


Abstract
Post-transcriptional modifications are added to ribosomal RNAs (rRNAs) to govern ribosome biogenesis and to fine-tune protein biosynthesis. In Escherichia coli and related bacteria, RlhA uniquely catalyzes formation of a 5-hydroxycytidine (ho5C) at position 2501 of 23S rRNA. However, the molecular and biological functions as well as the regulation of ho5C2501 modification remain unclear. We measured growth curves with the modification-deficient ΔrlhA strain and quantified the extent of the modification during different conditions by mass spectrometry and reverse transcription. The levels of ho5C2501 in E. coli ribosomes turned out to be highly dynamic and growth phase-dependent, with the most effective hydroxylation yields observed in the stationary phase. We demonstrated a direct effect of ho5C2501 on translation efficiencies in vitro and in vivo. High ho5C2501 levels reduced protein biosynthesis which however turned out to be beneficial for E. coli for adapting to oxidative stress. This functional advantage was small under optimal conditions or during heat or cold shock, but becomes pronounced in the presence of hydrogen peroxide. Taken together, these data provided first functional insights into the role of this unique 23S rRNA modification for ribosome functions and bacterial growth under oxidative stress.
INTRODUCTION
Post-transcriptional RNA modifications have gained increasing attention in recent years and have been recognized as a cellular mechanism to regulate RNA structure, function and stability (1,2). The physiological relevance of RNA modifications is reflected by the fact that their misregulation can have severe consequences (3,4). Ribosomal RNAs (rRNAs) are no exception since they have been shown to be decorated by numerous modifications. In Escherichia coli, there are 36 modification sites found on rRNA, 11 on the small subunit 16S rRNA and 25 on the large subunit 23S rRNA, the most common ones being the conversion of uridine to pseudouridine (Ψ) as well as ribose and nucleobase methylations (2). The modifications tend to be clustered around functionally important sites on the ribosome, such as the mRNA decoding center, the tRNA binding sites, the nascent peptide exit tunnel and the peptidyl transferase center (PTC). The specific functions of individual modifications, however, remain largely elusive. They have been implicated in proper ribosome biogenesis, assisting ribosome stability, interacting with ribosomal ligands such as tRNAs, as well as conveying antibiotic resistance against drugs targeting the ribosome (reviewed in (2,5)). Yet, no single or individual modification has been shown to be essential and phenotypes upon deletions of the modifications tend to be mild, fostering a view that post-transcriptional rRNA modifications serve more of a fine-tuning role in translational activity.
Unlike in eukaryotes and archaea, where rRNA modifications are predominantly added with the assistance of small nucleolar RNA–protein complexes (snoRNPs), E. coli utilizes protein-only enzymes that are site- or region-specific (5). In other words, for almost every single rRNA modification site a separate enzyme evolved. All but one rRNA modification enzyme of E. coli have been characterized with the one responsible for dihydrouridine formation at residue 2449 of the 23S rRNA remaining the sole unidentified enzyme. RlhA was the penultimate modifying enzyme described for E. coli rRNA modifications (6). It hydroxylates the nucleotide C2501 at position C5 to form ho5C2501 (Figure 1A). Nucleotide C2501 is in near proximity of the PTC and forms base-stacking interactions with the unmodified nucleotides G2446 and G2447 (Figure 1B). It is close to the nucleotides C2063 and A2451, both crucial components of the PTC (7–9).
Figure 1.
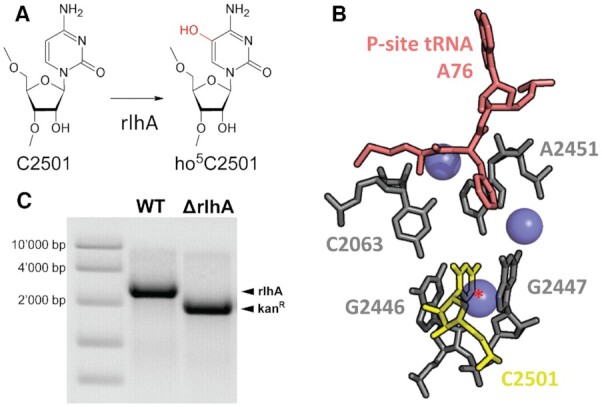
RlhA hydroxylates C2501 in close proximity to the peptidyl transferase center (PTC). (A) The cytosine at position 2501 of the E. coli 23S rRNA is hydroxylated at the C5 by the iron-sulfur protein RlhA (formerly ydcP). (B) Three-dimensional structure representation of the PTC architecture in bacteria including water molecules (PDB: 1VY4). C2501 inserts itself between two guanines (G2446, G2447) and is in close proximity to two PTC nucleotides (C2063, A2451) and the P-site tRNA (red). The site of hydroxylation at the C5 is marked with a red asterisk. (C) Ethidium bromide staining of colony PCR products representing the genomic rlhA region from wildtype (WT) and rlhA knockout (ΔrlhA) E. coli strains.
The identity of this modification was not described before 2011 (10), but the presence of a posttranscriptional modification at position C2501 was already suggested in 1993 based on a reverse transcription assay (11). However, subsequent ESI-MS analysis failed to identify any modification (12), likely due to the sub-stoichiometric and growth phase-dependent nature of this 16 Da modification (13). In E. coli, only 10–30% of ribosomes harbor the ho5C2501 modification in early exponential phase, which increases up to 70% in stationary phase (6,13). Additionally, RlhA activity was shown to rely on iron-sulfur cluster availability and the prephenate synthesis pathway (6). Finally, mutational studies identified four cysteines essential for RlhA action, with each single mutation leading to a complete loss of the catalytic activity, which led Kimura et al. to the conclusion that RlhA is most likely an iron-sulfur protein that uses prephenate as the hydroxyl group donor to produce ho5C2501 (6).
Interestingly, in Deinococcus radiodurans, an extremophilic bacterium highly resistant to radiation, desiccation, and oxidative stress (14), the hydroxylation of C2501 is stoichiometric and independent of the growth phase (10). The fact that this post-transcriptional modification is conserved in two evolutionary distant bacterial phyla (Proteobacteria and Deinococcus-Thermus) highlights that it is both important and ancient.
We therefore set out to first analyze ho5C2501 levels in ribosomes isolated from E. coli cells exposed to different stress conditions. Our quantitative data confirmed an enhanced hydroxylation of C2501 during stationary phase, but no rapid adjustment in modification levels to any of the tested stress conditions was observed. We showed, however, that the presence of the RlhA-catalyzed modification at C2501 is beneficial for E. coli grown under oxidative stress, possibly due to the upregulation of the DNA-protective protein Dps. Finally, we revealed a fine-tuning role of ho5C2501 for translation utilizing isolated and in vitro reconstituted ribosomes in translational assays. The presence of ho5C2501 slightly reduces the activity of ribosomes during in vitro translation but stimulates in vivo metabolic activities during oxidative stress. Taken together our results showed that hydroxylation of the C5 at 23S rRNA position 2501 fine-tunes translation and provides a growth benefit to E. coli during oxidative stress.
MATERIALS AND METHODS
Bacterial strains and media
The knockout strain ΔrlhA was obtained from the wildtype Escherichia coli K-12 strain BW25113 (Δ(araD-araB)567, ΔlacZ4787(::rrnB-3), λ–, rph-1, Δ(rhaD-rhaB)568, hsdR514) by replacement of the chromosomal rlhA gene by a kanamycin (Kan) resistance cassette according to a previously published protocol (16). Used primers can be found in Supplementary Table S1. All deletions were verified by colony PCR using the AccuPrime™ Taq DNA Polymerase High Fidelity kit from Invitrogen. All strains were transformed with either the empty vector pBbSc (17) carrying a chloramphenicol (CAM) resistance or a variant of the rescue vector pBbSc-rlhA described below. Dps overexpression was achieved by transforming the tetracycline (Tet) inducible pBbA2a-dps plasmid (see below) carrying an ampicillin (Amp) resistance into the ΔrlhA strain. E. coli strains were grown in LB-Miller (10 g NaCl per liter) medium with 30 μg/ml CAM added at 37°C if not specified otherwise.
Plasmid construction & PIPE cloning
Polymerase incomplete primer extension (PIPE) constitutes a restriction enzyme and ligation free method of cloning (18). Both the plasmid vectors pBbSc and pBbA2a as well as the rlhA and dps genomic DNA regions were amplified using the Invitrogen AccuPrime Taq DNA Polymerase High Fidelity kit with 20–50 ng DNA templates. An excess of PCR cycles were used (35 cycles), resulting in a small fraction of amplification products being incomplete. Primers for rlhA and dps amplification were designed to have 20 nt long 5′-extensions that are complementary to the vector primer pairs, ultimately allowing for base-pairing between incomplete amplification products from both reactions (see Supplementary Table S1 for all primers). For DNA transformation into XL-1 cells, vector and insert amplification products were mixed in a 1:10 ratio, respectively. Ligation and complementary repair of unpaired regions is performed by the bacterial cell. All cloned plasmids were checked by sequencing before transformation into CaCl2-competent E. coli cells. Mutations of the rlhA sequence (ATG start codon and C169A) were performed similarly using m-PIPE, which requires only one single PCR reaction with pBbSc-rlhA as template. Primers were designed to have complementary 5′-extensions with the desired base mutations.
Growth competition
Overnight seed cultures of 5 ml were prepared from a single colony of the wildtype or ΔrlhA strain. The next day, overnight cultures were re-diluted to an OD600 of 0.2 and 10 ml of each culture were mixed. Before incubation, an initial 50 μl sample was removed from the mixed culture (0 h) and diluted by a factor of 50,000 in LB + CAM (30 μg/ml) medium. 100 μl of the diluted sample were plated and incubated at 37°C for ∼20 h on both an LB + CAM (30 μg/ml) and an LB + Kan (25 μg/ml) plate for colony forming units (cfu) determination. To calculate the ratio of ΔrlhA to wildtype cells, cfu on the Kan plates (only ΔrlhA cells) were divided by the cfu on the CAM plates (total cells). Meanwhile, the 1:1 mixed culture of wildtype and ΔrlhA cells was incubated at 37°C and 220 rpm for 24 h. Afterwards, the culture was re-diluted 1:100 in 20 ml of fresh LB + CAM (30 μg/ml) medium. Next, a 200 μl sample was again removed from the re-diluted culture (24 h) and diluted by a factor of 12,500. Cfus were determined from the diluted sample as before and the re-diluted culture was once more incubated at 37°C and 220 rpm for 24 h. The procedure was then repeated two more times (48 and 72 h samples). Alternatively, the experiment was repeated with an initial 1:1 mixture of both the ΔrlhA and the rlhA complementation strain and 100 μl samples were taken to determine the ratio of rlhA to ΔrlhA cells by PCR. Briefly, the cells in the 100 μl samples were spun down and used as templates in a colony PCR against the insert region of the pBbSc plasmid using the OneTaq DNA Polymerase kit from NEB according to the manufacturer's protocol (annealing temperature: 62°C). PCR products were separated on a 1% agarose gel in 1× TAE. Primer sequences are found in Supplementary Table S1 (E.c.2501_prim_28 and E.c.2501_prim_29).
Sample preparation for SRM mass spectrometry and CMCT treatment
An overnight seed culture of 5 ml was prepared with a single colony of the investigated bacterial strain. The next day, overnight cultures were re-diluted 1:100 in 200 ml of LB + CAM (30μg/ml) and incubated under optimal conditions for 2 h, while growth was monitored by absorption (OD600). A first sample of 50 ml for total RNA isolation was taken before the addition of stress (OD600 ∼ 0.4). For cold and heat stress, cultures were put into an ice-cold or 50°C water bath for 5 min and then kept incubating at 25°C or 45°C, respectively. For oxidative stress, H2O2 was added to a final concentration of 2 mM. Under all conditions, another sample of 50 ml for total RNA isolation was taken 1 h after the addition of the stress and a sample of 5 ml for total RNA isolation was taken once cells reached OD600 ∼4.0. Cell samples for total RNA extraction were pelleted by centrifugation (5000 xg, 10 min, 4°C) and resuspended in 1x TEN (10 mM Tris–HCl pH 7.5, 1 mM EDTA, 100 mM NaCl). Total RNA was isolated with hot phenol (pH 4.3 ± 0.2, 67°C) containing 1% SDS and subsequent PCI treatment.
SRM mass spectrometry
23S rRNA was isolated from total RNA by gel electrophoresis on a 3% agarose gel. Isolated 23S rRNA was added to 50 U recombinant RNase T1 and 0.001 U Bacterial Alkaline Phosphatase per μg RNA in a final concentration of 110 mM ammonium acetate and incubated for 2 h at 37°C. Digestion reactions were then completely dried in a SpeedVac, and resuspended in mobile phase A (MPA, see below) to final concentration of 1 μg/ul in MS vials.
6 μg digested 23S rRNA were separated on a Thermo DNAPacTM RP column, 4 μm, 2.1 mm × 50 mm, on an Hitachi LC unit at a flow rate of 30 μl/min over a gradient of MPA (400 mM HFIP, 16 mM TEA in water, pH 7) and MPB (1:1 mixture of MPA and methanol). Digestion products were bound to the column in 2% MPB, eluted using a gradient from 2 to 18% MPB in 10 min, then to 22% in 1 min, followed by washing with 99% MPB for 5 min and re-equilibration. RNase T1 digestion products of interest eluted at approximately 15% MPB.
For mass spectrometry detection, the Thermo Scientific™ TSQ Quantiva triple quadrupole mass spectrometer and TSQ Quantiva Tune Application Software were used. Selected reaction monitoring (SRM) was performed in the negative ion mode over an m/z range of 500–1850 with a capillary temperature of 300°C, spray voltage of 3.44 kV and sheath gas 45 Arb, auxiliary gas 4 Arb, and sweep gas 2 Arb respectively. Monitoring was initially performed over the whole run, but later the time window was narrowed to 5–35 min when oligonucleotides of interest were observed to elute.
Parameters for SRM were established based on monoisotopic mass, electrospray series and CID fragments of the four oligoribonucleotide variants. We chose several characteristic w2–w5 and y2–y5, as well as some c2/5 CID fragment ions that unambiguously identified the different precursors (Supplementary Figure S1).
Peak intensity of SRM ion chromatograms was quantified in Xcalibur software using the Genesis algorithm. We calculated ho5C2501 frequency from the relative peak intensity of the observed modified and unmodified RNA fragments. For quantification we only considered RNA fragments also containing modification Cm2498, as these correspond to mature 50S ribosomes.
CMCT treatment and reverse transcription
10 μg of isolated total RNA was treated with 100 μg of CMCT (N-cyclohexyl-N’-β-(4-methylmorpholinium)ethylcarbodiimide p-tosylate, 100 mg/ml stock solution in water) in 1× CMCT buffer (40 mM B4K2O7 pH 7.9, 4 mM EDTA) at 37°C for 30 min. Treated RNA was precipitated and resuspended in 40 μl of 50 mM Na2CO3, pH 11.1 for hydrolysis at 37°C for 2 h. In a final volume of 10 μl, 500 ng of hydrolyzed RNA and 4 pmol of 32P 5′-labeled primer (Supplementary Table S1) were incubated in hybridization buffer (12.5 mM Tris–HCl, pH 8.4, 62.5 mM KCl) first 5 min at 92°C, followed by incubation at 42°C for 30 min. For reverse transcription, 10 μl of an extension mix containing 1 unit of AMV reverse transcriptase (Promega) and 800 μM of each dNTP in extension buffer (500 mM Tris-HCl, pH 8.4, 50 mM MgCl2, 50 mM DTT) were added and the mixture was incubated at 42°C for 30 min. Sequencing reactions were prepared analogous, but contained 500 ng of undigested total RNA in the hybridization mix and 200 μM dNTPs mix supplemented with 100 μM ddNTP in the extension mix. Reactions were stopped by the addition of 40 μl stop solution (4 M NH4AcO/20 mM EDTA) and 32P-labeled cDNA fragments were separated on a 15% polyacrylamide-urea gel and visualized using a Typhoon FLA 9500 phosphorimager.
Recovery and cell viability under oxidative stress
Overnight seed cultures of three biological replicates each from the rlhA and C169A strain were prepared as described above. The next day, overnight cultures were re-diluted 1:100 in 5 ml of LB + CAM (30 μg/ml) and incubated for 2 h under optimal conditions. OD600 of all cultures was measured and adjusted to the lowest value. 100 μl of each culture were diluted by a factor of 105 before plating 100 μl on selective LB plates to calculate initial cfu. Another 50 μl of adjusted cultures were added to a 96-well plate (Greiner, Flat Bottom Transparent Polystyrene) containing 50 μl of LB + CAM (30 μg/ml) with 2× of designated final concentrations of H2O2. Cultures were then incubated for up to 18 h in the Tecan M1000 PRO plate reader and OD600 was measured every 10 min. For cell viability, the experiment was prepared identically with H2O2 added to a final concentration of 5 mM After the addition, cells were incubated for 1 h at 37°C in the Tecan plate reader before 90 μl of the culture were diluted by a factor of 1111 and 200 μl were plated onto selective LB plates to calculate cfu after stress.
Ribosomal subunits preparation
Large and small subunits were isolated from exponentially growing E. coli cells by twice repeated 10–40% sucrose gradient purification according to previously published protocols (19,20). For the in vitro reconstitution of Thermus aquaticus 50S subunits, an atomic mutagenesis approach was implemented as previously described by our group (19,21). See Supplementary Figure S2 for the circularly permutated 23S rRNA (cp23S) and the RNA oligo containing either modified or unmodified C2501 used to fill the short sequence gap. Supplementary Table S1 contains the PCR primers used for cp23S rRNA template generation. As a positive control, canonical in vitro transcribed 23S rRNA was used for the in vitro reconstitution. As a negative control, in vitro reconstitution was performed using only cp23S rRNA without the addition of any synthetic oligo.
The ho5C modified building block for RNA synthesis was prepared according to reference (22) with exception of the final phosphitylation step: Precursor compound 5a (see (22), 250 mg, 0.29 mmol) was dissolved in anhydrous dichloromethane (1.85 ml) and treated with N,N-diisopropylethylamine (377 μl, 2.2 mmol) and 1-methylimidazole (11.5 μl, 0.14 mmol) at ambient temperatures. 2-Cyanoethyl-N,N-diisopropylchlorophosphoramidite (161 ul, 0.72 mmol) was added dropwise and the reaction was stirred for 3 h. Upon complete conversion of starting material, monitored via thin layer chromatography (TLC) (5% MeOH in CH2Cl2), the reaction mixture was quenched with methanol (200 μl), diluted with dichloromethane (45 ml), washed with saturated sodium bicarbonate solution and saturated sodium chloride solution. Organic phases were combined, dried over sodium sulfate and evaporated. The crude product was purified by column chromatography on silica gel (15 g of SiO2; 40–0% cyclohexane to 0–40% acetone in ethyl acetate) to yield the desired phosphoramidite 6 (see (22), 233 mg, 219 μmol; 76%) as white foam. Incorporation of the ho5C phosphoramidite 6 into RNA and deprotection was performed as described in reference (21).
In vitro translation
Translational activities of the isolated or in vitro reconstituted 50S subunits were tested on a single mRNA coding for the L12 protein of Methanococcus thermolithotrophicus or the Dps protein from E. coli as described in (19,21). To reduce the influence of pipetting errors when adding the 50S subunits to the translation mix, a master mix containing the 30S subunit as the limiting factor with only 0.25 equiv. of 30S per mol 50S added was prepared. Translation was performed at 37°C for 1 h, taking a sample for analysis every 15 min. The synthesized 35S-methionine-labeled proteins were separated on an 11% Tris-Tricine polyacrylamide gel and visualized using the Typhoon FLA 9500 phosphorimager.
Metabolic labeling
Translational activity was measured in vivo by incorporation of 35S-methionine into newly synthesized proteins. Therefore, both the rlhA and C169A strain were grown under optimal conditions to an OD600 ∼0.4 as described above. Once the desired OD600 value was reached, a sample of 1 ml was taken from the strains and 1 μl of l-35S-methionine (10 μCi/μl, Hartmann Analytic) was added. The sample was then incubated for an additional 10 min at 37°C before the cells were harvested and the synthesized proteins were analyzed on an 11% Tris-Tricine polyacrylamide gel as above. To test the translational activity under oxidative stress conditions, H2O2 was added to a final concentration of 2 mM to the OD600 ∼0.4 cultures and 1 ml samples for 35S-methionine incorporation were taken every 20 min after the addition.
Proteome analysis
Proteome analysis was performed in three biological replicates each of the rlhA and C169A strain at the Proteomics & Mass Spectrometry Core Facility (University of Bern). Therefore, 1 ml samples of exponentially growing cells (OD600 ∼0.4) were taken and washed three times with 1 ml of 1× PBS before mass spectrometry was performed as described in (23). Proteome analysis was performed using DEqMS (24). Proteins with a P-value <0.001 and log2 fold change >1 (upregulated) or log2 fold change <−1 (downregulated) were considered significantly altered.
DNA damage analysis
DNA damage was analyzed in the specified strains by isolation of plasmid DNA from exponentially growing cells (OD600 ∼0.4) before and after the addition of 2 mM H2O2. Therefore, 50 ml culture samples were taken at the marked time points and plasmid DNA was obtained using the Wizard® Plus SV Minipreps DNA Purification System (Promega). 1 μg of isolated plasmid DNA was separated on a 1% agarose gel and visualized by ethidium bromide. To induce the overexpression of Dps prior to the addition of the oxidative stress, 25 nM tetracycline in 50% ethanol was added to the cultures at the beginning of the exponential growth phase (OD600 ∼0.1).
RESULTS
RlhA catalyzes the hydroxylation of C2501 in 23S rRNA
The RlhA enzyme was previously reported as the sole source of the 23S rRNA modification ho5C2501 (6). To study the role of this modification in vivo, an rlhA knockout strain (ΔrlhA) was constructed by replacing the genomic locus with a kanamycin cassette in the wildtype strain BW25113 (WT) (16) (see Supplementary Figure S3A). The correct replacement of rlhA by the kanamycin cassette was controlled by PCR of genomic DNA (Figure 1C). Additionally, a complementation strain (rlhA) was constructed by cloning the rlhA genomic region into the low-copy pBbSc plasmid (17) (Supplementary Figure S3B, C). The rlhA gene possesses an unusual GUG start codon (25). To assess the relevance of this unusual start codon for RlhA expression levels, we constructed a strain carrying a canonical AUG start codon at the rlhA locus (referred to as AUG strain throughout).
Quantification of ho5C2501 by LC–MS/MS and primer extension assays
Previous methods to quantify ho5C2501 on 23S rRNA by LC–MS/MS relied on a DNA oligo-protected RNase A digestion to obtain the rRNA fragment of interest for subsequent analysis (6,13) (Supplementary Figure S1A). To circumvent this DNA oligo-dependent RNase protection step, we streamlined the method for direct access to rRNA fragments and subsequent LC–MS/MS in a selected reaction monitoring (SRM) mode (Supplementary Figure S1A). To this end, 23S rRNA isolated from cells grown either in early exponential or early stationary phase were digested with RNase T1. RNase T1 cuts RNA 3′ of G residues, resulting in specific and predictable 23S rRNA fragments (Supplementary Figure S1B, C). By using the SRM mode to only scan for specific m/z precursor and product pairs, the RNA fragment of interest can be quantitatively detected even in the presence of a large pool of unrelated sequences. After RNase T1 digestion, C2501 is found in a fragment spanning the 23S rRNA residues 2496–2502. This allowed us to utilize the neighboring ribose methylation Cm2498 as a positive control for mature ribosomes: Matching heptamer sequences with unmodified C2498 were considered to belong to nascent rRNA or not fully matured pre-50S (26) and were thus not considered in the quantification. The SRM method was set up to detect the two relevant fragments with either C2501 modified or unmodified on separate channels, initially identifying the precursor mass in MS1 and confirming the sequence in MS2 by scanning for select c, y, and w series product ions. Figure 2A illustrates typical changes in peak ratio for the two rRNA fragment variants isolated from exponentially growing cells and cells entering the stationary phase. Wildtype cells show an increase from around 30% of ho5C2501 modified ribosomes in exponential phase to around 80% in stationary phase, values comparable to previously published results (6,13) (Figure 2B). No modification was detected in ΔrlhA cells from either condition. The complementation strain (rlhA) on the other hand was overshooting wildtype levels to ∼60% of ribosomes modified already in exponential phase. Increased modification levels upon exogenous introduction of the rlhA gene have been observed before (6) and were further increased in the AUG strain in which the unusual GUG start codon normally found in rlhA (25) was mutated to a canonical AUG start codon, resulting in an almost complete modification already in exponential phase (Figure 2B).
Figure 2.

ho5C2501 levels can be quantified by both mass spectrometry and reverse transcription. (A) SRM LC–MS/MS chromatograms scanning for RNase T1-digested heptamer fragments of 23S rRNA residues 2496–2502, containing either ho5C2501 (upper panels) or C2501 (lower panels). Relative abundance is given within any individual sample. (B) Quantification of ho5C2501 levels in SRM LC–MS/MS chromatograms of 23S rRNA from the wildtype (WT), knockout (ΔrlhA), complementation (rlhA) and rlhA overexpressing (AUG) strains in exponential (OD600 ∼ 0.4) and in stationary phase (OD600 ∼ 4.0). Modification frequencies of ho5C2501 were calculated from the peak areas shown in (A), with the sum of peak areas for both fragments corresponding to 100%. Error bars indicate standard deviation of n = 2–3. The detection of residual modification in the ΔrlhA strain in stationary phase is likely a negligible contaminant during chromatography and is not significant, as it does not exceed the standard deviation. (C) Representative 15% polyacrylamide gel with cDNA generated from CMCT treated (+) and hydrolyzed total RNA samples isolated from the indicated bacterial strains in both exponential (0.4) and stationary phase (4.0). A buffer only, with no CMCT added, negative control (–) was included. A and G are ddNTP sequencing lanes that were prepared with untreated total RNA. Two other naturally occurring rRNA modifications are indicated (Cm2498 and Ψ2504), while position C2501 is marked in bold. A black arrowhead marks the expected site of the ho5C2501 stop upon CMCT-treatment and the open arrowhead shows the stop corresponding to Ψ2504. (D) Quantification of ho5C2501 signals in (C). Signal intensity of the WT exponential phase was set as 1 and the average intensity was calculated. Error bars indicate standard deviation of n = 3.
To verify the ho5C2501 quantification levels with an alternative and less time- and infrastructure-demanding approach, we utilized the chemical probing reagent CMCT on total RNA isolates. The CMCT-treated RNA was further hydrolyzed by alkaline pH followed by reverse transcription using radioactively labelled primers similar to published protocols (11). The approach is based on the observation that CMCT adducts at pseudouridines and apparently also at 5-hydroxycytidines are resistant to alkaline hydrolysis and thus result in a block of reverse transcription (27). As a consequence reverse transcription stops one nucleotide downstream of the CMCT-modified site. In Figure 2C, all CMCT treated samples displayed this band at position G2505, consistent with the known pseudouridine modification at position U2504, thus confirming the effectiveness of the detection method. Therefore, ho5C2501 modification levels were analyzed for all bacterial strains used in this study. The presence of the 5-hydroxy modification at residue C2501 results in an increased reverse transcription stop signal at residue G2502. By comparing samples taken from WT cells from early exponential and early stationary phase, the signal intensity of the primer extension stop increases ∼2-fold in the latter (Figure 2D), which is in a similar range as our formerly measured increase of ho5C2501 levels from 30% in exponential phase to 80% in stationary phase (Figure 2B). On the other hand, in the catalytically inactive rlhA mutant C169A, no signal above background could be detected (Figure 2C, lanes 9 and 10). Similar results as compared to the mass spectrometry analysis were also obtained for 23S rRNA isolated from the rlhA complementation and the rlhA overexpression (AUG) strains (Figure 2D).
Overall, we revealed very comparable quantitative results relative to earlier published methods for the detection of ho5C2501 levels utilizing both LC–MS/MS in the SRM mode and reverse transcriptase analysis of CMCT treated RNA (6,10,13).
RlhA-catalyzed hydroxylation of C2501 provides cells with a growth advantage
To compare the cellular fitness of strains expressing or lacking the rlhA gene, growth curves were performed in rich medium under optimal growth conditions (Figure 3A). A slight growth defect of the knockout strain compared to the wildtype strain could be observed, which was completely rescued in the complementation strain. To investigate if this phenotype was due exclusively to the modifying activity of RlhA and to exclude the possibility of a putative moonlighting activity of the enzyme, the rlhA gene on the rescue plasmid was mutated to a non-modifying version (C169A) according to previously published results (6). Cells complemented with the mutated rlhA displayed the same growth phenotype as the knockout strain during the first 3.5 h of incubation (Figure 3A), suggesting that the growth defect is only due to the lack of the ho5C2501 modification. Later, towards stationary phase, the C169A strain grew slightly slower compared to the ΔrlhA strain. Potentially, the presence of a catalytically inactive form of RlhA impedes either further ribosome biogenesis or interferes with other so far undescribed cellular functions of this protein. The RlhA overexpression strain (AUG), which carries ribosomes fully hydroxylated at C2501 already in exponential phase (Figure 2B), had the most significant growth defect (Figure 3A). This suggests that efficient growth requires the capability for dynamically regulating the extent of the ho5C2501 modification in a growth phase-dependent manner. The minor growth deficiency of the ΔrlhA strain was enhanced in growth competition experiments. In such assays, equal cell numbers (based on OD600 measurements) of the ΔrlhA and WT strains were mixed and subjected to several rounds of growth for 24 h into stationary phase followed by re-dilution into fresh rich medium (Figure 3B). Before each re-dilution, samples were taken from the culture to monitor the ratio of wildtype to knockout strain by plating in parallel on chloramphenicol plates (which allows growth of both WT and ΔrlhA strains) and kanamycin selection plates (where only the ΔrlhA cells can form colonies). A clear and steady increase of the WT population became apparent in the re-dilution cultures, demonstrating successful growth competition of cells expressing RlhA over the knockout strain (Figure 3E). A very similar trend was observed when the rlhA complementation strain was grown in competition to the ΔrlhA strain (Supplementary Figure S4A, B). To test whether the recovery from stationary phase was affected in general, both WT and ΔrlhA cells were grown to and kept in stationary phase for increasing amounts of time. Upon re-dilution into fresh rich medium, both strains displayed an initial lag phase that positively correlated with the age of the inoculum, followed by regular growth as observed before. No discernible increase of lag phases were observed for the ΔrlhA cells (Supplementary Figure S4C).
Figure 3.
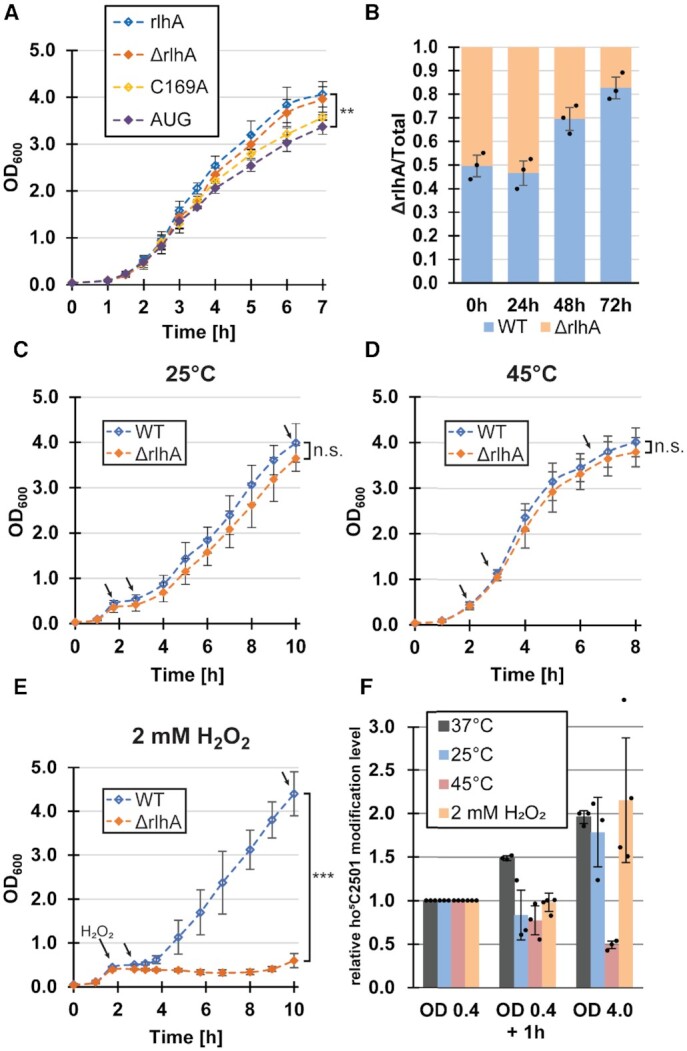
Cell growth and ho5C2501 modification levels under different stress conditions. (A) Average growth curves of the knockout cell line transformed with either an empty vector (ΔrlhA), the wildtype rlhA gene (rlhA), an inactive rlhA mutant (C169A), or the start codon mutated rlhA gene (AUG). All strains were incubated in LB medium at 37°C. Error bars indicate standard deviations (n = 3–6) and the significance was calculated for the last time point using paired Student's t-test (P = 0.008). (B) Average proportion of ΔrlhA cells grown in competition with wildtype cells (WT) after repeated 24 h incubation at 37°C, starting with an initial 1:1 mixture of both strains (0 h). The ratio of ΔrlhA cells was determined by comparison of cells growing on chloramphenicol plates (total) to cells growing on kanamycin selective plates (ΔrlhA). (C) Average growth curves of WT and ΔrlhA cell lines that were cold shocked and temperature shifted to 25°C after OD600 reached a value of 0.4 (∼2 h of incubation at 37°C). Error bars indicate standard deviation of n = 3 (n.s.; statistically not significant). Black arrows indicate time points when samples were taken for ho5C2501 level analysis by CMCT treatment and reverse transcription. (D) Same as in (C), but a heat shock was applied and the temperature was shifted to 45°C (n = 3). (E) Same as in (C), but H2O2 was added to a final concentration of 2 mM after OD600 reached a value of 0.4 and growth was continuously monitored at 37°C. Error bars indicate standard deviations (n = 4) and the significance was calculated using Student's t-test (***P < 0.005). In panels (C)–(E) black arrows indicate time points of sample removal for subsequent CMCT analyses to assess ho5C2501 levels. (F) Quantification of ho5C2501 levels in WT cells at the marked time points in panels (C)-(E) by reverse transcription of hydrolyzed CMCT-treated total RNA. ho5C2501 signal intensities of samples taken after 1 h of stress treatment (OD 0.4 + 1 h) and in stationary phase cultures (OD 4.0) were compared relative to the ho5C2501 signal intensities of samples taken after ∼2 h of incubation under unstressed conditions (OD 0.4). Additionally, ho5C2501 levels were analyzed in WT cells under optimal conditions without the addition of any stressor (37°C). The average relative signal intensity is displayed. Error bars indicate standard deviation of n = 3–4.
Taken together, these results show that E. coli cells expressing an active form of RlhA allowing a growth phase-dependent hydroxylation at C2501 have a minor growth advantage under unstressed conditions compared to cells lacking an active RlhA. Displaying only subtle phenotypes, especially under optimal conditions, is not surprising for the knockout of a single rRNA modifying enzyme and is consistent with previous observations (2). We therefore next proceeded to measuring ho5C2501 modification levels under different stress conditions to further investigate the function of this modification as a possible adaption to environmental cues.
ho5C2501 levels are not altered as a rapid response to different stress conditions
To test the role of the ho5C2501 modification as a potential adaption to different environmental conditions, WT and ΔrlhA cells were first grown in rich medium at 37°C. After 2 h under optimal conditions (OD600 of ∼0.4), a first sample was taken for analysis before the cultures were then exposed to either cold shock, heat shock, or oxidative stress. After 1 h of stress incubation, another sample was taken for analysis and growth was continued to be monitored (Figure 3C–E). In both the cold and heat shocked cultures, no significant growth differences between WT and ΔrlhA bacteria were observed (Figure 3C, D). However, a strikingly strong phenotype was observed under oxidative stress. After the addition of H2O2, both the WT and ΔrlhA strains immediately arrested growth as was reported before (28). After a lag phase of ∼2 h, the WT cells resumed growth and grew to normal stationary phase levels (Figure 3E), whereas the OD600 of the ΔrlhA cells even slightly decreased. It was not before 6 h after the addition of the hydrogen peroxide that the ΔrlhA cells showed signs of stress recovery. A final sample was withdrawn at all conditions once the cultures had reached stationary phase.
Next, ho5C2501 levels were analyzed in the collected samples utilizing CMCT treatment as described above. In all three tested stress conditions there was no significant change in ho5C2501 levels 1 h after stress induction (Figure 3F, Supplementary Figure S5). In contrast, when WT cells were kept incubating for 1 h at 37°C without any stress, modification levels steadily increased. In both the cold shock and oxidative stress conditions, there was no significant difference as compared to the unstressed cultures observable once they reached stationary phase. However, a clear reduction of ho5C2501 levels was observed for ribosomes in the stationary phase under heat shock stress conditions (Figure 3F).
ho5C2501 benefits cells under oxidative stress
The analysis of growth rates described above showed that the strongest difference between the WT and the ΔrlhA strains was observed under oxidative stress conditions (Figure 3E). We therefore first set out to quantify the observed lag phase upon addition of H2O2. To minimize the influence of secondary factors and to reduce the possibility that the observed phenotypes are due to a potential moonlighting function of RlhA, the ΔrlhA strain complemented with either a fully functional rlhA or its inactive single-point mutation version (C169A) was used in the following experiments. First, cells were grown for 2 h under optimal conditions as described above. Since a minor growth difference was observed already in the early exponential phase, OD600 was equalized before implementing the oxidative stress. To confirm equal starting amounts of viable cells, an initial sample was taken from both adjusted cultures, diluted and streaked out on rich-medium plates to calculate colony forming units (cfu) prior to the stress (Figure 4A). After the addition of 3–7 mM H2O2, growth in a 96 well plate was monitored (Figure 4B, C). Similar to previously published results (29), a H2O2 concentration-dependent prolonged lag phase was observed. As can be seen already at the endpoint of the experiment (Figure 4D), the complementation strain was able to tolerate higher concentrations of H2O2 than the strain expressing the catalytically inactive C169A mutant of RlhA. When both strains managed to mount an effective stress response to remove the H2O2 stressor, the C169A mutant strain generally recovered slower than the complementation strain. To better quantify this increase in lag time, a threshold value of OD600 = 0.125 was arbitrarily set onto the growth curves in Figure 4B and the time to reach this value was measured (Figure 4C). Exemplary for the difference between the two strains were the observations at 4 mM H2O2. While it took the complementation strain an average of 6 h to remove the stressor and to resume normal growth rates, only one of the three biological replicates of the inactive mutant strain resumed growth after 13 h. At 5 mM H2O2, still two out of three replicates of the complementation strain cultures were able to remove the stressor and resume growth, whereas none of the mutated strain cultures could recover. Therefore, it was investigated next whether cell viability of the arrested cells was affected differently under this condition of intense oxidative stress. To this end, cells were grown at optimal conditions for 2 h, re-diluted to equal starting cell counts, and H2O2 was then added to a final concentration of 5 mM. Cells were then incubated for 1 h at 37°C before taking another sample for cfu determination. Whereas the OD600 values were unchanged during this 1 h of incubation (Supplementary Figure S6A), viable cell counts were decreased significantly (P < 0.005) in both the complementation and the C169A mutant strains after the addition of hydrogen peroxide (Figure 4E, F). The mutant strain that completely lacks ho5C2501, however, displayed significantly less cfu/ml after 1 h of incubation compared to the complementation strain, demonstrating increased cell death under oxidative stress upon loss of a functional RlhA enzyme. Taken together, these results indicated that the presence of ho5C2501 in 23S rRNA is advantageous for the survival of E. coli cells upon addition of H2O2.
Figure 4.

ho5C2501 benefits cells under oxidative stress. (A) Equal cell numbers derived from strains either expressing active RlhA (rlhA) or the catalytically inactive RlhA mutant (C169A) were controlled by calculating average colony forming units (cfu) per milliliter before the addition of H2O2. Error bars represent standard deviation (n = 3). (B) Representative growth curves of a single biological replicate of rlhA (blue) and C169A (orange) in a 96 well plate after the addition of indicated amounts of H2O2. An arbitrary threshold line was drawn in green at OD600= 0.125 to measure lag time. (C) Quantification of lag time until the threshold of OD600= 0.125 was reached in (B). Bars represent average of the individual values that are marked with black points. (D) Endpoint of all three biological replicates of rlhA and C169A strains 18 h after the addition of 3–7 mM H2O2. Left column no H2O2 was added.(E) Representative example of colonies growing on selective plates before (t = 0) and after the addition of 5 mM H2O2 (t = 1 h). (F) Quantification of average cfu/ml before and after the addition of 5 mM H2O2. Standard deviation is marked by error bars and significance was calculated using Student's t-test (Pt = 0 = 0.1418, Pt = 1 h = 0.0003, n = 3).
ho5C2501 modification slows down translation in vitro
The nucleotide C2501 is located in close proximity to the PTC (Figure 1B), thus making it possible that the hydroxylation of this nucleobase might play a fine-tuning role in the catalytic center of the ribosome. To test this hypothesis, in vitro reconstituted ribosomes either carrying or lacking the hydroxylation at position C2501 were prepared. To this end, an atomic mutagenesis approach was employed (19), where the 23S rRNA was circularly permuted and in vitro transcribed with the new 5′-end at position G2523 and the new 3′-end at position C2483 (Supplementary Figure S2). The resulting gap from G2484–U2522 was filled with a chemically synthesized oligonucleotide with position C2501 either fully hydroxylated (ho5C2501) or not modified at all (C2501), providing us with large ribosomal subunits exclusively differing at this sole functional group. The reconstituted subunits were then assembled with native 30S subunits and subsequently used in an in vitro translation reaction. A clear reduction in translational activity was observed over the course of 1 h of incubation for ribosomes carrying the ho5C2501 modification (Figure 5A, B). Similar, albeit less pronounced effects were apparent when using in vivo-derived ribosomes isolated either from the ΔrlhA strain (and thus lacking ho5C2501 completely) or from the rlhA overexpressing strain (whose ribosomes were almost quantitatively carrying ho5C2501) (Figure 5C, D). These observations suggest that the other post-transcriptional modifications present in and around the PTC of in vivo-derived ribosomes can somewhat balance the effect of the single hydroxyl group at C2501 whereas the in vitro assembled 50S subunits are more sensitive to the absence/presence of ho5C2501. To investigate the role of ho5C2501 at elevated temperatures, conditions under which E. coli decreases the extent of hydroxylation at this residue (Figure 3F, Supplementary Figure S5), in vitro translation with fully modified ribosomes at 45°C was performed. Under these heat shock conditions the defect on protein biosynthesis was more pronounced as compared to 37°C (compare Figure 5C, D with Figure 5E, F). No obvious ribosome biogenesis defects were observed during the preparation of the ribosomal subunits (Supplementary Figures S7 and S10C, D). In sum, our results show that the ho5C2501 modification slightly, but reproducibly slows down protein synthesis during in vitro translation.
Figure 5.

ho5C2501 slows down translation in vitro. (A) Representative phoshporimager scan of in vitro translated 35S-labeled L12 protein (top) and coomassie stained proteins as loading control at the bottom. In vitro translations were performed with in vitro reconstituted 50S subunits utilizing an atomic mutagenesis approach with an unmodified (C2501) or modified (ho5C2501) RNA oligo. No oligo was added during 50S reconstitution as a negative control (no oligo). As a positive control, in vitro reconstitution was performed with in vitro transcribed canonical full-length 23S rRNA (+). (B) Quantification of average signal intensities at the marked time points with the signal intensity of C2501 (t = 60 min) set to 1. Error bars indicate standard deviation, significance was calculated using paired Student's t-test (P = 0.029, n = 4). (C) Same as in (A), but in vitro translation was performed at 37°C with in vivo-derived 50S subunits isolated from an unmodified knockout strain (ΔrlhA) and an almost quantitatively modified rlhA overexpression strain (AUG). (D) Quantification of average signal intensities at the marked time points with the signal intensity of ΔrlhA (t = 60 min) set to 1. Error bars indicate standard deviation, significance was calculated using paired Student's t-test (P = 0.042, n = 4). (E) Same as in (C), but in vitro translation was performed at 45°C. (F) Quantification of average signal intensities at the marked time points with the signal intensity of ΔrlhA (t = 60 min) set to 1. Error bars indicate standard deviation, significance was calculated using paired Student's t-test (P = 0.022, n = 4).
Metabolic labeling activities under oxidative stress recovered faster in cells with ribosomes harboring ho5C2501
To test translational activity in vivo, metabolic labelling by incorporation of 35S-methionine into newly translated proteins was performed in both the rlhA complementation and C169A mutant strains. Under unstressed conditions, and in support of the in vitro data (Figure 5), a minor reduction in translational activities was observed in the rlhA strain with ho5C2501-modified ribosomes (Figure 6A, lanes 1 and 6, Figure 6B). Next, metabolic labelling under oxidative stress conditions was investigated. Therefore, both the rlhA complementation and the C169A strains were stressed by addition of 2 mM H2O2 and samples were taken for metabolic labelling after increasing amounts of recovery time (Figure 6A, C). After 20 min of recovery, both strains showed no detectable translational activities yet. This is in line with findings from other groups showing that immediately after addition of H2O2, translation rates are severely inhibited (29). However, after prolonged recovery times, the rlhA complementation strain strongly outperformed the C169A strain, resulting in approximately twice the metabolic labelling activity after 80 min of recovery. These findings are in line with the findings of Figure 4 showing that the rlhA strain recovered better from H2O2 stress than the C169A strain.
Figure 6.

Metabolic labelling under oxidative stress conditions. (A) Representative phosphorimager scan of newly translated 35S-labeled proteins (top) and coomassie stained whole cell lysate as loading control at the bottom. The experiment was performed in the C169A strain lacking the ho5C2501 modification and in the rlhA complementation strain with ribosomes carrying the ho5C2501 modification. Samples for metabolic labelling were taken before the addition of 2 mM H2O2 (lanes 1 and 6) and every 20 min after the addition of the stress (lanes 2–5 and 7–10). (B) Quantification of average metabolic labelling activity before the addition of oxidative stress. Signal intensities of C169A (t = 0 min) were set to 1. Error bars indicate standard deviation, significance was calculated using paired Student's t-test (P = 0.109, n = 4). (C) Quantification of average metabolic labelling activities in recovering cells after the addition of 2 mM H2O2. Signal intensities of rlhA (t = 80 min) were set to 1. Error bars indicate standard deviation, significance was calculated using paired Student's t-test (P = 0.010, n = 4).
The DNA-protective protein Dps is downregulated in the absence of active RlhA
To investigate whether hydroxylation at 23S rRNA position C2501 had an effect on the expression of specific proteins, whole proteome analysis was performed on the rlhA complementation and C169A mutant strains. Protein samples were taken from both unstressed and H2O2-treated cells stressed for 60 min, a time point revealing significant differences in metabolic activities between the two strains (Figure 6A). Comparing unstressed and stressed C169A cells, it was evident that after 60 min, the cells were still mostly combating the oxidative stress, as demonstrated by the upregulation of mainly stress response genes, including several genes of the OxyR-regulon (Supplementary Figure S8). OxyR is a transcription factor regulating the H2O2 stress response in E. coli (29). In contrast, genes of several more GO-terms were upregulated in the stressed rlhA cells, indicative of an already recovered and adapted translational status as a result of a successful removal of the stressor (Supplementary Figure S8). Comparing both strains under unstressed conditions, only few proteins were found to be differentially expressed (Figure 7A). The most significantly upregulated protein in the rlhA complementation strain compared to the strain expressing the inactive RlhA mutant C169A was Dps (DNA protection during starvation protein). Dps can non-specifically bind and condensate DNA, resulting in a protective effect against a range of stresses, including oxidative stress (30–33). Additionally, it has been shown that Dps sequesters free Fe2+, thereby reducing the occurrence of the harmful Fenton reaction that produces highly destructive hydroxyl radicals in the presence of Fe2+ (34,35). Therefore, less DNA damage under oxidative stress conditions would be expected in E. coli strains with ho5C2501-modified ribosomes due to the protective effect of the upregulated Dps. To test this hypothesis, plasmid DNA damage was analyzed in strains expressing functional RlhA (WT and rlhA complementation cells) and compared to the ΔrlhA strain before and after the addition of 2 mM H2O2 (Figure 7B). As can be seen in all strains, a smeary and slowly migrating signal corresponding to nicked plasmid DNA increased upon addition of hydrogen peroxide (20 min). In both strains expressing functional RlhA, the nicked plasmid DNA signal disappeared upon further recovery (120 min) and only a sharp band corresponding to the supercoiled vector DNA was detectable. Interestingly, the kinetics of restoring plasmid DNA integrity (Figure 7B) and growth recovery from oxidative stress (Figure 3C) correlated well. In variance to the rlhA complementation and WT strains, the nicked plasmid DNA was never converted back into the supercoiled form in the ΔrlhA strain even after 120 min of recovery. To validate the protective effect of Dps on the plasmid DNA, a tetracycline-inducible Dps overexpression plasmid was introduced into the C169A cells, a strain that showed severely reduced Dps protein levels (Figure 7A). As can be seen in Figure 7C, induction of the Dps overexpression irrefutably protects plasmid DNA. No nicked plasmid DNA bands appeared after the addition of H2O2. In contrast, in the absence of Dps overexpression, plasmid DNA damage was still evident after prolonged recovery (Figure 7C). To substantiate this possible link between Dps levels and faster adaptation to oxidative stress, we ectopically overexpressed Dps in the C169A strain. Growth recovery and colony formation capacity was indeed enhanced at elevated Dps levels in the presence of ROS (Figures 7D, E and Supplementary Figure S6B). These findings are compatible with the hypothesis that high levels of the DNA-protective protein Dps, as seen in strains harbouring ribosomes with the ho5C2501 modification, result in increased resistance to oxidative stress due to less pronounced DNA damage.
Figure 7.

The DNA protective protein Dps is upregulated in strains that carry ho5C2501-modified ribosomes. (A) Volcano plot of the comparative proteome analysis of unstressed rlhA cells versus unstressed C169A cells. Proteins with a fold-change of at least ±2 and a P-value <0.001 were considered significantly altered (n = 3). (B) Isolated plasmids from unstressed cells (0 min) and from cells recovering from H2O2 stress (20 and 120 min) were separated on a 1% agarose gel to visualize oxidative damage to the plasmid DNA. Supercoiled plasmid DNA (empty vectors for WT and ΔrlhA strains or vector coding for the rlhA gene in the complementation strain) are indicated by filled arrowheads. An open arrowhead indicates nicked plasmid DNA. (C) Similar to (B), but instead a tetracycline inducible Dps overexpression plasmid was additionally added to the C169A strain (C169A + dps). (D) Average growth curves of the C169A + dps strain under oxidative stress conditions. The strain was incubated in LB medium at 37°C. After 1 h, Dps overexpression was either induced or not (+Tet and −Tet) and H2O2 was added to a final concentration of 2 mM after OD600 reached a value of 0.4 (∼2 h of incubation at 37°C; black arrow), followed by growth for another 6 h at 37°C. Error bars indicate standard deviations (n = 4) and the significance was calculated using Student's t-test (*P < 0.05, **P < 0.01). (E) Spot assay of tetracycline induced or uninduced C169A + dps cells 1 h after the treatment with 5 mM H2O2.
DISCUSSION
In this study we demonstrated that the nucleobase C5 hydroxylation of the 23S rRNA residue C2501 is catalyzed by RlhA in a growth phase-dependent manner in E. coli, confirming previously published results (6,10,13). While the ho5C2501 levels were low during exponential growth, they reached almost stoichiometric levels in stationary phase (Figure 2). No rapid adjustments of ho5C2501 levels were observed as a response to different stress conditions (Figure 3F). Instead, modification levels appeared to stagnate in the first hour of stress adaptation. An observation that could be explained by reduced de novo ribosome biogenesis as a consequence of the applied stressors (36–39). More interestingly, ho5C2501 levels were drastically reduced in stationary phase of heat-stressed E. coli cells (Figure 3D). Furthermore, ribosomes carrying stoichiometric levels of ho5C2501 possessed reduced in vitro translational activities at high temperatures as compared to 37°C (Figure 5C-F). Apparently, the hydroxylation of C2501 is detrimental under increased temperatures; an interpretation that seems to be supported by the fact that ribosomes isolated from the hyperthermophile T. thermophilus are completely devoid of this modification (10). T. thermophilus, a close relative of the fully modified D. radiodurans, is highly susceptible to radiation, desiccation, and oxidative stress (15). Taken together, these dissimilar modification patterns in T. thermophilus and D. radiodurans as a consequence of different growth temperatures are compatible with the idea of ho5C2501 playing a role in the adjustment of the cell's ribosomes to different environmental conditions.
Genomic deletion of rlhA resulted in only mild growth defects under optimal conditions and during temperature stress (Figure 3A, C, D). On the other hand, the presence of ho5C2501 was shown to provide a strong growth advantage during oxidative stress both in liquid cultures and in plating experiments (Figures 3E and 4). To gain more molecular insight into the contribution of ho5C2501 to ribosome functions, in vitro and in vivo translation assays were conducted with ribosomes either lacking this hydroxylation completely or carrying this 23S rRNA modification almost quantitatively. While ho5C2501 slightly but reproducibly inhibited in vitro protein biosynthesis (Figure 5), it clearly stimulated in vivo translation as assessed by metabolic labeling during H2O2 induced oxidative stress (Figure 6A, C). These seemingly contradicting results can be explained by the fact that translation rates need to be adequately adjusted to challenging environmental conditions and a rapid protein production does not necessarily benefit bacterial cells during harsh conditions. It is of utmost importance for an organism not to waste precious energy during stressful conditions and thus dimming translation rates (e.g. by elevating ho5C2501 levels) can represent a crucial advantage, especially during the early phase of stress encounters. These data also suggest that different levels of ho5C2501, as observed between exponential growth and stationary phase, can fine-tune the catalytic performance of the PTC.
The catalysis of peptide bonds is a well-concerted act involving functional groups of the 23S rRNA and P-site tRNA backbones, as well as coordinated water molecules (40). Beside these active site functional groups that likely promote transpeptidation in all ribosomes of all known organisms, post-transcriptional 23S rRNA modifications could potentially contribute a specific regulatory layer to PTC functions. In line with this assumption, the 23S rRNA modification m2A2503 in the nascent peptide exit tunnel has been shown to modulate the PTC by altering allosteric interactions within the ribosome (41). These rRNA modifications are not necessarily always beneficial for the cellular fitness, thus explaining why some of them are inducible and solely added upon specific environmental challenges. It was shown that the macrolide-inducible dimethylation of 23S rRNA residue A2058 by ErmC confers antibiotic resistance and alters the translatome by specifically allowing the synthesis of certain proteins (42). These findings demonstrate that 23S rRNA modifications levels can be dynamically altered based on a trade-off between competing cellular interests (i.e. effective translation rates versus resistance to stress).
From a chemical point of view, nucleobase oxidations confer an even more dramatic change in the electron density profile than methylations. If oxidative damage hits inner core PTC nucleobases, oxidations can have significant effects on the local electrochemical balance and thus can alter the translational performance of the ribosome as demonstrated in our previous study (21). The structural surrounding of the second-tier active site residue C2501 is essentially identical in ribosomes from D. radiodurans (C2501 fully hydroxylated), E. coli (C2501 dynamically hydroxylated in a growth phase-dependent manner) and T. thermophilus (C2501 completely unmodified), which suggests that the presence of the C5 hydroxylation does not lead to major rearrangements within the ribosome (Supplementary Figure S9). The same structural arrangement is also found in Saccharomyces cerevisiae and human ribosomes, both carrying a methylation at the corresponding position (m5C2870 and m5C4447, respectively) (Supplementary Figure S9) (43–45). Even if this oxidation does not affect overall PTC architecture, the hydroxylation of C2501 can still modulate the interactions of this universally conserved cytidine with neighboring residues. C2501 base-stacks between G2446 and G2447 thus fixing the cytosine in a defined position (Figure 1B). Judging from the X-ray structure, ho5C2501 could interact with the inner core PTC residues C2063 and A2451 as well as with A76 of the P-site tRNA (Figure 1B). The ribose 2′-OH groups of A2451 and A76 of P-site tRNA both play crucial roles in catalyzing peptide bond formation (8,46). C2063 on the other hand forms a highly conserved non-Watson-Crick base pair with A2450, which is required for processive translocation (9). Based on the local proximity to these key residues, ho5C2501 may allosterically interact with the active site to fine-tune peptide synthesis as a consequence to adaptation to environmental challenges. As the 5-OH moiety of ho5C2501 is not within hydrogen bonding distance to other residues, Kirpekar and coworkers suggested that the C5 hydroxyl group could coordinate a water molecule, which however is not visualized in the X-ray crystal structure (10). A later structure showed water within the PTC, but not within H-bonding distance to unmodified C2501 (40). As there are no structures available showing both ho5C2501 and active site waters, it cannot be excluded that they do interact. Theoretically, an ho5C2501 coordinated water could contribute to and affect the proton relay during catalysis of peptide bond formation.
How are ho5C2501 levels in E. coli ribosomes connected to cellular fitness? And how can the oxidation of a single nucleobase at position C2501 in close proximity to the PTC provide a selective advantage for bacteria in the presence of ROS? In our attempt to address these questions a whole proteome comparison was performed between cells either expressing or lacking the responsible modification enzyme RlhA during the adaptation phase to oxidative stress. The most significantly downregulated protein in the absence of the ho5C2501 was Dps (DNA protection during starvation protein) (Figure 7A). This protein has been demonstrated before to protect bacteria from various stressors, including ROS, by binding and compacting DNA (30–33). Interestingly, there is no Dps homologue found in T. thermophilus, while two Dps-related proteins are encoded in the D. radiodurans genome (15). We confirmed for E. coli cells that the lack of the ribosomal ho5C2501 modification is linked to low levels of Dps, resulting in decreased DNA integrity (Figure 7B). Therefore, the less compacted and thus more vulnerable DNA architecture towards ROS explains the rather severe growth disadvantage of E. coli deficient of functional RlhA and consequently lacking ho5C2501 (Figures 3E and 4B−F). Significantly, overexpression of Dps in a strain that lacks the ho5C2501 modification rescues DNA integrity (Figure 7C) and, more importantly, growth resumption during oxidative stress (Figure 7D, E). These experimental insights suggest a causal link between the presence of ho5C2501 in the ribosome, cellular Dps levels and stress adaptation in the presence of ROS. What remains to be understood, however, is the molecular basis why ribosomes devoid of ho5C2501 failed to specifically produce the Dps protein. One hypothesis is that the hydroxylation at C2501 in ribosomes is particularly important to translate the dps mRNA. To test this hypothesis, we performed in vitro translation assays utilizing dps mRNA transcripts as templates for ribosomes either lacking ho5C2501 or carrying essentially quantitative levels of this 23S rRNA modification. The results however did not demonstrate significant differences during in vitro translation under the applied conditions (Supplementary Figure S10A, B). Furthermore, polysome profiling and subsequent RT-qPCR or northern blot analysis did not reveal reduced dps mRNA loading to translating ribosomes in the ΔrlhA strain (Supplementary Figure S10C–F). In our view this does not exclude the possibility of a specialized translation requirement for the dps mRNA in vivo but demonstrates that the answer to the pivotal questions above is not trivial and therefore demands further dedicated research. What our study however did reveal is that a single 23S rRNA oxidation event at C2501 (by RlhA) protects E. coli cells from the physiological consequences of H2O2-triggered oxidative stress. As our findings show (Figure 3F), the C2501 modification levels are not changed as rapid response to oxidative stress encounter, as the hydroxylation of C2501 is performed during ribosome biogenesis. The production of new ribosomes is an energy-intensive and time-consuming process and thus not suited for a rapid response to environmental changes. Rather, the cell prepares the ribosomes through modification prior to stress encounter.
Taken together, our data suggest a dynamic regulatory mechanism of ho5C2501 modification that is determined by non-saturating rlhA expression levels and changing kinetics of ribosome biogenesis during different growth phases. The suboptimal GUG start codon gives rise to expression of a constant but relatively low level of RlhA protein in the cell. During rapid growth during exponential phase, ribosomes are synthesized at a fast pace, resulting in sub-stochiometric ho5C2501 modification. Lack of the modification is beneficial for rapid growth, allowing for faster protein synthesis. Once the cell reaches early stationary phase, priorities switch from rapid expansion to survival at the population level. In stationary phase ribosome biogenesis slows, allowing the low levels of RlhA present to modify more 23S rRNA. The resulting increase in ho5C2501 in ribosomes prepares the cell for challenging conditions (such as oxidative stress) by boosting protective protein factors like Dps, which is capable of sequestering free iron and protects DNA by compaction. The resulting lower translation rate of C2501-modified ribosomes is acceptable, because rapid protein synthesis is less important in stationary phase than during exponential growth. Thus, through the simple use of a non-optimal GUG start codon, E. coli is able to regulate the extent of a single rRNA modification close to the PTC. As a consequence the resulting proteome changes dynamically with growth phase, striking a balance between the priorities of rapid growth and preparedness to survival of oxidative stress.
DATA AVAILABILITY
The mass spectrometry proteomics data have been deposited to the ProteomeXchange Consortium via the PRIDE (47) partner repository with the dataset identifier PXD025875.
Supplementary Material
ACKNOWLEDGEMENTS
We would like to thank Christian Leumann and Pascal Küpfer for providing us initially with synthetic RNA carrying ho5C2501. For subsequent RNA synthesis we are grateful to Ronald Micura (University of Innsbruck). Furthermore, we acknowledge Pascal Krenger and Dario Bernasconi for experimental help during their bachelor thesis projects. We also thank Rob Ross for help with Quantiva instrumentation and Tutsomu Suzuki for providing the pBR-rlhA plasmid. Our thanks are extended to Robert Rauscher and Marina Cristodero for their valuable comments on the manuscript.
Notes
Present address: Jessica Willi, Department of Chemical and Biological Engineering, Northwestern University, Evanston, IL 60208, USA.
Contributor Information
Michel Fasnacht, Department of Chemistry, Biochemistry and Pharmaceutical Sciences, University of Bern, 3012 Bern, Switzerland; Graduate School for Cellular and Biomedical Sciences, University of Bern, 3012 Bern, Switzerland.
Stefano Gallo, Department of Chemistry, Biochemistry and Pharmaceutical Sciences, University of Bern, 3012 Bern, Switzerland; Graduate School for Cellular and Biomedical Sciences, University of Bern, 3012 Bern, Switzerland.
Puneet Sharma, Department of Chemistry, Biochemistry and Pharmaceutical Sciences, University of Bern, 3012 Bern, Switzerland.
Maximilian Himmelstoß, Institute of Organic Chemistry, Center for Molecular Biosciences Innsbruck, University of Innsbruck, 6020 Innsbruck, Austria.
Patrick A Limbach, Rieveschl Laboratories for Mass Spectrometry, Department of Chemistry, University of Cincinnati, OH, USA.
Jessica Willi, Department of Chemistry, Biochemistry and Pharmaceutical Sciences, University of Bern, 3012 Bern, Switzerland; Graduate School for Cellular and Biomedical Sciences, University of Bern, 3012 Bern, Switzerland; Rieveschl Laboratories for Mass Spectrometry, Department of Chemistry, University of Cincinnati, OH, USA.
Norbert Polacek, Department of Chemistry, Biochemistry and Pharmaceutical Sciences, University of Bern, 3012 Bern, Switzerland.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
Swiss National Science Foundation [31003A_188969 to N.P.]; P.S. was financed by the NCCR ‘RNA & Disease’ funded by the Swiss National Science Foundation; US National Institutes of Health [NIGMS R01 058843 to P.A.L.]. Funding for open access charge: Swiss National Science Foundation [31003A_188969].
Conflict of interest statement. The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
REFERENCES
- 1. Marbaniang C.N., Vogel J.. Emerging roles of RNA modifications in bacteria. Curr. Opin. Microbiol. 2016; 30:50–57. [DOI] [PubMed] [Google Scholar]
- 2. Sergiev P. V, Golovina A.Y., Prokhorova I. V, Sergeeva O. V, Osterman I.A., Nesterchuk M. V, Burakovsky D.E., Bogdanov A.A., Dontsova O.A.. Rodnina M.V., Wintermeyer W., Green R.. Modifications of ribosomal RNA: From enzymes to function. Ribosomes: Structure, Function, and Dynamics. 2011; Vienna: Springer Vienna; 97–110. [Google Scholar]
- 3. Frye M., Harada B.T., Behm M., He C.. RNA modifications modulate gene expression during development. Science. 2018; 361:1346–1349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Barbieri I., Kouzarides T.. Role of RNA modifications in cancer. Nat. Rev. Cancer. 2020; 20:303–322. [DOI] [PubMed] [Google Scholar]
- 5. Decatur W.A., Fournier M.J.. rRNA modifications and ribosome function. Trends Biochem. Sci. 2002; 27:344–351. [DOI] [PubMed] [Google Scholar]
- 6. Kimura S., Sakai Y., Ishiguro K., Suzuki T.. Biogenesis and iron-dependency of ribosomal RNA hydroxylation. Nucleic Acids Res. 2017; 45:12974–12986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Erlacher M.D., Lang K., Wotzel B., Rieder R., Micura Ronald, Polacek N.. Efficient ribosomal peptidyl transfer critically relies on the presence of the ribose 2′-OH at A2451 of 23S rRNA. J. Am. Chem. Soc. 2006; 128:4453–4459. [DOI] [PubMed] [Google Scholar]
- 8. Lang K., Erlacher M., Wilson D.N., Micura R., Polacek N.. The role of 23S ribosomal RNA residue A2451 in peptide bond synthesis revealed by atomic mutagenesis. Chem. Biol. 2008; 15:485–492. [DOI] [PubMed] [Google Scholar]
- 9. Chirkova A., Erlacher M.D., Clementi N., Zywicki M., Aigner M., Polacek N.. The role of the universally conserved A2450-C2063 base pair in the ribosomal peptidyl transferase center. Nucleic Acids Res. 2010; 38:4844–4855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Havelund J.F., Michael A., Giessing B., Hansen T., Rasmussen A., Scott L.G., Kirpekar F.. Identification of 5-hydroxycytidine at position 2501 concludes characterization of modified nucleotides in E. coli 23S rRNA. J. Mol. Biol. 2011; 411:529–536. [DOI] [PubMed] [Google Scholar]
- 11. Bakin A., Ofengand J.. Four newly located pseudouridylate residues in Escherichia coli 23S ribosomal RNA are all at the peptidyltransferase center: analysis by the application of a new sequencing technique. Biochemistry. 1993; 32:9754–9762. [DOI] [PubMed] [Google Scholar]
- 12. Kowalak J.A., Bruenger E., McCloskey J.A.. Posttranscriptional modification of the central loop of domain V in Escherichia coli 23 S ribosomal RNA. J. Biol. Chem. 1995; 270:17758–17764. [DOI] [PubMed] [Google Scholar]
- 13. Andersen T., Porse B.T., Kirpekar F.. A novel partial modification at C2501 in Escherichia coli 23S ribosomal RNA. RNA. 2004; 10:907–913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Mattimore V., Battista J.R.. Radioresistance of Deinococcus radiodurans: functions necessary to survive ionizing radiation are also necessary to survive prolonged desiccation. J. Bacteriol. 1996; 178:633–637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Omelchenko M. V., Wolf Y.I., Gaidamakova E.K., Matrosova V.Y., Vasilenko A., Zhai M., Daly M.J., Koonin E. V., Makarova K.S.. Comparative genomics of Thermus thermophilus and Deinococcus radiodurans: divergent routes of adaptation to thermophily and radiation resistance. BMC Evol. Biol. 2005; 5:57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Datsenko K.A., Wanner B.L.. One-step inactivation of chromosomal genes in Escherichia coli K-12 using PCR products. Proc. Natl. Acad. Sci. U.S.A. 2000; 97:6640–6645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Lee T.S., Krupa R.A., Zhang F., Hajimorad M., Holtz W.J., Prasad N., Lee S.K., Keasling J.D.. BglBrick vectors and datasheets: a synthetic biology platform for gene expression. J. Biol. Eng. 2011; 5:12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Klock H.E., Lesley S.A.. Doyle S.A. The polymerase incomplete primer extension (PIPE) method applied to high-throughput cloning and site-directed mutagenesis. High Throughput Protein Expression and Purification: Methods and Protocols. 2009; Totowa, NJ: Humana Press; 91–103. [DOI] [PubMed] [Google Scholar]
- 19. Erlacher M.D., Chirkova A., Voegele P., Polacek N.. Generation of chemically engineered ribosomes for atomic mutagenesis studies on protein biosynthesis. Nat. Protoc. 2011; 6:580–592. [DOI] [PubMed] [Google Scholar]
- 20. Khaitovich P., Tenson T., Kloss P., Mankin A.S.. Reconstitution of functionally active Thermus aquaticus large ribosomal subunits with in vitro-transcribed rRNA. Biochemistry. 1999; 38:1780–1788. [DOI] [PubMed] [Google Scholar]
- 21. Willi J., Küpfer P., Evéquoz D., Fernandez G., Katz A., Leumann C., Polacek N.. Oxidative stress damages rRNA inside the ribosome and differentially affects the catalytic center. Nucleic Acids Res. 2018; 46:1945–1957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Küpfer P.A., Leumann C.J.. Synthesis, base pairing properties and trans-lesion synthesis by reverse transcriptases of oligoribonucleotides containing the oxidatively damaged base 5-hydroxycytidine. Nucleic Acids Res. 2011; 39:9422–9432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Shankar V., Rauscher R., Reuther J., Gharib W.H., Koch M., Polacek N.. rRNA expansion segment 27Lb modulates the factor recruitment capacity of the yeast ribosome and shapes the proteome. Nucleic Acids Res. 2020; 48:3244–3256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Zhu Y., Orre L.M., Tran Y.Z., Mermelekas G., Johansson H.J., Malyutina A., Anders S., Lehtiö J.. DEqMS: a method for accurate variance estimation in differential protein expression analysis. Mol. Cell. Proteomics. 2020; 19:1047–1057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Grenier F., Matteau D., Baby V., Rodrigue S.. Complete genome sequence of Escherichia coli BW25113. Genome Announc. 2014; 2:e01038-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Purta E., O’Connor M., Bujnicki J.M., Douthwaite S.. YgdE is the 2′-O-ribose methyltransferase RlmM specific for nucleotide C2498 in bacterial 23S rRNA. Mol. Microbiol. 2009; 72:1147–1158. [DOI] [PubMed] [Google Scholar]
- 27. Ho N.W.Y., Gilham P.T.. Reaction of pseudouridine and inosine with N-cyclohexyl-N’-γ- (4-methylmorpholinium) ethylcarbodiimide. Biochemistry. 1971; 10:3651–3657. [PubMed] [Google Scholar]
- 28. Zhu M., Dai X.. Maintenance of translational elongation rate underlies the survival of Escherichia coli during oxidative stress. Nucleic Acids Res. 2019; 47:7592–7604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Fasnacht M., Polacek N.. Oxidative stress in bacteria and the central dogma of molecular biology. Front. Mol. Biosci. 2021; 8:392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Almiron M., Link A.J., Furlong D., Kolter R.. A novel DNA-binding protein with regulatory and protective roles in starved Escherichia coli. Genes Dev. 1992; 6:2646–2654. [DOI] [PubMed] [Google Scholar]
- 31. Wolf S.G., Frenkiel D., Arad T., Finkeil S.E., Kolter R., Minsky A.. DNA protection by stress-induced biocrystallization. Nature. 1999; 400:83–85. [DOI] [PubMed] [Google Scholar]
- 32. Ceci P., Cellai S., Falvo E., Rivetti C., Rossi G.L., Chiancone E.. DNA condensation and self-aggregation of Escherichia coli Dps are coupled phenomena related to the properties of the N-terminus. Nucleic Acids Res. 2004; 32:5935–5944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Nair S., Finkel S.E.. Dps protects cells against multiple stresses during stationary phase. J. Bacteriol. 2004; 186:4192–4198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Fenton H.J.H. Oxidation of tartaric acid in presence of iron. J. Chem. Soc., Trans. 1894; 65:899–910. [Google Scholar]
- 35. Zhao G., Ceci P., Ilari A., Giangiacomo L., Laue T.M., Chiancone E., Dennis Chasteen N.. Iron and hydrogen peroxide detoxification properties of DNA-binding protein from starved cells. A ferritin-like DNA-binding protein of Escherichia coli. J. Biol. Chem. 2002; 277:27689–27696. [DOI] [PubMed] [Google Scholar]
- 36. Zheng M., Wang X., Templeton L.J., Smulski D.R., LaRossa R.A., Storz G.. DNA microarray-mediated transcriptional profiling of the Escherichia coli response to hydrogen peroxide. J. Bacteriol. 2001; 183:4562–4570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Piersimoni L., Giangrossi M., Marchi P., Brandi A., Gualerzi C.O., Pon C.L.. De novo synthesis and assembly of rRNA into ribosomal subunits during cold acclimation in Escherichia coli. J. Mol. Biol. 2016; 428:1558–1573. [DOI] [PubMed] [Google Scholar]
- 38. Al Refaii A., Alix J.H.. Ribosome biogenesis is temperature-dependent and delayed in Escherichia coli lacking the chaperones DnaK or DnaJ. Mol. Microbiol. 2009; 71:748–762. [DOI] [PubMed] [Google Scholar]
- 39. René O., Alix J.H.. Late steps of ribosome assembly in E. coli are sensitive to a severe heat stress but are assisted by the HSP70 chaperone machine. Nucleic Acids Res. 2011; 39:1855–1867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Polikanov Y.S., Steitz T.A., Innis C.A.. A proton wire to couple aminoacyl-tRNA accommodation and peptide-bond formation on the ribosome. Nat. Struct. Mol. Biol. 2014; 21:787–793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Vázquez-Laslop N., Ramu H., Klepacki D., Kannan K., Mankin A.S.. The key function of a conserved and modified rRNA residue in the ribosomal response to the nascent peptide. EMBO J. 2010; 29:3108–3117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Gupta P., Sothiselvam S., Vázquez-Laslop N., Mankin A.S.. Deregulation of translation due to post-transcriptional modification of rRNA explains why erm genes are inducible. Nat. Commun. 2013; 4:1984. [DOI] [PubMed] [Google Scholar]
- 43. Maden B.E.H. Locations of methyl groups in 28 S rRNA of Xenopus laevis and man. Clustering in the conserved core of molecule. J. Mol. Biol. 1988; 201:289–314. [DOI] [PubMed] [Google Scholar]
- 44. Natchiar S.K., Myasnikov A.G., Kratzat H., Hazemann I., Klaholz B.P.. Visualization of chemical modifications in the human 80S ribosome structure. Nature. 2017; 551:472. [DOI] [PubMed] [Google Scholar]
- 45. Sharma S., Yang J., Watzinger P., Kötter P., Entian K.-D.. Yeast Nop2 and Rcm1 methylate C2870 and C2278 of the 25S rRNA, respectively. Nucleic Acids Res. 2013; 41:9062–9076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Schmeing T.M., Huang K.S., Kitchen D.E., Strobel S.A., Steitz T.A.. Structural insights into the roles of water and the 2′ hydroxyl of the P site tRNA in the peptidyl transferase reaction. Mol. Cell. 2005; 20:437–448. [DOI] [PubMed] [Google Scholar]
- 47. Perez-Riverol Y., Csordas A., Bai J., Bernal-Llinares M., Hewapathirana S., Kundu D.J., Inuganti A., Griss J., Mayer G., Eisenacher M.et al.. The PRIDE database and related tools and resources in 2019: improving support for quantification data. Nucleic Acids Res. 2019; 47:D442–D450. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The mass spectrometry proteomics data have been deposited to the ProteomeXchange Consortium via the PRIDE (47) partner repository with the dataset identifier PXD025875.