

Abstract
In recent decades, several viruses have resulted in large outbreaks with serious health, economic and social consequences. The current unprecedented outbreak of the new coronavirus, SARS-COV-2, necessitates intensive efforts for delivering effective therapies to eradicate such a deadly virus. Isatin is an opulent heterocycle that has been proven to provide tremendous opportunities in the area of drug discovery. Over the last fifty years, suitably functionalized isatin has shown remarkable and broad-spectrum antiviral properties. The review herein is an attempt to compile all of the reported information about the antiviral activity of isatin derivatives with an emphasis on their structure-activity relationships (SARs) along with mechanistic and molecular modeling studies. In this regard, we are confident that the review will afford the scientific community a valuable platform to generate more potent and cost-effective antiviral therapies based on isatin templates.
Keywords: Isatin; Indoline-2,3-dione; Viruses; SARS-COV-2; Antiviral drugs; Structure-activity relationships (SARs)
Introduction
Viral diseases are a worldwide threat affecting all human societies and contribute to significant threats to human health [1]. Viruses are the etiological agents in many epidemic and pandemic community outbreaks that result in substantial morbidity and mortality. In 2003, >800 deaths were reported due to the severe acute respiratory syndrome (SARS) epidemic originating from South China [2]. In 2012, acute respiratory viral disease called Middle East respiratory syndrome (MERS), caused by MERS coronavirus (MERS-COV), was reported in the Kingdom of Saudi Arabia with a > 35% mortality rate [3]. In late 2019, the latest coronavirus disease 2019 (COVID-19) pandemic emerged in China and has been a global concern since then. The novel severe acute respiratory syndrome coronavirus 2 (SARS-COV-2) has been confirmed as the causative agent of COVID-19 and has spread to over >200 territories, resulting in a significant number of fatalities (Table 1) [4, 5]. Moreover, many other viral diseases (Table 1) result in a significant number of deaths annually worldwide.
Table 1.
Common viral diseases with their estimated number of deaths
Vaccination and antiviral chemotherapy are considered the cornerstones of combating viral diseases [6]. Vaccination has been documented as a powerful tool in the eradication of fatal viral infections. In 1980, the smallpox vaccine played a vital role in eliminating the virus, and to date, several approved vaccines are available for fighting against >15 viral pathogens [7]. On the other hand, antiviral drug treatment is effective in controlling a wide range of viral infections, including HIV. Nevertheless, no antiviral drugs have been developed for many serious viral diseases, including the currently ongoing pandemic COVID-19. Moreover, many of the clinically used antiviral drugs have a wide range of shortcomings in terms of potency, selectivity and pharmacokinetic profiles [8]. In this regard, the high rate of viral strains resistant to the currently approved drugs together with the continuous emergence of new pathogenic viruses, exemplified by SARS-COV-2, dictates the development of new antiviral therapies. The most current popular approach in small molecule antiviral drug discovery is “one drug, multiple viruses”, which relies on the development of broad-spectrum antiviral agents (BSAAs) capable of inhibiting an array of human viruses [9].
Isatin (1H-indole-2,3-dione) (Fig. 1) is a privileged heterocyclic motif with a broad spectrum of biodynamic activities, such as anticancer [10], antitubercular [11, 12] antimalarial [13, 14] antibacterial [15] anticonvulsant [16] and antiviral [17, 18] activities. In addition, clinically successful isatin-based drugs, such as sunitinib, semaxanib and toceranib (Fig. 1), have been applied in the treatment of various diseases [19, 20].
Fig. 1.

Chemical structures of isatin, sunitinib, semaxanib and toceranib
The successful applications of isatin in clinical settings, the wide range of its pharmacological properties and different approaches of its possible structural manipulation have attracted the attention of medicinal chemists towards further elaboration of this opulent heterocycle [20, 21]. Accordingly, in the last few decades, there has been a surge of interest in the exploration of isatin derivatives against a wide range of pathogenic viruses. Despite the very enriched literature revealing the activity of isatin derivatives against a wide range of pathogenic viruses, not a single report about the antiviral activity of isatins was published until two years ago. The chemistry of isatins acting against only arboviruses was concisely reviewed by Gomes et al. in late 2018 [22]. To the best of our knowledge, no reports have comprehensively reviewed and discussed the antiviral activity of isatin derivatives. This review sheds light on recently reported antiviral activities of isatin-inspired compounds with the following objectives: (I) presentation of the most promising novel/new isatin-based antiviral compounds, (II) extensive discussion of their structure-activity relationships (SARs) with the aim of inspiring novel and creative design approaches, and (III) emphasizing future trends in the exploration of isatin scaffolds as promising antiviral cores.
Isatin derivatives as anti-HIV agents
In 2019, the WHO estimated that there were approximately 38 million HIV/AIDS cases globally, of which 690000 people died from HIV-related disorders [23]. Antiretroviral therapies (ART) have been documented to play a pivotal role in the treatment of HIV/AIDS patients [24]. However, the growing spread of resistance is the principal factor in ART failure, and hence, this necessitates the development of new anti-HIV drugs [25]. Motivated by the promising antiviral activities of fluoroquinolones and isatin derivatives, Pandeya et al. reported the anti-HIV activity of new norfloxacin-isatin Mannich bases [26]. Of the nine tested bases, compounds 1a and 1b (Fig. 2) were the most potent inhibitors of HIV-1 replication, with EC50 values of 11.3 and 13.9 µg/mL, respectively, with maximum protection ranging from 70–95%. With regard to SARs, installation of trimethoprim at C-3 and an electron-withdrawing group at C-5 of the isatin moiety appeared superior to the other substituents.
Fig. 2.
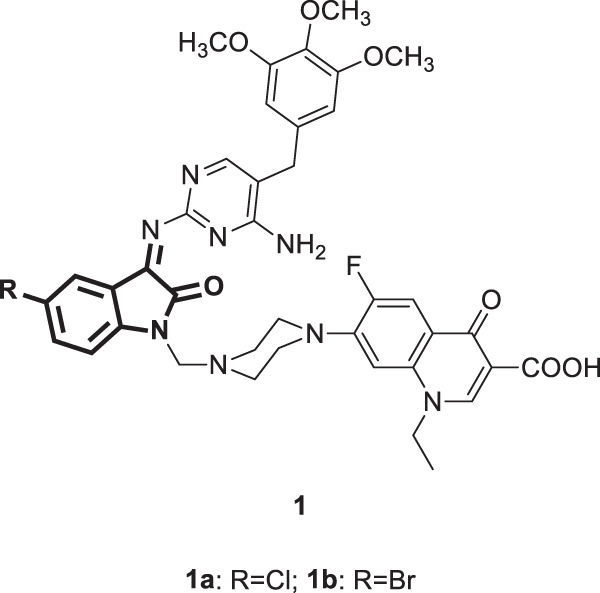
Chemical structures of norfloxacin-isatin Mannich bases
A set of isatin N-Mannich bases (Fig. 3) were synthesized by Pandeya et al. in 1999. Initially, the authors synthesized the Schiff bases of isatin with sulphadoxine, which were then converted into Mannich bases [27]. The anti-HIV activity of these bases was examined against HIV-1 and HIV-2 strains using the MTT assay. Compound 2a (Fig. 3) was identified as the most potent, exhibiting considerable activity up to 16% against both strains (EC50 more than 2 µg/mL). Regarding SARs, methyl-substituted compounds were less active than their unsubstituted counterparts, and morpholinomethyl substitution at position 1 of the isatin moiety appeared superior to the others.
Fig. 3.
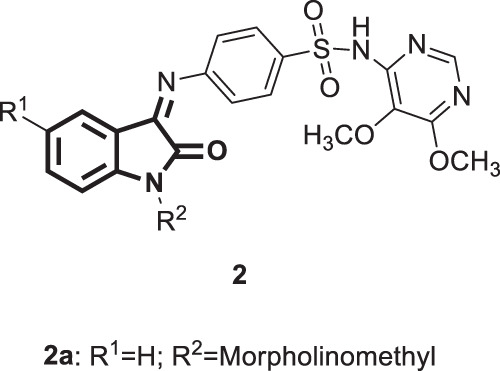
: Chemical structure of isatin N-Mannich bases
In an independent study, motivated by the reported potent antiviral activity of the thiosemicarbazone derivative methisazone [3] (Fig. 4), in the prophylaxis of smallpox [28], a series of Schiff and Mannich bases of isatin and thiosemicarbazide 4 (Fig. 4) was developed by Pandeya et al. [29]. The resultant compounds were screened for anti-HIV activity against HIV-1(III B) in MT-4 human cells. None of the investigated bases showed anti-HIV activity at a concentration level below their toxicity threshold. The authors attributed such a low activity to the relatively bulk hydrophobic p-chlorophenyl thiazolyl moiety at the end of the thiosemicarbazide group.
Fig. 4.
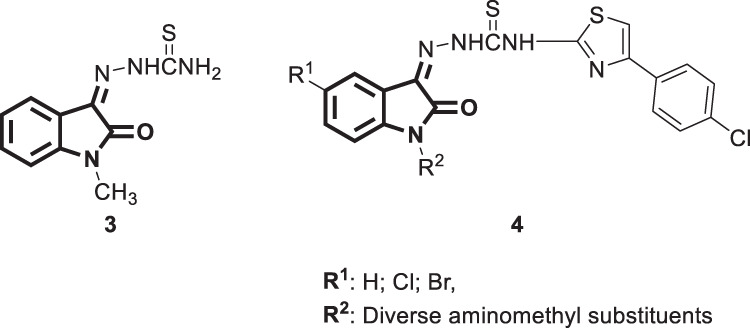
Chemical structures of Mannich bases of isatin and N-[4-(4’-chlorophenyl) thiazol-2-yl] thiosemicarbazide
Encouraged by the reported anti-HIV activity of quinazolinone scaffolds, Pandeya et al. [30] hybridized quinazoline derivatives with different isatins in an attempt to obtain new antiviral Schiff bases [5] (Fig. 5). The authors also derivatized the obtained Schiff bases adopting the Mannich reaction to yield their respective N-Mannich bases (Fig. 5). The resultant bases were examined for their antiviral activity against HIV-1 in human MT-4 cells. None of the tested compounds displayed any significant anti-HIV activity.
Fig. 5.
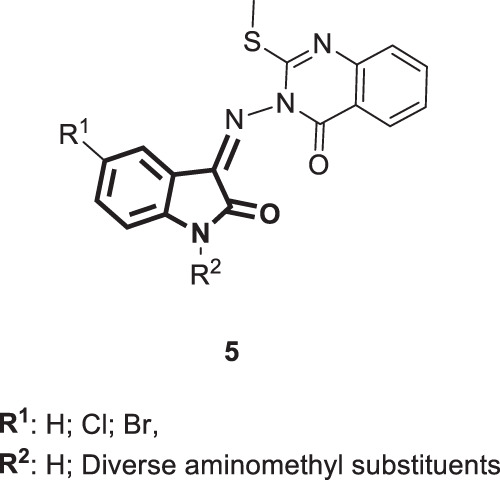
Chemical structures of the hybridized Schiff and Mannich bases
The two thiosemicarazone derivatives 6 and 7 (Fig. 6) were tested against HIV by Teitz et al. [31]. Their antiviral efficacy was concentration dependent, and the effective antiviral concentrations ranged from 0.17 to 2.04 µM for 6 and from 1.45 to 17.4 µM for 7. Viral inhibition by 50% was reported for 6 and 7 at concentrations of 0.34 and 2.9 µM, respectively. This inhibitory activity of these two thiosemicarbazones was ascribed to their ability to inhibit the synthesis of viral structural proteins. Therapeutic index values were determined for both compounds and found to be 20 and 30 for 6 and 7, respectively.
Fig. 6.
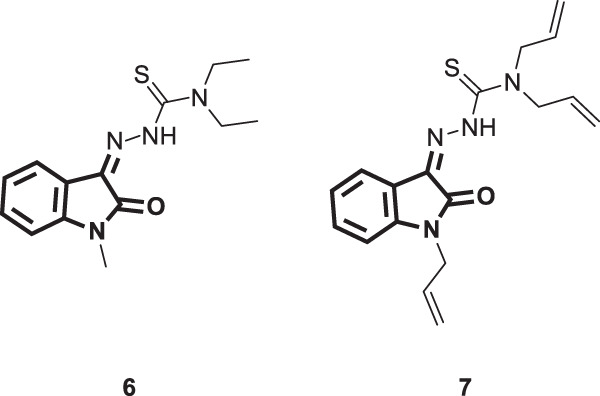
Chemical structures of thiosemicarazone derivatives
Sixty isatinyl thiosemicarbazone derivatives [32] (Fig. 7) were synthesized by Banerjee et al. in an attempt to obtain potent compounds for the treatment of HIV-TB coinfection [33]. Compounds 8a and 8b displayed promising activity against the replication of HIV-1 cells (EC50 1.69 and 4.18 µM, respectively). Better anti-HIV activity was associated with thiosemicarbazones with methoxy groups compared with hydroxy analogs. The authors attributed this to the involvement of the methoxy group in steric interactions with the aromatic residues of HIV-1 reverse transcriptase (RT), an essential enzyme in the replicative viral life cycle, which was further supported by in silico studies. The superior anti-HIV efficacy of 8a over 8b was ascribed to the establishment of hydrophobic interactions with the enzyme contributed by the chlorine atom at the para position of the phenyl piperazinyl methyl moiety and the methyl group that was present at C-5 of the isatin ring. Regarding SARs of the derivatives at C-5 of the isatin ring, higher activity was observed to be governed by the order of F = CH3 > Cl. Substitution by gatifloxacine, a fluoroquinolone, at N1 of the isatin moiety appeared superior to the other analogs. Furthermore, the 4-chlorophenyl piperazinyl methyl substituent at N1 was found to be better than the other aminomethyl groups. To gain insight into the possible mode of anti-HIV activity, 8a was examined for its capability to inhibit viral RT. Among the tested derivatives, compound 8a showed the highest inhibitory activity with an IC50 of 11.5 ± 1.5 µM.
Fig. 7.
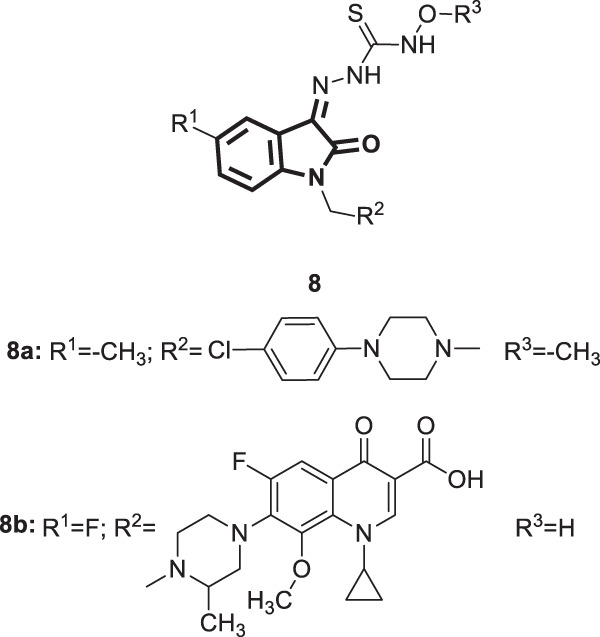
Chemical structures of isatinyl thiosemicarbazones 8 and the most potent compounds 8a and 8b
A set of aminopyrimidinimino isatin derivatives [34] (Fig. 8) was designed, synthesized and evaluated for their inhibitory activity on HIV-1 replication in MT-4 cells [35]. Compounds 9e, i, and l-o inhibited viral replication with EC50 values ranging from 12.1 to 62.1 µg/mL. Among these derivatives, compound 9l was the most potent, with a protection rate of >99% and SI > 13. Compound 9l showed inhibition of the HIV-1 RT enzyme (IC50 = 32.6 ± 6.4 µM). The anti-HIV activity of these derivatives did not correlate with their respective logP values. Antiviral and cytotoxicity assays against HCV showed analog 9k to be the most active, displaying 100% inhibition of viral replication with minimal cytotoxicity at 50 µg/mL. In addition, the synthesized compounds were tested for their in vitro antibacterial activity against 28 pathogenic bacteria, and the results revealed that all of the derivatives displayed mild to moderate activity against the tested bacteria. Compound 9e, which incorporates a 4-chlorophenyl piperazinomethyl moiety, was the best, and it was superior to lomefloxacin against 20 tested microorganisms. Moreover, primary screening against Mycobacterium tuberculosis strain H37Rv (at a concentration of 6.25 µg/mL) revealed compounds bearing a fluoroquinolone moiety (9 l–o) as the most potent as they completely inhibited mycobacterial growth, i.e., with MICs = 3.13 µg/mL.
Fig. 8.
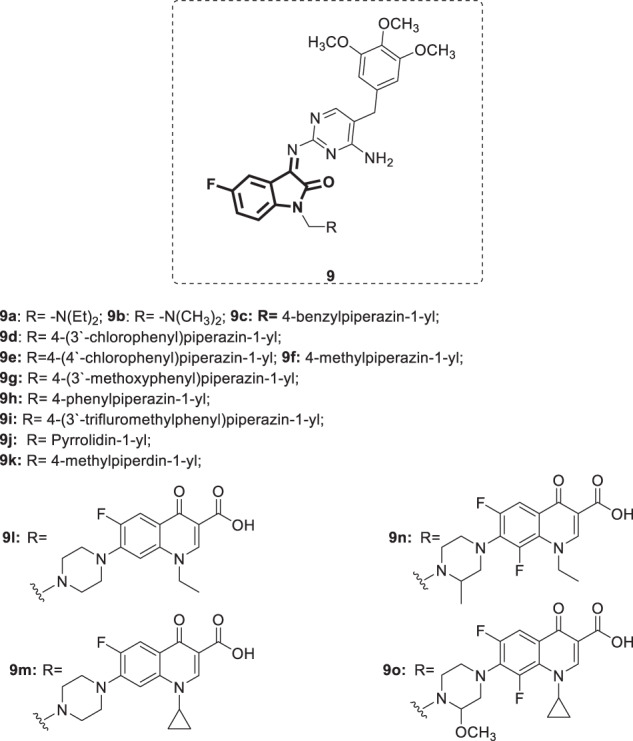
Chemical structures of aminopyrimidinimino isatin derivative 9
Based on reports defining the key structural features of nonnucleoside reverse transcriptase inhibitors (NNRTIs), a series of isatin β-thiosemicarbazone derivatives [36] (Fig. 9) was designed, synthesized and investigated for their anti-HIV activity in the CEM cell line [37]. The synthesized compounds demonstrated inhibition of viral replication with EC50 values ranging from 2.62 to >14.50 µM. Compounds 10c, f, and i were the most potent, with EC50 values in the range of 2.62–3.40 µM. Nevertheless, they were less potent than the positive control drug efavirenz. The other compounds were inactive below their respective toxicity thresholds.
Fig. 9.
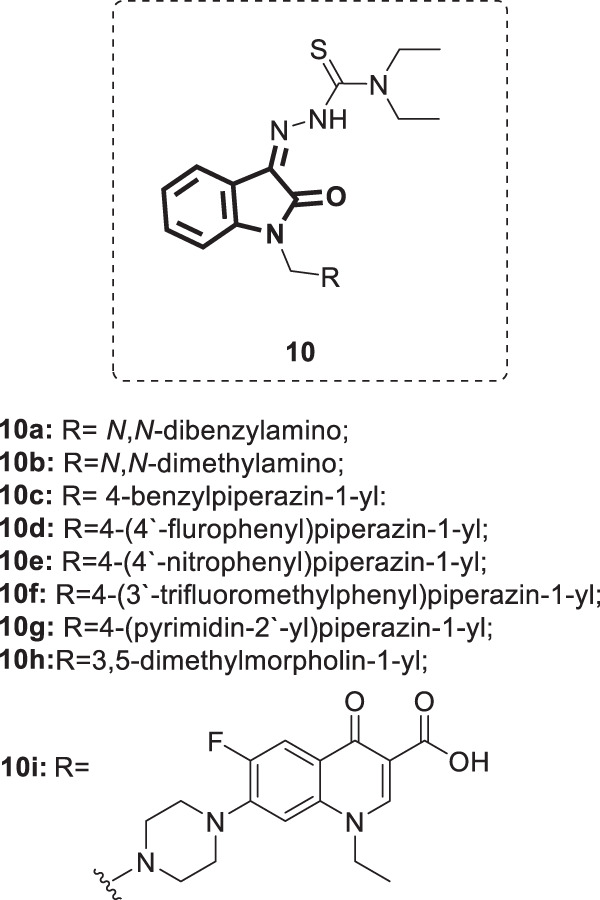
Chemical structures of isatin β-thiosemicarbazone derivatives
The highest activity and the least cytotoxicity were associated with compound 10f (EC50 = 2.62 µM and SI > 17.41). The superimposition technique indicated a good correlation between thiosemicarbazone and efavirenz with RMS values within the acceptable range. It is very difficult to draw a clear conclusion regarding SARs of these derivatives as the number of analogs is limited and the substitution pattern seems random.
Selvam et al. synthesized five Schiff‘ bases [6] (Fig. 10) by reacting isatin derivatives with sulfadimidine [38]. The obtained derivatives were evaluated for anti-HIV activity against both HIV-1 and HIV-2 in MT-4 cells using zidovudine as a positive control. The EC50 values for HIV-1 and HIV-2 inhibition were in the ranges of 8–15.3 µg/mL and 41.5–125 µg/mL, respectively. On the other hand, the standard drug had EC50 values of 1.2 and 0.62 ng/mL against HIV-1 and HIV-2, respectively. Cytotoxicity studies revealed that the toxic concentration of the investigated compounds was >125 µg/mL. While 11e showed the highest anti-HIV-2 activity (EC50 = 41.5 µg/mL), compounds 11a and e exhibited the most potent activity against HIV-1 (EC50 = 8 µg/mL). The SARs revealed that substitution by either electron-donating or electron-withdrawing groups at C-5 of the isatin motif was unfavorable to anti-HIV-1 activity and hence might not provide more potent derivatives. On the other hand, N-acetylation of position 1 in 11a increased the anti-HIV-2 activity 2-fold, showing promise for further molecular modification. The most active compounds were far less active than the reference drug; therefore, they still need to be modified.
Fig. 10.
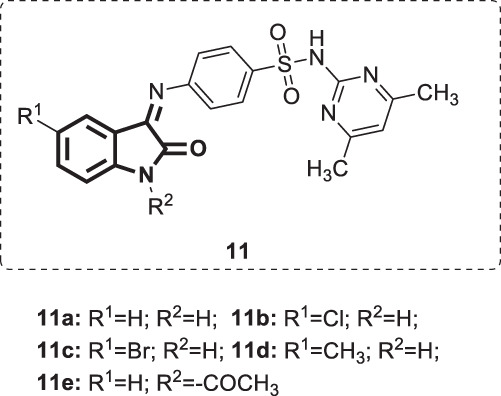
Chemical structures of isatin sulfadimidine Schiff’bases
Triazole is a structural subunit of some clinically successful antiviral drugs, such as ribavirin and taribavir, and could be useful in the development of more potent antiviral agents [39]. Accordingly, Schiff bases of isatin and aminotriazole 12 (Fig. 11) were designed and synthesized by Pandeya et al. [40]. Next, these bases were reacted with formaldehyde and various secondary amines to furnish the N-Mannich bases [8] (Fig. 11). Compounds 12a–c and 13a–i were tested for anti-HIV-1 activity in MT-4 cells. All bases were unable to inhibit the replication of HIV-1 at concentrations below the toxicity threshold (SI < 1).
Fig. 11.
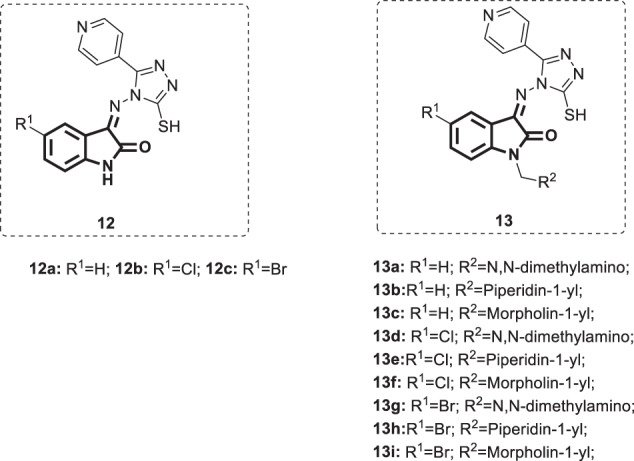
Chemical structures of Schiff bases 12 and Mannich bases 13 of isatin and (4‘-pyridyl)-4-amino-5-mercapto-4-(H)-1,2,4-triazole
Selvam et al. synthesized a series of isatine-sulfonamide hybrids [9] (Fig. 12), whose anti-HIV activity together with compound 11a,s was investigated against HIV-1 in MT-4 cells using zidovudine as a reference drug [41]. Compounds 14a–h were less active than compound 11a (EC50 = 13.6–116.06 µg/mL). Moreover, they showed very low inhibitory activity against HIV integrase (IC50 = 53.62–>250 µg/mL) compared with the positive control pyranodipyrimidine (IC50 = 0.03 µg/mL).
Fig. 12.
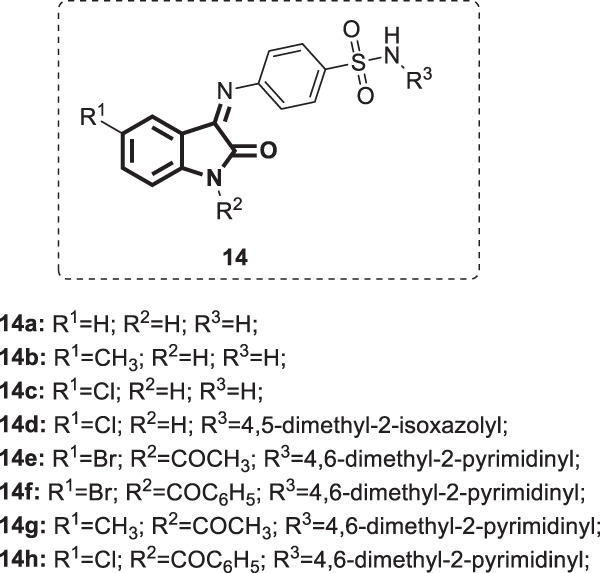
Chemical structures of isatin-sulfonamide derivatives
In continuation with their previous work [33], Sriram et al. designed and synthesized aminopyrimidinimino isatin hybrids [10] (Fig. 13) as a novel class of nonnucleoside reverse transcriptase inhibitors [42]. These hybrids were tested along with the standard drug nevirapine for their inhibitory activity on the replication of HIV-1 in both MT-4 and CEM cell lines. Although less active than nevirapine (EC50 = 0.1 μM), compounds 15c, l, and o were the most potent in the MT-4 cell lines, with EC50 values of 7.8, 5.6 and 7.6 μM, respectively, and maximum protection of 88.6-126% (SI > 12). The other compounds (15b, g, n, and p) showed protection in the range of 56–64.6% with SI values of 2–5. The SARs demonstrated that shortening of the two n-propyl groups in compound 15a (EC50 > 36 μM) to diethyl groups as in 15b (EC50 = 19.2 μM) did improve the activity. Replacement of the phenyl group attached to the piperazine in compound 15i (EC50 > 46 μM) with a pyridine group as in 15j (EC50 > 69 μM) did not boost the activity. In addition, introduction of the m-methoxyphenyl moiety as in 15g (EC50 = 22.6 μM) increased the potency 2-fold, while no effect was seen when the same group was introduced at ortho or para positions as in 15f and 15h, respectively. Replacement of the methoxy group in compound 15g by chloro or trifluoromethyl groups, as in 15d and 15k, respectively, resulted in a > 2-fold reduction in their potency. Substitution of the N-benzylpiperazine group in 15c with N-methylpiperazine as in 15e was detrimental to the activity, as was the replacement of the morpholin-1-yl group in 15l with pyrrolidin-1-yl in 15m. Preliminary screening of the synthesized compounds against HCV viral RNA replication at a 50 μg/mL concentration revealed compounds 15j and n–p as the most potent (inhibition of viral replication 80–84%) and least cytotoxic (cell growth >80%). Although compounds 15a, g, i, k, and m inhibited HCV viral replication in the range of 94–100%, they showed cytotoxicity, which necessitates their structural modification to obtain safer therapeutic candidates. These derivatives were also screened against M. tuberculosis strain H37Rv at a concentration of 50 μg/mL. The results showed that compounds bearing the fluoroquinolone motif (15n–p) were associated with the highest activity (complete inhibition of mycobacterial growth). The MICs of 15n–p were found to be 3.13 μg/mL, and they were free from cytotoxicity up to 62.5 μg/mL. Antibacterial activity evaluation of all of the synthesized compounds against 24 pathogenic bacteria revealed mild to moderate activity. Among these derivatives, 15n emerged as the most potent broad-spectrum chemotherapeutic agent active against HIV, HCV, M. tuberculosis and various pathogenic bacteria, making this compound eligible for further studies in the drug development process.
Fig. 13.

Chemical structures of aminopyrimidinimino isatin hybrids
In an independent study, Sriram et al. [43] synthesized a different set of aminopyrimidinimino isatin hybrids 16 (Fig. 14). In the next step, these hybrids were evaluated for their inhibitory activity on the replication of HIV-1 in both MT-4 and CEM cell lines. Compound 16n, bearing ciprofloxacin, was the most efficacious, with EC50 = 9.4 μg/mL, SI > 19 and a maximum protection of 101%. Compared with its analogs 9m (Fig. 8) and 15n (Fig. 13), compound 16n was more potent indicating the usefulness of -Cl substitution over those of -F or -Br. Compounds 16a–c, h, m, and p displayed maximum protection in the range of 59–95% with SI = 3. Relatively low activity was demonstrated by these derivatives in CEM cell lines (20–48%). The authors attributed such a low activity to the rapid rate of metabolism in the culture media. The SARs revealed that compound 16n bearing ciprofloxacin at N1 is twice as active as 16p, which carries lomefloxacin at the same position. Extension of diethyl groups in 16c to dibutyl groups in 16b improved the activity, as did their replacement with benzyl groups in 16a. A significant reduction in potency was observed when the 4-benzylpiperazin-1-yl group in 16d (>36.1 µg/mL) was replaced by a 4-methylpiperazin-1-yl group in 16i (>80 µg/mL). No increase in activity was found when the -Cl shifted from the meta position in 16e to the para position in 16f. Replacement of -Cl in 16f with -F in 16g resulted in a reduction in the anti-HIV activity, as did the insertion of a carbonyl group in 16f between the benzene ring and N4 of the piperazine ring to yield 16h. Replacement of 3′-Cl in 16e with a 3′-methoxy group in 16j did not enhance the potency. Shifting of the -CH3 group attached to the piperidine ring at position 2 in 16l (EC50 = 32.6 µg/mL) to position 4 as in 16m resulted in a > 2-fold reduction in activity (EC50 = 67.4 µg/mL). Investigation of all of the derivatives against HCV viral RNA replication at a 50 μg/mL concentration revealed compounds 16l, n, and o as the most potent (inhibition of viral replication 81–89%) and the least cytotoxic (cell growth>100%). Compounds 16e, f, and k were also effective in inhibiting HCV replication; however, they demonstrated significant cytotoxicity, which might hinder their further development. Similar to what has been reported by the same research group [42], compounds bearing the fluoroquinolone motif (16n–p) were associated with the highest antimycobacterial activity (100% inhibition of mycobacterial growth at 6.25 µg/mL). Further experiments revealed the MICs for compounds 16n and p to be 3.13 µg/mL; on the other hand, it was 1.56 µg/mL for 16o. Furthermore, compounds 16n–p were not cytotoxic to VERO cells up to 62.5 µg/mL. The antibacterial activity of compound 16 was evaluated against 24 pathogenic bacteria using a conventional agar dilution procedure. Generally, mild to moderate activity was observed; however, compounds 16np showed great potency among these derivatives. Therefore, compound 16n with broad spectrum antimicrobial activities could act as a lead molecule for further studies.
Fig. 14.

Chemical structures of aminopyrimidinimino isatin derivatives
Different isatin-lamivudine hybrids 17,18 (Fig. 15) were synthesized by Sriram et al. [44]. The obtained hybrids were evaluated against HIV-1 in a CEM cell line. Among them, five hybrids displayed potent antiviral activity. The results showed compound 17b to be equipotent to lamivudine with an EC50 of 0.0742 µM, CC50 of >200 µM and SI of >2100. The SARs demonstrated that the introduction of -F (17b and 18c and h) at the C-5 position of isatin boosted the activity (EC50 = 0.0742–1.16 µM) and resulted in less cytotoxic compounds (CC50 = 123–>200 µM), whereas -Cl (18b and e–g) was detrimental to activity (EC50 = 4.73–>12.5 µM) and increased toxicity (CC50 = EC50 = 4.73–12.5 µM), to the CEM cell lines. The Mannich bases [13] were less active than Schiff’s bases [12]. No significant difference in activity was observed between compounds bearing norfloxacin 18a–c and those incorporating ciprofloxacin 18d and e. Compounds 17 and 18 were evaluated for antimycobacterial activity against the M. tuberculosis H37Rv strain at a concentration of 6.25 µg/mL. While Schiff bases 17a and b moderately inhibited mycobacterial growth (56 and 82%, respectively), Mannich bases 18a–h displayed higher antimycobacterial activity (92–100% growth inhibition).
Fig. 15.

Chemical structures of isatin-lamivudine hybrids
In their quest to develop new anti-HIV therapeutics, Pawar et al. [45] designed various isatin analogs [14] (Fig. 16). Molecular docking analysis of these analogs was performed on five different crystal structures of RT complexed with five different ligands, namely, nevirapine, delaviridine, efavirenz, etravirine, and rilpivirine. The docking scores of 19a–c (Fig. 16) were comparable with those of these reference ligands, indicating their good binding affinity for this enzyme. The docking results revealed their potential binding in the NNRTI binding site in a similar mode to a known nonnucleoside inhibitor. H-bond interactions and hydrophobic interactions of isatin analogs with amino acid residues of the NNBP site seemed to play a major role. The ADME properties of the designed molecules were within the specified limits. With these promising in silico findings, the synthesis and biological testing of compounds 19a–c is highly recommended.
Fig. 16.

The designed isatin analogs
The isatin derivatives discussed here, such as compounds 6, 7, 8a, 10f and 17b, exhibited potent anti-HIV activity. The enriched SAR shown in Fig. 17 may provide new insight for further development of more potent candidates.
Fig. 17.

SAR of isatin derivatives with anti-HIV activity
Anti-SARS-COV isatin derivatives
Severe acute respiratory syndrome (SARS) is a viral atypical pneumonia associated with headache, high fever, myalgia and dizziness. Death may result from severe respiratory failure as a result of alveolar damage. Human coronavirus SARS-COV, belonging to the family Coronaviridae, was identified as the causative agent [46–48]. SARS originated in southern China in late 2002, and then the virus spread rapidly to more than 25 countries. During its initial outbreak, a high mortality rate (up to 15–19%) was reported [49]. With the emergence of the current COVID-19 pandemic, coronaviruses continue to represent a threat to humans; therefore, the possibility of SARS-COV reemergence cannot be overlooked. Despite the containment of the initial outbreaks via public health measures, the development of vaccines and antiviral agents for SARS-COV is essential to preparing for the next disease outbreak [50]. A wide range of antiviral agents capable of targeting SARS-COV have been identified; nonetheless, not a single approved antiviral drug is available for the treatment of SARS or SARS-like disease (MERS) [51]. As the SARS-COV main protease is considered an attractive therapeutic target owing to its viable role in the viral replication cycle, several studies have been conducted to develop isatin derivatives as potential SARS-COV main protease inhibitors [52–55]. In 2005, Chen et al. prepared a group of N-substituted isatin derivatives, compound 20 (Fig. 18), and their SARS-COV 3C-like protease inhibition activity was evaluated using fluorescence resonance energy transfer (FRET), which was further confirmed by HPLC analysis [54]. Compounds 20a–k showed IC50 values in the lower micromolar range (0.95–17.50 µM), among which the most promising were identified as 20e, f, and h (IC50 = 2, 0.98 and 0.95 µM, respectively).
Fig. 18.

Chemical structures of N-substituted isatin derivatives
The SARs (Fig. 19) revealed the superiority of the benzothiophenemethyl side chain over the other groups at position 1 of the isatin core. In addition, better activity was linked to substitution with electron-withdrawing groups such as 7-bromo (20f), 5-iodo (20h) or 7-nitro [20e] in the isatin moiety. On the other hand, electron-donating substituents such as OCH3, NH2 and CH3 were detrimental to the activity. Further studies identified 20h as a selective and potent inhibitor of SARS-COV protease versus other proteases, such as papain protease. A molecular modeling study demonstrated the ability of compound 20h to fit precisely into the SARS-COV protease binding pocket. Additionally, hydrophobic and electronic factors appeared to be key elements that determine the binding affinity. Therefore, compound 20h could act as a lead molecule for further investigation.
Fig. 19.

The SARs of N-substituted isatin derivatives as SARS-COV 3C-like protease inhibitors
In another work, a series of N-substituted 5-carboxamide-isatin derivatives [16] (Fig. 20) was designed by Zhou and his coworkers as potential SARS-COV 3C-like protease inhibitors [55]. The rationale behind their design was based on the potential hydrogen bonding that would result from the binding of the carboxamide group with Phe140 and His163 at the P1 site of the enzyme binding pocket. Meanwhile, the two carbonyl groups at C-2 and C-3 of the isatin core are involved in hydrogen bonds with the amino acid residues His41 and Cys145. The alkyl groups attached to N1 of the isatin moiety would soke into the hydrophobic S2 site of the binding pocket formed by Met49 and Met165. The enzyme activity was evaluated via colorimetric measurements of the protease activity, which was further confirmed by HPLC analysis. The results revealed that with the exception of 21e, compounds 21a–d and 21f exhibited moderate to excellent activities (IC50 = 0.37 ± 0.03–71 ± 6 µM). The most active compound 21f was >2.5-fold more potent than 20; thus, it could act as a lead molecule for further studies. Testing of 21f against other proteases revealed its selectivity towards SARS-COV 3C-like protease over the others. The SARs demonstrated that the introduction of the carboxamide group at the C-5 position of the isatin skeleton led to a greater binding affinity to the SARS-COV 3C-like protease. Replacement of this group with I, COOH or COOCH3 resulted in a significant reduction in the activity. Substitution at the N1 position by a bulkier hydrophobic group such as naphthalen-2-ylmethyl could enhance the activity, as evidenced by compound 21f. Replacement of the C-3 carbonyl oxygen by other groups, such as 4-methoxyphenylimino or 1-naphthylimino, yielded inactive compounds.
Fig. 20.
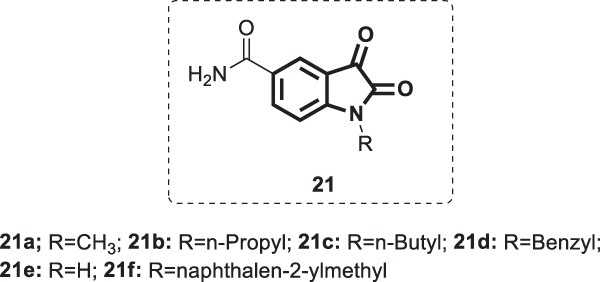
Chemical structure of N-substituted 5-carboxamide-isatin derivatives
Other groups of researchers designed and synthesized a series of 5-sulfonyl isatin derivatives 22 and 23 (Fig. 21) [52]. Then, these compounds were evaluated for their in vitro inhibition of SARS-COV 3C-like protease using fluorogenic substrate peptide. Compounds 22a–f showed promising inhibitory activity (IC50 = 1.18–12.66 µM) with 22d bearing a 4-methylpiperidinyl moiety, which displayed the greatest potency. The SARs suggested that the isatin core played a pivotal role in the activity, and the relative contribution of the substituents was piperidine > morpholine > piperazine. Within the group of compounds carrying a piperidine moiety, methyl substitution at position 4 of the piperidine ring is optimal for maximum activity. Modification of the isatin core, namely, alkylation at the N-1 position, was performed to examine the role of substitution at this position on the potency. Compounds 23a–k displayed moderate to excellent enzyme inhibition activity (IC50 = 1.04–17.82 µM). The SARs demonstrated that N-1 benzylation of 22d, the most active compound, yielded 23g, which is associated with very slight improvement in activity (IC50 = 1.04 µM). On the other hand, introduction of a β-naphthyl methyl group (23h; IC50 = 1.69 µM) slightly reduced the activity, while methylation at the same position resulted in a > 15-fold reduction in activity (23i; IC50 = 17.82 µM), demonstrating the key role of the specific size of the group in the activity. Functionalization of 22c (IC50 = 12.66 µM), bearing a morpholine moiety, with either a CH3 [23e] or benzyl (23f) group did not result in significant enhancement of the potency (IC50 = 9.91 and 13.86 µM, respectively), while β-naphthyl methyl group substitution resulted in a > 3-fold reduction in activity (IC50 = 39.87 µM). In silico studies conducted by the authors suggested modification of the R1 group with a simple six-membered ring alongside substitution at the N-1 position with a small group such as a methyl group for better activity. Overall, compounds 22a–f and 23a–k showed similar inhibitory activities to the 5-carboxamide substituted isatins (21a–f), with compounds 22d and 23g being the best and serving as ideal starting points for further studies.
Fig. 21.

Chemical structure of 5-sulfonyl isatin derivatives
In 2008, Selvam et al. screened compounds 11a–e together with compound 24 (Fig. 22) against SARS-COV in Huh 5-2 cells [56]. The results showed that compound 24 was the most effective (45% maximum protection at 125 µg/mL), and the other compounds were far less effective (0–22% maximum protection at 125 µg/mL). The authors claimed the importance of the C-3 sulfonamide group for optimum antiviral activity.
Fig. 22.
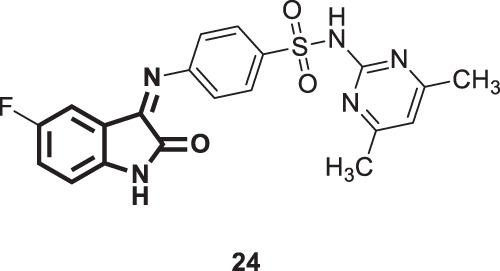
Chemical structure of compound 24
The isatin-inspired derivatives discussed here displayed promising SARS-COV inhibitory properties, and some of them could serve as potential lead compounds (20j, 21f and 23g). Nevertheless, studies aimed at exploring the isatin core to fight against SARS-COV are limited, and further studies are required. To this end, the structure-activity relationship (SAR) is presented in Fig. 23 for further rational development of isatin derivatives with higher potency against SARS-COV.
Fig. 23.

The SARs of isatin derivatives as SARS-COV inhibitors
Anti-SARS-COV-2 isatin derivatives
The outbreak of the ongoing COVID-19 pandemic was reported in Hubei Province, China, in December 2019, and the pandemic spread worldwide, leading to serious medical, economic, social and political impacts. A novel virus was later identified as the etiological agent and named severe acute respiratory syndrome coronavirus 2 (SARS-COV-2) [56, 57]. Despite the deleterious consequences of COVID-19 and the huge efforts expended on finding efficient therapies, no single drug has proven effective in the treatment of COVID-19 patients, and the currently adopted therapeutic strategies are either supportive or preventative [58, 59]. Thus, efficient drug discovery approaches should focus on developing new chemical entities that are capable of modulating targets that are critical to the virus life cycle. As SARS-COV-2 and SARS-COV strongly resemble each other, an increasing number of published works have grown around the theme of developing drugs based on their efficiency on SARS-COV [60]. The SARS-COV-2 main protease (also called 3C-like protease) plays a critical role in viral replication and transcription processes (Fig. 24), and its uniqueness to the virus renders it an ideal target for rational drug design [61–63]
Fig. 24.

The critical role of Mpro in the transcription and replication processes of the SARS-COV-2 life cycle. The open reading frames (ORF1a and ORF1ab) encode polyproteins (pp1a and pp1ab) that are subjected to proteolytic reactions catalyzed by two cysteine proteases, namely, the main protease (Mpro) and the papain-like protease (PLpro). Isatin derivatives could inhibit Mpro and thus restrain virus replication
Accordingly, a series of N-substituted isatin derivatives 25 (Fig. 25) has been designed and synthesized to act as potent inhibitors of the SARS-COV-2 main protease [64]. The enzyme inhibition activity of compounds 25 was evaluated using synthetic peptide-pNA as the substrate and tideglusib as a positive control. Compounds 25h, j, k, q, and w–ab showed inhibition activity >50% at 50 µM, while the positive control exhibited IC50 = 1.91 µM. Consistent with what has been reported by Zhou et al. in SARS-COV [55], maximum activity was associated with derivatives bearing carboxamide groups at C-5 of the isatin core (25v–z) (IC50 = 0.045–17.8 µM). Compounds 25y, z, and v were the most promising candidates (IC50 = 0.045, 0.047 and 0.053 µM, respectively). Compounds 25g–n and 25r–u were relatively less active or inactive against the SARS-COV-2 main protease. Considering the similarity between the main protease structures of coronaviruses, the absence of hydrogen bond-forming groups, such as carboxamides, at C-5 could explain the poor activity of the aforementioned derivatives, as previously reported by Zhou et al. [55]. Consistent with what is illustrated in Fig. 23, SAR studies indicated the necessity of bulky and rigid hydrophobic groups at the N-1 position for maximum activity against the SARS-COV-2 main protease. Bromination at C-6 of the naphthalene ring in 25y yielded 25z, which retained similar potency; on the other hand, introduction of an aryl group at the same position significantly diminished the activity, as did the insertion of a methylene group between the isatin ring and the carboxamide group (25a, and b). The similarities in SARs of compounds 25 as SARS-COV and SARS-COV-2 main protease inhibitors further prove their conservation of the same binding pocket; therefore, all reported inhibitors of SARS-COV could be explored as potential anti-SARS-COV-2 candidates.
Fig. 25.

Chemical structures of N-substituted isatin derivative 25
A molecular modeling study of the most active compound (25y) (Fig. 26) revealed hydrogen bond interactions between the carboxamide moiety as a donor with Asn142 and Glu166 and the two carbonyl groups at C-2 and C-3 with Cys145 as acceptors. In addition, the rigid hydrophobic naphthyl ring inserted into the hydrophobic binding tunnel formed by Met49 and Met165 in addition to the π- π stacking interaction with the His41 aromatic residue. Nearly 40-fold more potent than the positive control, compounds 25y, z, and v could serve as starting points for further drug development of potent anti-coronavirus drugs. To this end, future studies should focus on chemical optimization to minimize their high cytotoxicity and to enhance their cellular efficacy.
Fig. 26.
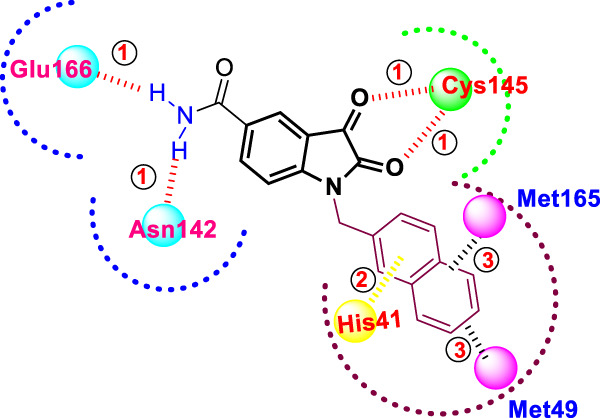
Molecular interaction of the N-substituted isatin derivative 25y with the SARS-COV-2 main protease binding pocket; bond 1 is an H-bond; 2 is a π-π stacking interaction; and 3 is a hydrophobic interaction
Badavath and his coworkers performed a molecular docking analysis on a series of heterocyclic derivatives in the binding site of the SARS-COV-2 main protease [65]. Among them, two isatin derivatives, 26 and 27 (Fig. 27), showed better interactions with the most important binding residues of the enzyme binding pocket with energies of <−10 kcal/mol. Furthermore, ADME analysis revealed that the properties of compounds 26 and 27 are within the specified limits.
Fig. 27.
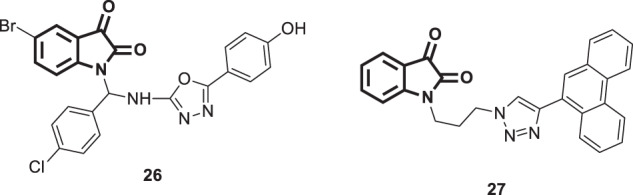
Chemical structures of the designed isatin derivatives 26 and 27 as potential SARS-COV-2 main protease inhibitors
Anti-arboviral isatin derivatives
Arboviruses are viruses associated with high morbidity and mortality, and the majority are zoonotic. The clinical picture of arboviral infections in humans ranges from silent infections to overt clinical diseases, including hemorrhagic fever, arthritis and encephalitis [66]. Most arboviruses belong to the following families: (i) Flaviviridae (dengue virus (DENV), Japanese encephalitis virus (JEV), West Nile virus (WNV) and Zika virus), (ii) Bunyaviridae (Crimean-Congo hemorrhagic fever and Rift Valley fever virus) and (iii) Togaviridae (chikungunya virus, Western equine encephalitis virus and Eastern equine encephalitis virus) [22, 67]. There is no licenced antiviral medicine for any of the arboviruses, and thus, the current therapeutic strategies are only supportive [67, 68]. Therefore, several studies have been performed to develop anti-arboviral drugs using the isatin core motivated by its broad spectrum of biological activities. These studies have been reviewed but briefly by Gomes et al. [22].
Anti-chikungunya virus isatin derivatives
Mishra and his coworkers synthesized a hybrid molecule (MBZM-N-IBT) [23] of 2-methyl benzimidazole and isatin-β-thiosemicarbazone (Fig. 28). This compound was evaluated for its anti-chikungunya virus activity [69]. Compound 28 displayed enhanced antiviral activity as it was able, through very short exposure, to significantly inhibit chikungunya virus replication in addition to diminishing viral RNA levels and consequently the expression of viral structural and nonstructural proteins (NSPs). In mammalian systems, the following events were identified: (i) a reduction in viral particle formation by >75%, (ii) decrease in viral RNA synthesis by >65% and (iii) reduction in viral protein levels by >97%. Moreover, compound 28 blunted the viral life cycle at the early and late stages, indicating multiple modes of antiviral action. Molecular modeling studies for compound 28 revealed strong binding affinities to the structural and nonstructural proteins of the chikungunya virus and related viruses. In silico pharmacokinetic prediction indicated favorable drug-like properties. Thus, compound 28 could represent an ideal starting point for further drug discovery steps. Nonetheless, a limitation of this study is that only a single compound was synthesized and investigated for activity against chikungunya virus. Given the promising activity exhibited by compound 28, further analogs are highly recommended to be synthesized and screened in the pursuit of more efficient therapies against this deadly pathogen.
Fig. 28.
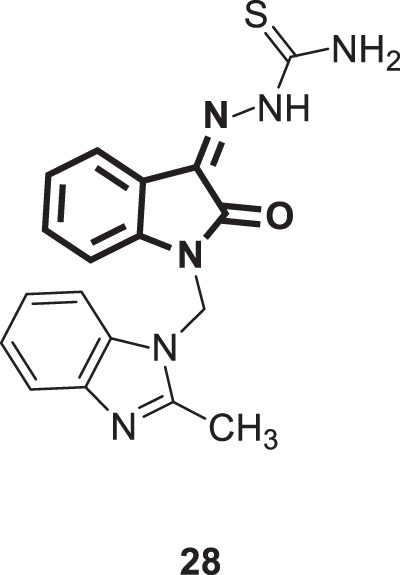
Chemical structure of MBZM-N-IBT [23]
Isatin derivatives against JEV, WNV and DENV
A set of new and reported 14 N-methylisatin-beta-thiosemicarbazone derivatives were tested in vitro against certain flaviviruses, namely, Japanese encephalitis virus (JEV), West Nile virus (WNV) and dengue virus-2 (DENV-2) [70]. Amongst them was SCH 16 (Fig. 29), whose exact identity is confusing because the authors drew a structure does not match the given chemical name as it lacks the thiosemicarbazone moiety. The given chemical name matches compound 10e, which was first reported by Bal et al. [37], and the synthetic route used supports the drawn structure therein. It appears that the authors mistakenly drew an incorrect structure for SCH 16 in their report, and thus in the current review it is presented as 10e. Compound 10e (the correct structure of SCH 16) fully inhibited JEV and WNV replication (IC50 = 16 and 4 µg/mL, respectively). Furthermore, in vivo studies conducted in a mouse model challenged intraperitoneally with LD50 of JEV revealed the ability of compound 10e at a 500 mg/kg dose to fully inhibit viral replication and consequently prevent mortality. Mechanistic studies suggested the inhibition of viral early protein translation as a potential mode of action for compound 10e. Thus, it could serve as a potential lead for the development of more potent antiviral drugs against JEV and WNV. No antiviral activity has been observed for this compound against DENV-2.
Fig. 29.
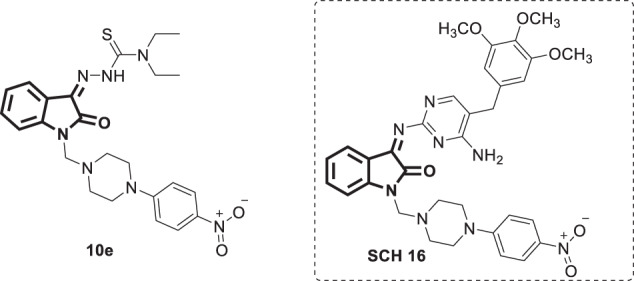
Chemical structure of N-methylisatin-β-thiosemicarbazone, 10e (SCH 16)
In an independent study [71], Sebastian and his coworkers performed experimental in vitro work to investigate whether the combination (in lower concentrations) of compound 10e (SCH 16) with mycophenolic acid, ribavirin or pentoxifylline could boost the activity against JEV. The rationale behind this is the ability of the aforementioned drugs to inhibit viral replication through interference at the late stage of the viral translation process. As compound 10e could influence viral early protein translation, its combination with drugs having different modes of action might offer a synergistic effect against JEV and could enhance the safety profile. The results of this study demonstrated that the combination of 10e with ribavirin or mycophenolic acid potentiated the efficacy. In contrast, antagonism was noticed for the combination of 10e with pentoxifylline. In addition to the potential direct anti-JEV effects expected from isatin derivatives discussed herein, Minami et al. described an additional therapeutic benefit of isatin (100 mg/kg per day for a week, intraperitoneal injection) in relieving JEV-induced parkinsonism in rats by increasing dopamine concentrations in the striatum [72].
Infection with WNV results in severe human disease, which particularly involves the CNS with clinical features similar to those of Parkinson’s disease [73]. Accordingly, Blázquez et al. [74] studied the potential effect of four antiparkinsonian agents, namely, isatin, L-dopa, selegiline, and amantadine, in restraining WNV multiplication. Substantial inhibition of viral replication was exhibited by isatin (IC50 = 0.78 mM). This, together with the ability of isatin to elevate dopamine concentration in the brain by virtue of its MAO inhibitory activity, renders it an ideal lead structure for further studies [75].
Dengue viruses (DENVs) are the etiological agents of the currently most common arthropod-borne viral disease in humans and are endemic in more than 100 countries worldwide. The lack of safe and effective vaccines and antiviral drugs limits the control measures to eradication of Aedes mosquito vectors [76]. Adopting high-throughput phenotypic screening, Zou et al. synthesized a series of spiropyrazolopyridones [24] (Fig. 30) as a new class of anti-DENV agents [77]. The enantiomers depicted in Fig. 30 were obtained by chiral HPLC resolution of their corresponding racemates. Compounds 29a–w showed promising antiviral activity against DENV-2 (EC50 = 0.006–85.3 µM), with compound 29q being the best. The SARs (summarized in Fig. 31) revealed that, as a general pattern, R enantiomers were more potent than S enantiomers. With regard to substitution at R1, parachlorobenzyl was needed for better activity, while meta- or orthochlorobenzyl (29c, and d) abolished the antiviral activity. Furthermore, for the para-benzyl substituted derivatives, electron-withdrawing groups such as -CF3 (29f–h; EC50 = 0.025, 0.021 and 3.9 µM, respectively) were favorable to the activity, whereas electron-releasing groups such as -OCH3 were detrimental (29e; EC50 = 6.2 µM). Furthermore, installation of a 3-chloropyridyl group at the R1 position was tolerable (29i; EC50 = 0.67 µM), and equipotency was retained by the R enantiomer (29a; EC50 = 42 nM, determined in the secondary viral titer reduction assay). In contrast, a significant drop in efficacy was reported when R1 was substituted by other groups, such as 2-chloropyridyl (29k; EC50 = 32.8 µM), 5-chloropyrimidinyl (29l; EC50 = 85.3 µM), cyclohexyl (29m; EC50 = 22.5 µM), phenyl (29n; EC50 = 32.5 µM), or p-chlorophenethyl (29o; EC50 = 7.2 µM). Reduction of either amide functionality (29v and w; EC50 = 4 and 1.1 µM, respectively) resulted in less potent compounds. N-methylation of either amide yielded derivatives without affecting the level of activity (29p and q; EC50 = 0.42 and 0.011 µM, respectively), indicating that neither NH is critical for optimum efficacy. With regard to R4 substitution, compound 29s, bearing a bromo group, still maintained substantial potency (EC50 = 0.05 µM). On the other hand, substitution by an electron-donating methoxy group yielded compound 29u (EC50 = 2.3 µM), which was >40-fold less potent than 29s. Following this, compound 29t, bearing the electron-withdrawing -CF3 group, diminished the potency by >20-fold relative to 29s, suggesting a far limited range of tolerated substituents at that position. Compound 29q, which was identified as the most potent among the spiropyrazolopyridones, exhibited poor physicochemical and pharmacokinetic properties, which impeded further stepping in the drug development process. However, compound 29a, although less potent, was identified as being the best in terms of physicochemical properties and in vivo pharmacokinetics profile. Therefore, compound 29a could represent a lead compound for further investigations.
Fig. 30.

Chemical structures of spiropyrazolopyridones
Fig. 31.

The SARs of spiropyrazolopyridones as anti-DENV-2
Based on the SAR studies illustrated in Fig. 31, further optimization efforts were made by Xu et al. [78]. The optimization approach focused mainly on the two amide groups of the indolone moiety to explore the chemical space to enhance the potency against all four serotypes of DENV. Accordingly, a set of substituted spiropyrazolopyridone analogs [25] (Fig. 32) were synthesized and evaluated against four DENV serotypes. Among these compounds, 30a (JMX0254) displayed the most potent and broad inhibitory activities, effective against DENV serotypes 1–3 (EC50 = 0.78, 0.16, and 0.035 μM, respectively). On the other hand, compounds 30b–h demonstrated relatively moderate to high anti-DENV activities with low micromolar to nanomolar potency against the four strains. Mechanistic studies proved that the dengue NS4B protein was the molecular target for these compounds. Furthermore, compound 30a showed an optimal in vivo pharmacokinetic profile accompanied by enhanced efficacy in the A129 mouse model. These encouraging results indicate its potential role as an anti-DENV drug candidate that dictates further drug development processes.
Fig. 32.
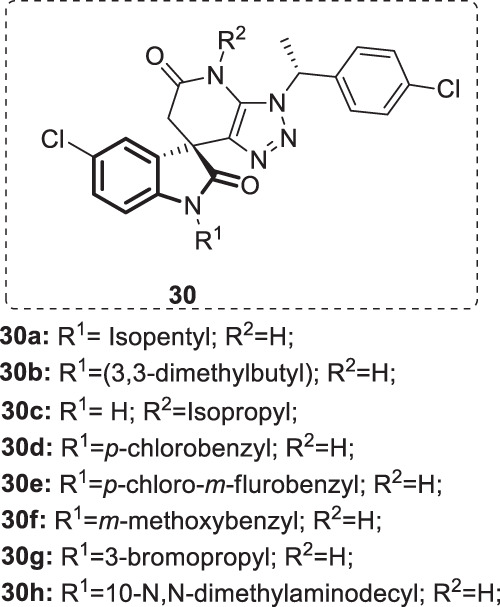
Chemical structures of substituted 4,6-dihydrospiro[(1–3)triazolo[4,5-b]pyridine-7,3′-indoline]-2′,5(3H)-dione analogs 30a-h
Anti-herpetic isatin derivatives
Herpes simplex viruses (HSVs) are double-stranded RNA viruses comprising HSV1 and HSV2, and they are the causative agents of serious health problems in humans worldwide. Both viruses belong to the alpha subfamily of the human herpesvirus family and share common structural features [79]. The principal characteristics of HSV infections are the high incidence and high recurrence rate [80]. The currently used drugs for HSV therapy are nucleoside derivatives, and among them, acyclovir is the most commonly used [81, 82]. However, the emergence of resistant strains and the prevalence of cross resistance among the strains dictate the development of new non-nucleoside-based efficient drugs [83]. Motivated by the reported therapeutic usefulness of isatin-β-thiosemicarbazones in an array of viral diseases, Kang et al. designed and synthesized a series of isatin-β-thiosemicarbazones [26–28] (Fig. 33) [84]. Utilizing the plaque reduction assay, these compounds were evaluated for antiviral activity against HSV-1 and HSV-2, and acyclovir was used as a positive control. Compounds 31–33 showed anti-HSV-1 and anti-HSV-2 activities in the ranges of 1.3–6.21 and 1.54–5.8 µM, respectively. Although less potent than acyclovir (IC50 = 0.3 µM), compound 33d, which bears a morpholine group, was identified as the most potent derivative against HSV-1 (IC50 = 1.3 µM) with cytotoxicity of >25 µM. Furthermore, it was able to inhibit HSV-2 at a concentration of 2.74 µM (IC50 of acyclovir was 1.27 µM). Replacement of morpholine with either aliphatic amines (dimethylamine, diethylamine, dipropylamine, N-methylamine, N-ethylamine or N-propylamine) or alicyclic amines such as piperdine or pyrrolidine significantly reduced the activity against HSV-1 (IC50 > 25 µM). Notably, compounds 33a, c, and d demonstrated potent and selective inhibition against HSV-2 (IC50 = 1.54, 5.8 and 3.14 µM, respectively). Interestingly, compound 31a exhibited promising activity against HSV-1 (IC50 = 2.97 µM) with minimal cytotoxicity (CC50 > 25 µM). Surprisingly, it was completely devoid of activity against HSV-2, indicating its selectivity against HSV-1. The SARs indicated that insertion of electron-releasing substituents (-CH3 and -OCH3) in the phenyl ring of compound 31a yielded derivatives with varied antiviral activity. The 3-methyl [31b], 3-methoxy [31c], and 4-methoxy [31d] derivatives demonstrated similar antiviral activity against HSV-1 (IC50 = 3.84, 2.64 and 2.20 µM, respectively). On the other hand, no anti-HSV activity was observed when the authors introduced these groups as 2-methyl, 4-methyl and 2-methoxy groups. Para substitution by electron-withdrawing groups (Cl or Br) delivered potent analogs against HSV-1 (IC50 = 2.69 and 3.29 µM, respectively). In addition, grafting of heterocyclic rings such as pyrazole (31g; IC50 = 6.21 µM) and morpholine (IC50 > 25 µM) at the para position reduced the efficacy against HSV-1. Furthermore, placement of -OCH3 or -OCF3 at C-5 of the isatin core of compound 31a resulted in 32a and b (IC50 = 5.01 and 5.9 µM, respectively), which were 2-fold less potent against HSV-1 than 31a. Substitution by other groups, such as F, Cl or Br, at the same position yielded totally inactive compounds, as did the N1 modification of the isatin ring by groups such as Me, Et, Ph or benzyl. As isatin-β-thiosemicarbazones have promising anti-herpetic action along with low cytotoxicity, this class of compounds provides a valid starting point for the discovery of more potent candidates. Therefore, the SARs of compounds 31–33 are compiled in Fig. 34 to aid in further rational development.
Fig. 33.

Chemical structures of isatin-β-thiosemicarbazones 31–33
Fig. 34.

The SARs of isatin-β-thiosemicarbazones as anti-HSV agents
Jarrahpour and coworkers designed and synthesized twelve new bis-Schiff bases of isatin (34 and 35) (Fig. 35) as broad-spectrum chemotherapeutic agents [85]. Then, the synthesized compounds were screened for antiviral activity against a panel of DNA and RNA viruses, including HSV-1 and HSV-2. None of these bases displayed notable antiviral activity, indicating their inability to restrain viral replication at a concentration that was ≥5-fold lower than the minimum cytotoxic concentration.
Fig. 35.
Chemical structures of bis-Schiff bases of isatin 34 and 36
Based on the potential therapeutic properties of nucleoside derivatives as effective medications in a vast range of viral diseases [86], a series of novel substituted isatin ribonucleosides [31] (Fig. 36) was synthesized by de Oliveira et al. [87]. Next, these compounds were evaluated for antiviral activity on HSV-1-infected cells. At a concentration of 50 µM, 36 exhibited mild to moderate viral replication inhibition in the range of 15–66% (calculated as % inhibition of virus yield) without cytotoxicity to Vero cells at the tested concentration. Among them, ribonucleoside 36f displayed the highest potency (66% inhibition of HSV-1 replication). Further screening of compounds 36 for their inhibitory activity against HIV-1 reverse transcriptase revealed complete inactivity.
Fig. 36.
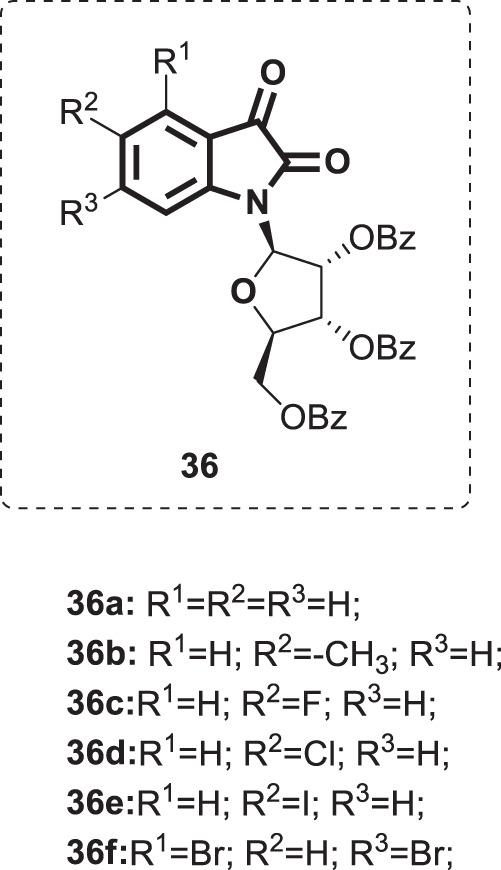
Chemical structures of substituted isatin ribonucleosides 36
Isatin derivatives against respiratory syncytial virus (RSV)
Human respiratory syncytial virus (RSV) is a negative-strand RNA virus and the etiological agent of severe acute lower respiratory tract infections in neonates and young children [88]. Those who are at most risk of developing severe clinical disease are under 50 days of age or those with cardiopulmonary comorbidities [89]. Although extensive studies have uncovered the details of the virus structural and functional features, there are very limited strategies for the prevention and treatment of RSV infection [90]. The current therapeutic strategy is restricted to aerosol administration of the nonspecific and teratogenic drug ribavirin. Thus, the development of efficient and selective RSV inhibitors is crucial [91]. Based on the structure of the potent RSV fusion inhibitor BMS-433771 (Fig. 37) [92], Sin et al. developed a train of benzimidazole-isatin oximes [33, 35, 37] (Figs. 37–39) that were envisioned as isosters for the bezimidazol-2-one motif inherent in BMS-433771 [93]. Following the selection of the optimal benzimidazole side chains (R2) from previous studies [94], the authors focused initially on optimization of the oxime substituent (R1). The synthesized benzimidazole-isatin oximes 37a–aa demonstrated excellent antiviral activity (EC50 = 5–100 nM). Compounds 37a, c, q, r, s, y, z, and aa were identified as the most potent derivatives relative to BMS-433771 (EC50 = 20 nM). Among them, benzimidazole-isatin oxime 37aa emerged as the most efficacious (EC50 = 5 nM). The SARs indicate a good correlation with that reported for the enzimidazole-2-one series [94, 95]. In this regard, small alkyl substitution at R1 is crucial for good cell permeability rather than potent antiviral activity, as evident from the activity of the parent compound 37a (EC50 = 10 nM). With the exception of 37i (EC50 = 96 nM), branching at the α- or β-position of the side chain appeared to be detrimental to the activity. Extension of the alkyl groups at the R1 position up to C5 furnished compounds that were 2-to-7-fold less potent than 37f (EC50 = 56 nM). Higher-level unsaturation at R1 exemplified by 37j and k resulted in improved antiviral activity (EC50s’ = 64 and 79 nM, respectively). Reasonable potency was observed upon insertion of a polar OH group at the β-position of compound 37k in conjunction with a nitrile moiety at the end of the chain (37l; EC50 = 65 nM). Incorporation of either neutral or acidic polar groups at the terminus of R1 offered potent antiviral analogs. Amongst the neutral polar groups are acetate (37m; EC50 = 62 nM) and amide (37n and o; EC50s’ = 26 and 45 nM, respectively). Derivatives bearing polar acidic groups such as 37p and q displayed enhanced potency (EC50s’ = 46 and 19 nM, respectively). Introduction of a Ph spacer between the oxime and the carboxylic acid moiety provided compounds with better activity (37r and s; EC50s’ = 16 and 16.5 nM, respectively). This could be extended to other polar, neutral substituents incorporated at the 4-position of the aryl ring, represented by compounds 37t–aa, which demonstrated excellent in vitro antiviral properties (EC50s’ = 5–51 nM).
Fig. 37.

Chemical structures of benzimidazole-isatin oximes 38
Fig. 39.

Chemical structures of benzimidazole-azaisatin oxime 39
The three nitrones 38a–c (Fig. 38) displayed moderate antiviral properties (EC50s’ = 230–898 nM).
Fig. 38.
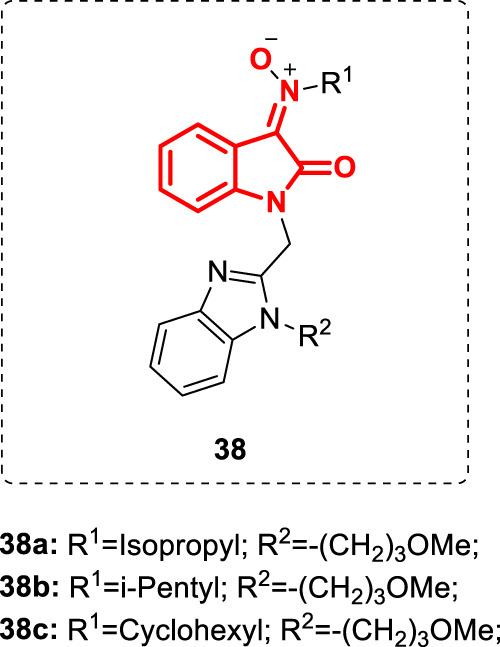
Chemical structures of nitrones 38a–c
Following this, the research was directed towards the optimization of pharmacokinetic properties. Compounds 37h and m were selected for further optimization because they exhibited good RSV inhibition (EC50 < 100 nM) associated with moderate lifetime and better cellular permeability. Consequently, attention was focused on the series of azaisatin oximes 39a–u (Fig. 38). The rationale behind this structural modification of 37h and m was to boost the metabolic stability through prevention of CYP-450-mediated aromatic hydroxylation at the isatin phenyl ring system while enhancing their water solubility. Compound 39 demonstrated RSV inhibition activity ranging from 7.3 nM–10.59 µM, with compound 39s being the best (EC50 = 7.3 nM). Compounds bearing fluoroethyl oxime moieties (39i, j, and n) had the best overall combination of efficacy, pharmacokinetic properties, cellular permeability and metabolic stability. Oral dosing of these analogs in the BALB/c mouse model of RSV infection revealed that 39n exhibited a similar pharmacokinetic profile to that of BMS-433771, although it was less effective in terms of antiviral efficacy. The SARs of benzimidazole-isatin oximes 37–39 are compiled in Fig. 40.
Fig. 40.

The SARs of benzimidazole-aza/isatin oximes as potent RSV inhibitors
Isatin derivatives against coxsackievirus
Coxsackieviruses (CVs) are enteroviruses that have been linked to fatal human diseases, including viral myocarditis and encephalitis, particularly in children and young adolescents [96]. Despite the serious morbidity and mortality that might arise from CVs, there is still no licenced drug that is effective for the treatment of acute CV infections [97, 98]. Thus, efforts have been made by Zhang et al. to identify novel CV inhibitors adopting a high-throughput screening approach [97]. In this context and encouraged by the potent antiviral activity of isatin derivatives, four isatin-based analogs 40 and 41 (Fig. 41) were synthesized and evaluated for their activity against coxsackievirus B3 replication (CVB3) using methisazone as a positive control. Among them, 40a emerged as the most active analog that showed a significant reduction in viral particle formation and consequently blunted the virus-induced apoptosis of the host cells. Furthermore, treatment of infected cells with 40a resulted in enhanced cell survival through upregulation of glucose-regulated protein 78 (GRP78), which is considered a novel antiviral mechanism in this regard. Therefore, compound 40a would be capable of inhibiting other picornaviruses, as they share the same machinery of protein synthesis initiation.
Fig. 41.
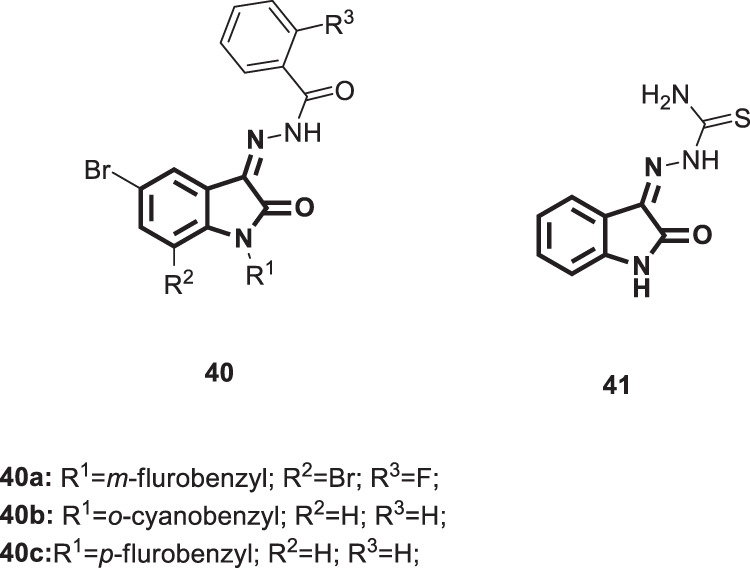
Chemical structures of anti-CVB3 isatin derivatives
Isatin derivatives as anti-poxvirus agents
Smallpox is a serious and fatal human disease caused by poxviruses that resulted in >300 million deaths worldwide during the 20th century [99]. Poxviruses are DNA viruses belonging to the family Orthopoxvirus that are capable of replicating in the cell cytoplasm [100]. Smallpox was eradicated globally due to a successful vaccination program, and no single case has been registered since 1978 [101]. Nevertheless, studies aimed at the development of vaccines and drugs are still in progress to offer protection against smallpox in the event that it is used as a biothreat agent [102]. The US Institute of Medicine has recommended the existence of at least two anti-poxvirus drugs mediating their action via two different modes of action to diminish the evolution of resistant strains [101]. Isatin derivatives such as methisazone [3] (Fig. 4) were shown 50 years ago to play a viable role as smallpox chemoprophylactic agents; however, their mode of action has not yet been identified [28, 103]. Methisazone has been reported to reduce disease morbidity and mortality when used prophylactically in susceptible contacts. However, the lack of therapeutic efficacy limits its use, and it is no longer manufactured as a drug substance [104]. In this regard, Pirrung and his coworkers followed a combinatorial method to optimize the isatin-β-thiosemicarbazone basic structure to enhance its antiviral activity [104]. Accordingly, a series of isatin-β-thiosemicarbazones was synthesized and examined for antiviral activity and cytotoxicity in vaccinia virus- and cowpox virus-infected human cells using methisazone [3] and cidofovir (a recently approved anti-poxvirus drug) as positive controls. Compounds 42a–d (Fig. 42) did inhibit cowpox virus plaque formation (EC50s’ = 6 ± 2.9–6.2 ± 1.6 µM with SI > 8) with greater potency than methisazone (EC50s’ = 14 ± 0.3 µM) and cidofovir (EC50s’ = 38 ± 1.9 µM). With regard to their activity against vaccinia virus plaque formation, these compounds exhibited enhanced efficacy (EC50s’ = 0.6 ± 6.8 µM with SI > 35) and were more potent than the standard drug cidofovir (EC50s’ = 27 ± 7.8 µM). In addition, compounds 42a, c, and d showed better activity in vaccinia virus plaque formation inhibition than methisazone (EC50s’ = 3.3 ± 3.2 µM). The SARs identified 5-bromoisatin as a key pharmacophore for better antiviral efficacy in the designed library. Compound 42d carrying acyclic secondary amine emerged as the most potent analog against both tested strains. As potent and much more selective anti-poxvirus isatin-β-thiosemicarbazone derivatives were identified, further drug discovery processes are warranted.
Fig. 42.
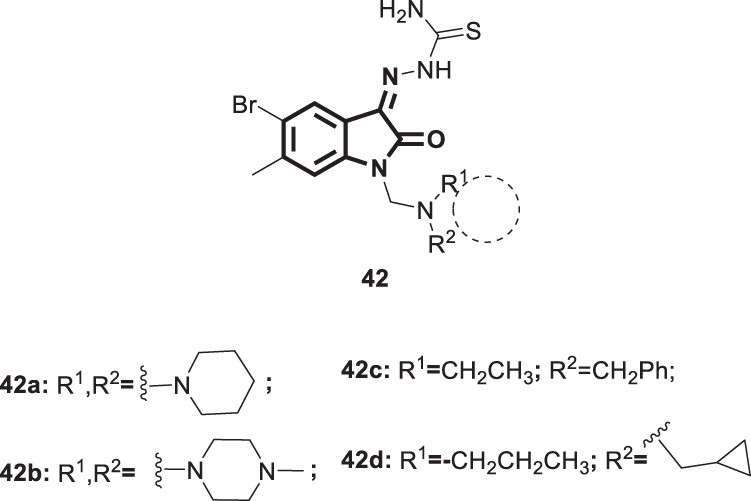
Chemical structures of isatin-β-thiosemicarbazone derivatives 41a–d
Isatin derivatives as anti-influenza virus agents
Influenza viruses belong to the family Orthomyxoviridae and are the main causative agents of upper respiratory tract infectious diseases and are associated with considerable morbidity and mortality rates in humans [105]. Vaccines and rapidly acting antiviral medications are the cornerstones of the prevention and treatment of influenza virus disease [106, 107]. The efficiency of vaccination is limited by continuous antigenic variation in the envelope glycoproteins of the virus [106]. Furthermore, the available anti-influenza virus drugs used for prevention and treatment are rather scarce, and the development of resistant strains represents a serious challenge [108]. Therefore, tremendous efforts are needed to develop new antivirals with improved therapeutic properties to overcome resistance to currently used drugs. In view of the broad-spectrum antiviral activities of isatin derivatives, Selvam et al. [109] evaluated the antiviral activity of compounds 11a–e (Fig. 10) against influenza A (H1N1, H3N2, and H5N1) and B viruses using ribavirin and oseltamivir as positive controls. Compounds 11a–e displayed similar potency associated with minimal cytotoxic effects (EC50s’ = 2.7–5.2 µg/mL against the H1N1 strain of influenza A), (EC50s’ = 13.8–26.0 µg/mL against the H3N2 strain of influenza), (EC50s’ = 3.1–6.3 µg/mL against the H5N1 strain of influenza), and (EC50s = 7.7–11.5 µg/mL against influenza B). Furthermore, these compounds were able to reduce the viral yield of the H1N1 strain by 90% at concentrations of 2–10 µg/mL. The efficacies of these compounds were similar to those of ribavirin but less than that of oseltamivir against all tested strains. Introduction of the F group at C-5 of compound 11a significantly reduced the potency against all tested strains, while other substituents such as Cl, Br or Me promoted the antiviral activity. On the other hand, N-acetylation of 11a yielded compound 11e with similar potency. Two bis-spirocyclic compounds 43 and 44 (Fig. 43) were synthesized by Kurbatov et al. [110]. Then, they were evaluated for their inhibitory activity against influenza virus A (H1N1) replication. Compound 44 demonstrated significant inhibitory activity (EC50 = 1.9 μg/mL) with higher cytotoxicity (CTD50 = 37 μg/mL), whereas compound 43 exhibited moderate inhibitory activity (EC50 = 26 μg/mL) associated with minimal cytotoxicity (CTD50 = 415 μg/mL).
Fig. 43.
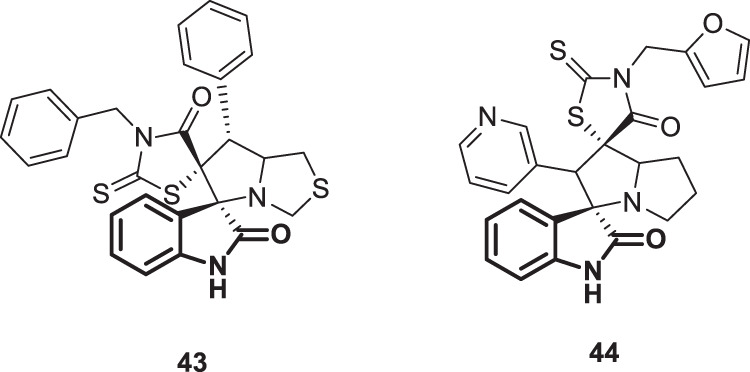
Chemical structures of bis-spirocyclic compounds 43 and 44
In another work, Karki and his coworkers synthesized a set of isatin-β-thiosemicarbazones (45a–j) [111]. Then, the antiviral activity was tested against a panel of viruses, including (i) influenza A virus H1N1 subtype, influenza A virus H3N2 subtype, and influenza B virus, in MDCK cell cultures; (ii) parainfluenza-3 virus (PI-3 V) feline corona virus (FIPV) and feline herpes virus in CRFK cell culture; (iii) HSV-1, HSV-2, vaccinia virus (VV), vesicular stomatitis virus (VSV), and herpes simplex virus-1 TK KOS ACVr (HSV-1 TK KOS ACVr) in HEL cell cultures; (iv) VSV, coxsackievirus B4 (CV-B4), and respiratory syncytial virus (RSV) in HeLa cell cultures; and (v) reovirus-1 (RV-1), sindbis virus (SV), CV-B4, and Punta Toro virus (PTV) in Vero cell cultures. Compounds 45b, 45c, 45d, and 45f (Fig. 44) displayed moderate antiviral activity against HEL cell cultures (EC50s’ = 11–20 µM), relative to the standard drugs (EC50s’ = 29, 10, >250, and >100 µM for brivudin, cidofovir, acyclovir, and ganciclovir, respectively). On the other hand, there were no evident antiviral effects of compounds 45a–j in the CRFK, MDCK, or Vero cell cultures.
Fig. 44.
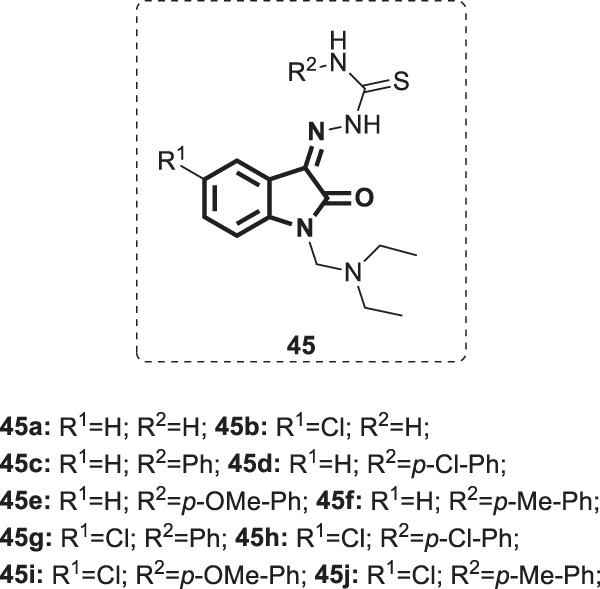
Chemical structures of isatin-β-thiosemicarbazones 45a–j
Anti-rhinovirus isatin derivatives
Human rhinoviruses (HRVs) are the principal causative agent of cold-like illnesses affecting mainly the upper respiratory tract [112]. Despite being of little direct medical impact, HRV infection is associated with serious economic implications in terms of missed work and redundant medical care [113]. HRV comprises three groups, designated A, B, and C, belonging to the genus Enterovirus and the family Picornaviridae. Efforts have been made to develop effective vaccination; however, the presence of a vast range of serotypes (>100) with genetic variation limits the development of efficient vaccines. Furthermore, there is no single clinically approved drug for the treatment of HRV infection, and the current therapeutic strategy is only supportive [112]. Thus, research has been accelerated to discover potent and selective anti-rhinovirus agents. Accordingly, Webber et al. adopted a structure-based drug discovery strategy together with molecular modeling and SAR to develop novel nonpeptidic rhinovirus 3C protease reversible inhibitors based on exploitation of the isatin core [114]. In this regard, a series of isatin derivatives, compound 46 (Fig. 45), was synthesized and examined for their potential inhibitory effects on purified HRV-14 3CP. Compounds bearing carboxamide groups at C-5 are far more potent (Ki = 0.002–0.051 µM) than those carrying other groups at the same position (Ki = 1.2 > 100 µM). In addition, compounds substituted at N-1 with larger alkyl groups are 2‒25-fold more potent than those bearing smaller alkyl groups. Compound 46s, bearing a benzothiophen group, was identified as the most potent analog (Ki = 0.002 ± 0.002 µM). From the earlier discussion along with the similarity between rhinovirus proteases and SARS-COV protease [54], the SARs shown in Fig. 23 could successfully be applied for compound 46. Further experiments revealed compounds 46 h, n, and s (Ki = 0.051, 0.011 and 0.002 µM, respectively), demonstrating higher selectivity for HRV-14 3CP than other proteases, such as chymotrypsin and cathepsin B. Unfortunately, the synthesized derivatives exhibited low in vitro antiviral potency (EC50s’ = > 5.6‒>100 µM) and were far less potent than the standard drug pirodavir (EC50 = 0.03 µM). The authors attributed this to potential problems related to physicochemical properties and cytotoxicity issues related to these compounds. Thus, further target-driven structural modification of these molecules could yield new antiviral drug candidates with broad spectrum antiviral properties.
Fig. 45.

Chemical structures of isatin derivatives 46a–aa
Isatin derivatives with anti-poliovirus activity
Poliovirus is a single-stranded RNA enterovirus of broad worldwide distribution, and it was a public health concern in the early 20th century. Although most infections are subclinical, only rare sporadic cases result in paralytic poliomyelitis [115, 116]. Due to the initiative launched by the WHO in 1988 regarding massive vaccination campaigns, near-total eradication of wild-type poliovirus has been achieved [117]. Research efforts to deliver potent antipoliovirus drugs are in progress in an attempt to obtain at least two antivirals with different modes of action [118]. Early efforts were made by Varma et al. in 1987 to discover new anti-poliovirus drugs based on isatin as the privileged core [119]. To that end, twelve isatin N-Mannich bases 47a–l (Fig. 46) were synthesized and examined for their potential inhibitory effects on the replication of poliovirus II. Compounds 47a–e, g, and j–l were active against poliovirus II when tested using the modified plaque suppression method. Furthermore, compound 47a displayed activity against HSV, measles, and parainfluenza-3 viruses. Further studies are required to investigate their exact EC50s’ and their potential molecular targets.
Fig. 46.
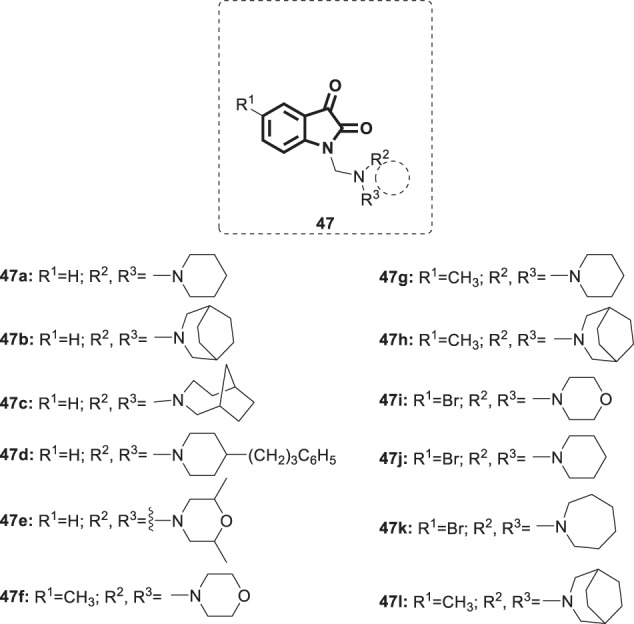
Chemical structures of isatin N-Mannich bases 47a–l
Isatin derivatives against vesicular stomatitis virus (VSV)
Vesicular stomatitis is an old zoonotic viral disease that primarily affects livestock and has the ability to infect humans who directly come in contact with infected animals in VSV endemic areas. Numerous investigations on the pathogenesis and immunology of VSV have not led to the development of efficient vaccines. To date, there is no specific antiviral therapy, and the therapeutic strategy is supportive [120]. Accordingly, a study was performed by Abbas and his coworkers to develop anti-VSV agents via exploration of the isatin ring system [121]. In this study, a series of fluorinated isatin derivatives, compounds 48 and 49 (Fig. 47), was synthesized and evaluated for their cytotoxicity and antiviral activity against VSV isolates using interferon as a positive control. Among compounds 48, the allyl group bearing analog 48c was identified as the most efficacious, with a log virus titer difference of 0.88. Replacement of the allyl group with a butyl group (47d) reduced the potency (log virus titer difference = 0.76). Compounds carrying groups such as Me, Et or Ph were less potent (log virus titer difference = 0.05–0.20). Within the group of compounds 49, analog 49a bearing a phenyl group displayed weak potency (log virus titer difference = 0.2). Introduction of the F group at the para position of the phenyl ring in compound 49a yielded the most active compound among series 49b (log virus titer difference = 0.9), which still lags behind the activity of interferon (log virus titer difference = 1.8). Conversely, replacement of the F group in 49b by an N,N-dimethylamino moiety at the same position (49c) reduced the activity (log virus titer difference = 0.68). In addition, the introduction of heterocyclic ring systems such as furan, thiophene or indoline-2-one at R1 provided analogs with reduced antiviral efficacy. Thus, compound 49b could be envisioned as the lead compound that dictates further structural optimization to attain more potent anti-VSV candidates.
Fig. 47.

Chemical structures of fluorinated isatin derivatives 47a–e and 48a–f
Future trends
In the present review, several isatin derivatives (Fig. 48) demonstrated excellent antiviral activity against a vast range of pathogenic viruses. The SARs described herein are enriched and, therefore, arouse innovative thoughts for designing novel and broad-spectrum antiviral analogs. In general, nonvoluminous electron-pulling groups at C-5 of the isatin ring together with the appropriate N-1 substitution are linked to better activity in most of the reported antiviral isatins. However, it is important to mention that several positions of the isatin ring, such as C-2, C-4, C-6 and C-7, have not been well explored and hence would obviously offer a valuable platform for designing prospective compounds. Replacement of the benzene ring in the isatin moiety by pyridine would improve pharmacokinetic properties, cellular permeability and metabolic stability. However, this replacement was found to reduce the antiviral efficacy, as exemplified by compound 39n. Therefore, future research aiming at the development of isatin-based antiviral analogs should focus on balancing different properties by following a stepwise optimization strategy. Furthermore, the essence of the isatin ring structural integrity in antiviral activity has not yet been examined and is therefore a subject of further investigation. The strategy of molecular hybridization has been recognized as a powerful tool in the construction of novel bioactive molecules [122]. Implementation of such a strategy has been found only in a single report in this review, which was concerned with the hybridization of the isatin moiety with the FDA-approved drug lamivudine [44]. The obtained hybrid molecules were highly potent (17b; EC50 = 0.0742 µM), signifying the potential role of molecular hybridization strategies in the future development of efficient antiviral therapeutics. The most potent anti-SARS-COV and anti-SARS-COV-2 compound was 25y (Fig. 48). It is a very potent main protease inhibitor of coronaviruses; meanwhile, it is very cytotoxic, which hinders its further development. Therefore, structural optimization is necessary by leveraging the ease of isatin ring synthetic modification. Insertion of imine, hydrazone, and thiosemicarbazone functionalities at C-3 might alleviate the cytotoxic properties of compound 25y. Considering the similarity between the rhinoviruses and coronaviruses main proteases, the same modification strategy applied for compound 25y could be adopted in 46s. Another attractive approach that could be employed in such cases is the prodrug strategy given that adequate hydrolytic liability is attained [123]. Compound 37aa (Fig. 48) is very active against RSV (EC50 = 5 nM); however, poor physicochemical and pharmacokinetic profiles blunt its further consideration in the drug development process. Adoption of drug delivery systems by combining compound 37aa with an appropriate carrier can offer protection from off-site metabolic biotransformation. Introduction of an electron-withdrawing group at C-5 could further enhance the potency of compound 10e (Fig. 48) against West Nile virus (WNV) (EC50 = 4 µg/mL). With regard to compound 42d, alkylation of the thiosemicarbazone amino group alongside the replacement of the C-5 Br by small groups such as the F group could enhance the potency. Finally, an attractive approach that could be considered to improve the activity and selectivity of the most potent antiviral isatins shown in Fig. 48 is metal complex formation. Metal ions offer an extra binding site to biomolecules, enabling further interactions, and they seem promising for the development of effective antiviral drugs with a unique mechanism of action. Thus, the future perspectives discussed herein would provide new insight for further rational designs of more potent analogs based on the isatin core.
Fig. 48.

The most potent antiviral isatins
Conclusion
In recent decades, the emergence of large viral outbreaks, exemplified by COVID-19, has already resulted in catastrophic impacts. This has motivated researchers worldwide to search for promising antiviral agents using diverse heterocycles. To this end, isatin, an indole analog, has been extensively studied. This review starkly highlights the recent developments of isatin derivatives as antiviral agents with emphasis on their SARs to guide further rational discovery of more potent analogs. In general, several candidates described herein have shown promising activity against different pathogenic viruses. For example, isatins 17b [44] and 25y [55, 64] were found to be equipotent or better than the relevant standards used in inhibiting HIV and SARS-COV-2, with IC50 values of 0.0742 and 0.045 µM, respectively. To develop an ideal antiviral agent, not only potency but also cytotoxicity should be considered. Some potent derivatives, reviewed herein, demonstrated low selectivity indices (SI), which hinder their further development. In addition, the physicochemical properties and pharmacokinetic profiles have been shown to be other drawbacks associated with some potent antiviral isatins. The use of 37 h, and m [93], for example, is limited by their metabolic instability and poor permeability albeit their potent antiviral efficacy (EC50 < 100 nM against RSV). In the current review, the SARs of isatin derivatives have been extensively discussed and presented to inspire novel and creative design approaches to eliminate the highlighted drawbacks. Isatin derivatives have been shown to display a wide range of biological activities. At first glance, this might not bode well for the drug development process, as it would mean that derivatives will potentially have many side effects. Nonetheless, fine tuning of isatin functionalization based on SARs would be useful in developing an ideal lead in this regard. Interesting research gaps have been identified, including (i) studies about the detailed mechanism of action, which could be useful in rational drug design and discovery processes; (ii) investigations of potent antiviral isatins on viruses sharing similar targets are very limited; and (iii) a lack of molecular modeling studies. Addressing these gaps has highlighted the way for future exploration of the isatin core as a promising antiviral core.
Compliance with ethical standards
Conflict of interest
The authors declare no competing interests.
Footnotes
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
References
- 1.Mishra S, Pandey A, Manvati S. Coumarin: an emerging antiviral agent. Vol. 6. Heliyon: Elsevier Ltd; 2020. p. e03217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Zhang MZ, Chen Q, Yang GF. A review on recent developments of indole-containing antiviral agents. Eur J Med Chem. 2015;89:421–41. doi: 10.1016/j.ejmech.2014.10.065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Azhar EI, Hui DSC, Memish ZA, Drosten C, Zumla A. The Middle East Respiratory Syndrome (MERS) Infect Dis Clin North Am. 2019;33:891–905. doi: 10.1016/j.idc.2019.08.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Munayco CV, Tariq A, Rothenberg R, Soto-Cabezas GG, Reyes MF, Valle A, et al. Early transmission dynamics of COVID-19 in a southern hemisphere setting: Lima-Peru: February 29th–March 30th, 2020. Infect Dis Model. 2020;5:338–45. doi: 10.1016/j.idm.2020.05.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.WHO. Coronavirus disease (COVID-19) pandemic. 2019. https://www.who.int/emergencies/diseases/novel-coronavirus-2019.
- 6.De Clercq E, Li G. Approved antiviral drugs over the past 50 years. Clin Microbiol Rev. 2016;29:695–747. doi: 10.1128/CMR.00102-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Graham BS. Advances in antiviral vaccine development. Immunological Rev. 2013;255:230–42. doi: 10.1111/imr.12098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.De Clercq E. Recent highlights in the development of new antiviral drugs. Curr Opin Microbiol. 2005;8:552–60. doi: 10.1016/j.mib.2005.08.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Andersen PI, Ianevski A, Lysvand H, Vitkauskiene A, Oksenych V, Bjørås M, et al. Discovery and development of safe-in-man broad-spectrum antiviral agents. Int J Infect Dis Int Soc Infect Dis. 2020;93:268–76. doi: 10.1016/j.ijid.2020.02.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Abdel-Aziz HA, Elsaman T, Al-Dhfyan A, Attia MI, Al-Rashood KA, Al-Obaid ARM. Synthesis and anticancer potential of certain novel 2-oxo-N’-(2-oxoindolin- 3-ylidene)-2H-chromene-3-carbohydrazides. Eur J Med Chem. 2013;70:358–63. doi: 10.1016/j.ejmech.2013.09.060. [DOI] [PubMed] [Google Scholar]
- 11.Aboul-Fadl T, Abdel-Aziz HA, Abdel-Hamid MK, Elsaman T, Thanassi J, Pucci MJ. Schiff bases of indoline-2,3-dione: potential novel inhibitors of mycobacterium tuberculosis (Mtb) DNA gyrase. Molecules. 2011;16:7864–79. doi: 10.3390/molecules16097864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Xu Z, Zhang S, Gao C, Fan J, Zhao F, Lv ZS, et al. Isatin hybrids and their anti-tuberculosis activity. Chin Chem Lett. 2017;28:159–67. doi: 10.1016/j.cclet.2016.07.032. [DOI] [Google Scholar]
- 13.Chiyanzu I, Clarkson C, Smith PJ, Lehman J, Gut J, Rosenthal PJ, et al. Design, synthesis and anti-plasmodial evaluation in vitro of new 4-aminoquinoline isatin derivatives. Bioorg Med Chem. 2005;13:3249–61. doi: 10.1016/j.bmc.2005.02.037. [DOI] [PubMed] [Google Scholar]
- 14.Thakur RK, Joshi P, Baranwal P, Sharma G, Shukla SK, Tripathi R, et al. Synthesis and antiplasmodial activity of glyco-conjugate hybrids of phenylhydrazono-indolinones and glycosylated 1,2,3-triazolyl-methyl-indoline-2,3-diones. Eur J Med Chem. 2018;155:764–71. doi: 10.1016/j.ejmech.2018.06.042. [DOI] [PubMed] [Google Scholar]
- 15.Guo H. Isatin derivatives and their anti-bacterial activities. Eur J Med Chem Elsevier Masson SAS. 2019;164:678–88. doi: 10.1016/j.ejmech.2018.12.017. [DOI] [PubMed] [Google Scholar]
- 16.Osman HM, Elsaman T, Yousef BA, Elhadi E, Ahmed AAE, Eltayib EM, et al. Schiff bases of isatin and adamantane-1-carbohydrazide:synthesis, characterization and anticonvulsant activity. J Chem. 2021;2021:6659156.. [Google Scholar]
- 17.Li W, Zhao SJ, Gao F, Lv ZS, Tu JY, Xu Z. Synthesis and in vitro anti-tumor, anti-mycobacterial and anti-HIV activities of diethylene-glycol-tethered bis-isatin derivatives. ChemistrySelect. 2018;3:10250–4. [Google Scholar]
- 18.Devale TL, Parikh J, Miniyar P, Sharma P, Shrivastava B, Murumkar P. Dihydropyrimidinone-isatin hybrids as novel non-nucleoside HIV-1 reverse transcriptase inhibitors. Bioorg Chem. 2017;70:256–66. doi: 10.1016/j.bioorg.2017.01.006. [DOI] [PubMed] [Google Scholar]
- 19.Xu Z, Zhao SJ, Lv ZS, Gao F, Wang Y, Zhang F, et al. Fluoroquinolone-isatin hybrids and their biological activities. Eur J Med Chem. 2019;162:396–406. doi: 10.1016/j.ejmech.2018.11.032. [DOI] [PubMed] [Google Scholar]
- 20.Varun S, Kakkar R. Isatin and its derivatives: a survey of recent syntheses, reactions, and applications. MedChemComm. 2019;10:351–68. doi: 10.1039/c8md00585k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Da Silva JFM, Garden SJ, Pinto AC. The chemistry of isatins: a review from 1975 to 1999. J Braz Chem Soc. 2001;12:273–324. [Google Scholar]
- 22.De Moraes Gomes PAT, Pena LJ, Leite ACL. Isatin derivatives and their antiviral properties against arboviruses: a review. Mini Rev Med Chem. 2018;19:56–62. doi: 10.2174/1389557518666180424093305. [DOI] [PubMed] [Google Scholar]
- 23.WHO. HIV/AIDS. 2020. https://www.who.int/news-room/fact-sheets/detail/hiv-aids.
- 24.Arts EJ, Hazuda DJ. HIV-1 antiretroviral drug therapy. Cold Spring Harbor Perspect Med. 2012;2:a007161.. doi: 10.1101/cshperspect.a007161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Zdanowicz MM. The pharmacology of HIV drug resistance. Am J Pharm Education. 2006;70:100.. doi: 10.5688/aj7005100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Pandeya SN, Sriram D, Nath G, De Clercq E. Synthesis, antibacterial, antifungal and anti-HIV activities of norfloxacin Mannich bases. Eur J Med Chem. 2000;35:249–55. doi: 10.1016/s0223-5234(00)00125-2. [DOI] [PubMed] [Google Scholar]
- 27.Pandeya SN, Yogeeswari P, Sriram D, De Clercq E, Pannecouque C, Witvrouw M. Synthesis and screening for anti-HIV activity of some N-Mannich bases of isatin derivatives. Chemotherapy. 1999;45:192–6. doi: 10.1159/000007182. [DOI] [PubMed] [Google Scholar]
- 28.Neyts J, De Clercq E. Therapy and short-term prophylaxis of poxvirus infections: Historical background and perspectives. Antivir Res. 2003;57:25–33. doi: 10.1016/s0166-3542(02)00197-3. [DOI] [PubMed] [Google Scholar]
- 29.Pandeya SN, Sriram D, Nath G, Declercq E. Synthesis, antibacterial, antifungal and anti-HIV activities of Schiff and Mannich basesderived from isatin derivatives and N-[4-(4’-chlorophenyl)thiazol-2-yl] thiosemicarbazide. Eur J Pharm Sci. 1999;9:25–31. doi: 10.1016/s0928-0987(99)00038-x. [DOI] [PubMed] [Google Scholar]
- 30.Pandeya SN, Sriram D, Nath G, De Clercq E. Synthesis, antibacterial, antifungal and anti-HIV evaluation of Schiff and Mannich bases of isatin derivatives with 3-amino-2-methylmercapto quinazolin-4(3H)-one. Pharm Acta Helv. 1999;74:11–7. doi: 10.1016/s0031-6865(99)00010-2. [DOI] [PubMed] [Google Scholar]
- 31.Teitz Y, Ronen D, Vansover A, Stematsky T, Riggs JL. Inhibition of human immunodeficiency virus by N-methylisatin-β4′:4′-diethylthiosemicarbazone and N-allylisatin-β-4′:4′-diallythiosemicarbazone. Antivir Res. 1994;24:305–14. doi: 10.1016/0166-3542(94)90077-9. [DOI] [PubMed] [Google Scholar]
- 32.WHO. Ebola Situation Reports. 2016. https://apps.who.int/ebola/ebola-situation-reports.
- 33.Banerjee D, Yogeeswari P, Bhat P, Thomas A, Srividya M, Sriram D. Novel isatinyl thiosemicarbazones derivatives as potential molecule to combat HIV-TB co-infection. Eur J Medicinal Chem. 2011;46:106–21. doi: 10.1016/j.ejmech.2010.10.020. [DOI] [PubMed] [Google Scholar]
- 34.Up to 650 000 people die of respiratory diseases linked to seasonal flu each year. 2017. https://apps.who.int/mediacentre/news/releases/2017/seasonal-flu/en/index.html.
- 35.Sriram D, Bal TR, Yogeeswari P. Design, synthesis and biological evaluation of novel non-nucleoside HIV-1 reverse transcriptase inhibitors with broad-spectrum chemotherapeutic properties. Bioorg Med Chem. 2004;12:5865–73. doi: 10.1016/j.bmc.2004.08.028. [DOI] [PubMed] [Google Scholar]
- 36.WHO. Global Strategy for dengue prevention and control, 2012–2020. 2020. https://www.who.int/denguecontrol/9789241504034/en/.
- 37.Bal TR, Anand B, Yogeeswari P, Sriram D. Synthesis and evaluation of anti-HIV activity of isatin β-thiosemicarbazone derivatives. Bioorg Med Chem Lett. 2005;15:4451–5. doi: 10.1016/j.bmcl.2005.07.046. [DOI] [PubMed] [Google Scholar]
- 38.Selvam P, Chandramohan M, De Clercq E, Witvrouw M, Pannecouque C. Synthesis and anti-HIV activity of 4-[(1,2-dihydro-2-oxo-3Hindol-3-ylidene) amino]-N(4,6-dimethyl-2-pyrimidinyl)-benzene sulfonamide and its derivatives. Eur J Pharmaceutical Sci. 2001;14:313–6. doi: 10.1016/s0928-0987(01)00197-x. [DOI] [PubMed] [Google Scholar]
- 39.Kharb R, Shahar Yar M, Chander, Sharma P. Recent advances and future perspectives of triazole analogs as promising antiviral agents. Mini Rev Med Chem. 2010;11:84–96. doi: 10.2174/138955711793564051. [DOI] [PubMed] [Google Scholar]
- 40.Pandeya SN, Sriram D, Nath G, De Clercq E. Synthesis, antibacterial, antifungal and anti-HIV evaluation of Schiff and Mannich bases of isatin and its berivatives with triazole. Arzneim Forsch Drug Res. 2000;50:55–9. doi: 10.1055/s-0031-1300164. [DOI] [PubMed] [Google Scholar]
- 41.Selvam P, Murugesh N, Chandramohan M, Debyser Z, Witvrouw M. Design, synthesis and antiHIV activity of novel isatine-sulphonamides. Indian J Pharm Sci. 2008;70:779–82. doi: 10.4103/0250-474X.49121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Sriram D, Ratan Bal T, Yogeeswari P. Aminopyimidinimo isatin analogues: design of novel non-nucleoside HIV-1 reverse transcriptase inhibitors with broadspectrum chemotherapeutic properties. J Pharm Pharm Sci. 2005;8:565–77. [PubMed] [Google Scholar]
- 43.Sriram D, Bal TR, Yogeeswari P. Synthesis, antiviral and antibacterial activities of isatin mannich bases. Medicinal Chem Res. 2005;14:211–28. [Google Scholar]
- 44.Sriram D, Yogeeswari P, Gopal G. Synthesis, anti-HIV and antitubercular activities of lamivudine prodrugs. Eur J Med Chem. 2005;40:1373–6. doi: 10.1016/j.ejmech.2005.07.006. [DOI] [PubMed] [Google Scholar]
- 45.Pawar VS, Lokwani DK, Bhandari SV, Bothara KG, Chitre TS, Devale TL, et al. Design, docking study and ADME prediction of Isatin derivatives as anti-HIV agents. Med Chem Res. 2011;20:370–80. [Google Scholar]
- 46.Vijayanand P, Wilkins E, Woodhead M. Severe acute respiratory syndrome (SARS): a review. Clin Med. 2004;4:152–60. doi: 10.7861/clinmedicine.4-2-152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Lee N, Hui D, Wu A, Chan P, Cameron P, Joynt GM, et al. A major outbreak of severe acute respiratory syndrome in Hong Kong. N Engl J Med. 2003;348:1986–94. doi: 10.1056/NEJMoa030685. [DOI] [PubMed] [Google Scholar]
- 48.Tong TR. Severe acute respiratory syndrome coronavirus (SARS-CoV) Perspect Med Virol. 2006;16:43–95. doi: 10.1016/S0168-7069(06)16004-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Drosten C, Günther S, Preiser W, van der Werf S, Brodt H-R, Becker S, et al. Identification of a novel coronavirus in patients with severe acute respiratory syndrome. N Engl J Med. 2003;348:1967–76. doi: 10.1056/NEJMoa030747. [DOI] [PubMed] [Google Scholar]
- 50.Grum-Tokars V, Ratia K, Begaye A, Baker SC, Mesecar AD. Evaluating the 3C-like protease activity of SARS-Coronavirus: recommendations for standardized assays for drug discovery. Virus Res. 2008;133:63–73. doi: 10.1016/j.virusres.2007.02.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Adedeji AO, Sarafianos SG. Antiviral drugs specific for coronaviruses in preclinical development. Curr Opin Virol. 2014;8:45–53. doi: 10.1016/j.coviro.2014.06.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Liu W, Zhu HM, Niu GJ, Shi EZ, Chen J, Sun B, et al. Synthesis, modification and docking studies of 5-sulfonyl isatin derivatives as SARS-CoV 3C-like protease inhibitors. Bioorg Medicinal Chem. 2014;22:292–302. doi: 10.1016/j.bmc.2013.11.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Selvam P, Murgesh N, Chandramohan M, De Clercq E, Keyaerts E, Vijgen L, et al. In vitro antiviral activity of some novel isatin derivatives against HCV and SARS-CoV viruses. Indian J Pharm Sci. 2008;70:91–4. doi: 10.4103/0250-474X.40339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Chen LR, Wang YC, Yi WL, Chou SY, Chen SF, Lee TL, et al. Synthesis and evaluation of isatin derivatives as effective SARS coronavirus 3CL protease inhibitors. Bioorg Medicinal Chem Lett. 2005;15:3058–62. doi: 10.1016/j.bmcl.2005.04.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Zhou L, Liu Y, Zhang W, Wei P, Huang C, Pei J, et al. Isatin compounds as noncovalent SARS coronavirus 3C-like protease inhibitors. J Medicinal Chem. 2006;49:3440–3. doi: 10.1021/jm0602357. [DOI] [PubMed] [Google Scholar]
- 56.Jean SS, Lee PI, Hsueh PR. Treatment options for COVID-19: the reality and challenges. J Microbiol Immunol Infect. 2020;53:436–43. doi: 10.1016/j.jmii.2020.03.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Zhou F, Yu T, Du R, Fan G, Liu Y, Liu Z, et al. Clinical course and risk factors for mortality of adult inpatients with COVID-19 in Wuhan, China: a retrospective cohort study. Lancet. 2020;395:1054–62. doi: 10.1016/S0140-6736(20)30566-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Abdalla AE, Xie J, Junaid K, Younas S, Elsaman T, Abosalif KOA, et al. Insight into the emerging role of SARS-CoV-2 nonstructural and accessory proteins in modulation of multiple mechanisms of host innate defense. Bosnian J Basic Med Sci. 2021;21:515–27. doi: 10.17305/bjbms.2020.5543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Pujari R, Thommana MV, Ruiz Mercedes B, Serwat A. Therapeutic options for COVID-19: a review. Cureus. 2020;12:e10480.. doi: 10.7759/cureus.10480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Dai W, Zhang B, Jiang XM, Su H, Li J, Zhao Y, et al. Structure-based design of antiviral drug candidates targeting the SARS-CoV-2 main protease. Science. 2020;368:1331–5. doi: 10.1126/science.abb4489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Liu X, Wang XJ. Potential inhibitors against 2019-nCoV coronavirus M protease from clinically approved medicines. J Genet Genom. 2020;47:119–21. doi: 10.1016/j.jgg.2020.02.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Cui W, Yang K, Yang H. Recent Progress in the Drug Development Targeting SARS-CoV-2 Main Protease as Treatment for COVID-19. Front Mol Biosci. 2020;7:616431. doi: 10.3389/fmolb.2020.616341.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Jin Z, Wang H, Duan Y, Yang H. The main protease and RNA-dependent RNA polymerase are two prime targets for SARS-CoV-2. Biochem Biophys Res Commun. 2021;538:63–71. doi: 10.1016/j.bbrc.2020.10.091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Liu P, Liu H, Sun Q, Liang H, Li C, Deng X, et al. Potent inhibitors of SARS-CoV-2 3C-like protease derived from N-substituted isatin compounds. Eur J Med Chem. 2020;206:112702. doi: 10.1016/j.ejmech.2020.112702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Badavath VN, Kumar A, Samanta PK, Maji S, Das A, Blum G, et al. Determination of potential inhibitors based on isatin derivatives against SARS-CoV-2 main protease (mpro): a molecular docking, molecular dynamics and structure-activity relationship studies. J Biomol Struct Dyn Taylor Francis. 2020;0:1–19. doi: 10.1080/07391102.2020.1845800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Endy TP. A García-Sastre, Mount Sinai School of Medicine, New York, NY, USA T P Endy, State University of New York, Syracuse, NY, USA. p. 313–21.
- 67.Cleton N, Koopmans M, Reimerink J, Godeke GJ, Reusken C. Come fly with me: review of clinically important arboviruses for global travelers. J Clin Virol Elsevier B V. 2012;55:191–203. doi: 10.1016/j.jcv.2012.07.004. [DOI] [PubMed] [Google Scholar]
- 68.Solomon T WR. Arthropod-borne viral encephalitides. In: Infections of the central nervous system. 3rd ed. Philadelphia: Lippincott Williams and Wilkins; 2004;205–30.
- 69.Mishra P, Kumar A, Mamidi P, Kumar S, Basantray I, Saswat T, et al. Inhibition of Chikungunya virus replication by 1-[(2-Methylbenzimidazol-1-yl) Methyl]-2-Oxo-Indolin-3-ylidene] Amino] Thiourea(MBZM-N-IBT) Sci Rep. 2016;6:1–13. doi: 10.1038/srep20122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Sebastian L, Desai A, Shampur MN, Perumal Y, Sriram D, Vasanthapuram R. N-methylisatin-beta-thiosemicarbazone derivative (SCH 16) is an inhibitor of Japanese encephalitis virus infection in vitro and in vivo. Virol J. 2008;5:1–12. doi: 10.1186/1743-422X-5-64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Sebastian L, Desai A, Yogeeswari P, Sriram D, Madhusudana SN, Ravi V. Combination of N-methylisatin-β-thiosemicarbazone derivative (SCH16) with ribavirin and mycophenolic acid potentiates the antiviral activity of SCH16 against Japanese encephalitis virus in vitro. Lett Appl Microbiol. 2012;55:234–9. doi: 10.1111/j.1472-765X.2012.03282.x. [DOI] [PubMed] [Google Scholar]
- 72.Minami M, Hamaue N, Hirafuji M, Saito H, Hiroshige T, Ogata A, et al. Isatin, an endogenous MAO inhibitor, and a rat model of Parkinson’s disease induced by the Japanese encephalitis virus. J Neural Transm Suppl. 2006;71:87–95. doi: 10.1007/978-3-211-33328-0_10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Kramer LD, Li J, Shi PY. West Nile virus. Lancet Neurol. 2007;6:171–81. doi: 10.1016/S1474-4422(07)70030-3. [DOI] [PubMed] [Google Scholar]
- 74.Blázquez AB, Martín-Acebes MA, Saiz JC. Inhibition of West Nile virus multiplication in cell culture by anti-parkinsonian drugs. Front Microbiol. 2016;7:1–10. doi: 10.3389/fmicb.2016.00296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Justo LA, Durán R, Alfonso M, Fajardo D, Faro LRF. Effects and mechanism of action of isatin, a MAO inhibitor, on in vivo striatal dopamine release. Neurochem Int. 2016;99:147–57. doi: 10.1016/j.neuint.2016.06.012. [DOI] [PubMed] [Google Scholar]
- 76.Tuiskunen Bäck A, Lundkvist Å. Dengue viruses – an overview. Infect Ecol Epidemiol. 2013;3:19839. doi: 10.3402/iee.v3i0.19839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Zou B, Chan WL, Ding M, Leong SY, Nilar S, Seah PG, et al. Lead optimization of spiropyrazolopyridones: a new and potent class of dengue virus inhibitors. ACS Medicinal Chem Lett. 2015;6:344–8. doi: 10.1021/ml500521r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Xu J, Xie X, Ye N, Zou J, Chen H, White MA, et al. Design, synthesis, and biological evaluation of substituted 4,6-dihydrospiro[[1,2,3]triazolo[4,5- b]pyridine-7,3′-indoline]-2′,5(3 H)-dione analogues as potent NS4B inhibitors for the treatment of dengue virus infection. J Med Chem. 2019;62:7941–60. doi: 10.1021/acs.jmedchem.9b00698. [DOI] [PubMed] [Google Scholar]
- 79.Xu X, Zhang Y, Li Q. Characteristics of herpes simplex virus infection and pathogenesis suggest a strategy for vaccine development. Rev Med Virol. 2019;29:1–12. doi: 10.1002/rmv.2054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Caumes E. Herpes simplex virus and varicella zoster virus infections in HIV- infected patients. Med et Maladies Infectieuses. 1998;28:245–52. [Google Scholar]
- 81.Balfour HHJ. Antiviral drugs. N. Engl J Med. 1999;340:1255–68. doi: 10.1056/NEJM199904223401608. [DOI] [PubMed] [Google Scholar]
- 82.Whitley RJ. Herpes simplex virus infection. Semin Pediatr Infect Dis. 2002;13:6–11. doi: 10.1053/spid.2002.29752. [DOI] [PubMed] [Google Scholar]
- 83.Field AK, Biron KK. “The end of innocence” revisited: resistance of herpesviruses to antiviral drugs. Clin Microbiol Rev. 1994;7:1–13. doi: 10.1128/cmr.7.1.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Kang IJ, Wang LW, Hsu TA, Yueh A, Lee CC, Lee YC, et al. Isatin-β-thiosemicarbazones as potent herpes simplex virus inhibitors. Bioorg Medicinal Chem Lett. 2011;21:1948–52. doi: 10.1016/j.bmcl.2011.02.037. [DOI] [PubMed] [Google Scholar]
- 85.Jarrahpour A, Khalili D, De Clercq E, Salmi C, Brunel JM. Synthesis, antibacterial, antifungal and antiviral activity evaluation of some new bis-Schiff bases of isatin and their derivatives. Molecules. 2007;12:1720–30. doi: 10.3390/12081720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Seley-radtke KL, Yates MK. Since January 2020 Elsevier has created a COVID-19 resource centre with free information in English and Mandarin on the novel coronavirus COVID- 19. The COVID-19 resource centre is hosted on Elsevier Connect, the company’ s public news and information. Antivir Res. 2018;154:66–86. [Google Scholar]
- 87.De Oliveira MRP, Torres JC, Garden SJ, Dos Santos CVB, Rocha Alves T, Pinto AC, et al. Synthesis and antiviral evaluation of isatin ribonucleosides. Nucleosides Nucleotides Nucleic Acids. 2002;21:825–35. doi: 10.1081/NCN-120016510. [DOI] [PubMed] [Google Scholar]
- 88.Meanwell NA, Krystal M. Respiratory syncytial virus: Recent progress towards the discovery of effective prophylactic and therapeutic agents. Drug Discov Today. 2000;5:241–52. doi: 10.1016/s1359-6446(00)01500-2. [DOI] [PubMed] [Google Scholar]
- 89.Greensill J, McNamara PS, Dove W, Flanagan B, Smyth RL, Hart CA. Human metapneumovirus in severe respiratory syncytial virus bronchiolitis. Emerg Infect Dis. 2003;9:372–5. doi: 10.3201/eid0903.020289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Griffiths C, Drews SJ, Marchant DJ. Respiratory syncytial virus: Infection, detection, and new options for prevention and treatment. Clin Microbiol Rev. 2017;30:277–319. doi: 10.1128/CMR.00010-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Yu KL, Zhang Y, Civiello RL, Kadow KF, Cianci C, Krystal M, et al. Fundamental structure-activity relationships associated with a new structural class of respiratory syncytial virus inhibitor. Bioorg Medicinal Chem Lett. 2003;13:2141–4. doi: 10.1016/s0960-894x(03)00383-4. [DOI] [PubMed] [Google Scholar]
- 92.Yu KL, Sin N, Civiello RL, Wang XA, Combrink KD, Gulgeze HB, et al. Respiratory syncytial virus fusion inhibitors. Part 4: optimization for oral bioavailability. Bioorg Medicinal Chem Lett. 2007;17:895–901. doi: 10.1016/j.bmcl.2006.11.063. [DOI] [PubMed] [Google Scholar]
- 93.Sin N, Venables BL, Combrink KD, Gulgeze HB, Yu KL, Civiello RL, et al. Respiratory syncytial virus fusion inhibitors. Part 7: structure-activity relationships associated with a series of isatin oximes that demonstrate antiviral activity in vivo. Bioorg Medicinal Chem Lett. 2009;19:4857–62. doi: 10.1016/j.bmcl.2009.06.030. [DOI] [PubMed] [Google Scholar]
- 94.Wang XA, Cianci CW, Yu KL, Combrink KD, Thuring JW, Zhang Y, et al. Respiratory syncytial virus fusion inhibitors. Part 5: optimization of benzimidazole substitution patterns towards derivatives with improved activity. Bioorg Medicinal Chem Lett. 2007;17:4592–8. doi: 10.1016/j.bmcl.2007.05.102. [DOI] [PubMed] [Google Scholar]
- 95.Yu KL, Zhang Y, Civiello RL, Trehan AK, Pearce BC, Yin Z, et al. Respiratory syncytial virus inhibitors. Part 2: Benzimidazol-2-one derivatives. Bioorg Medicinal Chem Lett. 2004;14:1133–7. doi: 10.1016/j.bmcl.2003.12.072. [DOI] [PubMed] [Google Scholar]
- 96.Sin J, Mangale V, Thienphrapa W, Gottlieb RA, Feuer R. Recent progress in understanding coxsackievirus replication, dissemination, and pathogsenesis. Virology. 2015;484:288–304. doi: 10.1016/j.virol.2015.06.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Zhang HM, Dai H, Hanson PJ, Li H, Guo H, Ye X, et al. Antiviral activity of an isatin derivative via induction of PERK-Nrf2-mediated suppression of cap-independent translation. ACS Chem Biol. 2014;9:1015–24. doi: 10.1021/cb400775z. [DOI] [PubMed] [Google Scholar]
- 98.Yang YJ, Liu JN, Pan XD. Synthesis and antiviral activity of lycorine derivatives. J Asian Nat Products Res Taylor Francis. 2020;22:1188–96. doi: 10.1080/10286020.2020.1844674. [DOI] [PubMed] [Google Scholar]
- 99.Thèves C, Biagini P, Crubézy E. The rediscovery of smallpox. Clin Microbiol Infect. 2014;20:210–8. doi: 10.1111/1469-0691.12536. [DOI] [PubMed] [Google Scholar]
- 100.Moss B. Poxvirus DNA replication. Cold Spring Harb Perspect Biol. 2013;5:1–12. doi: 10.1101/cshperspect.a010199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Meyer H, Ehmann R, Smith GL. Smallpox in the post-eradication Era. Viruses. 2020;12:1–11. doi: 10.3390/v12020138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Smallpox. 2021. https://www.cdc.gov/smallpox/.
- 103.Heiner GG, Fatima N, Russell PK, Haase AT, Ahmad N, Mohammed N, et al. Field trials of methisazone as a prophylactic agent against smallpox. Am J Epidemiol. 1971;94:435–49. doi: 10.1093/oxfordjournals.aje.a121340. [DOI] [PubMed] [Google Scholar]
- 104.Pirrung MC, Pansare SV, Das Sarma K, Keith KA, Kern ER. Combinatorial optimization of isatin-β-thiosemicarbazones as anti-poxvirus agents. J Medicinal Chem. 2005;48:3045–50. doi: 10.1021/jm049147h. [DOI] [PubMed] [Google Scholar]
- 105.Te Velthuis AJW, Fodor E. Influenza virus RNA polymerase: Insights into the mechanisms of viral RNA synthesis. Nat Rev Microbiol. 2016;14:479–93. doi: 10.1038/nrmicro.2016.87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Mayer D, Molawi K, Martínez-Sobrido L, Ghanem A, Thomas S, Baginsky S, et al. Identification of cellular interaction partners of the influenza virus ribonucleoprotein complex and polymerase complex using proteomic-based approaches. J Proteome Res. 2007;6:672–82. doi: 10.1021/pr060432u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Ye N, Chen H, Wold EA, Shi PY, Zhou J. Therapeutic potential of spirooxindoles as antiviral agents. ACS Infect Dis. 2016;2:382–92. doi: 10.1021/acsinfecdis.6b00041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Das K. Antivirals targeting influenza a virus. J Medicinal Chem. 2012;55:6263–77. doi: 10.1021/jm300455c. [DOI] [PubMed] [Google Scholar]
- 109.Selvam P, Murugesh N, Chandramohan M, Sidwell RW, Wandersee MK, Smee DF. Anti-influenza virus activities of 4-[(1,2-dihydro-2-oxo-3H-indol-3- ylidene)amino]-N-(4,6-dimethyl-2-pyrimidin-2-yl)benzenesulphonamide and its derivatives. Antiviral Chem Chemother. 2006;17:269–74. doi: 10.1177/095632020601700504. [DOI] [PubMed] [Google Scholar]
- 110.Kurbatov SV, Zarubaev VV, Karpinskaya LA, Shvets AA, Kletsky ME, Burov ON, et al. Synthesis and antiviral activity of bis-spirocyclic derivatives of rhodanine. Russian Chem Bull. 2014;63:1130–6. [Google Scholar]
- 111.Karki SS, Kulkarni AA, Kumar S, Veliyath SK, De Clercq E, Balzarini J. Synthesis and biological evaluation of 2-(5-substituted-1-((diethylamino) methyl)-2-oxoindolin-3-ylidene)-N-substituted-hydrazinecarbothioamides. Medicinal Chem Res. 2013;22:2014–22. doi: 10.1007/s00044-012-0184-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Jacobs SE, Lamson DM, Kirsten S, Walsh TJ. Human rhinoviruses. Clin Microbiol Rev. 2013;26:135–62. doi: 10.1128/CMR.00077-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Turner RB. Rhinovirus: more than just a common cold virus. J Infect Dis. 2007;195:765–6. doi: 10.1086/511829. [DOI] [PubMed] [Google Scholar]
- 114.Webber SE, Tikhe J, Worland ST, Fuhrman SA, Hendrickson TF, Matthews DA, et al. Design, synthesis, and evaluation of nonpeptidic inhibitors of human rhinovirus 3C protease. J Medicinal Chem. 1996;39:5072–82. doi: 10.1021/jm960603e. [DOI] [PubMed] [Google Scholar]
- 115.Melnick JL. Current status of poliovirus infections. Clin Microbiol Rev. 1996;9:293–300. doi: 10.1128/cmr.9.3.293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Minor P. The polio endgame. Hum Vaccin Immunother. 2014;10:i–iii. doi: 10.4161/21645515.2014.981115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Blondel B, Colbère-Garapin F, Couderc T, Wirotius A, Guivel-Benhassine F. Poliovirus, pathogenesis of poliomyelitis, and apoptosis. Curr Top Microbiol Immunol. 2005;289:25–56. doi: 10.1007/3-540-27320-4_2. [DOI] [PubMed] [Google Scholar]
- 118.McKinlay MA, Collett MS, Hincks JR, Steven Oberste M, Pallansch MA, Okayasu H, et al. Progress in the development of poliovirus antiviral agents and their essential role in reducing risks that threaten eradication. J Infect Dis. 2014;210:S447–53. doi: 10.1093/infdis/jiu043. [DOI] [PubMed] [Google Scholar]
- 119.Varma RS, Nobles WL. Antiviral, antibacterial, and antifungal activities of isatin N‐mannich bases. J Pharm Sci. 1975;64:881–2. doi: 10.1002/jps.2600640539. [DOI] [PubMed] [Google Scholar]
- 120.Letchworth GJ, Rodriguez LL, Del cbarrera J. Vesicular stomatitis. Vet J. 1999;157:239–60. doi: 10.1053/tvjl.1998.0303. [DOI] [PubMed] [Google Scholar]
- 121.Abbas SY, Farag AA, Ammar YA, Atrees AA, Mohamed AF, El-Henawy AA. Synthesis, characterization, and antiviral activity of novel fluorinated isatin derivatives. Monatshefte fur Chem. 2013;144:1725–33. doi: 10.1007/s00706-013-1034-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.He ZX, Gong YP, Zhang X, Ma LY, Zhao W. Pyridazine as a privileged structure: an updated review on anticancer activity of pyridazine containing bioactive molecules. Eur J Med Chem [Internet] 2021;209:112946. doi: 10.1016/j.ejmech.2020.112946. [DOI] [PubMed] [Google Scholar]
- 123.Ferraz de Paiva RE, Vieira EG, Rodrigues da Silva D, Wegermann CA, Costa Ferreira AM. Anticancer compounds based on isatin-derivatives: strategies to ameliorate selectivity and efficiency. Front Mol Biosci. 2021;7:1–24. doi: 10.3389/fmolb.2020.627272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.UNAIDS DATA 2019. 2019. https://www.unaids.org/en/resources/documents/2019/2019-UNAIDS-data.
- 125.WHO. Global hepatitis report. 2017. https://www.who.int/hepatitis/publications/global-hepatitis-report2017/en/.