

ABSTRACT
HIV-1 virion production is driven by Gag and Gag-Pol (GP) proteins, with Gag forming the bulk of the capsid and driving budding, while GP binds Gag to deliver the essential virion enzymes protease, reverse transcriptase, and integrase. Virion GP levels are traditionally thought to reflect the relative abundances of GP and Gag in cells (∼1:20), dictated by the frequency of a −1 programmed ribosomal frameshifting (PRF) event occurring in gag-pol mRNAs. Here, we exploited a panel of PRF mutant viruses to show that mechanisms in addition to PRF regulate GP incorporation into virions. First, we show that GP is enriched ∼3-fold in virions relative to cells, with viral infectivity being better maintained at subphysiological levels of GP than when GP levels are too high. Second, we report that GP is more efficiently incorporated into virions when Gag and GP are synthesized in cis (i.e., from the same gag-pol mRNA) than in trans, suggesting that Gag/GP translation and assembly are spatially coupled processes. Third, we show that, surprisingly, virions exhibit a strong upper limit to trans-delivered GP incorporation; an adaptation that appears to allow the virus to temper defects to GP/Gag cleavage that may negatively impact reverse transcription. Taking these results together, we propose a “weighted Goldilocks” scenario for HIV-1 GP incorporation, wherein combined mechanisms of GP enrichment and exclusion buffer virion infectivity over a broad range of local GP concentrations. These results provide new insights into the HIV-1 virion assembly pathway relevant to the anticipated efficacy of PRF-targeted antiviral strategies.
IMPORTANCE HIV-1 infectivity requires incorporation of the Gag-Pol (GP) precursor polyprotein into virions during the process of virus particle assembly. Mechanisms dictating GP incorporation into assembling virions are poorly defined, with GP levels in virions traditionally thought to solely reflect relative levels of Gag and GP expressed in cells, dictated by the frequency of a −1 programmed ribosomal frameshifting (PRF) event that occurs in gag-pol mRNAs. Herein, we provide experimental support for a “weighted Goldilocks” scenario for GP incorporation, wherein the virus exploits both random and nonrandom mechanisms to buffer infectivity over a wide range of GP expression levels. These mechanistic data are relevant to ongoing efforts to develop antiviral strategies targeting PRF frequency and/or HIV-1 virion maturation.
KEYWORDS: Gag, Gag-Pol, HIV, PRF, cis-acting RNA element, protease, reverse transcription, ribosomal frameshift, virion, virus assembly
INTRODUCTION
Retroviruses encode cis-acting RNA structural elements that regulate key stages of viral replication, including but not limited to transcription, splicing, RNA nuclear export, translation, RNA genome dimerization, and genome packaging (1–3). A well-studied example is the human immunodeficiency virus type 1 (HIV-1) programmed ribosomal frameshift (PRF) regulatory element that controls the synthesis of Gag and Gag-Pol (GP) virion proteins from a single viral unspliced gag-pol mRNA (4–10). Gag forms the largest percentage of the HIV-1 virion and is expressed as an ∼55-kDa precursor polyprotein (Pr55) cleaved into the subunits matrix (MA; p17), capsid (CA; p24), nucleocapsid (NC; p7), and late domain (p6) and two spacer peptides (SP1 and SP2) (11, 12). GP is larger (Pr160) and consists of identical MA, CA, SP1, and NC subunit domains fused to a linker protein (p6*) upstream of subunits encoding the viral enzymes protease (PR), reverse transcriptase (RT), and integrase (IN) (13). Gag expression is sufficient to generate virus-like particles even in the absence of all other viral proteins. However, GP incorporation into virions is essential to virion infectivity because it delivers PR, which mediates cleavage of Gag and GP during capsid maturation, RT, which reverse transcribes the viral RNA genome to form double-stranded DNA (dsDNA) following capsid delivery into target cells, and IN, which integrates the dsDNA proviral genome intermediate into host cell chromatin. GP incorporation levels need to be tightly controlled, based on prior studies showing that GP overexpression negatively impacts capsid maturation steps (14–17) and the stability of packaged RNA genome dimers (14).
With the exception of spumaviruses, which express Gag and Pol from independent mRNAs, all other known retroviruses use translational recoding of a single gag-pol transcript to generate Gag and GP, employing either frameshifting (FS) or codon read-through mechanisms (18). For HIV-1, the PRF element regulates FS frequency through the activities of two proximal cis-acting regulatory sequences: a conserved heptanucleotide “slippery” sequence (UUUUUUA) (SS) located upstream of a strong (ΔG = −21.8 kcal/mol) 11-bp RNA stem-loop positioned 164 bases prior to the gag stop codon (8, 19). A consensus model posits that ribosome stalling at the stem-loop leads to −1 FS events at the SS ∼5 to 10% of the time, so that Gag and GP are synthesized at an approximate ratio of 20:1 (Gag to GP) (4, 9, 20). Increasing the local thermodynamic stability of the PRF stem-loop enhances FS frequency both in vitro (19) and in cells (21), and we showed previously that stem-loop modifications increasing FS frequency as little as 2-fold can yield reductions in viral infectivity of >80% (21). Accordingly, the PRF element may represent a viable target for antiviral intervention. Indeed, cell-permeant small molecules designed to bind the stem-loop and increase FS frequency have been shown to reduce viral infectivity in cell culture (22).
The incorporation of Gag and GP into virus particles is traditionally viewed as a stochastic process, wherein the ratio of GP to Gag found in virions roughly equals that found in cells, with GP recruited to virions through random associations with Gag and the viral RNA scaffold (21, 23, 24). To better understand how altering PRF frequency affects virion infectivity, here we studied the assembly characteristics of mutant HIV-1 viruses engineered to either increase or abolish frameshifting. Using these viruses, we demonstrated that GP incorporation levels are, in fact, governed by nonstochastic mechanisms, with GP being moderately enriched in virions relative to cells and, more strikingly, much more efficiently incorporated into virions when cogenerated with Gag in cis (i.e., from the same gag-pol mRNA) than in trans (i.e., when Gag and GP originate from separate gag and GP mRNAs). Unexpectedly, we also discovered that virions impose an ∼2-fold saturation ceiling on GP incorporation when GP is delivered in trans, a feature that helps the virus to preserve low-level infectivity even when cytoplasmic concentrations of GP are exceptionally high.
RESULTS
Increasing PRF stem-loop stability enhances GP synthesis and incorporation into virions.
Because our previous work showed that increasing FS frequency results in significant decreases to virion infectivity (21), the initial goal of this study was to more precisely define the levels of PRF and virion GP incorporation that would be needed to abrogate HIV-1 replication in the context of an antiviral strategy (Fig. 1 and 2). To this end, we focused on a subset of FS mutants at either end of the frameshifting spectrum: M1, which we had previously shown to exhibit increased FS frequency due to stronger local base-pairing interactions at the base of the PRF stem-loop, and −SS, a mutant that encodes two U-to-C mutations within the slippery sequence 5′ to the PRF stem-loop, thereby abolishing frameshifting entirely (Fig. 1A) (21).
FIG 1.

Increasing PRF element stem-loop stability results in an increase in GP production and enriched levels of GP associated with virions. (A) Representation of the wild-type (WT) HIV-1 gag-pol mRNA frameshift (FS) site secondary structure and that of PRF mutants –SS and M1. M1 was engineered to exhibit elevated stem-loop stability, with ΔGLocal being the predicted free energy of the first 3 bp of the WT and M1 stem-loops. (B) WT, M1, and −SS Gag and GP expression and incorporation into virus particles. HEK293T cells generating WT or the indicated PRF element mutant viruses were cultured in the presence of the protease inhibitor saquinavir (10 μM) to prevent Gag and GP processing. Cell lysates and virions were harvested at 48 h. Gag and GP were detected by Western blotting using anti-p24Gag primary antiserum and infrared-labeled secondary antibodies. HSP90 was detected as a loading control. (C) FS frequency was defined as the ratio of cell-associated GP to Gag (orange bars). GP incorporation frequency into virions was defined as the ratio of virion-associated GP to Gag (blue bars). Fold changes in GP/Gag ratios for the indicated conditions are indicated by black lines, with comparisons derived from three independently performed biological replicates. Error bars represent the standard deviations of the means. *, P < 0.05, **, P < 0.001, and ***, P < 0.0001. P values were derived using Student’s two-tailed t test for comparisons as indicated in the figure, except for −SS P values, which were derived from comparisons of FS frequency and GP incorporation levels to the WT values.
FIG 2.

Virus complementation reveals a nonlinear relationship between GP expression and viral infectivity at suboptimal levels of GP. (A) Infectivity of virions produced by WT, M1, and −SS reporter viruses. Viral GFP expression was quantified and normalized to that of WT virus (set to 100%). (B) Schematic of viral 2-color complementation assay wherein WT or PRF mutant GFP reporter viruses were coexpressed in cells generating a Rev− HIV-1 RFP reporter virus encoding a WT PRF element. “FSE” refers to the RNA frameshift regulatory element. (C) Summary of infectious yields of PRF mutant viruses produced alone or coproduced with WT RFP reporter virus in HEK239T cells expressed at a 1:1 ratio. Pseudotyped virus particles were assayed by infecting HEK293T cells (an example is shown) prior to measuring per-well fluorescence intensity relative to the WT control. (D) Data from panel C shown in graphical format. Error bars represent standard deviations of the means derived from three independent biological replicates. (E) Predicted cellular GP/Gag ratios plotted against cumulative relative infectivity, with measurements derived from panels A and D. The dashed green line indicates that a GP/Gag ratio of 0.5 yields 90% overall (GFP plus RFP) infectivity for the −SS+WT condition, close to that of WT virus alone. In contrast, the dashed red line indicates a that a GP/Gag ratio of more than 2-fold yields less infectivity (52%) for the M1+WT condition than WT. Comparisons were derived from three independently performed biological replicates. *, P < 0.05; **, P < 0.001; ***, P < 0.0001; n.s., nonsignificant (for comparisons of M1 and −SS to the corresponding WT using Student’s two-tailed t test [A and D]).
To establish baseline levels of FS frequency and GP incorporation into virions, green fluorescent protein (GFP) reporter viruses bearing wild-type (WT), M1, and −SS frameshift sites were transfected into HEK293T cells treated with the protease inhibitor saquinavir, and pelleted virions and lysates were harvested at 48 h posttransfection. Saquinavir prevents Gag and GP proteolytic cleavage, allowing direct measurements of precursor levels using quantitative immunoblotting (Fig. 1B). FS frequency and GP incorporation frequency were measured as GP/Gag ratios relative to WT virus for cells (Fig. 1C, orange bars) or virions (Fig. 1C, blue bars), respectively. The WT FS frequency was 6.8% (±1.7; n = 3) (Fig. 1C, orange bars), similar to prior studies (4, 9, 20), while virion GP incorporation frequency was 22.5% (±2.4; n = 3) (Fig. 1C, blue bars), indicating that GP/Gag ratios were enriched 3.6-fold in WT virions relative to cells (Fig. 1C, WT; compare orange and blue bars). In comparison, M1 exhibited a markedly higher FS frequency of 22% relative to WT (±4.35, n = 3) (Fig. 1C, orange bars), which corresponded to virion GP incorporation levels of 34.5% (±3.71; n = 3) (Fig. 1C, blue bars). Accordingly, M1 virus exhibited a 3.2-fold-greater frameshift frequency than WT (Fig. 1C, compare M1 and WT orange bars), although M1 virions exhibited only a 1.6-fold enrichment of GP incorporation frequency (Fig. 1C, M1, compare orange and blue bars). As anticipated, the FS mutant −SS produced GP-deficient virus particles (Fig. 1B and C).
A “weighted Goldilocks” scenario: HIV-1 infectivity is more tolerant of subphysiological levels of GP than when GP is in excess.
To better define the relationship between cellular GP expression levels and viral infectivity, we generated WT, M1, and −SS GFP reporter viruses pseudotyped using vesicular stomatitis virus G protein (VSV-G) and used the supernatants to infect HEK293T target cells (Fig. 2A). Relative to WT, we observed significantly lower infectivity (∼5-fold compared to WT) based on reporter gene (GFP) expression for M1 (18% ± 0.88%; n = 3) (Fig. 2A), consistent with our prior study (21). We note that the M1 PRF mutations caused a small number of amino acid changes in Gag and GP (see details in reference 21 and Materials and Methods). However, these changes were in regions of the proteins (Gag p1 and GP p6*) previously shown to accommodate changes without affecting Gag or GP assembly function (21, 25–27). As expected, −SS virions, which lacked GP due to the frameshift-blocking −SS mutation, were not infectious (Fig. 2A).
We next tested the effects of modulating cellular GP/Gag ratios by coexpressing WT, M1, or −SS GFP virus genomes at a 1:1 ratio with WT PRF reporter virus expressing red fluorescent protein (RFP) instead of GFP (Fig. 2; schematic depiction of virus constructs is in Fig. 2B). We hypothesized that WT RFP virus would improve the infectivity of both M1 and −SS GFP viruses by either reducing (with M1) or increasing (with −SS) the relative cell-associated GP/Gag ratio. In this experiment, the WT RFP virus was also mutated to inactivate the viral rev gene, needed to activate unspliced and partially spliced viral RNA nuclear export, so that it would be expressed only if coexpressed in the same cell with a complementing GFP virus genome supplying Rev in trans (28). The VSV-G-pseudotyped virions resulting from these cotransfections were used to infect HEK293T cells, which were fixed at 48 h postinfection (hpi), and GFP, RFP, or both combined were measured to assess relative levels of infectivity (Fig. 2C and D).
As anticipated, coexpressing M1 GFP and WT RFP virus genomes (M1+WT) yielded higher cumulative infectivity (51.7% ± 2.8%, n = 3) than M1 alone (Fig. 2B to D), which was consistent with a cellular GP/Gag ratio of ∼2-fold higher being needed to achieve an ∼50% drop to infectivity (Fig. 2E, red dashed line). Interestingly, when infectivity yields from independent expression of the mutant M1 GFP and WT RFP viral genomes were compared to the coexpression results, M1 (GFP) infectivity was only moderately improved by coexpressing the WT viral genome (∼18% [Fig. 2A, M1] to 35.2% ± 9.86%; n = 3 [Fig. 2D, M1, green bar]), and WT (RFP) infectivity was only moderately decreased (to 68.1% ± 14.32%; n = 3 [Fig. 2D, WT, middle red bar]). We interpreted this result as indicating that while excess GP generated by M1 is deleterious to both viruses, it had a bigger net negative effect on M1 genomes potentially due to its operating in cis (addressed further in Fig. 3 and 4).
FIG 3.

Suboptimal activity of GP supplied in trans suggests a cis preference for GP incorporation into virions. (A) Schematic of GPOnly HIV-1 RFP reporter virus and altered slippery sequence that places pol in the 0 reading frame. “FSRE” refers to the RNA frameshift regulatory element. (B) Western blot analysis of Gag and GP expression confirmed that the GPOnly RFP virus generated GP and not Gag, and at higher levels than GP derived from WT transcripts. Total Gag plus GP protein expression levels were similar for WT and GPOnly transcripts. Note that GP expression alone did not produce virus particles. Hairlines indicate sites where bands from a single immunoblot were spliced together to improve data presentation, but with no further modifications. (C) Effects of GP on viral infectivity and Gag processing when coexpressed with WT, −SS, or M1 viruses. HEK293T cells were transfected with plasmids encoding WT, –SS, or M1 GFP reporter viruses (1,500 ng input plasmid) in the presence or absence of increasing amounts of GPOnly RFP reporter virus plasmid (25, 50, 100, 150, 200, and 300 ng). Viral infectivity was determined as for Fig. 2, with values shown normalized to WT virus production, for three independently performed biological replicates. Both GFP (green) and RFP (red) output is shown for each condition, with error bars representing the standard deviations of the means for both GFP and RFP infectivity, normalized to WT. Cell lysates and virions were also probed for p24Gag by Western blotting as described for Fig. 1B. Hairlines indicate sites where bands from a single immunoblot were spliced together to improve data presentation, but with no further modifications. (D) Virion p24Gag levels were quantified from the experiment reported in panel C and plotted against the corresponding total relative infectivity measurements (GFP and RFP) for each coexpression scenario. Pearson correlation coefficients were determined for each condition. (E to G) Plots depicting expected infectivity (dashed blue lines) based on expected per-transcript GP/Gag ratios (derived as shown in Table 1) and the predictive model in Fig. 2E, relative to actual observed infectivity (orange lines; measured in panel C) for WT, −SS, and M1 coexpression with GPOnly, respectively. Error bars represent the standard deviations of the means for the three biological replicates.
FIG 4.

Protease inhibitor treatment confirms cis preference and reveals a strong upper limit to trans-mediated delivery of Gag-Pol into virions. (A) HEK293T cells cultured in the presence of 10 μM saquinavir were transfected to express WT, M1, or −SS viruses (1,500 ng plasmid) in the absence or presence of increasing levels of GPOnly virus (25, 100, or 500 ng plasmid). Cell lysates and virions were harvested at 48 h and probed for Gag and GP levels by quantitative Western blotting as for Fig. 1B. All values were normalized to the WT-only condition (black dashed line). The apparent limit of trans GP incorporation is indicated by the red dashed line. Hairlines indicate sites where bands from a single immunoblot were spliced together to improve data presentation, but with no further modifications. (B) Bar graph showing the fold change in GP incorporation efficiencies calculated from GP band intensities for virions divided by GP associated with cells, from three independent biological replicates, showing greater incorporation of GP when made in cis by WT and M1 viruses. Error bars represent the standard deviations of the means. *, P < 0.05; **, P < 0.001; ***, P < 0.0001 (for comparisons to WT using Student’s two-tailed t test).
Also as expected, 1:1 coexpression of −SS GFP virus genomes with WT RFP virus genomes improved −SS virus infectivity. Remarkably, however, this rescue was nearly complete (90.2% ± 1.33%; n = 3), despite the predicted GP/Gag ratio of only 50% relative to the WT-alone control (Fig. 2C and D). When coexpressed, both the −SS (GFP) and WT (RFP) genomes exhibited relatively high levels of infectivity (83.4% ± 20.4% for −SS and 96.9% ± 18.1% for WT, respectively; n = 3) (Fig. 2C and D). This result demonstrated that GP is efficiently recruited to virions packaging −SS genomes in trans even when intracellular levels of GP are relatively low.
Using these data, we plotted predicted cellular GP/Gag ratios relative to cumulative infectivity from Fig. 2A and D in Fig. 2E, illustrating a nonlinear “weighted Goldilocks” scenario for cellular GP levels wherein “too little” GP (e.g., a predicted 50% level of GP/Gag in cells compared to WT GP/Gag levels set to 100%) (Fig. 2E, green dashed lines) is well tolerated by the virus, while “too much” GP (e.g., an ∼2-fold GP/Gag increase; Fig. 2E, red dashed lines) is more detrimental (−SS+WT compared to M1+WT conditions [Fig. 2E]).
Suboptimal effects of trans-delivered GP on infectivity suggest a cis preference for GP incorporation into virions.
We next sought to test our “weighted Goldilocks” model by more carefully controlling levels of GP expressed in cells using a Gag-Pol-only (GPOnly) RFP reporter virus, wherein we abolished frameshifting by placing the pol coding region directly into the 0 reading frame (Fig. 3A). As expected, the GPOnly virus yielded exclusive production of GP (Fig. 3B) and, interestingly, did not produce virus particles when expressed alone, potentially reflecting GP’s large size relative to Gag and its lacking the p6Gag late domain necessary to recruit components of the cellular endosomal sorting complex required for transport (ESCRT) machinery, which mediates virus particle release (Fig. 3B) (29–32).
We coexpressed the GPOnly virus at increasing levels with WT, M1, and −SS GFP viruses, with relative infectivity measured as described for Fig. 2 (Fig. 3C and D; Table 1). As expected, increasing levels of GP expressed in trans resulted in a dose-dependent decrease in WT GFP infectivity, and coexpression with the GPOnly virus markedly improved the infectivity of −SS GFP virus, which confirmed GP functionality (Fig. 3C).
TABLE 1.
Summary of predicted and observed infectivity measurements for the GP titration experiment reported in Fig. 3
| Plasmid(s) | Amt (ng) | Relative expected in-cell GP/Gag ratioa | Infectivity (% GFP) |
|
|---|---|---|---|---|
| Predictedb | Observedc | |||
| WT | 1,500 | 1.00 | 100 | 100.0 |
| WT:GPOnly | 1,500:25 | 1.22 | 92.5 | 94.6 |
| 1,500:50 | 1.44 | 83 | 85.5 | |
| 1,500:100 | 1.86 | 63 | 75.4 | |
| 1,500:150 | 2.25 | 47.7 | 67.9 | |
| 1,500:200 | 2.62 | 35.5 | 63.4 | |
| 1,500:300 | 3.29 | 16 | 43.2 | |
| −SS | 1,500 | 0.00 | 0 | 3.1 |
| −SS:GPOnly | 1,500:25 | 0.24 | 46 | 10.2 |
| 1,500:50 | 0.47 | 87 | 28.9 | |
| 1,500:100 | 0.92 | 101 | 56.4 | |
| 1,500:150 | 1.34 | 87.5 | 77.2 | |
| 1,500:200 | 1.74 | 69 | 80.8 | |
| 1,500:300 | 2.48 | 39.5 | 88.3 | |
| M1 | 1,500 | 3.24 | 18 | 20.5 |
| M1:GPOnly | 1,500:25 | 3.42 | 12 | 29.5 |
| 1,500:50 | 3.60 | 7 | 28.1 | |
| 1,500:100 | 3.95 | 0 | 33.2 | |
| 1,500:150 | 4.28 | 0 | 31.5 | |
| 1,500:200 | 4.59 | 0 | 24.3 | |
| 1,500:300 | 5.15 | 0 | 20.7 | |
Calculated based on the ratios of viral constructs expressed and per transcript frameshift frequency ratios measured from Fig. 1.
Predicted from complementation-based modeling in Fig. 2E, utilizing calculated expected GP/Gag ratios. GFP viruses (WT, −SS, or M1) were expressed from plasmids cotransfected with increasing amounts of GPOnly virus and/or empty vector control plasmid to a total DNA concentration of 1,800 ng.
Measured from Fig. 3C. All values were normalized to the WT-alone condition.
Unexpectedly, expressing increasing amounts of GPOnly virus with M1 virus did not lead to any further decrease in M1 infectivity; that remained in the range of 20 to 30% of WT even at what we estimated would be extraordinarily high (>4-fold relative to WT [Table 1]) predicted cellular GP/Gag ratios (Fig. 3C and G). Anti-p24Gag immunoblot confirmed that these effects were not attributable to the GPOnly virus affecting the efficiency of virus particle assembly, considering that virion-associated p24Gag levels were similar for M1 and WT virions across the full range of GP expression levels (Fig. 3C). Further, virion p24Gag levels correlated well only with infectivity for −SS virus coexpressed with low levels of GP, as would be expected due to GPOnly virus being able to complement the −SS virus’s lack of protease activity (Fig. 3C; correlation analysis is in Fig. 3D). Accordingly, we could explain this result only by hypothesizing that HIV-1 virion infectivity is partially buffered against the effects of trans-delivered GP, perhaps by limiting GP incorporation levels in virions. Consistent with this hypothesis, GPOnly coexpressed with WT or −SS virus in trans also exhibited less-than-expected effects on either loss (WT) or gain (−SS) of virion infectivity based on predicted GP/Gag ratios (Fig. 3C, E, and F; predicted ratios are given in Table 1).
Protease inhibitor treatment confirms a cis preference for GP incorporation into virions and reveals an upper limit to trans-delivered GP.
To more directly test for a cis preference, we measured the levels of cis- versus trans-mediated GP incorporated into virions in the absence of proteolytic cleavage, directly quantifying Gag and GP precursor levels in cells and virions under saquinavir treatment after coexpressing WT, M1, or −SS with increasing amounts of GPOnly virus (Fig. 4A).
Remarkably, GP/Gag ratios in virions for any coexpression condition never exceeded an ∼2-fold virion incorporation upper limit, suggesting a ceiling for GP incorporation where only a limited amount of trans-delivered GP can be incorporated into assembling virions before saturation (Fig. 4A, compare lane 4 to lane 1), always lower than the cis-mediated level of GP incorporation achieved by M1 even in the absence of any coexpressed GP (Fig. 4A, compare lane 5 to lane 4). Moreover, increasing GPOnly expression had little or no effect on GP levels in M1 virions (Fig. 4A, lanes 5 to 8), thus likely explaining why M1 viruses did not lose infectivity when GP was overexpressed (Fig. 3C and G). Interestingly, we noted that particularly high levels of GPOnly expression caused some trans-mediated repression of Gag synthesis in cells (e.g., in Fig. 4A, compare Pr55Gag lane 4 to lane 1 for cell lysates). However, these effects would not be anticipated to account for any differences in GP/Gag ratios in virions.
That HIV-1 exhibits a trans GP incorporation ceiling was most clearly illustrated when increasing levels of GPOnly virus were expressed with −SS virus, wherein GP levels could never achieve those observed for M1 virions, even when GP was expressed in marked excess (Fig. 4A; compare lanes 10, 11, and 12 to lane 5; the red dotted line indicates the apparent trans upper limit). Comparing virion GP levels to in-cell GP levels under cis versus trans conditions further supported the notion that GP is much more efficiently incorporated into virions when expressed in cis than in trans (Fig. 4B; in particular, compare WT and M1 without GPOnly conditions to −SS with GPOnly coexpression).
Taken together, this analysis reinforced (i) that efficient GP enrichment into virions is highly favored under cis conditions relative to trans and (ii) that HIV-1 is capable of preventing the incorporation of trans-delivered GP to virions should GP be expressed at extraordinarily high levels.
Excessive cis-mediated GP incorporation into virions causes premature Gag/GP cleavage and loss of Pol subunits RT and IN from virions.
The observation that M1 virus maintained ∼20% infectivity even in the presence of a marked excess of GP prompted us to attempt to better define the mechanistic basis for the M1 infectivity defect in the first place. We had previously shown that M1 and other PRF mutant viruses exhibiting elevated FS frequency cause changes to Gag cleavage efficiency reflected in a decrease in virion-associated Pr55/p24 ratios, consistent with excessive protease activity (21). To evaluate the impact of excess GP on M1 virion genesis over time, we compared WT and M1 virus Gag/GP virion production over 48 h using anti-p24Gag immunoblotting. We observed an ∼2-fold decrease in M1 Gag Pr55/p24 ratios relative to WT in harvested virions even at the earliest time of virion detection (24 h posttransfection), consistent with the idea that M1’s relatively high levels of GP cause premature Gag proteolytic processing (Fig. 5A, compare the quantification of Pr55/p24 at 24 h [lanes 4 and 5] and 48 h [lanes 7 and 8] in the virion blot).
FIG 5.

Excessive cis-mediated GP incorporation into virions causes premature Gag/GP cleavage and loss of Pol subunits RT and IN from virions. (A) Time course analysis of HIV-1 virions produced from HEK293T cells generating WT or M1 GFP reporter virus at 12, 24, and 48 h posttransfection. Gag cleavage patterns over the experimental time course were analyzed by Western blotting, and blots were probed using anti-p24Gag antisera. Comparison of lanes 4 to 5 and 7 to 8 illustrates the effects of the M1 PRF in increasing GP levels in cells and Gag cleavage kinetics in M1 virions relative to WT virions. (B and D) HEK293T cells generating WT, −SS, or M1 reporter virus were cultured in the absence (vehicle control) or presence of saquinavir, with virions harvested at 48 h posttransfection. GP incorporation was measured by Western blot using either anti-reverse transcriptase (RT) primary antisera (B) or anti-integrase (IN) primary antisera (D). (C and E) Relative levels of GP, RT subunits p51 and p66 (C), or IN (E) in virions for −SS and M1 relative to WT vehicle control or saquinavir treatment (n = 3). Error bars represent the standard deviations of the means. *, P < 0.05; **, P < 0.001; ***, P < 0.0001 (for comparisons of M1 and −SS to the appropriate WT control set using Student’s two-tailed t test).
To subsequently evaluate the impact of premature cleavage on the status of crucial Pol products, we also probed cells and virions to detect RT and IN at the 48-h time point using antisera specific for either protein, with virions prepared in the presence or absence of saquinavir (Fig. 5B and D). Remarkably, this experiment revealed that despite elevated (∼1.4-fold) levels of GP polyprotein incorporation (Fig. 5C and E, gray bars), M1 virions contained smaller amounts of RT and IN (∼10% less) than WT virions when generated in the presence of an active protease (Fig. 5B and D, compare lanes 3 and 1; quantified in Fig. 5C and E, blue and orange bars). This net loss of GP products (IN and RT) in M1 virions (Fig. 5B to E) might most easily be explained by a dropout of a subset of IN and RT products occurring due to premature or aberrant GP cleavage events, occurring either during or after the process of virion assembly. Taken together, these data suggested that Gag and GP cleavage events resulting from excess GP in M1 virus may account for at least some of the loss of infectivity observed for the M1 mutant (Fig. 2A).
A GP incorporation ceiling may limit defects to reverse transcription.
Considering that IN and RT were lost from M1 virions and because high levels of GP were previously shown to affect RNA dimer stability (14), we subsequently measured levels of viral RNA associated with M1 virions as well as the capacity of these virions to complete reverse transcription using quantitative real-time PCR (qRT-PCR) (Fig. 6). We observed no differences in the abundance of unspliced viral RNA in association with M1 virions compared to WT, in the presence or absence of saquinavir, indicating that M1’s infectivity defects are unlikely to reflect a defect in net viral RNA genome packaging (Fig. 6A). In contrast, assessment of RT products in infected target cells revealed significant decreases to the levels of early, late, and two-long-terminal-repeat (2LTR) circle DNA RT products, indicating that M1 virions are highly defective in the process of reverse transcription (Fig. 6B). As expected, no RT products were observed for FS mutant −SS, which did not express GP. We were surprised, however, to detect a moderate but reproducible ∼50% decrease in the efficiency of viral RNA packaging by the −SS virus (Fig. 6A, −SS alone [black and gray bars]).
FIG 6.
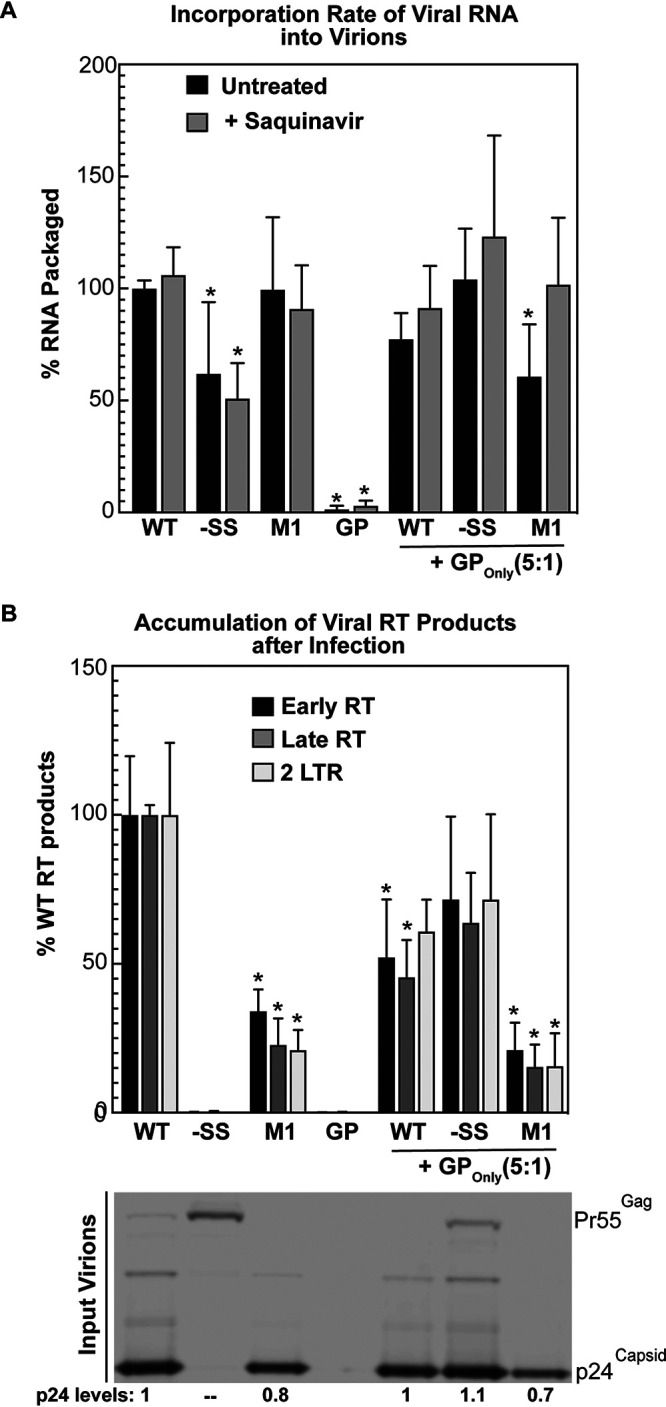
Infectivity defects observed in virions with increased GP/Gag ratios are attributed to impaired reverse transcription but not RNA packaging. (A) WT or FS mutants were expressed in the absence (untreated [black bars]; ANOVA, P < 2 × 10−15) or presence (treated [gray bars]; ANOVA, P < 8 × 10−11) of saquinavir in HEK293T cells either alone or coexpressed with GPOnly at a ratio of 5:1 (WT or FS mutant:GPOnly). Full-length unspliced RNA harvested from virions and cell lysates was extracted and quantified by qRT-PCR. RNA packaging efficiency was determined by comparison of in-virion to in-cell (supernatant/cell) RNA relative to WT. *, P < 0.01 for comparison of each condition to the appropriate WT control set using one-way ANOVA. (B) Pseudotyped virus particles harvested from cells expressing WT or FS reporter viruses either alone or coexpressed with GPOnly were assayed for production of reverse transcription products by infecting HEK293T cells. Infected cells were harvested and probed for early RT (black bars; ANOVA, P < 3 × 10−25), late RT (dark gray bars; ANOVA, P < 3 × 10−27), and 2LTR (light gray bars; ANOVA, P < 8 × 10−18) reverse transcription products by qRT-PCR. Virions were probed for p24Gag by immunoblotting to ensure infection of equivalent virions between conditions. Error bars represent the standard deviations of the means. *, P <P 0.001 for comparison of each condition to the appropriate WT control set using one-way ANOVA.
We also sought to better understand the loss of infectivity we had observed for WT virus in the presence of elevated levels of trans-delivered GP (Fig. 3), measuring RNA packaging and RT efficiency for WT, M1, or −SS viruses coexpressed with GPOnly virus at plasmid ratios of WT or FS mutants to GPOnly of 5:1 (Fig. 3C and 4A and B). Similar to M1, excess GP incorporated into virions did not affect genome packaging (Fig. 6A) but impacted the process of reverse transcription (Fig. 6B; compare WT+GP and M1+GP to WT and M1 alone). As expected, coexpressing GPOnly with −SS virus led to a nearly complete rescue to the accumulation of RT products (Fig. 6B, −SS+GP), correlating well with the rescue of infectivity that we had previously observed for −SS virus complemented with GPOnly in trans (Fig. 3C). Moreover, GPOnly coexpression eliminated the decrease in RNA packaging we had observed for −SS virus expressed alone, potentially indicating a role for GP in the genome packaging process.
Taken together, these results demonstrated that excess incorporation of GP into virions for these conditions impaired reverse transcription, either at the early RT step or potentially before early RT takes place. Accordingly, the GP incorporation ceiling illustrated in Fig. 4 may have evolved to help the virus maintain infectivity through reducing the likelihood of losing essential Pol products (e.g., RT and IN) from virions, as well as to buffer potential deleterious effects of excess GP on capsid integrity affecting reverse transcription.
DISCUSSION
Incorporation of viral enzymes is essential to the production of mature, infectious HIV-1 virions. Here, we exploited HIV-1 PRF mutants to determine how much frameshifting and GP incorporation can be tolerated by HIV-1 virions prior to losing infectivity. We demonstrate that unlike what is generally assumed, GP incorporation into virions is not an entirely stochastic (random) process; with GP being enriched in virions relative to cells (Fig. 1) and much more efficiently incorporated into virions when expressed in cis than when provided in trans (Fig. 4). Interestingly, GP expression levels and viral infectivity exhibited a nonlinear or “weighted Goldilocks” relationship, with virions retaining a high degree of infectivity at subphysiological levels of GP incorporation and losing infectivity to a greater extent when GP incorporation levels were amplified (Fig. 2). However, GP incorporation into virions never exceeded an ∼2-fold upper limit (Fig. 4), with virions retaining low level infectivity (∼20% of WT) even at remarkably elevated levels of cellular GP/Gag (Fig. 3 and 4).
Regarding mechanisms, foundational studies by Shehu-Xhilaga et al., among others, previously demonstrated that GP expressed either in cis or in trans is readily incorporated into virions (14, 17, 33, 34) and that this process is mediated through interactions between Gag and GP CA domains (35–38) and may be further facilitated by the NC-bound RNA scaffold (17, 23, 24). We built on these studies by expressing GPOnly in trans and at variable levels in the context of WT and FS mutant extremes; allowing us to more finely tease apart the efficiency of GP incorporation at increasing levels in cis (from M1) or in trans (from GPOnly). Our results suggest that nonstochastic mechanisms both (i) promote GP enrichment into virions at subphysiological GP levels and (ii) limit GP incorporation into virions when GP levels are in marked excess. A potential implication of these GP “buffering” mechanisms is that drugs designed to either downregulate or upregulate FS frequency (10, 22) may have difficulty completely abrogating viral replication in the context of a monotherapy, as demonstrated by the M1 virus, which retains low-level (∼20%) infectivity despite the virions carrying what may be saturated levels of GP (Fig. 4A).
We also provide direct evidence for a cis preference of GP incorporation into virions by showing that while trans-delivered GP is incorporated into virions and can rescue the infectivity of −SS viruses (Fig. 3C and F), its efficiency of incorporation is weak relative to that of GP made in cis (as demonstrated for the hyperframeshifting mutant M1 [Fig. 3 and 4]). We propose that this strong cis preference reflects GP and Gag undergoing more favorable local interactions in the cytoplasm soon or immediately after being synthesized from a common pool of gag-pol mRNAs (Fig. 1 and 7A). It is established that Gag (and presumably GP) form low-order multimers (dimers and trimers) in the cytosol prior to membrane binding (39, 40). Previously, a “stepwise” model was proposed wherein Gag-RNA preassembly complexes form in the cytosol and grow progressively in molecular mass over time (41–44). Alternatively, a more recent study by Deng et al. indicated that most of the Gag in the cytosol is associated with ribosomal complexes and not committed to formation of assembly intermediates until after membrane binding (45). Regardless, our results provide strength to the argument that translation and virus particle assembly are coupled, semicompartmentalized processes.
FIG 7.

Summary of findings. (A) Compartmentalized cis Gag-GP interactions promote efficient cis delivery of GP to virions. PRF element mutant M1 exhibits ∼3-fold-greater frameshifting than WT virus, resulting in levels of GP incorporation efficiency that can never be achieved by GP expressed in trans (i.e., from a separate gag-pol mRNA). (B) Our data also reinforce that GP is enriched in virions relative to cell lysates and show that HIV-1 will accommodate only an ∼2-fold increase in GP incorporation. Accordingly, the virus has evolved a mechanism to restrict excessive incorporation of GP into virions, likely providing it with a means to buffer per-virion infectivity. (C) M1 particles are less infectious, at least in part because overframeshifting results in premature Gag and GP proteolytic processing in budding virions and impairs the process of reverse transcription.
As to where in the cell these interactions are first initiated, two recent studies utilizing the SunTag labeling technique on the Gag polyprotein (46, 47) showed that translating HIV-1 gag-pol mRNAs exhibit dynamic movements and can be found throughout the cell cytoplasm, with Lyon et al. directly resolving bursts of nonlocalized translational frameshifting (46). Our group and others have similarly shown in multiple studies that translationally competent gag-pol mRNAs fill the cytoplasm long prior to the onset of virion assembly (47–51). We also recently demonstrated that artificially tethering gag-pol mRNAs to nonnatural sites in the cell (e.g., vesicles or the actin cytoskeleton) relocalizes Gag and biases virus particle assembly to occur at these nonnatural sites (52). Thus, we propose that Gag-GP interactions are initiated throughout the cytosol in association with translating gag-pol mRNAs, but with a bias toward sites of assembly triggered by Gag-membrane interactions initiated at the latest stages of replication. Further high-resolution imaging studies performed over the course of infection will be required to generate a more complete understanding of Gag and GP coordinate trafficking.
Conversely, we observed HIV-1 virions to exhibit a strong upper limit to excess GP incorporation when GP was delivered in trans, with WT particles unable to incorporate more than ∼2-fold the wild-type level of GP and no additional GP tolerated by M1 virions that we speculate are GP saturated (Fig. 4). We thus hypothesize that HIV-1, and possibly other retroviruses, evolved to buffer GP incorporation levels through an evolutionarily conserved −1 PRF translational control coupled to compartmentalized GP-Gag interactions (see model depiction in Fig. 7A and B). While the mechanistic bases for the GP incorporation ceiling and GP saturation are still unclear, a prior study by Haraguchi et al. demonstrated that full-length GP is inefficiently trafficked to membranes, a defect remedied, at least in part, by truncation of the C-terminal domain of Pol to include only PR (53). Elevated GP expression was also shown in earlier studies to promote GP-GP homodimerization, causing premature protease activation in the cytoplasm (16, 54). GP’s lack of a p6 late domain needed for virion membrane abscission was suggested to affect the efficiency of virus particle release (29, 35, 55) and protease activation (17, 56) when GP is overexpressed. While neither defect (GP autoprocessing or GP-linked restriction of particle release) was overtly detected in our coexpression system (Fig. 3 and 5), M1 virion cleavage profiles (Fig. 5) did suggest that at least part of the loss of M1 infectivity was due to premature processing of Gag and GP during budding, consistent with a need for tight control of GP incorporation levels (57, 58). These results further support prior data from Bendjennat and Saffarian, wherein impaired or slowed budding of viral particles led to premature GP activation and loss of GP cleavage products from bud sites (58).
In our hands, M1’s increased levels of GP did not negatively impact CA levels but significantly reduced levels of virion-associated IN and RT (Fig. 5). It seems unlikely that these relatively mild changes to RT and IN (∼10% decrease relative to WT) account for the entirety of infectivity loss observed for mutant M1 (∼80% decrease relative to WT), with the rest potentially being due to other mechanisms such as genome dimer destabilization as previously put forth by Shehu-Xhilaga et al. (14). Indeed, we observed that excess GP in M1 virus did not affect RNA packaging but significantly impaired reverse transcription (Fig. 6). We thus propose that in the context of native infection, HIV-1 is adapted to prevent excess incorporation of trans-delivered GP into single assembling virions using its ceiling mechanism, thereby reducing the odds of losing core virion components (see model depiction in Fig. 7C) and endangering reverse transcription fidelity. Additional efforts to resolve and localize Gag, GP, and other components of M1 virions may further shed light on exactly how excess GP is incorporated into virions and causes virion defects.
MATERIALS AND METHODS
Plasmids and cell culture.
Human embryonic kidney 293T (HEK293T) cells and human HeLa cervical cancer cells were obtained from the American Type Culture Collection (ATCC, Manassas, VA, USA). Cells were cultured in Dulbecco’s modified Eagle medium supplemented with 10% fetal bovine serum, 1% penicillin-streptomycin, and 1% l-glutamine. All cells were maintained at 37°C, 50% humidity, and 5% CO2. Full-length HIV-1 proviral plasmids were derived from a pNL4-3 molecular clone wherein both the env and vpr reading frames contained inactivating point mutations and further modified to express a GFP reporter in place of nef (59). WT or PRF element mutant (−SS and M1) versions of the reporter virus were generated as previously described (21). Mutations in −SS did not affect the Gag coding region. M1 encoded three nonsynonymous mutations within the p1 spacer region of Gag (I438M, W439G, and G447R) and two nonsynonymous mutations within the transframe p6* peptide of Gag-Pol (L6G and R14P). These mutations did not affect Gag’s or GP’s ability to assemble virus particles (Fig. 1B and D) (21), consistent with previous reports showing Gag and GP to accommodate changes to the coding sequence in these regions (25–27). pBluescript was used as an empty vector control, and pVSV-G was used for pseudotyping virions for infectivity assays. A plasmid encoding full-length Gag-Pol (with pol moved into the 0 reading frame), designated the GP-only (GPOnly) RFP reporter virus, was generated using primers designed for site-directed mutagenesis and overlapping PCR to incorporate mutations into pNL4-3 backbone using SpeI and AgeI restriction sites (Fig. 3A). The Rev− HIV-1 RFP reporter virus (Fig. 2B) was created as previously described (60). For viruses used for coexpression assays described for Fig. 2 and 3, the GFP reporter in the nef gene was replaced with a red fluorescent protein (mCherry, but referred to as RFP here) using NotI and XhoI restriction enzyme cut sites. All plasmids were verified using restriction digestion (New England Biosciences, Ipswich, MA, USA) and DNA sequencing (Functional Biosciences, Inc., Madison, WI, USA).
Retroviral assembly and infectivity assays.
HEK293T cells were plated as a monolayer in 6-well tissue culture dishes (Genesee Scientific, San Diego, CA, USA) prior to transfection. For each well, 2 μg of plasmids encoding either WT or PRF element mutant versions of HIV-1 reporter virus were transfected into cells using polyethylenimine (PEI; Polysciences Inc., Warrington, PA, USA) and Gibco Opti-MEM (Thermo Fisher Scientific, Waltham, MA, USA). Twenty-four hours posttransfection, each well was supplemented with fresh medium. For experiments in which total uncleaved Gag and Gag-Pol levels were to be measured, at 24 h posttransfection, medium was replaced with fresh medium supplemented with either a 10 μM concentration of saquinavir, an HIV protease inhibitor (61), or dimethyl sulfoxide (DMSO) (vehicle control). For GP incorporation assembly assays, 1.5 μg of WT or frameshift mutant versions of NL4-3 were cotransfected with increasing levels of GPOnly or pBluescript, totaling 0.5 μg of DNA. For experiments in which harvested virions were used for single-cycle infectivity assays, WT or frameshift site mutant assembly assays were cotransfected with 1:10 mass per transfection pVSV-G plasmid DNA. HeLa or HEK293T cells were plated into a subconfluent monolayer in 24-well glass-bottom dishes (Eppendorf, Hamburg, Germany). Prior to infection, medium in each well was replaced with fresh medium supplemented with 2 mg/ml of Polybrene to enhance viral titer. Three hundred fifty microliters of viral supernatants produced from the above assembly assays was added to each well. Twenty-four hours postinfection (hpi), medium within each well was replaced with fresh medium. Infectivity assays were carried out until 48 hpi. Due to each PRF mutant or combination exhibiting variability in Gag cleavage efficiency and GP incorporation levels, equal volumes of viral supernatants were used in each infectivity assay, with virion levels being monitored by anti-p24Gag immunoblotting (for example, see Fig. 3C and D and 6B).
Immunofluorescence and immunoblotting.
For all infectivity assays, at 48 hpi, cells were washed with phosphate-buffered saline (PBS) and fixed with 4% paraformaldehyde in PBS for 15 min, and nuclei were stained with DAPI (4′,6-diamidino-2-phenylindole) for 5 min. After fixation and staining, all samples were maintained in PBS prior to imaging. For all immunoblotting experiments, cells and supernatants were harvested 48 h posttransfection, except for time course analyses, where cells and viral supernatants were harvested at 6, 12, 24, and 48 h posttransfection. Harvested cells were lysed in radioimmunoprecipitation assay (RIPA) buffer (10 mM Tris-HCl [pH 7.5], 150 mM NaCl, 1 mM EDTA, 0.1% SDS, 1% Triton X-100, 1% sodium deoxycholate) and passed through a 26.5-gauge needle. Total protein concentrations from cell lysates were measured using a DC protein assay (Bio-Rad, Hercules, CA, USA), and equal amounts of proteins, per condition, were added to a 1:1 ratio of 2× dissociation buffer (DB) (62.5 mM Tris-HCl [pH 6.8], 10% glycerol, 2% sodium dodecyl sulfate [SDS], 10% β-mercaptoethanol) and boiled prior to SDS-PAGE. Supernatants were passed through a 0.45-μm filter, and virions were centrifuged for 2 h at 21,130 × g and pelleted through a 20% (wt/vol) sucrose cushion. Pelleted virions were subsequently lysed in RIPA buffer and mixed with DB, and equal volumes of pelleted virion lysates were analyzed for each condition.
SDS-PAGE was performed on both lysate and virion samples, where up to 15 μl of samples was loaded into 10% polyacrylamide gels. Each gel was run at 120 V for 70 min and incubated for 30 min in transfer buffer prior to transfer of proteins onto a nitrocellulose membrane (0.2-μm pore size). Each membrane was blocked for 1 h in 1.5% milk in PBS and 0.1% Tween (PBS-T) and incubated in the following primary antibodies: 1:500 dilution of the monoclonal anti-p24Gag antibody (produced from hybridoma 183-H12-5C) obtained from the NIH AIDS Reagent Program, Division of AIDS, NIAID, NIH (Bethesda, MD, USA; from Bruce Chesebro) (62), a 1:2,000 dilution of polyclonal anti-reverse transcriptase (RT) antibody (Ab63911; Abcam, Cambridge, UK), a 1:500 dilution of monoclonal anti-integrase (IN) antibody (2C11) (no. 7374) obtained from the NIH AIDS Reagent Program, Division of AIDS, NIAID, NIH (Bethesda, MD, USA; from Dag E. Helland) (63), and a 1:3,000 dilution of anti-heat shock protein 90 (HSP90) antibody (H-114 [catalog no. sc-7947]; Santa Cruz Biotechnology, Dallas, TX, USA). Following incubation in primary antibodies, membranes were washed with PBS-T, incubated for 2 h with a 1:10,000 dilution of anti-mouse or anti-rabbit immunoglobulin secondary antibodies conjugated to the infrared fluorophore IRDye680 or IRDye800 (LiCor Biosciences, Lincoln, NE, USA), washed with PBS-T, and imaged using a LiCor Fx Odyssey. All immunofluorescence and immunoblotting experiments were performed using samples from three independent biological replicates, defined as separate sets of samples generated on different days.
Microscopy and image analysis.
Imaging experiments were performed using a Nikon Ti-Eclipse inverted wide-field microscope (Nikon Corp., Minato, Tokyo, Japan) and a 20× Plan Apo lens objective (NA, 0.75) as previously described (60). Image acquisition for each condition was performed using an Orca-Flash4.0 C11440 digital complementary metal oxide semiconductor (CMOS) camera (Hamamatsu Photonics, Skokie, IL, USA) and Nikon NIS Elements software (v4.13.04). Each image was collected using differential interference contrast (DIC), and the following fluorescence excitation/emission (nanometer ranges) filter sets: 325 to 375/435 to 485 (blue fluorescent protein [BFP]), 490 to 510/520 to 550 (yellow fluorescent protein [YFP]), and 565 to 590/590 to 650 (mCherry). For each condition, four fields of view (FOV) were acquired with a 20× Plan Apo lens objective and 50% light intensity. All images were processed and analyzed using Fiji/ImageJ2 software (64). Each FOV was processed in Fiji as follows: background was subtracted using a rolling-ball radius of 50 pixels prior to measurement of integrated density (area × mean gray value) or raw integrated density (the sum of the values of all pixels within an image) for each channel (mCherry, GFP, and BFP) imaged. Per condition, cells within each FOV were used to calculate the relative fluorescence units (RFU) through measurement of the average integrated density or average raw integrated density of mCherry-positive cells or GFP-positive cells (infected cells) normalized to total number of cells (DAPI − nucleus stain).
Analysis of viral RNA packaging.
Cells (1 × 106) were transfected with PEI as described above for retrovirus assembly assays. At 24 h posttransfection, 150 μl of medium was removed and treated with 1 ml of Benzonase (Sigma, St. Louis, MO) for 30 min at 37°C to remove DNA. RNA was harvested using the Direct-zol Plus RNA miniprep kit (Zymo Research), following the manufacturer’s instructions. Similarly, RNA was isolated from the transfected cells using the Direct-zol Plus RNA miniprep kit (Zymo Research). Viral RNA was analyzed from the supernatant and cells by quantitative real-time reverse transcription-PCR using the iTAQ universal probe one-step kit (Bio-Rad) with a primer set designed to pick up viral transcripts and not proviral DNA (65): universal transcript using the forward primer P9501 (CAGATGCTGCATATAAGCAGCTG), the universal reverse primer 10T20 (TTTTTTTTTTTTTTTTTTTTTGAAGCACTC), and a universal transcript probe (CCTGTACTGGGTCTCTCTGG). A standard curve was derived using a gene block containing the amplicon to determine the absolute number of molecules present. The ratio of RNA in virions versus cellular transcripts was calculated and reported as percentage of transcripts produced from transfections with a WT HIV-1 genome. For comparisons with three or more groups, a one-way analysis of variance (ANOVA) was performed using Microsoft Excel. If this determined that a statistically significant difference existed within the group (P < 0.05), a further post hoc test was done using Tukey’s honestly significant difference test (HSD) in Excel (66, 67). If only two groups were compared, Student’s t test in Excel was used. ANOVA results and relevant Tukey’s HSD and t test comparisons are shown in the figures, unless P was >0.05 for the ANOVA, in which case only the ANOVA results are shown.
Analysis of reverse transcription products.
Cells (1 × 106) were infected with VSV-G pseudotyped VLPs. At 24 h postinfection, total DNA was harvested using the DNeasy kit from Qiagen. Quantitative real-time PCR was performed using the SsoFast probe kit (Bio-Rad). For early RT products, we used primers as previously described (68): ert2f (GTGCCCGTCTGTTGTGTGAC), ert2r (GGCGCCACTGCTAGAGATTT), and ert2 probe (CTAGAGATCCCTCAGACCCTTTTAGTCAGTGTGG). For late RT and 2LTR, we used primers that were described previously (69) but modified as necessary for analyzing the NL4-3 strain as follows: HIV-1 late forward MH531 (TGTGTGCCCGTCTGTTGTGT), rev MH532v (GAGTCCTGCGTCGAGAGATC), late RT probe (CAGTGGCGCCCGAACAGGGA), HIV-1 2LTR Fwd primer (AACTAGGGAACCCACTGCTTAAG), HIV-1 2LTR Rev primer (TCCACAGATCAAGGATATCTTGTC), and HIV-1 2LTR probe (ACACTACTTTGAGCACTCAAGGCAAGCTTT). A standard curve was derived using a gene block (Integrated DNA Technologies) containing the amplicon to determine the absolute number of molecules present, and total reverse transcription products in each sample were calculated and compared to infection with WT HIV-1 genomes. p24Gag Western blots were used to ensure infection with equivalent numbers of virions. Statistical analyses were performed as described above for analysis of viral RNA packaging.
ACKNOWLEDGMENTS
The following reagents were obtained through the NIH AIDS Reagent Program, Division of AIDS, NIAID, NIH: HIV-1 p24 hybridoma (183-H12-5C) from Bruce Chesebro (62), HIV-1 IN monoclonal antibody (2C11) from Dag E. Helland (catalog no. 7374) (63), and saquinavir.
This research was supported by NIH grants R01AI110221 (N.M.S.), P01CA022332 (N.M.S., P.A.), R35GM118131 (S.E.B.), and T32GM008349 (B.E.B.); NSF graduate research fellowships DGE1747503 (B.E.B.) and DGE-1256269 (J.T.B.); a Shaw Scientist Award from the Greater Milwaukee Foundation (N.M.S.); and student fellowships from the University of Wisconsin-Madison (J.R.K.) and Wisconsin Alumni Research Foundation (J.T.B.).
Contributor Information
Nathan M. Sherer, Email: nsherer@wisc.edu.
Viviana Simon, Icahn School of Medicine at Mount Sinai.
REFERENCES
- 1.Watts JM, Dang KK, Gorelick RJ, Leonard CW, Bess JW, Jr, Swanstrom R, Burch CL, Weeks KM. 2009. Architecture and secondary structure of an entire HIV-1 RNA genome. Nature 460:711–716. 10.1038/nature08237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.D'Souza V, Summers MF. 2005. How retroviruses select their genomes. Nat Rev Microbiol 3:643–655. 10.1038/nrmicro1210. [DOI] [PubMed] [Google Scholar]
- 3.Kuzembayeva M, Dilley K, Sardo L, Hu WS. 2014. Life of psi: how full-length HIV-1 RNAs become packaged genomes in the viral particles. Virology 454-455:362–370. 10.1016/j.virol.2014.01.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Jacks T, Power MD, Masiarz FR, Luciw PA, Barr PJ, Varmus HE. 1988. Characterization of ribosomal frameshifting in HIV-1 gag-pol expression. Nature 331:280–283. 10.1038/331280a0. [DOI] [PubMed] [Google Scholar]
- 5.Parkin NT, Chamorro M, Varmus HE. 1992. Human immunodeficiency virus type 1 gag-pol frameshifting is dependent on downstream mRNA secondary structure: demonstration by expression in vivo. J Virol 66:5147–5151. 10.1128/JVI.66.8.5147-5151.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Baril M, Dulude D, Gendron K, Lemay G, Brakier-Gingras L. 2003. Efficiency of a programmed -1 ribosomal frameshift in the different subtypes of the human immunodeficiency virus type 1 group M. RNA 9:1246–1253. 10.1261/rna.5113603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Staple DW, Butcher SE. 2003. Solution structure of the HIV-1 frameshift inducing stem-loop RNA. Nucleic Acids Res 31:4326–4331. 10.1093/nar/gkg654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Staple DW, Butcher SE. 2005. Solution structure and thermodynamic investigation of the HIV-1 frameshift inducing element. J Mol Biol 349:1011–1023. 10.1016/j.jmb.2005.03.038. [DOI] [PubMed] [Google Scholar]
- 9.Brierley I, Dos Ramos FJ. 2006. Programmed ribosomal frameshifting in HIV-1 and the SARS-CoV. Virus Res 119:29–42. 10.1016/j.virusres.2005.10.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Brakier-Gingras L, Charbonneau J, Butcher SE. 2012. Targeting frameshifting in the human immunodeficiency virus. Expert Opin Ther Targets 16:249–258. 10.1517/14728222.2012.665879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Freed EO. 2015. HIV-1 assembly, release and maturation. Nat Rev Microbiol 13:484–496. 10.1038/nrmicro3490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Sundquist WI, Krausslich HG. 2012. HIV-1 assembly, budding, and maturation. Cold Spring Harb Perspect Med 2:a006924. 10.1101/cshperspect.a006924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Frankel AD, Young JA. 1998. HIV-1: fifteen proteins and an RNA. Annu Rev Biochem 67:1–25. 10.1146/annurev.biochem.67.1.1. [DOI] [PubMed] [Google Scholar]
- 14.Shehu-Xhilaga M, Crowe SM, Mak J. 2001. Maintenance of the Gag/Gag-Pol ratio is important for human immunodeficiency virus type 1 RNA dimerization and viral infectivity. J Virol 75:1834–1841. 10.1128/JVI.75.4.1834-1841.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Gan X, Gould SJ. 2012. HIV Pol inhibits HIV budding and mediates the severe budding defect of Gag-Pol. PLoS One 7:e29421. 10.1371/journal.pone.0029421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Haraguchi H, Sudo S, Noda T, Momose F, Kawaoka Y, Morikawa Y. 2010. Intracellular localization of human immunodeficiency virus type 1 Gag and GagPol products and virus particle release: relationship with the Gag-to-GagPol ratio. Microbiol Immunol 54:734–746. 10.1111/j.1348-0421.2010.00276.x. [DOI] [PubMed] [Google Scholar]
- 17.Yu FH, Huang KJ, Wang CT. 2020. HIV-1 mutant assembly, processing and infectivity expresses Pol independent of Gag. Viruses 12:54. 10.3390/v12010054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hill M, Tachedjian G, Mak J. 2005. The packaging and maturation of the HIV-1 Pol proteins. Curr HIV Res 3:73–85. 10.2174/1570162052772942. [DOI] [PubMed] [Google Scholar]
- 19.Mouzakis KD, Lang AL, Vander Meulen KA, Easterday PD, Butcher SE. 2013. HIV-1 frameshift efficiency is primarily determined by the stability of base pairs positioned at the mRNA entrance channel of the ribosome. Nucleic Acids Res 41:1901–1913. 10.1093/nar/gks1254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Korniy N, Goyal A, Hoffmann M, Samatova E, Peske F, Pohlmann S, Rodnina MV. 2019. Modulation of HIV-1 Gag/Gag-Pol frameshifting by tRNA abundance. Nucleic Acids Res 47:5210–5222. 10.1093/nar/gkz202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Garcia-Miranda P, Becker JT, Benner BE, Blume A, Sherer NM, Butcher SE. 2016. Stability of HIV frameshift site RNA correlates with frameshift efficiency and decreased virus infectivity. J Virol 90:6906–6917. 10.1128/JVI.00149-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Hilimire TA, Bennett RP, Stewart RA, Garcia-Miranda P, Blume A, Becker J, Sherer N, Helms ED, Butcher SE, Smith HC, Miller BL. 2016. N-Methylation as a strategy for enhancing the affinity and selectivity of RNA-binding peptides: application to the HIV-1 frameshift-stimulating RNA. ACS Chem Biol 11:88–94. 10.1021/acschembio.5b00682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Khorchid A, Halwani R, Wainberg MA, Kleiman L. 2002. Role of RNA in facilitating Gag/Gag-Pol interaction. J Virol 76:4131–4137. 10.1128/jvi.76.8.4131-4137.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Huang Y, Khorchid A, Wang J, Parniak MA, Darlix JL, Wainberg MA, Kleiman L. 1997. Effect of mutations in the nucleocapsid protein (NCp7) upon Pr160(gag-pol) and tRNA(Lys) incorporation into human immunodeficiency virus type 1. J Virol 71:4378–4384. 10.1128/JVI.71.6.4378-4384.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Hill MK, Shehu-Xhilaga M, Crowe SM, Mak J. 2002. Proline residues within spacer peptide p1 are important for human immunodeficiency virus type 1 infectivity, protein processing, and genomic RNA dimer stability. J Virol 76:11245–11253. 10.1128/jvi.76.22.11245-11253.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.de Marco A, Heuser AM, Glass B, Krausslich HG, Muller B, Briggs JA. 2012. Role of the SP2 domain and its proteolytic cleavage in HIV-1 structural maturation and infectivity. J Virol 86:13708–13716. 10.1128/JVI.01704-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Leiherer A, Ludwig C, Wagner R. 2009. Uncoupling human immunodeficiency virus type 1 Gag and Pol reading frames: role of the transframe protein p6* in viral replication. J Virol 83:7210–7220. 10.1128/JVI.02603-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Henderson BR, Percipalle P. 1997. Interactions between HIV Rev and nuclear import and export factors: the Rev nuclear localisation signal mediates specific binding to human importin-beta. J Mol Biol 274:693–707. 10.1006/jmbi.1997.1420. [DOI] [PubMed] [Google Scholar]
- 29.Mergener K, Facke M, Welker R, Brinkmann V, Gelderblom HR, Krausslich HG. 1992. Analysis of HIV particle formation using transient expression of subviral constructs in mammalian cells. Virology 186:25–39. 10.1016/0042-6822(92)90058-w. [DOI] [PubMed] [Google Scholar]
- 30.Garrus JE, von Schwedler UK, Pornillos OW, Morham SG, Zavitz KH, Wang HE, Wettstein DA, Stray KM, Cote M, Rich RL, Myszka DG, Sundquist WI. 2001. Tsg101 and the vacuolar protein sorting pathway are essential for HIV-1 budding. Cell 107:55–65. 10.1016/s0092-8674(01)00506-2. [DOI] [PubMed] [Google Scholar]
- 31.Demirov DG, Ono A, Orenstein JM, Freed EO. 2002. Overexpression of the N-terminal domain of TSG101 inhibits HIV-1 budding by blocking late domain function. Proc Natl Acad Sci USA 99:955–960. 10.1073/pnas.032511899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Demirov DG, Orenstein JM, Freed EO. 2002. The late domain of human immunodeficiency virus type 1 p6 promotes virus release in a cell type-dependent manner. J Virol 76:105–117. 10.1128/jvi.76.1.105-117.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Hill MK, Hooker CW, Harrich D, Crowe SM, Mak J. 2001. Gag-Pol supplied in trans is efficiently packaged and supports viral function in human immunodeficiency virus type 1. J Virol 75:6835–6840. 10.1128/JVI.75.15.6835-6840.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Shehu-Xhilaga M, Lee JY, Campbell S, Marshall JA, Crowe SM, Mak J. 2002. Overexpression and incorporation of GagPol precursor does not impede packaging of HIV-1 tRNA(Lys3) but promotes intracellular budding of virus-like particles. J Biomed Sci 9:697–705. 10.1007/BF02254998. [DOI] [PubMed] [Google Scholar]
- 35.Smith AJ, Srinivasakumar N, Hammarskjold ML, Rekosh D. 1993. Requirements for incorporation of Pr160gag-pol from human immunodeficiency virus type 1 into virus-like particles. J Virol 67:2266–2275. 10.1128/JVI.67.4.2266-2275.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Huang M, Martin MA. 1997. Incorporation of Pr160(gag-pol) into virus particles requires the presence of both the major homology region and adjacent C-terminal capsid sequences within the Gag-Pol polyprotein. J Virol 71:4472–4478. 10.1128/JVI.71.6.4472-4478.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Wills JW, Craven RC. 1991. Form, function, and use of retroviral gag proteins. AIDS 5:639–654. 10.1097/00002030-199106000-00002. [DOI] [PubMed] [Google Scholar]
- 38.Takagi S, Momose F, Morikawa Y. 2017. FRET analysis of HIV-1 Gag and GagPol interactions. FEBS Open Bio 7:1815–1825. 10.1002/2211-5463.12328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Kutluay SB, Bieniasz PD. 2010. Analysis of the initiating events in HIV-1 particle assembly and genome packaging. PLoS Pathog 6:e1001200. 10.1371/journal.ppat.1001200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Hendrix J, Baumgartel V, Schrimpf W, Ivanchenko S, Digman MA, Gratton E, Krausslich HG, Muller B, Lamb DC. 2015. Live-cell observation of cytosolic HIV-1 assembly onset reveals RNA-interacting Gag oligomers. J Cell Biol 210:629–646. 10.1083/jcb.201504006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Barajas BC, Tanaka M, Robinson BA, Phuong DJ, Chutiraka K, Reed JC, Lingappa JR. 2018. Identifying the assembly intermediate in which Gag first associates with unspliced HIV-1 RNA suggests a novel model for HIV-1 RNA packaging. PLoS Pathog 14:e1006977. 10.1371/journal.ppat.1006977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Reed JC, Molter B, Geary CD, McNevin J, McElrath J, Giri S, Klein KC, Lingappa JR. 2012. HIV-1 Gag co-opts a cellular complex containing DDX6, a helicase that facilitates capsid assembly. J Cell Biol 198:439–456. 10.1083/jcb.201111012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Lingappa JR, Reed JC, Tanaka M, Chutiraka K, Robinson BA. 2014. How HIV-1 Gag assembles in cells: putting together pieces of the puzzle. Virus Res 193:89–107. 10.1016/j.virusres.2014.07.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Dooher JE, Schneider BL, Reed JC, Lingappa JR. 2007. Host ABCE1 is at plasma membrane HIV assembly sites and its dissociation from Gag is linked to subsequent events of virus production. Traffic 8:195–211. 10.1111/j.1600-0854.2006.00524.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Deng Y, Hammond JA, Pauszek R, Ozog S, Chai I, Rabuck-Gibbons J, Lamichhane R, Henderson SC, Millar DP, Torbett BE, Williamson JR. 2021. Discrimination between functional and non-functional cellular Gag complexes involved in HIV-1 assembly. J Mol Biol 433:166842. 10.1016/j.jmb.2021.166842. [DOI] [PubMed] [Google Scholar]
- 46.Lyon K, Aguilera LU, Morisaki T, Munsky B, Stasevich TJ. 2019. Live-cell single RNA imaging reveals bursts of translational frameshifting. Mol Cell 75:172–183.E9. 10.1016/j.molcel.2019.05.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Chen J, Liu Y, Wu B, Nikolaitchik OA, Mohan PR, Chen J, Pathak VK, Hu WS. 2020. Visualizing the translation and packaging of HIV-1 full-length RNA. Proc Natl Acad Sci USA 117:6145–6155. 10.1073/pnas.1917590117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Pocock GM, Zimdars LL, Yuan M, Eliceiri KW, Ahlquist P, Sherer NM. 2017. Diverse activities of viral cis-acting RNA regulatory elements revealed using multicolor, long-term, single-cell imaging. Mol Biol Cell 28:476–487. 10.1091/mbc.E16-08-0612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Pocock GM, Becker JT, Swanson CM, Ahlquist P, Sherer NM. 2016. HIV-1 and M-PMV RNA nuclear export elements program viral genomes for distinct cytoplasmic trafficking behaviors. PLoS Pathog 12:e1005565. 10.1371/journal.ppat.1005565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Behrens RT, Aligeti M, Pocock GM, Higgins CA, Sherer NM. 2017. Nuclear export signal masking regulates HIV-1 Rev trafficking and viral RNA nuclear export. J Virol 91:e02107-16. 10.1128/JVI.02107-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Jouvenet N, Simon SM, Bieniasz PD. 2009. Imaging the interaction of HIV-1 genomes and Gag during assembly of individual viral particles. Proc Natl Acad Sci USA 106:19114–19119. 10.1073/pnas.0907364106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Becker JT, Sherer NM. 2017. Subcellular localization of HIV-1 gag-pol mRNAs regulates sites of virion assembly. J Virol 91:e02315-16. 10.1128/JVI.02315-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Haraguchi H, Noda T, Kawaoka Y, Morikawa Y. 2012. A large extension to HIV-1 Gag, like Pol, has negative impacts on virion assembly. PLoS One 7:e47828. 10.1371/journal.pone.0047828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Karacostas V, Wolffe EJ, Nagashima K, Gonda MA, Moss B. 1993. Overexpression of the HIV-1 gag-pol polyprotein results in intracellular activation of HIV-1 protease and inhibition of assembly and budding of virus-like particles. Virology 193:661–671. 10.1006/viro.1993.1174. [DOI] [PubMed] [Google Scholar]
- 55.Park J, Morrow CD. 1992. The nonmyristylated Pr160gag-pol polyprotein of human immunodeficiency virus type 1 interacts with Pr55gag and is incorporated into viruslike particles. J Virol 66:6304–6313. 10.1128/JVI.66.11.6304-6313.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Yu FH, Chou TA, Liao WH, Huang KJ, Wang CT. 2015. Gag-Pol transframe domain p6* is essential for HIV-1 protease-mediated virus maturation. PLoS One 10:e0127974. 10.1371/journal.pone.0127974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Krausslich HG. 1991. Human immunodeficiency virus proteinase dimer as component of the viral polyprotein prevents particle assembly and viral infectivity. Proc Natl Acad Sci USA 88:3213–3217. 10.1073/pnas.88.8.3213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Bendjennat M, Saffarian S. 2016. The race against protease activation defines the role of ESCRTs in HIV budding. PLoS Pathog 12:e1005657. 10.1371/journal.ppat.1005657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Adachi A, Gendelman HE, Koenig S, Folks T, Willey R, Rabson A, Martin MA. 1986. Production of acquired immunodeficiency syndrome-associated retrovirus in human and nonhuman cells transfected with an infectious molecular clone. J Virol 59:284–291. 10.1128/JVI.59.2.284-291.1986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Aligeti M, Behrens RT, Pocock GM, Schindelin J, Dietz C, Eliceiri KW, Swanson CM, Malim MH, Ahlquist P, Sherer NM. 2014. Cooperativity among Rev-associated nuclear export signals regulates HIV-1 gene expression and is a determinant of virus species tropism. J Virol 88:14207–14221. 10.1128/JVI.01897-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Vella S, Floridia M. 1998. Saquinavir. Clinical pharmacology and efficacy. Clin Pharmacokinet 34:189–201. 10.2165/00003088-199834030-00002. [DOI] [PubMed] [Google Scholar]
- 62.Chesebro B, Wehrly K, Nishio J, Perryman S. 1992. Macrophage-tropic human immunodeficiency virus isolates from different patients exhibit unusual V3 envelope sequence homogeneity in comparison with T-cell-tropic isolates: definition of critical amino acids involved in cell tropism. J Virol 66:6547–6554. 10.1128/JVI.66.11.6547-6554.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Nilsen BM, Haugan IR, Berg K, Olsen L, Brown PO, Helland DE. 1996. Monoclonal antibodies against human immunodeficiency virus type 1 integrase: epitope mapping and differential effects on integrase activities in vitro. J Virol 70:1580–1587. 10.1128/JVI.70.3.1580-1587.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Schindelin J, Arganda-Carreras I, Frise E, Kaynig V, Longair M, Pietzsch T, Preibisch S, Rueden C, Saalfeld S, Schmid B, Tinevez JY, White DJ, Hartenstein V, Eliceiri K, Tomancak P, Cardona A. 2012. Fiji: an open-source platform for biological-image analysis. Nat Methods 9:676–682. 10.1038/nmeth.2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Shan L, Rabi SA, Laird GM, Eisele EE, Zhang H, Margolick JB, Siliciano RF. 2013. A novel PCR assay for quantification of HIV-1 RNA. J Virol 87:6521–6525. 10.1128/JVI.00006-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Yandell BS. 1997. Practical data analysis for designed experiments. Chapman & Hall, London, United Kingdom. [Google Scholar]
- 67.Bruce JW, Bracken M, Evans E, Sherer N, Ahlquist P. 2021. ZBTB2 represses HIV-1 transcription and is regulated by HIV-1 Vpr and cellular DNA damage responses. PLoS Pathog 17:e1009364. 10.1371/journal.ppat.1009364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Munk C, Brandt SM, Lucero G, Landau NR. 2002. A dominant block to HIV-1 replication at reverse transcription in simian cells. Proc Natl Acad Sci USA 99:13843–13848. 10.1073/pnas.212400099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Butler SL, Hansen MS, Bushman FD. 2001. A quantitative assay for HIV DNA integration in vivo. Nat Med 7:631–634. 10.1038/87979. [DOI] [PubMed] [Google Scholar]