

Abstract
trans-Cyclooctenes (TCOs) are essential partners for the fastest known bioorthogonal reactions, but current synthetic methods are limited by poor diastereoselectivity. Especially hard to access are hydrophilic TCOs with favorable physicochemical properties for live cell or in vivo experiments. Described is a new class of TCOs, ‘a-TCOs’, that is prepared in high yield via stereocontrolled 1,2-additions of nucleophiles to trans-cyclooct-4-enone, which itself was prepared on large scale in two steps from 1,5-cyclooctadiene. Computational transition state models rationalize the diastereoselectivity of 1,2-additions to deliver a-TCO products, which were also shown to be more reactive than standard TCOs and less hydrophobic than even a trans-oxocene analog. Illustrating the favorable physicochemical properties of a-TCOs, a fluorescent TAMRA derivative in live HeLa cells was shown to be cell-permeable through intracellular Diels-Alder chemistry and to washout more rapidly than other TCOs.
Keywords: trans-cyclooctene, tetrazine, diastereoselective, hydrophilic, bioorthogonal
Graphical Abstract
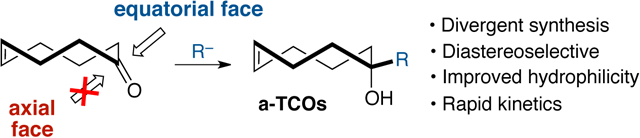
Bottleneck removed! Readily accessible trans-cyclooct-4-enone undergoes diastereoselective additions with a broad range of nucleophiles to give access to a-TCOs, a family of trans-cyclooctenes with high reactivity and improved physiochemical properties.
Introduction
Bioorthogonal reactions are a class of rapid, selective reactions that can proceed efficiently and selectively in biological systems without interfering with biological functional groups.[1] Bioorthogonal chemistry has enabled a deeper understanding of native biological processes and expanded the frontiers of chemical biology through innovations in nuclear medicine,[2] drug delivery,[3] and biomaterials.[4] The tetrazine ligation with trans-cyclooctenes (TCOs) has been at the forefront of bioorthogonal methodologies due to rapid reaction kinetics that generally exceed k2 104 M−1s−1.[5]
TCO derivatives are synthesized by a photochemical flow-method under singlet sensitized conditions, driving an otherwise unfavorable isomeric ratio in favor of the trans-isomer via selective metal complexation to Ag(I).[6] Flow photoisomerization has been carried out on scales as large as 20 grams,[7] and protocols have been developed where flow is mimicked by periodically stopping irradiation and capturing TCO product by filtering through AgNO3-silica and resubjecting the filtrate to photoisomerization (254 nm).[8] While this method has been used to synthesize a large number of different TCO derivatives,[9] the production of diastereomers due to the planar chirality of the alkene presents a bottleneck for the majority of TCO syntheses.
For example, the most frequently utilized TCO derivatives are the axial and equatorial diastereomers of 5-hydroxy-trans-cyclooctene, which are produced through photoisomerization of 5-hydroxy-cis-cyclooctene in 72%.[6] Derivatives of the axial diastereomer have emerged as especially useful, as they are an order of magnitude more reactive than equatorial diastereomers,[5,10] can promote fluorogenic effects for cell imaging,[11] and have been employed in ‘click-to-release’ strategies for bioorthogonal uncaging.[3a,3b] However, photoisomerization produces the equatorial:axial isomers in 2.2:1 ratio, and thus produces ≤24% of the axial diastereomer. A modified procedure using Ag(I) sulfonated silica gel slightly improves the yield of the axial diastereomer to ≤27%.[12] Even for the equatorial diastereomer, the need to separate diastereomers represents a limitation for material throughput. For other TCO derivatives, chromatographic separation of diastereomers can be very difficult and not feasible by flash chromatography.[13] One solution to the diastereomer issue has been to utilize s-TCO— a highly strained dienophile prepared from the meso compound precursor.[14] s-TCO is most reactive dienophile known, and is suitable for applications as a probe molecule in bioorthogonal chemistry,[3e,15] but due to alkene isomerization, s-TCO is often unsuitable as a reporter where more prolonged cellular incubation is required.[16] Currently, several TCO derivatives are available for purchase, but they are very expensive. A general and diastereoselective synthesis of TCO derivatives could greatly increase the availability of these useful compounds for chemical biology research.
In addition to stereoselectivity, hydrophobicity has been a longstanding issue with TCOs where non-specific binding and extensive wash-out protocols in live cell assays are undesirable consequences.[17] Recently, heterocyclic trans-cyclooctenes with backbone oxygen atoms have been shown to have higher hydrophilicity and improved physiochemical properties for cellular and in vivo imaging applications.[13,18] However, a drawback for these oxo-TCOs are lengthy syntheses that produce diastereomers that can be separated only with difficulty.
Described here is a diastereoselective method for synthesizing TCOs with favorable physiochemical properties in high yield and useful scale through the stereocontrolled additions of nucleophiles to trans-cyclooct-4-enone 2 (Figure 1). As 2 lacks a stereocenter, it therefore can be prepared on large scale using flow photochemistry from cis-cyclooct-4-enone (1) without the complication of diastereoselectivity in the photoisomerization step encountered previously. The rigid structure of 2 distinguishes the faces of the ketone, and computation was used to predict that nucleophilic additions to 2 take place exclusively from the equatorial-face of the ketone to produce axial products as single diastereomers. By engaging a range of nucleophiles, ketone 2 serves as a general platform for preparing a range of functionalized axial-5-hydroxy-trans-cyclooctene (‘a-TCO’) analogs. The method provides improved access to new compounds as well as known TCO derivatives required for in vivo radiochemistry,[10] click-to-release chemistry,[2a,3a,3e] and sulfenic acid detection in live cells.[19] An a-TCO derivative was shown to be more reactive than both axial and equatorial diastereomers of 5-hydroxy-trans-cyclooctene as well as oxo-TCO in cycloadditions with tetrazines. As a demonstration of the favorable physicochemical properties of a-TCOs, a fluorescent TAMRA derivative was shown to be cell-permeable by demonstrating intracellular Diels-Alder reactions in live cells and was shown to washout of HeLa cells more rapidly than even a hydrophilic oxo-TCO analog.
Figure 1.
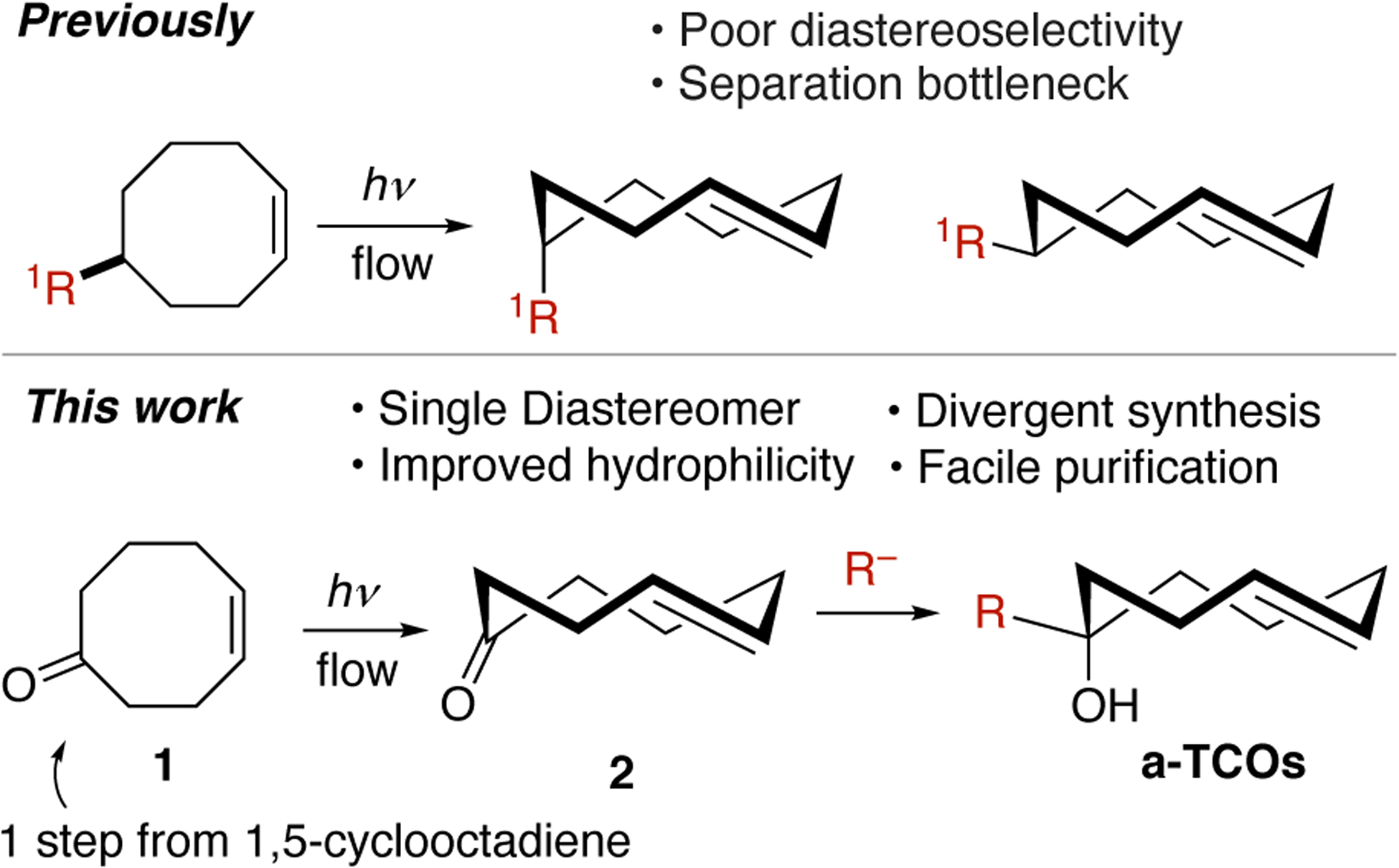
Previous approach to trans-cyclooctene syntheses compared to general, divergent, and diastereoselective platform described in this paper.
Results and Discussion
Ketone 1 was prepared simply in a single step by the Wacker oxidation (5 mol% Pd(OAc)2, AcOH, benzoquinone) of 1,5-cyclooctadiene 3 on multigram scale (Figure 2A).[20] Alternately, 1 can be prepared in a single step by Dess-Martin oxidation of 5-hydroxy-cis-cyclooctene.19 Photochemical isomerization of 1 was conducted using the general photochemical flow method.[6,9] Using a small cartridge and bed of capture silica, ketone 2 was prepared in 62% yield and at a rate of ~150 mg/h, and in a typical workflow, ketone 2 was prepared in 2.5 g batches.
Figure 2.
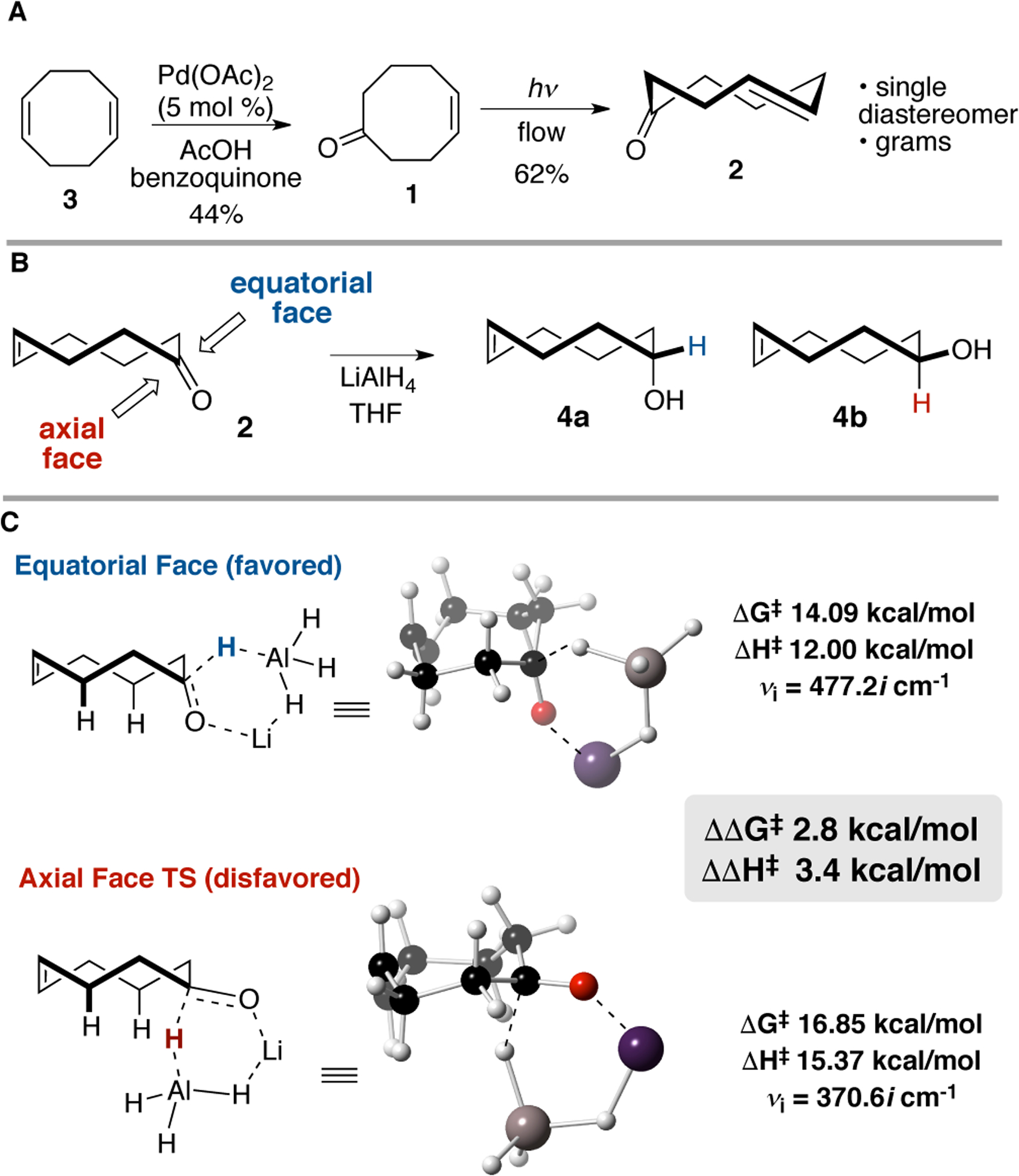
(A) Synthesis of cis-cyclooct-4-enone 1 and trans-cyclooct-4-enone 2. (B) Reaction of trans-cyclooct-4-enone 2 with LiAlH4. Nucleophilic addition can occur to the equatorial or axial face of 2 producing diastereomers 4a and 4b, respectively. (B) Transition state calculations predict nucleophilic addition to equatorial face would be favored over the axial face.
Computation was used to predict if nucleophilic addition to TCO 2 would be diastereoselective (Figure 2B). It was reasoned that 1,3-diaxial interactions in the lowest energy ‘crown’ conformation of the parent TCO would favor addition to the equatorial face by nucleophiles, akin to the facial selectivity of conformationally biased cyclohexanones.[21] The barriers for the reaction of LiAlH4 with 2 were calculated at the M06L/6–311+G(d,p) level using a SCRF solvent model for THF (Figure 2C). The calculated barriers relative to the pre-reaction complex for the equatorial attack by hydride are ΔG‡ 14.09 kcal mol−1 and ΔH‡ 12.00 kcal mol−1. The barrier is significantly lower than that calculated for axial attack (ΔΔG‡ 2.8 kcal/mol and ΔΔH‡ 3.4 kcal/mol). Encouraged by these computational predictions, we experimentally investigated additions of nucleophiles to TCO 2 (Scheme 1A). In agreement with the computational data, nucleophilic addition of hydride occurred exclusively to the equatorial face of 2 to produce 4a. While LiAlH4 gave 4a in only 67% yield due partial alkene isomerization, NaBH4 provided 4a as a single diastereomer in 90% yield with no isomerization. Derivatives of axial alcohol 4a are especially useful for their rapid kinetics[10] and their utility in click-to-release chemistry,[3e] but previous syntheses of 4a were low yielding (≤27%) and required separation from major diastereomer 4b.[6,12] The improved route described here is short, selective and more scalable.
Scheme 1.
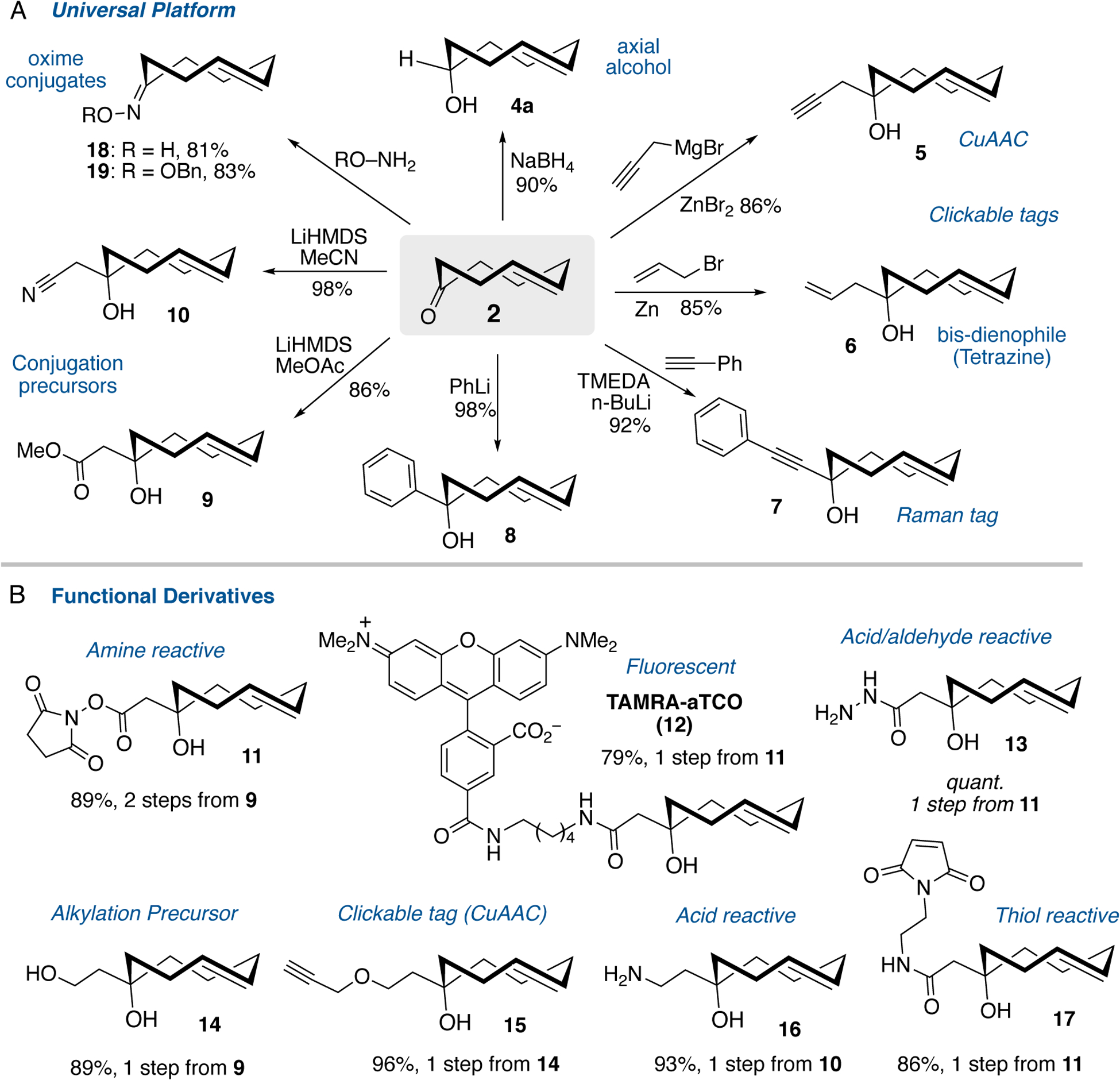
(A) Nucleophilic addition reactions of trans-cyclooct-4-enone (2) serves as a universal platform for the diastereoselective synthesis of a-TCOs as well as oxime conjugates. (B) Functional derivatives readily available from conjugation precursors 9 and 10
Nucleophilic additions with a diverse array of nucleophiles were preformed to create a library of a-TCOs (Scheme 1). Compound 5 bearing a bioorthogonal alkyne tag was recently developed for the capture of cellular protein sulfenic acids via transannular thioetherification with subsequent proteomic analysis enabled by a CuAAC-chemistry workflow.[19] We note that 5 (and other a-TCOs) are still suitable for selective bioorthogonal chemistry as they only modify sulfenic acids at relatively high TCO concentration (generally ≥500 μM), but not under the conditions typically used in intracellular bioorthogonal chemistry (generally ≤10 μM). In the previous study, 5 was synthesized by direct photoisomerization in only 6% yield and required separation from two isomers. As shown in Scheme 1A, compound 5 can be prepared in 86% yield as a single diastereomer by adding propargyl magnesium bromide to ketone 2. Similarly, compound 6 bearing a simple alkene tag can be constructed in 85% yield and as a single diastereomer by the addition of allyl zinc bromide to ketone 2. Like trans-cyclooctenes, simple α-olefins can function as dienophiles in tetrazine ligation but with much slower kinetics, providing handles for potential sequential bioorthogonal chemistry applications.[22] Organolithium nucleophiles generated in-situ were used to synthesize TCOs 7-8. Illustrating the ability to introduce a tag that may be useful for Raman spectroscopy and imaging,[23] TCO 7 bearing a phenylacetylene group was synthesized by the addition of lithium phenyl acetylene to 2 in 92% yield. TCO 8 was synthesized similarly by the addition of phenyl lithium to 2 in 98% yield and serves as a model reaction for nucleophilic addition of aryl groups to TCO 2.
The diastereoselective additions of 2 with methyl α-lithioacetate or lithioacetonitrile provided a straightforward path to introduce handles for conjugation via amide bond or ether formation. The reaction of ketone 2 with LiHMDS/methyl acetate produced ester 9 in 86% yield. Similarly, the combination of 2 with LiHMDS and acetonitrile produced nitrile 10 in 98% yield. Hydrolysis of 9 with trimethyltin hydroxide[24] followed by DIC-mediated coupling with N-hydroxysuccinimide gave NHS ester 11 in 89% yield (Scheme 1B). Alternately, using LiOH gave 11 in 46% yield after DIC coupling. Through amide coupling, 11 gave the fluorescent TAMRA-conjugate 12 with favorable physiochemical properties, and 11 was also combined with hydrazine monohydrate to give hydrazide 13 with a potential handle for aldehyde conjugation.[25] TCO 9 also was combined with LiAlH4 to provide diol 14 in 89% yield which was next used in a Williamson ether synthesis with NaH/propargyl bromide to give 96% yield of propargyl ether 15. Compound 15 serves a more stable alternative to alkyne-tagged TCO 6, which is found to polymerize if not stored in solution. Amine 16 was synthesized by LiAlH4 reduction of TCO nitrile 10 in 93% yield to introduce an acid reactive conjugation handle. Maleimide 17 was prepared though amide coupling of TCO 11 with N-(2-aminoethyl)maleimide in 86% yield. The yield of 17 is significantly higher than what was observed in previous preparations of TCO-maleimides from activated nitrophenylcarbonates.[14] Furthermore, oxime conjugates 18 and 19 could be prepared by the direct condensation of the corresponding hydroxylamines in the presence of pyridine (Scheme 1A). In summary, ketone 2 can serve as a readily prepared, central intermediate for the diastereoselective preparation of a range of hydrophilic a-TCO conjugates.
a-TCO derivatives also display rapid kinetics in Diels-Alder reactions compared to most other TCOs. The kinetics for the reaction of a-TCO derivative 14 toward a 3,6-dipyridyl-s- tetrazinyl succinamic acid derivative 20 were measured by stopped flow kinetics at 25 °C in 95:5 PBS:MeOH (Figure 3A). With a second-order rate constant of 150,000 ± 8000 M−1s−1, a- TCO is more than twice as reactive toward 20 as the axial isomer of 5-hydroxy-trans-cyclooctene (4a, 70,000 ± 1800 M−1s−1), and nearly 7-times more reactive than equatorial isomer 4b (22,400 ± 40 M−1s−1).[5] The faster kinetics are likely due an increase in olefinic strain for a-TCO due to steric effect of geminal substitution in the 8-membered ring backbone. Likely for the same reason, similar rate accelerations have been observed for more sterically encumbered derivatives of 4a.[10] a-TCO 14 is also more reactive than oxo-TCO,[13] and the conformationally strained, bicyclic d-TCO is only 2.2-times more reactive than 14.[5] As shown in Figure 3B, a-TCO derivatives are also calculated to have improved physiochemical properties relative to other TCO derivatives. While both oxo-TCO and d-TCO were previously introduced as less hydrophobic bioorthogonal reagents, the methylamine conjugate of a-TCO is calculated to have even a lower cLogP value.
Figure 3.
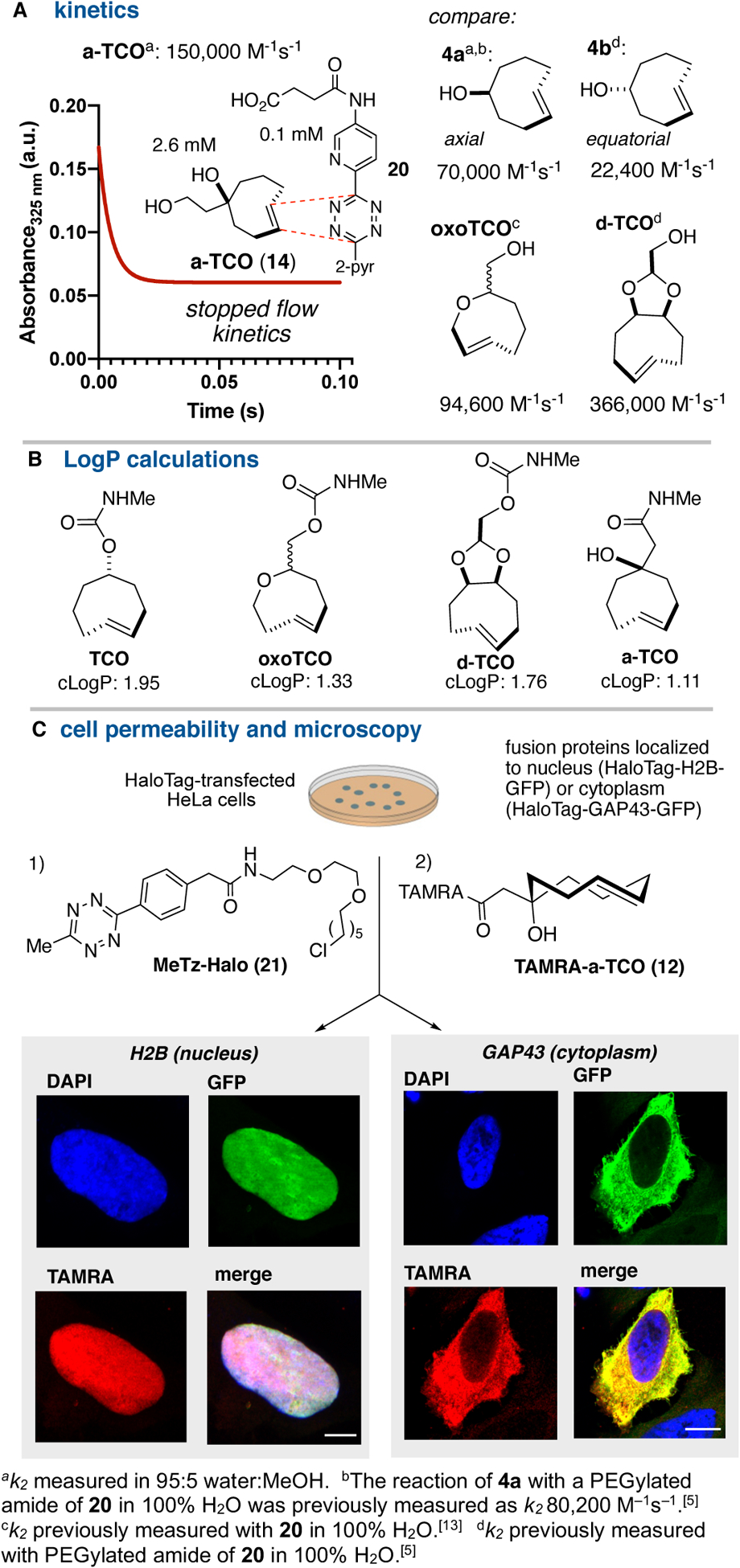
(A) Stopped flow kinetics under pseudo-first order conditions were used to determine second order rate constants for the reactions of tetrazine 20 with 14, allowing comparison to less reactive 4a and 4b. (B) cLogP calculations for a series of analogs of TCO, oxoTCO, d-TCO, and a-TCO illustrate the improved hydrophilicity of a-TCO conjugates. (C) Cell permeability was demonstrated by incorporation of MeTz-Halo (21) into HeLa cells transfected with either H2B-HaloTag-GFP (nuclear) or GAP43-HaloTag-GFP (cytoplasmic), followed by labeling with TAMRA-a-TCO (1 μM). Confocal microscopy images of transfected cells labeled with TAMRA-a-TCO show subcellular colocalization of GFP and TAMRA fluorescence, consistent with selective intracellular labeling. Scale bars for H2B and GAP43 labeling are 5 μM and 10 μM, respectively.
The stability of a-TCO 14 is very similar to that of axial-5-hydroxy-trans-cyclooctene 4a, which is used broadly for applications in bioorthgonal chemistry. In MeOD, 35 mM solutions of both 14 and 4a are >99% stable after 1 week at room temperature. After 24 h at room temperature in D2O-PBS (pD 7.4), 33 mM solutions of 14 and 4a display 90% and 85% stability, respectively. In D2O-PBS containing 25 mM mercaptoethanol, 49% of both 14 and 4 remained after 20 h.
The fluorescent conjugate TAMRA-a-TCO 12 was shown to be cell permeable through selective bioorthogonal reaction inside live cells using the HaloTag self-labeling platform (Figure 3C). Here, cells are transfected with a GFP-HaloTag construct fused to a protein that controls subcellular localization, and then labeled by a tetrazine-HaloTag ligand 21. Upon subsequent reaction with fluorescent TAMRA-a-TCO, conjugation is expected only in those cells that express the HaloTag fusion protein, and co-localization of GFP and TAMRA fluorescence is expected. As shown in Figure 3C, HeLa cells were transfected with either HaloTag-H2B-GFP (nucleus) or HaloTag-GAP43-GFP (cytoplasm), labeled with MeTz-Halo 21 (10 μM), washed and then treated with TAMRA-a-TCO (1 μM) for 30 min, at which point the TCO reagent was chased by a non-fluorescent tetrazine, and the cells were fixed and imaged. As shown in Figure 3C, for both nuclear and cytoplasmic targets, selective colocalization of the TAMRA signal with GFP was observed in both cases.
The advantage of the increased hydrophilicity for a-TCO conjugates was demonstrated by comparing the washout times for fluorescent derivatives in mammalian cells. While TCO-derivatives offer rapid labeling kinetics, a consideration for labeling by fluorophore-tagged TCOs has been the background fluorescence due to non-covalent cellular binding that can be ameliorated only by extended washout times. More hydrophilic oxo-[13] and dioxo-trans-cyclooctenes[18a] with backbone oxygens offer improvements, but are difficult to synthesize and, for dioxo-TCO, display reduced Diels-Alder kinetics. As shown in Figure 4, we prepared the fluorescent conjugates TAMRA-TCO, TAMRA-oxo-TCO and TAMRA-a-TCO and compared their cellular washout times to unconjugated TAMRA. Thus, HeLa cells were incubated for 30 min with TAMRA- washed three times with DPBS, and then cell media was exchanged after 10, 40 and 120 minutes. After each wash, cells were imaged live by fluorescence microscopy with illumination at 531 nm and fixed-intensity across all samples. Widefield images of the cells after 3 washes are shown in Figure 4B; images after the earlier and later washings are shown in Figure S10. Background fluorescence was quantified by dividing total fluorescence by the number of cells in each image (Figure 4C). For TAMRA-TCO, cells are markedly fluorescent after 3 washings, and still display significant background after washing for 2 hours. The background is improved with TAMRA-oxo-TCO and especially with TAMRA-a-TCO, which after initial 3x wash shows an 85% reduction in background fluorescence relative to TAMRA-TCO. After 2 hours, washout of TAMRA-a-TCO is essentially complete with background equivalent to TAMRA itself, whereas TAMRA-TCO and TAMRA-oxo-TCO both still display residual fluorescence even after 2 hours.
Figure 4.
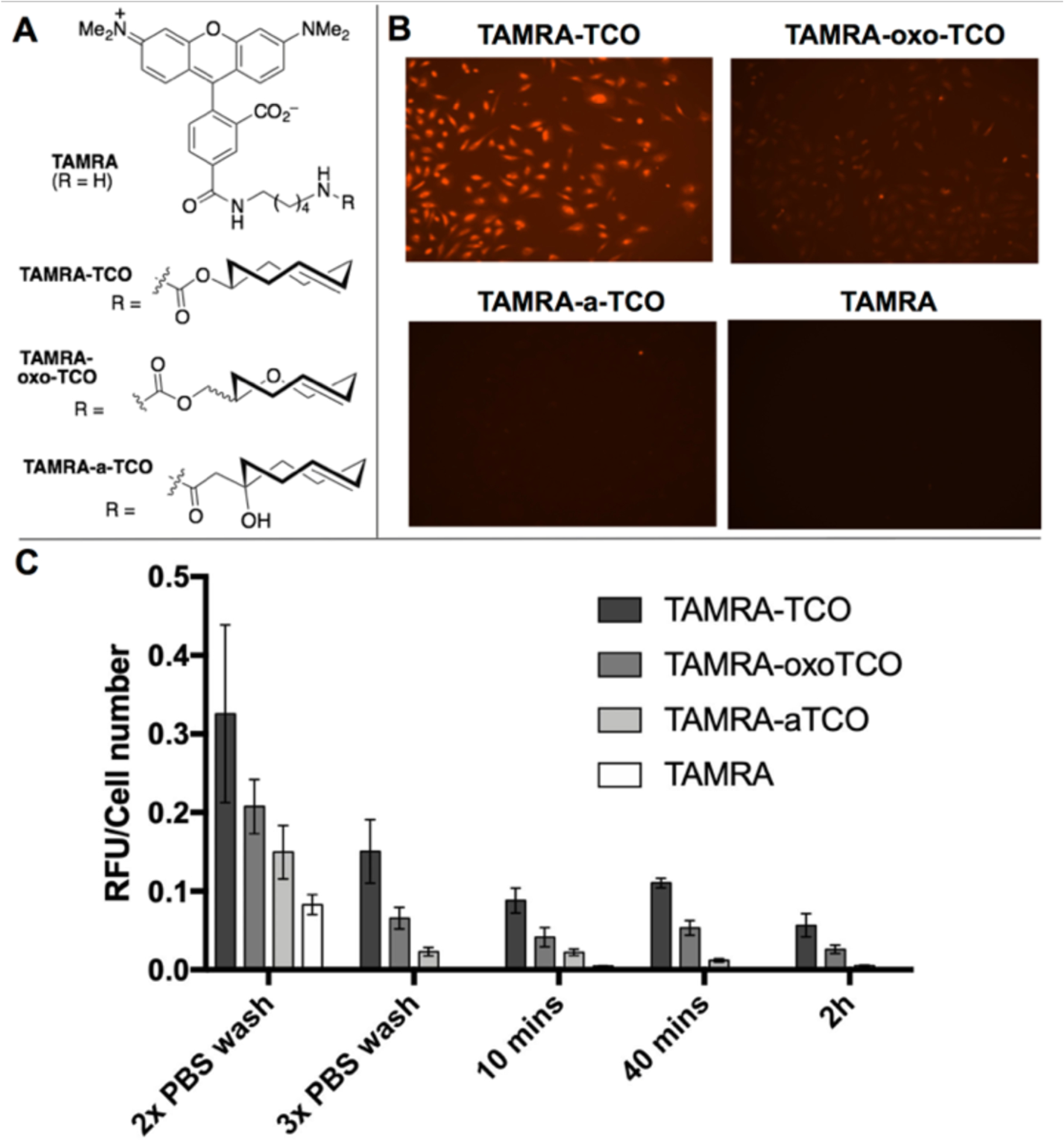
(A) Structures of TAMRA and conjugates with TCO, oxo-TCO and a-TCO. (B,C) HeLa cells were incubated for 30 min with TAMRA-dyes, and cells were initially washed three times with PBS, and then cell media was exchanged after 10, 40 and 120 minutes. After each wash, cells were imaged live by fluorescence microscopy with illumination at 531 nm and with fixed-intensity across all samples. (B) Widefield images cells after 3 washes. (C) Comparison of background fluorescence across all experiments, quantified by dividing total fluorescence by the number of cells in each image.
Conclusion
In summary, a-TCOs are a class of trans-cyclooctenes with favorable physiochemical properties that can be prepared in high yield through the stereocontrolled additions of nucleophiles to trans-cyclooct-4-enone (2), a trans-cyclooctene that can be prepared on large scale in two steps from 1,5-cyclooctadiene. Computation was used to rationalize diastereoselectivity of 1,2-additions to deliver a-TCO products. The strategy can be applied to the synthesis of a range of usefully functionalized a-TCOs with high yield, selectivity. a-TCOs were also shown to be more reactive than standard TCOs and less hydrophobic than even hydrophilic oxo-TCO analogs. As a demonstration of the favorable physicochemical properties of a-TCOs, a fluorescent TAMRA derivative was shown to be cell-permeable by demonstrating intracellular Diels-Alder chemistry in live cells and to washout of HeLa cells more rapidly and completely than TCO and oxo-TCO analogs.
Supplementary Material
Acknowledgements
This work was supported by NIH GM132460 and Pfizer. Instrumentation was supported by NIH awards P20GM104316, P30GM110758, S10RR026962, and S10OD016267 and NSF awards CHE-0840401, CHE-1229234, and CHE-1048367. J. R. and A. J. were supported as CBI fellows through NIH T32-GM1333395.
Footnotes
Supporting information for this article is given via a link at the end of the document.
References
- [1].a) Sletten EM, Bertozzi CR, Angew. Chem. Int. Ed 2009, 48, 6974–6998; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Sletten EM, Bertozzi CR, Acc. Chem. Res 2011, 44, 666–676; [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Ramil CP, Lin Q, Chem Commun 2013, 49, 11007–11022; [DOI] [PMC free article] [PubMed] [Google Scholar]; d) King M, Wagner A, Bioconjug. Chem 2014, 25, 825–839; [DOI] [PMC free article] [PubMed] [Google Scholar]; e) Lang K, Chin JW, ACS Chem. Biol 2014, 9, 16–20; [DOI] [PMC free article] [PubMed] [Google Scholar]; f) Patterson DM, Nazarova LA, Prescher JA, ACS Chem. Biol 2014, 9, 592–605; [DOI] [PMC free article] [PubMed] [Google Scholar]; g) Vrabel M, Carell T, Cycloadditions in Bioorthogonal Chemistry, Springer International Publishing, Switzerland, 2016; [DOI] [PMC free article] [PubMed] [Google Scholar]; h) Qin LH, Hu W, Long YQ, Tetrahedron Lett. 2018, 59, 2214–2228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].a) Rossin R, Robillard MS, Curr. Opin. Chem. Biol 2014, 21, 161–169; [DOI] [PubMed] [Google Scholar]; b) Meyer JP, Adumeau P, Lewis JS, Zeglis BM, Bioconjug. Chem 2016, 27, 2791–2807. [DOI] [PubMed] [Google Scholar]
- [3].a) Versteegen RM, Rossin R, ten Hoeve W, Janssen HM, Robillard MS, Angew. Chem. Int. Ed 2013, 52, 14112–14116; [DOI] [PubMed] [Google Scholar]; b) Li J, Jia S, Chen PR, Nat. Chem. Biol 2014, 10, 1003–1005; [DOI] [PubMed] [Google Scholar]; c) Carlson JCT, Mikula H, Weissleder R, J. Am. Chem. Soc 2018, 140, 3603–3612; [DOI] [PubMed] [Google Scholar]; d) Ji XY, Pan ZX, Yu BC, De la Cruz LK, Zheng YQ, Ke BW, Wang BH, Chem. Soc. Rev 2019, 48, 1077–1094; [DOI] [PubMed] [Google Scholar]; e) van Onzen A, Versteegen RM, Hoeben FJM, Filot IAW, Rossin R, Zhu T, Wu J, Hudson PJ, Janssen HM, Ten Hoeve W, Robillard MS, J. Am. Chem. Soc 2020, 142, 10955–10963. [DOI] [PubMed] [Google Scholar]
- [4].Brown TE, Anseth KS, Chem. Soc. Rev 2017, 46, 6532–6552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Darko A, Wallace S, Dmitrenko O, Machovina MM, Mehl RA, Chin JW, Fox JM, Chem. Sci 2014, 5, 3770–3776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Royzen M, Yap GPA, Fox JM, J. Am. Chem. Soc 2008, 130, 3760–3761. [DOI] [PubMed] [Google Scholar]
- [7].Rossin R, van Duijnhoven SM, Ten Hoeve W, Janssen HM, Kleijn LH, Hoeben FJ, Versteegen RM, Robillard MS, Bioconjug. Chem 2016, 27, 1697–1706. [DOI] [PubMed] [Google Scholar]
- [8].a) Devaraj NK, Upadhyay R, Haun JB, Hilderbrand SA, Weissleder R, Angew. Chem. Int. Ed 2009, 48, 7013–7016; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Schoch J, Staudt M, Samanta A, Wiessler M, Jaschke A, Bioconjug. Chem 2012, 23, 1382–1386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Pigga JE, Fox JM, Isr. J. of Chem 2020, 60, 207–218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Rossin R, van den Bosch SM, Ten Hoeve W, Carvelli M, Versteegen RM, Lub J, Robillard MS, Bioconjug. Chem 2013, 24, 1210–1217. [DOI] [PubMed] [Google Scholar]
- [11].Vazquez A, Dzijak R, Dracinsky M, Rampmaier R, Siegl SJ, Vrabel M, Angew. Chem. Int. Ed 2017, 56, 1334–1337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Darko A, Boyd SJ, Fox JM, Synthesis 2018, 50, 4875–4882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Lambert WD, Scinto SL, Dmitrenko O, Boyd SJ, Magboo R, Mehl RA, Chin JW, Fox JM, Wallace S, Org. Biomol. Chem 2017, 15, 6640–6644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Taylor MT, Blackman ML, Dmitrenko O, Fox JM, J. Am. Chem. Soc 2011, 133, 9646–9649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Murrey HE, Judkins JC, Am Ende CW, Ballard TE, Fang Y, Riccardi K, Di L, Guilmette ER, Schwartz JW, Fox JM, Johnson DS, J. Am. Chem. Soc 2015, 137, 11461–11475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Fang Y, Judkins JC, Boyd SJ, Am Ende CW, Rohlfing K, Huang Z, Xie Y, Johnson DS, Fox JM, Tetrahedron 2019, 75, 4307–4317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Uttamapinant C, Howe JD, Lang K, Beranek V, Davis L, Mahesh M, Barry NP, Chin JW, J. Am. Chem. Soc 2015, 137, 4602–4605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].a) Kozma E, Nikic I, Varga BR, Aramburu IV, Kang JH, Fackler OT, Lemke EA, Kele P, Chembiochem 2016, 17, 1518–1524; [DOI] [PubMed] [Google Scholar]; b) Wang M, Vannam R, Lambert WD, Xie Y, Wang H, Giglio B, Ma X, Wu Z, Fox J, Li Z, Chem Commun 2019, 55, 2485–2488; [DOI] [PubMed] [Google Scholar]; c) Liang D, Wu K, Tei R, Bumpus TW, Ye J, Baskin JM, PNAS 2019, 116, 15453–15462. [DOI] [PubMed] [Google Scholar]
- [19].Scinto SL, Ekanayake O, Seneviratne U, Pigga JE, Boyd SJ, Taylor MT, Liu J, Am Ende CW, Rozovsky S, Fox JM, J. Am. Chem. Soc 2019, 141, 10932–10937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Aakermark B, Larsson EM, Oslob JD, J. Org. Chem 1994, 59, 5729–5733. [Google Scholar]
- [21].Neufeldt SR, Jimenez-Oses G, Comins DL, Houk KN, J. Org. Chem 2014, 79, 11609–11618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].a) Niederwieser A, Spate AK, Nguyen LD, Jungst C, Reutter W, Wittmann V, Angew. Chem. Int. Ed 2013, 52, 4265–4268; [DOI] [PubMed] [Google Scholar]; b) Lee YJ, Kurra Y, Yang Y, Torres-Kolbus J, Deiters A, Liu WR, Chem Commun 2014, 50, 13085–13088; [DOI] [PubMed] [Google Scholar]; c) Wang WW, Zeng Y, Wu B, Deiters A, Liu WR, ACS Chem. Biol 2016, 11, 1973–1981. [DOI] [PubMed] [Google Scholar]
- [23].Yamakoshi H, Dodo K, Palonpon A, Ando J, Fujita K, Kawata S, Sodeoka M, J. Am. Chem. Soc 2012, 134, 20681–20689. [DOI] [PubMed] [Google Scholar]
- [24].Nicolaou KC, Estrada AA, Zak M, Lee SH, Safina BS, Angew. Chem. Int. Ed 2005, 44, 1378–1382. [DOI] [PubMed] [Google Scholar]
- [25].Dirksen A, Dawson PE, Bioconjug. Chem 2008, 19, 2543–2548. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.