

Abstract
Background:
Multi-cancer tests offer screening for multiple cancers with one blood draw, but the potential population impact is poorly understood.
Methods:
We formulate mathematical expressions for expected numbers of (1) individuals exposed to unnecessary confirmation tests (EUC), (2) cancers detected (CD), and (3) lives saved (LS) given test performance, disease incidence and mortality, and mortality reduction. We add colorectal, liver, lung, ovary, and pancreatic cancer to a test for breast cancer, approximating prevalence at ages 50, 60, or 70 using incidence over the next 5 years and mortality using corresponding probabilities of cancer death over 15 years in the Surveillance, Epidemiology, and End Results registry.
Results:
EUC is overwhelmingly determined by specificity. For a given specificity, EUC/CD is most favorable for higher-prevalence cancers. Under 99% specificity and sensitivities as published for a 50-cancer test, EUC/CD is 1.1 for breast+lung versus 1.3 for breast+liver at age 50. Under a common mortality reduction associated with screening, EUC/CD is most favorable when the test includes higher-mortality cancers (e.g., 19.9 for breast+lung versus 30.4 for breast+liver at age 50 assuming a common 10% mortality reduction).
Conclusions:
Published multi-cancer test performance suggests a favorable tradeoff of EUC to CD, yet the full burden of unnecessary confirmations will depend on the post-test work-up protocol. Harm-benefit tradeoffs will be improved if tests prioritize more prevalent and/or lethal cancers for which curative treatments exist.
Impact:
The population impact of multi-cancer testing will depend not only on test performance but also on disease characteristics and efficacy of early treatment.
Keywords: Early detection, incidence-based mortality, lives saved, multi-cancer, screening, unnecessary confirmation tests
Introduction
The advent of liquid biopsy technology has ushered in a new era of cancer diagnostics. Assays to detect circulating cell-free DNA supplemented by protein and methylation signatures promise to dramatically alter the landscape of cancer surveillance and early detection. In particular, the possibility of multi-cancer early detection, where a single blood sample is interrogated for multiple cancers, is attracting a great deal of attention (1,2).
Several multi-cancer tests are in development, each harnessing different features of the circulating tumor DNA. Liu et al present a test using targeted methylation analysis of circulating cell-free DNA that in principle detects and localizes more than 50 cancer types, and quantify its sensitivity for a pre-specified set of 12 cancers (3). Cohen et al present diagnostic performance of a test using circulating DNA and protein biomarkers (4); Lennon et al identify 10 cancer types using an updated version of the test (5). And Cristiano et al present a test that uses fragmentation patterns of cell-free DNA across the genome along with mutation-based cell-free DNA, estimating the diagnostic performance of this test across 7 cancer types (6).
Multi-cancer testing offers the potential for improved diagnosis of cancers where tests already exist, such as mammograms in women with dense breasts (7,8). It is less invasive than some existing tests, such as colonoscopies for colorectal cancer. Further, multi-cancer tests include cancers for which tests do not currently exist, either because the search for biomarkers with adequate performance has been unsuccessful or because disease prevalence is extremely low (9). Aggregating diagnosis of these cancers into a single test could increase the combined prevalence to an acceptable level for population screening. There may also be an economic advantage to doing a single test rather than a series of individual tests.
At this point however, many questions exist about the likely impact of multi-cancer testing on population benefits, such as cancer deaths prevented, and harms, such as unnecessary imaging tests or biopsies (1). These outcomes depend critically on disease prevalence at the time of the test, mortality in the absence of the test, and the ability to treat potentially fatal disease if detected early.
This study investigates how the potential benefits and harms of a single-occasion multi-cancer test depend on test and disease characteristics. We define a multi-cancer test as a test that screens for a specified set of target cancers. We first explore the simple setting of hypothetical two-cancer tests to establish a conceptual foundation for thinking about outcomes in the multi-cancer setting. We then examine more realistic tests involving up to six cancers with implications for more general multi-cancer tests. Our analysis creates a framework for quantifying the population impact of multi-cancer tests while pointing to criteria for the number and type of cancers to include.
Methods
Overview
Suppose that we have a test for a single cancer (cancer A) and our goal is to evaluate a test that includes two cancers (cancers A and B). We assume that the test produces an assessment of whether cancer is present and indicates a tissue of origin (TOO). We further assume that individuals do not have both cancers present concurrently at the time of the test. We present results for hypothetical two-cancer tests and show how they depend on test performance and cancer characteristics. We then present results for breast cancer (cancer A) and colorectal, liver, lung, ovary, or pancreatic cancer (cancer B), evaluating the measures of harm and benefit as we sequentially build up to six cancers based on published characteristics of an existing multi-cancer test (3). We select these cancers to include both common (breast, lung) and rare (ovary, liver) cancers as well as cancers with particularly poor prognosis if detected at advanced stage (ovary, pancreatic). For all multi-cancer tests, we produce results for single-occasion testing at ages 50, 60, or 70 to explore age dependence of the outcomes.
Test performance
The sensitivity of a multi-cancer test has two components: (1) the test sensitivity, which is the probability the test returns a cancer signature given that a targeted cancer is present, and (2) the probability of correct localization for each targeted cancer. For cancer A, the test sensitivity is the probability that the test returns a cancer signature when cancer A is present. Both the test sensitivity and the probability of correct localization may differ across cancers. We note that, while our discussion refers to sensitivity in general terms, it is the sensitivity to detect early-stage tumors that counts. Published multi-cancer studies have shown considerably poorer sensitivities for early compared with advanced-stage tumors (4).
We define the marginal sensitivity for a target cancer as the probability that the test returns a cancer signal and correctly localizes the target cancer when it is present. The marginal sensitivity of a multi-cancer test for cancer A can be written:
| (1) |
where PA(T+) is the sensitivity of the multi-cancer test, i.e., the probability the test returns a cancer signal given cancer A is present, and LA(T+) is the probability of correctly localizing the TOO as A given that the multi-cancer test returns a cancer signal (T+) and cancer A is in fact present. With k targeted cancers there are k marginal sensitivities. An early-stage version of the marginal sensitivity can be defined that specifies the test sensitivity as the probability the test returns a cancer signal if an early-stage cancer is present with the correct localization probability defined accordingly.
We define the specificity of a multi-cancer test as the probability the test returns a “no cancer” signature when none of the targeted cancers is present. If a cancer outside of the target set is detectable by the test and produces a cancer signature, this would be considered a false-positive result. This implies that the specificity of a multi-cancer test will vary with the specified set of target cancers.
Outcome metrics
In single-cancer testing, a key metric used to evaluate harm-benefit is the positive predictive value (PPV). In this setting, 1/PPV is often cited as the number of biopsies required per cancer detected, and 1/PPV−1 the number of unnecessary biopsies per cancer detected (assuming that all positive tests are followed by a biopsy). Thus, for example, a PPV of 1/3 corresponds to 2 unnecessary biopsies per cancer detected, and a PPV of 1/2 corresponds to 1 unnecessary biopsy per cancer detected. The PPV has proven to be a useful metric even though it depends on disease prevalence and therefore may not generalize from one population to another.
In single-cancer testing, each individual is subject to one biopsy at most, so the unnecessary biopsies arise solely from false positive tests, i.e., from individuals without cancer. In multi-cancer testing, however, unnecessary confirmation tests may occur in individuals who have one of the cancers included in the test, when the multi-cancer test correctly returns a cancer signal but incorrectly identifies the TOO. In this case, a confirmation test performed to verify that cancer is present in the putative TOO would constitute an unnecessary confirmation test. Thus, in multi-cancer testing, unnecessary confirmation tests arise from individuals without any of the targeted cancers as well as from those whose tumor is incorrectly localized.
For a two-cancer test, we can directly formulate the expected number of screened individuals potentially exposed to unnecessary confirmation (EUC) as:
| (2) |
where N is the number of individuals tested, ρA and ρB are the prevalence of cancers Aand B, respectively, and Sp is the specificity of the test. The first two terms in square brackets reflect the correct return of a cancer signal but incorrect localization, and the third term reflects a false positive. In practice, if the specificity is even modestly below 100% and the prevalence of each cancer is not high, the third term will dominate. We note that EUC reflects persons exposed to unnecessary confirmation tests and not the actual number of such tests performed.
The expected number of cancers detected depends on the marginal sensitivity of the multi-cancer test to detect each cancer and, in the setting of two cancers, is given by:
| (3) |
The formulation of CD reflects the expected number of people diagnosed with cancer following confirmation precipitated by the multi-cancer test.
We project the expected lives saved by assuming a disease-specific mortality reduction for each targeted cancer that is detected early and applying this value to the cumulative risk of disease-specific death without the test. The mortality reduction due to any screening test is a complex function of the interaction between screening test performance, screening protocol, and disease natural history. Rather than explicitly modeling these interactions for each cancer, we assume a value for the mortality reduction among cancers detected by the test. Specifically, we write the expected number of lives saved as:
| (4) |
where mA and mB are probabilities of cancer-specific death for cancers A and B, respectively, in the absence of the screening test, and RA and RB are the corresponding mortality reductions among those detected by the test. Implicit in this formula is the understanding that lives saved will be limited to cases that are prevalent at the time of the test. For a given mortality reduction factor, this formulation automatically increases the expected lives saved in proportion to the marginal sensitivity; higher marginal sensitivity will lead to more lives saved. We note that the mortality reduction factor need not be the same for all cancers, and for a given cancer it may depend on whether marginal sensitivity is specified for all cancers or for early-stage cancers.
General expressions for UC, CD, and LS for a k-cancer test are given in the Supplementary Methods and Materials (available online). In practice, all three outcomes depend on the protocol for confirmation testing, including how to proceed if confirmation testing of the putative TOO returns a negative result. In addition to the absolute outcomes, we also examine EUC/CD/ and EUC/LS as measures of harm-to-benefit tradeoffs.
Evaluation of hypothetical and more realistic multi-cancer tests
We first consider a two-cancer test that screens for (A) breast cancer and (B) colorectal, liver, lung, ovary, or pancreatic cancer among women in the United States. We base sensitivities and correct localization probabilities on estimates from Liu et al for cancers in stages I-III (3) and specificity (99%) on their estimate (99.3%). Similar values for specificity have been reported in other studies (4,6). Because a multi-cancer test for fewer cancers than the number considered in that study (n=50) may have a lower specificity, we also examine a lower specificity (97%) in a sensitivity analysis.
Age-specific prevalence of cancer is based on incidence rates observed in the Surveillance, Epidemiology, and End Results (SEER) registry over the years 2000–2002 inclusive (10). We assume that cancers diagnosed within the 5-year age groups 50–54, 60–64, and 70–74 years are prevalent at ages 50, 60, and 70 years, respectively, and thus could potentially be detected by a multi-cancer test offered at these ages. The prevalence at age 60, for example, is based on the number of diagnoses at ages 60–64 divided by the population at age 60, which we approximate using one-fifth of the population ages 60–64.
At any given age, screening cannot affect mortality among cancers diagnosed prior to that age. Therefore, we use SEER incidence-based mortality, which permits calculation of the probability of diagnosis at a specific age (e.g., age 50–54) and subsequent cancer-specific death over a specified time interval (11). We estimate 15-year incidence-based mortality and apply the assumed mortality reduction to cancers detected early to project expected lives saved. We restrict the time interval for mortality to 10 years in a sensitivity analysis.
For screening benefit, we consider the consensus benefit estimate of 20% mortality reduction across breast cancer screening trials (12) as an upper bound since we are modeling only single-occasion screening and the trials all evaluated serial screening. We denote the mortality reduction among women randomized to screening by . This is not the same as the mortality reduction among women whose breast cancers were detected early by screening (denoted RA above). If we assume that detectability of a latent cancer is independent of the mortality risk of that cancer in the absence of screening, then we can write:
| (5) |
For example, if the marginal sensitivity of a test to detect breast cancer (MSA) is 60%, then a 20% mortality reduction among women randomized to screening () corresponds to a 33% mortality reduction among women with cancers detected early by the test (RA). We adopt this formulation as a transparent device to link marginal sensitivity with deaths prevented by screening. In practice, latent tumors that are detected may be more advanced and have poorer baseline survival than those not detected, and thus this assumption may produce optimistic projections of lives saved.
Since we are modeling only single-occasion testing, we use a 10% mortality reduction associated with screening () for each cancer, but also examine how results change under mortality reductions of 5% and 20%. For MSA equal to 60%, for example, these values correspond toRA=16% , RA=8% (, and RA=33% . In a sensitivity analysis, we use for three high-prevalence cancers for which screening tests currently exist and for three low-prevalence cancers to explore how increasing the benefit for low-prevalence cancers impacts the relative ordering of harm-benefit measures across the six target cancers.
Online calculator
To provide direct access to the proposed framework, we created an online calculator. Users can configure a multi-cancer test with up to 50 cancers for any combination of sex, race (All races or Black), screening age (50, 60, or 70 years); input test specificity; and input test sensitivity, localization probability, and mortality reduction associated with early detection for each targeted cancer. The interface calculates the three outcomes formulated in this paper, and results can be downloaded for external analysis.
Results
A hypothetical two-cancer test
Figure 1 shows EUC and CD for a hypothetical two-cancer test under specified diagnostic characteristics and prevalence settings. The values used for prevalence are low in keeping with the estimates derived from the SEER registry for the cancers examined in the more realistic analyses. Projected values for a wider range of settings are shown in Supplementary Table 1 (available online).
Figure 1.
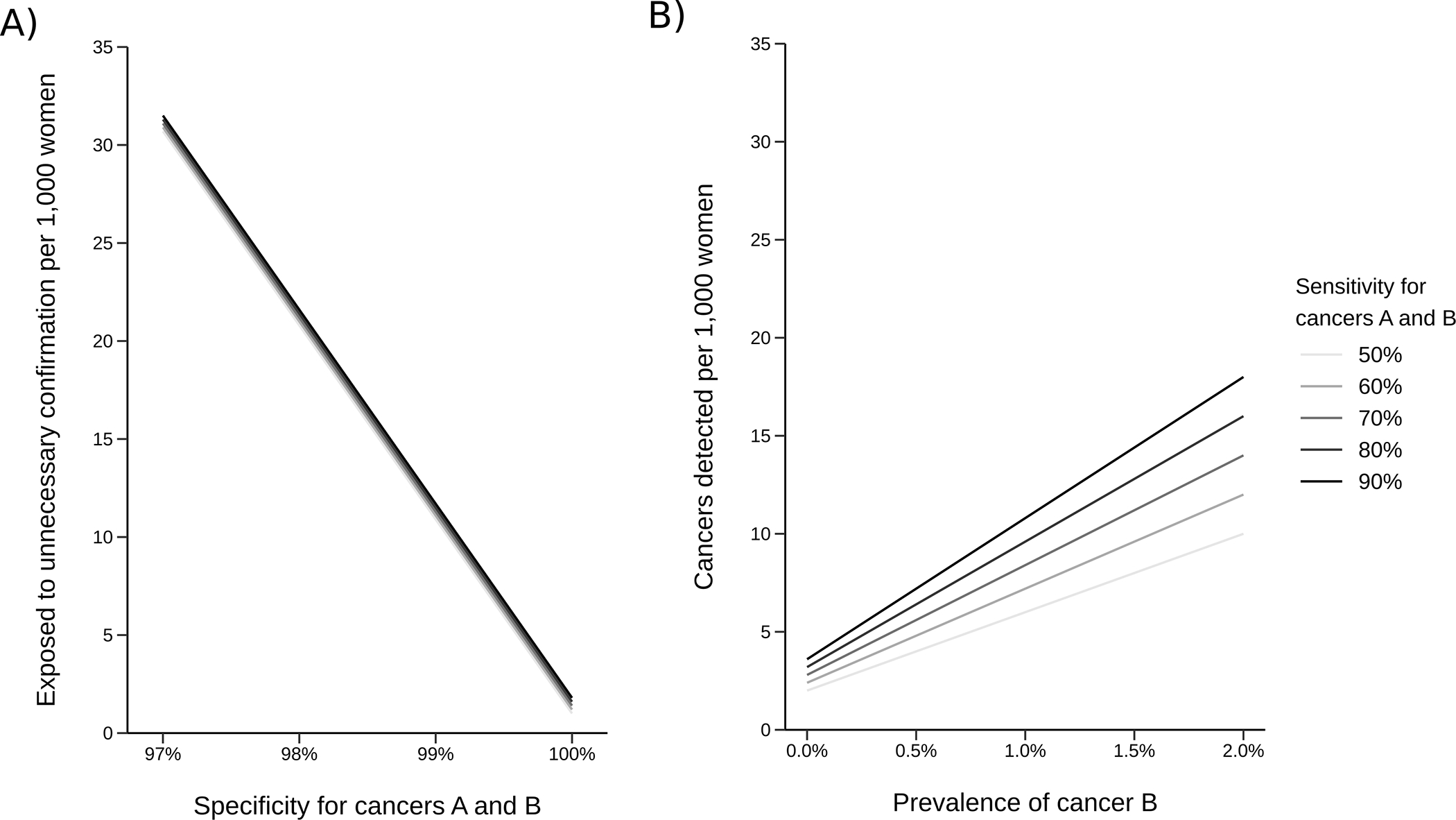
Hypothetical outcomes of a two-cancer test per 1,000 individuals: A) expected number of individuals potentially exposed to unnecessary confirmation tests given sensitivity and specificity for both cancers and B) expected cancers detected given sensitivity for both cancers and prevalence of cancer B. Both outcomes assume the prevalence of cancer A is 0.5% and the localization probability for both cancers is 80%.
Results show that EUC is driven primarily by the specificity of the test. Its dependence on sensitivity is minor; its dependence on prevalence and localization accuracy (Supplementary Figure 1) are also small. CD increases with the prevalence of cancer B and the sensitivity of the test, leading to lower EUC/CD for higher-prevalence cancers. While the results are intuitive, the message is that harm-benefit tradeoffs of multi-cancer testing depend on multiple factors pertaining to the cancers included in addition to the characteristics of the test.
A more realistic test for up to six cancers
Table 1 gives age-specific 5-year risks of cancer diagnosis, which we use to approximate prevalence for the cancers considered. As expected, breast and lung cancer have the highest prevalence while liver cancer has the lowest. All prevalence estimates are less than 2.5%, and they are less than 0.3% for liver, ovary, and pancreatic cancers. Table 2 summarizes sensitivities and localization probabilities from Liu et al (3) for these cancers. The highest marginal sensitivity is for colorectal cancer; the lowest is for liver cancer. This table also shows mortality reductions associated with early detection that correspond to 10% mortality reductions associated with screening for each target cancer.
Table 1.
Five-year net cumulative incidence and 15-year incidence-based mortality for selected cancers based on diagnoses in the Surveillance, Epidemiology, and End Results program in 2000–2002.
| Tissue of origin | Age, y | 5-year probability of diagnosis, % | 15-year probability of death, % |
|---|---|---|---|
| Breast | 50 | 1.24 | 0.32 |
| 60 | 1.89 | 0.66 | |
| 70 | 2.23 | 1.29 | |
| Colorectal | 50 | 0.23 | 0.10 |
| 60 | 0.52 | 0.26 | |
| 70 | 1.04 | 0.71 | |
| Liver | 50 | 0.02 | 0.01 |
| 60 | 0.05 | 0.04 | |
| 70 | 0.09 | 0.08 | |
| Lung | 50 | 0.25 | 0.20 |
| 60 | 0.83 | 0.71 | |
| 70 | 1.52 | 1.35 | |
| Ovary | 50 | 0.11 | 0.07 |
| 60 | 0.20 | 0.15 | |
| 70 | 0.25 | 0.21 | |
| Pancreas | 50 | 0.04 | 0.03 |
| 60 | 0.12 | 0.10 | |
| 70 | 0.26 | 0.24 |
Table 2.
Test sensitivity, localization probability, and implied marginal sensitivity of multi-cancer test for selected stage I-III cancers based on Liu et al. The implied mortality reduction among cases detected early corresponding to a 10% mortality reduction associated with single-occasion screening is also shown.
| Tissue of origin | Test sensitivity, % | Localization probability, % | Marginal sensitivity, % | Mortality reduction, % |
|---|---|---|---|---|
| Breast | 64 | 96 | 61 | 16 |
| Colorectal | 74 | 97 | 72 | 14 |
| Lung | 59 | 92 | 54 | 18 |
| Ovary | 67 | 96 | 64 | 16 |
| Pancreas | 78 | 79 | 62 | 16 |
| Liver | 68 | 72 | 49 | 20 |
The 5 two-cancer tests (breast cancer plus one of the other cancers) are associated with similar EUC (Supplementary Table 2), but they show clear patterns of higher EUC/CD when prevalence of the candidate second cancer is lower (Figure 2). As expected, EUC/LS is an order of magnitude greater than EUC/CD. Both harm-to-benefit measures improve with screening age and are most favorable when the second cancer is lung cancer, which has the highest prevalence and mortality (except at age 50 years when EUC/CD is most favorable for breast+colorectal cancer). These features lead to lung cancer having the highest CD and LS at ages 60 and 70 despite the marginal sensitivity for lung cancer being relatively low. Based on these metrics, the test for breast+lung cancer is optimal among the two-cancer tests considered at these ages. Similar patterns are observed under a more modest screening benefit; Figure 2 and Supplementary Table 2 also show how EUC/LS changes when the mortality reduction associated with screening is 5%, 10%, or 20%.
Figure 2.
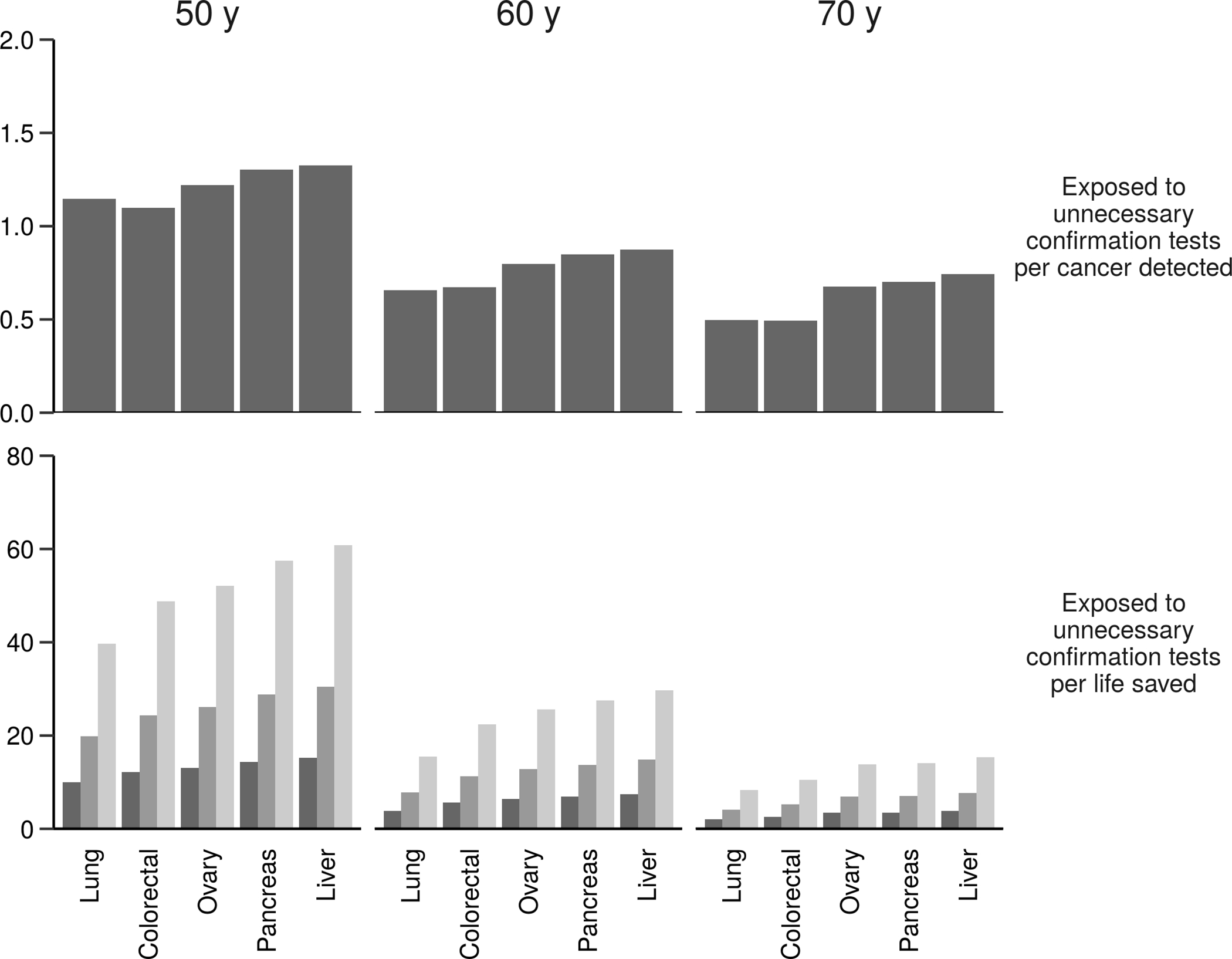
Expected number of women exposed to unnecessary confirmation tests per cancer detected (top row) and per life saved (bottom row) associated with adding specific cancers to an existing test for breast cancer at specific screening ages (columns) assuming specificity of the two-cancer test is 99%, 15-year incidence-based mortality from the Surveillance, Epidemiology, and End Results program, and the mortality reduction associated with screening for each cancer is 5% (light bars), 10% (medium bars), or 20% (dark bars).
Similar patterns are observed when adding a third cancer to a breast+lung cancer test (Figure 3 and Supplementary Table 3). Once again, EUC is similar across tests considered, and the optimal candidate has the highest prevalence and mortality, in this case colorectal cancer. Building up to a six-cancer test in this way yields the projected outcomes shown in Table 3. At all ages EUC/CD is below 1, indicating a favorable tradeoff of numbers exposed to unnecessary confirmation and cancers detected, likely due to the high values used for sensitivity and the relatively high prevalence of cases combined across cancers.
Figure 3.
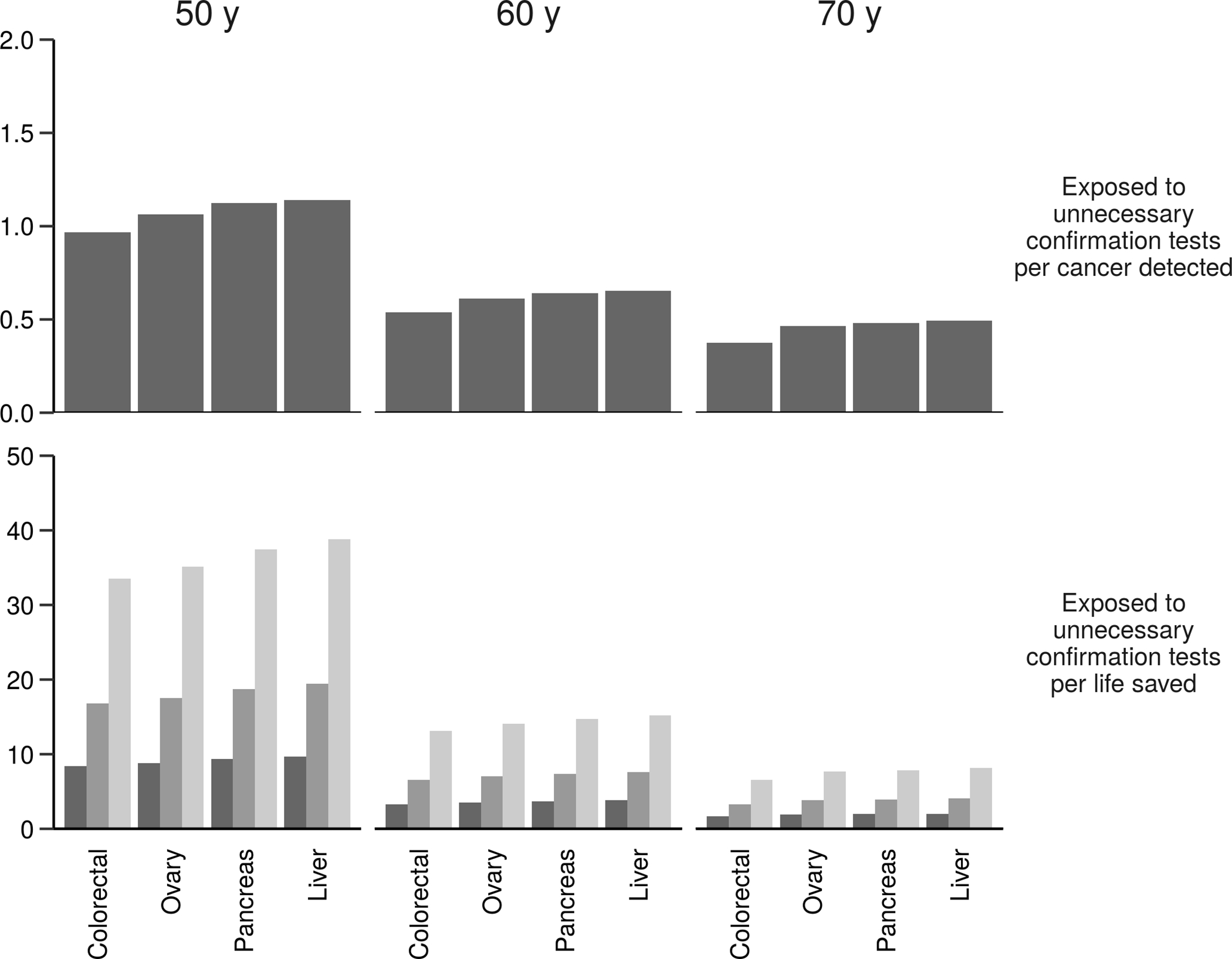
Expected number of women exposed to unnecessary confirmation tests per cancer detected (top row) and per life saved (bottom row) associated with adding specific cancers to an existing test for breast and lung cancers at specific screening ages (columns) assuming specificity of the three-cancer test is 99%, 15-year incidence-based mortality from the Surveillance, Epidemiology, and End Results program, and the mortality reduction associated with screening for each cancer is 5% (light bars), 10% (medium bars), or 20% (dark bars).
Table 3.
Expected number of women potentially exposed to unnecessary confirmation tests (EUC), expected cancers detected (CD), expected lives saved (LS), and harm-benefit tradeoffs EUC/CD and EUC/LS per 1,000 women tested with a multi-cancer test for breast, colorectal, liver, lung, pancreatic, and ovary cancers assuming 99% specificity and a common 10% mortality reduction associated with screening for each cancer.
| Screening age, y | EUC, n | CD, n | LS, n | EUC/CD | EUC/LS |
|---|---|---|---|---|---|
| 50 | 10.4 | 11.7 | 0.7 | 0.9 | 14.2 |
| 60 | 11.0 | 22.1 | 1.9 | 0.5 | 5.7 |
| 70 | 11.6 | 33.1 | 3.9 | 0.4 | 3.0 |
Lowering test specificity from 99% to 97% triples EUC/LS for any two-cancer test without altering the relative ordering of this tradeoff across candidate second cancers (Supplementary Figure 2). In contrast, improving the mortality reduction associated with screening for low-prevalence cancers from 10% to 50% alters the ordering of the two-cancer tests in terms of harm-benefit tradeoffs. Under these settings, the breast+ovary two-cancer test is associated with lower EUC/LS than the breast+lung cancer test for 50- and 60-year-old women (Supplementary Figure 3). Restricting attention from 15- to 10-year probabilities of cancer death (Supplementary Figure 4) does not alter the original patterns in the harm-benefit measures.
An online calculator that permits users to configure a multi-cancer test for a range of patient populations and test characteristics for selected target cancers is available at http://mced-calculator.fredhutch.org.
Discussion
Multi-cancer tests represent a major technological advance in cancer early detection, but their impact on clinically and policy-relevant outcomes remains to be determined. In this study we present a quantitative framework that formalizes how test performance and disease features combine to produce population harm and benefit. We use this framework to address the question of how to compose a multi-cancer test, building up from one to six cancers on the basis of harm-benefit tradeoffs. In practice, detectable signatures apply to a large number of cancers that will automatically be identifiable by the test, thus the question of how to specify a multi-cancer test may translate into whether to remove, rather than add, cancers. We note, however, that this is only a conceptual difference, as cancer signatures associated with less favorable harm-benefit tradeoffs for additional work-up can be excluded at the outset, and a bottom-up approach can be taken to configure a sustainable multi-cancer test.
A framework to clarify multi-cancer testing outcomes is necessitated by the novelty of the multi-cancer testing technology and the ways in which it alters the foundational metrics of diagnostic performance. In addition to multiple components of test sensitivity, we must also reconsider the standard definition of specificity in the setting of a multi-cancer test. In single-cancer testing, a truly negative test is taken to imply that the individual being tested does not have that cancer. In multi-cancer testing for a specified set of cancers, a truly negative test implies that the person being tested does not have one of the cancers in the target set. In practice, specificity may depend on the number of cancers in the test, with a test targeting a smaller set of cancers potentially having poorer specificity than a test targeting a larger set of cancers. However, including fewer cancers in the test may reduce the complexity of subsequent work-up and the actual number of confirmation tests performed. The number and type of cancers included must necessarily consider this tradeoff. At this point, the optimal protocol to confirm the result of a multi-cancer test is an open question.
Our analysis considers a limited set of outcome measures and approximates them based on information currently available plus a few transparent assumptions. Precise projection of any of these measures would require information about disease natural history and screening test performance in a prospective setting. This information would be also needed to explore other measures of benefit and harm, such as overdiagnosis, and to extrapolate from single-occasion to serial testing protocols.
The primary measure of harm in this study was unnecessary confirmation testing. In the absence of an established protocol for confirmation testing after a multi-cancer test, we only quantify persons exposed to unnecessary confirmation tests and not the number or cost of these tests, which may vary in terms of their invasiveness and accuracy. These features, as well as their implications for patient quality of life, should be included in a full accounting of the burden of unnecessary confirmation testing.
Our results are subject to limitations that stem mostly from the uncertainty about the natural history of different cancers and the need to use available and/or simplified inputs to fill out our harm-benefit framework. First, diagnostic performance estimates were sourced from a published multi-cancer testing study (3). These performance estimates were not prospective; they were derived from patients with and without a diagnosed cancer. In a prospective setting of a healthy population, we might expect lower sensitivity, particularly for early-stage tumors. Second, we approximated disease prevalence at the time of the test by the incidence within 5-year age groups and baseline mortality by the risk of cancer-specific death over the next 15 years among individuals diagnosed at those ages. We acknowledge that, particularly for some cancers known or suspected to have longer latencies, the underlying prevalence at the time of the test could be higher than that assumed. Conversely, for cancers with shorter latencies and poorer baseline survival, the 15-year interval for baseline mortality might be too long. Using a shorter interval for all cancers did not produce different decisions about which cancers to include, although as expected the associated harm-benefit tradeoff was less favorable. Ideally, we would want to project the prevalence of early-stage cancer at the time of the test, but this would require additional data and modeling.
We used published cancer screening trials to provide a benchmark for how disease-specific mortality might be reduced by multi-cancer early detection. These trials all included serial screening in the intervention group, while we model one-time testing. We use the observed mortality reduction as an upper bound for the mortality reduction associated with screening, which we convert to a mortality reduction among cases detected early by the test. This conversion is based on an assumption that detectability of a latent tumor is independent of its risk of disease-specific mortality. This assumption permits the lives saved to increase with the test sensitivity but will potentially produce an optimistic assessment of deaths prevented.
The extent to which multi-cancer tests might prevent disease-specific deaths is still highly uncertain and depends on how early they can reliably identify potentially fatal tumors as well as the efficacy of early treatment, which varies across cancers. The mortality benefit also will be affected by how the tests are used in practice, whether alongside or instead of existing tests, such as those for breast, colorectal, and lung cancer. By utilizing current estimates of prevalence and mortality for these cancers, we modeled multi-cancer tests used alongside existing tests. This motivated our sensitivity analysis, which assumes a less pronounced mortality benefit among currently-screened-for cancers than among non-screened-for cancers.
While our analysis is designed to address primary questions about the population impact of multi-cancer testing, it also raises many more. Beyond metrics for harm and benefit and how to reliably approximate them, questions remain about which cancers to include and how best to prioritize confirmation testing. A consensus about these matters will be needed before we can compare the different multi-cancer testing products currently under development. There are also important questions about the place of multi-cancer tests alongside established early detection modalities and how frequently the tests should be offered.
In conclusion, while multi-cancer testing may offer the possibility of screening for many cancers, its population impact depends on characteristics of both the cancers and the test as well as the potential benefit of early detection. The primary goal of this study has been to clarify in a quantitative framework the link between these features and clinically relevant population health outcomes. A key lesson from previous population screening tests is that the consequences of any early detection approach go far beyond test performance. Much more work is needed to determine harm-benefit tradeoffs of multi-cancer tests, as well as whether and how to deploy multi-cancer tests to reduce the population cancer burden.
Supplementary Material
Acknowledgements:
We acknowledge helpful comments on previous drafts from Drs. Noel S. Weiss and Scott D. Ramsey.
Role of the funder: The funding agencies had no role in the design of the study; the collection, analysis, or interpretation of the data; the writing of the manuscript; or the decision to submit the manuscript for publication.
Funding
This work was supported in part by the National Cancer Institute at the National Institutes of Health (grant number R50 CA221836 to R.G.) and the Rosalie and Harold Rea Brown Endowed Chair (R.E.).
Footnotes
Conflicts of interest:
Dr. Etzioni reported receiving personal fees from Grail outside the submitted work. Dr. Etzioni also holds shares in Seno Medical. The other authors declare no potential conflicts of interest.
Data Availability
Reproducible code used to generate all tables and figures in this article is available at Code Ocean https://codeocean.com/capsule/9054015/tree/v3
References
- 1.Beer TM. Novel blood-based early cancer detection: diagnostics in development. Am J Manag Care 2020;26(14 Suppl):S292–S9 doi 10.37765/ajmc.2020.88533. [DOI] [PubMed] [Google Scholar]
- 2.Srivastava S, Hanash S. Pan-Cancer Early Detection: Hype or Hope? Cancer Cell 2020;38(1):23–4 doi 10.1016/j.ccell.2020.05.021. [DOI] [PubMed] [Google Scholar]
- 3.Liu MC, Oxnard GR, Klein EA, Swanton C, Seiden MV, Consortium C. Sensitive and specific multi-cancer detection and localization using methylation signatures in cell-free DNA. Ann Oncol 2020;31(6):745–59 doi 10.1016/j.annonc.2020.02.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Cohen JD, Li L, Wang Y, Thoburn C, Afsari B, Danilova L, et al. Detection and localization of surgically resectable cancers with a multi-analyte blood test. Science 2018;359(6378):926–30 doi 10.1126/science.aar3247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Lennon AM, Buchanan AH, Kinde I, Warren A, Honushefsky A, Cohain AT, et al. Feasibility of blood testing combined with PET-CT to screen for cancer and guide intervention. Science 2020;369(6499) doi 10.1126/science.abb9601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Cristiano S, Leal A, Phallen J, Fiksel J, Adleff V, Bruhm DC, et al. Genome-wide cell-free DNA fragmentation in patients with cancer. Nature 2019;570(7761):385–9 doi 10.1038/s41586-019-1272-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Carney PA, Miglioretti DL, Yankaskas BC, Kerlikowske K, Rosenberg R, Rutter CM, et al. Individual and combined effects of age, breast density, and hormone replacement therapy use on the accuracy of screening mammography. Ann Intern Med 2003;138(3):168–75 doi 10.7326/0003-4819-138-3-200302040-00008. [DOI] [PubMed] [Google Scholar]
- 8.von Euler-Chelpin M, Lillholm M, Vejborg I, Nielsen M, Lynge E. Sensitivity of screening mammography by density and texture: a cohort study from a population-based screening program in Denmark. Breast Cancer Res 2019;21(1):111 doi 10.1186/s13058-019-1203-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Croswell JM, Ransohoff DF, Kramer BS. Principles of cancer screening: lessons from history and study design issues. Semin Oncol 2010;37(3):202–15 doi 10.1053/j.seminoncol.2010.05.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.National Cancer Institute. SEER*Stat Database: Incidence - SEER 18 Regs Research Data + Hurricane Katrina Impacted Louisiana Cases, Nov 2018 Sub (2000–2016) <Katrina/Rita Population Adjustment>. 2019.
- 11.National Cancer Institute. SEER*Stat Database: Incidence-Based Mortality - SEER 18 Regs (Excl Louisiana) Research Data, Nov 2018 Sub (2000–2016) <Katrina/Rita Population Adjustment>. 2019.
- 12.Independent U.K. Panel on Breast Cancer Screening. The benefits and harms of breast cancer screening: an independent review. Lancet 2012;380(9855):1778–86 doi 10.1016/S0140-6736(12)61611-0. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Reproducible code used to generate all tables and figures in this article is available at Code Ocean https://codeocean.com/capsule/9054015/tree/v3