

Abstract
Background:
Mesalamine, 5-aminosalicylic acid (5-ASA), is a potent antioxidant and is known to enhance peroxisome proliferator–activated receptor γ activity in the intestine. Our previous studies suggested reduced Phosphoinositide 3-Kinase (PI3K)/β-catenin signaling as a mechanism for 5-ASA chemoprevention in chronic ulcerative colitis (CUC). We now hypothesize that 5-ASA mediates changes in intestinal epithelial cell (IEC) reactive oxygen species during colitis to affect phosphatase and tensin homolog (PTEN), PI3K, and β-catenin signaling.
Methods:
Here, we examined effects of 5-ASA on oxidant-induced cell signaling pathways in HT-29 cells, IECs from mice, and biopsy tissue from control and CUC patients. Samples were selected to control for inflammation between untreated and 5-ASA–treated CUC patients.
Results:
Direct evaluation of IEC in H2O2-stimulated whole colonic crypts indicated that 5-ASA reduces reactive oxygen species levels in lower crypt IECs where long-lived progenitor cells reside. Analysis of biopsies from patient samples revealed that 5-ASA increases expression of the antioxidant catalase in CUC patients. Also, 5-ASA increased nuclear peroxisome proliferator–activated receptor γ protein and target gene expression. Data showed 5-ASA–induced peroxisome proliferator–activated receptor γ DNA binding to the PTEN promoter (chromatin immunoprecipitation) and reduced both phosphorylated and oxidized (inactive) PTEN protein levels. Analysis of patient samples revealed 5-ASA that also reduced levels of active phosphorylated Akt in inflamed colitis tissue. Reduced PI3K/Akt signaling and expression of β-catenin target genes in 5-ASA–treated CUC patients additionally suggests enhanced PTEN activity as well.
Conclusions:
Therefore, 5-ASA reduces CUC-induced reactive oxygen species in colonic progenitor cells and enhances PTEN activity, thus attenuating PI3K/Akt signaling. These data suggest that the antioxidant properties of 5-ASA may be the predominant mechanism for 5-ASA chemoprevention.
Keywords: chronic ulcerative colitis, mesalamine, PPARγ, intestinal epithelial cells, ROS
Chronic ulcerative colitis (CUC) is an inflammatory bowel disease characterized by mucosal inflammation, ulceration, and bleeding. CUC can be distinguished from infectious colitis by the presence of crypt architectural distortion with crypt branching and dropout. These features suggest that chronic inflammation targets the intestinal epithelium. In addition, severe chronic inflammation increases the risk for colorectal cancer in patients with inflammatory bowel disease.1–4 The primary treatment for mild to moderate ulcerative colitis (UC) is mesalamine or 5-aminosalicylic acid (5-ASA). 5-ASA exerts anti-inflammatory effects through reduction of cyclooxygenase 2,5 inducible nitric oxide synthase,6 interleukin 8, tumor necrosis factor,5,7–9 and nuclear factor κB activation.10–12 Studies concur that 5-ASA reduces cancer risk in CUC.13–15 Potential mechanisms suggested for mesalamine chemoprevention in CUC include its property as a potent oxygen radical scavenger.16,17 Furthermore, antitumor effects are linked to its antiproliferative and proapoptotic effects on epithelial cells as exhibited in vitro12,18 and in vivo in mice19 and patients.20
Recent studies suggest that 5-ASA activation of the nuclear receptor peroxisome proliferator–activated receptor γ (PPARγ) may explain several 5-ASA effects related to chemoprevention.21,22 PPARγ is expressed in adipocytes, colon epithelial cells, and to a lesser extent in macrophages.23 On ligand binding, PPARγ associates with retinoid X receptor to initiate transcription at PPAR response elements in target genes. PPARγ targets include those involved in glucose and lipid metabolism and adipocyte and epithelial differentiation.24 Target genes expressed in colon epithelial cells include kruppel-like factor 4, 3-hydroxy-3-methylglutaryl-CoA synthase (HMGCS2), keratin 20, and phosphatase and tensin homolog (PTEN).25,26 The PPARγ gene has a PPAR response element and is therefore itself a target of PPARγ transcriptional activity. Synthetic ligands for PPARγ such as rosiglitazone and pioglitazone increase insulin sensitivity in diabetes27 and provide therapeutic benefits in UC.28,29 Studies suggest an increase in cytoplasmic PPARγ and a loss of nuclear PPARγ in malignancy.30 Data also support a direct role for PPARγ in tumorigenesis by the frequent detection of loss-of-function mutations in many cancers.31
Aside from its role as a transcription factor, PPARγ also performs nongenomic functions in the cytosol such as binding to β-catenin to regulate the ubiquitination and enhance the degradation of β-catenin.32,33 Wnt signaling stabilizes β-catenin, allowing its nuclear translocation.34 Nuclear β-catenin associates with T-cell factor 4 (TCF4)35 and induces transcription of genes linked to cellular processes such as proliferation (c-myc36), differentiation (G-protein coupled receptor 4937) and adhesion (matrix metalloproteinase 738). Using a site-specific antibody for β-catenin phosphorylated at serine 552 by Akt (P-β-catenin552), we reported that P-β-cateninS552 levels increased in UC patients and were attenuated in 5-ASA–treated patients.39 These findings indicate that 5-ASA impairs Wnt/β-catenin activation in colitis, a key signaling pathway in normal, inflamed, and dysplastic epithelial function.
PTEN is the primary negative regulator of Phosphoinositide 3-Kinase (PI3K) signaling as it prevents activation of the downstream effector Akt.40 PTEN is inactivated by oxidation by H2O2 and through phosphorylation in the PDZ domain.41,42 Schwab et al43 reported that 5-ASA–mediated PTEN upregulation is abolished in dominant-negative PPARγ mutant cells. In the present study, we investigated the relationship between PPARγ, β-catenin, and PI3K/PTEN pathways, which potentially converge to impact the dysregulation of epithelial intestinal stem cells (ISC) and progenitor cells (PCs) during the development of inflammation-associated colon cancer. Investigation of the mechanism of 5-ASA chemoprevention not only inform therapeutic strategies for the treatment of UC patients but also help define the relationship between inflammation and cancer.
MATERIALS AND METHODS
Human Colonic Specimens and Colitis Scores
Human biopsies were obtained from patients 18 years or older undergoing diagnostic or surveillance colonoscopy in the Gastrointestinal Clinic at Northwestern Memorial Hospital. No patients were pregnant, had a history of small or large bowel surgery, bleeding diathesis, or coagulopathy. No control or CUC patients were on corticosteroids, immunomodulators, or biologic agents and were only on mesalamine agents when indicated. Ulcerative Colotis Disease Activity Index (UCDAI), or Sutherland index, is determined by stool frequency, rectal bleeding, mucosal appearance, and the physician’s rating of disease activity.44 Untreated (active) CUC patients underwent diagnostic colonoscopy for increased symptoms (mean UCDAI score of 7.8 ± 1.6). All patient materials were approved by Northwestern University Office for the Protection of Human Subjects. Hematoxylin and eosin staining was performed to determine inflammatory scores by 2 independent investigators in a blinded fashion as previously described.45 Inflammatory scores in human biopsies were determined from 0 to 3 based on increasing severity of mononuclear infiltration, active cryptitis, and epithelial ulceration with immunohistochemistry analysis performed on chronically inflamed adjacent mucosa.
Animals
C57BL/6J (B6) were purchased from Jackson Laboratory (Bar Harbor, ME). Mice were maintained in the barrier facility at Northwestern University Center for Comparative Medicine, and all animal experiments were approved by the Animal Care and Usage Committee of Northwestern University. Mice were between 6 and 12 weeks old for all experiments.
Cell Culture and Activation
HT-29 cells were purchased from American Type Culture Collection and cultured in Dulbecco’s modified Eagle’s medium (Cellgro, Manassas, VA) supplemented with 10% fetal bovine serum (Life Technologies, Grand Island, NY) and 1% Pen/Strep solution (Cellgro). Preceding each experiment, cells were grown to 80% confluence and then serum starved overnight. The following day the cell culture supernatant was removed and saved (referred to as conditioned media). Before stimulation, cells were pretreated with 30 mM 5-ASA (pH of 7.4) for 1 hour. Cells were then stimulated with 50:50 serum replete media and conditioned media with or without 30 mM 5-ASA (pH 7.4). Cells were harvested at 3 hours for chromatin immunoprecipitation (ChIP), 4 hours for RNA, and 24 hours for flow cytometry.
Chromatin Immunoprecipitation Assay
Cells were grown and stimulated as described above followed by fixation to a final concentration of 1% formaldehyde for 10 minutes. ChIP was then performed using a modified protocol from Millipore (Billerica, MA). Briefly, after fixation, 10% glycine was added for 5 minutes to stop the cross-linking, cells were washed twice with phosphate buffer solution (PBS), and scraped in ice-cold PBS plus 0.5 mM phenylmethylsulfonyl fluoride. Cells were pelleted and resuspended in lysis buffer (1% sodium dodecyl sulfate (SDS), 10 mM EDTA, 50 mM Tris-HCL; pH 8.1) for 10 minutes and sonicated to produce DNA fragments approximately 500 base pair. Debris was cleared by centrifugation, and the supernatant was diluted in ChIP dilution buffer (0.01% SDS. 1.1% Triton X-100, 1.2 mM EDTA, 16.7 mM Tris-HCL, 167 mM NaCl, and a Roche [Indianapolis, IN] complete protease inhibitor tablet; pH 8.1). The sample was precleared using a salmon sperm (Life Technologies)/Protein A/G agarose (Santa Cruz, Santa Cruz, CA) slurry. Beads were removed by centrifugation. Input sample was removed, saved, and remaining supernatant was incubated with 2 μg antibodies to PPARγ (Santa Cruz) or PolII (transcription control, Santa Cruz) plus IgG (control) for 12 hours. Salmon sperm (Life Technologies)/Protein A/G agarose slurry was added for 2 hours to precipitate the immune complexes. Samples were centrifuged to collect the beads, washed once in low salt buffer, once in high salt buffer, once in LiCl buffer, and twice in 1X TE. Complexes were eluted in 1% SDS/0.1M NaHCO3 and treated with RNaseA. Cross-linking (including for the input sample) was reversed using 5 M NaCl at 65 degrees (overnight) followed by treatment with proteinase K. DNA was extracted with phenol/chloroform and precipitated with ethanol. PCR was then performed using primers for glyceraldehyde-3-phosphate dehydrogenase and PTEN.
Laser Capture Microdissection and Real-Time Quantitative RT-PCR
Epithelial cells were captured from formalin-fixed paraffin-embedded tissues by laser capture microdissection (LCM) using the VERITAS system (Arcturus Bioscience, Mount View, CA). RNA was isolated and reverse transcribed using the paradise whole transcript reverse-transcribed reagent system (Arcturus). Quality of each sample was determined with the paradise sample quality assessment kit (Arcturus).
Total RNA was isolated using the RNeasy Mini Kit (Qiagen, Germantown, MD) from RNAlater preserved biopsy specimens or HT-29 cell lines. Total RNA was also purified from Allprotect preserved human samples using the AllPrep DNA/RNA/Protein mini kit (Qiagen). Complementary DNA was synthesized using the high capacity complementary DNA reverse transcription kit (Applied Biosystems, Foster City, CA). qRT-PCR was performed using the Power SYBR green PCR master mix (Applied Biosystems) on the ABI 7500 real-time PCR system. Primers were designed to span exon junctions using Primer Express software 3.0 (Applied Biosystems) based on nucleotide sequences from the NCBI data bank (see Table, Supplemental Digital Content 1, http://links.lww.com/IBD/A187). For each sample, the Ct value for target genes and glyceraldehyde-3-phosphate dehydrogenase (internal reference) was determined. Fold increases were calculated using the ddCt method.
Protein Extraction and Western Blot Analysis
Nuclear fractions were obtained from frozen biopsies by passing cells in 50 mM Tris-HCl pH 7.4, 100 mM NaCl, 0.01% digitonin, plus protease and phosphatase inhibitors through a 27-gauge needle followed by 5 minutes on ice and 5-minute centrifugation at 16,000g at 4°C. Next, the resulting pellet was resuspended in 50 mM Tris-HCl pH 7.4, 100 mM NaCl, 0.2% Triton X-100, plus protease, and phosphatase inhibitors followed by 30 minutes on ice and 5-minute centrifugation at 16,000g at 4°C. This time the resulting pellet was resuspended in 50 mM Tris-HCl pH 7.4, 100 mM NaCl, 0.5% DDM (n-dodecyl β-D-maltoside; Life Technologies), 0.1% benzonase, plus protease, and phosphatase inhibitors followed by 30 minutes at room temperature and 5-minute centrifugation at 16,000g at 4°C. The final pellet consists of the nuclear and cytoskeletal subcellular fractions.
Total protein was extracted from Allprotect Tissue Reagent (Qiagen)–preserved biopsies according to the manufacturer’s instructions. For oxidized protein determination, frozen biopsies were homogenized in radio-immunoprecipitation assay buffer. The addition of iodoacetamide (Sigma, St Louis, MO) prevents proteins already in reduced conformation from forming disulfide bonds, therefore allowing discrimination of oxidized from reduced proteins run under non-reduced conditions. Proteins were separated by SDS-polyacrylamide gel electrophoresis and transferred to polyvinylidene difluoride membrane (Millipore). The membranes were blocked with protein-free T20 blocking buffer (Pierce, Rockford, IL) and incubated with primary antibodies specific for PPARγ, PTEN, P-PTEN, and P-Akt (Cell Signaling); P-PPARγ (Santa Cruz); and β-actin (Sigma); and TATA box binding protein (Abcam, Cambridge, MA) as a loading control followed by the corresponding anti-mouse or anti-rabbit secondary antibody (Thermo Scientific, Rockford, IL) at room temperature. Proteins were detected by chemiluminescence (West Pico or West Dura Kits; Pierce). Densitometry was measured in Photoshop, and fold increases were calculated by normalizing to actin and determining the relative increase compared with control samples.
Mouse Colon Crypt Isolation and Detection of Reactive Oxygen Species
The peritoneum of anesthetized mice was opened so that nicks could be cut in the rectum and near the cecum. The colons were flushed with 20 mL of Hanks’ Balanced Salt Solution (37°C, pH 7.5, Ca+ and Mg+ free). The thoracic cavity was next opened, and 30 mL of 30 mM EDTA+ in Hanks’ Balanced Salt Solution was perfused via the left ventricle in 3 minutes and the vena cava was cut. The colon was next dissected from the mouse, inside out, and placed in a tube with 1 mM EDTA+ in Hanks’ Balanced Salt Solution (wash buffer) with 10 mM dithiothreitol and vortexed for 10 seconds followed by a 10-minute incubation at room temperature. Next, the tissue was moved into new wash buffer and incubated for 10 minutes at 37°C. The tissue was transferred into mini bead-beater tubes (Biospec, Bartlesville, OK) with new wash buffer, shaken with a mini bead-beater (2500 revolutions per minute, 20 seconds), and crypts settled to the bottom of the tubes set on ice.
Whole crypts were resuspended in RPMI plus reactive oxygen species (ROS) detection Reagent (Enzo) with or without 20 mM 5-ASA (pH 7.4) for 1 hour at 37°C. H2O2-treated samples were given 50 mM H2O2 for 5 minutes before washing. Crypts were overlayed with Cygel (Biostatus) plus DRAQ5 (Cell Signaling) according to the manufacturer’s instructions and imaged on a Zeiss LSM 510 confocal microscope (Northwestern University Cell Imaging Facility). One mouse was used for each ex vivo experiment, and 5–6 crypts were analyzed per condition. The experiment was repeated 3 times.
Flow Cytometry
HT-29 cells were cultured and stimulated as described above. Single cell suspensions were fixed immediately after trypsin detachment from petri dishes with an equal volume of prewarmed Phosflow Fix Buffer I (BD, Franklin Lakes, NJ) and incubated at 37°C for 10 minutes. Cells were washed with PBS, pelleted, and permeabilized with Phosflow Perm Buffer III and then incubated for 30 minutes at 4°C. Cells were then washed twice followed by staining with p-Akt PE (BD) and Ki-67 FITC (BD). For ROS detection, mouse colon epithelial cell crypts were isolated as described above followed by incubation in 1.2 U dispase (BD) and 25 μg DNase (Sigma) plus MgCl for 3 minutes, 37°C. Single cell suspensions were incubated with mouse Fc Block (Miltenyi, Auburn, CA) followed by CD45 and CD44 surface staining for 30 minutes on ice. After washing, samples were resuspended in PBS plus ROS Detection Reagent (Enzo, Farmingdale, NY) for 1 hour at 37°C. After 30 minutes, some samples received 20 mM 5-ASA for the remaining time. H2O2 was added to the indicated samples in the final 5 minutes. As a control for 5-ASA quenching, 5-ASA was also added to H2O2-treated tubes immediately before flow cytometry. There was no significant difference in fluorescence between H2O2 alone and H2O2 plus 5-ASA–added post-ROS detection reagent staining (data not shown). All other samples were washed and resuspended in PBS plus Dapi for dead cell exclusion. For each experiment, cells were collected on an FACSCantoII (BD) and analyzed using Flowjo (Treestar, Ashland, OR).
Statistical Analysis
P values were calculated by Graphpad Prism software using analysis of variance followed by a Tukey posttest for comparison of more than 2 data sets. Significance (*) refers to P values <0.05 between CUC and 5-ASA. A Student’s t test was performed when 2 groups were compared.
RESULTS
5-ASA Impairs ROS Levels in PCs
Given that 5-ASA is a potent oxygen radical scavenger, we tested its effect on mouse crypt intestinal epithelial cell (IEC) exposed to hydrogen peroxide. Intracellular ROS stress was measured using an ROS detection reagent (see Materials and Methods). Examination of crypts treated with H2O2 indicates that ROS levels increased in regions corresponding to proliferating progenitor transit amplifying cells immediately above the crypt base. Quantification of intact crypt images suggested 5-ASA treatment–attenuated ROS levels (Fig. 1B, C). To quantify the effects of 5-ASA on intracellular ROS, IECs were analyzed by flow cytometry using CD44 as a marker of lower ISC/PC.46 Data in Figure 1D indicate that ROS levels were highest in CD44+ cells. Furthermore, data show that 5-ASA reduced ROS levels by over 40% compared with H2O2-treated control IEC (Fig. 1E). Together, the findings suggest that 5-ASA reduces oxidant stress in populations of lower crypt PC.
FIGURE 1.
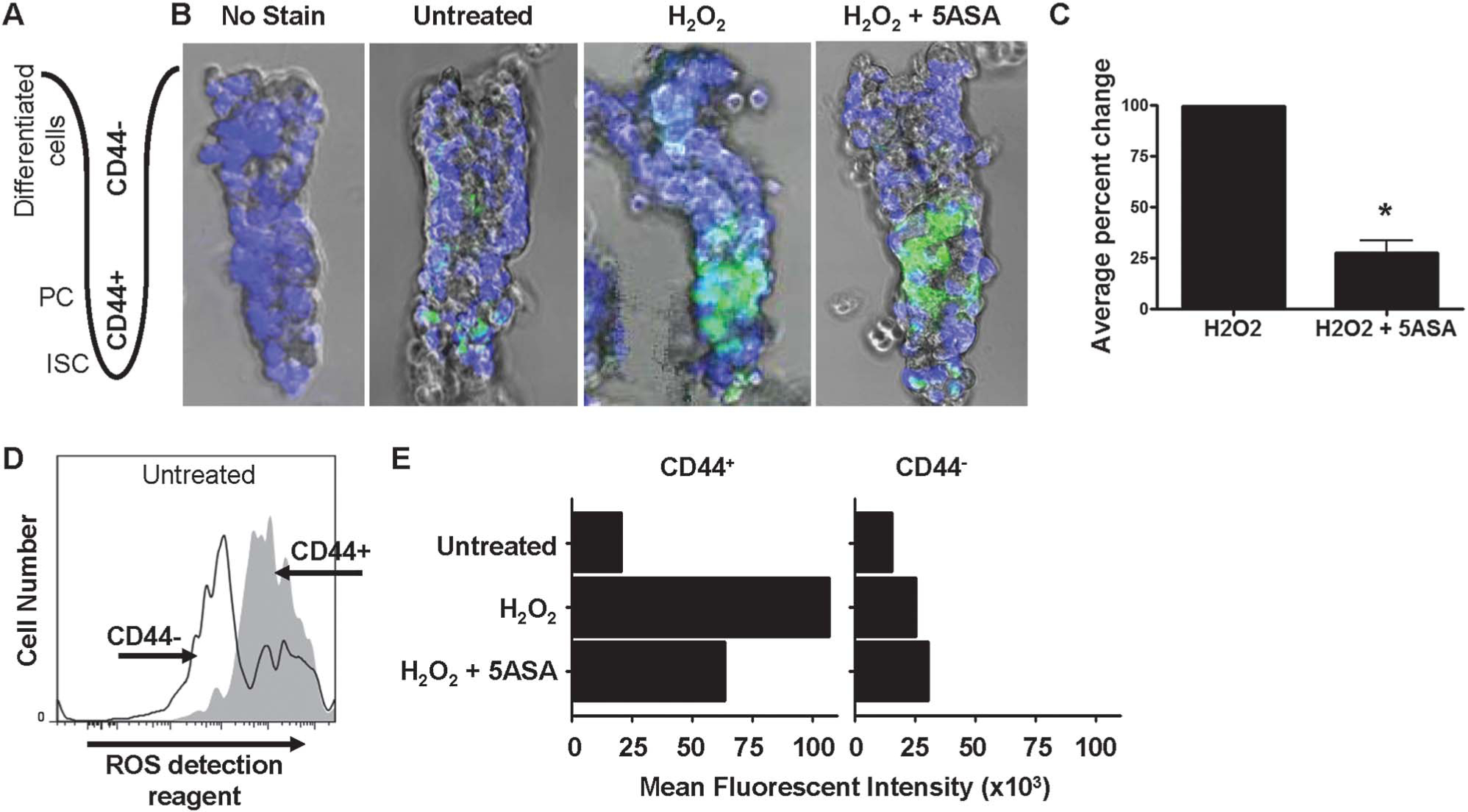
Basal ROS is elevated in intestinal PC. A, Cartoon of colon crypt showing the orientation of the intact crypts that were isolated from mice and imaged by live-cell fluorescent and light microscopy where CD44− differentiated IECs are at the top and CD44+ ISC/PC are at the bottom. B, Images of intact colonic crypts treated with H2O2 with or without 5-ASA stained with the ROS detection reagent (green) and DRAQ5 (blue pseudocolor) to stain all nuclei. Some crypts were not stained with the ROS detection reagent or treated with H2O2 as controls. C, Percent change in the relative fluorescent intensity of ROS detection reagent staining between H2O2 with or without 5-ASA. Crypt images were analyzed in ImageJ, and calculations were performed using peak values in the lower crypt. Average value of 3 independent experiments is shown (*P < 0.05). D, Flow cytometry data showing the number of ROS detection reagent–stained mouse colon IEC. Live, CD45−, CD44+ cells were gated on to determine the mean fluorescent intensity of each cell population, as shown in E of ROS detection reagent–stained cells untreated or treated with H2O2, with or without 5-ASA.
5-ASA Increases Antioxidant Gene Expression in Ulcerative Colitis
To examine the effect of 5-ASA on genes involved in oxidant stress, biopsies were collected from untreated and 5-ASA–treated CUC patients. To control for tissue inflammation, samples from untreated patients were compared with those refractory to mesalamine treatment. This approach permits interrogation of mesalamine-induced changes independent of anti-inflammatory effects.39 We assessed inflammatory scores by hematoxylin and eosin and disease activity by the UCDAI index (see Materials and Methods and Fig. A and B, Supplemental Digital Content 2, http://links.lww.com/IBD/A188). Only patients with scores greater than 2 for inflammation and 7 for UCDAI were included in this study. Catalase is an enzyme responsible for the breakdown of H2O2. Catalase is also a well-known PPARγ target gene.47 We therefore investigated catalase gene regulation in colitis patients by qRT-PCR. Of note, catalase messenger RNA (mRNA) levels were 50% higher in 5-ASA–treated colitis patients compared with untreated CUC-positive controls (Fig. 2A). Conversely, expressions of GPX1 and GPX2, 2 other key enzymes responsible for the decomposition of H2O2 in the intestine, were not affected by 5-ASA (Fig. 2B). Expressions of ROS-enhancing enzymes, NOX1 and Duoxa2, were elevated in CUC with no apparent effect of 5-ASA (Fig. 2B). Notably, Duoxa2 averaged nearly 100-fold higher in both groups of colitis patients. Together, these data suggest that 5-ASA enhances expression of catalase, a major antioxidant gene, while not directly affecting expression of genes involved in mediating IEC oxidant stress in CUC.
FIGURE 2.
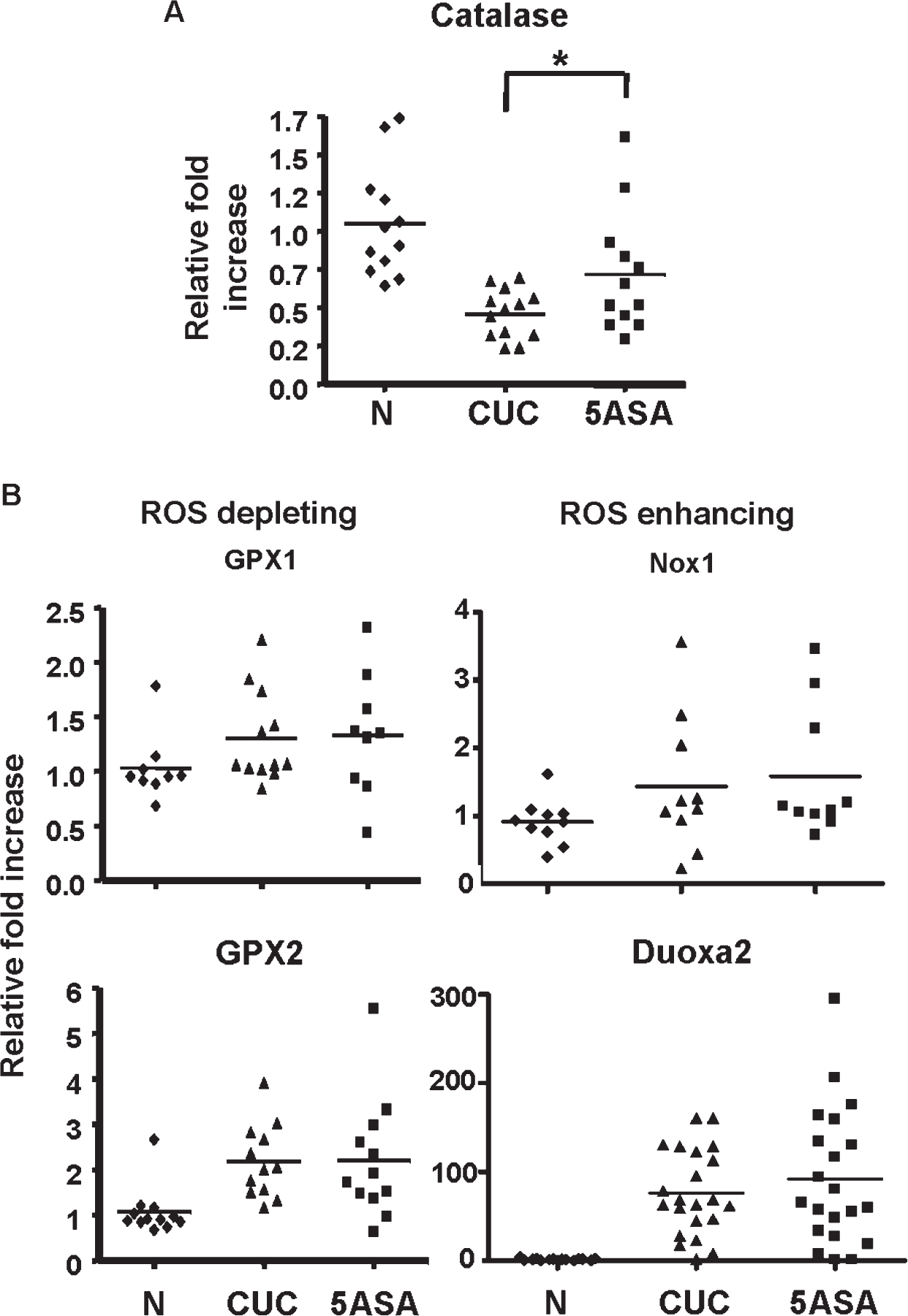
5-ASA alters ROS gene expression in CUC patients. Colon biopsies were taken during endoscopy from normal patients (N) or active CUC patients with (5-ASA) or without (CUC) 5-ASA. Only CUC patients with comparative UCDAI and H&E inflammatory scores were used in these experiments (see Fig., Supplemental Digital Content 2, http://links.lww.com/IBD/A188). qRT-PCR was performed on mRNA from whole biopsy tissue for the ROS gene catalase (A) and several ROS depleting and enhancing genes (B). * indicate where a significant change was seen with 5-ASA treatment. n ≥ 10 in each group. H&E, hematoxylin and eosin.
5-ASA Increases PPARγ Transcriptional Activity
Given that 5-ASA is a known PPARγ agonist, we determined whether 5-ASA altered levels of PPARγ inactivated by mitogen-activated protein kinase phosphorylation at Serine 82/112 (P-PPARγ). Analysis of protein extracts from patient biopsies shows that 5-ASA reduced P-PPARγ levels an d increased nuclear accumulation of PPARγ in colitis patients (Fig. 3A and see Fig. A, Supplemental Digital Content 3, http://links.lww.com/IBD/A189). These data led us to interrogate expression of PPARγ target genes in patient biopsies (Fig. 3B). Data show that 5-ASA increased mRNA levels for PPARγ targets, kruppel-like factor 4, HMGCS2, and keratin 20 by over 25% compared with levels detected in patients with untreated active colitis. Results also revealed that 5-ASA induced expression of PPARγ mRNA. Significant changes in PTEN mRNA expression were not detected (see Fig. B, Supplemental Digital Content 3, http://links.lww.com/IBD/A189). Together, these data indicate that mesalamine promotes PPARγ transcriptional activity independent of its anti-inflammatory properties in human inflammatory bowel disease.
FIGURE 3.
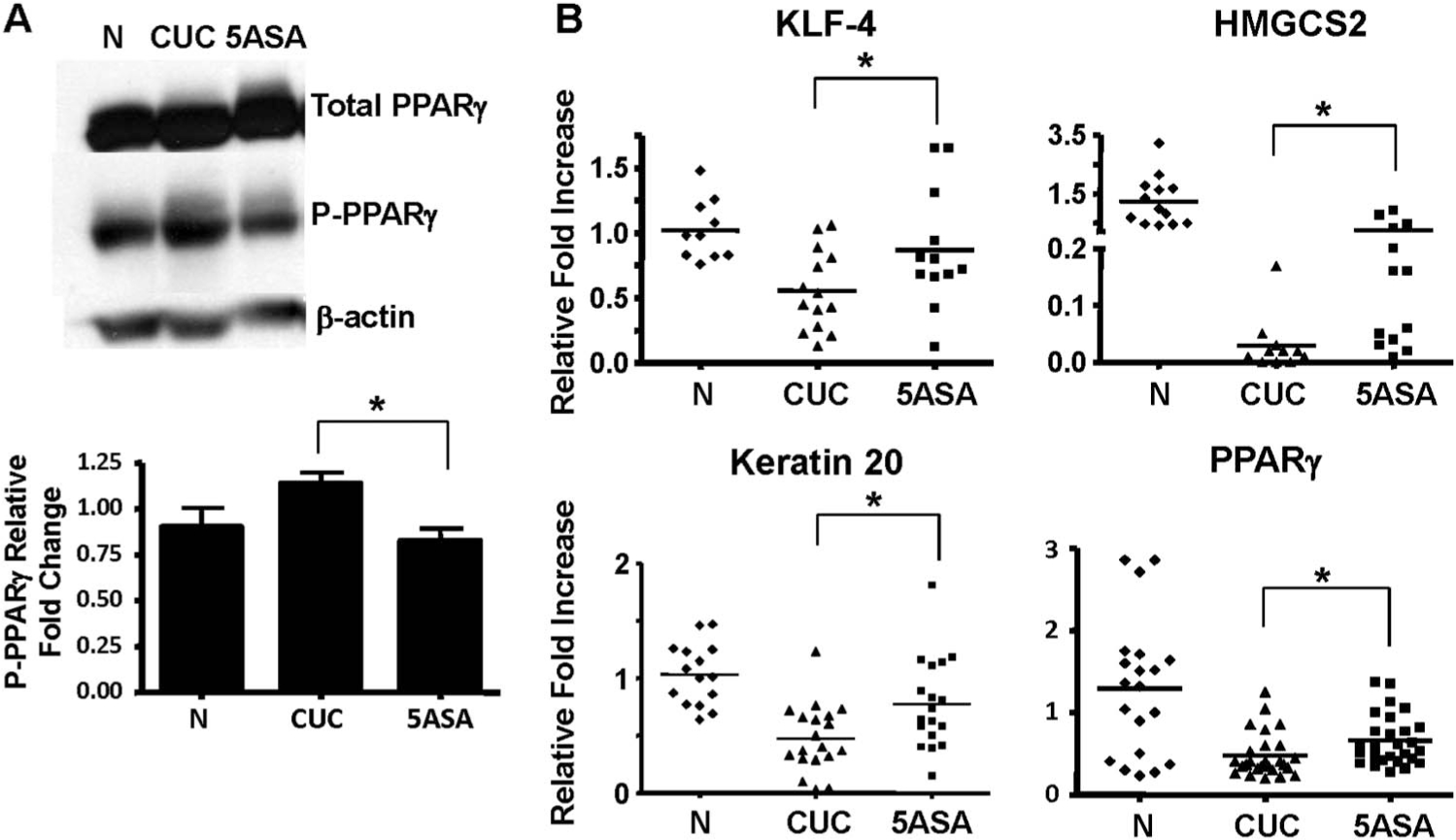
PPARγ activity is impaired in CUC and improved with 5-ASA. A, WB analysis of total PPARγ and transcriptionally inactive P-PPARγ from whole cell extracts from biopsies obtained from normal, active CUC, and 5-ASA–treated CUC patients (top). Densitometry of western blots performed on 5 individual patients per group indicates the decrease in P-PPARγ detection in CUC with 5-ASA compared to untreated patients (normalized to β-actin) is statistically significant. B, qRT-PCR of PPARγ target genes comparing normal and 5-ASA–untreated versus 5-ASA–treated CUC patients. * indicate significant changes in mRNA levels in the CUC patient groups. WB, Western blot.
5-ASA–Induced PPARγ Increases PTEN Transcription and Function
To further examine regulation of PPARγ target genes by 5-ASA, mRNA levels were analyzed in HT-29 cells after serum repletion. Data in Figure 4A show that 5-ASA treatment of cells enhanced transcription of kruppel-like factor 4 and keratin 20 mRNA with minimal changes detected for PTEN (Fig. 4A). We therefore examined whether 5-ASA affected PPARγ regulation of PTEN transcription by ChIP assay. ChIP revealed that 5-ASA increased binding of PPARγ to the PTEN promoter (Fig. 4B). The failure to detect a clear increase in PTEN mRNA (Fig. 4A) suggests that beyond 5-ASA–induced PPARγ binding to the PTEN promoter, other pathways regulate PTEN mRNA levels in HT-29 cells.
FIGURE 4.
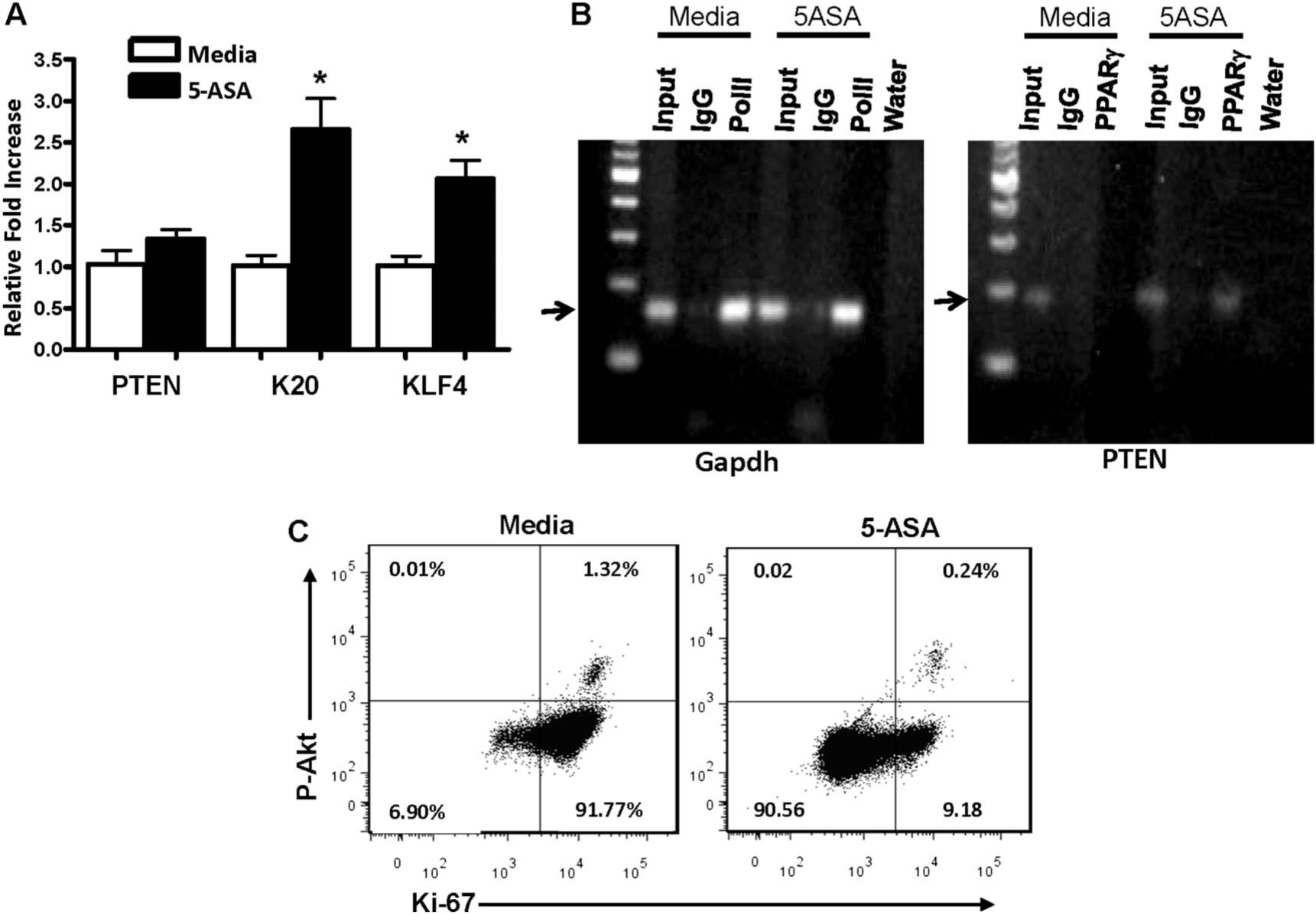
PTEN transcription and function is induced by 5-ASA–mediated PPARγ transcriptional activation in vitro. Serum-starved HT-29 cells were stimulated by the reintroduction of serum plus Conditioned Media before lysing cells untreated or treated with 5-ASA. A, qRT-PCR quantifying mRNA expression of the PPARγ target genes shown. B, Images of agarose gels showing results from ChIP assays performed on lysates from the cells stimulated with complete media alone or with 5-ASA using the antibody indicated at the top of each lane followed by PCR amplification of either glyceraldehyde-3-phosphate dehydrogenase or PTEN-specific primers. C, Results from flow cytometry analysis of cells to determine the number of proliferating (Ki-67 positive) HT-29 cells with active phosphorylated Akt (P-Akt) after release from serum starvation by media alone or with 5-ASA treatment.
Because PTEN is the major negative regulator of PI3K/Akt signaling and Akt activity is a critical factor in cell proliferation and survival in IECs,48 we examined active P-Akt levels in HT-29 cells. Following 24 hours of 5-ASA treatment of HT-29 cells, cells were stained with antibodies directed against P-Akt (active) and the proliferation marker Ki-67. Flow cytometry revealed that 5-ASA reduced proliferation (93%–9.42%) and the proportion of cells with active Akt signaling by >50% (Fig. 4C). Together, the data were consistent with the notion that 5-ASA increases PPARγ-mediated PTEN and reduces Akt signaling and ultimately cell proliferation in HT-29 cells.
5-ASA Reduces PTEN Oxidation and Increases Activity
To further examine the possibility that 5-ASA affected PTEN signaling, patient biopsies were examined for total and phosphorylated (inactive) PTEN. Human biopsies examined by Western blot revealed an average 28% reduction in P-PTEN in tissue from 5-ASA–treated CUC compared with untreated active CUC (Fig. 5A). Additionally, we found a significant amount of PTEN protein in untreated patients in the inactive oxidized conformation, whereas PTEN in 5-ASA patients favored the active reduced conformation (Fig. 5B). Further analysis indicates that 5-ASA increased the relative levels (ratio) of activated (reduced) compared with inactivated (oxidized) PTEN (Fig. 5B). These data correlated with an increase in nuclear PPARγ levels (see Fig. A, Supplemental Digital Content 3, http://links.lww.com/IBD/A189). To determine whether posttranslational changes in PTEN regulation affected Akt signaling, human tissue was investigated for Akt activity by Western blot. P-Akt levels were 45% lower in biopsies from 5-ASA–treated patients compared with untreated CUC (Fig. 5C). To address the possibility that the decreased Akt was simply due to decreased PI3K receptor activation. We measured mRNA for several representative growth factors implicated in the enhanced PI3K signaling seen in UC (Insulin-like growth factor-1, connective tissue growth factor, and amphiregulin) and determined that there was no significant difference between the treated and untreated patients (see Fig., Supplemental Digital Content 4, http://links.lww.com/IBD/A190). Together, these data indicate that 5-ASA affects PTEN by increasing the relative amounts of active/inactive PTEN. Thus, the data are consistent with the conclusion that 5-ASA decreases Akt activation through alterations in PTEN protein conformation.
FIGURE 5.
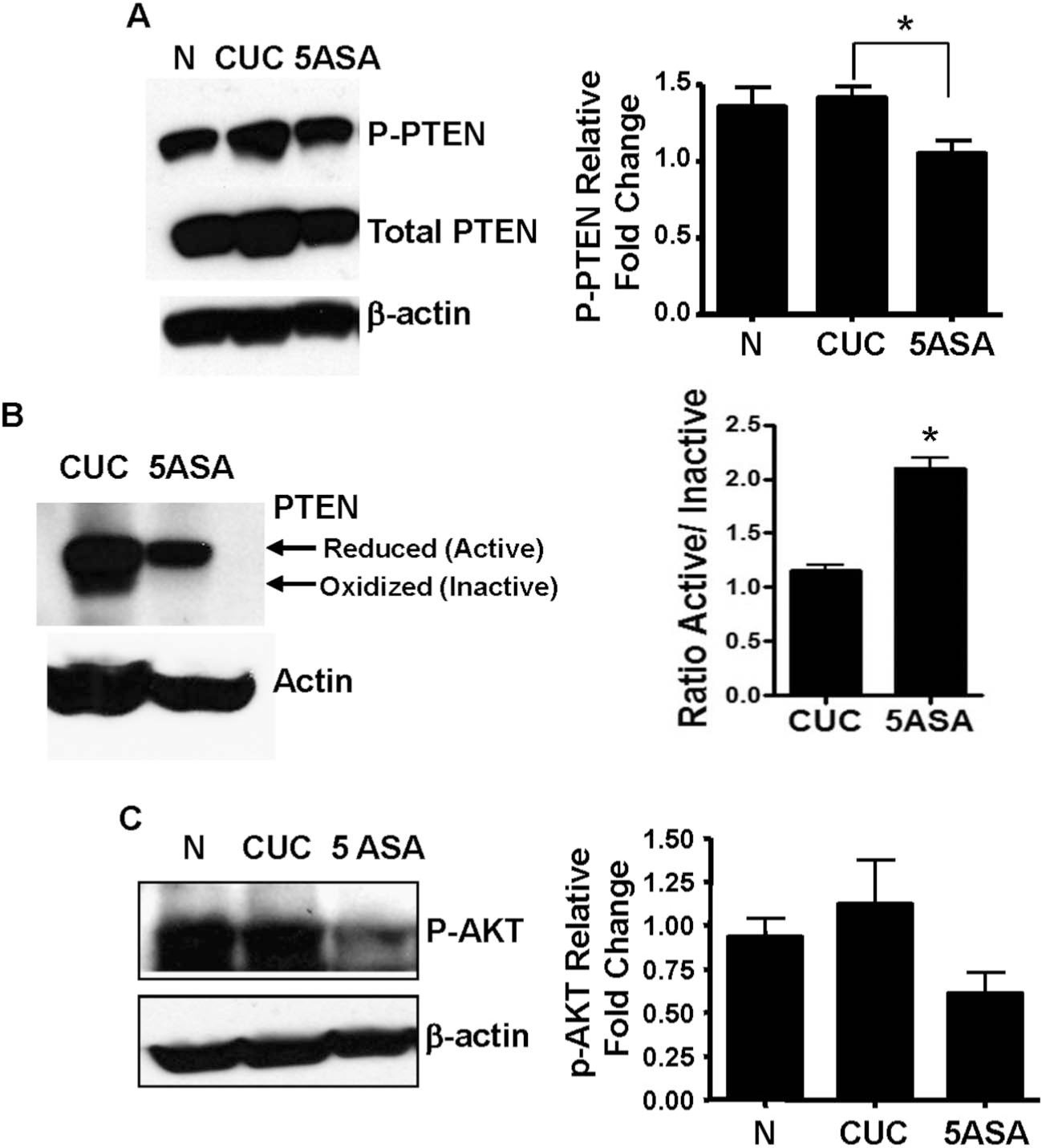
5-ASA–treated patients display reduced levels of PTEN inactivation. A, Protein extracts from N, CUC, and 5-ASA–treated human biopsy specimens were analyzed by WB for total and phosphorylated (inactivated) PTEN. Relative fold changes and statistical significance (*) were determined by densitometry from 6 patients per group relative to total actin. B, The oxidative status of PTEN was analyzed under nonreducing conditions by electrophoresis of lysates from CUC and 5-ASA–treated patients followed by WB. The bar graph (right) indicates mean ratios of active (reduced)/inactive (oxidized) PTEN in untreated CUC and 5-ASA–treated CUC. C, Akt activation was assessed by WB for P-Akt and densitometry of results from 7 patients per group relative to total actin. Actin serves as a loading control for all experiments. WB, Western blot. * indicate statistical significance.
5-ASA Inhibits β-catenin Signaling in CUC Specimens From Patients
We previously reported that 5-ASA reduces epithelial P-β-catenin552 levels in colitis patients.39 Additionally, we found that PI3K was a key upstream component to the increased Wnt/β-catenin signaling seen in colitis.49 Thus, effects detected here on PTEN and Akt activation (Fig. 5) suggest that 5-ASA may reduce β-catenin–induced gene transcription by virtue of its effect on PI3K signaling. To determine if 5-ASA reduces WNT/β-catenin target gene expression in epithelial cells, mRNA expression was analyzed in whole biopsies and by LCM. Analysis revealed that 5-ASA treatment significantly decreased c-myc, G-protein coupled receptor 49 and matrix metalloproteinase 7 transcription compared with untreated CUC patients (Fig. 6A). qRT-PCR results from epithelial-enriched LCM mRNA preparation paralleled results from whole tissue experiments (Fig. 6B). Purity of our dissections is represented in Fig., Supplemental Digital Content 5, http://links.lww.com/IBD/A191. These data extend prior findings39 and support the conclusion that patients on 5-ASA have decreased epithelial β-catenin signaling.
FIGURE 6.
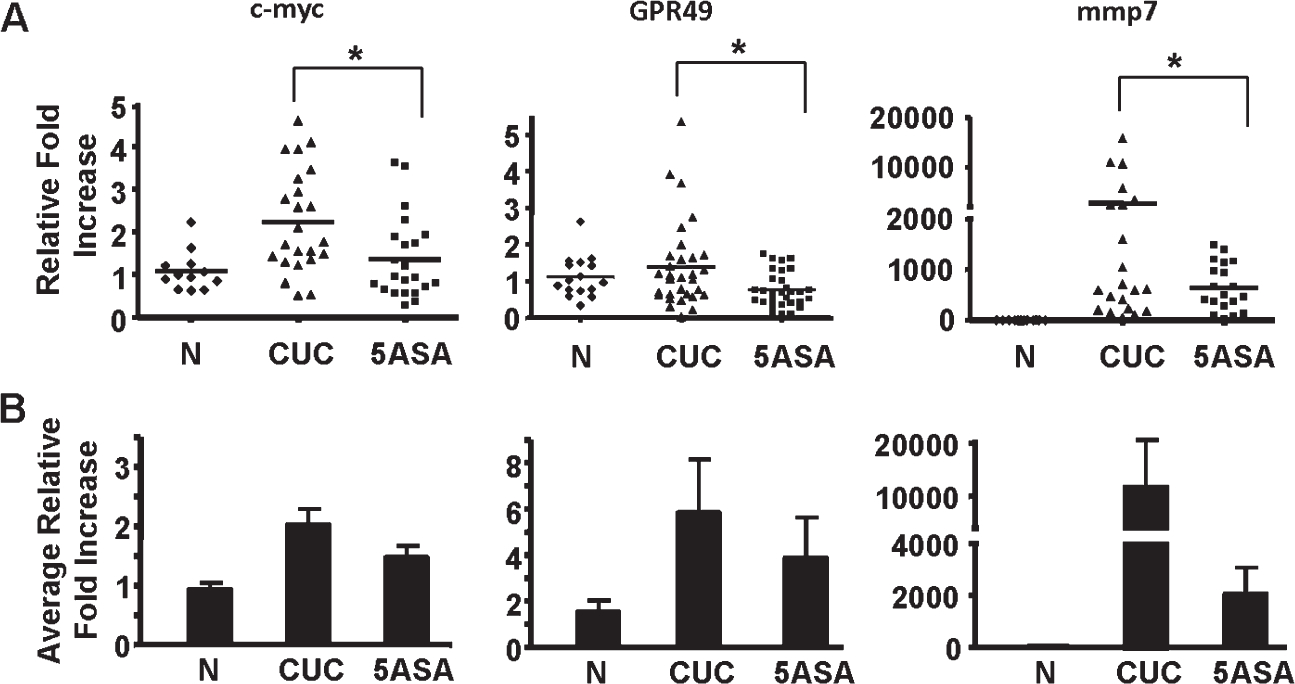
β-catenin signaling is decreased in 5-ASA–treated CUC patients independent of inflammation. qRT-PCR was performed to examine relative fold increases in mRNA expression of β-catenin target genes in normal, CUC, and 5-ASA colon biopsy specimens collected from human patients. mRNA was examined from whole biopsy tissue specimens (A) or LCM-extracted epithelial cells (B). * indicate where a significant decrease was seen with 5-ASA treatment. n ≥ 7 in each group.
DISCUSSION
Others and we have observed that inflammation in human and rodent models of colitis expands populations of proliferating ISC/PC in intestinal crypts.39,49–51 Given that data in the central nervous system indicate that ROS modulates the balance of self-renewal and differentiation,52,53 we considered the possibility that 5-ASA may impact signaling pathways that regulate these events. We found that ROS levels were higher in epithelial cells in the lower third of colonic crypts, which are also high in levels of CD44. Treatment of crypts with 5-ASA reduced levels of PC ROS. The overall reduction in oxidant stress by 5-ASA was supported by reduced oxidized PTEN (as discussed below). In addition, we detected increased levels of the antioxidant catalase in biopsies from 5-ASA–treated CUC patient. Previous reports had found that 5-ASA preferentially provided antioxidant activity within the membrane.54 Data presented here suggest that the antioxidant properties extend to sources of ROS within the cytosol. It is at this level that we propose that 5-ASA influences critical cell signaling events.
Data here indicate that 5-ASA impacts the function of PPARγ by multiple mechanisms. Previous results show that 5-ASA specifically binds to the PPARγ receptor, thus providing an agonist effect.55 Studies here help elucidate the downstream effects of this interaction. 5-ASA enhanced nuclear PPARγ protein levels in CUC patient samples and diminished mitogen-activated protein kinase phosphorylation–phosphorylated PPARγ. As phosphorylation by mitogen-activated protein kinase phosphorylation interferes with PPARγ trancriptional activity,56 these data provide an additional mechanism for how 5-ASA may increase the transcriptional activity of PPARγ. The enhanced transcriptional activity of PPARγ was supported by detection of increased mRNA for PPARγ target genes in 5-ASA–treated patients (Fig. 2B). ChIP assay confirmed that 5-ASA induced PPARγ transactivation of the PTEN promoter. The PPARγ-independent transcription detected in untreated HT-29 cells was likely due to other transcription factors such as p53, which also plays a role in PTEN regulation. It is possible that 5-ASA–induced PPARγ transcriptional activity enhanced PPARγ protein levels, given that PPARγ mRNA was also elevated in 5-ASA–treated CUC patients. Thus, we propose that 5-ASA treatment leads to modification of epithelial PPARγ expression, protein phosphorylation, and DNA binding that in combination increases target gene expression.
Absorption of active 5-ASA beyond the intestinal mucosa is limited and has potent antioxidant properties,57 making 5-ASA an ideal agent for chemoprevention of epithelial cancers. Our prior data39 indicated that 5-ASA reduced levels of Akt-phosphorylated β-catenin. We interpreted these results as 5-ASA attenuates ISC activation in CUC. Data here provide additional mechanistic explanation for 5-ASA–mediated reduction in PI3K signaling. Given that PTEN is a major negative regulator of PI3K, and a target of PPARγ, we consider that 5-ASA–mediated changes in PTEN activity are a potential link between PPARγ and PI3K signaling pathways. Our data demonstrate that 5-ASA–induced PPARγ directly binds to the PTEN promoter. Analysis of patient samples indicates that that 5-ASA decreased inactive PTEN. In addition, the antioxidant properties of 5-ASA increased relative levels of active PTEN by reducing the amount of oxidized PTEN (Figs. 4A, B). We provide further evidence of increased PTEN activity as determined by a substantial decrease in P-Akt in 5-ASA–treated patient samples and HT-29 cells. Together, these data support the notion that PTEN is a central mediator of 5-ASA effects on cell signaling.
Data presented here help explain the usefulness of mesalamine in chemoprevention of colitis associated cancer. Barker et al58 indicate that colorectal cancer begins in stem and PCs that reside in the crypt base. Examinations here reveal that cells just above the base of the crypt are susceptible to oxidant stress (Fig. 1). Although not formally tested here, we believe these cells to be long-lived progenitor populations derived from crypt base ISC. This view fits with the paradigm proposed by Clarke and colleagues that stem cells are protected by high levels of antioxidant enzymes whereas PC display more ROS.59 Data here indicate mesalamine reduces ROS levels in PC of the lower crypt. In prior publications, we reported that acute and chronic forms of inflammation induce β-catenin activation within PC populations.39,49 Our findings, together with data in this article, support the model that 5-ASA decreases PTEN oxidation, thereby attenuating PI3K and β-catenin signaling in PC. In several models of carcinogenesis, cancer stem cells are thought to be derived from activated (and mutated) PC.60 We conclude that effects described here allow mesalamine to attenuate PC activation and crypt IEC responses in chronic colitis. Given that colitis associated cancer likely originates in ISC and PC populations, these observations provide an attractive explanation for how mesalamine reduces neoplastic transformation in colitis.
Supplementary Material
ACKNOWLEDGMENTS
The authors would like to acknowledge Josette Ragheb Williams, MD, for her help in consenting patients, collecting biopsy specimens, and determining patient scores for this study. Supported by the National Institutes of Health (R01DK-054778 and R01AI-6171702 to T.A. Barrett). Research reported in this publication was supported by the National Institute of Diabetes and Digestive and Kidney Diseases of the National Institutes of Health under Award Number R01DK095662. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
Footnotes
The authors have no conflicts of interest to disclose.
Supplemental digital content is available for this article. Direct URL citations appear in the printed text and are provided in the HTML and PDF versions of this article on the journal’s Web site (www.ibdjournal.org).
REFERENCES
- 1.Bernstein CN, Blanchard JF, Kliewer E, et al. Cancer risk in patients with inflammatory bowel disease: a population-based study. Cancer. 2001;91: 854–862. [DOI] [PubMed] [Google Scholar]
- 2.Ekbom A, Helmick C, Zack M, et al. Ulcerative colitis and colorectal cancer. A population-based study. N Engl J Med. 1990;323:1228–1233. [DOI] [PubMed] [Google Scholar]
- 3.Karlen P, Lofberg R, Brostrom O, et al. Increased risk of cancer in ulcerative colitis: a population-based cohort study. Am J Gastroenterol. 1999;94:1047–1052. [DOI] [PubMed] [Google Scholar]
- 4.Tang J, Sharif O, Pai C, et al. Mesalamine protects against colorectal cancer in inflammatory bowel disease. Dig Dis Sci. 2010;55:1696–1703. [DOI] [PubMed] [Google Scholar]
- 5.Stolfi C, Fina D, Caruso R, et al. Cyclooxygenase-2-dependent and-independent inhibition of proliferation of colon cancer cells by 5-aminosalicylic acid. Biochem Pharmacol. 2008;75:668–676. [DOI] [PubMed] [Google Scholar]
- 6.Kennedy M, Wilson L, Szabo C, et al. 5-aminosalicylic acid inhibits iNOS transcription in human intestinal epithelial cells. Int J Mol Med. 1999;4: 437–443. [DOI] [PubMed] [Google Scholar]
- 7.Subramanian S, Rhodes JM, Hart CA, et al. Characterization of epithelial IL-8 response to inflammatory bowel disease mucosal E. coli and its inhibition by mesalamine. Inflamm Bowel Dis. 2008;14:162–175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kaiser GC, Yan F, Polk DB. Mesalamine blocks tumor necrosis factor growth inhibition and nuclear factor kappaB activation in mouse colonocytes. Gastroenterology. 1999;116:602–609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Stenson WF, Lobos E. Sulfasalazine inhibits the synthesis of chemotactic lipids by neutrophils. J Clin Invest. 1982;69:494–497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Bantel H, Berg C, Vieth M, et al. Mesalazine inhibits activation of transcription factor NF-kappaB in inflamed mucosa of patients with ulcerative colitis. Am J Gastroenterol. 2000;95:3452–3457. [DOI] [PubMed] [Google Scholar]
- 11.Egan LJ, Mays DC, Huntoon CJ, et al. Inhibition of interleukin-1-stimulated NF-kappaB RelA/p65 phosphorylation by mesalamine is accompanied by decreased transcriptional activity. J Biol Chem. 1999;274:26448–26453. [DOI] [PubMed] [Google Scholar]
- 12.Kim YH, Kim MH, Kim BJ, et al. Inhibition of cell proliferation and invasion in a human colon cancer cell line by 5-aminosalicylic acid. Dig Liver Dis. 2009;41:328–337. [DOI] [PubMed] [Google Scholar]
- 13.Nielsen OH, Munck LK. Drug insight: aminosalicylates for the treatment of IBD. Nat Clin Pract Gastroenterol Hepatol. 2007;4:160–170. [DOI] [PubMed] [Google Scholar]
- 14.Rubin DT, LoSavio A, Yadron N, et al. Aminosalicylate therapy in the prevention of dysplasia and colorectal cancer in ulcerative colitis. Clin Gastroenterol Hepatol. 2006;4:1346–1350. [DOI] [PubMed] [Google Scholar]
- 15.Velayos FS, Terdiman JP, Walsh JM. Effect of 5-aminosalicylate use on colorectal cancer and dysplasia risk: a systematic review and metaanalysis of observational studies. Am J Gastroenterol. 2005;100:1345–1353. [DOI] [PubMed] [Google Scholar]
- 16.Ahnfelt-Ronne I, Nielsen OH, Christensen A, et al. Clinical evidence supporting the radical scavenger mechanism of 5-aminosalicylic acid. Gastroenterology. 1990;98:1162–1169. [DOI] [PubMed] [Google Scholar]
- 17.Gionchetti P, Guarnieri C, Campieri M, et al. Scavenger effect of sulphasalazine (SASP), 5-aminosalicylic acid (5-ASA), and olsalazine (OAZ). Gut. 1990;31:730–731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Luciani MG, Campregher C, Fortune JM, et al. 5-ASA affects cell cycle progression in colorectal cells by reversibly activating a replication checkpoint. Gastroenterology. 2007;132:221–235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Brown WA, Farmer KC, Skinner SA, et al. 5-aminosalicyclic acid and olsalazine inhibit tumor growth in a rodent model of colorectal cancer. Dig Dis Sci. 2000;45:1578–1584. [DOI] [PubMed] [Google Scholar]
- 20.Bus PJ, Nagtegaal ID, Verspaget HW, et al. Mesalazine-induced apoptosis of colorectal cancer: on the verge of a new chemopreventive era? Aliment Pharmacol Ther. 1999;13:1397–1402. [DOI] [PubMed] [Google Scholar]
- 21.Parenti S, Ferrarini F, Zini R, et al. Mesalazine inhibits the beta-catenin signalling pathway acting through the up-regulation of mu-protocadherin gene in colo-rectal cancer cells. Aliment Pharmacol Ther. 2010;31:108–119. [DOI] [PubMed] [Google Scholar]
- 22.Rousseaux C, Lefebvre B, Dubuquoy L, et al. Intestinal antiinflammatory effect of 5-aminosalicylic acid is dependent on peroxisome proliferator-activated receptor-gamma. J Exp Med. 2005;201:1205–1215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Sarraf P, Mueller E, Jones D, et al. Differentiation and reversal of malignant changes in colon cancer through PPARgamma. Nat Med. 1998;4:1046–1052. [DOI] [PubMed] [Google Scholar]
- 24.Gupta RA, Sarraf P, Mueller E, et al. Peroxisome proliferator-activated receptor gamma-mediated differentiation: a mutation in colon cancer cells reveals divergent and cell type-specific mechanisms. J Biol Chem. 2003;278:22669–22677. [DOI] [PubMed] [Google Scholar]
- 25.Gupta RA, Brockman JA, Sarraf P, et al. Target genes of peroxisome proliferator-activated receptor gamma in colorectal cancer cells. J Biol Chem. 2001;276:29681–29687. [DOI] [PubMed] [Google Scholar]
- 26.Patel L, Pass I, Coxon P, et al. Tumor suppressor and anti-inflammatory actions of PPARgamma agonists are mediated via upregulation of PTEN. Curr Biol. 2001;11:764–768. [DOI] [PubMed] [Google Scholar]
- 27.Yki-Jarvinen H. Thiazolidinediones. N Engl J Med. 2004;351:1106–1118. [DOI] [PubMed] [Google Scholar]
- 28.Lewis JD, Lichtenstein GR, Deren JJ, et al. Rosiglitazone for active ulcerative colitis: a randomized placebo-controlled trial. Gastroenterology. 2008;134:688–695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Liang HL, Ouyang Q. A clinical trial of combined use of rosiglitazone and 5-aminosalicylate for ulcerative colitis. World J Gastroenterol. 2008;14:114–119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Burgermeister E, Seger R. MAPK kinases as nucleo-cytoplasmic shuttles for PPARgamma. Cell Cycle. 2007;6:1539–1548. [DOI] [PubMed] [Google Scholar]
- 31.Sarraf P, Mueller E, Smith WM, et al. Loss-of-function mutations in PPAR gamma associated with human colon cancer. Mol Cell. 1999;3:799–804. [DOI] [PubMed] [Google Scholar]
- 32.Fujisawa T, Nakajima A, Fujisawa N, et al. Peroxisome proliferator-activated receptor gamma (PPARgamma) suppresses colonic epithelial cell turnover and colon carcinogenesis through inhibition of the beta-catenin/T cell factor (TCF) pathway. J Pharmacol Sci. 2008;106:627–638. [DOI] [PubMed] [Google Scholar]
- 33.Liu J, Wang H, Zuo Y, et al. Functional interaction between peroxisome proliferator-activated receptor gamma and beta-catenin. Mol Cell Biol. 2006;26:5827–5837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Taketo MM. Wnt signaling and gastrointestinal tumorigenesis in mouse models. Oncogene. 2006;25:7522–7530. [DOI] [PubMed] [Google Scholar]
- 35.Hurlstone A, Clevers H. T-cell factors: turn-ons and turn-offs. EMBO J. 2002;21:2303–2311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.He TC, Sparks AB, Rago C, et al. Identification of c-MYC as a target of the APC pathway. Science. 1998;281:1509–1512. [DOI] [PubMed] [Google Scholar]
- 37.Barker N, van Es JH, Kuipers J, et al. Identification of stem cells in small intestine and colon by marker gene Lgr5. Nature. 2007;449:1003–1007. [DOI] [PubMed] [Google Scholar]
- 38.Crawford HC, Fingleton BM, Rudolph-Owen LA, et al. The metalloproteinase matrilysin is a target of beta-catenin transactivation in intestinal tumors. Oncogene. 1999;18:2883–2891. [DOI] [PubMed] [Google Scholar]
- 39.Brown JB, Lee G, Managlia E, et al. Mesalamine inhibits epithelial beta-catenin activation in chronic ulcerative colitis. Gastroenterology. 2010; 138:595–605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Stambolic V, Suzuki A, de la Pompa JL, et al. Negative regulation of PKB/Akt-dependent cell survival by the tumor suppressor PTEN. Cell. 1998;95:29–39. [DOI] [PubMed] [Google Scholar]
- 41.Vazquez F, Grossman SR, Takahashi Y, et al. Phosphorylation of the PTEN tail acts as an inhibitory switch by preventing its recruitment into a protein complex. J Biol Chem. 2001;276:48627–48630. [DOI] [PubMed] [Google Scholar]
- 42.Lee S-R, Yang K-S, Kwon J, et al. Reversible inactivation of the tumor suppressor PTEN by H2O2. J Biol Chem. 2002;277:20336–20342. [DOI] [PubMed] [Google Scholar]
- 43.Schwab M, Reynders V, Loitsch S, et al. PPARgamma is involved in mesalazine-mediated induction of apoptosis and inhibition of cell growth in colon cancer cells. Carcinogenesis. 2008;29:1407–1414. [DOI] [PubMed] [Google Scholar]
- 44.Sutherland LR, Martin F, Greer S, et al. 5-Aminosalicylic acid enema in the treatment of distal ulcerative colitis, proctosigmoiditis, and proctitis. Gastroenterology. 1987;92:1894–1898. [DOI] [PubMed] [Google Scholar]
- 45.Truelove SC, Richards WC. Biopsy studies in ulcerative colitis. Br Med J. 1956;1:1315–1318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.van de Wetering M, Sancho E, Verweij C, et al. The β-catenin/TCF-4 complex imposes a crypt progenitor phenotype on colorectal cancer cells. Cell. 2002;111:241–250. [DOI] [PubMed] [Google Scholar]
- 47.Girnun GD, Domann FE, Moore SA, et al. Identification of a functional peroxisome proliferator-activated receptor response element in the rat catalase promoter. Mol Endocrinol. 2002;16:2793–2801. [DOI] [PubMed] [Google Scholar]
- 48.Sheng H, Shao J, Townsend CM Jr, et al. Phosphatidylinositol 3-kinase mediates proliferative signals in intestinal epithelial cells. Gut. 2003;52: 1472–1478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Lee G, Goretsky T, Managlia E, et al. Phosphoinositide 3-kinase signaling mediates beta-catenin activation in intestinal epithelial stem and progenitor cells in colitis. Gastroenterology. 2010;139:869–881, 881.e1–e9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Dahan S, Roda G, Pinn D, et al. Epithelial: lamina propria lymphocyte interactions promote epithelial cell differentiation. Gastroenterology. 2008;134:192–203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Humphries A, Graham TA, McDonald SA. Stem cells and inflammation in the intestine. Recent Results Cancer Res. 2011;185:51–63. [DOI] [PubMed] [Google Scholar]
- 52.Tsatmali M, Walcott EC, Crossin KL. Newborn neurons acquire high levels of reactive oxygen species and increased mitochondrial proteins upon differentiation from progenitors. Brain Res. 2005;1040:137–150. [DOI] [PubMed] [Google Scholar]
- 53.Smith J, Ladi E, Mayer-Pröschel M, et al. Redox state is a central modulator of the balance between self-renewal and differentiation in a dividing glial precursor cell. Proc Natl Acad Sci. 2000;97:10032–10037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Pearson DC, Jourd’heuil D, Meddings JB. The anti-oxidant properties of 5-aminosalicylic acid. Free Radic Biol Med. 1996;21:367–373. [DOI] [PubMed] [Google Scholar]
- 55.Dubuquoy L, Rousseaux C, Thuru X, et al. PPARγ as a new therapeutic target in inflammatory bowel diseases. Gut. 2006;55:1341–1349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Burns KA, Vanden Heuvel JP. Modulation of PPAR activity via phosphorylation. Biochim Biophys Acta. 2007;1771:952–960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Sandborn WJ, Hanauer SB. Systematic review: the pharmacokinetic profiles of oral mesalazine formulations and mesalazine pro-drugs used in the management of ulcerative colitis. Aliment Pharmacol Ther. 2003;17:29–42. [DOI] [PubMed] [Google Scholar]
- 58.Barker N, Ridgway RA, van Es JH, et al. Crypt stem cells as the cells-of-origin of intestinal cancer. Nature. 2009;457:608–611. [DOI] [PubMed] [Google Scholar]
- 59.Diehn M, Cho RW, Lobo NA, et al. Association of reactive oxygen species levels and radioresistance in cancer stem cells. Nature. 2009;458:780–783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Reya T, Morrison SJ, Clarke MF, et al. Stem cells, cancer, and cancer stem cells. Nature. 2001;414:105–111. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.