2.89] | 10.5 | 0.99 | 0.99 | 0.99 [0.46‐1.94] |
| Low platelets | 15 | 0.01 | 4.16 × 10−2 | 0.58 [0.37‐0.87] | 21.5 | 0.79 | 0.89 | 0.95 [0.63‐1.41] | 32.2 | 1.51 × 10−5 | 3.00 × 10−4 | 2.35 [1.59‐3.47] | 13.7 | 0.08 | 0.28 | 0.58 [0.3‐1.04] |
| Leucopenia | 54 | 0.01 | 4.51 × 10−2 | 1.49 [1.1‐2.1] | 49.3 | 0.69 | 0.89 | 0.93 [0.67‐1.3] | 47.8 | 0.84 | 0.89 | 1.03 [0.73‐1.46] | 31.6 | 2.00 × 10−3 | 2.16 × 10−2 | 0.48 [0.3‐0.76] |
| Lymphopenia | 40.7 | 0.3 | 0.37 | 1.18 [0.86‐1.64] | 41.1 | 0.54 | 0.89 | 1.1 [0.79‐1.54] | 38.9 | 0.72 | 0.85 | 1.1 [0.75‐1.51] | 25.3 | 8.00 × 10−3 | 5.28 × 10−2 | 0.51 [0.31‐0.83] |
| Hemolytic anemia | 4 | 0.016 | 0.05 | 0.41 [0.18‐0.81] | 9.1 | 0.35 | 0.81 | 1.33 [0.72‐2.38] | 11.1 | 0.05 | 0.14 | 1.83 [1‐3.24] | 6.3 | 0.63 | 0.91 | 0.8 [0.3‐1.8] |
| Hematological criterium | 69.9 | 0.19 | 0.25 | 1.26 [0.89‐1.78] | 68 | 0.61 | 0.89 | 0.91 [0.64‐1.3] | 71.1 | 0.12 | 0.21 | 1.34 [0.93‐1.96] | 51.6 | 2.00 × 10−3 | 2.16 × 10−2 | 0.5 [0.32‐0.78] |
| Vascular manifestations | ||||||||||||||||
| Any vascular event | 25.2 | 0.003 | 2.94 × 10−2 | 0.57 [0.4‐0.83] | 29.2 | 0.42 | 0.81 | 1.16 [0.8‐1.69] | 41.7 | 3.00 × 10−3 | 0.02 | 1.74 [1.2‐2.5] | 31.6 | 0.42 | 0.84 | 0.82 [0.5‐1.32] |
| Any arterial event | 16.4 | 0.02 | 0.06 | 0.6 [0.38‐0.92] | 18.3 | 0.24 | 0.80 | 1.31 [0.83‐2.02] | 25.6 | 0.11 | 0.21 | 1.41 [0.92‐2.16] | 23.2 | 0.72 | 0.91 | 0.90 [0.51‐1.55] |
| Ischemic heart disease | 9.3 | 0.45 | 0.50 | 0.81 [0.45‐1.4] | 9.6 | 0.19 | 0.80 | 1.48 [0.81‐2.64] | 10.6 | 0.79 | 0.88 | 0.92 [0.50‐1.63] | 12.6 | 0.82 | 0.91 | 0.92 [0.44‐1.82] |
| Ischemic cerebrovascular disease | 8.9 | 0.17 | 0.24 | 0.68 [0.39‐1.16] | 9.1 | 0.83 | 0.89 | 1.1 [0.6‐1.84] | 14.4 | 0.12 | 0.21 | 1.5 [0.89‐2.5] | 11.6 | 0.72 | 0.91 | 0.88 [0.42‐1.71] |
| Venous thromboembolism | 13.3 | 0.08 | 0.14 | 0.67 [0.42‐1.03] | 15.1 | 0.64 | 0.89 | 0.9 [0.57‐1.39] | 25 | 1.00 × 10−3 | 1.40 × 10−2 | 1.97 [1.29‐2.98] | 13.7 | 0.29 | 0.64 | 0.71 [0.37‐1.29] |
Abbreviations: β2GP1, β2 glycoprotein I; ANA, antinuclear antibody; CI, confidence interval; CL, cardiolipin; FDR, false discovery rate; OR, odds ratio; RNP, ribonucleoprotein; SLE, systemic lupus erythematosus.
Swedish patients (N = 720).
P value for the cluster as binary term in the logistic regression model.
Benjamini and Yekutieli (51) step‐up FDR control.
ORs and 95% CIs for the predictable variable: cluster (a binary term). In the logistic regression models, the clinical variable was considered the response variable, the predictable variable was the cluster status, and age and sex were included as covariables in the model.
Healthy individuals (n = 3654) matched by ethnicity were considered controls in the genetic association analysis. Controls were either derived from the Swedish epidemiological investigation of rheumatoid arthritis study (17) (n = 3186) or hospital employees and medical students from Columbus, Ohio (13) (n = 468). The ethical board at Ohio State University and the Stockholm regional ethics board approved the study, and all participants gave informed written consent to participation. Neither patients nor the public were involved in the design, conduct, reporting, or dissemination plans of our research.
Determination of antibody status
All autoantibodies were analyzed in the 911 samples using the same methods and instruments at Karolinska University Hospital (Table 2). Antinuclear antibody (ANA) screening, including antibodies to double‐stranded DNA (dsDNA), nucleosomes, ribonucleoprotein (RNP) 68, RNPA, Smith (Sm), Sm/RNP, anti‐Sjögren syndrome antigen A (SSA)/Ro52, anti‐SSA/Ro60, and anti‐Sjögren syndrome antigen B (SSB)/La, was analyzed in serum by BioPlex 2200 (Bio‐Rad Laboratories). The specific ANAs were calibrated according to the specifications of the manufacturer.
Antibodies against cardiolipin (anti‐cardiolipin [CL] immunoglobulin G [IgG]/immunoglobulin M [IgM]) and β2glycoproteinI (anti‐ β2 glycoprotein I [β2GP1] IgG) were analyzed in the serum of patients by ELISA (Orgentec), with aPL cutoff levels for positivity corresponding to the 99th percentile of the normal population (18).
Detection of cytokines
The levels of 14 cytokines in circulation were evaluated in 446 available patients’ samples from the Karolinska University Hospital (IFN‐g, IP‐10/CXCL10, TNF‐α, MCP‐1/CCL2, IL‐10, IL‐8, IL‐15, MIP‐1β/CCL4, IL‐6, IL‐16, IL‐23, IFN‐a, IL‐17, and IFN‐λ) (details can be found in supplementary material).
Genotyping
HLA‐DRB1‐typing was performed either by sequence‐specific primer polymerase chain reaction assay (DR low‐resolution kit; Olerup SSP) (19) or by direct Sanger sequencing (Beijing Genomics Institute).
Statistical analyses
Unsupervised cluster analysis was implemented using the Gower distance matrix (20), followed by partition around medoids cluster calculation (21). A silhouette index was used for a number of cluster validations (21), and the visualization was performed by applying the t‐distributed stochastic neighbor embedding method (22). Only patients with SLE who were positive for at least one autoantibody were included in the cluster analyses (n = 743); patients who were negative for all 13 autoantibodies were assigned to Cluster/Subgroup 4 (n = 168) (Figure 1A). We refer to these clusters as disease subgroups in this study. The details of the statistical analyses can be found in the supplementary information. Briefly, logistic regression was used to estimate the relation between genetic or clinical variables and each subgroup, using sex and age at inclusion as covariables. The respective P values, odds ratios (ORs), and 95% confidence intervals (CIs) were also obtained. The Cochran‐Mantel‐Haenszel test was used to calculate the OR for meta‐analyses or combined OR (ORcombined) and 95% CI for the stratified genetic association analysis of the Swedish and the Ohio cohorts. Additionally, survival analysis was implemented to evaluate whether there is a relation between the age of the patients and the subgroups. Kruskal‐Wallis and Dunn’s tests were used to evaluate differences between cytokine levels and disease activity scores among the subgroups. A false discovery rate (FDR) of 5% was assumed (FDR P value [FDRp]). The analyses were performed using R v3.6.3, R v4.0.2 (23), and PLINK v1.9 (24) software.
Figure 1.
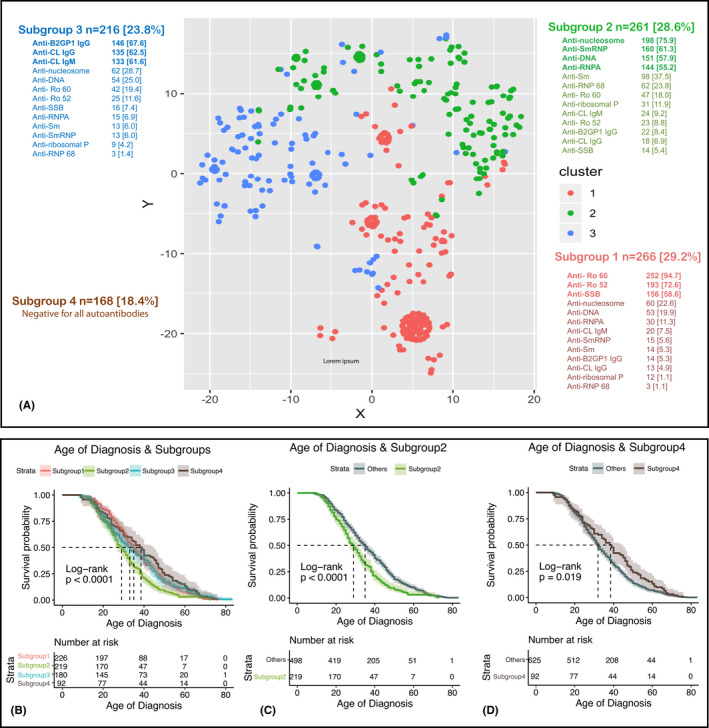
A, t‐distributed stochastic neighbor embedding plot representing the subgroups of patients with systemic lupus erythematosus identified by an unsupervised analysis using 13 autoantibodies. B‐D, Survival plots for the age at disease diagnosis for all the subgroups (B), Subgroup 2 compared with the rest of the patients (C), and Subgroup 4 compared with the rest of patients (D). β2GPI, β2 glycoprotein I; CL, cardiolipin; dsDNA, double stranded deoxyribonucleic acid; Ig, immunoglobulin; RNP, ribonucleoprotein; Sm, Smith; SSA/B, Sjögren’s syndrome antigen A/B.
RESULTS
Thirteen autoantibodies define four SLE clusters
The studied cohorts and the distribution of the positivity for autoantibodies are presented in Tables 1 and 2. Cluster analysis based on the autoantibody status grouped the patients with SLE into four subgroups (Figure 1A and supplementary information). An autoantibody was considered dominant in a subgroup if it was present in at least 50% of patients in that subgroup. Subgroup 1 (29.3%) is dominated by anti‐SSA/Ro60/Ro52 and anti‐SSB/La; Subgroup 2 (28.7%) is dominated by anti‐nucleosome, anti‐SmRNP, anti‐dsDNA, and anti‐RNPA; Subgroup 3 (23.8%) is dominated by aPL (anti‐CL/IgG/IgM and anti‐β2GPI/IgG); and Subgroup 4, (18.2%) comprises patients who were negative for all 13 studied autoantibodies at inclusion. Notably, we observed that the age at SLE diagnosis and age at inclusion in this study differ significantly among subgroups (Figure 1B, Supplementary Figure 1A, Supplementary Table 1 and supplementary information). Particularly, the patients from Subgroup 2 were diagnosed at a younger age (median = 29 years, 95% CI = 27‐31 years) (Supplementary Table 1 and Figure 1C), whereas patients from Subgroup 4 had the highest age at diagnosis (median = 38.5 years, 95% CI = 32‐45 years) (Supplementary Table 1 and Figure 1D). No significant differences regarding sex distribution across subgroups were observed (logistic regression P = 0.65). However, the percentage of men was highest for Subgroup 2 (13%), lowest for Subgroup 4 (7.3%), and similar for Subgroups 1 (11.7%) and 3 (12%).
HLA‐DRB1 associations: case–control and case‐only analyses
We evaluated the relationship between the HLA‐DRB1 alleles and the four SLE subgroups by comparing each subgroup with healthy controls (HCs), as well as comparing each subgroup with the rest of the patients with SLE.
Case–control analyses
The HLA‐DRB1*03 and *15 alleles were confirmed as risk factors for SLE when compared with all the patients to HCs (ORcombined = 2.3, 95% CI = 2.1‐2.4, FDRp = 3E‐32 and ORcombined = 1.4, 95% CI = 1.2‐1.5, FDRp = 4E‐5, respectively) (Figure 2A and Supplementary Table 2).
Figure 2.
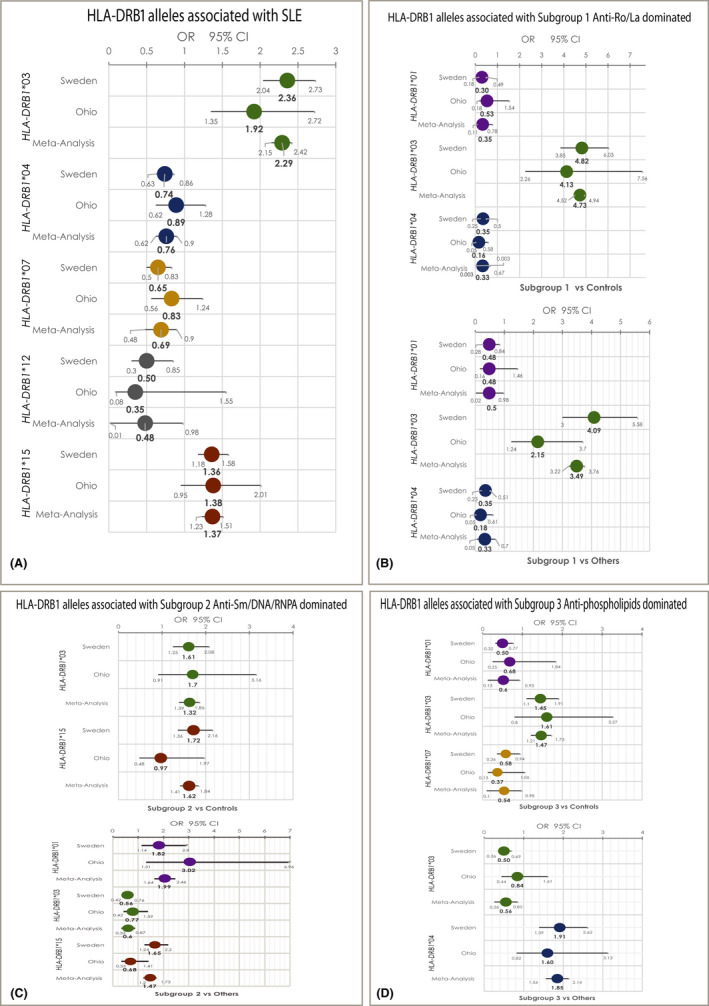
Forest plots for the statistically significant HLA‐DRB1 alleles’ associations with systemic lupus erythematosus (SLE) in three of the four identified subgroups. (A) Significantly associated HLA‐DRB1 alleles with patients with SLE compared with control subjects. (B) Significantly associated HLA‐DRB1 alleles with Subgroup 1, dominated by anti‐SSA/Ro52/Ro60/SSB positivity, when compared with control subjects (upper panel) and when compared with other patients with SLE (lower panel). (C) Significantly associated HLA‐DRB1 alleles with Subgroup 2, dominated by anti‐nucleosome/SmRNP/DNA/RNPA, when compared with control subjects (upper panel) and when compared with other patients with SLE (lower panel). (D) Significantly associated HLA‐DRB1 alleles with Subgroup 3, dominated by anti‐β2GPI‐IgG/aCL‐IgG/IgM positivity, when compared with control subjects (upper panel) and when compared with other patients with SLE (lower panel). No significant associations between HLA‐DRB1 alleles and Subgroup 4, patients with negative status for the 13 studied autoantibodies, were detected. RNP, ribonucleoprotein.
We found that the HLA‐DRB1*03 allele is a risk factor for each of the Subgroups 1, 2, and 3 when compared with HCs. However, the effect size for this association is higher for Subgroup 1 (anti‐SSA/Ro‐SSB/La dominated). The ORcombined for the HLA‐DRB1*03 presence in Subgroup 1 compared with HCs was 4.7 (95% CI = 4.5‐4.9, FDRp = 7.8E‐47), whereas for Subgroups 2 and 3 it was less than 1.65, with no overlap between CIs (Figures 2B‐D and supplementary Tables 3‐5).
The HLA‐DRB1*15 allele was a risk factor only for Subgroup 2 when compared with HC (ORcombined = 1.6, 95% CI = 1.4‐1.8, FDRp = 1.7 × 10−4) (Figure 2C and supplementary Tables 3‐6). Additionally, the frequency of the HLA‐DRB1*01 allele was lower in Subgroups 1 and 3 compared with the controls (ORcombined = 0.33, 95% CI = 0.11‐0.78, FDRp = 3.7 × 10−6 and OR = 0.52, 95% CI = 0.13‐0.9, FDRp = 0.02, respectively) (Figures 2B and 2D and supplementary Tables 3 and 5). The HLA‐DRB1*04 allele was associated as a protective factor for Subgroup 1, as was the HLA‐DRB1*07 allele for Subgroup 3, compared with HC (ORcombined = 0.33, 95% CI = 0.003‐0.67, FDRp = 4.7E‐10 and ORcombined = 0.54, 95% CI = 0.1‐0.98, FDRp = 0.02, respectively) (Figures 2B and 2D and supplementary Tables 3 and 5). No significant associations were detected between HLA‐DRB1 alleles and Subgroup 4 (Supplementary Table 6).
Case‐only analyses
We compared the HLA‐DRB1 allele frequencies between the four subgroups of patients with SLE. The HLA‐DRB1*03 allele was more frequent only in Subgroup 1 compared with the rest of the patients with SLE (Figure 2B and supplementary Tables 3‐6). A negative association was observed for this allele with Subgroups 2 and 3 (Figures 2C and 2D and supplementary Tables 4 and 5), reflecting the high frequency of the HLA‐DRB1*03 allele in Subgroup 1. The HLA‐DRB1*15 allele was more frequent only in Subgroup 2 compared with other patients (Figure 2C and supplementary Tables 3‐6), which is consistent with the association observed in the case–control design analysis. The HLA‐DRB1*01 allele emerged as a more frequent factor for Subgroup 2 compared with the rest of the patients with SLE (Figure 2C and Supplementary Table 4). In contrast, the HLA‐DRB1*01 allele was less frequent in Subgroup 1 compared with the rest of the patients (Figure 2B and Supplementary Table 3). In a similar fashion, the HLA‐DRB1*04 was more frequent in Subgroup 3 and appeared to be underrepresented in Subgroup 1 in comparison with the other patients with SLE (Figures 2B and 2D and supplementary Tables 3 and 5). No significant associations were observed between the HLA‐DRB1 alleles and the autoantibody‐negative Subgroup 4 (Supplementary Table 6).
Cytokine evaluations
To address further immunological SLE heterogeneity, we measured several soluble mediators of the immune system in 446 plasma or serum samples from patients with SLE. Our analyses revealed differences between the four subgroups for the levels of eight cytokines (IFN‐γ, IP‐10/CXCL10, TNF‐α, MCP‐1/CCL2, IL‐10, IL‐8, IL‐15, and MIP1‐ß/CCL4) (Table 4 and Supplementary Table 7). Most differences were due to the lowest levels of cytokines in Subgroup 4 and highest levels in Subgroup 2. We observed that IFN‐γ levels were significantly higher in Subgroup 2 compared with Subgroups 3 and 4. Additionally, IL‐10 levels were higher in Subgroup 2 compared with Subgroups 1, 3, and 4. IP‐10/CXCL10 levels were significantly higher in patients from Subgroup 1 compared with Subgroup 3. There was a trend for higher levels of MIP1‐ß/CCL4 in patients from Subgroup 3 when compared with Subgroups 1 and 2. These data point towards distinct cytokine/interferon profiles in the defined SLE subgroups.
Table 4.
Relations between cytokine levels in plasma of Swedish patients with SLE and autoantibody subgroups
| Cytokine | Cluster | Mean | Median (1st‐3rd Quartile), pg/ml | Kruskal‐Wallis Test (df = 3) | Dunn Test | |||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Cluster 1 a (n = 140; 31.4%) | Cluster 2 b (n = 137; 30.7%) | Cluster 3 c (n = 114; 25.6%) | ||||||||||
| χ2 | P Value d | FDRp e | P Value d | FDRp e | P Value d | FDRp e | P Value d | FDRp e | ||||
| IFN‐γ | 1 | 22.0 | 13.3 (8.21‐23.8) | 25.77 | 1.10 × 10−5 | 1.50 × 10−4 | ‐ | ‐ | ‐ | ‐ | ‐ | ‐ |
| 2 | 34.3 | 13.8 (7.81‐28.9) | ‐ | ‐ | ‐ | 0.455 | ‐ | ‐ | ‐ | ‐ | ‐ | |
| 3 | 32.5 | 9.84 (5.83‐18.1) | ‐ | ‐ | ‐ | 0.002 | 0.003 | 0.002 | 0.003 | ‐ | ‐ | |
| 4 f | 12.8 | 7.25 (4.48‐10.9) | ‐ | ‐ | ‐ | 1.00 × 10−5 | 1.00 × 10−4 | 1.00 × 10−5 | 1.00 × 10−4 | 0.043 | 0.052 | |
| IP‐10/CXCL10 | 1 | 1947 | 808 (548‐1571) | 23.57 | 3.10 × 10−5 | 2.20 × 10−4 | ‐ | ‐ | ‐ | ‐ | ‐ | ‐ |
| 2 | 1686 | 858 (447‐1681) | ‐ | ‐ | ‐ | 0.17 | ‐ | ‐ | ‐ | ‐ | ‐ | |
| 3 | 1502 | 610 (423‐1614) | ‐ | ‐ | ‐ | 0.019 | 0.028 | 0.123 | ‐ | ‐ | ‐ | |
| 4 f | 862 | 469 (337‐729) | ‐ | ‐ | ‐ | 8.00 × 10−7 | 8.00 × 10−6 | 1.00 × 10−5 | 1.00 × 10−4 | 0.002 | 0.004 | |
| TNF‐α | 1 | 5.53 | 4.55 (3.35‐6.06) | 20.82 | 1.10 × 10−4 | 5.40 × 10−4 | ‐ | ‐ | ‐ | ‐ | ‐ | ‐ |
| 2 | 5.84 | 5.24 (3.39‐6.54) | ‐ | ‐ | ‐ | 0.122 | ‐ | ‐ | ‐ | ‐ | ‐ | |
| 3 | 5.91 | 4.49 (3.23‐6.21) | ‐ | ‐ | ‐ | 0.475 | ‐ | 0.152 | ‐ | ‐ | ‐ | |
| 4 f | 4.68 | 3.01 (2.26‐4.36) | ‐ | ‐ | ‐ | 1.00 × 10−4 | 4.00 × 10−4 | 9.60 × 10−6 | 9.00 × 10−5 | 2.00 × 10−4 | 3.00 × 10−4 | |
| MCP‐1/CCL2 | 1 | 152 | 113 (90.6‐168) | 19.22 | 2.50 × 10−4 | 8.60 × 10−4 | ‐ | ‐ | ‐ | ‐ | ‐ | ‐ |
| 2 | 154 | 122 (82‐151) | ‐ | ‐ | ‐ | 0.262 | ‐ | ‐ | ‐ | ‐ | ‐ | |
| 3 | 160 | 118 (85.7‐160) | ‐ | ‐ | ‐ | 0.44 | ‐ | 0.325 | ‐ | ‐ | ‐ | |
| 4 f | 98 | 85.7 (68.3‐108) | ‐ | ‐ | ‐ | 1.00 × 10−5 | 1.00 × 10−4 | 2.00 × 10−4 | 3.00 × 10−4 | 1.00 × 10−4 | 2.00 × 10−4 | |
| IL‐10 | 1 | 1.30 | 0.67 (0.67‐1.13) | 16.77 | 7.90 × 10−4 | 2.20 × 10−3 | ‐ | ‐ | ‐ | ‐ | ‐ | ‐ |
| 2 | 2.35 | 0.81 (0.67‐1.85) | ‐ | ‐ | ‐ | 0.0082 | 0.0246 | ‐ | ‐ | ‐ | ‐ | |
| 3 | 1.61 | 0.67 (0.67‐1.34) | ‐ | ‐ | ‐ | 0.4581 | ‐ | 0.0163 | 0.0195 | ‐ | ‐ | |
| 4 f | 0.83 | 0.67 (0.67‐0.73) | ‐ | ‐ | ‐ | 0.0128 | 0.0257 | 2.00 × 10−5 | 2.00 × 10−4 | 0.0131 | 0.0197 | |
| IL‐8 | 1 | 8.81 | 5.48 (3.49‐8.97) | 13.27 | 4.10 × 10−3 | 9.60 × 10−3 | ‐ | ‐ | ‐ | ‐ | ‐ | ‐ |
| 2 | 9.03 | 6 (3.31‐10.3) | ‐ | ‐ | ‐ | 0.265 | ‐ | ‐ | ‐ | ‐ | ‐ | |
| 3 | 9.35 | 5.33 (3.18‐9.13) | ‐ | ‐ | ‐ | 0.262 | ‐ | 0.113 | ‐ | ‐ | ‐ | |
| 4 f | 4.91 | 3.6 (2.4‐5.11) | ‐ | ‐ | ‐ | 9.00 × 10−4 | 0.003 | 2.00 × 10−4 | 0.001 | 0.006 | 0.012 | |
| IL‐15 | 1 | 3.52 | 3.06 (2.44‐3.85) | 9.87 | 0.02 | 0.03 | ‐ | ‐ | ‐ | ‐ | ‐ | ‐ |
| 2 | 3.54 | 3 (2.34‐4.10) | ‐ | ‐ | ‐ | 0.459 | ‐ | ‐ | ‐ | ‐ | ‐ | |
| 3 | 3.97 | 3.07 (2.29‐4.81) | ‐ | ‐ | ‐ | 0.498 | ‐ | 0.46 | ‐ | ‐ | ‐ | |
| 4 f | 2.94 | 2.45 (2.12‐2.94) | ‐ | ‐ | ‐ | 0.002 | 0.012 | 0.003 | 0.006 | 0.003 | 0.008 | |
| MIP‐1β/CCL4 | 1 | 86.6 | 68.5 (51.4‐105) | 10.09 | 0.02 | 0.03 | ‐ | ‐ | ‐ | ‐ | ‐ | ‐ |
| 2 | 93.2 | 72.7 (50.5‐107) | ‐ | ‐ | ‐ | 0.38 | ‐ | ‐ | ‐ | ‐ | ‐ | |
| 3 | 105.0 | 84 (55.2‐121) | ‐ | ‐ | ‐ | 0.025 | 0.076 | 0.049 | 0.059 | ‐ | ‐ | |
| 4 f | 76.1 | 60. 1(47.7‐78.1) | ‐ | ‐ | ‐ | 0.048 | 0.071 | 0.031 | 0.062 | 0.001 | 0.006 | |
Abbreviations: β2GP1, β2 glycoprotein I; CL, cardiolipin; FDR, false discovery rate; RNP, ribonucleoprotein; SLE, systemic lupus erythematosus; sm, Smith; TNF, tumor necrosis factor
Only significant results are shown here; the full table is disclosed in the Supplementary Table 7.
Swedish patients with SLE (n = 446).
Cluster 1 (anti‐Ro/La).
Cluster 2 (anti‐nucleosome/Sm/DNA/RNP).
Cluster 3 (anti‐β2GP1/CL IgG/CL–IgM).
Nominal P Value.
Benjamini and Yekutieli (51) step‐up FDR control.
Cluster 4 (negative for 13 autoantibodies); n = 55 (12.3%).
Clinical manifestations
Specific clinical manifestations among the defined subgroups were investigated (Table 3). Nephritis is more common in individuals from Subgroup 2 (OR = 1.61, 95% CI = 1.1‐2.3, FDRp = 0.12), although the corrected P value did not reach significance. Conversely, discoid skin lesions, photosensitivity, and leucopenia are more frequent in Subgroup 1 patients (OR = 1.71, 95% CI = 1.2‐2.5, FDRp = 0.03; OR = 1.68, 95% CI = 1.2‐2.4, FDRp = 0.03; and OR = 1.49, 95% CI = 1.1‐2.1, FDRp = 0.04, respectively). Thrombocytopenia and vascular manifestations were more frequent in individuals from Subgroup 3 (OR = 2.35, 95% CI = 1.6‐3.5, FDRp = 3 × 10−4 and OR = 1.74, 95% CI = 1.2‐2.5, FDRp = 0.02, respectively). No clinical manifestations were overrepresented in Subgroup 4; on the contrary, leukopenia and lymphopenia were less frequent in this group. These analyses reveal that the defined SLE subgroups bear differential patterns of clinical manifestations.
Disease activity and organ damage
Using the data available for 446 patients with SLE, we further investigated whether the disease subgroups also differ in disease activity and organ damage. The omnibus test demonstrated that there are differences for SLEDAI and SDI but not for SLAM (Supplementary Table 8). Furthermore, differences were observed when the subgroups were compared with one another; SLEDAI scores were higher in Subgroup 2 and Subgroup 3 compared with Subgroup 1 (FDRp = 1 × 10−4 and 0.018, respectively). SDI scores were higher in Subgroup 3 compared with Subgroup 2 (FDRp = 0.016), and a similar trend was observed when Subgroup 3 was compared with Subgroup 1 (FDRp = 0.097).
DISCUSSION
Our study demonstrates that a subdivision of patients with SLE into four subgroups, based on the profile of 13 commonly measured SLE‐related autoantibodies, reveals differences across the groups regarding genetic background, age of disease onset, cytokine profile, clinical manifestations, disease activity, and organ damage characteristics (Figure 3).
Figure 3.
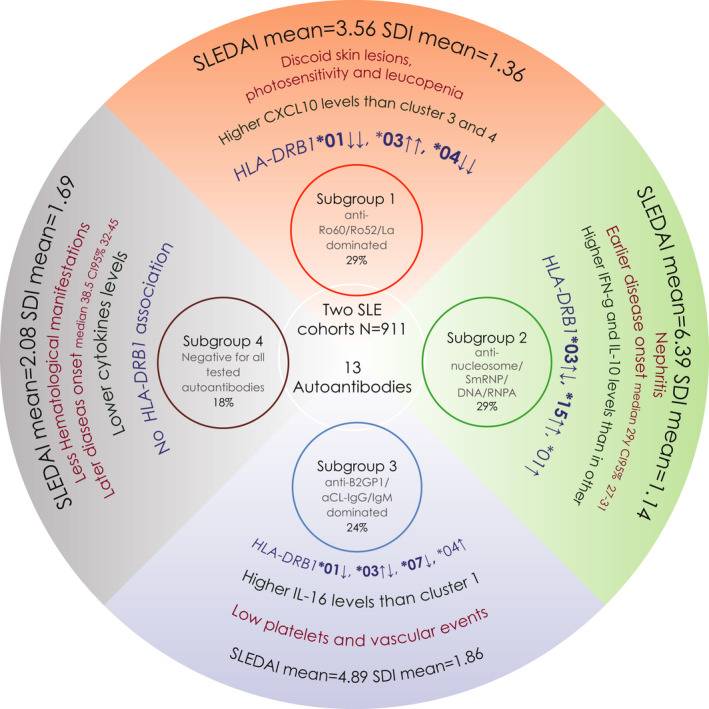
Representation of the four systemic lupus erythematosus (SLE) disease subgroups identified in the present study. The HLA‐DRB1 alleles in bold text represent significant associations observed in the cases‐versus‐controls analysis; regular text represents significant associations observed in the cases versus cases analysis. The arrows pointing upwards symbolize a risk association, whereas the arrows pointing downwards symbolize a protective association. When two arrows appear, it means that the association between that subgroup with the given allele was observed in both types of analyses (ie, cases versus controls and cases versus cases). The HLA‐DRB1*03 allele was significantly associated with Subgroups 1, 2, and 3 for both types of analyses (ie, cases versus controls and cases versus cases); therefore, the first arrow closed to that allele symbolize the direction of the association in the cases‐versus‐controls analysis, the second arrow indicates the direction of the association observed in the cases versus cases analysis. Note that the arrows have the same direction only for Subgroup 1. Remarkably, Subgroup 2 is characterized by core SLE features; Subgroups 1 and 3 have convincing similarities to the primary Sjögren's syndrome and the primary antiphospholipid syndrome (pAPS), respectively; and Subgroup 4 is a milder version with fewer autoantibodies and no HLA‐DRB1 association. On the basis of these observations, we suggest that the autoantibody profile can be used to classify the presently diagnosed patients with SLE into several more homogeneous subgroups, and this approach should be considered when designing future therapeutic trials. SDI, Systemic Lupus International Collaborating Clinics/American College of Rheumatology Damage Index; SLEDAI, SLE Disease Activity Score.
Our results demonstrate differential associations between HLA‐DRB1 alleles and the identified SLE subgroups (Figure 2). These observations are in line with the known relationships between specific HLA genetic variants and the occurrence of autoantibodies and subgroups in several autoimmune diseases (8, 9, 10, 11). The strong association between the HLA‐DRB1*03 allele and Subgroup 1 further illustrates the known association between the HLA‐DRB1*03 allele and anti‐SSA/Ro52/Ro60‐SSB/La autoantibodies (25), which dominate this group. The association between the HLA‐DRB1*15 allele and SLE is here primarily driven by Subgroup 2, which is dominated by anti‐nucleosome/SmRNP/dsDNA/RNPA antibodies. The relationship between this allele and anti‐dsDNA was previously reported (25), but this result points to novel associations between the HLA‐DRB1*15 allele and anti‐nucleosome and anti‐SmRNP antibodies. These observations suggest that different HLA alleles may have a pivotal impact on the development of autoantibody profiles in SLE, indicating diverse pathogenic mechanisms for the subgroups, including the involvement of antigen‐specific T cells.
We propose that it may be more logical to divide SLE into several distinct disease subsets or diagnostic entities as an alternative to improve common criteria for SLE. This view is supported by the present study, which, for the first time, relates autoantibody‐defined SLE subgroups to different HLA‐DRB1 allele, and by previously published studies performed in different populations (26, 27, 28, 29).
Our observations are partially similar to those of To et al (29), who studied seven autoantibodies in a multiethnic cohort. They identified three clusters that resemble ours, including one with SSA‐Ro/SSB‐La, one with Sm/RNP, and one with aPL. The main difference between their study and ours is that dsDNA autoantibodies were present both in the SSA‐Ro/SSB‐La and the aPL clusters. Concordant with our results, vascular manifestations were associated with the aPL group, and nephritis was more frequent in the Sm/RNP group. Recently it was shown that anti‐nucleosome and anti‐dsDNA are strongly associated with lupus nephritis in patients with SLE from Egypt (30), supporting our observation in relation to renal involvement. Similarly, Artim‐Esen et al (26) evaluated seven autoantibodies in a single‐center cohort from Istanbul. They identified five clusters, with striking similarities to our study and to that by To et al. Despite different methodologies, three large studies, including the present one, comprising a total of 3120 patients with SLE, demonstrate the existence of SLE subgroups characterized by anti‐SSA‐Ro/SSB‐La, anti‐Sm/RNP and aPL antibodies. Importantly, the present study adds new dimensions to this view by incorporating genetics, cytokines, and disease activity profiles. These subgroups may differ in their pathogenic mechanisms. For instance, we previously reported that patients with SLE who were grouped by SSA‐Ro/SSB‐La or aPL antibodies exhibit different proteomic profiles and natural IgM antibodies targeting phosphorylcholine (31, 32). Notably, two of the observed subgroups resemble other diagnostic entities. The anti‐SSA‐Ro/SSB‐La subgroup has many similarities with primary Sjögren's syndrome, especially regarding autoantibodies and a known strong genetic association with the HLA‐DRB1*03 allele. It is of note that the genetic association with HLA‐DRB1*03 is restricted to the SSA/SSB‐positive subgroup of primary Sjögren's syndrome (10). On the other hand, the anti‐β2GPI/anti‐CL subgroup shares features with the primary antiphospholipid syndrome (pAPS). Though less studied, the HLA‐DRB1*04 allele has been linked to pAPS (33, 34). The present antiphospholipid syndrome (APS) criteria (18) and a study by Unlu et al (35) indicate that clinical symptoms in patients with primary and secondary APS do not essentially differ, which hint a close relationship between Subgroup 3 and pAPS.
Patients in Subgroup 2 of this study were younger at disease onset than patients in other subgroups (Figures 1A and 1B). Young age has been reported to be associated with nephritis (36, 37), and this pattern was also present in our data (hazard ratio of 1.4, 95% CI = 1.2‐1.6, for having nephritis in a survival test adjusted for age at diagnosis; median age of 27 years, 95% CI = 25‐30, for patients with SLE and nephritis, in contrast to 35.5 years, 95% CI = 33‐38, for patients without nephritis). Lupus nephritis was more common in Subgroup 2, though it was of borderline significance because age was added as a covariable, which was a significant component of the model (P = 1.5 × 10−5). We believe that the patients in Subgroup 2 constitute the core of what is traditionally referred to as SLE. Common manifestations in this disease subgroup, such as the presence of nephritis, anti‐dsDNA, and complement consumption, are highly weighted in the SLEDAI score (14), likely explaining the association between high SLEDAI‐2K scores and Subgroup 2. Both IFN‐γ and IL‐10 are known to be upregulated in SLE (38, 39, 40, 41), and elevated IFN‐γ activity preceded SLE onset by several years (42). The highest IFN‐γ levels were observed in patients from Subgroup 2, confirming reports of a positive association with nephritis and dsDNA antibodies (32, 39). Probably, these patients may share the molecular signatures described as the “IFN‐high” SLE subset (43) and the “interferon cluster” (44). Furthermore, IFN‐γ–expressing cells were frequent and stained intensely in renal tissue from patients with pediatric lupus nephritis, especially in cases with proliferative nephritis (class III and IV) (45). IL‐10 levels were also highest in individuals from Subgroup 2, corroborating associations with active lupus nephritis (39, 40). In line with these observations, a T‐cell subset, which produces high levels of both IFN‐γ and IL‐10, was recently detected both in blood and renal tissue from patients with active lupus nephritis (38).
β2GPI is considered the main antigen for aPL, and the presence of β2GPI recognizing T cells in both SLE and primary APS suggests a role for HLA‐DRB1 genes, as reviewed by Rauch et al (46). Several prospective clinical studies have identified aPL as a risk factor for vascular events (47, 48), a major cause of morbidity and premature mortality in SLE (49, 50). Early identification of aPL‐positive patients belonging to Subgroup 3 is therefore important because preventive interventions will hopefully reduce the heavy vascular burden. We therefore suggest that early characterization of autoantibody profiles could be a helpful tool for clinical prognosis and the design of treatment strategies in SLE.
Patients in Subgroup 4, older at diagnosis and characterized by the absence of the investigated autoantibodies and HLA‐DRB1 associations, had generally milder disease with fewer hematological manifestations and lower levels of circulating proinflammatory cytokines. Although not significant, the frequencies of serositis (52.6%) and arthritis (85.3%) were higher compared with those of the other subgroups (Table 3). Most of these patients (~96%; Table 3) were ever‐positive for ANA. Patients from Subgroup 4 could also be positive for noninvestigated ANA specificities, or they could transiently have been positive for the investigated autoantibodies.
Our study has several strengths, including the large and well‐characterized patient samples from two distinct areas. All autoantibodies were measured by the same well‐standardized methods in one laboratory, and the cytokine measurements and evaluation of clinical manifestations were addressed at the same time point as the autoantibody determination. These SLE subgroups should nevertheless be seen as clusters in a continuous gradient of genetic, immunological, and clinical manifestations. Such continuum is laborious to infer but may be reflected in a correlation matrix among autoantibodies (Supplementary Figure 3) or through gene expression and methylation signatures, as recently attempted (44). For example, nephritis is more frequent in Subgroup 2 (46.1%,); although not significant, it also appears to be increased in Subgroup 3 (38.9%) compared with Subgroup 1 (23.4%) and Subgroup 4 (33.7%) (Table 3).
It is a limitation of our study that only European white individuals were included, so our results cannot be generalized to other genetic ancestries. Also, all measurements were performed in a cross‐sectional design, though some antibodies, especially antibodies targeting dsDNA and nucleosomes, are known to vary over time.
To conclude, we demonstrate that, on the basis of the autoantibody profile, four SLE subphenotypes can be identified. These groups differ regarding genetic predisposition as well as clinical and laboratory characteristics. Replication studies in other genetic ancestries, with a longitudinal design, including incident cases, evaluating additional relevant data domains (eg, all known SLE genetic risk factors and relevant tissue and cell transcriptomics) are necessary before our results can be used in a clinical context. Still the current results are important, in line with previous studies and clinical experience, and they may influence future delineations and treatment decisions for patients with SLE.
AUTHOR CONTRIBUTIONS
All authors were involved in drafting the article or revising it critically for important intellectual content, and all authors approved the final version to be published. Drs. Diaz‐Gallo, Svenungsson, and Padyukov had full access to all the data in the study and take responsibility for the integrity of the data and the accuracy of the data analysis.
Study conception and design
Diaz‐Gallo, Lundström, Padyukov, Svenungsson.
Acquisition of data
Oke, Lundström, Elvin, Ling Wu, Eketjäll, Zickert, Gustafsson, Jönsen, Leonard, Birmingham, Nordmark, Bengtsson, Rönnblom, Gunnarsson, Yu, Padyukov, Svenungsson.
Analysis and interpretation of data
Diaz‐Gallo, Padyukov, Svenungsson.
Supporting information
Supplementary Material
ACKNOWLEDGMENTS
The authors thank all the donors, patients, and control subjects for contributing to this study. They also thank Janet Ahlberg for language editing.
Supported by the Swedish Research Council–Ventenskapsrådet (2014‐33867, 2018‐02535, 2018‐02399), the King Gustaf V 80th Birthday Fund–Stiftelsen Konung Gustaf V:s 80‐årsfond (FAI‐2017‐0390, FAI‐2018‐0518, FAI‐2019‐0597, and FAI‐2018‐0478), the Swedish Heart‐Lung Foundation–Hjärt‐Lungfonden (20170257), Stockholm County Council–Stockholms Läns Landsting (20170038), the Swedish Rheumatism Association–Reumatikerförbundet (R‐739631, R‐861801, R‐932138, and R‐930005), Ulla och Gustaf af Uggla stiftelse (2018‐02670 and 2020‐02395), Swedish Society of Medicine, and the Ingegerd Johansson Donation–Svenska Läkaresällskapet (SLS‐713911 and SLS‐936449).
Drs. Padyukov and Svenungsson contributed equally and have the right to list their name last in their CV or similar.
Susanna Eketjäll is an employee of Astra‐Zeneca, and Astra‐Zeneca provided kits for analyses of cytokine levels. However, Astra‐Zeneca did not take part in design, data analyses of this study or writing of this manuscript. No other disclosures relevant to this article were reported.
REFERENCES
- 1. Diaz‐Gallo LM, Martin J. Common genes in autoimmune diseases: a link between immune‐mediated diseases. Expert Rev Clin Immunol 2012;8:107–9. [DOI] [PubMed] [Google Scholar]
- 2. Allen ME, Rus V, Szeto GL. Leveraging heterogeneity in systemic lupus erythematosus for new therapies. Trends Mol Med 2021;27:152–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Tan EM, Cohen AS, Fries JF, Masi AT, McShane DJ, Rothfield NF, et al. The 1982 revised criteria for the classification of systemic lupus erythematosus. Arthritis Rheum 1982;25:1271–7. [DOI] [PubMed] [Google Scholar]
- 4. Petri M, Orbai AM, Alarcon GS, Gordon C, Merrill JT, Fortin PR, et al. Derivation and validation of the Systemic Lupus International Collaborating Clinics classification criteria for systemic lupus erythematosus. Arthritis Rheum 2012;64:2677–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Aringer M, Costenbader K, Daikh D, Brinks R, Mosca M, Ramsey‐Goldman R, et al. 2019 European League Against Rheumatism/American College of Rheumatology Classification Criteria for Systemic Lupus Erythematosus. Arthritis Rheumatol 2019;71:1400–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Hom G, Graham RR, Modrek B, Taylor KE, Ortmann W, Garnier S, et al. Association of systemic lupus erythematosus with C8orf13‐BLK and ITGAM‐ITGAX. N Engl J Med 2008;358:900–9. [DOI] [PubMed] [Google Scholar]
- 7. Castano‐Rodriguez N, Diaz‐Gallo LM, Pineda‐Tamayo R, Rojas‐Villarraga A, Anaya JM. Meta‐analysis of HLA‐DRB1 and HLA‐DQB1 polymorphisms in Latin American patients with systemic lupus erythematosus. Autoimmun Rev 2008;7:322–30. [DOI] [PubMed] [Google Scholar]
- 8. Padyukov L, Silva C, Stolt P, Alfredsson L, Klareskog L. A gene‐environment interaction between smoking and shared epitope genes in HLA‐DR provides a high risk of seropositive rheumatoid arthritis. Arthritis Rheum 2004;50:3085–92. [DOI] [PubMed] [Google Scholar]
- 9. Gorlova O, Martin JE, Rueda B, Koeleman BP, Ying J, Teruel M, et al. Identification of novel genetic markers associated with clinical phenotypes of systemic sclerosis through a genome‐wide association strategy. PLoS Genet 2011;7:e1002178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Gottenberg JE, Busson M, Loiseau P, Cohen‐Solal J, Lepage V, Charron D, et al. In primary Sjogren's syndrome, HLA class II is associated exclusively with autoantibody production and spreading of the autoimmune response. Arthritis Rheum 2003;48:2240–5. [DOI] [PubMed] [Google Scholar]
- 11. Thorlacius GE, Hultin‐Rosenberg L, Sandling JK, Bianchi M, Imgenberg‐Kreuz J, Pucholt P, et al. Genetic and clinical basis for two distinct subtypes of primary Sjogren's syndrome. Rheumatology (Oxford) 2021;60:837–48. 10.1093/rheumatology/keaa367 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Lundstrom E, Gustafsson JT, Jonsen A, Leonard D, Zickert A, Elvin K, et al. HLA‐DRB1*04/*13 alleles are associated with vascular disease and antiphospholipid antibodies in systemic lupus erythematosus. Ann Rheum Dis 2013;72:1018–25. [DOI] [PubMed] [Google Scholar]
- 13. Yang Y, Chung EK, Wu YL, Savelli SL, Nagaraja HN, Zhou B, et al. Gene copy‐number variation and associated polymorphisms of complement component C4 in human systemic lupus erythematosus (SLE): low copy number is a risk factor for and high copy number is a protective factor against SLE susceptibility in European Americans. Am J Hum Genet 2007;80:1037–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Gladman DD, Ibanez D, Urowitz MB. Systemic lupus erythematosus disease activity index 2000. J Rheumatol 2002;29:288–91. [PubMed] [Google Scholar]
- 15. Liang MH, Socher SA, Roberts WN, Esdaile JM. Measurement of systemic lupus erythematosus activity in clinical research. Arthritis Rheum 1988;31:817–25. [DOI] [PubMed] [Google Scholar]
- 16. Gladman D, Ginzler E, Goldsmith C, Fortin P, Liang M, Urowitz M, et al. The development and initial validation of the Systemic Lupus International Collaborating Clinics/American College of Rheumatology damage index for systemic lupus erythematosus. Arthritis Rheum 1996;39:363–9. [DOI] [PubMed] [Google Scholar]
- 17. Stolt P, Bengtsson C, Nordmark B, Lindblad S, Lundberg I, Klareskog L, et al. Quantification of the influence of cigarette smoking on rheumatoid arthritis: results from a population based case‐control study, using incident cases. Ann Rheum Dis 2003;62:835–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Miyakis S, Lockshin MD, Atsumi T, Branch DW, Brey RL, Cervera R, et al. International consensus statement on an update of the classification criteria for definite antiphospholipid syndrome (APS). J Thromb Haemost 2006;4:295–306. [DOI] [PubMed] [Google Scholar]
- 19. Olerup O, Zetterquist H. HLA‐DR typing by PCR amplification with sequence‐specific primers (PCR‐SSP) in 2 hours: an alternative to serological DR typing in clinical practice including donor‐recipient matching in cadaveric transplantation. Tissue Antigens 1992;39:225–35. [DOI] [PubMed] [Google Scholar]
- 20. Gower JC. A general coefficient of similarity and some of its properties. Biometrics 1971;27:857. 10.2307/2528823. [DOI] [Google Scholar]
- 21. Kaufman L, Rousseeuw PJ. Partitioning around medoids (Program PAM). In: Kaufman L, Rousseeuw PJ. Finding groups in data: an introduction to cluster analysis. Wiley Online Library; 1990. 10.1002/9780470316801.ch2. [DOI] [Google Scholar]
- 22. Van der Maaten L, Hintton G. Visualizing data using t‐SNE. Journal of Machine Learning Research 2008;9:2579–605. [Google Scholar]
- 23. Team RC . R: a language and environment for statistical computing. 2020. URL: https://www.r‐project.org/.
- 24. Purcell S, Neale B, Todd‐Brown K, Thomas L, Ferreira MA, Bender D, et al. PLINK: a tool set for whole‐genome association and population‐based linkage analyses. Am J Hum Genet 2007;81:559–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Galeazzi M, Sebastiani GD, Morozzi G, Carcassi C, Ferrara GB, Scorza R, et al. HLA class II DNA typing in a large series of European patients with systemic lupus erythematosus: correlations with clinical and autoantibody subsets. Medicine (Baltimore) 2002;81:169–78. [DOI] [PubMed] [Google Scholar]
- 26. Artim‐Esen B, Cene E, Sahinkaya Y, Ertan S, Pehlivan O, Kamali S, et al. Cluster analysis of autoantibodies in 852 patients with systemic lupus erythematosus from a single center. J Rheumatol 2014;41:1304–10. [DOI] [PubMed] [Google Scholar]
- 27. Li PH, Wong WH, Lee TL, Lau CS, Chan TM, Leung AM, et al. Relationship between autoantibody clustering and clinical subsets in SLE: cluster and association analyses in Hong Kong Chinese. Rheumatology (Oxford) 2013;52:337–45. [DOI] [PubMed] [Google Scholar]
- 28. Pacheco Y, Barahona‐Correa J, Monsalve DM, Acosta‐Ampudia Y, Rojas M, Rodriguez Y, et al. Cytokine and autoantibody clusters interaction in systemic lupus erythematosus. J Transl Med 2017;15:239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. To CH, Petri M. Is antibody clustering predictive of clinical subsets and damage in systemic lupus erythematosus? Arthritis Rheum 2005;52:4003–10. 10.1002/art.21414. [DOI] [PubMed] [Google Scholar]
- 30. Elsayed SA, Mohafez OMM. Autoantibodies spectrum in lupus nephritis in a cohort of Egyptian patients: relation to disease activity and prognostic value. Egypt. Rheumatol Rehabil 2020;47. [Google Scholar]
- 31. Gronwall C, Hardt U, Gustafsson JT, Elvin K, Jensen‐Urstad K, Kvarnstrom M, et al. Depressed serum IgM levels in SLE are restricted to defined subgroups. Clin Immunol 2017;183:304–15. [DOI] [PubMed] [Google Scholar]
- 32. Idborg H, Zandian A, Sandberg AS, Nilsson B, Elvin K, Truedsson L, et al. Two subgroups in systemic lupus erythematosus with features of antiphospholipid or Sjogren's syndrome differ in molecular signatures and treatment perspectives. Arthritis Res Ther 2019;21:62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Galeazzi M, Sebastiani GD, Tincani A, Piette JC, Allegri F, Morozzi G, et al. HLA class II alleles associations of anticardiolipin and anti‐beta2GPI antibodies in a large series of European patients with systemic lupus erythematosus. Lupus 2000;9:47–55. [DOI] [PubMed] [Google Scholar]
- 34. Sebastiani GD, Iuliano A, Cantarini L, Galeazzi M. Genetic aspects of the antiphospholipid syndrome: An update. Autoimmun Rev 2016;15:433–9. [DOI] [PubMed] [Google Scholar]
- 35. Unlu O, Erkan D, Barbhaiya M, Andrade D, Nascimento I, Rosa R, et al. The impact of systemic lupus erythematosus on the clinical phenotype of antiphospholipid antibody‐positive patients: results from the antiphospholipid syndrome alliance for clinical trials and international clinical database and repository. Arthritis Care Res (Hoboken) 2019;71:134–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Mak A, Mok CC, Chu WP, To CH, Wong SN, Au TC. Renal damage in systemic lupus erythematosus: a comparative analysis of different age groups. Lupus 2007;16:28–34. [DOI] [PubMed] [Google Scholar]
- 37. Stahl‐Hallengren C, Jonsen A, Nived O, Sturfelt G. Incidence studies of systemic lupus erythematosus in Southern Sweden: increasing age, decreasing frequency of renal manifestations and good prognosis. J Rheumatol 2000;27:685–91. [PubMed] [Google Scholar]
- 38. Caielli S, Veiga DT, Balasubramanian P, Athale S, Domic B, Murat E, et al. A CD4(+) T cell population expanded in lupus blood provides B cell help through interleukin‐10 and succinate. Nat Med 2019;25:75–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Idborg H, Eketjäll S, Pettersson S, Gustafsson JT, Zickert A, Kvarnström M, et al. TNF‐α and plasma albumin as biomarkers of disease activity in systemic lupus erythematosus. Lupus Sci Med 2018;5:e000260. 10.1136/lupus-2018-000260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Koenig KF, Groeschl I, Pesickova SS, Tesar V, Eisenberger U, Trendelenburg M. Serum cytokine profile in patients with active lupus nephritis. Cytokine 2012;60:410–6. [DOI] [PubMed] [Google Scholar]
- 41. Oke V, Gunnarsson I, Dorschner J, Eketjall S, Zickert A, Niewold TB, et al. High levels of circulating interferons type I, type II and type III associate with distinct clinical features of active systemic lupus erythematosus. Arthritis Res Ther 2019;21:107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Munroe ME, Lu R, Zhao YD, Fife DA, Robertson JM, Guthridge JM, et al. Altered type II interferon precedes autoantibody accrual and elevated type I interferon activity prior to systemic lupus erythematosus classification. Ann Rheum Dis 2016;75:2014–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Cui M, Li T, Yan X, Wang C, Shen Q, Ren H, et al. Blood Genomics Identifies Three Subtypes of Systemic Lupus Erythematosus: “IFN‐High”, “NE‐High”, and “Mixed”. Mediators Inflamm 2021;2021:6660164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Barturen G, Babaei S, Catala‐Moll F, Martinez‐Bueno M, Makowska Z, Martorell‐Marugan J, et al. Integrative analysis reveals a molecular stratification of systemic autoimmune diseases. Arthritis Rheumatol 2021;73:1073–85. [DOI] [PubMed] [Google Scholar]
- 45. Yazici MU, Orhan D, Kale G, Besbas N, Ozen S. Studying IFN‐gamma, IL‐17 and FOXP3 in pediatric lupus nephritis. Pediatr Nephrol 2014;29:853–62. [DOI] [PubMed] [Google Scholar]
- 46. Rauch J, Salem D, Subang R, Kuwana M, Levine JS. β2‐glycoprotein i‐reactive t cells in autoimmune disease. Front Immunol 2018;9:2836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Gustafsson J, Gunnarsson I, Börjesson O, Pettersson S, Möller S, Fei GZ, et al. Predictors of the first cardiovascular event in patients with systemic lupus erythematosus ‐ a prospective cohort study. Arthritis Res Ther 2009;11:R186. 10.1186/ar2878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Toloza SM, Uribe AG, McGwin G Jr, Alarcon GS, Fessler BJ, Bastian HM, et al. Systemic lupus erythematosus in a multiethnic US cohort (LUMINA). XXIII. Baseline predictors of vascular events. Arthritis Rheum 2004;50:3947–57. [DOI] [PubMed] [Google Scholar]
- 49. Bernatsky S, Boivin JF, Joseph L, Manzi S, Ginzler E, Gladman DD, et al. Mortality in systemic lupus erythematosus. Arthritis Rheum 2006;54:2550–7. [DOI] [PubMed] [Google Scholar]
- 50. Gustafsson JT, Simard JF, Gunnarsson I, Elvin K, Lundberg IE, Hansson LO, et al. Risk factors for cardiovascular mortality in patients with systemic lupus erythematosus, a prospective cohort study. Arthritis Res Ther 2012;14:R46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Benjamini Y, Yekutieli D. The control of the false discovery rate in multiple testing under dependency. Ann Stat 2001;29:1165–88. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Material