

Abstract
SARS-CoV-2, which causes the current pandemic of respiratory illness, is evolving continuously and generating new variants. Nevertheless, most of the sequence analyses thus far focused on nucleotide substitutions despite the fact that insertions and deletions (indels) are equally important in the evolution of SARS-CoV-2. In this study, we analyzed 1,099,664 high-quality sequences of SARS-CoV-2 genomes to re-construct the evolutionary and epidemiological histories of indels. Our analysis revealed 289 circulating indel types (237 deletion and 52 insertion types, each represented by more than ten genomic sequences), among which eighteen were recurrent indel types, each represented by more than 500 genome sequences. Although indels were identified across the entire genome, most of them were identified in nsp6, S, ORF8, and N genes, among which ORF8 indel types had the highest frequencies of frameshift. Geographical and temporal analyses of these variants revealed a few alterations of dominant indel types, each accompanied by geographic expansion to different countries and continents, which resulted in the fixation of several types of indels in the field, including the current variants of concern. Evolutionary and structural analyses revealed that indels involving S N-terminal domain regions were linked to the 3/4 variants of concern, resulting in significantly altered S protein that might contribute to the selective advantage of the corresponding variant. In sum, our study highlights the important role of insertions and deletions in the evolution and spread of SARS-CoV-2.
Keywords: SARS-CoV-2, deletions, insertions, evolution, molecular epidemiology
1. Introduction
A new type of betacoronavirus causing severe respiratory disease was identified in December 2019 (Zhou et al. 2020), which was later officially named as SARS-CoV-2 by the International Committee on Taxonomy of Viruses (NC_045512), and the disease it causes was named as coronavirus disease 2019 (COVID-19) by the World Health Organization (WHO) (Gorbalenya et al. 2020). The virus has since spread rapidly across the globe, causing recurrent epidemics in many countries and regions around the world. As of 24 September 2021, SARS-CoV-2 was circulating in 223 countries or regions, with more than 233,278,752 cases and 4,774,507 deaths reported thus far (Dong, Du, and Gardner 2020).
Like other ssRNA(+) viruses, SARS-CoV-2 is prone to genomic variation, including the substitution, insertions, and deletions. Substitutions have been intensively studied in relation to changes in the structure and/or function of the viral proteins, which in turn result in altered virulence, antigenic properties, or transmissibility of the virus (Hou et al. 2020; Plante et al. 2020). Based on substitutions, viruses were divided into more than 1,593 Pango lineages with shared sequence identity, phylogenetic relationships, and temporal and geographic structure (Rambaut et al. 2020). Several lineages defining substitutions N501Y and E484K cause amino acid changes that strengthened the binding of the receptor binding domain of S to the ACE2 receptor, making the variants 70 per cent more contagious than the predecessor lineage (Davies et al. 2021; Khan et al. 2021). Furthermore, among these lineages, WHO identified, based on transmissibility, pathogenicity and the impact on vaccines, several SARS-CoV-2 variants of concern (VOCs), including Alpha (B.1.1.7), Beta (B.1.351), Gamma (P.1), and Delta (B.1.617.2), and variants of interest (VOIs), including Epsilon (B.1.429 + B.1.427), Zeta (P.2), Eta (B.1.525), Theta (P.3), Iota (B.1.526), Kappa (B.1.617.1), Lambda (C.37), and Mu (B.1.621).
Nevertheless, despite intensive research on substitution, the role of insertions and deletions (indels) has not been systematically investigated. A few indels were identified within the VOCs. For example, B.1.1.7 strain contains two S protein deletions (del69-70HV and del145Y) in the N-terminal domain (NTD) (Shen et al. 2021); B.1.351 contains del242–244 in the S protein NTD domain; and B.1.1.7 and B.1.351 contained del145Y and del242–244, respectively, that render resistance to most NTD-directed monoclonal antibodies by previous strains (Wang et al. 2021b). Other reported indels included nsp1 del241-243 (Benedetti et al. 2020), nsp2 del268 (Bal et al. 2020), ORF6 34-nt deletion (Quéromès et al. 2021), and ORF7a 81-nt deletion (Holland et al. 2020), and, amongst others, most of these indels resulted in virus progenies that have been spread to other patients. Interestingly, the largest indel reported so far is the 382-nucleotide deletion (∆382) in the ORF8 region, which appeared in Singapore in January and February of 2020 (Su et al. 2020), which terminated the translation of ORF8 at positions 28,229. It may result in a milder infection, which makes it suitable for design of attenuated vaccine (Young et al. 2020; Zinzula 2021). There are also reports that the del675-679 in S protein may restrict virus replication in Vero cells at the late phase (Liu et al. 2020).
Importantly, rapid genome sequencing and online sharing of SARS-CoV-2 genomes by public health and research teams worldwide had provided us invaluable insights into the ongoing evolution and epidemiology of the virus, as well as the global variations during the pandemic, and thus played an important role in virus surveillance and its eventual mitigation and control. The Global Initiative on Sharing All Influenza Data (GISAID) (Shu and McCauley 2017) database contains a large number of COVID-19 genome sequences, which make it possible to analyze and trace the sequence variation and evolution of SARS-CoV-2 on a global scale such that any variants with altered pathogenicity or antigenic properties can be promptly identified.
In order to systematically characterize the indels of SARS-CoV-2, we performed a genome-wide indels analyses on 1,031,249 complete-genome sequences of the SARS-CoV-2 collected from more than 166 countries or regions. Our analyses revealed the frequencies, genome distributions, and molecular characteristics of all indels that are circulating in the field. For highly prevalent indels, we further characterized the temporal and spatial dynamics and evolutionary histories of the corresponding variants. And structure and functional impacts of relevant proteins were subsequently evaluated.
2. Results
2.1. A general characterization of circulating SARS-CoV-2 genomic indels
Compared to the prototype strain, a total of 3,854 types of deletions and 891 types of insertions were detected among the 1,031,249 SARS-CoV-2 genome sequences (Tables S1 and S2). Among these, only 237 deletion types and 52 insertion types were regarded as ‘circulating’ genomic variant based on our criterion for ‘circulating’ indels that exist in more than ten genomic sequences. And the numbers were reduced to seventy deletion types and twenty insertion types if we set the threshold of detection frequencies higher at 50 (Fig. 1). Among the 237 types of deletions, 34.18 per cent (81/237) caused frameshift and 65.82 per cent (156/237) did not (Fig. 1A). On the other hand, a total of twenty-five frameshifts (48.08 per cent) and twenty-seven non-frameshift (51.92 per cent) were observed in the fifty-two insertions types (Fig. 1B). Generally, insertion occurs less frequently than deletion, with the ratio of deletion:insertion as 31.33:1, and the ratio of non-frameshift indels:frameshift indels as 3.89:1. The frameshift frequency of deletion mutation in the genes coding for accessory proteins (37.97 per cent) is significantly higher than that of non-structural proteins (0.3 per cent) and structural proteins (0.28 per cent). The latter genes are essential for viral propagation, and the indels detected in these regions may represent sequencing errors and dead-end genomic products. Strikingly, the sequence counts for frameshift indels in ORF8 were one or two orders of magnitude higher than other protein-coding genes (Fig. 1C).
Figure 1.
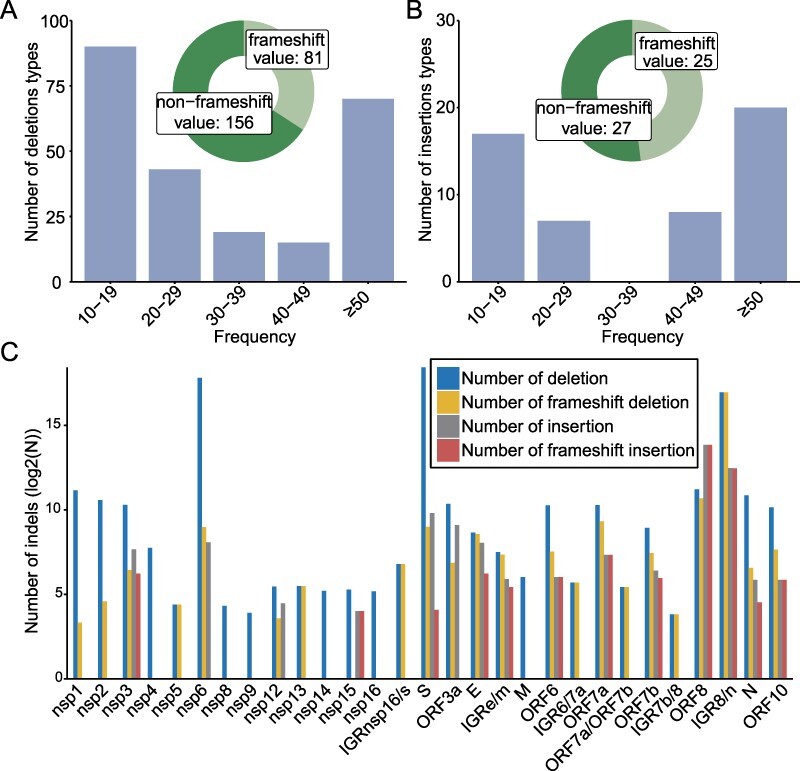
Overview of indels within global SARS-CoV-2 genomes. The number of deletion (A) and insertion types (B) and the proportion of indels causing frameshift were described. (C) Distribution of deletions and insertions on different SARS-CoV-2 proteins. The N represents the number of indels sequences.
2.2. Genome-wide diversity of indels
We further characterized the distribution of indels on SARS-CoV-2 genomes with the exception of 5ʹ and 3ʹ UTR as these regions are prone to have sequencing errors (Fig. 2). For deletions, circulating forms were detected in all protein-coding genes with the exception of non-structural proteins nsp7, nsp10, and nsp11, which contained no deletion. On the other hand, higher frequencies of deletions were detected in structural genes encoding the spike protein (n = 32), N protein (n = 18), as well as non-structural protein genes, such as nsp3 (n = 29), and accessory genes ORF7a (n = 27), ORF8 (n = 26), and OFR3a (n = 22) (Fig. 2A). Deletions with the most successful epidemiological outcome were detected in nsp6 pos_11288-9 (22.12 per cent of 1,031,249 sequences), S pos_21765-6 (21.41 per cent), S pos_21991-3 (12.22 per cent), and intergenic region IGR8/n pos_28271-1 (12.32 per cent). The most common lengths for deletions are 3 nt (36.71 per cent), 1 nt (18.14 per cent), 6 nt (11.81 per cent), 9 nt (10.55 per cent), and 2 nt (5.06 per cent). The 1-nt and 2-nt indels were mostly detected in the non-coding regions and accessory proteins, which do not disrupt the translation of viral proteins essential for viral replication. Interestingly, our data also revealed a number of large-fragment deletions (>50 nt in length), which is mainly identified in the genes encoding accessory proteins ORF7a and ORF8 (Fig. 2A).
Figure 2.
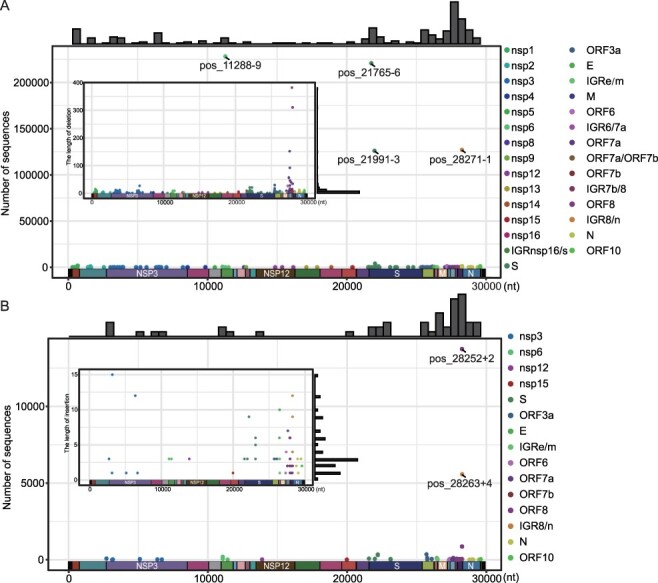
The distribution of indels on SARS-CoV-2 genome. The sequence number (outer plot) and length (inner plot) of deletion (A) and insertion (B) types were described. The histograms above the outer plots show the frequencies of indel types along the entire genome. The deletion types associated with each gene is marked with different colors.
As for insertions, they were discovered in thirteen genes, with most types discovered in ORF8 gene (n = 8), followed by spike gene (n = 7), and nsp3 gene (n = 6) (Fig. 2B). The three types of insertions with most genome sequences were all associated with ORF8, including pos_28252+2 (1.33 per cent), ORF8 pos_28255+2 (0.08 per cent), and IGR8/n pos_28263+4 (0.54 per cent). And the most common insertion length included 3 nt (32.69 per cent), 1 nt (19.23 per cent), and 2 nt (15.38 per cent).
2.3. Temporal and spatial dynamics of highly prevalent genomic variants of SARS-CoV-2
We characterized the molecular epidemiological features of fifteen deletion and three insertion types with more than 500 sequences (Tables S1 and S2), which we named as recurrent deletion or insertion types (RDT or RIT) (Table 1). Interestingly, indel types with the highest frequencies included pos_11288-9 (n = 228,125), pos_21765-6 (n = 220,758), pos_21991-3 (n = 126,048), and pos_28271-1 (n = 127,020), located in nsp6, S, and IGR8/n regions, respectively.
Table 1.
Recurrent deletion or insertion types (RDT or RIT) for SARS-CoV-2.
| Name | Region | Start position and indels nucleotidesa | Indels of nucleotides | Frequency |
|---|---|---|---|---|
| RDT-nsp1 | nsp1 | pos_686-9 | AAGTCATTT | 1,771 |
| RDT-nsp2-1 | nsp2 | pos_1598-6 | GGTCTT | 528 |
| RDT-nsp2-2 | nsp2 | pos_1605-3 | ATG | 854 |
| RDT-nsp6 | nsp6 | pos_11288-9 | TCTGGTTTT | 228,125 |
| RDT-S-1 | S | pos_21765-6 | TACATG | 220,758 |
| RDT-S-2 | S | pos_21991-3 | TTA | 126,048 |
| RDT-S-3 | S | pos_22029-6 | AGTTCA | 3,931 |
| RDT-S-4 | S | pos_22189-3 | TAT | 556 |
| RDT-S-5 | S | pos_22281-9 | CTTTACTTG | 887 |
| RDT-ORF3a | ORF3a | pos_26155-3 | GTT | 631 |
| RDT-ORF6 | ORF6 | pos_27205-3 | TTT | 1,014 |
| RDT-ORF8 | ORF8 | pos_28254-1 | A | 865 |
| RDT-ORF8/N | IGR8/n | pos_28271-1 | A | 127,020 |
| RDT-N | N | pos_28278-3 | CTG | 1,201 |
| RDT-ORF10 | ORF10 | pos_29582-6 | TTTCCG | 930 |
| RIT-ORF8-1 | ORF8 | pos_28252+2 | TG | 13,744 |
| RIT-ORF8-2 | ORF8 | pos_28255+2 | TC | 861 |
| RIT-ORF8/N | IGR8/n | pos_28263+4 | AACA | 5,582 |
Note: IGR8/n, the intergenic region between ORF8 and N gene; athe number following the ‘-’ and ‘+’ signs indicates the number of deleted and inserted nucleotides, respectively.
To reveal epidemiological dynamics, we mapped the distributions of RDT or RIT through time and across different geographical locations (Fig. 3). For RDTs, the earliest occurrence (i.e. RDT-nsp1) appeared in the USA and Canada in January 2020, but its abundance level remained low (<0.5 per cent) since then (Fig. 3A). Between January and October 2020, other eleven earlier RDTs began to emerge, most of which had spread to multiple countries and continents. And among these, the RDT-nsp2-2 appeared in thirty-three countries, reaching 1.7 per cent of total sequences in March 2020. Nevertheless, these RDTs all disappeared or diminished significantly in numbers by the end of 2020. Indeed, they were gradually replaced by the RDT-nsp6, RDT-S-1, RDT-S-2 and RDT-ORF8/N, which emerged in Feb 2020, appearing in Netherlands and Portugal initially and later spreading to more than eighty countries to become the dominant (>10 per cent) types in the field (Fig. 3A, Fig. S1). As of 29 May 2021, all six continents contained these four variants, with the most abundant type, namely RDT-nsp6, reaching 22 per cent of total sequences.
Figure 3.
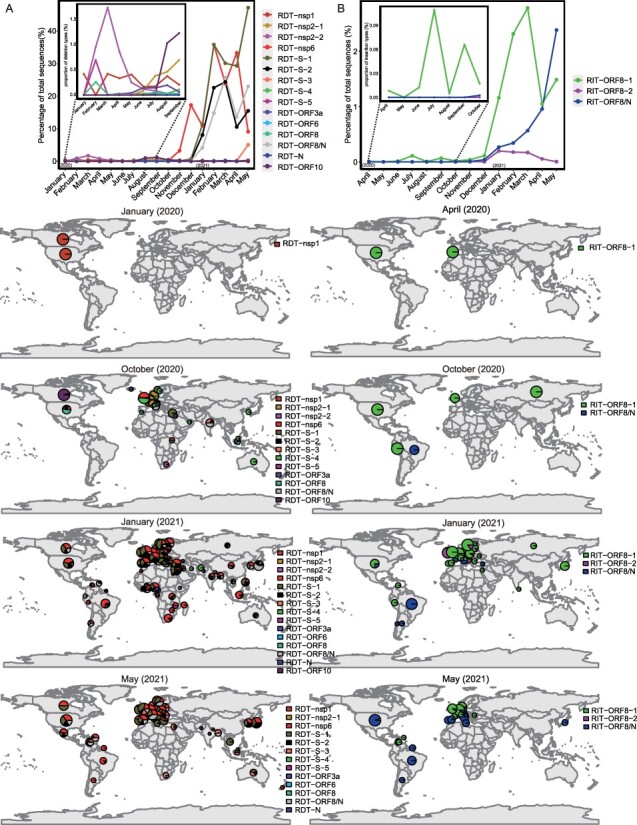
Temporal and spatial dynamics of the RDTs and RITs. The temporal (upper panel) and geographic (lower panel) distribution of RDTs (A) and RITs (B). For temporal distribution, the sequence counts of RDT and RIT are normalized against total sequences counts in each month. For geographic distributions, the size of the circle/pie chart is proportional to the log10(N + 1) transformation of the total sequence count and, therefore, reflects the size of sampling. For clarity, the geographic distributions are shown for January (2020), April (2020), October (2020), January (2021), and May (2021) months, whereas a more complete temporal and geographic change can be found in Figures S1 and S2 for RDT and RIT, respectively.
As for RITs, the earliest type, namely RIT-ORF8-1, appeared in April 2020 in Spain and USA and later spread to forty-six countries and six continents, with the highest prevalence recorded in Mar 2021 (Fig. 3B). Other two types, RIT-ORF8/N and RIT-ORF8-2, appeared in October 2020 and November 2020, respectively. In the field, RIT-ORF8-1 remained the most dominant insertion type until May 2021, when the proportion of RIT-ORF8/N (2.4 per cent) exceeded that of RIT-ORF8-1 (1.5 per cent). As of 29 May 2021, RIT-ORF8/N had spread to thirty-three countries and five continents, and its population was still expanding (Fig. S2).
2.4. Phylogenetic analysis of SARS-CoV-2 genomes based on indel mutations
We further analyzed the evolutionary history of RDTs and RITs by mapping them onto a phylogenetic tree that described the major circulating lineages (Fig. 4). Generally, there were strong associations between the circulating lineages defined by nucleotide substitutions and recurrent indel variants (Fig. 4). For example, eight RDTs and two RITs were associated with B.1.1.7 (Alpha variant) (Fig. 4), which was also labeled as a VOC by WHO. The other VOCs contained one or two RDTs or RITs, among which RDT-S-1, RDT-S-2 were identified in B.1.1.7 (Alpha variant), RDT-S-3 were identified in B.1.617.2 (Delta variant), and RDT-S-5 were identified in B.1.351 (Beta variant). Interestingly, majority of these RDTs or RITs identified in VOCs were associated with S proteins. On the other hand, among the five VOIs registered by WHO, only one (i.e. B.1.525 (Eta)) contained recurrent indel variants, which are RDT-ORF6 and RDT-N, located in ORF6 and N genes, respectively (Fig. 4). For the rest of lineages, we identified indel types in S, namely RDT-S-4, pos_22205+9, and pos_22206+9, within B.1.36, B.1.214.2, and A.2.5, respectively. Among these, pos_22205+9 and pos_22206+9 were not defined as RIT because their associated sequences, 305 and 348, respectively, were below the 500 threshold. Interestingly, more than half of the RDT and RIT defined in this study appeared with more than one occurrence, in multiple and paraphyletic lineages (Fig. 4, Table S3), suggesting multiple and independent occurrence of these indel types.
Figure 4.
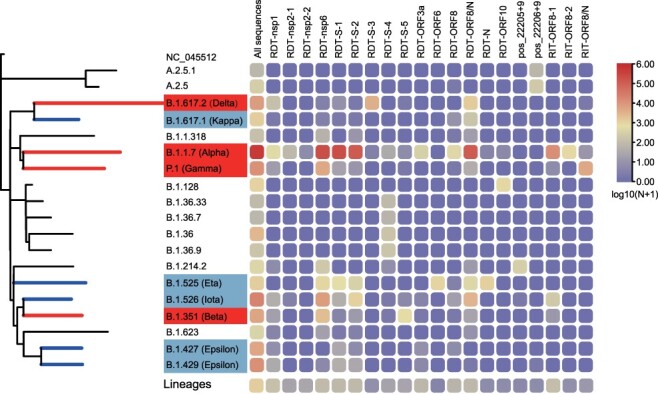
Distribution of RDT and RIT in Pango lineage. The phylogenetic tree is constructed using the nextstrain augur pipeline and based on a set of reference sequences downloaded from GISAID. For the definition of variants of concern (red) and variants of interest (CornflowerBlue), please refer to the official website of WHO (https://www.who.int/en/activities/tracking-SARS-CoV-2-variants/). The number of RDTs and RITs shown in the heatmap are transformed using (log(N + 1)).
2.5. Structural modeling of spike glycoprotein with recurrent indels
We next evaluated the impact of major types of indels on Spike protein structure and function. Specifically, sequence comparisons revealed that RDT-S-1, RDT-S-2, RDT-S-4, and RDT-S-5 caused the deletion of 69–70HV, 145Y, 210I, and 241–243LLA from S proteins, RDT-S-3 caused the replacement of EFR with G at 156–158aa, pos_22289-6 caused deletion at 242–243LA, and pos_22205+9 and pos_22206+9 caused insertions at 215 (TDR) and 215 (AAGY) (Fig. 5A). Interestingly, all major indel types identified here occurred at the NTD domain of S protein. The impact of these indels were subsequently evaluated by PROVEAN software, which suggested ins215TDR as ‘deleterious’ and resulted in decreased protein stability (score −2.999, cutoff = −2.5), whereas others are ‘Neutral’. Furthermore, structural modeling revealed changes in 3D structures after the introduction of indels, which were reflected in NTD loop region and β-pleated sheet (Fig. 5B1–B8). Specifically, the del210I caused the loss of 2.5 Å of hydrogen bonds between I210 and F186 (Fig. 5B4); the del241-243LLA caused the loss of 2.7 Å, 2.7 Å, and 2.8 Å between L241, A243, and G103, T240, and I100 (Fig. 5B5); the insertion of ins215TDR caused 215-TDR217 to form 3.1 Å, 2.8 Å, 3.3 Å, 3.5 Å, and 3.2 Å hydrogen bonds with Y269, T29, and Y91 (Fig. 5B7), and ins215AAGY causes Y218 to form 3.1 Å, 2.8 Å, and 3.0 Å of hydrogen bonds with Y91, T95, and A267 (Fig. 5B8). Importantly, these deletions and insertions of spike glycoprotein may impede the function of the loop region and the N-terminal and C-terminal ends of the β-pleated sheet.
Figure 5.
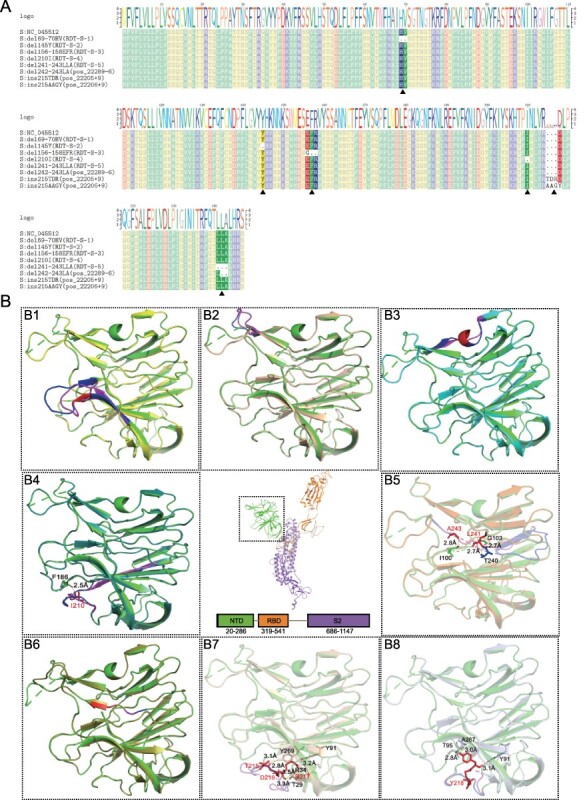
Structural analysis of spike glycoprotein with recurrent indels. (A) The multiple sequences alignments display of RDT-S-1, RDT-S-2, RDT-S-3, RDT-S-4, RDT-S-5, pos_22289-6, pos_22205+9, pos_22206+9. (B) Tertiary structure of the recurrent indels of spike glycoprotein. B1–B8 are different S protein NTD indels tertiary structure align with template. (B1–B8) del69-70HV (yellow), del145Y (wheat), del156-158EFR (cyan), del210I (forest), del241-243LLA (orange), del242-243LA (splitpea), ins215TDR (light wheat), ins215AAGY (silver) and align with template PDB: 7CWU (green). Deletion or insertion area (red), amino acid change region (pink), corresponding normal area by amino acid change region (blue).
3. Discussion
Our study examined 1,031,249 complete-genome sequences of SARS-CoV-2 collected from across the world and revealed a remarkable number of indels across the entire genome of the virus. Our result demonstrated that insertions and deletions, like nucleotide substitutions, are important driving forces that contribute to the diversity of SARS-CoV2 viruses, some of which have selective advantages such that they were later fixed and became dominant types in the field (Aleem, Akbar Samad, and Slenker 2021). For example, it has been demonstrated that the deletion RDT-S-1 in Alpha variant B.1.1.7 resulted in increased spike infectivity (Meng et al. 2021). The deletion RDT-S-2 in Alpha variant B.1.1.7 resulted in increased spike escape neutralization mediated by mAbs targeting the antigenic supersite (Zost et al. 2020; McCallum et al. 2021). On the other hand, the most dominant variant currently circulating in the field (>35.36 per cent of sequences as of 29 May 2021) with a significantly higher R0 (Liu and Rocklov 2021), namely Delta variant (B.1.617.2), contained a recurrent deletion type RDT-S-3 located at S protein and resulted in immune escape (McCallum et al. 2021). Collectively, the presence of more than one RDTs in three out of four major VOCs identified so far revealed that indels are highly relevant of the emergence of variant with altered biological or antigenic properties.
Interestingly, a number of indel types revealed in this study cause frameshift and disruption of the corresponding ORFs. These frameshift indels were mostly located in the genes encoding accessory proteins and much less frequently in both structural and non-structural genes (n < 20). A high occurrence rate is observed in indel types within ORF8. Previous studies have shown that ORF8 is subject to high substitution rate and less selective constraint (Pereira 2020; Tang et al. 2020). Therefore, ORF8 is an indel hotspot and most likely non-essential for the survival of SARS-CoV-2, although it has been suggested that the ORF8 product was probably involved in the regulation of host immune system (Zinzula 2021). Indeed, a deletion of as large as 382 nt in ORF8 has been reported, which resulted in not only the survival and dominance of the strain within patient but also the subsequent spread to Singapore (Young et al. 2020) and Taiwan, China (Gong et al. 2020). On the other hand, frameshift indels of structural and non-structural protein-coding genes were mostly deleterious. Their occurrences are probably due to accidental selection of a damaged or less viable genome as the template for PCR amplification. Alternatively, it might be simply due to sequencing error, which occurred frequently when it contains repeats of single nucleotide. For example, a deletion type (i.e. pos_11083-1) that causes the disruption of nsp6 protein was identified from 492 sequences from Denmark, the USA, Poland, and the UK. However, the position where deletion occurred follows an eight nucleotide poly(T) stretch, suggesting that it is more likely to be sequencing artifact than a naturally occurred and circulating deletion type.
Compared to other genes, the indels at the spike gene are of particular concern because many of them were RDTs or RITs that were associated with VOCs, which had significantly different antigenic properties or transmission dynamics compared to the prototype strains such that they replaced previous circulating strains to be the most dominant variants in the field (Torjesen 2021). Two mechanisms have been proposed for the selective advantage of indels within the S genes. First, it could result in significantly altered epitopes, which subsequently causes immune escape (Zost et al. 2020; McCallum et al. 2021). It has been demonstrated under experimental conditions that del60-75, del139-146, del210-212, and del242-248 S proteins, which were at the NTD epitopes for monoclonal antibodies, resulted in immune escape [22]. Interestingly, RDT-S-3 and RDT-S-5 are also located within the interaction zone of S1-targeting mAb 4A8 (Chi et al. 2020) and S2X333 (McCallum et al. 2021), suggesting their potential roles in immune escape. Another mechanism that renders selective advantage of the virus is that the indels within S protein might cause increase in infectivity. One study that focused on the H69/V70 deletion of the Alpha variant revealed that it increases S1/S2 cleavage and results in higher spike infectivity (Meng et al. 2021). Nevertheless, more data are required to demonstrate whether spike protein-associated RDTs and RITs identified in this study (i.e. RDT-S-2, RDT-S-3, RDT-S-4, RDT-S-5, pos_22205+9, and pos_22206+9) are relevant for spike infectivity.
There are several limitations in our investigation. Due to the fact that a large number of patients with COVID-19 disease have not been sequenced, the sequences included in our study did not fully reflect the SARS-CoV-2 diversity in countries and regions with less genomic sequencing. In addition, despite our effort to rule out indels that were resulted from sequencing artifacts, it is possible that some of the circulating type of indels are due to sequencing errors (i.e. the frameshift indels of ORF1ab), although the number of such occurrence is most likely very low.
4. Material and methods
4.1. Data collection and processing
As of 29 May 2021, a total of 1,099,664 high-quality SARS-CoV-2 genome sequences were downloaded from the GISAID website (Shu and McCauley 2017) before filtering low-quality sequences by the following options: (1) complete; (2) high coverage; (3) and low coverage excl. To do unbiased genomic variation analysis, we did further filtering and deleted those sequences with more than fifty consecutive N bases (50 Ns). Following the QC steps, 1,031,249 sequences were included in the study, which were sampled from 166 countries or regions, including the USA (n = 271,494), the UK (n = 244,460), Germany (n = 83,160), Denmark (n = 69,766), and Sweden (n = 39,418), amongst others (Table S4).
4.2. Genomic indels analysis
Genomic indels were defined based on the genome of prototype SARS-CoV-2 strain identified from Wuhan, namely Wuhan-Hu-1 (NC_045512.2) (Wu et al. 2020). Multiple sequences alignments were performed using the progressive method (FFT-NS-2) implemented in MAFFT (version 7.4) (Katoh et al. 2002). The whole-genome indels analysis was carried out using the pipeline implemented in the CoVa software (version 0.2) (Young et al. 2020). Indels that appeared in 5ʹ and 3ʹ UTRs were excluded from the analyses. Seqtk program (https://github.com/lh3/seqtk) was used to extract the genome sequences with indels and subjected to a second CoVa to remove false positives. These steps were repeated two or three times before the final manual inspection of the alignment involving major types of indels.
For each reliable indel identified, the naming follows the pattern ‘gene pos_genomic position ± length’ to indicate the gene and genomic position of occurrence, whether they are insertions or deletions, as well as how many bases are involved, which was exemplified by S pos_21765-6, ORF8 pos_28252+2.
4.3. Phylogenetic analyses
We used nextstrain augur tool (Hadfield et al. 2018) for phylogenetic analyses, which contained SARS-CoV-2 pango lineage reference strains in the GISAID database that described the major historical and current genomic variants defined by fixed nucleotide substitutions. We then map the indel information to these major lineages using pango nomenclature program (Rambaut et al. 2020). All of the modifications were implemented by the iTOL software (Letunic and Bork 2019).
4.4. Structural prediction and analysis
SARS-CoV-2 S proteins were aligned using mafft software and visualized with texshade software (Beitz 2000). The structural models for spike proteins with indels were constructed using the computer-guided homology modeling method implemented in SWISS-MODEL online server (Waterhouse et al. 2018) using Cryo-EM structure of SARS-CoV-2 spike proteins trimer (PDB ID: 7CWU) (Wang et al. 2021a), the prototype S protein, as the template. The similarity between all sequences and the template were greater than 99.45 per cent, GMQE (Global Model Quality Estimate) were greater than 0.67, and QMEANDisCo Global were 0.72 ± 0.05. The visualization of modeled structure were carried out by PyMOL (Schrodinger, LLC. (2015), The PyMOL Molecular Graphics System, Version 1.8.) in here or UCSF chimera software (Pettersen et al. 2004). Prediction of potential impact on biological function was carried out by PROVEAN (Protein Variation Effect Analyzer) (Choi and Chan 2015). To understand the implications of the amino acid indels in the mutants, we constructed the hydrogen bond changes by PyMOL software in the three-dimensional structure of the indels region.
Supplementary Material
Acknowledgement
We are grateful to the submitters from many laboratories for the sequences in the GISAID database.
Contributor Information
Xue Liu, Centre for Infection and Immunity Study (CIIS), School of Medicine (Shenzhen), Shenzhen Campus of Sun Yat-sen University, Sun Yat-sen University, Shenzhen, Guangdong 518107, China.
Liping Guo, Centre for Infection and Immunity Study (CIIS), School of Medicine (Shenzhen), Shenzhen Campus of Sun Yat-sen University, Sun Yat-sen University, Shenzhen, Guangdong 518107, China.
Tiefeng Xu, Centre for Infection and Immunity Study (CIIS), School of Medicine (Shenzhen), Shenzhen Campus of Sun Yat-sen University, Sun Yat-sen University, Shenzhen, Guangdong 518107, China.
Xiaoyu Lu, Centre for Infection and Immunity Study (CIIS), School of Medicine (Shenzhen), Shenzhen Campus of Sun Yat-sen University, Sun Yat-sen University, Shenzhen, Guangdong 518107, China.
Mingpeng Ma, Centre for Infection and Immunity Study (CIIS), School of Medicine (Shenzhen), Shenzhen Campus of Sun Yat-sen University, Sun Yat-sen University, Shenzhen, Guangdong 518107, China.
Wenyu Sheng, Centre for Infection and Immunity Study (CIIS), School of Medicine (Shenzhen), Shenzhen Campus of Sun Yat-sen University, Sun Yat-sen University, Shenzhen, Guangdong 518107, China.
Yinxia Wu, Centre for Infection and Immunity Study (CIIS), School of Medicine (Shenzhen), Shenzhen Campus of Sun Yat-sen University, Sun Yat-sen University, Shenzhen, Guangdong 518107, China.
Hong Peng, Centre for Infection and Immunity Study (CIIS), School of Medicine (Shenzhen), Shenzhen Campus of Sun Yat-sen University, Sun Yat-sen University, Shenzhen, Guangdong 518107, China.
Liu Cao, Centre for Infection and Immunity Study (CIIS), School of Medicine (Shenzhen), Shenzhen Campus of Sun Yat-sen University, Sun Yat-sen University, Shenzhen, Guangdong 518107, China.
Fuxiang Zheng, Centre for Infection and Immunity Study (CIIS), School of Medicine (Shenzhen), Shenzhen Campus of Sun Yat-sen University, Sun Yat-sen University, Shenzhen, Guangdong 518107, China.
Siyao Huang, Centre for Infection and Immunity Study (CIIS), School of Medicine (Shenzhen), Shenzhen Campus of Sun Yat-sen University, Sun Yat-sen University, Shenzhen, Guangdong 518107, China.
Zixiao Yang, Centre for Infection and Immunity Study (CIIS), School of Medicine (Shenzhen), Shenzhen Campus of Sun Yat-sen University, Sun Yat-sen University, Shenzhen, Guangdong 518107, China.
Jie Du, Centre for Infection and Immunity Study (CIIS), School of Medicine (Shenzhen), Shenzhen Campus of Sun Yat-sen University, Sun Yat-sen University, Shenzhen, Guangdong 518107, China.
Mang Shi, Centre for Infection and Immunity Study (CIIS), School of Medicine (Shenzhen), Shenzhen Campus of Sun Yat-sen University, Sun Yat-sen University, Shenzhen, Guangdong 518107, China.
Deyin Guo, Centre for Infection and Immunity Study (CIIS), School of Medicine (Shenzhen), Shenzhen Campus of Sun Yat-sen University, Sun Yat-sen University, Shenzhen, Guangdong 518107, China.
Supplementary data
Supplementary data is available at Virus Evolution online.
Funding
This study was supported by the National Natural Science Foundation of China (#32041002), Shenzhen Science and Technology Program (KQTD20180411143323605, JSGG20200225150431472 and KQTD20200820145822023), Guangdong Zhujiang Talents Program (#2016LJ06Y540), Guangdong Province ‘Pearl River Talent Plan’ Innovation and Entrepreneurship Team Project (2019ZT08Y464), and National Key Research and Development Program of China.
Conflict of interest
The authors have declared no competing interests.
References
- Aleem A., Akbar Samad A. B., and Slenker A. K. (2021) Emerging Variants of SARS-CoV-2 and Novel Therapeutics against Coronavirus (COVID-19). Treasure Island, FL: StatPearls Publishing. [PubMed] [Google Scholar]
- Bal A. et al. (2020) ‘Molecular Characterization of SARS-CoV-2 in the First COVID-19 Cluster in France Reveals an Amino Acid Deletion in Nsp2 (Asp268del)’, Clinical Microbiology and Infection, 26: 960–2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beitz E. (2000) ‘TeXshade: Shading and Labeling of Multiple Sequence Alignments Using LaTeX2e’, Bioinformatics, 16: 135–9. [DOI] [PubMed] [Google Scholar]
- Benedetti F. et al. (2020) ‘Emerging of a SARS-CoV-2 Viral Strain with a Deletion in nsp1’, Journal of Translational Medicine, 18: 329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chi X. et al. (2020) ‘A Neutralizing Human Antibody Binds to the N-terminal Domain of the Spike Protein of SARS-CoV-2’, Science, 369: 650–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi Y., and Chan A. P. (2015) ‘PROVEAN Web Server: A Tool to Predict the Functional Effect of Amino Acid Substitutions and Indels’, Bioinformatics, 31: 2745–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davies N. G. et al. (2021) ‘Estimated Transmissibility and Impact of SARS-CoV-2 Lineage B.1.1.7 In England’, Science, 372: eabg3055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dong E., Du H., and Gardner L. (2020) ‘An Interactive Web-Based Dashboard to Track COVID-19 in Real Time’, The Lancet Infectious Diseases, 20: 533–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gong Y.-N. et al. (2020) ‘SARS-CoV-2 Genomic Surveillance in Taiwan Revealed Novel ORF8-Deletion Mutant and Clade Possibly Associated with Infections in Middle East’, Emerging Microbes and Infections, 9: 1457–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gorbalenya A. E. et al. (2020) ‘The Species Severe Acute Respiratory Syndrome-Related Coronavirus: Classifying 2019-nCoV and Naming It SARS-CoV-2’, Nature Microbiology, 5: 536–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hadfield J. et al. (2018) ‘Nextstrain: Real-Time Tracking of Pathogen Evolution’, Bioinformatics, 34: 4121–3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holland L. A. et al. (2020) ‘An 81-Nucleotide Deletion in SARS-CoV-2 ORF7a Identified from Sentinel Surveillance in Arizona (January to March 2020)’, Journal of Virology, 94: e00711–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hou Y. J. et al. (2020) ‘SARS-CoV-2 D614G Variant Exhibits Efficient Replication Ex Vivo and Transmission in Vivo’, Science, 370: 1464–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katoh K. et al. (2002) ‘MAFFT: A Novel Method for Rapid Multiple Sequence Alignment Based on Fast Fourier Transform’, Nucleic AcidsResearch, 30: 3059–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khan A. et al. (2021) ‘Higher Infectivity of the SARS-CoV-2 New Variants Is Associated with K417N/T, E484K, and N501Y Mutants: An Insight from Structural Data’, Journal of Cellular Physiology, 236: 7045–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Letunic I., and Bork P. (2019) ‘Interactive Tree Of Life (iTOL) v4: Recent Updates and New Developments’, Nucleic Acids Research, 47: W256–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Y., and Rocklov J. (2021) ‘The Reproductive Number of the Delta Variant of SARS-CoV-2 Is Far Higher Compared to the Ancestral SARS-CoV-2 Virus’, Journal ofTravelMedicine, 28: taab124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Z. et al. (2020) ‘Identification of Common Deletions in the Spike Protein of Severe Acute Respiratory Syndrome Coronavirus 2’, Journal of Virology, 94: e00790–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCallum M. et al. (2021) ‘N-terminal Domain Antigenic Mapping Reveals a Site of Vulnerability for SARS-CoV-2’, Cell, 184: 2332–47 e16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meng B. et al. (2021) ‘Recurrent Emergence of SARS-CoV-2 Spike Deletion H69/V70 and Its Role in the Alpha Variant B.1.1.7’, Cellreports, 35: 109292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pereira F. (2020) ‘Evolutionary Dynamics of the SARS-CoV-2 ORF8 Accessory Gene’, Infection Genetics and Evolution, 85: 104525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pettersen E. F. et al. (2004) ‘UCSF Chimera? A Visualization System for Exploratory Research and Analysis’, Journal of Computational Chemistry, 25: 1605–12. [DOI] [PubMed] [Google Scholar]
- Plante J. A. et al. (2021) ‘Spike Mutation D614G Alters SARS-CoV-2 Fitness’, Nature, 592: 116–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quéromès G. et al. (2021) ‘Characterization of SARS-CoV-2 ORF6 Deletion Variants Detected in a Nosocomial Cluster during Routine Genomic Surveillance, Lyon, France’, Emerging Microbes and Infections, 10: 167–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rambaut A. et al. (2020) ‘A Dynamic Nomenclature Proposal for SARS-CoV-2 Lineages to Assist Genomic Epidemiology’, Nature Microbiology, 5: 1403–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen L. et al. (2021) ‘Rapidly Emerging SARS-CoV-2 B.1.1.7 Sub-lineage in the United States of America with Spike Protein D178H and Membrane Protein V70L Mutations’, Emerging Microbes and Infections, 10: 1293–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shu Y., and McCauley J. (2017) ‘GISAID: Global Initiative on Sharing All Influenza Data – From Vision to Reality’, Eurosurveillance, 22: 30494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Su Y. C. F. et al. (2020) ‘Discovery and Genomic Characterization of a 382-Nucleotide Deletion in ORF7b and ORF8 during the Early Evolution of SARS-CoV-2’, mBio, 11: e01610–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang X. L. et al. (2020) ‘On the Origin and Continuing Evolution of SARS-CoV-2’, National Science Review, 7: 1012–23. National Science Review 2020, 1012–23. doi: 10.1093/nsr/nwaa036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Torjesen I. (2021) ‘Covid-19: Delta Variant Is Now UK’s Most Dominant Strain and Spreading through Schools’, BMJ, 373: n1445. <https://www.bmj.com/content/373/bmj.n1445.long>. [DOI] [PubMed] [Google Scholar]
- Wang N. et al. (2021a) ‘Structure-based Development of Human Antibody Cocktails against SARS-CoV-2’, CellResearch, 31: 101–3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang P. et al. (2021b) ‘Antibody Resistance of SARS-CoV-2 Variants B.1.351 and B.1.1.7’, Nature, 593: 130–5. [DOI] [PubMed] [Google Scholar]
- Waterhouse A. et al. (2018) ‘SWISS-MODEL: Homology Modelling of Protein Structures and Complexes’, Nucleic Acids Research, 46: W296–303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu F. et al. (2020) ‘A New Coronavirus Associated with Human Respiratory Disease in China’, Nature, 579: 265–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Young B. E. et al. (2020) ‘Effects of a Major Deletion in the SARS-CoV-2 Genome on the Severity of Infection and the Inflammatory Response: An Observational Cohort Study’, The Lancet, 396: 603–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou P. et al. (2020) ‘A Pneumonia Outbreak Associated with a New Coronavirus of Probable Bat Origin’, Nature, 579: 270–3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zinzula L. (2021) ‘Lost in Deletion: The Enigmatic ORF8 Protein of SARS-CoV-2’, Biochemical and Biophysical Research Communications, 538: 116–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zost S. J. et al. (2020) ‘Potently Neutralizing and Protective Human Antibodies against SARS-CoV-2’, Nature, 584: 443–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.