

Abstract
Background
Ovarian clear cell carcinoma (OCCC) is a rare ovarian cancer histotype that tends to be resistant to standard platinum-based chemotherapeutics. We sought to better understand the role of DNA methylation in clinical and biological subclassification of OCCC.
Methods
We interrogated genome-wide methylation using DNA from fresh frozen tumors from 271 cases, applied non-smooth non-negative matrix factorization (nsNMF) clustering, and evaluated clinical associations and biological pathways.
Results
Two approximately equally sized clusters that associated with several clinical features were identified. Compared to Cluster 2 (N=137), Cluster 1 cases (N=134) presented at a more advanced stage, were less likely to be of Asian ancestry, and tended to have poorer outcomes including macroscopic residual disease following primary debulking surgery (p-values <0.10). Subset analyses of targeted tumor sequencing and immunohistochemical data revealed that Cluster 1 tumors showed TP53 mutation and abnormal p53 expression, and Cluster 2 tumors showed aneuploidy and ARID1A/PIK3CA mutation (p-values <0.05). Cluster-defining CpGs included 1,388 CpGs residing within 200 bp of the transcription start sites of 977 genes; 38% of these genes (N=369 genes) were differentially expressed across cluster in transcriptomic subset analysis (p-values <10−4). Differentially expressed genes were enriched for six immune-related pathways, including interferon alpha and gamma responses (p-values < 10−6).
Conclusions
DNA methylation clusters in OCCC correlate with disease features and gene expression patterns among immune pathways.
Impact
This work serves as a foundation for integrative analyses that better understand the complex biology of OCCC in an effort to improve potential for development of targeted therapeutics.
Keywords: Clinical epidemiology, Biomarkers of disease, Epigenomics, Tumor Clustering, Gynecologic neoplasms
Introduction
Ovarian clear cell carcinoma (OCCC) remains an enigmatic histotype of epithelial ovarian cancer (EOC) [1]. When diagnosed at an advanced stage, it has a worse outcome than the more common high-grade serous histotype [2–4], and it tends to present at a younger age, showing a poorer response to platinum-based therapy, the mainstay treatment for EOC. As reviewed previously [5, 6], relatively small studies suggest that OCCC possesses some singularly unique features. Like endometrioid EOC, it can arise from endometriotic lesions; OCCC is generally TP53 wild-type with recurrent somatic mutations in PIK3CA and ARID1A and with a relatively low frequency of structural rearrangements [7–10]. Although we and others have shown that tumor DNA methylation profiles differ between OCCC and other histotypes [10–12], methylation profiles among OCCC tumors have not been comprehensively evaluated.
OCCC with ARID1A mutations display dysregulation of chromatin remodeling [9] and frequently overexpress HNF1B via hypomethylation which has been reported to be associated with a methylated phenotype [12]. Mismatch repair deficiency, resulting from DNA mismatch repair gene mutation or hypermethylation has also been reported in OCCC, albeit at a relatively low frequency and predominantly in older patients [13]. In addition to the paucity of studies on methylation and OCCC, there have been few gene expression studies. Upregulation of genes in the IL6-STAT3-HIF and glycogen pathways suggest a response to persistent oxidative stress and inflammation [14, 15]. Tan et al. [16] described two groups of OCCC: a mesenchymal-like subtype, with increased proliferation, tumor-infiltrating lymphocytes (TILs), and poorer outcome, and an epithelial-like tumor subtype which presented earlier in stage and with mutations in SWI/SNF genes. The tumor microenvironment may also contribute to an immune-suppressive state, suggesting a role for immunotherapeutics such as checkpoint inhibitors [17]. PD-L1 expression is common in OCCC (~45%) and is more common in more advanced disease [18], supporting the tenet of an immunosuppressive microenvironment. OCCCs are known to express hypoxia-related genes [19] which also influence the tumor microenvironment and potentially T cell responses. Despite such promising avenues, it has been challenging to find ideal molecular targets [20].
Epigenome-wide OCCC studies have generally been limited in sample size, with the largest being less than 20 cases [10, 11, 21]. To date, none have had comparable statistical power for evaluation of genome-wide DNA methylation in the context of other genomic and clinical features on the scale of The Cancer Genome Atlas (TCGA) high-grade serous EOC study [9, 22]. In this paper, we evaluate the hypothesis that epigenomic profiling of a relatively large collection of OCCC tumors can identify subclasses that may provide biological insight and show distinct clinical behavior patterns.
Materials and Methods
Study Participants
Clinical data and chemo-naïve fresh frozen tumor material were examined from women diagnosed with invasive OCCC and enrolled into research studies from the following sites: Memorial Sloan Kettering Cancer Center Gynecology Tissue Bank (New York NY, USA) [23], Mayo Clinic (Rochester MN, USA) [10], University of Cambridge (England) [24], Cedars-Sinai Medical Center (Los Angeles CA, USA) [25], University of Pittsburgh (Pittsburgh PA, USA), Gynaecological Oncology Biobank (GynBiobank) at Westmead Hospital (Sydney, Australia) [26], University of Edinburgh (Scotland) [27], Canadian Ovarian Experimental Unified Resource (COEUR, Canada) [28, 29], Brigham and Women’s Hospital (Boston MA, USA) [30], and University of Pennsylvania (Philadelphia PA, USA) [31]. Participants provided written informed consent to IRB-approved protocols. To confirm histotype, tumor sections were reviewed by an expert gynecological pathologist (MK) using Napsin A, p53, and WT1 immunohistochemical data [32].
DNA Methylation Arrays
Following bisulfite modification, Illumina Infinium MethylationEPIC Beadchips were run on DNA samples arrayed on three 96-well plates including extracted DNA from tissues containing >70% tumor from 239 participants, eight laboratory control DNAs (Human Methylated and Non-Methylated Control DNA Set; Catalogue #D5014, Zymo Research), and four participant duplicates using a standard operating procedure based on the Illumina protocol. Following scanning, intensity data were imported into the Genome Studio Methylation Module for analysis. Data were normalized, and detection p-values (reflecting the likelihood that the signal is distinguishable from the internal negative controls) were calculated for each CpG; call rate reflects the percentage of CpGs detected [33, 34]. Sample-independent controls included those for bisulfite conversion allowing identification of those with incomplete conversion; positive and negative controls were included to determine whether any probes should be excluded due to poor performance. The methylation status of the target CpG sites was determined by comparing the ratio of fluorescent signal from the methylated allele to the sum of the fluorescent signals from both methylated and unmethylated alleles, the beta value. These values per CpG range from 0 (unmethylated) to 1 (fully methylated). Laboratory controls and participant duplicates indicated excellent assay performance (e.g., r2=0.99 for beta values of participant duplicates). Nine participant samples showed poor performance and were excluded, including eight with call rate <95% and one outlier for median methylation intensity; no samples revealed sex error or mean or median detection p-value >0.05. For quality control (QC), CpGs probes were excluded if they were located at a SNP location, failed in more than 10% of samples, were located on the Y chromosome, were determined to be cross-reactive, or overlap genetic variants [33][34]; this resulted in 707,744 probes passing QC on the EPIC array. For an additional 41 patients from the Mayo Clinic, published data derived from the Illumina Infinium HumanMethylation450k Beadchip were used [10], and CpG probes were analyzed which overlap the EPIC and 450K datasets after QC. In combination, the resulting analytical set consisted of 344,914 CpG probes and 271 cases.
Methylation Clusters and Annotation
We analyzed 344,914 CpGs representing the intersection of CpG sites included on the Illumina Infinium HumanMethylation450k and MethylationEPIC Beadchips. Data were normalized separately for the two platforms, and batch correction across the two platforms were performed via COMBAT [33]. On the 1% most variable CpGs as defined by median absolute deviation (3,450 probes), we evaluated three clustering methods (Brunet [35], Lee [36], and non-smooth non-negative matrix factorization, nsNMF [37], as implemented in the R package ‘NMF’ [https://cran.r-project.org/web/packages/NMF/index.html]). Consensus clustering with 100 runs was performed, and nsNMF resulted in the most stable cluster assignment and was chosen as the most appropriate method. The optimal number of clusters was determined by cophenetic correlation coefficient assessment [35] which showed the largest drop at two clusters over the span of two to seven clusters. We implemented 2,000 bootstrap samples to estimate confidence intervals (CI) for the cophenetic coefficient for k=2 through k=7 clusters [38]. As shown in the cophenetic correlation plots along with the consensus map, estimates were highly reproducible with k=2 (with narrow CI) and highly variable for k>2 (wide CIs), emphasizing the reasoning of k=2 as the optimal number of clusters for subsequent analysis (Figure 1). Feature extraction was used to determine which CpGs had the greatest impact on the derived clusters (2,437 CpGs). Cluster 1 CpGs were hyper-methylated in Cluster 1 versus Cluster 2 and vice versa. We characterized CpGs with annotation derived from lllumina Corporation (San Diego, CA) and Ensembl (v78, GRCh38), limiting to CpGs in loci likely to be cis regulatory regions, defined as within 5′ UTR and 200 base pairs (bp) of a transcriptional start site (TSS). Gene set enrichment analysis (GSEA) of methylation data (differentially methylated genes) was used to assess the extent of enrichment of cluster-defining CpGs within cancer hallmark gene sets [39] using the Bioconductor package ‘missMethyl’, which was designed specifically for methylation data [40].
Figure 1. nsNMF DNA methylation Clustering Based on 271 OCCC Tumors.

(A) The estimated cophenetic correlation coefficient is plotted against the number of clusters (Rank), for k=2 through k=7, along with the corresponding 95% confidence interval that is empiricially calculated from 2,000 bootstrap samples. (B) For k=2 through k=7, the plot of the consensus matrix displays the agreement in cluster group across 100 clustering runs (red indicates 1=agreement, blue indicates 0=disagreement).
Association Testing
For the clinical characteristics, somatic mutation, and immunohistochemical features described below, association testing used a Kruskal-Wallis test for quantitative measures and Pearson’s chi-square test for categorical variables, unless any cell count was less than five, in which case Fisher’s exact testing with simulated p-value based on 2,000 replicates was used. Exploratory analyses examined larger number of clusters and excluded Illumina Infinium HumanMethylation450k Beadchip data.
Clinical Characteristics.
We examined associations between cluster and baseline clinical features including age at diagnosis (continuous), stage (early, advanced), study continent (North America, Europe, Australia), self-reported race (white non-Hispanic, Asian, other), extent of residual disease following primary debulking surgery (no macroscopic, macroscopic), presence of adjacent endometriosis (yes, no), menopause status (postmenopausal, pre/perimenopausal), and prior endometriosis (yes, no).
Somatic Mutation Data.
We also examined the association between cluster and somatic mutation derived from targeted DNA screening on a subset of 234 (87%) OCCC tumors across study sites with adequate tissue. DNA sequencing used a custom Nimblegen capture-based panel which of 166 putative OCCC driver genes based on pilot studies and COSMIC Cancer Gene Census5. Median coverage was 539x. Raw sequence data were aligned to the human genome (NCBI build 37) using BWA with variant calling for single nucleotide variants via Mutect2, Strelka, and Caveman and insertions/deletions using Pindel, Mutect2, and Strelka. Mutations were classified as pathogenic based upon their annotation in OncoKB8, frequency of occurrence in COSMIC and our combined OCCC database of previously published sequencing data, predicted pathogenicity based on PolyPhen9 and SIFT10, and literature review. Analyzed features included aneuploidies (continuous number of chromosomal or chromosomal arm level events), microsatellite instability (MSI) score (continuous) [41], single gene somatic mutation status (for ARID1A, TP53, PIK3CA, BRCA1, BRCA2), paired gene somatic mutation status (for ARID1A/PIK3CA), and a hierarchical somatic mutation classification (ARID1A mutation with one other mutation in PIK3CA, PIK3R1, KRAS, PPP2R1A, SPOP, or TERT [Group A]; multiple ARID1A mutations with one other mutation in PIK3CA or PIK3R1 [Group B]; single ARID1A mutation [Group C]; multiple ARID1A mutations without mutations in PIK3CA or PIK3R1 [Group D]; mutation in PIK3CA, PIK3R1, KRAS, PPP2R1A, SPOP, or TERT [Group E]; TP53 mutation without mutations in ARID1A or SMARCA4 [Group F]; SMARCA4 mutation not in combination with a mutation described above [Group G]; and remaining tumors [Group H]). Tumor mutation burden or mutation number was calculated as the sum of the presence or absence of a mutation in all targetted genes.
Tumor Immunohistochemistry.
On small subsets of up to 38 cases, immunohistochemical data was used to evaluate association of tumor methylation cluster with levels of CD8+ TILs (negative [none], low [1–2 per field], moderate [3–19 per field], high [≥20 per field]) [42] and protein expression categories for ARID1A (absent [internal control retained], present, subclonal loss [distinct area of absence with internal control and presence in the same core]), HNF1β (absent, any score less than score 2, diffuse [>50%] at least moderate intensity) and p53 (complete absence with internal control, wildtype pattern [variable intensity 1–90% of nuclei], overexpression [strong intensity >90% of nuclei]) [32].
Outcome Analyses
Overall survival analyses were restricted to a subset of 253 cases with vital status and survival time data from date of diagnosis and allowed for left truncation with censoring at five years from diagnosis. As covariates, we included race (white non-Hispanic, Asian, other), study continent (North America, Europe, Australia), and age at diagnosis (continuous and quadratic, assigned as site median for three cases), and we stratified by disease stage (FIGO stage I+II, III+IV, unknown), and extent of residual disease (no macroscopic, macroscopic, unknown). Proportionality of hazards was examined using Schoenfeld residuals. In addition, contingency analysis was done on cluster with primary treatment response (complete response, partial response, stable disease, progressive disease) and vital status at five years using chi-square testing. Analyses were also conducted for progression-free survival available on a subset of 248 cases.
Gene Expression Analyses
To further understand CpGs of interest, we used tumor RNA sequence data on a subset of N=116 OCCC patients across multiple contributing sites with sufficient tissue available for total RNA extraction. RNA-Seq libraries were prepared using poly(A) enrichment with sequencing of 100 bp paired-end libraries on Illumina’s HiSeq at a targeted depth of 40 million reads per sample. Alignment using STAR (version STAR_2.5.1b) against the reference genome hg38 (GENCODE v26). Reads were summarized using featureCounts (version 1.5.0-p1). Because gene expression data can often be skewed, a van der Waerden rank transformation [43] was applied. We assessed the differential gene expression between tumor methylation clusters using a moderated t-test as implemented in the R package ‘limma’, with a false discovery rate (FDR) threshold of 0.05 to correct for multiple testing. To assess pathway enrichment of genes differentially expressed by feature cluster, GSEA was performed with cancer hallmark gene sets [39] using ‘goseq’ R Bioconductor package [44]. For CpGs driving feature clusters and within likely cis regulatory regions as defined above, we assessed the correlation between tumor methylation and cis gene expression using a generalized linear model, with gene expression as the response variable and CpG methylation beta value as the predictor variable.
Results
Study Participants and Methylation Clustering
A total of 271 women from ten study sites were included in this large-scale analysis of genome-wide OCCC tumor DNA methylation data (Table 1); key characteristics did not vary by study site other than race; Asian ancestry was more common in participants from GynBiobank at Westmead Hospital (31%) and Memorial Sloan Kettering Cancer Center Gynecology Tissue Bank (23%) than other study sites. Thirty-five percent of participants were diagnosed at advanced stage (FIGO III, IV), 23% reported prior endometriosis, and, following primary therapy, 37% were deceased at last follow-up within five years.
Table 1.
Characteristics of 271 Participants with Invasive Ovarian Clear Cell Carcinoma
| N (%) | |
|---|---|
|
| |
| Study site (country) | |
| Memorial Sloan Kettering Cancer Center (USA) | 64 (24%) |
| Mayo Clinic (USA) | 56 (21%) |
| Cedars Sinai Medical Center (USA) | 31 (11%) |
| University of Cambridge (United Kingdom) | 27 (10%) |
| University of Pittsburgh (USA) | 23 (9%) |
| Westmead Hospital (Australia) | 22 (8%) |
| Edinburgh (United Kingdom) | 20 (7%) |
| COEUR (Canada) | 13 (5%) |
| Brigham and Women’s Hospital (USA) | 9 (3%) |
| University of Pennsylvania (USA) | 6 (2%) |
| Race, self-reported | |
| White non-Hispanic | 180 (86%) |
| Asian | 25 (12%) |
| Black | 4 (2%) |
| Other/Unknown | 62 |
| Age at diagnosis | |
| Mean (Range), N | 58.1 (31–88), 268 |
| FIGO stage | |
| Early (I, II) | 166 (65%) |
| Advanced (III, IV) | 91 (35%) |
| Unknown | 14 |
| Tumor primary site | |
| Ovary | 209 (88%) |
| Omentum | 4 (2%) |
| Pelvis | 4 (2%) |
| Peritoneum | 3 (1%) |
| Fallopian tube | 1 (<1%) |
| Other | 17 (7%) |
| Unknown | 33 |
| Residual disease | |
| No macroscopic disease | 181 (76%) |
| Macroscopic disease | 57 (24%) |
| Unknown | 33 |
| Prior endometriosis, self-reported | |
| Yes | 33 (23%) |
| No | 108 (77%) |
| Unknown | 130 |
| Primary therapy outcome | |
| Complete response | 157 (80%) |
| Partial response | 7 (4%) |
| Stable disease | 9 (5%) |
| Progressive disease | 23 (12%) |
| Unknown | 75 |
| Progression within five years | |
| Yes | 137 (55%) |
| No | 111 (45%) |
| Unknown | 23 |
| Time to progression among progressors, months | |
| Mean (Range), N | 18.8 (0.03–59.7), 137 |
| Time to last follow-up among non-progressors, months | |
| Mean (Range), N | 51.0 (0.40–60.0), 111 |
| Vital status at five years | |
| Alive | 159 (63%) |
| Deceased | 94 (37%) |
| Unknown | 18 |
| Time to last follow-up among living, months | |
| Mean (Range), N | 45.7 (0.40–60.0), 159 |
COEUR, Canadian Ovarian Experimental Unified Resource; FIGO, International Federation of Gynecology and Obstetrics; residual disease following primary debulking surgery.
nsNMF clustering of the 1% most variable CpGs (3,450 CpGs) intersecting Illumina Infinium HumanMethylation450k and MethylationEPIC Beadchips resulted in 134 (49%) OCCC cases in methylation Cluster 1 and 137 (51%) OCCC in Cluster 2. A heat map using the 2,437 CpGs contributing most significantly to the clustering (as determined by the feature extraction) is shown in Supplemental Figure 1A, and the basis matrix (matrix W or the metagenes) heat map is shown in Supplemental Figure 1B. Among the CpGs contributing to clustering, a total of 1,388 reside within 200 bp of TSS of a gene (N=977 genes); these genes that did not fall into particular cancer hallmark pathways [39]. Clustering was consistent when excluding Illumina Infinium HumanMethylation450k Beadchip data.
Clinical, Tumor Mutation, and Immunohistochemical Associations
Table 2 shows clinical, molecular, and pathologic covariates that differed by methylation cluster at p<0.10 (full results in Supplemental Table 1). Cluster 1 included OCCC that tended to be TP53 mutation positive (p<0.001), have abnormal p53 protein expression (p<0.013), be of advanced FIGO stage (p=0.022), and have macroscopic residual disease. Cluster 2 tumors were more likely to be ARID1A mutation positive (p<0.001), PIK3CA (p<0.001) mutation positive, of Asian race, and early stage (p=0.022) with increased total aneuploidy (p<0.001). While ARID1A and PIK3CA mutations were common in these OCCC tumors (47%, 43% respectively in this study), we expected TP53 mutations to be less common (~11–13%), yet found them in 20% of cases overall and significantly more in Cluster 1 cases (31%). These particular cases were re-reviewed to confirm their histology. Supplementary Figure 2 shows the distribution across tumors of the presence of mutations in the five genes that define the mutation clusters (ARID1A, PIK3CA, TP53, BRCA1, BRCA2).
Table 2.
Distributions of Selected Demographic, Clinical, and Molecular Characteristics by DNA Methylation Cluster (p<0.10)
| Cluster 1 (N=134) | Cluster 2 (N=137) | P value | |
|---|---|---|---|
|
| |||
| Illumina Infinium Methylation BeadChip | 0.042 | ||
| MethylationEPIC | 120 (90%) | 110 (80%) | |
| HumanMethylation450k | 14 (10%) | 27 (20%) | |
| Self-reported race | 0.012 | ||
| White non-Hispanic | 95 (92%) | 85 (80%) | |
| Asian | 6 (6%) | 19 (18%) | |
| Black | 2 (2%) | 2 (2%) | |
| Missing/other | 31 | 31 | |
| FIGO stage | 0.022 | ||
| Early (I, II) | 73 (57%) | 93 (72%) | |
| Advanced (III, IV) | 54 (42%) | 37 (28%) | |
| Unknown | 7 | 7 | |
| Residual disease | 0.065 | ||
| No macroscopic | 85 (71%) | 96 (81%) | |
| Macroscopic | 35 (29%) | 22 (19%) | |
| Unknown | 14 | 19 | |
| p53 expression | 0.013 | ||
| Wild type pattern: variable intensity 1–90% of nuclei | 13 (72%) | 22 (100%) | |
| Complete absence with internal control | 2 (11%) | 0 | |
| Overexpression, strong intensity >90% of nuclei | 3 (17%) | 0 | |
| Unknown | 116 | 115 | |
| TP53 mutation | <0.001 | ||
| Yes | 37 (31%) | 10 (9%) | |
| No | 82 (69%) | 105 (91%) | |
| Unknown | 15 | 22 | |
| ARID1A mutation | <0.001 | ||
| Yes | 38 (32%) | 73 (63%) | |
| No | 81 (68%) | 42 (37%) | |
| Unknown | 15 | 22 | |
| PIK3CA mutation | 0.002 | ||
| Yes | 39 (33%) | 62 (54%) | |
| No | 80 (67%) | 53 (46%) | |
| Unknown | 15 | 22 | |
| ARID1A/PIK3CA mutation | <0.001 | ||
| Yes/yes | 18 (15%) | 48 (42%) | |
| Yes/no | 20 (17%) | 25 (22%) | |
| No/yes | 21 (18%) | 14 (12%) | |
| No/no | 60 (50%) | 28 (24%) | |
| Unknown | 15 | 22 | |
| Total aneuploidy | <0.001 | ||
| Mean (range) | 7.1 (0–27) | 11.1 (0–28) | |
| Unknown | 15 | 22 | |
| Somatic mutation group | <0.001 | ||
| ARID1A mutation with one other mutation in PIK3CA, PIK3R1, KRAS, PPP2R1A, SPOP, or TERT (Group A) | 21 (18%) | 24 (21%) | |
| Multiple ARID1A mutations with one other mutation in in PIK3CA or PIK3R (Group B) | 7 (6%) | 33 (28%) | |
| Single ARID1A mutation (Group C) | 8 (7%) | 3 (3%) | |
| Multiple ARID1A mutations without mutations in PIK3CA or PIK3R1 (Group D) | 2 (2%) | 13 (11%) | |
| Mutation in PIK3CA, PIK3R1, KRAS, PPP2R1A, SPOP, or TERT (Group E) | 29 (24%) | 27 (23%) | |
| TP53 mutation without mutations in ARID1A or SMARCA4 (Group F) | 28 (24%) | 4 (4%) | |
| SMARCA4 mutation (Group G) | 6 (5%) | 2 (2%) | |
| Undefined (Group H) | 18 (15%) | 9 (8%) | |
| Unknown | 15 | 22 | |
| Vital status | 0.01 | ||
| Alive | 69 (55%) | 90 (70%) | |
| Deceased | 56 (45%) | 38 (30%) | |
| Unknown | 9 | 9 | |
| Median survival, months | 58.7 | NA | 0.01 |
| Time to progression among progressors, months; mean (range), N | 16.6 (0.03–50.3), 68 | 20.9 (0.16–59.7), 69 | 0.07 |
Kruskal-Wallis sum test was used for categorical tests, unless any cell less than five, then Pearson’s chi-squared test with simulated p-value based on 2000 replicates used; Fisher’s exact test used for quantitative measures; FIGO, International Federation of Gynecology and Obstetrics; total aneuploidy: number of chromosomal or chromosomal arm level events.
Consistent with single gene mutation results, multi-gene mutation groups were found to associate with methylation clusters (p<0.001; Table 2). Cluster 1 tumors tended to be mutation Group F (TP53 mutation positive with no mutation in ARID1A or SMARCA4), mutation Group C (have a single ARID1A mutation,) or mutation Group G (SMARCA4 mutations). Cluster 2 tumors were more likely to be mutation Group B (multiple ARID1A mutations with a mutation in PIK3CA, PIK3RA1, KRAS, PPP2R1A, SPOP, or TERT), or mutation Group D (multiple ARID1A mutations without mutations in PIK3CA or PIK3RA1). No association was seen between Clusters and study continent, age at diagnosis, menopause status, history of or presence of endometriosis, MSI score, extent of whole genome duplication or tumor mutation burden (Supplemental Table 1). The distributions of the clinical and molecular features significantly differing in Cluster 1 and Cluster 2 are shown in Figure 2 and an overview of these characteristics for each methylation cluster is provided in Figure 3.
Figure 2. Alignment of Tumor and Clinical Features by DNA Methylation Clusters in 271 OCCC Tumors.
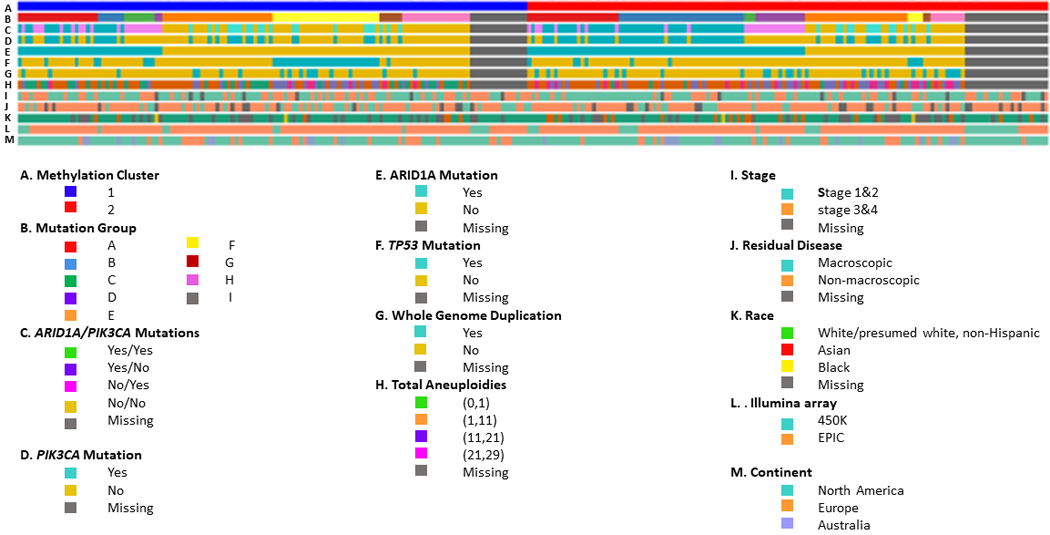
Columns represent samples with those on the left indicating (A) Methylation Cluster 1 tumors and those on the right Methylation Cluster 2 tumors, (B) mutation group (see methods), (C) ARID1A/PIK3CA mutations (yes/yes, yes/no, no/yes, no/no, missing), (D) PIK3CA mutation (yes, no, missing), (E) ARID1A mutation (yes, no, missing), (F) TP53 mutation (yes, no, missing), (G) WGD, whole-genome duplication (yes, no, missing), (H) total aneuploidy (none, 1–10, 11–20, 21–29, missing), (I) stage (early, advanced, missing), (J) residual disease (macroscopic, no macroscopic, missing), (K) race (White non-Hispanic, Asian, Black, missing), (L) Illumina Infinium Beadchip (HumanMethylation450, Methylation EPIC), (M) continent (North America, Europe, Australia). Samples are ordered based on hierarchical clustering which used nsNMF two-cluster analysis, although the dendrogram is suppressed.
Figure 3. Overview of Characteristics of DNA Methylation Clusters.

Methylated genes with decreased gene expression are shown, along with molecular and clinical characteristics of OCCC samples within each methylation-based cluster.
Clinical Outcomes
Vital status at five years was also associated with methylation clusters, with 55% of Cluster 1 cases and 70% of Cluster 2 cases alive at time of follow-up (Table 2, p=0.01); similarly, time to disease progression was shorter on average by 4.3 months in Cluster 1 cases compared to Cluster 2 cases (Table 2, p=0.07). Consistent with these observations and published literature on ARID1A- and PIK3CA-mutant OCCC, univariate analysis of overall survival time revealed an apparent association with Cluster 2 having longer survival (Supplemental Figure 3, Cluster 1 v Cluster 2 HR 1.70, 95% CI 1.13 – 2.57; p=0.015). However, the proportional hazard assumption for the cluster association was violated with an attenuation of risk difference towards five years (p=0.037). Covariate adjustment for age, continent, and race with stratification by stage and residual disease attenuated the estimated cluster-associated risk (Cluster 1 v Cluster 2, HR 1.48, 95% CI 0.97 – 2.27; p= 0.067); proportional hazards remained in violation (p=0.027). Subset analyses of cases by disease stage, suggested that methylation cluster may associate with overall survival time only among women diagnosed at advanced stage. However, as proportional hazards again were violated, survival analysis results should be considered suggestive at most and larger studies with time-dependent analyses are needed. There was no association between methylation cluster and primary therapy outcome (partial, stable disease, progressive disease, no evidence of disease).
Transcriptomic Analyses
From among the 1,388 cluster-defining CpGs that lie within 200 bp of transcription start sites (N=977 genes, from methylation clustering above), we further analyzed 971 CpGs mapped to 700 genes in RNA sequence data. At the CpG level, among the cluster-driving CpGs determined from the feature extraction, we observed cis correlations between methylation and gene expression at 113 CpGs (46 CpGs for Cluster 1 and 67 for Cluster 2; Q value < 10−4, Supplemental Table 2). Among the top cluster-driving CpGs (top 100 from feature extraction), the most statistically significant were thirteen CpGs associated with decreased expression in twelve genes (AP2A2, ACKR2, RXFP1, CTH, CEP44, R11–141M3.5, ANKS4B, FAM149A, LMF1, TNS2, WIPF1, ATOH8) (Supplementary Figure 4).
In a subset of 116 cases with RNA sequence data, we also evaluated differential RNA expression by methylation cluster (Supplemental Table 2) and expression at 5,854 genes had an FDR <0.05. At the gene level, among 977 genes with 1,388 cluster-defining CpGs residing within 200 bp of transcription start sites, we found that 369 genes (38%) were differentially expressed across methylation clusters (p < 10−4, Supplemental Table 3). Through GSEA, we observed that these 369 differentially expressed genes were significantly overrepresented in nine of the 50 hallmark gene sets [39]. Six of the pathways (67%) are categorized as immune-related including inflammation, and interferon alpha and gamma responses (Table 3). Genes contributing to these pathways that were significantly differently expressed across clusters are provided in Supplemental Table 4 and include the non-receptor tyrosine kinase JAK2, complement factor H, and toll-like receptor 2. No enrichment of differentially expressed genes was seen in gene sets related to other pathways, cellular components, or functions [39].
Table 3.
Hallmark Gene Sets Over-represented Among Genes that were Differentially Expressed across OCCC Methylation Cluster 1 and Cluster 2
| Process Category | Description (Gene Set ID) | N (%) | P-value |
|---|---|---|---|
|
| |||
| Immune | Interferon gamma response (17) | 107 (55%) | 1.32 × 10−11 |
| Development | Epithelial mesenchymal transition (6) | 105 (54%) | 1.02 × 10−9 |
| Immune | Allograft rejection (13) | 93 (53%) | 9.66 × 10−9 |
| Immune | Interferon alpha response (16) | 55 (57%) | 1.80 × 10−7 |
| Immune | Inflammation (19) | 88 (47%) | 1.59 × 10−5 |
| Immune | Complement cascade (15) | 85 (46%) | 7.53 × 10−5 |
| Signaling | KRAS signaling, downregulated genes (43) | 82 (44%) | 6.08 × 10−4 |
| DNA damage | UV response: downregulated genes (11) | 63 (44%) | 7.91 × 10−3 |
| Immune | IL6 STAT3 signaling during acute phase response (18) | 37 (46%) | 6.10 × 10−3 |
P<10−2; N (%) represent number of genes differentially expressed by OCCC methylation Cluster 1 v Cluster 2 and % of genes in each overall gene set.
Discussion
We report on examination of genome-wide tumor methylation in 271 women with OCCC, a collaborative effort involving ten institutions across five countries. Clustering algorithms were applied to discern whether there existed methylation subgroups with distinct clinical, molecular, or prognostic characteristics. Quantitative molecular analyses sought to highlight pathways that may bridge epigenomic and clinical associations.
Comparing diagnostic results of three clustering approaches revealed nsMNF with rank k=2 to be the most stable method. Subsequent nsNMF methylation clustering of tumors produced two broad groups: Cluster 1 with ARID1A/PIK3CA mutations, early stage and aneuploidy, and Cluster 2 with TP53 mutations, later stage and residual disease. Mutational cluster analysis revealed that ARID1A multiple mutations were almost exclusively in Cluster 2. ARID1A deficiency impairs DNA double strand break repair [21] and limits chromatin access, impairing interferon expression and promoting an immunosuppressive environment [45].
While OCCC tumors are thought to have low levels of genomic instability, a recent study [11] reported moderate levels of chromosomal gains and losses in OCCC. In the present study, we note that those OCCC with ARID1A/PIK3CA mutations had higher levels of chromosomal aneuploidy, while TP53 mutations were more common than previously reported and at more advanced stages of disease [22]. That Cluster 2 with multiple ARID1A mutations appears to be ARID1A deficient may explain the greater genomic instability associated with this cluster, as assessed by aneuploidies.
Pathway analyses provide support for a role of immune related pathways in OCCC, from the tumor microenvironment or tumor cells themselves. Looking at genes identified by clustering analyses based on gene expression in OCCC previously [14, 15, 16], little overlap was seen in genes significantly associated with methylation clusters with those reported in the Anglelsio et al [15] study, nor with the cytokine genes examined in Yanajhara et al [15], although the number of OCCC were relatively small in these two studies. Tan et al [16], reported gene expression in 222 ovarian clear cell carcinomas, noting two clusters, epithelial-like and mesenchymal-like. However, there was little overlap between those genes and those in our gene expression associated with methylation clustering.
Strengths of this study include utilization of the largest OCCC sample size to date, use of multiple study sites, consideration of genome-wide epigenomics, and incorporation of tumor molecular results where possible. Analysis of outcome differences between the methylation clusters suggested improved prognosis in Cluster 2, but this was complicated by potential changes in survival relationships over time. Although modeling prognostic analyses will require further consideration of potentially time-dependent survival patterns in larger patient collections, current results suggest that immune-related methylation factors may provide an avenue for focused development of potential future therapeutics. A potential weakness of this report is the difference in resolution between the two methylation arrays. However, we found that results were consistent when analyses restricted to Illumina Infinium MethylationEPIC BeadChip (85% of cases). Because sample size was limited in subset analyses presented here, more complete somatic data is needed to further clarify relationships between tumor mutations, DNA methylation, gene expression, and proteomics in OCCC. Greater overall sample size with follow-up data will allow also enable appropriate statistical evaluation of a variety of interactions and improved assessment of overall and progression-free survival. As the most extensive OCCC methylation study to date, this study represents a foundation on which to build upon for future clinical, molecular, and epidemiologic investigation.
Supplementary Material
Acknowledgements
Brigham and Women’s Hospital: Foundation for Women’s Cancer as part of the Reproductive Scientist Development Program, Honorable Tina Brozman Foundation, Minnesota Ovarian Cancer Alliance, Deborah and Robert First Family Foundation, Greg and Peggy Strakosch, the Saltonstall Foundation, the Potter Foundation, and the Brigham Ovarian Cancer Research Fund.
Cedars Sinai Medical Center: The work was supported in part by the American Cancer Society SIOP-06-258-01-COUN (K. Lawrenson).
Mayo Clinic: R21-CA222867 (Goode, E.L., Cunningham, J.M.), R01-CA248288 (Goode, E.L.), and P50-CA136393 (Goode, E.L..).
Memorial Sloan Kettering Cancer Center: This work was funded in part the National Cancer Institute Cancer Center Core Grant No. P30-CA008748 (MSK: B. Weigelt, J. Konner, E. Papaemmanuil). B. Weigelt is funded in part by Cycle for Survival and Breast Cancer Research Foundation grants.
University of Edinburgh: We thank the Nicola Murray Foundation for their generous support of the laboratory and the NRS Lothian Bioresource volunteers for their participation. We also acknowledge the NHS Trusts and staff for their contribution to this work.
University of Pittsburgh: NIH SPORE in ovarian cancer (P50 CA228991, E. Elishaev, F. Mogduno), the Penn Medicine Translational Center of Excellence in Ovarian Cancer, the Dr. Miriam and Sheldon G. Adelson Medical Research Foundation, Tina’s Wish Foundation, and the Claneil Foundation.
University of Pittsburgh: This project used the UPMC Hillman Cancer Center and Tissue and Research Pathology/Pitt Biospecimen Core shared resource which is supported in part by award P30CA047904 (F. Modugno).
Westmead Hospital: We thank all the women who participated in the GynBiobank, and gratefully acknowledge the Departments of Gynaecological Oncology, Medical Oncology and Anatomical Pathology at Westmead Hospital, Sydney. The Gynaecological Oncology Biobank at Westmead (GynBiobank), a member of the Australasian Biospecimen Network-Oncology group, was funded by the National Health and Medical Research Council of Australia (ID 310670 & ID 628903, A. Difazio) and the Cancer Institute NSW (12/RIG/1-17 & 15/RIG/1–16, A. Difazio).
Financial Disclosures: CG discloses research funding (AstraZeneca, Aprea, Nucana, Tesaro and Novartis) and honoraria/consultancy fees (Roche, AstraZeneca, MSD, Tesaro, Nucana, Clovis, Foundation One, Cor2Ed, and Sierra Oncology) and is named on issued/pending patents related to predicting treatment response in ovarian cancer outside the scope of the work described here. ADeF discloses research funding and honoraria (AstraZeneca) outside the scope of the work described here.
Footnotes
All other authors declare no conflicts of interest.
References
- 1.Iida Y, Okamoto A, Hollis RL, Gourley C, Herrington CS, Clear cell carcinoma of the ovary: a clinical and molecular perspective. Int J Gynecol Cancer, 2020. 0: p. 1–12. [DOI] [PubMed] [Google Scholar]
- 2.Kurman RJ, Origin and molecular pathogenesis of ovarian high-grade serous carcinoma. Ann Oncol, 2013. 24 Suppl 10: p. x16–21. [DOI] [PubMed] [Google Scholar]
- 3.Worley MJ, Welch RW, Berkowitz RS, Ng S-W, Endometriosis-associated ovarian cancer: a review of pathogenesis. Int J Mol Sci, 2013. 14(3): p. 5367–79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Mabuchi ST, Sugiyama, and Kimura T, Clear cell carcinoma of the ovary: molecular insights and future therapeutic perspectives. J Gynecol Oncol, 2016. 27(3): p. e31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Del Carmen MG, Birrer M, and Schorge JO, Clear cell carcinoma of the ovary: A review of the literature. Gynecol Oncol, 2012. 126(3): p. 481–490. [DOI] [PubMed] [Google Scholar]
- 6.Ayhan A, Kuhn E, Wu R-C, Ogawa H, Bahadirli-Talbott A, Mao T-L, et al. , CCNE1 copy-number gain and overexpression identify ovarian clear cell carcinoma with a poor prognosis. Mod Pathol, 2017. 30(2): p. 297–303. [DOI] [PubMed] [Google Scholar]
- 7.Anglesio MS, Bashahati A, Wang YK, Senz J, Ha G, Yang W, et al. , Multifocal endometriotic lesions associated with cancer are clonal and carry a high mutation burden. J Pathol, 2015. 236(2): p. 201–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Wiegand KC, Shah SP, Al-Agha OM, Zhao Y, Tse K, Zeng T, et al. , ARID1A mutations in endometriosis-associated ovarian carcinomas. N Engl J Med 2010. 363(16): p. 1532–1543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Jones S, Wang T-L, Shih L-M, Nakayama K, Roden R, Glas R, et al. , Frequent mutations of chromatin remodeling gene ARID1A in ovarian clear cell carcinoma. Science, 2010. 330(6001): p. 228–231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Cicek MS, Koestler DC, Fridley BL, Kalli KR, Armasu SM, Larson MC et al. , Epigenome-wide ovarian cancer analysis identifies a methylation profile differentiating clear-cell histology with epigenetic silencing of the HERG K+ channel. Hum Mol Genet, 2013. 22(15): p. 3038–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Engqvist H, Parris TZ, Biermann J, Werner Ronnerman E, Larsson P, Sundflet K. et al. , Integrative genomics approach identifies molecular features associated with early-stage ovarian carcinoma histotypes. Sci Rep, 2020. 10(1): p. 7946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Shen H, Fridley BL, Song H, Lawrenson K, Cunningham JM, Ramus SJ, et al. , Epigenetic analysis leads to identification of HNF1B as a subtype-specific susceptibility gene for ovarian cancer. Nat Commun, 2013. 4: p. 1628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Leskela S, Romero I, Cristobal E, Perez-Mies B, Rosa-Rosa JM, Gutierrez-Pecharroman A, et al. , Mismatch Repair Deficiency in Ovarian Carcinoma: Frequency, Causes, and Consequences. Am J Surg Pathol, 2020. 44(5): p. 649–656. [DOI] [PubMed] [Google Scholar]
- 14.Anglesio MS, George J, Kulbe H, Friedlander M, Rischin D, Lemech C, et al. , IL6-STAT3-HIF signaling and therapeutic response to the angiogenesis inhibitor sunitinib in ovarian clear cell cancer. Clin Cancer Res, 2011. 17(8): p. 2538–48. [DOI] [PubMed] [Google Scholar]
- 15.Yanaihara N, Angeliso MS, Ochiai K, Hirata Y, Saito M, Nagata C, et al. , Cytokine gene expression signature in ovarian clear cell carcinoma. Int J Oncol, 2012. 41(3): p. 1094–100. [DOI] [PubMed] [Google Scholar]
- 16.Tan TZ, Ye J, Yee CV, Lim D, Ngoi NY, Tan DSP, et al. , Analysis of gene expression signatures identifies prognostic and functionally distinct ovarian clear cell carcinoma subtypes. EBioMedicine, 2019. 50: p. 203–210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Oda K, Hamanishi J, Matsuo K, Hasegawa K. Genomics to immunotherapy of ovarian clear cell carcinoma: Unique opportunities for management. Gynecol Oncol, 2018. 151(2): p. 381–389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Zhu J, Wen H, Bi R, Wu X, Prognostic value of programmed death-ligand 1 (PD-L1) expression in ovarian clear cell carcinoma. J Gynecol Oncol, 2017. 28(6): p. e77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ji JX, Wang YK, Cochrane DR, Huntsman DG, Clear cell carcinomas of the ovary and kidney: clarity through genomics. J Pathol, 2018. 244(5): p. 550–564. [DOI] [PubMed] [Google Scholar]
- 20.Amano T, Chano T, Yoshino F, Kimura F, Maurkami T, Current Position of the Molecular Therapeutic Targets for Ovarian Clear Cell Carcinoma: A Literature Review. Healthcare (Basel), 2019. 7(3). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Shen J, Peng Y, Wei L, Zhang W, Yang L, Lan L, et al. , ARID1A Deficiency Impairs the DNA Damage Checkpoint and Sensitizes Cells to PARP Inhibitors. Cancer Discov, 2015. 5(7): p. 752–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Friedlander ML, Russell K, Millios S, Gatalica Z, Bender R, Voss A, Molecular profiling of clear cell ovarian cancers: identifying potential treatment targets for clinical trials. Int J Gynecol Cancer, 2016. 26(4): p. 648–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Phelan CM, Kuchenbaecker KB, Tyrer J, Kar SP, Lawrenson K, Winham SJ, Dennis J, et al. , Identification of 12 new susceptibility loci for different histotypes of epithelial ovarian cancer. Nat Genet, 2017. 49(5): p. 680–691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Gounaris I. and Brenton JD, Molecular pathogenesis of ovarian clear cell carcinoma. Future Oncol, 2015. 11(9): p. 1389–405. [DOI] [PubMed] [Google Scholar]
- 25.Gusev A, Lawrenson K, Lin X, Lyra PC Jr, Kar S, Vavra KC, et al. , A transcriptome-wide association study of high-grade serous epithelial ovarian cancer identifies new susceptibility genes and splice variants. Nat Genet, 2019. 51(5): p. 815–823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Garsed DW, Alsop K, Fereday S, Emmanuel C, Kennedy KJ, Etemadmoghadam D, et al. , Homologous Recombination DNA Repair Pathway Disruption and Retinoblastoma Protein Loss Are Associated with Exceptional Survival in High-Grade Serous Ovarian Cancer. Clin Cancer Res, 2018. 24(3): p. 569–580. [DOI] [PubMed] [Google Scholar]
- 27.Irodi A, Rye T, Churchman M, Bartos C, Mackean M, Nussey F, et al. , Patterns of clinicopathological features and outcome in epithelial ovarian cancer patients: 35 years of prospectively collected data. BJOG, 2020. [DOI] [PubMed] [Google Scholar]
- 28.Le Page C, Rahimi K, Kobel M, Tonin PN, Meunier L, Portelance L, et al. , Characteristics and outcome of the COEUR Canadian validation cohort for ovarian cancer biomarkers. BMC Cancer, 2018. 18(1): p. 347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Le Page C, Kobel M, de Ladurantaye M, Rahimi K, Madore J, Babinsky S, et al. , Specimen quality evaluation in Canadian biobanks participating in the COEUR repository. Biopreserv Biobank, 2013. 11(2): p. 83–93. [DOI] [PubMed] [Google Scholar]
- 30.Elias KM, Emori MM, Westerling T, Long H, Budina-Kolomets A, Li F, et al. , Epigenetic remodeling regulates transcriptional changes between ovarian cancer and benign precursors. Jci Insight, 2016. 1(13). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Coetzee SG, Shen HC, Hazellette DJ, Lawrenson K, Kuchenbaecker K,Tyrer J. et al. , Cell-type-specific enrichment of risk-associated regulatory elements at ovarian cancer susceptibility loci. Human Molecular Genetics, 2015. 24(13): p. 3595–3607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kobel M, Kalloger SE, Lee S, Duggan MA, Kelemen L, Prentice L, et al. , Biomarker-based ovarian carcinoma typing: a histologic investigation in the ovarian tumor tissue analysis consortium. Cancer Epidemiol Biomarkers Prev, 2013. 22(10): p. 1677–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Johnson WE, Li C, Rabinovic A, Adjusting batch effects in microarray expression data using empirical Bayes methods. Biostatistics, 2007. 8(1): p. 118–27. [DOI] [PubMed] [Google Scholar]
- 34.Pidsley R, Zotenko E, Peters TJ, Lawrence MG, Risbridger GP, Molloy P, et al. , Critical evaluation of the Illumina MethylationEPIC BeadChip microarray for whole-genome DNA methylation profiling. Genome Biol, 2016. 17(1): p. 208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Brunet JP, Tamayo P, Golub TR, Mesirov JP, Metagenes and molecular pattern discovery using matrix factorization. Proc Natl Acad Sci U S A, 2004. 101(12): p. 4164–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Lee DD and Seung HS, Learning the parts of objects by non-negative matrix factorization. Nature, 1999. 401(6755): p. 788–91. [DOI] [PubMed] [Google Scholar]
- 37.Pascual-Montano A, Carazo JM, Kochi K, Lehmann D, Nonsmooth nonnegative matrix factorization (nsNMF). IEEE Trans Pattern Anal Mach Intell, 2006. 28(3): p. 403–15. [DOI] [PubMed] [Google Scholar]
- 38.Bodelon C, Killian JK, Sampson JN, Anderson WF, Matsuno R, Brinton LA, et al. , Molecular Classification of Epithelial Ovarian Cancer Based on Methylation Profiling: Evidence for Survival Heterogeneity. Clin Cancer Res, 2019. 25(19): p. 5937–5946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Liberzon A, Birger C, Thorvaldsdottir H, Ghandi M, Mesirov JP, Tamayo P, The Molecular Signatures Database (MSigDB) hallmark gene set collection. Cell Syst, 2015. 1(6): p. 417–425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Phipson B. and Matsimovic J. missMethyl: Analysing Illumina HumanMethylation BeadChip Data. Available from: https://www.bioconductor.org/packages/devel/bioc/vignettes/missMethyl/inst/doc/missMethyl.html#gene-ontology-analysis.
- 41.Niu B, Ye K, Zhang Q, Lu C, Xie M, McLellan MD, et al. , MSIsensor: microsatellite instability detection using paired tumor-normal sequence data. Bioinformatics, 2014. 30(7): p. 1015–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Goode EL, Block MS, Kalli KR, Vierkant RA, Chen W, Fogarty ZC, et al. , Dose-Response Association of CD8+ Tumor-Infiltrating Lymphocytes and Survival Time in High-Grade Serous Ovarian Cancer. JAMA Oncol, 2017. 3(12): p. e173290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.van der Waerden B, Order tests for the two-sample problem and their power. . Proc Koninklijke Nederlandse Akademie van Wetenschappen, 1952. 55: p. 453–558. Ser B. [Google Scholar]
- 44.Young MD, Wakefield MJ, Smyth GK, Oshlack A, Gene ontology analysis for RNA-seq: accounting for selection bias. Genome Biol, 2010. 11(2): p. R14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Li J, Wang W, Zhang Y, Cieslik M, Guo J, Tan M, et al. , Epigenetic driver mutations in ARID1A shape cancer immune phenotype and immunotherapy. J Clin Invest, 2020. 130(5): p. 2712–2726. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.