

Abstract
Antibody-drug conjugates (ADCs) have recently gained traction in the biomedical community due to their promise for human therapeutics and an alternative to chemotherapy for cancer. Crucial metrics for ADC efficacy, safety, and selectivity are their drug-antibody ratios (DARs). However, DAR characterization (i.e., determining the average number of conjugated drugs on the antibody) through analytical methods remains challenging due to the heterogeneity of drug conjugation as well as the numerous post-translational modifications possible in the monoclonal antibody. Herein, we report on the use of high-resolution ion mobility spectrometry separations in structures for lossless ion manipulations coupled to mass spectrometry (SLIM IMS-MS) for the rapid and simultaneous characterization of the drug load profile (i.e., stoichiometric distribution of the number of conjugated drugs present on the mAb), determination of the weighted average DAR in both the heavy and light chains of a model antibody-drug conjugate, and calculation of the overall DAR of the ADC. After chemical reduction of the ADC and a subsequent 31.5 m SLIM IMS separation, the various drug-bound antibody species could be well resolved for both chains. We also show significantly higher resolution separations were possible for these large ions with SLIM IMS as compared to ones performed on a commercially available (1 m) drift tube IMS-MS platform. We expect high-resolution SLIM IMS separations will augment the existing toolbox for ADC characterization, particularly to enable the rapid optimization of DAR for a given ADC and thus better understand its potential toxicity and potency.
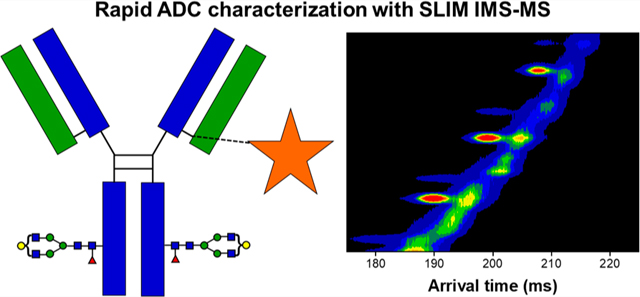
Graphical Abstract
With the growing interest from the biopharmaceutical industry to develop new classes of human therapeutics, monoclonal antibodies (mAbs) have recently received much attention.1,2 Unfortunately, the first-generation of mAbs proved to be ineffective, potentially due to their lack of specificity for a given antigen.3,4 To circumvent these bottlenecks, a new class of derivatized antibodies, termed antibody-drug conjugates (ADCs), has been developed. ADCs consist of a monoclonal antibody that is covalently modified (conjugated) with a cytotoxic drug (payload), usually through a lysine- or cysteine-based chemical linker.3–5 Initial results have been promising, demonstrating that ADCs provide increased selectivity, efficacy, and safety over alternative chemotherapeutic methods or other first generation mAbs.3–6 The chemotherapeutic advantages of ADCs are hypothesized to be due to the histological selectivity of the antibody for a given antigen, in conjunction with the potential pharmacokinetic properties of the payload (i.e., drug conjugate) and its respective chemical linker.3–10
Critical to evaluating the toxicity and efficacy of ADCs is the characterization of their drug-to-antibody ratios (DARs) or average number of drugs conjugated to mAb.4–6,10,11 This has been so much of an issue that the FDA has declared the use of novel analytical techniques for mAb and ADC characterization a top priority.6 However, DAR determination remains analytically challenging owing to the heterogeneous distribution of drug molecules conjugated along the monoclonal antibody (i.e., the drug load profile), as well as the complexities due to the many possible mAb post-translational modifications (e.g., glycosylation, deamidation, or oxidation).2,12–17 Chromatographic approaches (e.g., size exclusion chromatography, SEC; hydrophobic interaction chromatography, HIC; reversed-phase liquid chromatography, RPLC), as well as 2D-LC methods, remain the norm for ADC characterization.13,14,18,19 Unfortunately, such chromatographic approaches are typically very low in throughput due to long separation and column regeneration times; additionally, some useful mobile phases preclude online coupling to mass spectrometry (MS). Native MS-based methods have gained considerable traction for their ability to preserve noncovalent interactions and thus intact ADC measurements and information on the array of possible drug conjugate species.20–23 Unfortunately, both native MS and chromatographic approaches provide little information on how drug conjugation or its DAR affects the structure of the mAb and, for example, whether the number and location of bound drugs alters selectivity or efficacy.
Ion mobility spectrometry, especially when coupled to mass spectrometry (i.e., IMS-MS), has emerged as a powerful postionization gas-phase separation technique that complements existing LC-MS-based methods.24,25 In IMS, ions are separated in a buffer gas based on their gas-phase structures/shapes and mass-to-charge (m/z); however, for large macromolecules, the dependence on mass is much weaker.25 IMS-MS has already shown promise for various omics-based applications26–31 both as a standalone technique and when coupled to chromatographic separations or MS imaging modalities. Only recently has IMS-MS been utilized to study mAbs and profile the drug load distributions of ADCs, demonstrating good agreement for DAR characterization between IMS-MS and MS.22,23,32,33 It is also important to note that a recent study utilizing IMS-MS and collision-induced unfolding revealed subtle structural differences in mAbs conjugated with biotin.23 Additionally, the use of field asymmetric ion mobility spectrometry (FAIMS) has been reported for the rapid characterization of the heavy and light chains of a reduced mAb.34
However, IMS separations have not been integrated into the pharmaceutical pipeline for ADC analysis, largely due to insufficient IMS resolution of, for example, conformational or positional differences based on drug conjugation.21–23,33 Higher resolution IMS separations are attractive for potentially a better understanding on how covalent drug conjugation can alter the structure of an ADC and thus affect its viability as a biopharmaceutical product.
Recently, we have developed high-resolution IMS separations utilizing traveling waves (TW) in structures for lossless ion manipulations (SLIM) coupled to mass spectrometry (i.e., SLIM IMS-MS).35–39 SLIM IMS separations benefit from long serpentine paths and the option to route ions to even longer path lengths through serpentine ultralong paths with extended routing (SUPER) multipass capabilities.38 These advancements have provided greatly increased resolving power (Rp) of IMS measurements (>1000), especially as compared to commercially available IMS platforms, as well as benefiting from the ability to inject “in-SLIM” larger ion populations (2 to 3 orders of magnitude increased charge capacity).35,38 SLIM IMS-MS has previously shown utility in providing high-resolution separations of various peptides, carbohydrates, lipids, metabolites, isotopologues, isotopomers, and other biomolecules.40–51
Herein we report on the use of SLIM IMS-MS to enable the rapid and simultaneous characterization and separation of drug payload species on both the heavy and light chains of a model antibody–drug conjugate. We demonstrate that SLIM IMS-based separations provide higher resolution of ADC species as compared to a commercial 1 m drift tube (DT) IMS-MS platform. Furthermore, our calculated results of the summed, average, drug–antibody ratio for this model ADC are in good agreement with previous literature. We anticipate that SLIM IMS-MS-based measurements will provide new insights on how drug conjugation alters the structural properties of ADCs and thus help provide insights into their efficacy and selectivity as biotherapeutics.
EXPERIMENTAL SECTION
Reagents and Experimental Conditions.
The antibody–drug conjugate standard (SigmaMAb dansyl-cadavarine-SMCC; SigmaMAb ADC mimic), Amicon Ultra 0.5 mL centrifugal filters with a molecular weight cutoff of 10 000 Da, and tris(2-carboxyethyl)phosphine hydrochloride (TCEP) reducing agent were purchased from Sigma-Aldrich (Milwaukee, WI). The 13.5 m total path length TW SLIM SUPER IMS-MS platform used in these experiments (Figure S2) has been previously described.35–38 A pressure of 10 and 1.4 Torr, both nitrogen gas, was optimized for the high-pressure ion funnel and in the SLIM chamber, respectively, for all SLIM IMS separations. Pressures were controlled with needle valves (Swagelok, Solon OH) and measured using a convectron gauge. Square traveling waves of 15 V (0-peak) amplitude at 120 m/s velocity were used in all experiments and generated using a custom power supply (GAA Custom Engineering, LLC, Benton City, WA). Nanoelectrospray ionization conditions (3.5 kV) at flow rates of 0.5 μL/min were used for direct infusion of the chemically reduced ADC. Ions were introduced through a 750 μm i.d. heated capillary (130 °C) into a series of funnels (high pressure ion funnel and ion funnel trap) prior to entering the SLIM IMS serpentine path. Ions were accumulated in-SLIM for 250 ms, as previously described,40,44 by pulsing appropriate voltages in the ion funnel trap. Data was acquired with a U1084A 8-bit ADC digitizer (Keysight Technologies, Santa Rosa, CA). See Supporting Information for the SLIM IMS-MS voltages used. DTIMS-MS experiments were performed using an Agilent 6560 platform with an electric field of 16 V/cm at a typical pressure of 4 Torr nitrogen. For a complete list of DTIMS voltages used, see Supporting Information. The observed signal intensities with nanoESI direct infusion for 4 Torr nitrogen in DTIMS-MS were similar to those with 1.4 Torr nitrogen in the SLIM IMS-MS. For both DTIMS-MS and SLIM IMS-MS, separations were summed for 5 min to construct the extracted arrival time distributions.
Chemical Reduction and Sample Preparation for Direct Infusion.
To perform chemical reduction and thus break the disulfide bonds of the intact ADC, a 2.5 mg/mL solution of the ADC standard and 50 mM TCEP was prepared with 0.1% formic acid in LC-MS grade water and incubated at 55 °C for 60 min. Following reduction, a series of buffer exchange steps were performed to remove residual salts prior to direct infusion nanoESI. Briefly, ammonium acetate was added to the ADC-TCEP mixture to a final concentration of 50 mM at a final volume of 0.5 mL adjusted with 0.1% formic acid. Centrifugation with a 10 000 Da molecular weight cutoff filter (MWCO) at 14 000 rpm for 7 min was performed. This ammonium acetate addition and centrifugation step was performed twice. Subsequently, centrifugation was performed three more times but with adjusting the final volume to 0.5 mL only with 0.1% formic acid (i.e., no ammonium acetate was added). After three more centrifugation steps, the MWCO filter was inverted and centrifuged at 1000 rpm for 2 min to ensure that the concentrated sample was retained. The final product was diluted to ∼0.1 mg/mL concentration in 50/50 acetonitrile/water with 0.1% formic acid (classic denaturing conditions) and was subjected to nanoelectrospray direct infusion with both SLIM IMS-MS and DTIMS-MS platforms.
Data Processing.
For the SLIM IMS-MS experiments, data were acquired with home-built acquisition software and visualized with our unified ion mobility format (UIMF) viewer (URL: https://github.com/PNNL-Comp-Mass-Spec/UIMF-Viewer). For the DTIMS-MS experiments, data were acquired with the Agilent MassHunter workstation and visualized in the Agilent IMS-MS browser. Arrival time distributions were extracted for the m/z of the heavy and light chain species for each observed drug payload (i.e., payload species) for both SLIM IMS-MS and DTIMS-MS measurements and imported into Origin Pro 2017. A 10-point moving average smoothing was performed for all the extracted arrival time distributions from both instrument platforms. Weighted average DARs for the heavy and light chains were calculated as previously described,21,33 where IMS peak areas for each drug payload species (n) observed (0 to n) were extracted with Origin Pro 2017 (eq 1). The summed DAR of the overall ADC (heavy plus light chain) was estimated by eq 2.53
| (1) |
| (2) |
RESULTS AND DISCUSSION
Comparing the Separation of Reduced Heavy Chain Drug Payload Species Using DTIMS-MS and SLIM IMS-MS.
A key goal of this study was to simultaneously characterize how drug load can potentially affect the gas-phase structure of both the heavy and light chain antibody–drug conjugates, the resolution achievable with varying the number of drugs bound, and the ability to quantify DAR with SLIM IMS-MS. It was unclear if the ∼668 Da cysteine-conjugated drug mimic (Figure 1) would cause any observable mobility differences compared to the naked antibody (i.e., 0 binding drug payload) as well as with increasing drug payload (e.g., 1, 2, or 3 binding drug molecules). Table 1 shows the molecular weights for the intact antibody, reduced light chains, and reduced, glycosylated (G0F, G1F, and G2F), heavy chains. Direct infusion of the reduced ADC under denaturing conditions revealed a broad charge state distribution for both the heavy and light chains, with most intense charge states being z = 50 and z = 24, respectively. After 250 ms of in-SLIM ion accumulation and 4.5 m of SLIM IMS separation, the heavy and light chains were well resolved from one another, as evident from their distinct IMS trendlines in the 2D IMS-MS plot (Figure 2A). Although the m/z’s of the highly charged heavy chain and the lesser charged light chain overlap to some extent, the light chain ions have higher mobilities (i.e., are faster) than those of heavy chain ions with similar m/z values. These distinct heavy and light chain trendlines are less evident in the 2D DTIMS-MS plot (Figure 2B) due to its lower resolution IMS separations. Otherwise, only minor differences are observed when comparing the MS of the respective SLIM IMS and DTIMS separations and which are primarily attributable to the lower signal intensities with the DTIMS and perhaps a minor shift in the charge state distributions due to the longer times scale of the SLIM IMS separation. Unsurprisingly, the reduced heavy chain ions yielded a much more complex arrival time distribution trendline than its light chain counterpart, which we largely attribute to its three potential glycan decorations (an addition of a D-galactose moiety increases the total molecular weight by only ∼162 Da, which at z = 50 increases its m/z by ∼3), as well as having a much higher overall charge state with closer IMS peak spacing (i.e., smaller mobility differences among neighboring charge states and glycan attachments (Figure 3). Also observed from this 2D IMS-MS data, and the annotated mass spectra in Figure 3, was that the drug payload distribution for the light chain ranged from 0 to 1 bound, whereas the drug payload distribution for the heavy chain ranged from 0 to 3 bound. It is important to mention that no unbound drug was detected in either experiment, indicating that the gentle ion source and transmission conditions do not induce undesired fragmentation in either DTIMS or SLIM IMS separations. For additional annotations of the observed light chain and glycosylated heavy chain species, which were subjected to further SLIM IMS-MS separations, see Figure 3. (See Supporting Information for the exact m/z values observed.)
Figure 1.

Depiction of the model antibody–drug conjugate used in all experiments. The heavy chain is colored in blue, while the light chain is colored in green. The disulfide bonds in the hinge region are highlighted in black color. The structure of the drug mimic (∼668 Da) that is covalently linked to the monoclonal antibody through cysteine conjugation is also provided. Note: this figure only portrays a single site occupancy, whereas several sites can potentially be occupied. The three potential glycan attachments (G0F, G1F, and G2F) are shown utilizing the commonly accepted graphical symbols,52 where G0F has 0 galactose moieties, G1F has 1 galactose moiety, and G2F has 2 galactose moieties.
Table 1.
List of Theoretical Intact Molecular Weights for the Reduced Light Chain and Reduced, Glycosylated, Heavy Chains
| portion of ADC | molecular weight (Da) |
|---|---|
| intact ADC (G0F and 0 drugs) | 146658 |
| light chain | 22942.2 |
| heavy chain (G0F glycan) | 50403.2 |
| heavy chain (G1F glycan) | 50565.3 |
| heavy chain (G2F glycan) | 50727.5 |
Figure 2.

2D IMS-MS plot of the reduced antibody–drug conjugate after a 4.5 m SLIM IMS separation (A) and 1 m DTIMS separation (B). The light chain exhibits an overall lower charge state distribution compared to its heavy chain counterpart, as evidenced by less closely spaced IMS-MS peaks. Arrows illustrate selected m/z regions for both the heavy and light chains, where annotated mass spectra are shown in Figure 3. We attribute the “noisier” background in DTIMS to be from lower, overall, signal intensity compared to SLIM IMS.
Figure 3.

Annotated mass spectra of the glycosylated heavy chain for the z = 50 charge state (A) and of light chain for charge states z = 20 to z = 25 (B) after 4.5 m of SLIM IMS separation. The G2F glycan decoration is not observed at drug payloads of 2 and 3. For a full table of the m/z values for both the heavy and light chains, see Supporting Information.
Initially, we examined how SLIM IMS separations could characterize and separate the varying number of bound drug molecules on the heavy chain of this ADC. We also compared the SLIM IMS-MS analyses with those using a commercially available 1 m DTIMS-MS platform. As reported previously, additional passes through the SLIM serpentine path typically enable higher resolution measurements.38 However, because most previous SLIM IMS separations have focused on smaller molecules,40–46 it was unclear how such larger and more highly charged ions would behave during SLIM IMS separations extending over several hundred milliseconds due to e.g., stability of their structures, potential for charge transfer, increased peak broadening/diffusion, and overall ion confinement. Indeed, both the heavy and light chains were transmitted without significant or observable losses over several passes (Figure S3 in Supporting Information). In future work we plan to utilize size exclusion chromatography to provide reduced light and heavy chain sample fractions to increase signal-to-noise (S/N) with the direct infusion nanoESI SLIM IMS-MS measurements and thus permit longer, and higher resolution, IMS separations.
To examine the potential for rapid characterization of the heavy and light chain ADC mixture, we utilized 31.5 m SLIM IMS separations (using two additional passes) which provided a sufficient mobility range, as well as both increased IMS resolution and higher measurement S/N compared to DTIMS. We note that at this total IMS path length, the less intense G2F glycan decoration was not observed for each, 0 to 3, payload species with sufficient intensity and was omitted from the extracted arrival time distributions (see annotations in Figure 3). Figure 4 illustrates the separation of the heavy chain with 0 to 3 drug payloads for both the G0F and G1F glycan decorations for the most intense charge state (z = 50). Figure 4 shows the much higher resolution measurements using 31.5 m SLIM IMS separations compared to with 1 m DTIMS. Resolution between each payload species can be calculated according to eq 3, where ta and tb are the arrival times for the two species (a,b) and fwhma and fwhmb are their respective peak widths at half-maximum. The prefactor of 1.18 comes from the fact that a peak at fwhm has a width of 2.354σ, and thus an average baseline width of two peaks (a,b) would yield 1.18. IMS resolving power can be calculated according to eq 4, where t represents the arrival time and Δt represents its width at fwhm.
Figure 4.

Comparison of IMS separation for the G0F glycan attachment at various drug–antibody ratios for the heavy chain of the Sigma ADC standard at z = 50 between a 1 m DTIMS-MS platform (A) and a 31.5 m SLIM IMS-MS platform (B) and the G1F glycan attachment between DTIMS-MS (C) and SLIM IMS-MS (D). Average DARs are calculated using eq 1.
| (3) |
| (4) |
For the G0F glycan decoration on the heavy chain, a resolution of 0.07 was observed between the 0 and 1 drug payload species in the DTIMS and a resolution of 0.23 in the SLIM IMS. Between the 0 and 3 drug payload species, a resolution of 0.27 was observed in the DTIMS and 0.74 in the SLIM IMS. Collectively in our experiments, we observed an average DTIMS resolving power of ∼25 and a SLIM IMS resolving power of ∼75 and, which based upon previous work with small ions, is limited by the species being studied rather than the IMS platform. We also noted a lower intensity potential additional substructure at the front of each DTIMS peak in our SLIM IMS measurements. However, after 31.5 m, this feature appears as a more diffuse elevated baseline component of the main SLIM IMS peak. As briefly indicated later, we tentatively attribute this to a minor contribution of charge transfer during the SLIM IMS separation.
From this cross-platform IMS-MS comparison we can make several observations: (1) the 31.5 SLIM IMS separations provide much higher resolution for drug payloads ranging from 0 to 3 on the heavy chain, for both the G0F and G1F glycan decoration, as compared to a commercially available 1 m DTIMS-MS platform; (2) the calculated weighted average DAR for the heavy chain (eq 1) at this charge state is similar for both the G0F and G1F for the respective SLIM IMS-MS or DTIMS-MS measurements; (3) the calculated weighted average DAR from these two glycosylation states is also similar for the DTIMS and SLIM IMS data. For the SLIM IMS-MS results of the heavy chain at a charge state of z = 50, an average DAR was calculated to be 1.41 ± 0.03 for the G0F species and 1.41 ± 0.04 for the G1F species. For calculated drug load distributions of the heavy chain at this charge state, see Supporting Information. These results show that SLIM IMS-MS-based separations provide higher resolution than DTIMS for the reduced heavy chain of an ADC.
Characterization of Drug Payloads on the Reduced Light Chain Using SLIM IMS-MS.
We now turn our attention to the smaller, and lesser charged, reduced light chain to determine if we could resolve the various drug payload species. Figures 2 and 3 show the expected greater light chain intensities compared to that of the heavy chain and primarily attributed to the fewer charge states detected (most intense charge states z = 20 to z = 25), the lack of glycan attachments, and lower drug loading. We subjected (concurrently and rapidly; 5 min of total experiment time) the most intense reduced light chain charge states to 31.5 m SLIM IMS separations for measurements to characterize the drug load distribution in this ADC standard. Figure 5 shows the arrival time distributions 0 and 1 loaded drugs on the reduced light chain of the ADC for the six most intense charge states after a 31.5 m SLIM IMS separation. All arrival time distributions were normalized to the most intense m/z peak (e.g., 1 loaded drug at z = 24). Compared to its heavy chain counterpart, the light chain drug load species are much better resolved. Figure S4 show only minor differences in relative MS intensities between 4.5 and 31.5 m SLIM IMS separations (i.e., 0 and 2 passes). We globally observe the 0 and 1 drug payload species of the light chain to be well resolved from one another after 31.5 m of SLIM IMS separation.
Figure 5.

Extracted arrival time distributions for 0 and 1 drug load to the reduced light chain of the ADC, at charge states ranging from z = 20 to z = 25, after a 31.5 m SLIM IMS separation.
The arrival time distributions from Figure 5 also appear to indicate some small variability of the DAR based upon the charge state surveyed (i.e., the peak area ratios for 0 and 1 loaded drugs changes for different charge states). This observation has been previously reported21 for an intact ADC under native ESI-MS conditions, and indeed this also applies for the reduced light chain of our Sigma mAb ADC mimic. We hypothesize that the slightly different charge state envelopes are due to small structural changes or a potential additional charge site for the ADC compared to its unconjugated (0 drug payload) counterparts. We emphasize that the multiple IMS peaks observed (e.g., in the case of 1 drug payload at z = 20) yield indistinguishable mass spectra (the arrival time distributions are constructed by extracting only the corresponding m/z slice/window). The calculated weighted average DAR values for each of the charge states shown in Figure 5 are provided in Table 2. This significant difference in calculated total DAR between nearby charge states indicates that care must be taken when comparing overall, average, DAR values obtained by MS- or IMS-MS-based methods. Future work will probe whether traveling wave-based IMS separations54,55 may influence the observed charge state distribution (e.g., charge state reduction due to charge transfer or ions of a given mobility range may not being well resolved, confined, and/or transmitted under certain TW conditions). In this study, we noted no significant difference between the TW SLIM IMS and the DTIMS separation ones other than IMS resolution and IMS resolving power, as indicated by similar relative DARs (Figure 3). Our observed IMS resolving powers are nearly double those previously reported for ions of similar size and molecular weight.56 We are further exploring the use of native SLIM IMS-MS-based experiments56 on various ADCs with a range of potential drug conjugates.
Table 2.
Calculated Weighted Average DARs for the Reduced Light Chain at Various Charge States Based on an Average of Triplicate Trials and One Standard Deviation
| charge state | DAR |
|---|---|
| 25 | 0.76 ± 0.02 |
| 24 | 0.78 ± 0.05 |
| 23 | 0.77 ± 0.04 |
| 22 | 0.67 ± 0.02 |
| 21 | 0.65 ± 0.01 |
| 20 | 0.62 ± 0.04 |
CONCLUSIONS
Herein we have reported the utility of SLIM IMS-MS-based separations to rapidly and simultaneously characterize drug conjugation on both the reduced light and reduced, glycosylated, heavy chains and to determine their respective DARs as well as the overall DAR for an antibody–drug conjugate. The calculated DAR values for both the G0F and G1F glycan decoration at z = 50 were in good agreement between DTIMS-MS and SLIM IMS-MS. For the analyses of the reduced heavy chain, we observed that 31.5 m total path length SLIM IMS separations provided much higher resolution of the various drug payload species than a commercially available 1 m DTIMS-MS platform. For the reduced light chain, we observed that the 0 and 1 drug loads onto the mAb could also be resolved by the SLIM IMS separations. Future work is needed to understand the origin of the multiple IMS peaks observed at a drug payload of 1 in the light chain and determine if they could possibly have any effect on ADC efficacy as measured by bioassays. Additionally, from a clinical perspective, the ability to rapidly assess, and optimize, DAR for a given ADC without the need for LC separations would greatly increase throughput.
The weighted average of the DARs from Table 2, along with the calculated DARs for the G0F and G1F heavy chain in Figure 4 (z = 50), result in a summed average DAR for this model ADC of 4.29 ± 0.04 (eq 2), in good agreement with previous work57,58 (∼4.3), as well as that provided by Sigma-Aldrich (4.04). The reduced light chain data provided DAR values that varied slightly with charge state, an observation we tentatively attribute to the conjugated drug mimic slightly altering the gas-phase structure of the ADC as compared to the unconjugated mAb. While some minor charge state dependency is evident for calculated DARs, our results are in good agreement with previous work and provide an initial proof-of-concept demonstration for the ability to rapidly characterize the drug load profile and determine an average DAR for an ADC. Future work will more fully explore the variance and uncertainties related to DAR calculations as a function of the charge state distribution sampled, payload and linker, and any potential impacts of SLIM IMS separation conditions used.
Regardless of the noted minor DAR variations, our results clearly demonstrate the utility of SLIM IMS-MS-based analyses for providing rapid (in ∼5 min total measurement time) simultaneous characterization of both reduced light and heavy chains of an antibody–drug conjugate. We envision that such high-resolution SLIM IMS separations will provide additional insights on ADCs due to their combined sensitivity for both mass and 3D structural changes and provide an important addition to the existing analytical toolbox. We are working to assess other ADCs with varying drug conjugates (i.e., different payloads and linkers) as well as to perform these measurements under native MS conditions with intact ADCs. Such studies, in conjunction with existing methodologies, will aid in providing insights into attributes such as potency, toxicity, and stability of ADCs in the context of their role as human therapeutics and antigen binding.
Supplementary Material
ACKNOWLEDGMENTS
Portions of this work were funded by support from the National Institute of General Medical Sciences (P41 GM103493-10) and NIH National Cancer Institute (R33 CA217699). Experiments were performed at the W. R. Wiley Environmental Molecular Sciences Laboratory (EMSL) at Pacific Northwest National Laboratory (PNNL). PNNL is a DOE national scientific user facility and operated by Battelle (contract DE-AC05-76RL01830). The authors also acknowledge the ongoing collaboration between Janssen Research & Development and Pacific Northwest National Laboratory.
Footnotes
ASSOCIATED CONTENT
Supporting Information
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acs.analchem.9b05209.
Overlaid arrival time distributions for the reduced light chain, varying glycosylations for the reduced heavy chain, example DAR calculation, m/z values, and voltages used (PDF)
Complete contact information is available at: https://pubs.acs.org/10.1021/acs.analchem.9b05209
The authors declare no competing financial interest.
Contributor Information
Gabe Nagy, Biological Sciences Division, Pacific Northwest National Laboratory, Richland, Washington 99354, United States.
Isaac K. Attah, Biological Sciences Division, Pacific Northwest National Laboratory, Richland, Washington 99354, United States
Christopher R. Conant, Biological Sciences Division, Pacific Northwest National Laboratory, Richland, Washington 99354, United States
Weijing Liu, Environmental Molecular Sciences Laboratory, Pacific Northwest National Laboratory, Richland, Washington 99352, United States.
Sandilya V.B. Garimella, Biological Sciences Division, Pacific Northwest National Laboratory, Richland, Washington 99354, United States
Harsha P. Gunawardena, Janssen Research &Development, The Janssen Pharmaceutical Companies of Johnson & Johnson, Spring House, Pennsylvania 19477, United States.
Jared B. Shaw, Environmental Molecular Sciences Laboratory, Pacific Northwest National Laboratory, Richland, Washington 99352, United States.
Richard D. Smith, Biological Sciences Division, Pacific Northwest National Laboratory, Richland, Washington 99354, United States.
Yehia M. Ibrahim, Biological Sciences Division, Pacific Northwest National Laboratory, Richland, Washington 99354, United States.
REFERENCES
- (1).Singh S; et al. Curr. Clin. Pharmacol. 2018, 13 (2), 85–99. [DOI] [PubMed] [Google Scholar]
- (2).Beck A; Wagner-Rousset E; Ayoub D; Van Dorsselaer A; Sanglier-Cianférani S. Anal. Chem. 2013, 85 (2), 715–736. [DOI] [PubMed] [Google Scholar]
- (3).Diamantis N; Banerji U. Br. J. Cancer 2016, 114 (4), 362–367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (4).Nejadmoghaddam MR; Minai-Tehrani A; Ghahremanzadeh R; Mahmoudi M; Dinarvand R; Zarnani AH Avicenna J. Med. Biotechnol 2019, 11 (1), 3–23. [PMC free article] [PubMed] [Google Scholar]
- (5).Dan N; Setua S; Kashyap VK; Khan S; Jaggi M; Yallapu MM; Chauhan SC Pharmaceuticals 2018, 11 (2), 32–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (6).Saber H; Leighton JK Regul. Toxicol. Pharmacol. 2015, 71 (3), 444–452. [DOI] [PubMed] [Google Scholar]
- (7).Lu J; Jiang F; Lu A; Zhang G. Int. J. Mol. Sci. 2016, 17, 561–582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (8).McCombs JR; Owen SC AAPS J. 2015, 17 (2), 339–351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (9).Hedrich WD; Fandy TE; Ashour HM; Wang H; Hassan HE Clin. Pharmacokinet. 2018, 57 (6), 687–703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (10).Sun X; Ponte JF; Yoder NC; Laleau R; Coccia J; Lanieri L; Qiu Q; Wu R; Hong E; Bogalhas M; Wang L; Dong L; Setiady Y; Maloney EK; Ab O; Zhang X; Pinkas J; Keating TA; Chari R; Erickson HK; Lambert JM Bioconjugate Chem. 2017, 28 (5), 1371–1381. [DOI] [PubMed] [Google Scholar]
- (11).Tang Y; Tang F; Yang Y; Zhao L; Zhou H; Dong J; Huang W. Sci. Rep. 2017, 7 (1), 7763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (12).Atmanene C; Wagner-Rousset E; Malissard M; Chol B; Robert A; Corvaïa N; Dorsselaer AV; Beck A; Sanglier-Cianférani S. Anal. Chem. 2009, 81 (15), 6364–6373. [DOI] [PubMed] [Google Scholar]
- (13).Källsten M; Hartmann R; Artemenko K; Lind SB; Lehmann F; Bergquist J. Analyst 2018, 143 (22), 5487–5496. [DOI] [PubMed] [Google Scholar]
- (14).Neupane R; Bergquist J. Eur. J. Mass Spectrom. 2017, 23 (6), 417–426. [DOI] [PubMed] [Google Scholar]
- (15).Rao C; Rangan VS; Deshpande S. Bioanalysis 2015, 7 (13), 1561–1564. [DOI] [PubMed] [Google Scholar]
- (16).Wagh A; Song H; Zeng M; Tao L; Das TK mAbs 2018, 10 (2), 222–243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (17).Wakankar A; Chen Y; Gokarn Y; Jacobson FS mAbs 2011, 3 (2), 161–172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (18).Chen T; Chen Y; Stella C; Medley CD; Gruenhagen JA; Zhang KJ Chromatogr. B: Anal. Technol. Biomed. Life Sci. 2016, 1032, 39–50. [DOI] [PubMed] [Google Scholar]
- (19).Goyon A; Kim MT-J; Dai L; Cornell C; Jacobson F; Guillarme D; Stella C. Anal. Chem. 2019, 91 (23), 14896–14903. [DOI] [PubMed] [Google Scholar]
- (20).Botzanowski T; Erb S; Hernandez-Alba O; Ehkirch A; Colas O; Wagner-Rousset E; Rabuka D; Beck A; Drake PM; Cianférani S. mAbs 2017, 9 (5), 801–811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (21).Debaene F; Bœuf A; Wagner-Rousset E; Colas O; Ayoub D; Corvaïa N; Van Dorsselaer A; Beck A; Cianférani S. Anal. Chem. 2014, 86 (21), 10674–10683. [DOI] [PubMed] [Google Scholar]
- (22).Marcoux J; Champion T; Colas O; Wagner-Rousset E; Corvaïa N; Van Dorsselaer A; Beck A; Cianférani S. Protein Sci. 2015, 24 (8), 1210–1223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (23).Tian Y; Lippens JL; Netirojjanakul C; Campuzano IDG; Ruotolo BT Protein Sci. 2019, 28 (3), 598–608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (24).Chen Z; Glover MS; Li L. Curr. Opin. Chem. Biol. 2018, 42, 1–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (25).Gabelica V; Marklund E. Curr. Opin. Chem. Biol. 2018, 42, 51–59. [DOI] [PubMed] [Google Scholar]
- (26).Morrison KA; Clowers BH TrAC, Trends Anal. Chem. 2019, 119, 115625. [Google Scholar]
- (27).Sans M; Feider CL; Eberlin LS Curr. Opin. Chem. Biol. 2018, 42, 138–146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (28).Zheng X; Smith FB; Aly NA; Cai J; Smith RD; Patterson AD; Baker ES Anal. Bioanal. Chem. 2019, 411 (19), 4673–4682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (29).Kyle JE; Zhang X; Weitz KK; Monroe ME; Ibrahim YM; Moore RJ; Cha J; Sun X; Lovelace ES; Wagoner J; Polyak SJ; Metz TO; Dey SK; Smith RD; Burnum-Johnson KE; Baker ES Analyst 2016, 141 (5), 1649–1659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (30).Eldrid C; Ujma J; Kalfas S; Tomczyk N; Giles K; Morris M; Thalassinos K. Anal. Chem. 2019, 91 (12), 7554–7561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (31).Ujma J; Ropartz D; Giles K; Richardson K; Langridge D; Wildgoose J; Green M; Pringle SJ Am. Soc. Mass Spectrom. 2019, 30 (6), 1028–1037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (32).Debaene F; Wagner-Rousset E; Colas O; Ayoub D; Corvaïa N; Van Dorsselaer A; Beck A; Cianférani S. Anal. Chem. 2013, 85 (20), 9785–9792. [DOI] [PubMed] [Google Scholar]
- (33).Huang RYC; Deyanova EG; Passmore D; Rangan V; Deshpande S; Tymiak AA; Chen GJ Am. Soc. Mass Spectrom. 2015, 26 (10), 1791–1794. [DOI] [PubMed] [Google Scholar]
- (34).Melani RD; Srzenti-ç K; Gerbasi VR; McGee JP; Huguet R; Fornelli L; Kelleher NL mAbs 2019, 11 (8), 1351–1357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (35).Deng L; Garimella SVB; Hamid AM; Webb IK; Attah IK; Norheim RV; Prost SA; Zheng X; Sandoval JA; Baker ES; Ibrahim YM; Smith RD Anal. Chem. 2017, 89 (12), 6432–6439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (36).Deng L; Ibrahim YM; Baker ES; Aly NA; Hamid AM; Zhang X; Zheng X; Garimella SVB; Webb IK; Prost SA; Sandoval JA; Norheim RV; Anderson GA; Tolmachev AV; Smith RD Chemistry Select 2016, 1 (10), 2396–2399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (37).Deng L; Ibrahim YM; Hamid AM; Garimella SV; Webb IK; Zheng X; Prost SA; Sandoval JA; Norheim RV; Anderson GA; Tolmachev AV; Baker ES; Smith RD Anal. Chem. 2016, 88 (18), 8957–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (38).Deng L; Webb IK; Garimella SVB; Hamid AM; Zheng X; Norheim RV; Prost SA; Anderson GA; Sandoval JA; Baker ES; Ibrahim YM; Smith RD Anal. Chem. 2017, 89 (8), 4628–4634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (39).Garimella SV; Hamid AM; Deng L; Ibrahim YM; Webb IK; Baker ES; Prost SA; Norheim RV; Anderson GA; Smith RD Anal. Chem. 2016, 88 (23), 11877–11885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (40).Chouinard CD; Nagy G; Webb IK; Garimella SVB; Baker ES; Ibrahim YM; Smith RD Anal. Chem. 2018, 90 (18), 11086–11091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (41).Chouinard CD; Nagy G; Webb IK; Shi T; Baker ES; Prost SA; Liu T; Ibrahim YM; Smith RD Anal. Chem. 2018, 90 (18), 10889–10896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (42).Nagy G; Attah IK; Garimella SVB; Tang K; Ibrahim YM; Baker ES; Smith RD Chem. Commun. 2018, 54, 11701–11704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (43).Nagy G; Chouinard CD; Attah IK; Webb IK; Garimella SVB; Ibrahim YM; Baker ES; Smith RD Electrophoresis 2018, 39 (0), 3148–3155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (44).Nagy G; Kedia K; Attah IK; Garimella SVB; Ibrahim YM; Petyuk VA; Smith RD Anal. Chem. 2019, 91 (7), 4374–4380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (45).Nagy G; Veličković D; Chu RK; Carrell AA; Weston DJ´; Ibrahim YM; Anderton CR; Smith RD Chem. Commun. 2019, 55 (3), 306–309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (46).Wojcik R; Nagy G; Attah IK; Webb IK; Garimella SVB; Weitz KK; Hollerbach A; Monroe ME; Ligare MR; Nielson FF; Norheim RV; Renslow RS; Metz TO; Ibrahim YM; Smith RD Anal. Chem. 2019, 91 (18), 11952–11962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (47).Wojcik R; Webb IK; Deng L; Garimella SV; Prost SA; Ibrahim YM; Baker ES; Smith RD Int. J. Mol. Sci. 2017, 18 (1), 183–194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (48).Zheng X; Deng L; Baker ES; Ibrahim YM; Petyuk VA; Smith RD Chem. Commun. 2017, 53 (56), 7913–7916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (49).Ben Faleh A; Warnke S; Rizzo TR Anal. Chem. 2019, 91 (7), 4876–4882. [DOI] [PubMed] [Google Scholar]
- (50).Warnke S; Ben Faleh A; Pellegrinelli RP; Yalovenko N; Rizzo TR Faraday Discuss. 2019, 217, 114–125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (51).Warnke S; Ben Faleh A; Scutelnic V; Rizzo TR J. Am. Soc. Mass Spectrom. 2019, 30, 2204–2211. [DOI] [PubMed] [Google Scholar]
- (52).Varki A; Cummings RD; Aebi M; Packer NH; Seeberger PH; Esko JD; Stanley P; Hart G; Darvill A; Kinoshita T; Prestegard JJ; Schnaar RL; Freeze HH; Marth JD; Bertozzi CR; Etzler ME; Frank M; Vliegenthart JFG; Lutteke T; Perez S; Bolton E; Rudd P; Paulson J; Kanehisa M; Toukach P; Aoki-Kinoshita KF; Dell A; Narimatsu H; York W; Taniguchi N; Kornfeld S. Glycobiology 2015, 25 (12), 1323–1324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (53).Wagner-Rousset E; Janin-Bussat M-C; Colas O; Excoffier M; Ayoub D; Haeuw J-F; Rilatt I; Perez M; Corvaïa N; Beck A. mAbs 2014, 6 (1), 173–184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (54).Shvartsburg AA; Smith RD Anal. Chem. 2008, 80 (24), 9689–99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (55).Giles K; Williams JP; Campuzano I. Rapid Commun. Mass Spectrom. 2011, 25 (11), 1559–1566. [DOI] [PubMed] [Google Scholar]
- (56).Allen SJ; Eaton RM; Bush MF Anal. Chem. 2016, 88 (18), 9118–9126. [DOI] [PubMed] [Google Scholar]
- (57).Hai ME.; Schuste RJ.; McCar KS. Poster presentation in Proceedings of the American Society for Mass Spectrometry National Conference, 2019. [Google Scholar]
- (58).Nguyen JM; Smith J; Rzewuski S; Legido-Quigley C; Lauber MA mAbs 2019, 11 (8), 1358–1366. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.