

Abstract
Background:
Gut microbial alterations have been linked to chronic liver disease and hepatocellular carcinoma (HCC). The role of the oral microbiome in liver cancer development has not been widely investigated.
Methods:
Bacterial 16S rRNA sequences were evaluated in oral samples from 90 HCC cases and 90 controls who were a part of a larger U.S. case-control study of HCC among patients diagnosed from 2011-2016.
Results:
The oral microbiome of HCC cases showed significantly reduced alpha diversity compared to controls (Shannon p=0.002; Simpson p = 0.049), and beta diversity significantly differed (weighted Unifrac p=0.004). The relative abundance of 30 taxa significantly varied including Cyanobacteria, which was enriched in cases compared to controls (p=0.018). Cyanobacteria was positively associated with HCC (odds ratio 8.71, 95% CI 1.22 – 62.00, p = 0.031) after adjustment for age, race, birthplace, education, smoking, alcohol, obesity, type 2 diabetes, HCV, HBV, fatty liver disease, aspirin use, other NSAID use, laboratory batch, and other significant taxa. When stratified by HCC risk factors, significant associations of Cyanobacteria with HCC were exclusively observed among individuals with negative histories of established risk factors as well as females and college graduates. Cyanobacterial genes positively associated with HCC were specific to taxa producing microcystin, the hepatotoxic tumor promotor, and other genes known to be up-regulated with microcystin exposure.
Conclusions:
Our study provides novel evidence that oral Cyanobacteria may be an independent risk factor for HCC.
Impact:
These findings support future studies to further examine the causal relationship between oral cyanobacteria and HCC risk.
Keywords: hepatocellular carcinoma, Cyanobacteria, microbiome, cyanotoxins, microcystins
Introduction.
The incidence of liver cancer has been steadily increasing in the United States with rates nearly tripling over the past 3 decades.(1,2) Annually, nearly 42,000 U.S. residents are diagnosed with liver cancer, and over 80% of patients die of the disease within 5 years.(1) The rising incidence of hepatocellular carcinoma (HCC), the most common primary liver cancer, has been largely attributed to the growing prevalence of chronic hepatitis C (HCV) and, more recently, metabolic conditions including obesity, type 2 diabetes, and non-alcoholic fatty liver disease (NAFLD).(3) Nonetheless, the growing burden of HCC suggests the influence of unrecognized factors.
Gut microbial alterations have been linked to chronic liver disease and HCC and may involve a number of mechanisms including the production of DNA-damaging metabolites, activation of toll-like receptor signaling and tumor necrosis factor-α production, alteration of bile and choline availability, metabolism of ethanol to acetaldehyde, proinflammatory effects, and the promotion of insulin resistance.(4-6) There is evidence that gut microbial changes observed in liver disease may originate in the oral cavity through regular passage of bacteria from the mouth to the intestine.(4) Over half of gut bacteria enriched in cirrhosis patients are typically found in the oral cavity (4), suggesting mouth-to-gut microbial translocation. Reductions in oral commensal bacteria and increases in pathogenic species have been observed in patients with decompensated cirrhosis.(7) Passage of oral bacteria to distal organs may also be facilitated in periodontal disease, a major manifestation of oral dysbiosis, in which accumulated bacteria and bacterial metabolites can travel through the bloodstream to other sites.(8) Perturbations of the oral microbiome, including specific pathogenic bacteria, have been associated with cancers of the head and neck, esophagus, and pancreas.(9-11) Periodontal disease has been linked to chronic liver disease and liver cancer risk.(12-14) However, the role of the oral microbiome in liver cancer development has not been widely investigated. The objective of our study was to evaluate the relationship of the oral bacterial microbiome with HCC.
Materials and Methods.
The study population was comprised of 90 HCC cases and 90 controls with oral samples obtained from a larger population-based case-control study of 673 patients with HCC and 1,166 controls. Details of the study have been previously described.(15) Patients with HCC included males and females aged 35–84 years diagnosed from 2011-2016 identified through the National Cancer Institute Surveillance, Epidemiology, and End Results (SEER) cancer registries in Connecticut, operated by Yale Cancer Center, and in New Jersey, managed by the Rutgers Cancer Institute of New Jersey, and through the Liver Transplant Center at Columbia University Irving Medical Center. Control subjects were adult males and females without cancer selected from the general population through random digit dialing in Connecticut and New Jersey. Written informed consent was obtained from study subjects and the investigation was approved by the institutional review boards at each recruitment site. The study was conducted in accordance with the U.S. Common Rule.
Risk factor information was collected, including demographic data, anthropometry, cigarette smoking, alcohol use, medical history (viral hepatitis, type 2 diabetes, fatty liver disease, cirrhosis), and regular use of aspirin and other non-steroidal anti-inflammatory drugs (NSAIDs). A total of 501 cases and 989 controls provided self-collected saliva/oral fluid samples using the Saliva Self-Collection Kit (OG-250, DNA Genotek, Ottawa, Ontario, Canada) and the OraSure® oral specimen collection device (OraSure Technologies, Inc., Bethlehem, PA, USA) according to the manufacturers’ instructions. Total DNA was extracted from saliva/oral fluid samples using commercial reagents. These samples were used for HCV assessment and genomic analyses. Residual DNA samples were utilized for the present study. Ninety HCC cases were selected to include approximately equal representation by race/ethnicity including (non-Hispanic) Whites, Blacks, and Asians, and Hispanics. Ninety controls were selected with approximate frequency matching to cases by sex and race/ethnicity.
16S rRNA sequencing.
Oral DNA samples (5 ng/μl) were amplified in PCR reactions targeting the V3-V5 (16) region of the bacterial 16S ribosomal RNA (rRNA) gene. The initial PCR utilized custom primers ligated with overhang Illumina adapter sequences (Integrated DNA Technologies, Inc. Coralville, IA, USA). Amplicons were purified using Agencourt AMPure XP beads (Beckman Coulter, Brea, CA, USA). To allow for multiplexing, a second PCR was run to attach unique indices to each end of 16S amplicons using the Nextera XT Index Kit (Illumina, San Diego, CA, USA). Bar-coded amplicons were purified, quantified by Nanodrop 2000 (ThermoFisher, Waltham, MA, USA), normalized to equal concentrations, pooled, and checked on agarose gels for library sizes. Bar-coded libraries (50ul of 100 ng/μl) were assayed by 2 x 300 paired-end sequencing using the 600-cycle v3 kit on the MiSeq platform (Illumina) at the University of Hawaii.
Bioinformatic and statistical analyses.
Sequencing data were processed using QIIME2 v.2020.2.(17) Raw sequences were demultiplexed and trimmed of primers followed by denoising and removal of chimeras using DADA2.(18) Filtered sequences were clustered into amplicon sequence variants, and representative sequences were aligned to construct phylogenetic trees. Taxonomic classification was optimized by extracting reads from the Greengenes 13.8 reference database specific to V3-V5. The naive Bayes classifier was trained against this reference database and taxonomy assigned to representative sequences using the trained classifier. The microbial database of the National Center for Biotechnology Information (NCBI) (19) was utilized for additional sequence characterization.
SAS version 9.4 (SAS Institute, Carey, NC, USA) was used for statistical analyses of data. Tests with p<0.05 were considered to be significant. The oral microbiomes of HCC cases and controls were compared by diversity indices and taxa composition. Taxa composition was evaluated by relative abundance, which was estimated based on the number of 16S sequences assigned to a taxon as a proportion of the total sequences in a sample. Alpha diversity was measured by Shannon, Simpson, Chao1, Faith’s Phylogenetic Diversity Whole Tree, and Operational Taxonomic Unit indices. Beta diversity was evaluated by principal coordinate analysis and UniFrac distances with comparisons between groups measured by PERMANOVA.
Bacterial taxa relative abundance was evaluated as both categorical and continuous variables using the Chi-square, Wilcoxon Two-Sample and Kruskal–Wallis tests. Low/high levels of taxa were defined using cut-offs based on the mean relative abundance of controls. Univariate logistic regression with Firth bias correction (20) was used to model the association of individual taxa (low/high levels) with HCC status. Taxa significantly associated with HCC were each individually evaluated in multivariate models which included established liver cancer risk factors and other factors significantly differing between HCC cases and controls. Taxa remaining significant in multivariate analyses were jointly evaluated in a final multivariate model. (Cirrhosis was not included as a covariate in any models as it is an immediate clinical precursor to HCC. Household income, which was missing from more than 10% of the study population, was excluded from the analyses.)
The bacterial functional gene composition was inferred using PICRUSt2.(21) Metagenomic comparisons were made utilizing the Kruskal–Wallis tests and the Benjamini-Hochberg procedure to account for false discovery rate (FDR) due to multiple testing. Correlation of bacterial taxa and gene composition employed the Spearman correlation coefficient.
Results.
Compared to controls, HCC cases were older and comprised larger proportions of non-Whites, non-U.S. born individuals, and those with a high school education or lower (Table 1). HCV was the major risk factor among HCC cases with nearly 58% affected. In addition to HCV, other liver cancer risk factors more predominant in cases compared to controls included hepatitis B (HBV), cigarette smoking, type 2 diabetes, fatty liver disease, and cirrhosis. A history of regular aspirin and other NSAID use were less prevalent in cases compared to controls. Obesity (body mass index ≥30 kg/m2) and alcohol consumption did not significantly vary between cases and controls.
Table 1.
Characteristics of Hepatocellular Cases and Controls
| Cases (n=90) | Controls (n=90) | Cases (n=90) | Controls (n=90) | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| No. | % | No. | % | p value1 | No. | % | No. | % | p value1 | |||
| Sex | BMI | |||||||||||
| Male | 66 | 73.3 | 55 | 61.1 | 0.081 | <30 kg/m2 | 72 | 84.7 | 64 | 72.3 | 0.055 | |
| Female | 24 | 26.7 | 35 | 38.9 | ≥30 kg/m2 | 13 | 15.3 | 24 | 27.3 | |||
| Age group | Cigarette smoking | |||||||||||
| <60 | 31 | 34.4 | 45 | 51.7 | 0.02 | No | 27 | 30 | 55 | 62.5 | <0.0001 | |
| 60+ | 59 | 65.6 | 42 | 48.3 | Yes | 63 | 70 | 33 | 37.5 | |||
| Race/Ethnicity | Alcohol consumption | |||||||||||
| White | 20 | 22.2 | 41 | 45.6 | 0.003 | No | 28 | 31.1 | 38 | 43.2 | 0.096 | |
| Black | 20 | 22.2 | 21 | 23.3 | Yes | 62 | 68.9 | 50 | 56.8 | |||
| Hispanic | 20 | 22.2 | 13 | 14.4 | Type 2 diabetes | |||||||
| Other | 30 | 33.3 | 15 | 16.7 | No | 50 | 56.2 | 71 | 80.7 | 0.001 | ||
| Birthplace | Yes | 39 | 43.8 | 17 | 19.3 | |||||||
| U.S. | 53 | 58.9 | 64 | 75.3 | 0.021 | Fatty liver disease | ||||||
| Non-U.S. | 37 | 41.1 | 21 | 24.7 | No | 50 | 56.2 | 71 | 80.7 | 0.001 | ||
| Marital status | Yes | 39 | 43.8 | 17 | 19.3 | |||||||
| Single/never married | 15 | 17.2 | 9 | 10.3 | 0.29 | Cirrhosis | ||||||
| Married | 50 | 57.5 | 50 | 57.5 | No | 70 | 79.6 | 88 | 100 | <0.0001 | ||
| Separated/divorced | 17 | 19.5 | 17 | 19.5 | Yes | 18 | 20.4 | 0 | 0 | |||
| Widowed | 5 | 5.8 | 11 | 12.6 | Aspirin use | |||||||
| Education | No | 64 | 75.3 | 53 | 60.2 | 0.034 | ||||||
| High school or lower | 41 | 46.1 | 8 | 9.3 | <0.0001 | Yes | 21 | 24.7 | 35 | 39.8 | ||
| College | 37 | 41.6 | 48 | 55.8 | Other NSAID use | |||||||
| Graduate school | 11 | 12.4 | 30 | 34.9 | No | 80 | 94.1 | 72 | 81.8 | 0.013 | ||
| Viral hepatitis | Yes | 5 | 5.9 | 16 | 19.2 | |||||||
| Negative | 25 | 27.8 | 86 | 95.6 | <0.0001 | |||||||
| HBV | 13 | 14.4 | 0 | 0 | ||||||||
| HCV | 48 | 53.3 | 4 | 4.4 | ||||||||
| HBV and HCV | 4 | 4.4 | 0 | 0 | ||||||||
Chi Square test
Missing values: Age (3 controls); birthplace (5 controls); marital status (3 cases, 3 controls); education (1 case; 4 controls); BMI (5 cases, 2 controls; smoking (2 controls); alcohol (2 controls); type 2 diabetes (1 case, 2 controls); fatty liver disease (2 cases, 2 controls); cirrhosis (2 controls); aspirin (5 cases, 2 controls); other NSAIDs (5 cases, 2 controls)
In order to ascertain biases in the study population, their characteristics were compared to that of the overall case-control study population previously described.(15) The study population subset differed from the overall study population with respect to race/ethnicity, marital status, and birthplace. Compared to the overall case study population, the case subset included larger proportions of non-Whites (28% vs. 78%, p<0.0001), and those born outside of the U.S. (17% vs. 41%, p<0.0001). Relative to the overall control study population, the control subset included larger proportions of non-Whites (7% vs. 54%, p<0.0001), those born outside of the U.S. (8% vs. 25%, p<0.0001), and a smaller proportion of married individuals (67% vs. 58%, p= 0.03). Liver cancer risk factors and other medical history (including fatty liver, cirrhosis, aspirin, other NSAID) did not significantly differ in the subset of 90 cases and 90 controls compared to the overall study population. An exception was viral hepatitis status: Relative to the overall case study population, the case subset included larger proportions of individuals with HBV (3% vs. 14%, p<0.0001); compared to the overall control study population, the control subset included a larger proportion of individuals with a history of HCV (1% vs. 4%, p = 0.0035).
Oral DNA samples yielded a total of 32,972,048 demultiplexed bacterial sequences with an average of 184,201 sequences per sample. After quality filtering, 10,687 representative sequences were yielded with a mean length of 561 base pairs per sequence. Taxonomic assignment identified a total of 13 phyla, 122 genera, and 83 species in oral samples.
Compared to controls, HCC cases showed significantly reduced alpha diversity based on Shannon diversity (5.84 and 6.23, respectively; p = 0.002), Simpson diversity (0.96 and 0.97, respectively; p = 0.049) (Figure 1), and observed Operational Taxonomic Units (OTU) (175.68 and 213.58, respectively; p <0.0001). Beta diversity significantly differed between cases and controls (unweighted and weighted Unifrac p=0.01 and p=0.004) (Figure 2).
Figure 1.
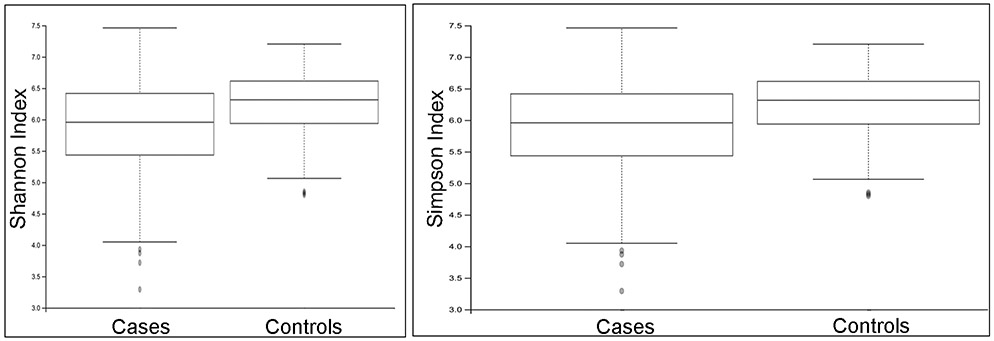
Oral microbiome showed significantly reduced alpha diversity in HCC cases compared to controls: a) Shannon diversity index 5.84 and 6.23, respectively, p = 0.002; b) Simpson diversity index 0.96 and 0.97, respectively, p = 0.049.
Figure 2.

Oral microbiome beta diversity in cases (red) and controls (blue) based on weighted Unifrac p=0.004
Thirty taxa were significantly enriched or reduced in HCC. At the phylum level, Cyanobacteria, was 2.3-fold higher in HCC cases and Tenericutes 0.74-fold lower compared to controls. Seven genera and 1 species were enriched (ranging from 1.06 – 35 -fold higher) in HCC cases relative to controls and 12 genera and 8 species were reduced (ranging from 0.27 – 0.93 -fold).
The 30 taxa differentially enriched or reduced in HCC were individually evaluated in multivariate models which included sex, age group (<60, ≥60 years), race/ethnicity (White, Black, Hispanic, Asian and Others), education (high school or lower, college or higher), birthplace (U.S., non-U.S.), HBV, HCV, alcohol, smoking, obesity, type 2 diabetes, fatty liver disease, aspirin use, other NSAID use, and laboratory batch. Five taxa remained significant in multivariate analyses including Cyanobacteria, Chryseobacterium, Neisseria elongata, Shuttleworthia satelles, Veillonella dispar. These 5 taxa were jointly evaluated in the final model. In the final model, only Cyanobacteria remained independently, positively associated with HCC (adjusted OR 8.71, 95% CI 1.22 – 62.00, p = 0.031) after adjustment for age, race, birthplace, education, smoking, alcohol, obesity, type 2 diabetes, HCV, HBV, fatty liver disease, aspirin use, other NSAID use, laboratory batch, and other significant taxa (Table 2 and Figure 3). HCV, type 2 diabetes, and a ≤high school education were also each independently, positively associated with HCC.
Table 2.
Association of Risk Factors with Hepatocellular Carcinoma
| Adjusted Odds ratio1 |
95% Confidence Interval |
P value | ||
|---|---|---|---|---|
| Cyanobacteria (high vs. low)2 | 8.71 | 1.22 | 62.00 | 0.031 |
| Chryseobacterium (high vs. low)2 | 17.27 | 0.36 | 838.81 | 0.151 |
| Neisseria elongata (high vs. low)2 | 0.19 | 0.02 | 1.69 | 0.136 |
| Shuttleworthia satelles (high vs. low)2 | 0.23 | 0.05 | 1.12 | 0.070 |
| Veillonella dispar (high vs. low)2 | 0.30 | 0.08 | 1.09 | 0.067 |
| Sex: Male (vs Female) | 1.77 | 0.41 | 7.68 | 0.447 |
| Age: ≥60 (vs. <60 years) | 2.28 | 0.62 | 8.41 | 0.216 |
| Education: high school or lower (vs. college or higher) | 8.66 | 1.75 | 42.77 | 0.008 |
| Race: Black (vs. White) | 5.27 | 0.07 | 397.84 | 0.637 |
| Hispanic (vs. White) | 3.27 | 0.37 | 28.88 | 0.939 |
| Other race (vs. White) | 0.23 | 0.003 | 19.76 | 0.748 |
| Birthplace: U.S. (vs. non-U.S.) | 0.32 | 0.05 | 2.29 | 0.256 |
| Fatty liver disease (yes vs. no) | 15.95 | 0.93 | 274.29 | 0.056 |
| Aspirin use (yes vs. no) | 0.52 | 0.12 | 2.25 | 0.384 |
| Non-steroidal anti-inflammatory drug use (yes vs. no) | 2.02 | 0.29 | 14.19 | 0.481 |
| Cigarette smoking (yes vs. no) | 2.85 | 0.70 | 11.66 | 0.146 |
| Excess alcohol consumption (yes vs. no) | 0.71 | 0.17 | 2.88 | 0.629 |
| Body mass index (≥30 kg/m2 vs. <30 kg/m2) | 0.31 | 0.07 | 1.39 | 0.124 |
| Type 2 diabetes (yes vs. no) | 5.18 | 1.21 | 22.28 | 0.027 |
| Hepatitis C (yes vs. no) | 55.14 | 10.78 | 282.07 | <.0001 |
| Hepatitis B (yes vs. no) | 8.43 | 0.54 | 131.22 | 0.128 |
| Laboratory batch | 0.24 | 0.00 | 17.78 | 0.514 |
Adjusted for all variables listed
Defined using cut-offs based on the mean relative abundance in controls
Figure 3.

Enrichment of Cyanobacteria in HCC cases (0.42%) compared to controls (0.19%), p=0.018. Mean relative abundance depicted in horizontal line.
The relationship of Cyanobacteria with HCC was further examined stratified by individual risk factors. The association of Cyanobacteria with HCC varied by diabetes, alcohol, obesity, HCV, HBV, fatty liver disease, aspirin use, and other NSAID use with significant associations observed for those with a negative history of each risk factor but not among those with a positive history (Figure 4). The relationships also varied by sex and education with significant associations limited to females and college graduates. The joint effects of Cyanobacteria and these risk factors were evaluated in separate models using interaction terms and none were significant with the exception of sex (p=0.035). In the full, multivariate model, the joint effect of Cyanobacteria and sex with HCC risk was not significant (p= 0.062).
Figure 4.

Association of Cyanobacteria with HCC risk by etiology: Significant associations observed for females, college graduates, and those with a negative history of HBV, HCV, type 2 diabetes, alcohol, obesity, fatty liver disease, aspirin use, and other NSAID use. (*No observation)
Interrogation of individual 16S sequences classified as Cyanobacteria in the NCBI database yielded 114 candidate species from 65 genera representing marine, freshwater, and terrestrial Cyanobacteria. Inferred metagenomic analyses identified bacterial genes correlated with Cyanobacteria (Table 3). Four of these bacterial genes were highly significantly correlated with Cyanobacteria (r = 0.99, p<0.0001), indicating that these genes were specifically from Cyanobacteria, and also significantly enriched in HCC cases (FDR p <0.05) all-trans-8'-apo-beta-carotenal 15,15'-oxygenase, coenzyme F420 hydrogenase, magnesium-protoporphyrin IX monomethyl ester (oxidative) cyclase, and plastoquinol--plastocyanin reductase. Nitric oxide dioxygenase was also significantly enriched in HCC cases (FDR p <0.05) and correlated with Cyanobacteria (r= 0.62, p <0.0001). Stearoyl-CoA 9-desaturase (SCD1), which did not differ between cases and controls, was correlated with Cyanobacteria (r=0.57, p <0.0001).
Table 3.
Bacterial Genes: Correlation with Cyanobacteria and Enrichment in HCC Cases
| Bacterial gene1 | Correlation with Cyanobacteria |
Enriched in HCC Cases (FDR p <0.0.05) |
|
|---|---|---|---|
| r 2 | p value | Cases | |
| All-trans-8'-apo-beta-carotenal 15,15'-oxygenase | 0.99 | <.0001 | X |
| Coenzyme F420 hydrogenase | 0.99 | <.0001 | X |
| Magnesium-protoporphyrin IX monomethyl ester (oxidative) cyclase | 0.99 | <.0001 | X |
| Plastoquinol--plastocyanin reductase | 0.99 | <.0001 | X |
| Nitric oxide dioxygenase | 0.62 | <.0001 | X |
| Stearoyl-CoA 9-desaturase | 0.57 | <.0001 | |
| S-(hydroxymethyl)glutathione dehydrogenase | 0.36 | <.0001 | |
| Catalase peroxidase | 0.34 | <.0001 | |
| 3-hydroxyisobutyrate dehydrogenase | 0.33 | <.0001 | |
| 4-hydroxyphenylpyruvate dioxygenase | 0.32 | <.0001 | |
| Sorbitol-6-phosphate 2-dehydrogenase | 0.30 | <.0001 | |
| 3-alpha(or 20-beta)-hydroxysteroid dehydrogenase | 0.30 | <.0001 | |
| Cholesterol oxidase | 0.29 | 0.0001 | |
| L-lysine N(6)-monooxygenase (NADPH) | 0.28 | 0.0001 | X |
| Pyrimidine monooxygenase | 0.27 | 0.0002 | X |
| Cyclohexanone monooxygenase | 0.26 | 0.0004 | X |
| Alkanesulfonate monooxygenase | 0.25 | 0.0006 | |
| Precorrin-3B synthase | 0.25 | 0.0007 | |
| 4-hydroxymandelate oxidase | 0.25 | 0.0009 | X |
| Protocatechuate 3,4-dioxygenase | 0.24 | 0.001 | X |
| Decaprenylphospho-beta-D-erythro-pentofuranosid-2-ulose 2-reductase | 0.24 | 0.0012 | |
| Gluconate 2-dehydrogenase (acceptor) | 0.24 | 0.0012 | |
| Alcohol dehydrogenase (azurin) | 0.24 | 0.0012 | |
| Decaprenylphospho-beta-D-ribofuranose 2-dehydrogenase | 0.23 | 0.0016 | |
| D-galactose 1-dehydrogenase | 0.23 | 0.0016 | X |
| Alcohol dehydrogenase | 0.23 | 0.0023 | |
| 5,6-dimethylbenzimidazole synthase | 0.23 | 0.0023 | |
| Quinoprotein glucose dehydrogenase (PQQ, quinone) | 0.23 | 0.0023 | X |
| 4-phosphoerythronate dehydrogenase | −0.23 | 0.0021 | |
90 bacterial genes were significantly correlated with Cyanobacteria; those with r <0.23 or < −0.23 are not shown.
Spearman correlation
To assess factors influencing microbial composition independent of liver cancer, controls were separately evaluated. Among controls, alpha diversity was higher in males than in females (Shannon diversity p = 0.019; Simpson diversity p= 0.01 Observed OTU p=0.006). Beta diversity (weighted Unifrac) varied between Whites and non-Whites (p=0.037) and by HCV status (p=0.018). Cyanobacteria relative abundance did not vary by HCC risk factors among controls.
Discussion.
Our study provides unique preliminary evidence linking HCC to oral dysbiosis, including reduced microbial richness and evenness and changes in the abundance of specific taxa. Most notably, we report for the first time that oral Cyanobacteria may play a role in the development of HCC. Cyanobacteria, which comprised less than 1% of the overall oral microbiome, was positively associated with HCC after controlling for established liver cancer risk factors and other taxa enriched or reduced in HCC. Interestingly, when further examined individually by key risk factors and other characteristics, the association of Cyanobacteria with HCC was observed exclusively among those with a negative history of diabetes, alcohol, obesity, HCV, HBV, and fatty liver disease, aspirin use, and other NSAID use as well as females and college graduates. Aspirin was previously found to be inversely associated with HCC in our larger study population (15) and in other studies.(22,23) Lower education, which was more prevalent in HCC cases in the larger study population, remained a significant risk factor for HCC in the present analyses. The incidence of HCC is 2-4 fold lower in females than males (1) and the basis of this disparity is not entirely understood. Among controls, oral bacterial diversity was significantly higher in males indicating underlying microbial differences between the sexes. Nonetheless, the relative abundance of Cyanobacteria did not vary by sex or by HCC risk factors when evaluated among controls, indicating that differences in Cyanobacteria levels alone do not account for the etiologic variation in the association of Cyanobacteria and HCC. The relationship of Cyanobacteria with HCC specifically in the absence of known risk factors underscores the possibility that oral Cyanobacteria may be an unrecognized independent risk factor for HCC. While the multifactorial etiology of HCC in the U.S. has been well characterized, there is evidence that a substantial proportion of patients diagnosed with HCC have no underlying risk factors. In a cohort of over 11,000 HCC cases diagnosed in the U.S. in 2000-2014, 24% had no known etiology.(24)
Cyanobacteria are photosynthetic, gram-negative bacteria that occur in all terrestrial and aquatic ecosystems.(25,26) Cyanobacteria include over 2,600 species which produce a wide range of secondary metabolites, including toxins targeting the liver, nervous system, skin, and gastrointestinal tract. (25,26) Three cyanotoxins have recognized or putative tumor-promoting properties in the liver including microcystin, nodularin, and cylindrospermopsin.(25) The candidate Cyanobacteria species identified in oral samples included known producers of all three of these cyanotoxins including of the genera Anabaena, Anabaenopsis, Aphanizomenon, Cylindrospermopsis, Dolichospermum, Fischerella, Lyngbya, Nodularia, Nostoc, Oscillatoria, Planktothrix, Raphidiopsis, and Synechococcus.(25,27,28)
Microcystins, the most common and well-characterized of the cyanotoxins, are cyclic peptides produced by multiple species and include more than 80 variants.(26) Microcystins are classified as group 2B carcinogens, or possibly carcinogenic to humans.(26) Microcystins function as tumor promotors, primarily through inhibition of protein phosphatases 1 and 2A, resulting in excessive phosphorylation of intermediate filaments and microfilaments and damage to the cytoskeleton of hepatocytes.(29)
Importantly, our findings specifically implicate microcystins in the association of Cyanobacteria with HCC risk. Two of the bacterial genes highly correlated with Cyanobacteria, all-trans-8'-apo-beta-carotenal 15,15'-oxygenase, which is involved in retinal biosynthesis (30), and plastoquinol-plastocyanin reductase, a key enzyme in photosynthesis, were also significantly enriched in HCC. These genes are specifically found in microcystin-producing genera, including Nostoc and Synechococcus (28,31), which were among the candidate Cyanobacteria identified in oral samples.
Cyanobacteria are particularly abundant in nutrient-rich bodies of water where they can form blooms, which increases the risk of human exposure.(25) Blooms containing toxin-producing Cyanobacteria are found in fresh, brackish, and marine waters globally and are increasing in the U.S. and worldwide due to changes in global temperatures combined with eutrophication of water bodies.(25) The evidence linking cyanotoxins to HCC in human populations have been largely limited to ecologic studies. Studies in China have observed increased risk of liver cancer in communities with microcystin-contaminated drinking water sources.(32,33) Using remote sensing, Cyanobacteria blooms were documented in water bodies in over 60% of U.S. counties and significant clusters of NAFLD death were linked to bloom-rich coastal areas.(34) Cyanobacteria blooms have also been linked to elevated liver cancer mortality in parts of Europe.(35) In one of the only studies to directly evaluate the association of cyanotoxins as measured in human samples with liver cancer risk, a hospital-based case-control study in China observed serum microcystin to be an independent risk factor for HCC.(36)
We can only speculate on the source of oral Cyanobacteria in our study population comprised of residents of three Northeast U.S. coastal states. Human exposure to cyanobacteria toxins primarily occurs through oral ingestion of contaminated drinking water and, to a lesser extent, dermal contact and inhalation of aerosolized toxin during recreational and other water activities.(26,37) The possibility of drinking water as a source of Cyanobacteria hepatotoxins in our study population cannot be ruled out. Cyanobacteria and cyanotoxins are not regulated in the U.S. although a few states have developed guidelines for microcystins in drinking water.(37) The World Health Organization (WHO) has recommended an upper limit of 1 ng/ml for microcystins in drinking water designated for human use.(38) Cyanotoxins have been detected in U.S. drinking water sources. In a survey of 33 U.S. lake and reservoir water sources, microcystins were detected in the majority of raw water samples, albeit nearly all at levels under the WHO limit. Cyanobacteria toxins can also affect irrigation sources, resulting in contamination of soil and agricultural products.(39) Exposure may also occur through contaminated seafood including fish and crustaceans.(28,37)
The ability of oral bacteria and their metabolites to access the liver is consistent with evidence that oral bacteria can be translocated to the gut and other distal organs; moreover, this process may be exacerbated in individuals with advanced liver disease as well as those with periodontal disease.(4,7,8) The premise that oral Cyanobacteria can lead to exposure of the liver to cyanotoxins is supported by our recent work showing that oral exposure to environmental sources of Cyanobacteria can result in both local and systemic levels of cyanotoxins.(40) In Guam, a U.S. Pacific Island territory, Cyanobacteria was found to be the predominant bacteria in locally-grown Areca catechu nuts and Piper betle leaves(40), plants which are used in betel nut chewing practiced throughout parts of Asia and the Pacific.(41,42) Oral Cyanobacteria relative abundance was 90-fold higher in current chewers compared to former/never chewers.(40) Importantly, we also detected microcystins/nodularins and cylindrospermopsin in both saliva and plant samples. In an independent sample, we detected these hepatotoxins at varying levels in saliva, sera, and urine of both chewers and non-chewers suggesting more widespread environmental exposure.(43)
Our study findings provide some evidence of indirect effects of microcystin on HCC development through the dysregulation of lipid metabolism. Aberrant lipid metabolism, including alterations in fatty acid synthesis, has been linked to the pathogenesis of HCC.(44) A number of animal studies have demonstrated that oral ingestion of microcystins, including chronic low-dose exposure, in the context of preexisting NAFLD, can exacerbate progression to the more severe form, non-alcoholic steatohepatitis.(45-48) We observed the bacterial gene, SCD1, a key enzyme in fatty acid metabolism, to be highly correlated with Cyanobacteria. In animal models, microcystin exposure influences the expression of SCD1 and other lipogenic genes leading to hepatic lipid accumulation.(49) SCD1 is overexpressed in a number of cancers including HCC.(50) Nonetheless, SCD1 in oral bacteria did not significantly differ between cases and controls, underscoring the complexity of the interplay of bacterial and host genes.
Our findings are bolstered by the population-based design of the overall study, which covered three U.S. metropolitan areas. Nonetheless, our purposeful sample selection to achieve comparable numbers by race likely resulted in a study subset not entirely representative of the larger study population including greater proportions of non-U.S-born individuals and those with a history of HBV— possibly reflecting birthplace in regions of the world with endemic HBV. Our study findings are also limited by potential reverse causality given the case-control study design with oral samples collected after cancer diagnosis. Presumably, exposure to tumor-promoting effects of cyanotoxins would have occurred in the years prior to liver cancer diagnosis. Ideally, our findings should be confirmed in larger prospective studies measuring local and systemic levels of cyanotoxins in prediagnostic biospecimens. No information was available on periodontal history, residential history, sources of drinking water, produce and seafood, or exposure to bodies of water. We were unable to definitively ascertain the oral Cyanobacteria species given the large number of candidate taxa.
Our study provides the first evidence that oral Cyanobacteria may be an independent risk factor for HCC, possibly through the direct tumor-promoting effects of microcystins and other hepatotoxins and their dysregulating influence on lipid metabolism. Our findings have important implications for current climate-related increases in Cyanobacteria overgrowth and its potential impact on liver cancer risk in the U.S. and worldwide. Future studies are necessary to further examine the causal relationship between oral cyanobacteria and HCC risk.
Acknowledgements.
H. Yu received grant R01CA138698. H. Yu, L. Mishra, Li, and K. Shetty received grant U01CA230690. A.M. Stroup received contract 75N91021D00009. The New Jersey Department of Health received cooperative agreement NU5U58DP006279-02-00. The authors acknowledge the Pacific Island Partnership for Cancer Health Equity (PIPCHE), which provided support for prior research which served as the basis for the methods and resources used in the current study. PIPCHE is supported by the National Cancer Institute of the National Institutes of Health under Award Numbers U54CA143727 and U54CA143728.
Financial Support:
This work was supported by funding from the National Cancer Institute (Grant Nos. R01CA138698 and U01CA230690); institutional funding provided to the University of Hawaii Cancer Center; the New Jersey State Cancer Registry supported by the National Program of Cancer Registries of the Centers for Disease Control and Prevention under cooperative agreement NU5U58DP006279-02-00 awarded to the New Jersey Department of Health, the Surveillance, Epidemiology, and End Results program of the National Cancer Institute under contract 75N91021D00009 awarded to the Rutgers Cancer Institute of New Jersey, and the State of New Jersey.
Footnotes
Data Sharing. The complete 16S sequencing dataset is available through the National Center for Biotechnology Information (biosample accession number SAMN18915539).
The authors declare no potential conflicts of interest.
References.
- 1.Howlader N NA, Krapcho M, Miller D, Brest A, Yu M, Ruhl J, Tatalovich Z, Mariotto A, Lewis DR, Chen HS, Feuer EJ, Cronin KA (eds). . SEER Cancer Statistics Review, 1975-2017, National Cancer Institute. Bethesda, MD, https://seer.cancer.gov/csr/1975_2017/, based on November 2019 SEER data submission, posted to the SEER web site, April 2020. [Google Scholar]
- 2.Njei B, Rotman Y, Ditah I, Lim JK. Emerging trends in hepatocellular carcinoma incidence and mortality. Hepatology 2015;61(1):191–9 doi 10.1002/hep.27388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.McGlynn KA, Petrick JL, El-Serag HB. Epidemiology of Hepatocellular Carcinoma. Hepatology 2021;73 Suppl 1:4–13 doi 10.1002/hep.31288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Qin N, Yang F, Li A, Prifti E, Chen Y, Shao L, et al. Alterations of the human gut microbiome in liver cirrhosis. Nature 2014;513(7516):59–64 doi 10.1038/nature13568. [DOI] [PubMed] [Google Scholar]
- 5.Boursier J, Diehl AM. Implication of gut microbiota in nonalcoholic fatty liver disease. PLoS Pathog 2015;11(1):e1004559 doi 10.1371/journal.ppat.1004559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Minemura M, Shimizu Y. Gut microbiota and liver diseases. World J Gastroenterol 2015;21(6):1691–702 doi 10.3748/wjg.v21.i6.1691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Bajaj JS, Betrapally NS, Hylemon PB, Heuman DM, Daita K, White MB, et al. Salivary microbiota reflects changes in gut microbiota in cirrhosis with hepatic encephalopathy. Hepatology 2015;62(4):1260–71 doi 10.1002/hep.27819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Li X, Kolltveit KM, Tronstad L, Olsen I. Systemic diseases caused by oral infection. Clin Microbiol Rev 2000;13(4):547–58 doi 10.1128/cmr.13.4.547-558.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Fan X, Alekseyenko AV, Wu J, Peters BA, Jacobs EJ, Gapstur SM, et al. Human oral microbiome and prospective risk for pancreatic cancer: a population-based nested case-control study. Gut 2018;67(1):120–7 doi 10.1136/gutjnl-2016-312580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Hayes RB, Ahn J, Fan X, Peters BA, Ma Y, Yang L, et al. Association of Oral Microbiome With Risk for Incident Head and Neck Squamous Cell Cancer. JAMA Oncol 2018;4(3):358–65 doi 10.1001/jamaoncol.2017.4777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Peters BA, Wu J, Pei Z, Yang L, Purdue MP, Freedman ND, et al. Oral Microbiome Composition Reflects Prospective Risk for Esophageal Cancers. Cancer Res 2017;77(23):6777–87 doi 10.1158/0008-5472.CAN-17-1296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Yang B, Petrick JL, Abnet CC, Graubard BI, Murphy G, Weinstein SJ, et al. Tooth loss and liver cancer incidence in a Finnish cohort. Cancer Causes Control 2017;28(8):899–904 doi 10.1007/s10552-017-0906-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Thistle JE, Yang B, Petrick JL, Fan JH, Qiao YL, Abnet CC, et al. Association of tooth loss with liver cancer incidence and chronic liver disease mortality in a rural Chinese population. PLoS One 2018;13(9):e0203926 doi 10.1371/journal.pone.0203926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Helenius-Hietala J, Suominen AL, Ruokonen H, Knuuttila M, Puukka P, Jula A, et al. Periodontitis is associated with incident chronic liver disease-A population-based cohort study. Liver Int 2019;39(3):583–91 doi 10.1111/liv.13985. [DOI] [PubMed] [Google Scholar]
- 15.Shen Y, Risch H, Lu L, Ma X, Irwin ML, Lim JK, et al. Risk factors for hepatocellular carcinoma (HCC) in the northeast of the United States: results of a case-control study. Cancer Causes Control 2020;31(4):321–32 doi 10.1007/s10552-020-01277-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Baker GC, Smith JJ, Cowan DA. Review and re-analysis of domain-specific 16S primers. J Microbiol Methods 2003;55(3):541–55 doi 10.1016/j.mimet.2003.08.009. [DOI] [PubMed] [Google Scholar]
- 17.Bolyen E, Rideout JR, Dillon MR, Bokulich NA, Abnet CC, Al-Ghalith GA, et al. Reproducible, interactive, scalable and extensible microbiome data science using QIIME 2. Nat Biotechnol 2019;37(8):852–7 doi 10.1038/s41587-019-0209-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Callahan BJ, McMurdie PJ, Rosen MJ, Han AW, Johnson AJ, Holmes SP. DADA2: High-resolution sample inference from Illumina amplicon data. Nat Methods 2016;13(7):581–3 doi 10.1038/nmeth.3869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Federhen S The NCBI Taxonomy database. Nucleic Acids Res 2012;40(Database issue):D136–43 doi 10.1093/nar/gkr1178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Heinze G, Schemper M. A solution to the problem of separation in logistic regression. Stat Med 2002;21(16):2409–19 doi 10.1002/sim.1047. [DOI] [PubMed] [Google Scholar]
- 21.Douglas GM, Maffei VJ, Zaneveld JR, Yurgel SN, Brown JR, Taylor CM, et al. PICRUSt2 for prediction of metagenome functions. Nat Biotechnol 2020;38(6):685–8 doi 10.1038/s41587-020-0548-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Petrick JL, Sahasrabuddhe VV, Chan AT, Alavanja MC, Beane-Freeman LE, Buring JE, et al. NSAID Use and Risk of Hepatocellular Carcinoma and Intrahepatic Cholangiocarcinoma: The Liver Cancer Pooling Project. Cancer Prev Res (Phila) 2015;8(12):1156–62 doi 10.1158/1940-6207.CAPR-15-0126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Sahasrabuddhe VV, Gunja MZ, Graubard BI, Trabert B, Schwartz LM, Park Y, et al. Nonsteroidal anti-inflammatory drug use, chronic liver disease, and hepatocellular carcinoma. J Natl Cancer Inst 2012;104(23):1808–14 doi 10.1093/jnci/djs452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Brar G, Greten TF, Graubard BI, McNeel TS, Petrick JL, McGlynn KA, et al. Hepatocellular Carcinoma Survival by Etiology: A SEER-Medicare Database Analysis. Hepatol Commun 2020;4(10):1541–51 doi 10.1002/hep4.1564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Buratti FM, Manganelli M, Vichi S, Stefanelli M, Scardala S, Testai E, et al. Cyanotoxins: producing organisms, occurrence, toxicity, mechanism of action and human health toxicological risk evaluation. Arch Toxicol 2017;91(3):1049–130 doi 10.1007/s00204-016-1913-6. [DOI] [PubMed] [Google Scholar]
- 26.International Agency for Research on Cancer Working Group. Ingested nitrate and nitrite, and cyanobacterial peptide toxins. IARC monographs on the evaluation of carcinogenic risks to humans. Volume 94. Lyon, France: International Agency for Research on Cancer; 2010. [PMC free article] [PubMed] [Google Scholar]
- 27.Cires S, Alvarez-Roa C, Wood SA, Puddick J, Loza V, Heimann K. First report of microcystin-producing Fischerella sp. (Stigonematales, Cyanobacteria) in tropical Australia. Toxicon 2014;88:62–6 doi 10.1016/j.toxicon.2014.06.010. [DOI] [PubMed] [Google Scholar]
- 28.Jakubowska N, Szelag-Wasielewska E. Toxic picoplanktonic cyanobacteria--review. Mar Drugs 2015;13(3):1497–518 doi 10.3390/md13031497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Fujiki H, Suganuma M. Tumor promoters--microcystin-LR, nodularin and TNF-alpha and human cancer development. Anticancer Agents Med Chem 2011;11(1):4–18 doi 10.2174/187152011794941163. [DOI] [PubMed] [Google Scholar]
- 30.Kloer DP, Ruch S, Al-Babili S, Beyer P, Schulz GE. The structure of a retinal-forming carotenoid oxygenase. Science 2005;308(5719):267–9 doi 10.1126/science.1108965. [DOI] [PubMed] [Google Scholar]
- 31.Ahrazem O, Gomez-Gomez L, Rodrigo MJ, Avalos J, Limon MC. Carotenoid Cleavage Oxygenases from Microbes and Photosynthetic Organisms: Features and Functions. Int J Mol Sci 2016;17(11) doi 10.3390/ijms17111781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Yu SZ. Primary prevention of hepatocellular carcinoma. J Gastroenterol Hepatol 1995;10(6):674–82 doi 10.1111/j.1440-1746.1995.tb01370.x. [DOI] [PubMed] [Google Scholar]
- 33.Yu S, Zhao N, Zi X. [The relationship between cyanotoxin (microcystin, MC) in pond-ditch water and primary liver cancer in China]. Zhonghua Zhong Liu Za Zhi 2001;23(2):96–9. [PubMed] [Google Scholar]
- 34.Zhang F, Lee J, Liang S, Shum CK. Cyanobacteria blooms and non-alcoholic liver disease: evidence from a county level ecological study in the United States. Environ Health 2015;14:41 doi 10.1186/s12940-015-0026-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Svircev Z, Drobac D, Tokodi N, Vidovic M, Simeunovic J, Miladinov-Mikov M, et al. Epidemiology of primary liver cancer in Serbia and possible connection with cyanobacterial blooms. J Environ Sci Health C Environ Carcinog Ecotoxicol Rev 2013;31(3):181–200 doi 10.1080/10590501.2013.824187. [DOI] [PubMed] [Google Scholar]
- 36.Zheng C, Zeng H, Lin H, Wang J, Feng X, Qiu Z, et al. Serum microcystin levels positively linked with risk of hepatocellular carcinoma: A case-control study in southwest China. Hepatology 2017;66(5):1519–28 doi 10.1002/hep.29310. [DOI] [PubMed] [Google Scholar]
- 37.United States Environmental Protection Agency. Drinking Water Health Advisory for the Cyanobacterial Microcystin Toxins. June 2015. [Google Scholar]
- 38.World Health Organization. Guidelines for drinking-water quality: fourth edition incorporating the first addendum WHO, Geneva, Switzerland, pp. 344–346. Guidelines for drinking-water quality: fourth edition incorporating the first addendum. Geneva, Switzerland: World Health Organization 2017. 344–6 p. [PubMed] [Google Scholar]
- 39.Drobac D, Tokodi N, Kiprovski B, Malencic D, Vazic T, Nybom S, et al. Microcystin accumulation and potential effects on antioxidant capacity of leaves and fruits of Capsicum annuum. J Toxicol Environ Health A 2017;80(3):145–54 doi 10.1080/15287394.2016.1259527. [DOI] [PubMed] [Google Scholar]
- 40.Hernandez BY, Zhu X, Sotto P, Paulino Y. Oral exposure to environmental cyanobacteria toxins: Implications for cancer risk. Environ Int 2021;148:106381 doi 10.1016/j.envint.2021.106381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Paulino YC, Novotny R, Miller MJ, Murphy SP. Areca (Betel) Nut Chewing Practices in Micronesian Populations. Hawaii J Public Health 2011;3(1):19–29. [PMC free article] [PubMed] [Google Scholar]
- 42.International Agency for Research on Cancer Working Group. A review of human carcinogens. Part E: Personal habits and indoor combustions. IARC monographs on the evaluation of carcinogenic risks to humans. Volume 100E. Lyon, France: International Agency for Research on Cancer; 2009. [Google Scholar]
- 43.Hernandez BY ZX, Sotto P, Paulino Y. Areca nut/betel quid chewing, oral cyanobacteria, and exposure to cyanotoxins. American Association for Cancer Research Conference on The Microbiome, Viruses, and Cancer. 2020; Orlando, Florida, U.S.A. [Google Scholar]
- 44.Sangineto M, Villani R, Cavallone F, Romano A, Loizzi D, Serviddio G. Lipid Metabolism in Development and Progression of Hepatocellular Carcinoma. Cancers (Basel) 2020;12(6) doi 10.3390/cancers12061419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.He J, Li G, Chen J, Lin J, Zeng C, Chen J, et al. Prolonged exposure to low-dose microcystin induces nonalcoholic steatohepatitis in mice: a systems toxicology study. Arch Toxicol 2017;91(1):465–80 doi 10.1007/s00204-016-1681-3. [DOI] [PubMed] [Google Scholar]
- 46.Lad A, Su RC, Breidenbach JD, Stemmer PM, Carruthers NJ, Sanchez NK, et al. Chronic Low Dose Oral Exposure to Microcystin-LR Exacerbates Hepatic Injury in a Murine Model of Non-Alcoholic Fatty Liver Disease. Toxins (Basel) 2019;11(9) doi 10.3390/toxins11090486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Clarke JD, Dzierlenga A, Arman T, Toth E, Li H, Lynch KD, et al. Nonalcoholic fatty liver disease alters microcystin-LR toxicokinetics and acute toxicity. Toxicon 2019;162:1–8 doi 10.1016/j.toxicon.2019.03.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Albadrani M, Seth RK, Sarkar S, Kimono D, Mondal A, Bose D, et al. Exogenous PP2A inhibitor exacerbates the progression of nonalcoholic fatty liver disease via NOX2-dependent activation of miR21. Am J Physiol Gastrointest Liver Physiol 2019;317(4):G408–G28 doi 10.1152/ajpgi.00061.2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Lei H, Song Y, Dong M, Chen G, Cao Z, Wu F, et al. Metabolomics safety assessments of microcystin exposure via drinking water in rats. Ecotoxicol Environ Saf 2021;212:111989 doi 10.1016/j.ecoenv.2021.111989. [DOI] [PubMed] [Google Scholar]
- 50.Pope ED 3rd, , Kimbrough EO, Vemireddy LP, Surapaneni PK, Copland JA 3rd, Mody K. Aberrant lipid metabolism as a therapeutic target in liver cancer. Expert Opin Ther Targets 2019;23(6):473–83 doi 10.1080/14728222.2019.1615883. [DOI] [PMC free article] [PubMed] [Google Scholar]