

Abstract
Background
Interventions that induce ketosis simultaneously lower blood glucose and the explanation for this phenomenon is unknown. Additionally, the glucose‐lowering effect of acute ketosis is greater in people with type 2 diabetes (T2D). On the contrary, L‐alanine is a gluconeogenic substrate secreted by skeletal muscle at higher levels in people with T2D and infusing of ketones lower circulating L‐alanine blood levels. In this study, we sought to determine whether supplementation with L‐alanine would attenuate the glucose‐lowering effect of exogenous ketosis using a ketone ester (KE).
Methods
This crossover study involved 10 healthy human volunteers who fasted for 24 h prior to the ingestion of 25 g of d‐β‐hydroxybutyrate (βHB) in the form of a KE drink (ΔG®) on two separate visits. During one of the visits, participants additionally ingested 2 g of L‐alanine to see whether L‐alanine supplementation would attenuate the glucose‐lowering effect of the KE drink. Blood L‐alanine, L‐glutamine, glucose, βHB, free fatty acids (FFA), lactate and C‐peptide were measured for 120 min after ingestion of the KE, with or without L‐alanine.
Findings
The KE drinks elevated blood βHB concentrations from negligible levels to 4.52 ± 1.23 mmol/L, lowered glucose from 4.97 ± SD 0.39 to 3.77 ± SD 0.40 mmol/L, and lowered and L‐alanine from 0.56 ± SD 0.88 to 0.41 ± SD 0.91 mmol/L. L‐alanine in the KE drink elevated blood L‐Alanine by 0.68 ± SD 0.15 mmol/L, but had no significant effect on blood βHB, L‐glutamine, FFA, lactate, nor C‐peptide concentrations. By contrast, L‐alanine supplementation significantly attenuated the ketosis‐induced drop in glucose from 28% ± SD 8% to 16% ± SD 7% (p < .01).
Conclusions
The glucose‐lowering effect of acutely elevated βHB is partially due to βHB decreasing L‐alanine availability as a substrate for gluconeogenesis.
Keywords: alanine, diabetes, d‐β‐hydroxybutyrate, exogenous ketosis, gluconeogenesis, hyperglycaemia, hypoglycaemia, ketone bodies
This was crossover study involved two separate visits for 10 healthy human volunteers who fasted for 24 h prior to the ingestion of 25 g of d‐β‐hydroxybutyrate (βHB) monoester (ΔG®). During one of the visits, participants ingested 2 g of L‐alanine. Blood L‐alanine, L‐glutamine, glucose, βHB, free fatty acids (FFA), lactate and C‐peptide were measured. Demonstrating that ketosis induces hypoglycemia, in part, by limiting L‐alanine availability for gluconeogenesis.
1. INTRODUCTION
In the fed state, the human brain relies almost exclusively on glucose as an energy source. However, in the fasted state or during carbohydrate scarcity, the human body has two adaptive mechanisms: (i) Gluconeogenesis, which promotes the catabolism of muscle tissue and (ii) Ketogenesis, which produces primarily d‐β‐hydroxybutyrate (βHB) from fat stores and provides an alternative fuel substrate for the brain. This latter option fuels the energy expensive brain, while at the same time preserving valuable lean tissue. 1
Interestingly, L‐alanine is the amino acid with the largest concentration changes during prolonged starvation 2 and the infusion of ketone bodies lowers L‐alanine blood levels more than any other amino acid. 3 In fact, L‐alanine acts as an intermediary metabolite in the transfer of pyruvate from the skeletal muscle to the liver 4 and, while it is far from being the most prominent gluconeogenic substrate, is particularly relevant for hepatic gluconeogenesis. 5
On the contrary, in type 2 diabetes (T2D), excessive gluconeogenesis underlies hyperglycemia. 6 , 7 , 8 Thus, one could predict that βHB, by inhibiting gluconeogenesis, 9 could help manage hyperglycemia in T2D. Consistent with this hypothesis, ketogenic diets have been shown to effectively reverse T2D 10 , 11 and, while most of the benefit of ketogenic diets in T2D likely comes from carbohydrate reduction, it is not yet clear to what degree βHB itself might play a supporting therapeutic role. In fact, in both animals and humans, exogenous βHB are alone sufficient to decrease blood glucose. 12 , 13 , 14 , 15
Even further, it has been known for decades that ketone bodies reduce blood glucose even in people uncapable of producing insulin and that L‐alanine is likely involved in this mechanism. 3
On this regard, indirect evidence suggests that ketone bodies limit the secretion of L‐alanine by reducing skeletal muscle glycolysis and, consequently, by limiting the availability of intramuscular pyruvate. 3 , 16
The development of an enantiomerically pure medical‐grade exogenous ketone ester (KE) for human consumption, following on the possibility that βHB could itself benefit persons with T2D, has inspired interest in the exploration of exogenous ketosis for T2D. 17 Thus, it is plausible that exogenous ketones decrease blood glucose by reducing L‐alanine availability for gluconeogenesis and that this effect could help improve glucose control. 18 This study sought to test this hypothesis by investigating whether L‐alanine supplementation attenuates the glucose‐lowering effect of exogenously induced ketosis.
2. METHODS
2.1. Ethics
This study was pre‐registered at ISRCTN16169021 and received Ethics approval by the East of England—Cambridge South Research Ethics Committee on 18 February 2019 (Reference 18/EE/0115). All participants gave their written informed consent.
This study was conducted in accordance with the guidelines set forth by the International Conference on Harmonisation Guidelines for Good Clinical Practice and the Declaration of Helsinki. 19 Potentially pregnant participants were excluded.
2.2. Statistical analysis
We based our expected glucose difference (0.9 mmol/L) in what was reported as the maximal glucose difference between low and high doses of exogenous ketones in a similar setting. 15 Sample size was calculated to detect a blood glucose difference of at least 0.9 mmol/L between two groups with eight measurements, and f effect size of 0.5 from a standard deviation of 0.9 within each group (measured but not reported in the final publication 15 ), a statistical power of 80% and an alpha probability of 0.05. As a result, 10 participants per group were needed. To achieve a sex balanced cohort, five men and 5 women were recruited, and all completed the study. Sample size calculations were performed using G*Power (F tests – ANOVA: Repeated measures, between factors) version 3.1. 20
All statistical tests and analysis were performed using Excel® (Microsoft). Data, presented as means ± standard deviations (SD), were analysed using a trial × time, repeated measures ANOVA with replication and was adjusted with Bonferroni's correction for multiple comparisons. Differences were considered significant at p < .006 (equivalent to 0.05/8 after a Bonferroni correction for keeping a 0.05 significance threshold). Since baseline blood concentrations of L‐alanine and L‐glutamine have large interpersonal variations due to body composition, comparisons were made using the percentual change from baseline.
2.3. Materials and procedures
10 healthy participants engaged in a randomized crossover experiment in which they came to the laboratory on two occasions after a 24 h fast. On one visit, participants drank a mix 25 g of the ΔG® KE ((R)‐3‐ hydroxy butyl (R)‐3‐hydroxybutyrate, provided by TdeltaS Ltd.) diluted in 25 ml water. On the other visit, participants consumed the same drink with an additional 2g of L‐alanine (provided by Hard Eight Nutrition LLC) to match skeletal muscle release of L‐alanine per 2 h. 6 After the ingestion of the drinks, all subjects were asked to remain seated or laying in a semi‐Fowler position. They were allowed to perform quiet activities such as reading or watching movies.
After ingestion, the KE monoester bond is cleaved by esterases in the gut wall, yielding βHB and butanediol in equal amounts. Both are absorbed into the portal circulation and the latter is taken up by the liver, where it is converted to βHB by alcohol dehydrogenase. βHB leaves hepatocytes via monocarboxylate transporters. Pharmacokinetic studies have shown that, in the fasted and resting states, the βHB monoester can induce ketosis for 3–4 h, with a peak at ~1 h. 21
Blood samples were collected through a venous cannula at baseline and for 120 min. After collection, samples kept on ice were centrifuged for 10 min at 2,897.86 g. The plasma component was aliquoted into Eppendorf tubes, and these were stored at −80°C until later analysis. Frozen samples were thawed at room temperature for ≈30 min before biochemical analyses were undertaken.
Glucose, βHB, lactate and free fatty acids were assayed using a commercial semi‐automated bench‐top analyser (ABX Pentra). L‐alanine, L‐glutamine and human C‐peptide were measured using kits (by Abcam: ab83394, ab145659, and ab178641, respectively).
3. RESULTS
3.1. Participant anthropometric characteristics
Ten healthy participants, five males and five females, were enrolled in this crossover trial. All participants were of normal weight (mean BMI 23.2 kg/m2 ± 2.4), and mean age was 40 years ± 16 years. Two of our five female participants were postmenopausal. One of the remaining three had been using a hormonal intrauterine device for the last 18 months at the time of enrolment, one had been using a subdermal contraceptive implant for the last 6 months at the time of enrolment and both visits of the other female participant happened during her early follicular phase.
Table 1 summarizes anthropometric characteristics of our participants. Ingestion of the KE drink was well‐tolerated by all participants and none presented symptomatic hypoglycaemia.
TABLE 1.
Participant characteristics
| Sex | 5 (F), 5 (M) |
|---|---|
| Age (years) | 40 (16) range: 19–70 |
| Height (cm) | 176 (15) range: 154–209 |
| Weight (kg) | 73 (18) range: 49–105 |
| BMI (kg/m2) | 23.2 (2.4) range: 19–27 |
Data are expressed as mean (Standard deviation).
3.2. Blood L‐alanine, L‐glutamine and glucose concentration changes after ketone ester ingestion, with and without L‐alanine supplementation
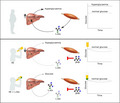
In the absence of L‐alanine supplementation, inducing acute ketosis with the KE raised blood βHB concentration to 4.52 ± 1.24mmol/L and L‐glutamine by 9% ± 4% (absolute change from 1.09 to 1.22 ± 0.04 mmol/L).
By contrast, exogenous ketosis lowered blood L‐alanine by 26% ± 9.3% (absolute change from 0.56 to 0.44 ± 0.05 mmol/L) and blood glucose by 28% ± 8% (absolute change from 4.97 to 3.77 ± 0.41 mmol/L). Compared to baseline, changes in βHB, glucose and alanine, but not glutamine, were significantly different (p < .01).
In the presence of L‐alanine supplementation, inducing ketosis raised blood βHB concentration to 4.64 ± 1.35 mmol/L and L‐glutamine by 10% ± 6% (absolute change from 1.12 to 1.26 ± 0.05 mmol/L). The intra‐assay coefficient of all samples was <10%.
As expected, L‐alanine supplementation led to a net increase in L‐alanine by 16% (absolute change from 0.54 to 0.68 ± 0.05 mmol/L), thus maintaining L‐alanine availability for gluconeogenesis (Figure 1). Correspondingly, blood glucose decreased by a lesser amount, only by 17% ± 5% (absolute change from 5.01 to 4.03 ± 0.32 mmol/L). Again, changes in βHB, glucose and L‐alanine, but not L‐glutamine, were significant (p < .01).
FIGURE 1.
Blood L‐alanine, L‐glutamine and βHB concentration changes after inducing acute ketosis with and without L‐alanine supplementation. Data are presented as the mean ± SD (n = 10 participants, measurements by triplicate). As expected, Alanine supplementation raised blood Alanine throughout the trial
3.3. L‐alanine supplementation attenuated the glucose‐lowering effect of acute ketosis
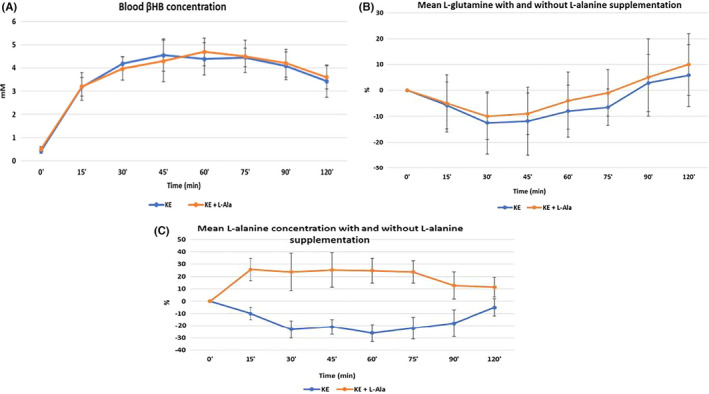
2 g L‐alanine significantl reduced the magnitude of the glucose‐lowering effect of exogenous ketosis and glucose levels were consistently lower at all time points when L‐alanine was supplemented (Figure 2).
FIGURE 2.
L‐alanine supplementation attenuated the glucose‐lowering effect of ketosis. Data are presented as mean concentration changes (n = 10 participants, measurements by triplicate). Y‐axis = per cent change from baseline of blood glucose concentration. X‐axis = Time (minutes) after drinking 25 of βHB ketone monoester. Mean change after baseline was analysed with a two‐way ANOVA (trial × time) and found to be statistically significant between treatments (KE vs. KE + L‐alanine) statistically significant differences (p < .006). Glucose data were tested for normality using a Shapiro‐Wilk test
3.4. L‐alanine supplementation did not impact changes in blood lactate, free fatty acids (FFA) and C‐peptide during acute ketosis
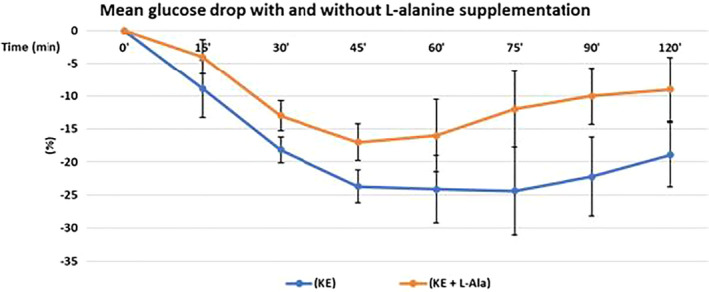
Compared to baseline, FFA concentrations were significantly lower at most time points 30 min after KE ingestion. C‐peptide was moderately elevated in both groups at the 30 min time point. Blood lactate, FFA and C‐peptide concentrations were similar at all time points after ingestion of KE regardless of L‐alanine supplementation, as in Figure 3.
FIGURE 3.
Blood lactate, free fatty acids (FFA) and C‐peptide during acute ketosis with and without L‐alanine. Mean change after baseline was analysed with a two‐way (trial × time) ANOVA. Statistical significance after a Bonferroni correction was defined as p < .006
3.5. Correlation between the dose of ketone monoester with maximal βHB concentration and glucose change in the blood
Since all participants consumed 25 ml of the ketone monoester, they differed in the effective dose of ketone monoester they received, ranging between 242 and 491 mg/kg. As expected, βHB maximal blood concentration correlated with the dose of ketone monoester (R 2 = .61 p < .01). Interestingly, the correlation between the magnitude of the glucose change with the dose of ketone monoester was very comparable (R 2 = .59 vs R 2 = .61, respectively) suggesting changes in blood glucose were proportional to and mostly driven by the consumption of the ketone monoester. Figure [Link], [Link] illustrates these results.
4. DISCUSSION
These data suggest that a decrease in L‐alanine availability contributes to the glucose‐lowering effect of acute ketosis. However, it is also clear that the glucose‐lowering effect of acute ketosis is a result of multiple mechanisms.
4.1. The glucose‐alanine cycle
During prolonged fasting, L‐alanine blood levels decrease more than those of any other amino acid, largely because of hepatic uptake of L‐alanine to fuel gluconeogenesis. 2 This phenomenon prompted the discovery of the glucose‐alanine cycle, in which pyruvate is transformed into L‐alanine (pyruvate in disguise) via transamination in skeletal muscle. L‐alanine is released to the bloodstream, taken up by the liver, and transformed back into pyruvate to fuel gluconeogenesis. 4 Thus, any reduction in intramuscular pyruvate would result in lower L‐alanine levels in the blood and less gluconeogenesis.
Ketone oxidation decreases intramuscular pyruvate via multiple mechanisms: (i) First, ketone oxidation reduces glycolysis and pyruvate in skeletal muscle 16 and (ii) second, ketone body oxidation increases acetyl CoA levels. 22 Acetyl CoA is an allosteric activator of pyruvate carboxylase, thus, driving the conversion of pyruvate to oxaloacetate. 23 (iii) In addition to decreasing pyruvate for transamination into L‐alanine, ketosis also directly decreases L‐alanine production by reducing skeletal muscle protein breakdown. 24
Furthermore, in the liver, L‐alanine supports gluconeogenesis. L‐alanine allosterically inhibits the liver isozyme of pyruvate kinase, the enzyme responsible for the last step of glycolysis. 25 Thus, L‐alanine restriction would favour glycolysis over gluconeogenesis. L‐alanine is also a potent glucagon secretion agonist, 26 and glucagon stimulates gluconeogenesis. Even further, the blood concentration of glucagon also drops after the ingestion of βHB salts after an overnight fast; 27 however, it does not in the postabsorptive state. 28
4.2. The glucose‐lowering effect of ketosis is context dependent and pleiotropic
As observed in this study, the ability of exogenously induced ketosis to reduce circulating L‐alanine as fuel for gluconeogenesis cannot account for the entire glucose‐lowering effect of KE ingestion. Other mechanisms are at play.
Infusion and islet studies have shown that exogenous ketosis can stimulate insulin secretion. 29 , 30 However, even while we observed a transient C‐peptide spike at 30 min after KE ingestion, it should be noted that this does not necessarily translate into an equal magnitude, one‐to‐one, spike in systemic circulating insulin due to insulin liver extraction. What is more, even if we assume this C‐peptide change correlates with an equal magnitude insulin change, the latter would not be large or long enough to account for the entire glucose‐lowering effect observed.
There is also evidence that a decrease in gluconeogenesis and hepatic glucose output, rather than an increase in insulin‐mediated glucose clearance from the blood, accounts for most of the glucose‐lowering effect of ketosis. 9 For example, exogenous ketone infusions in patients with type 1 diabetes demonstrate that acute ketosis lowers blood glucose 3 even in the absence of insulin. Thus, it is unlikely that the minor insulinogenic effect of exogenous ketosis accounts for most of the glucose‐lowering effect KE ingestion.
The glucose‐lowering effect of acute ketosis is also not restricted to the fasting state. Inducing acute ketosis lowers the postprandial glycaemic curve after a dextrose challenge without inducing significant differences in insulin secretion. 31 One of the proposed mechanisms is that βHB‐mediated inhibition of lipolysis 31 depletes the blood supply of fatty acids, driving an increase in glucose uptake. However, this has not been observed after the ingestion of niacin, which also blocks lipolysis by acting on the same receptor as βHB and has no effect on glycaemia. 32
Additionally, as mentioned before, it is also worth highlighting that acute ketosis lowered glucose in postabsorptive humans despite not lowering glucagon. 28 As gluconeogenesis contributes to elevations in blood glucose in the postabsorptive state, βHB‐induced attenuation of gluconeogenesis could also explain (at least partially) the postprandial attenuation of the glycaemic curve that has been previously observed.
It is also worth noting that glycerol is a gluconeogenic substrate. However, evidence suggests that, in the fasting humans, L‐alanine, and lactate, not glycerol, are the major gluconeogenic substrates. Additionally, limiting FFA availability could also contribute to the observed glucose‐lowering effect of KEs by reducing β‐oxidation and, therefore, the availability of reducing equivalents, high energy phosphates and acetyl‐CoA (which, as mentioned before, allosterically regulates pyruvate carboxylation 23 ). Nonetheless, since ketosis also reduces the availability of glycerol and FFA by inhibiting lipolysis, 33 ketosis could be potentially beneficial for individuals in whom gluconeogenesis is pathologically elevated 27 regardless of which substrate is the most important contributor to gluconeogenesis.
Importantly, it is unlikely that this is a KE‐specific effect. While the hepatic conversion of butanediol derived from ketone ester into βHB would alter hepatocyte NAD+/NADH balance (therefore altering endogenous glucose production), studies comparing the ingestion of equimolar quantities of ketone salts and ketone esters (that lack a butanediol component) demonstrate glucose‐lowering effects of similar magnitudes. 15
In contrast, a study in people with type 2 diabetes and using sodium acetoacetate infusions with and without somatostatin concluded that ketone bodies can suppress elevated hepatic glucose output independently of changes in insulin secretion, only when the concomitant stimulation of glucagon secretion is prevented. 34
Even when the physiological effects of exogenous acetoacetate are not comparable to those of exogenous βHB (because they have opposite effects on the NAD/NADH couple), since alanine is a potent glucagon secretion agonist, βHB‐induced reduction would also yield less glucagon secretion.
Figure 4 summarizes some of the mechanisms whereby exogenously induced acute ketosis could lower blood glucose concentration.
FIGURE 4.
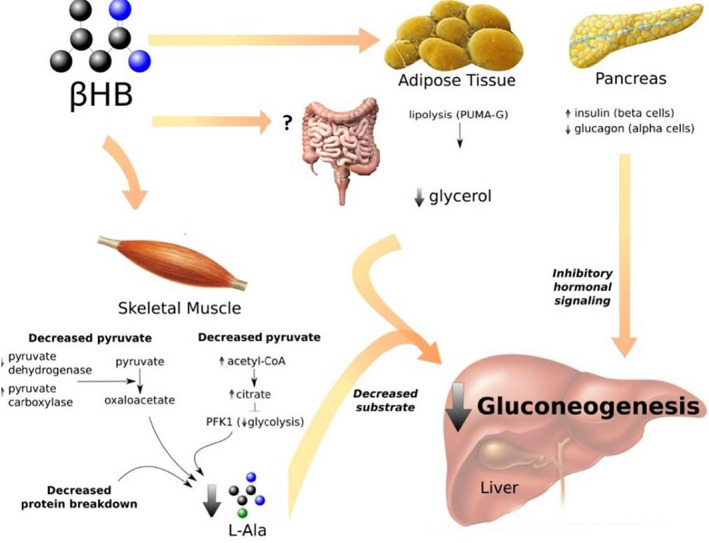
Mechanisms whereby ketosis lowers blood glucose. In skeletal muscle: βHB increases the concentration of acetyl CoA, 22 which inhibits pyruvate dehydrogenase and activates pyruvate carboxylase. 23 As a result, more pyruvate is transformed into oxaloacetate. Additionally, the rise in acetyl CoA inhibits phosphofructokinase‐1 (PFK1), downregulating glycolysis and, therefore, decreasing pyruvate production. 45 Through these two mechanisms, pyruvate levels are reduced and there is less pyruvate available for transamination into L‐alanine. βHB also decreases protein breakdown, further reducing L‐alanine release. 16 In adipose tissue: βHB inhibits lipolysis via de PUMA‐G receptor, 33 reducing the release of glycerol, a minor gluconeogenic substrate. In the pancreas: βHB promotes insulin release by the beta cells. 30 , 46 Furthermore, there is less L‐alanine to stimulate glucagon release by the alpha cells. 26 The decrease in gluconeogenic substrates (L‐alanine and glycerol) and inhibitory hormonal signalling (increase in insulin/glucagon ratio; and L‐alanine allosterically inhibits pyruvate kinase 25 ) cause a decrease in gluconeogenesis by the liver (and also possibly other peripheral tissues, like the intestines, that can perform gluconeogenesis). The net result of acute ketosis is a small increase in peripheral glucose uptake, due to a small increase in insulin, and a larger decrease in glucose release by gluconeogenic tissues
4.3. Limitations of this study
First, the relative contribution of the different gluconeogenic organs (liver, kidneys and intestine) and of the different gluconeogenic substrates (lactate, glycerol, alanine and glutamine) varies depending on whether a person is in the fed or fasted state. 35 , 36 Since we hypothesized Alanine's role was due to its potential influence on gluconeogenesis, we chose to evaluate the effects of acute ketosis after fasting for 24 h because the relative contribution of gluconeogenesis to endogenous glucose production is 47 ± 49% after 14 h and 67 ± 41% after 22 h. 37 , 38 However, there is evidence suggesting that, in the context of short‐term fasting, alanine is the most important contributor via liver gluconeogenesis. 5 , 39 , 40 , 41 , 42
Additionally, our sample size is likely underpowered to detect specific time point differences between both interventions. However, finding these difference falls beyond the scope of our work since we are interested in the overall effect of Alanine on the glucose reduction curve and not so much in its effect on a particular time point along that curve.
Since all participants in this study were fasted for control purposes, we cannot extrapolate these insights to other metabolic states where there are different relative contributions from the kidneys and the intestines. Future studies may choose to investigate the same question in postprandial participants or using labelled substrates.
Additionally, while unlikely, an unexpected glucose‐lowering interaction effect between L‐alanine supplementation and exogenously induced ketosis cannot be entirely ruled out. However, other studies have observed the opposite (an increment in hepatic glucose production and ketosis downregulation after Alanine supplementation). 43 Nonetheless, a future study should include an independent L‐alanine arm to compare the impact of L‐alanine on glycaemia in the absence of ketosis. Additional limitations (since we did not measure them) and informative measurements for future studies are nitrogen balance and glucagon.
Finally, it has been proposed that glycerol is the most relevant gluconeogenic substrate. 44 Even if this is true, since acute ketosis also reduces the availability of glycerol, the overall conclusion of ketosis being potentially therapeutic for individuals in whom gluconeogenesis is pathologically elevated holds true.
5. CONCLUSION
In fasting healthy humans, exogenous ketosis results in a reduction in L‐alanine availability to fuel gluconeogenesis and that this contributes, at least partially, to the glucose‐lowering effects of acute ketosis. These data provide insight about the potential mechanisms behind the elsewhere reported benefits of ketogenic interventions for patients living with type 2 diabetes. More research is needed to confirm these findings.
CONFLICT OF INTERESTS
The intellectual property covering the uses of ketone bodies and ketone esters are owned by BTG Plc, Oxford University Innovation Ltd and the US National Institutes of Health. Professor Kieran Clarke, as an inventor, will receive a share of the royalties under the terms prescribed by each institution. Professor Clarke is a director of TdeltaS Ltd, a company spun out of the University of Oxford to develop products based on the science of ketone bodies in human nutrition. The other authors declare that they have no competing financial interests or personal relationships that could have influenced the work reported in this paper.
AUTHOR CONTRIBUTION
Adrian Soto‐Mota: Conceptualization (lead); Data curation (lead); Formal analysis (lead); Investigation (lead); Methodology (lead); Software (lead); Visualization (lead); Writing‐original draft (lead). Nicholas G. Norwitz: Formal analysis (supporting); Visualization (supporting); Writing‐original draft (supporting). Rhys D. Evans: Conceptualization (supporting); Formal analysis (supporting); Investigation (supporting); Methodology (supporting); Supervision (equal); Writing‐original draft (equal). Kieran Clarke: Conceptualization (equal); Formal analysis (equal); Investigation (equal); Methodology (equal); Resources (equal); Supervision (equal); Writing‐original draft (equal).
Supporting information
Fig S1A
Fig S1B
ACKNOWLEDGMENTS
The authors thank Vicky Ball for her diligent support. Dr. Adrian Soto‐Mota thanks the Consejo Nacional de Ciencia y Tecnología (CONACYT, Mexico) and the Instituto Nacional de Ciencias Médicas y Nutrición Salvador Zubirán (FUNSAED, Mexico) for their financial support.
Soto‐Mota A, Norwitz NG, Evans RD, Clarke K. Exogenous d‐β‐hydroxybutyrate lowers blood glucose in part by decreasing the availability of L‐alanine for gluconeogenesis. Endocrinol Diab Metab.2022;5:e300. doi: 10.1002/edm2.300
Funding information
Adrian Soto‐Mota is funded by CONACYT and INCMSNZ. The KE drinks were provided by TdeltaS Ltd, Oxford.
DATA AVAILABILITY STATEMENT
Data described in the manuscript, code book and analytic code will be made available upon request pending approval by TdeltaS Ltd.
REFERENCES
- 1. Cahill GF, George FC. Fuel metabolism in starvation. Ann Rev Nutr. 2006;26:1‐22. [DOI] [PubMed] [Google Scholar]
- 2. Felig P, Owen OE, Wahren J, Cahill GF. Amino acid metabolism during prolonged starvation. J Clin Invest. 1969;48:584‐594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Sherwin RS, Hendler RG, Felig P. Effect of diabetes mellitus and insulin on the turnover and metabolic response to ketones in man. Diabetes. 1976;25:776‐784. [DOI] [PubMed] [Google Scholar]
- 4. Felig P, Pozefsk T, Marlis E, Cahill GF. Alanine: key role in gluconeogenesis. Science. 1970;167:1003‐1004. [DOI] [PubMed] [Google Scholar]
- 5. Stumvoll M, Meyer C, Perriello G, Kreider M, Welle S, Gerich J. Human kidney and liver gluconeogenesis: Evidence for organ substrate selectivity. Am J Physiol Endocrinol Metab. 1998;274:E817‐E826. [DOI] [PubMed] [Google Scholar]
- 6. Consoli A, Nurjhan N, Reilly JJ, Bier DM, Gerich JE. Mechanism of increased gluconeogenesis in noninsulin‐dependent diabetes mellitus. Role of alterations in systemic, hepatic, and muscle lactate and alanine metabilism. J Clin Invest. 1990;86:2038‐2045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Consoli A, Nurjhan N. Contribution of gluconeogenesis to overall glucose output in diabetic and nondiabetic men. Ann Med. 1990;22:191‐195. [DOI] [PubMed] [Google Scholar]
- 8. Magnusson I, Rothman DL, Katz LD, Shulman RG, Shulman GI. Increased rate of gluconeogenesis in type II diabetes mellitus. A 13C nuclear magnetic resonance study. J Clin Invest. 1992;90:1323‐1327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Beylot M, Chassard D, Chambrier C, et al. Metabolic effects of a D‐β‐hydroxybutyrate infusion in septic patients. Crit Care Med. 1994;22:1091‐1098. [DOI] [PubMed] [Google Scholar]
- 10. Hallberg SJ, McKenzie AL, Williams PT, et al. Effectiveness and safety of a novel care model for the management of type 2 diabetes at 1 year: an open‐label, non‐randomized. Controlled study. Diabetes Ther. 2018;9:583‐612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Lim EL, Hollingsworth KG, Aribisala BS, Chen MJ, Mathers JC, Taylor R. Reversal of type 2 diabetes: normalisation of beta cell function in association with decreased pancreas and liver triacylglycerol. Diabetologia. 2011;54:2506‐2514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Nakatani T, Yasuda K, Ozawa K, Tobe T. Changes in blood glucose levels in relation to blood ketone body ratio following hypertonic glucose infusion in 70% hepatectomized rabbits. Eur Surg Res. 1984;16:303‐311. [DOI] [PubMed] [Google Scholar]
- 13. Felts PW, Crofford B, Park CR. Effect of infused ketone bodies on glucose utilization in the dog. J Clin Invest. 1964;43:638‐646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Muller MJ, Paschen U, Seitz HJ. Effect of ketone bodies on glucose production and utilization in the miniature pig. J Clin Invest. 1984;74:249‐261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Stubbs BJ, Cox PJ, Evans RD, et al. On the metabolism of exogenous ketones in humans. Front Physiol. 2017;8:848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Wu G, Thompson JR. The effect of ketone bodies on alanine and glutamine metabolism in isolated skeletal muscle from the fasted chick. Biochem J. 1988;255:139‐144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Soto‐Mota A, Norwitz NG, Clarke K. Why a D‐β‐hydroxybutyrate monoester? Biochem Soc Transac. 2020;48:51‐59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Soto‐Mota A, Norwitz NG, Evans R, Clarke K, Barber TM. Exogenous ketosis in patients with type 2 diabetes: Safety, tolerability and effect on glycaemic control. Endocrinol Diabetes Metab. 2021;4(3):1‐7. 10.1002/edm2.264 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. WMA . WMA DECLARATION OF HELSINKI – ETHICAL PRINCIPLES FOR Scientific Requirements and Research Protocols. World Medical Association 29–32 (2013).
- 20. Faul F, Erdfelder E, Buchner A, Lang A‐G. Statistical power analyses using G*Power 3.1: tests for correlation and regression analyses. Behav Res Methods. 2009;41(4):1149‐1160. [DOI] [PubMed] [Google Scholar]
- 21. Clarke K, Tchabanenko K, Pawlosky R, et al. Kinetics, safety and tolerability of (R)‐3‐hydroxybutyl (R)‐3‐hydroxybutyrate in healthy adult subjects. Regul Toxicol Pharmacol. 2012;63:401‐408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Sato K, Kashiwaya Y, Keon CA, et al. Insulin, ketone bodies, and mitochondrial energy transduction. FASEB J. 1995;9:651‐658. [DOI] [PubMed] [Google Scholar]
- 23. Jitrapakdee S, St Maurice M, Rayment I, Cleland WW, Wallace JC, Attwood PV. Structure, mechanism and regulation of pyruvate carboxylase. Biochem J. 2008;413:369‐387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Cox PJ, Kirk T, Ashmore T, et al. Nutritional ketosis alters fuel preference and thereby endurance performance in athletes. Cell Metab. 2016;24:256‐268. [DOI] [PubMed] [Google Scholar]
- 25. Van Veelen CWM, Rijksen G, Vlug AMC, Staal GEJ. Correlation between Alanine inhibition of pyruvate kinase and composition of K‐M hybrids. Clin Chim Acta. 1981;110:113‐120. [DOI] [PubMed] [Google Scholar]
- 26. Müller WA, Faloona GR, Unger RH. The effect of alanine on glucagon secretion. J Clin Invest. 1971;50:2215‐2218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Svart M, Rittig N, Pedersen SB, Jessen N, Møller N. Oral 3‐hydroxybutyrate ingestion decreases endogenous glucose production, lipolysis, and hormone‐sensitive lipase phosphorylation in adipose tissue in men: a human randomized, controlled, crossover trial. Diabetic Med. 2021;38:1‐8. [DOI] [PubMed] [Google Scholar]
- 28. Mikkelsen KH, Seifert T, Secher NH, Grøndal T, van Hall G. Systemic, cerebral and skeletal muscle ketone body and energy metabolism during acute hyper‐D‐β‐hydroxybutyratemia in post‐absorptive healthy males. J Clin Endocrinol Metab. 2015;100:636‐643. [DOI] [PubMed] [Google Scholar]
- 29. Balasse E, Ooms H, Lambilliotte J. Evidence for a stimulatory effect of ketone bodies on insulin secretion in man. Horm Metab Res. 1970;2:371‐372. [DOI] [PubMed] [Google Scholar]
- 30. Biden TJ, Taylor KW. Effects of ketone bodies on insulin release and islet‐cell metabolism in the rat. Biochem J. 1983;212:371‐377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Myette‐Côté É, Caldwell HG, Ainslie PN, Clarke K, Little JP. A ketone monoester drink reduces the glycemic response to an oral glucose challenge in individuals with obesity: a randomized trial. Am J Clin Nutr. 2019;110:1491‐1501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Wang W, Basinger A, Neese RA, Christiansen M, Hellerstein MK. Effects of nicotinic acid on fatty acid kinetics, fuel selection, and pathways of glucose production in women. Am J Physiol Endocrinol Metab. 2000;279:E50‐E59. [DOI] [PubMed] [Google Scholar]
- 33. Taggart AKP, Kero J, Gan X, et al. (D)‐β‐hydroxybutyrate inhibits adipocyte lipolysis via the nicotinic acid receptor PUMA‐G. J Biol Chem. 2005;280:26649‐26652. [DOI] [PubMed] [Google Scholar]
- 34. Henry RR, Brechtel G, Lim KH. Effects of ketone bodies on carbohydrate metabolism in non‐insulin‐dependent (type II) diabetes mellitus. Metabolism. 1990;39:853‐858. [DOI] [PubMed] [Google Scholar]
- 35. Chiasson JL, Liljenquist JE, Sinclair Smith BC, Lacy WW. Gluconeogenesis from alanine in normal postabsorptive man. Intrahepatic stimulatory effect of glucagon. Diabetes. 1975;24:574‐584. [DOI] [PubMed] [Google Scholar]
- 36. Chiasson JL, Atkinson RL, Cherrington AD, et al. Effects of insulin at two dose levels on gluconeogenesis from alanine in fasting man. Metab Clin Exp. 1980;29:810‐818. [DOI] [PubMed] [Google Scholar]
- 37. Landau BR, Wahren J, Chandramouli V, Schumann WC, Ekberg K, Kalhan SC. Contributions of gluconeogenesis to glucose production in the fasted state. J Clin Invest. 1996;98:378‐385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Landau BR, Fernandez CA, Previs SF, et al. A limitation in the use of mass isotopomer distributions to measure gluconeogenesis in fasting humans. Am J Physiol Endocrinol Metab. 1995;269:E18‐E26. [DOI] [PubMed] [Google Scholar]
- 39. Sarabhai T, Roden M. Hungry for your alanine: When liver depends on muscle proteolysis. J Clin Invest. 2019;129:4563‐4566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Shah AM, Wondisford FE. Tracking the carbons supplying gluconeogenesis. J Biol Chem. 2020;295:14419‐14429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Martin G, Ferrier B, Conjard A, et al. Glutamine gluconeogenesis in the small intestine of 72 h‐fasted adult rats is undetectable. Biochem J. 2007;401:465‐473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Petersen KF, Dufour S, Cline GW, Shulman GI. Regulation of hepatic mitochondrial oxidation by glucose‐alanine cycling during starvation in humans. J Clin Invest. 2019;129:4671‐4675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Genuth SM. Effects of oral alanine administration in fasting obese subjects. Metabolism. 1973;22:927‐937. [DOI] [PubMed] [Google Scholar]
- 44. Wang Y, Kwon H, Su X, Wondisford FE. Glycerol not lactate is the major net carbon source for gluconeogenesis in mice during both short and prolonged fasting. Mol Metab. 2020;31:36‐44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Usenik A, Legiša M. Evolution of allosteric citrate binding sites on 6‐ phosphofructo‐1‐kinase. PLoS One. 2010;5:e15447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Beylot M, Khalfallah Y, Riou JP, Cohen R, Normand S, Mornex R. Effects of ketone bodies on basal and insulin‐stimulated glucose utilization in man. J Clin Endocrinol Metab. 1986;63:9‐15. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Fig S1A
Fig S1B
Data Availability Statement
Data described in the manuscript, code book and analytic code will be made available upon request pending approval by TdeltaS Ltd.