

Abstract
Leptomeningeal anastomoses are small distal anastomotic vessels also known as pial collaterals in the brain. These vessels redirect blood flow during an occlusion and are important for stroke treatment and outcome. Pial collaterals have unique hemodynamic forces and experience significantly increased luminal flow and shear stress after the onset of ischemic stroke. However, there is limited knowledge of how pial collaterals respond to flow and shear stress, and whether this response is altered in chronic hypertension. Using an in vitro system, pial collaterals from normotensive and hypertensive rats (n=6–8/group) were isolated and luminal flow was induced with intravascular pressure maintained at 40 mmHg. Collateral lumen diameter was measured following each flowrate in the absence or presence of pharmacologic inhibitors and activators. Collaterals from male and female Wistar rats dilated similarly to increased flow (2 μl/min: 58.4±18.7% vs. 67.9±7.4%; p=0.275), and this response was prevented by inhibition of the transient receptor potential vanilloid type 4 channel, as well as inhibitors of nitric oxide and intermediate-conductance calcium-activated potassium channels, suggesting shear stress-induced activation of this pathway was involved. However, the vasodilation was significantly impaired in hypertensive rats (2 μl/min: 17.7±7.7%), that was restored by inhibitors of reactive oxygen species and mimicked by angiotensin II. Thus, flow- and shear stress-induced vasodilation of pial collaterals appears to be an important stimulus for increasing collateral flow during large vessel occlusion. Impairment of this response during chronic hypertension may be related to poorly engaged pial collaterals during ischemic stroke in hypertensive subjects.
Keywords: leptomeningeal anastomoses, flow, shear stress, hypertension, angiotensin II
Graphical Abstract
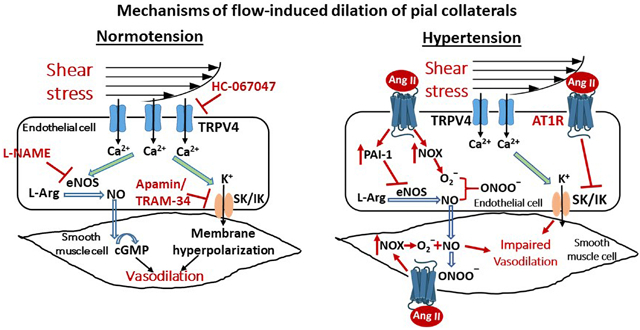
Introduction
Stroke is the second leading cause of death worldwide and the leading cause of devastating disability (1). Currently, the only effective treatment for acute ischemic stroke is rapid restoration of blood flow with tissue plasminogen activator (tPA) and/or clot removal by mechanical thrombectomy. However, the majority of stroke patients are excluded from recanalization therapies due to time-limitations of tPA or the lack of salvageable tissue, also known as the penumbra (2). The ischemic penumbra is a peri-infarct region with constrained blood supply in which neuronal depolarization has not yet occurred; metabolic activity is maintained but electrical activity is impaired, making it potentially salvageable (3). During large vessel occlusion, leptomeningeal anastomoses (LMAs) importantly redirect cerebral blood flow from non-occluded territories to the occluded area to perfuse the penumbra and limit infarct expansion (4). Clinical studies have demonstrated that good pial collateral status is a critical determinant of a smaller infarct and favorable long-term stroke outcome, making the function of LMAs an important therapeutic target (5, 6).
In contrast to nonanastomotic or terminal pial arterioles, LMAs experience unique hemodynamic forces, including low, bidirectional shear stress and flow, low myogenic tone and have no perivascular innervation (7, 8). Immediately after the onset of large vessel occlusion or middle cerebral artery occlusion (MCAO), there is a large increase in retrograde blood flow through pial collaterals. Animal studies have shown that pial collateral flow velocity increases 10–20 times in response to MCAO in rodents; shear stress increases 15-fold after ligation of middle cerebral artery (MCA) in normotensive mice (9). Physiological shear stress is the tangential frictional force in the endothelial cells due to blood flow that is a potent vasodilatory force. Flow and shear stress promotes production and release of endothelial vasodilatory factors such as nitric oxide (NO) and prostacyclin, thereby regulating flow (10). In contrast, low shear stress leads to endothelial dysfunction (11, 12). The large increase in flow and shear stress in pial collaterals during large vessel occlusion that is created due to the large pressure differential between the occluded and patent vascular territories suggests these biomechanical forces are important regulators of LMA diameters and collateral flow.
Luminal flow and shear stress can cause vasodilation through several mechanisms, including activation of transient receptor potential (TRP) vanilloid 4 (TRPV4) channels. TRPV4 is a nonselective cation channel sensitive to mechanical forces, including shear stress (13). Activation of TRPV4 channels promotes Ca2+ influx in the endothelium that activates small- and intermediate- conductance Ca2+-activated K+ (SK/IK) channels, leading to endothelial hyperpolarization and vasodilation (14). Additionally, increased endothelial cell Ca2+ in response to shear stress activates endothelial NO synthase (eNOS) and increases NO levels (15). However, it is unknown if LMAs express TRPV4 channels, or have the capacity to dilate in response to luminal flow and shear stress at levels that occur during large vessel occlusion. In the current study, we hypothesized that increased flow and shear stress would be a potent vasodilator of LMAs and investigated mechanisms by which this would occur, including activation of TRPV4 channels, SK/IK channels, and NO.
It is well-established that chronic hypertension, a prominent co-morbidity of stroke patients, increases the susceptibility of the brain to injury. One mechanism by which hypertension impacts stroke outcome is through poor collateral flow and low amounts of penumbral tissue. Experimental studies have shown in models of chronic hypertension that LMAs are vasoconstricted and there is rapid evolution of penumbra to infarct (7, 16, 17). In addition, endothelium-dependent vasodilation is impaired in various forms of experimental hypertension, and in primary and secondary forms of human hypertension (18). Decreased activity of NOS, increased production of superoxide ions, decreased generation of endothelium-derived hyperpolarizing factors have all been shown to contribute to the impairment of endothelium-dependent vasodilation (18). In addition, endothelial dysfunction associated with chronic hypertension is prevented with angiotensin converting enzyme (ACE) inhibitors, including improving impaired flow-dependent vasodilation in hypertensive patients (19), suggesting an important role for angiotensin II (Ang II) in impaired flow-mediated dilation. Given these findings, we also hypothesized that flow-mediated dilation of LMAs would be impaired in chronic hypertension. We therefore measured flow- and shear stress-induced dilation in LMAs from spontaneously hypertensive rats (SHR) compared to normotensive Wistar rats, and investigated mechanisms of impaired vasodilation, including the role of Ang II, NOS, nicotinamide adenine dinucleotide phosphate oxidase (NADPH oxidase or NOX)-generated superoxide, and plasminogen activator inhibitor-1 (PAI-1).
Material and Methods
Animals
Experiments were conducted using adult 16–28 week old male and female Wistar rats, and male spontaneously hypertensive rats (SHRs) from Charles River (Wilmington, MA, USA). Animals were housed in the Animal Care Facility at the University of Vermont, an Association for Assessment and Accreditation of Laboratory Animal Care-accredited facility. Animals were kept on a 12-h light/dark cycle and allowed free access to food and water. All animal procedures were approved by the Institutional Animal Care and Use Committee at the University of Vermont and complied with the National Institutes of Health guidelines for care and usage of laboratory animals. Results of experiments complied with ARRIVE guidelines. The data that support the findings of this study are available from the corresponding author upon reasonable request.
Experimental system
Figure 1A shows the diagram of the system for studying flow and shear stress in LMAs. Before animal euthanasia, resistance of proximal and distal cannulas (R1 and R2) were measured based on Ohm’s law (R= ΔP/Q) (20). A syringe pump (Kent Scientific, Torrington, CT, USA) was used to deliver a specific flow rate (0–2 μl/min), proximal and distal pressure (P1 and P2) were adjusted to maintain 40 mmHg of intravascular pressure. Shear stress was calculated by the Hagen-Poiseuille’s formula (τ = 4ηQ/πr3), with the assumptions that the vessel was cylindrical, flow was steady and laminar, and the fluid was constant viscosity and behaved as a Newtonian fluid (21, 22). See the Supplemental material for details.
Figure 1.
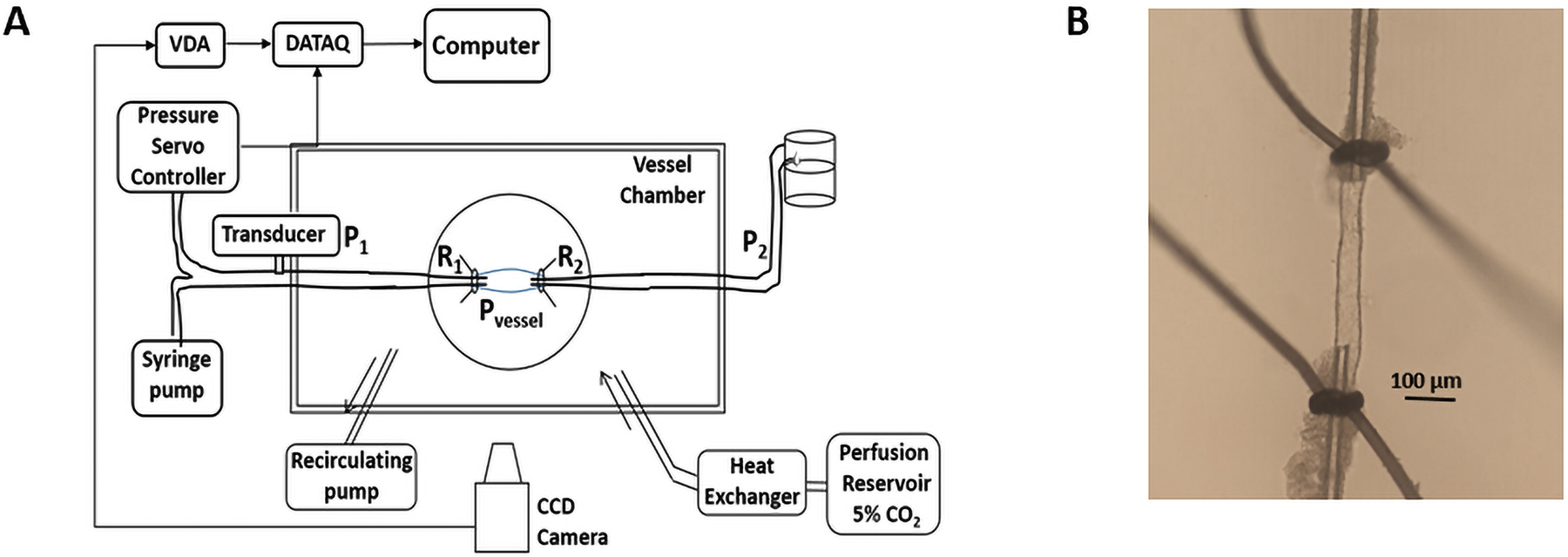
Methodology used for studying flow and shear stress in LMAs. A, Schematic image showing the system for studying flow and shear stress in LMAs. R1: proximal cannula resistance, R2: distal cannula resistance, P1: proximal pressure, P2: distal pressure, Pvessel: intravascular pressure of LMA, VDA: video dimension analyzer. B, LMA isolated, both ends mounted on glass cannulas within an arteriograph chamber and pressurized.
Experimental protocols
LMAs were studied isolated and pressurized, as we have previously done (7). To investigate the underlying mechanisms, flow-induced responses of LMAs were measured in the absence and presence of pharmacological agonists and antagonists. See Supplemental materials for details.
Drugs and solutions
Details on Drugs and solutions are available in the online-only Supplemental material.
Data calculations and statistical analysis
Data are presented as mean ± standard deviation. Data were analyzed using repeated measures analysis of variance (ANOVA) with Bonferroni post-hoc test for multiple comparisons. The D’Agostino-Pearson Omnibus normality test and ANOVA were performed using GraphPad Prism (version 8.0; San Diego, CA). Differences were considered statistically significant at P<0.05. See figure legends for detailed statistical analysis.
Results
Flow-induced vasodilation in LMAs from normotensive male and female Wistar rats
LMAs from male and female Wistar rats had similar active lumen diameters (at 40 mmHg: 46±10 μm vs 37±6 μm, P>0.05) and myogenic tone (40 mmHg: 29.3±4.8% vs 28.3±13.3%, P>0.05). As shown in the Supplemental Figure S1A, LMAs from male Wistar rats dilated to increased flow, and the vasodilation was consistent when repeated three times. In addition, reactivity of the vasodilatory response was similar for each repeated measure (Supplemental Figure S1B). Similarly, LMAs from female Wistar rats showed equivalent vasodilation to increased flow, which was repeatable three times (Supplemental Figure S1C and S1D). The average LMA vasodilatory reactivity from the three repeated measures is shown in Figure 2A and 2B and compared between male and female Wistar rats. LMAs from both male and female Wistar rats dilated similarly to increased flow. Figure 2C shows increased shear stress induced as flow rate increased, and there was no statistical difference between male and female Wistar rats. Figure 2D shows representative tracings of flow-induced vasodilation of LMAs from male and female Wistar rats. As there was no sex difference in flow-mediated response of LMAs, subsequent experiments were conducted using male rats only.
Figure 2.
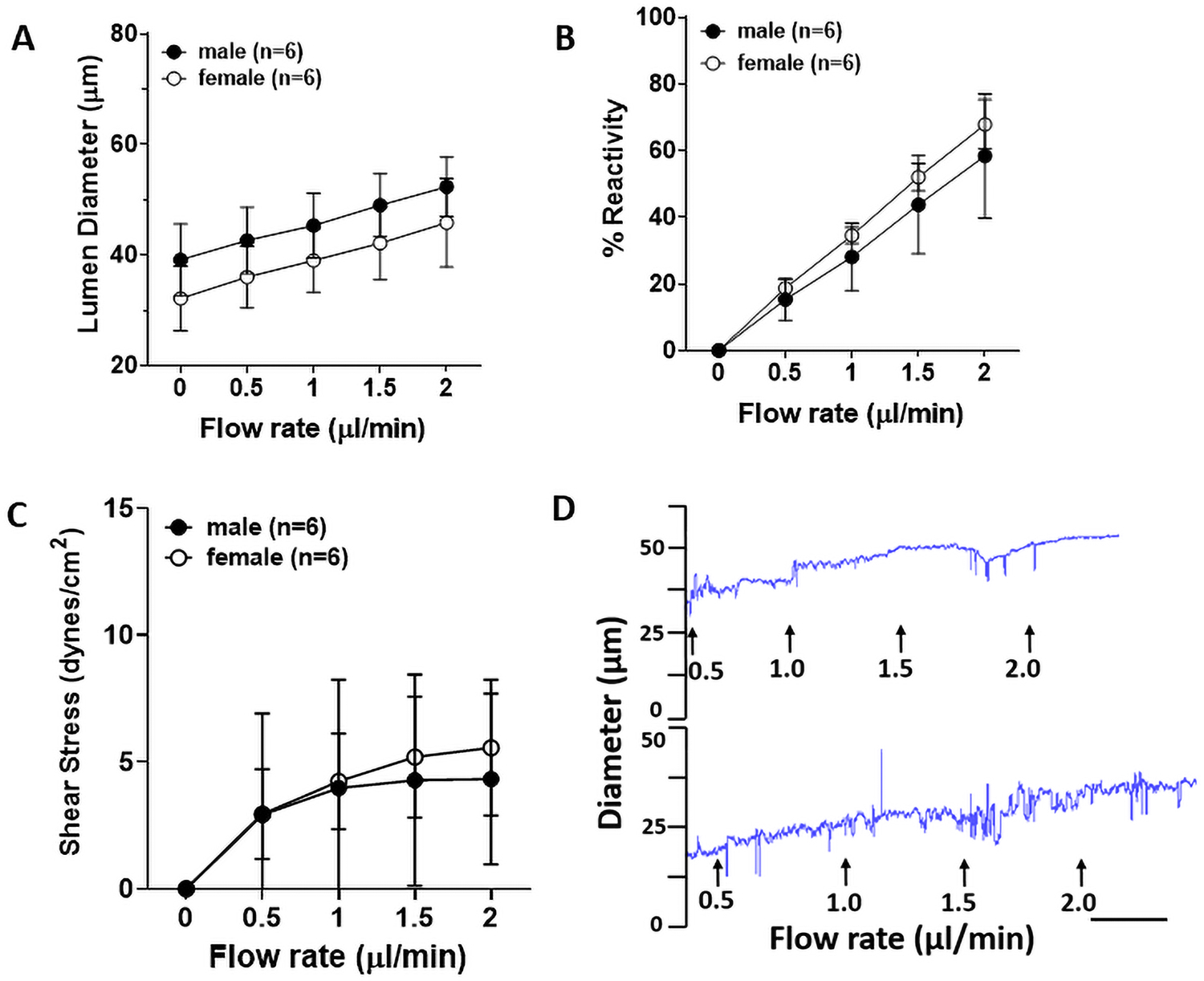
Flow-induced vascular responses in LMAs from normotensive male and female Wistar rats. A, Graph showing flow-induced lumen diameter increase in LMAs from male and female Wistar rats (n=6/group). LMAs from male and female Wistar rats had similar lumen diameter increase as the flow rate increased., Graph showing reactivity of flow-induced vasodilation in LMAs from male and female Wistar rats. LMAs from male and female Wistar rats showed similar % reactivity increase as the flow rate increased. C, Graph showing flow-induced shear stress in LMAs from both male and female Wistar rats. LMAs from male and female Wistar rats had similar luminal shear stress increase as the flow rate increased. D, Representative tracings of flow-induced vasodilatory response of LMAs from male (upper) and female (lower tracing) Wistar rats. Scale bar = 3 min. Data were analyzed by unpaired t-test.
Mechanism of flow-induced vasodilation in LMAs from male rats
We hypothesized that increased shear stress in LMAs activate TRPV4 channels, which may subsequently increase activity of NOS and SK/IK channels causing flow-induced vasodilation. To test this, we first determined if LMAs functionally express TRPV4 channels using a pharmacological approach. As shown in the Supplemental Figure S2, isolated and perfused LMAs dilated to increased concentration of TRPV4 agonist GSK101. These results indicate that TRPV4 channels are functionally expressed in LMAs.
Next, we studied flow-induced vasodilation in LMAs with or without treatment of TRPV4 antagonist HC-067047. Treatment with HC-067047 significantly reduced flow-induced vasodilation compared to control LMAs without HC-067047, indicating that TRPV4 channels were involved in flow-mediated (Figure 3A). To determine if the flow-induced vasodilation was dependent on SK or IK channels, we treated the isolated LMAs with either apamin, a specific inhibitor of SK channels, or TRAM-34, an inhibitor of IK channels. As shown in Figure 3B, TRAM-34 treatment, but not apamin, significantly prevented flow-induced dilation in LMAs, indicating flow-mediated vasodilation was dependent on IK channels, but not SK. The role of NO was also studied using the NOS inhibitor L-NAME. Figure 3C shows that L-NAME treatment significantly impaired flow-induced vasodilation in LMAs, demonstrating a role for NO in flow-induced dilation as well. A summary diagram of these mechanisms of flow-induced dilation is shown in Figure 3D.
Figure 3.
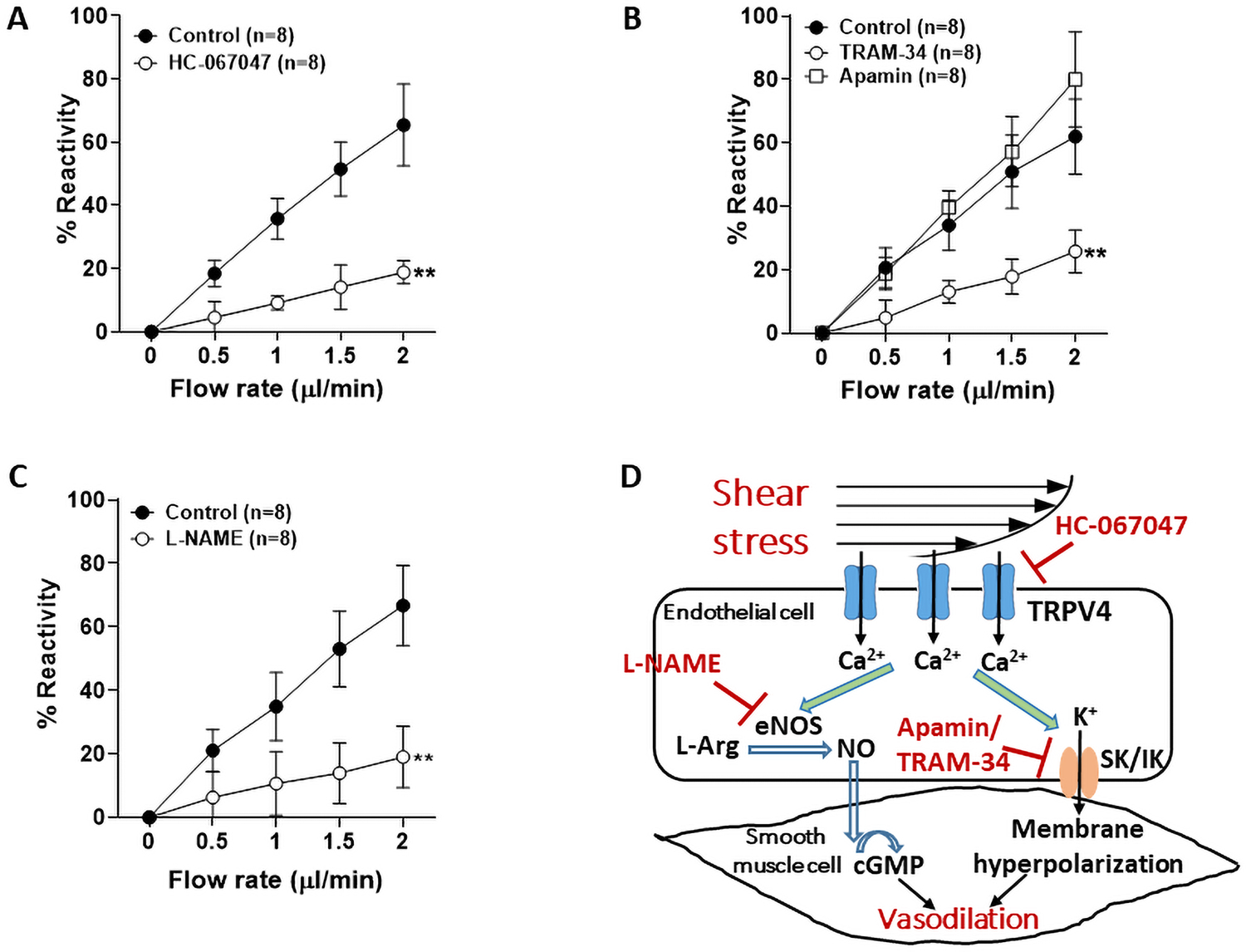
Mechanisms of flow-induced vasodilation in LMAs in normotensive male rats (n=8/group). A, Flow-induced vasodilation in LMAs with or without treatment of HC-067047 (10−6 M), a TRPV4 antagonist. Treatment with HC-067047 significantly decreased flow-induced vasodilation in LMAs. **P<0.01 vs. control group at all flow rates. B, Flow-induced vasodilation in LMAs with or without treatment of IK channel inhibitor TRAM-34 (10−6 M), or SK channel inhibitor apamin (3×10−7 M). Treatment with IK channel inhibitor significantly attenuated flow-induced vasodilation. **P<0.01 vs. control and apamin groups at all flow rates. C, Flow-induced vasodilation in LMAs with or without treatment of NOS inhibitor L-NAME (10−3 M). Treatment with L-NAME significantly diminished flow-induced vasodilation in LMAs. **P<0.01 vs. control group at all flow rates. Data were analyzed with repeated measures ANOVA followed by post-hoc Bonferroni test for multiple comparisons. D, Diagram of the pathways of flow-induced vasodilation in LMAs tested. Shear stress activates endothelial TRPV4 channels and causes Ca2+ influx, which subsequently increases activity of eNOS and NO production. NO diffuses freely to smooth muscle cells and activates soluble guanylate cyclase, which synthesizes cGMP and causes smooth muscle relaxation and subsequent dilation. Ca2+ influx also activates SK and/or IK channels and cause endothelium-dependent vasodilation. L-Arg: L-arginine, cGMP: cyclic guanosine monophosphate.
Flow-induced vasodilation was impaired in LMAs from hypertensive rats
To understand whether chronic hypertension changed the flow-induced response in LMAs, we isolated LMAs from SHRs and induced flow. As shown in Supplemental Figure S3A and S3B, isolated LMAs from SHRs constricted to increased intravascular pressure and showed considerable myogenic tone. When flow was induced, LMA lumen diameter increased little and this response was consistent when repeated 3 times (Supplemental Figure S3C and S3D).
Next, we compared the flow-induced response of LMA between Wistar rats and SHRs. As shown in Figure 4A, LMAs from SHRs were significantly smaller, and had a significantly diminished vasodilatory response to induced flow. Figure 4B shows that LMAs from SHRs had significantly impaired vasodilation and % reactivity compared to LMAs from Wistar rats. Figure 4C shows the calculated shear stress of LMAs as flow rate increased, and LMAs from SHRs had significantly higher shear stress at flow rates of 1–2 μl/min compared to Wistar rats. A representative tracing of flow-induced vascular response in LMA from SHRs was shown in Figure 4D.
Figure 4.
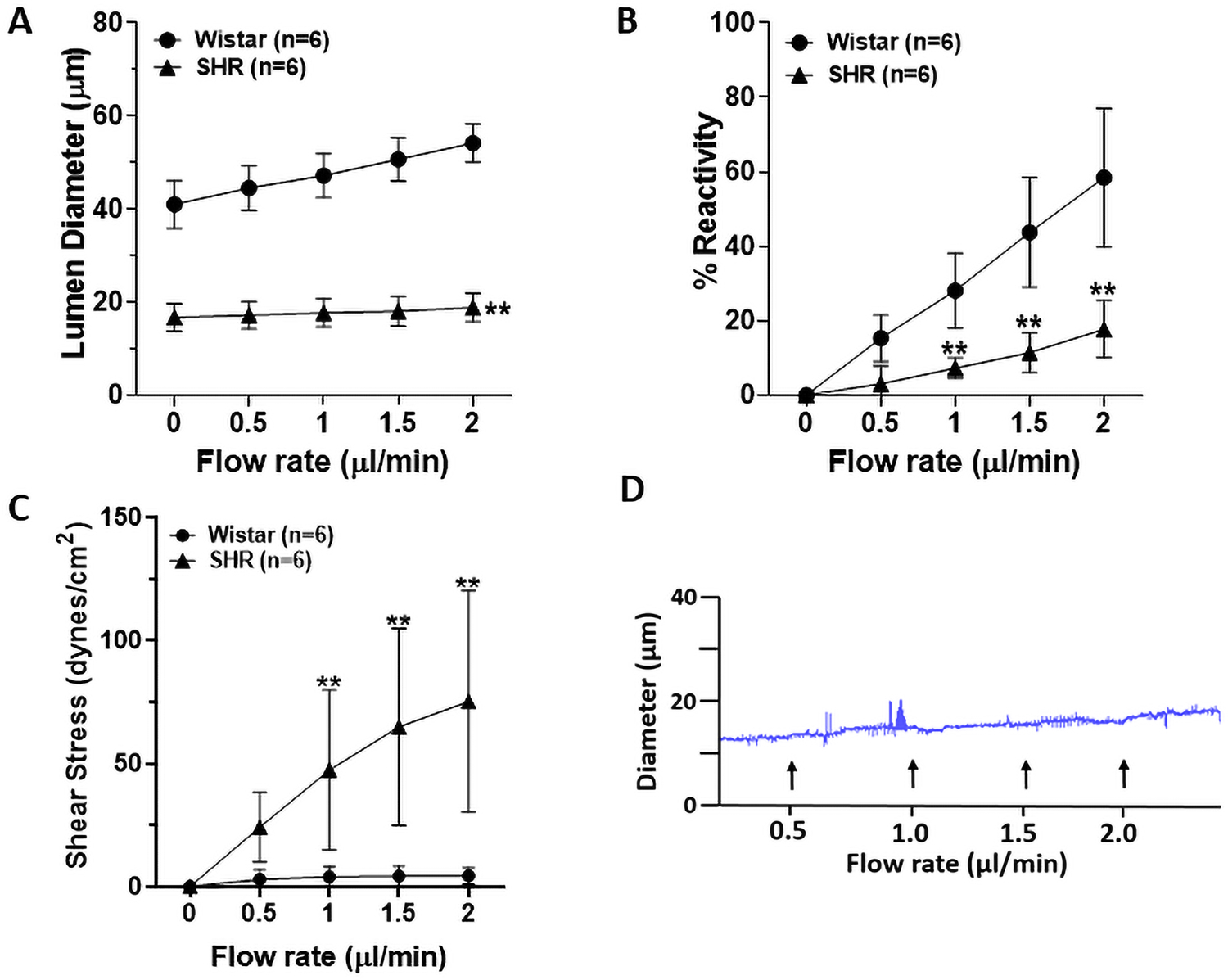
Flow- and shear stress-induced vascular responses in LMAs from male Wistar and SHRs (n=6/group). A, Graph showing lumen diameter changes in response to increased flow of LMAs from Wistar rats and SHRs. Flow-induced vascular responses were impaired in LMAs from SHRs compared to Wistar rats. **P<0.01 vs Wistar at all flow rates. B, Graph showing reactivity and flow-induced vasodilatory response of LMAs from Wistar rats and SHRs. Compared to LMAs from Wistar rats, flow-induced vasodilation was significantly diminished in LMAs from SHRs. **P<0.01 vs Wistar at all flow rates. C, Graph showing flow-induced shear stress in LMAs from both Wistar rats and SHRs. Flow-induced increase of shear stress was significantly greater in LMAs from SHRs. **P<0.01 vs Wistar. D, Representative tracing of flow-induced vascular response in LMA from SHRs. Data were analyzed with repeated measures ANOVA followed by post-hoc Bonferroni test for multiple comparisons.
Effect of Ang II and AT1Rs on flow-induced dilation in LMAs
One of the major mechanisms of vascular dysfunction during chronic hypertension involves Ang II activation of the AT1R (23). We therefore investigated the role of Ang II and AT1R activation in impaired flow-induced dilation. Figure 5A shows flow-induced dilation of isolated LMAs from Wistar rats treated with Ang II with or without AT1R blocker candesartan. Treatment with Ang II significantly impaired flow-induced vasodilation compared to LMAs at control condition, and this was restored with candesartan treatment. Figure 5B shows calculated shear stress with increased flow rate. Ang II treatment significantly elevated induced shear stress, and candesartan treatment restored shear stress to the control level. Figure 5C shows a representative tracing of impaired flow-induced vasodilation of LMAs with Ang II treatment, which was restored with addition of candesartan.
Figure 5.
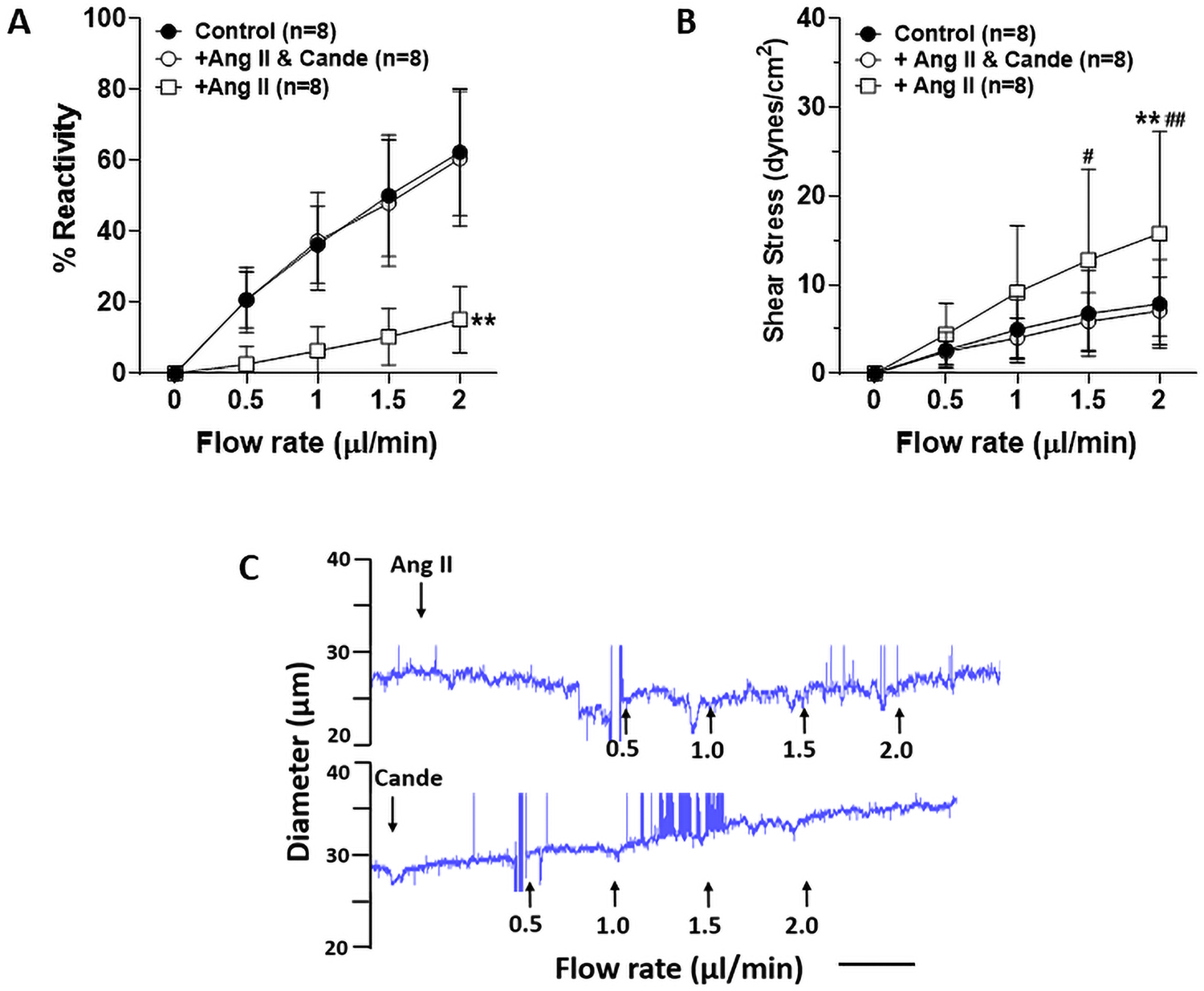
Effect of Ang II on flow-induced vasodilation in LMAs from male Wistar rats (n=8/group). A, Graph showing flow-induced vasodilation in LMAs with treatment of Ang II (10−5 M) or candesartan (Cande, 10−5 M). Ang II treatment significantly decreased flow-induced vasodilatory response which was restored by candesartan treatment. **P<0.01 vs Ang II & Cande and control groups at all flow rates. B, Graph showing flow-induced shear stress in LMAs with treatment of Ang II or candesartan, Ang II treatment significantly increased luminal shear stress induced by flow which was restored by candesartan treatment. **P<0.01 vs control group; #P<0.05, ##P<0.01 vs Ang II & Cande group. C, Representative tracings of flow-induced vascular response in LMA with treatment of Ang II (upper) or candesartan (lower tracing). Scale bar = 3 min. Data were analyzed with repeated measures ANOVA followed by post-hoc Bonferroni test for multiple comparisons.
To further investigate underlying mechanisms of impaired flow-induced vasodilation during chronic hypertension, LMAs were isolated from SHRs and the same flow rate was induced with various pharmacological treatment. LMAs were vasoconstricted and had significantly impaired flow-induced dilation and therefore we did not repeat experiments done in LMAs from Wistar rats. This was because TRAM-34 and L-NAME impaired flow-induced dilation to a similar extent as in SHR. We therefore chose an approach that investigated potential pathological pathways known to be increased in hypertension that also may have impaired flow-mediated dilation. As shown in Figure 6A, LMAs from SHRs under control conditions had only 12.4±3.5% dilatory response at 2 μl/min of flow. Treatment with apocynin to inhibit NOX restored the flow-mediated response (49.2±21.2%). Similarly, NS309 which activates SK/IK channels also restored the response (42.6±9.1%). Lastly, TM5441 was given to inhibit PAI-1 also significantly restored the impaired flow-induced vasodilation (39.8±11.1%). Apocynin, NS309 and TM5441 are all endothelium-dependent vasodilators, and the treatment dilated LMAs (15.1±4.4, 18.7±9.4%, 21.3±13.8% respectively) prior to the flow-induced vasodilation (concentrations of inhibitors were chosen that only produced a small dilation). Figure 6B shows the calculated shear stress of LMAs from SHRs to induced flow was reduced with apocynin, NS309 and TM5441 treatment. To determine if the increased flow-induced vasodilatory response was due to this initial small dilation and not the specific inhibitor, we used diltiazem, a smooth muscle cell-dependent vasodilator. Figure 6C shows that despite a similar initial vasodilation (18.8±5.0%), diltiazem treatment did not restore the impaired flow-induced vasodilation in LMA from SHRs. Figure 6D shows the pathway tested for mechanisms by which AngII might be impairing flow-mediated dilation. Via activating AT1R, elevated Ang II levels could impair SK/IK channel activity and increase PAI-1 expression in endothelial cells (24, 25). Additionally, elevated Ang II levels produce more superoxide ions, which quickly react with NO and reduce the bioavailability of NO (26).
Figure 6.
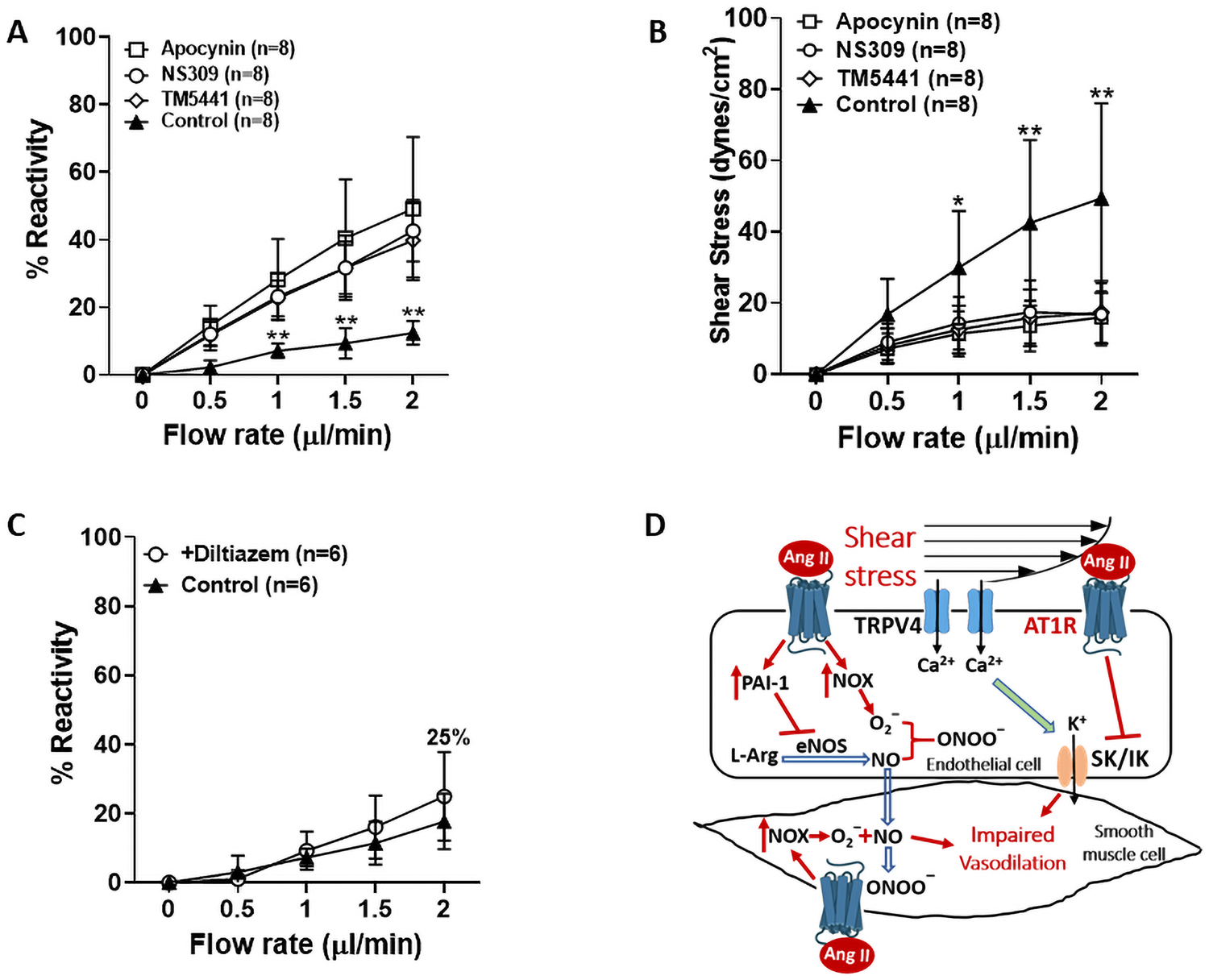
Mechanisms of impaired flow-induced vasodilation in LMAs from male SHRs (n=6–8/group). A, Graph showing flow-induced LMA vascular response with or without treatment of apocynin (10−4 M), NS309 (10−6 M) or TM5441 (10−5 M). Apocynin, NS309 and TM5441 treatments all restored the impaired flow-induced vasodilation in LMAs from SHRs. **P<0.01 vs Apocynin, NS309 and TM5441 groups. B, Graph showing flow-induced shear stress in LMAs with or without treatment of apocynin, NS309 or TM5441. Apocynin, NS309 and TM5441 treatment all restored the greater luminal shear stress induced by increased flow rates in LMAs from SHRs. *P<0.05, **P<0.01 vs Apocynin, NS309 and TM5441 groups C, Graph showing diltiazem treatment (3×10−7 M) did not change flow-induced LMA vascular response. D, Summary diagram showing the pathways of impaired flow-induced vasodilation in LMAs tested. Via activating AT1R, increased levels of Ang II impairs SK/IK channel activity and increases PAI-1 endothelial cells. In addition, elevated Ang II levels increase NOX activity to produce more superoxide ions that react with NO and reduce its bioavailability. Taken together, elevated Ang II levels during hypertension impairs flow-induced vasodilation in LMAs. Ang II: angiotensin II, AT1R: angiotensin II type 1 receptor, TRPV4: transient receptor potential vanilloid 4, PAI-1: plasminogen activator inhibitor-1, NOX: NADPH oxidase, L-Arg: L-arginine, eNOS: endothelial nitric oxide synthase, NO: nitric oxide, SK/IK: small- and intermediate- conductance Ca2+ sensitive K+ channels.
Discussion
In the current study, we showed that LMAs from normotensive Wistar rats dilated to increased flow and shear stress, similarly in males and females. Shear stress appeared to activate TRPV4 channels, which may subsequently increase activity of NOS and IK channels, thus causing flow-induced dilation in LMAs. On the contrary, flow- and shear stress-induced vasodilation was significantly impaired in LMAs from SHRs. We found that elevated levels of Ang II diminished flow- and shear stress-induced vasodilation in LMAs via activating AT1R. Additionally, the impaired flow-induced LMA vasodilation during chronic hypertension was potentially due to decreased activity of SK/IK channels, overexpression of PAI-1 and/or reactive oxygen species (ROS) that impaired NO bioavailability. These results indicate therapies that enhance collateral flow may effectively sustain the penumbra during ischemic stroke by further dilating LMAs under normotensive conditions. However, LMAs have impaired dilatory response in hypertension and may be poorly engaged in collateral therapies. Understanding the underlying mechanisms by which LMAs dilate to induced flow may lead to more effective therapies to enhance pial collateral flow.
Our study found that LMAs from normotensive rats dilated to increased flow, and the vasodilation was consistent when repeated three times. Flow- and shear stress-induced vascular responses have been studied previously in cerebral arteries and arterioles. For example, in rabbit MCAs, intraluminal flow caused relaxation of rabbit MCAs that was reduced by endothelium denudation or inhibiting NOS with L-NAME endothelium (27). In a study by Wellman et al., flow-induced dilation was inhibited by barium chloride, suggesting that flow-induced shear stress activated endothelial inwardly rectifying K+ channels, thus leading to increased synthesis and release of NO (28). Although LMAs are different vascular segments compared to MCAs, our findings showed similar mechanisms of flow-induced vasodilation. When treated with NOS inhibitor L-NAME, the flow-induced dilation in LMAs was significantly attenuated, indicating a role for NO in flow-induced vasodilation. We also found that inhibition of IK, but not SK channels reduced the flow-mediated dilation, suggesting that it is the increase in TRPV4-induced cell Ca2+ in endothelium that both activates NOS and IK channels to promote vasodilation.
TRPV4 has been shown in previous studies to be involved in flow-induced vasorelaxation (29,30). Hartmannsgruber et al. showed that shear stress-induced cerebral arterial vasodilation was eliminated in TRPV4 knockout mice (30). In endothelial cells of MCAs, activation of TRPV4 channels promotes Ca2+ influx across the luminal and abluminal face of the endothelium (31). Increased intracellular Ca2+ in endothelial cells causes subsequent activation of SK and/or IK channels, thus leading to endothelium-dependent hyperpolarization and vasodilation (14). Additionally, Ca2+ influx in response to mechanical or chemical stimulation has been shown to regulate endothelial functions, such as synthesis of endothelial NOS and NO levels (15). In the current study, we showed that treatment with TRPV4 antagonist HC-067047 significantly reduced flow-induced vasodilation, further demonstrating that TRPV4 channels were involved in flow- and shear stress-induced vasodilation. Thus, TRPV4 appears to be a primary sensor of shear stress that activates several downstream pathways to promote vasodilation.
Despite evidence showing flow- and shear stress-induced vasodilation in cerebral vasculature (27, 28, 32, 33), some studies have reported contradictory findings. Madden et al. isolated cat MCAs and reported flow-induced depolarization and constriction at 100 mmHg (34). In addition, the constriction was independent of endothelium but was through mechanisms involving free radicals and tyrosine kinase. Similarly, Bryan et al. demonstrated that integrins containing the β3-chain were involved in shear stress-induced constrictions in rat MCAs and penetrating arterioles (21); with increasing rates of shear stress, Ca2+ in vascular smooth muscle cells increased 60–70 nM that could be regulated by integrins (35). These contrary findings to the current study may be related to the level of arterial tone or shear stress. In other in vitro studies in which vessels were kept at constant pressure, constriction or dilation to flow seemed to depend on the flowrate. At 60 mmHg, rat cerebral arterioles dilated at low flow rates (≤ 10 μl/min) and constricted at higher flows (>10 μl/min) (36), whereas piglet MCAs at 20 mmHg constricted at low flowrates (0.077 to 0.152 ml/min) and dilated at higher ones (0.212 to 1.60 ml/min) (37). In the current study, we attempted to model shear stress and flow rates that have been measured during MCAO and found LMAs dilate.
We also found that flow- and shear stress-induced vasodilation was significantly impaired in LMAs from SHRs. Multiple studies have reported impaired flow-mediated vasodilation in hypertensive patients or the model of hypertension in peripheral vascular beds, such as gracilis muscle arterioles (38), epicardial coronary artery (19), and arteries from gluteal fat biopsies (39). The impaired vasodilation in those studies was largely due to impaired endothelial NO activity. Additionally, impaired TRPV4 functions have been found in hypertension and therefore may partially contribute to the impaired flow-induced vasodilation (29). Ca2+ influx through TRPV4 channels in endothelial cells of mesenteric arteries were shown to activate IK channels to induce subsequent vasodilation (29). These highly efficient TRPV4-IK signaling modules were disrupted in a mouse model of Ang II-induced hypertension, resulting in a significant reduction in endothelium-dependent vasodilation (29). Our data showed that Ang II treatment significantly diminished flow- and shear stress-induced vasodilation in LMAs from Wistar rats, and this was restored with candesartan treatment. In addition, the impaired flow-induced LMA vasodilation in SHRs was restored by pharmacological agents that target Ang II-mediated vascular effects, including the PAI-1 inhibitor TM5441 and the NOX inhibitor. The restoration of flow-mediated dilation by these compounds could be due to restoring NO through increasing bioavailability or enhanced NOS activity (40, 41). However, the selectivity of apocynin used was such that it may have scavenged hydrogen peroxide. Thus, further studies are needed to better understand these pathways, including their interactions.
There are some limitations to this study. First, we used an in vitro system to model flow and shear stress and measure vasodilation. This is a powerful technique that allowed us to keep intravascular pressure constant while changing flow rates. We chose to use an intravascular pressure of 40 mmHg for both Wistar and SHR. However, pressures in LMAs may be different in the different strains. In addition, PO2 and PCO2 were kept physiological in the bath (~90 and 40 mmHg, respectively). However, LMAs have little flow and are likely to experience lower levels of blood gases that may change their reactivity. Second, although we did not see sex differences in flow-mediated dilation, it is possible females have different mechanisms of dilation to flow than males. In addition, the response of LMAs from females to hypertension may be different. Third, we investigated the role of Ang II in LMAs from Wistar rats as proof-of-principal that it could mimic what we measured in LMAs from SHR. The potent vasoconstriction of LMAs from SHR that also had impaired flow-induced dilation precluded us from inhibiting TRPV4, NO and SK/IK channels that we used in Wistar LMAs because all these compounds also caused vasoconstriction likely because they are already inhibited or impaired in SHR. We chose instead to target pathways known to be affected in hypertension (and Ang II) that could also impair vasodilation.
Perspectives:
The present study found that LMAs from male and female Wistar rats dilated similarly to increased flow- and shear stress, and this response was mediated by TRPV4-induced NO and IK channel activation. These results may provide some basis for targeting TRPV4 and its downstream signaling mediators to improve flow- and shear stress-induced vasodilation in LMAs to enhance collateral flow. On the contrary, the vasodilation was significantly impaired in SHRs. Inhibiting pathological pathways that are elevated in hypertension, including NADPH oxidase (NOX) and PAI-1, restored flow-induced dilation in LMAs from SHRs, suggesting that targeting these pathways could improve collateral perfusion in chronic hypertension.
Supplementary Material
Novelty and Significance:
What Is New? We show that flow and shear stress caused potent vasodilation of pial collaterals that was impaired in hypertension and by Ang II.
What is Relevant? Hypertension is associated with poor collateral flow and decreased amount of salvageable tissue during large vessel occlusion. Understanding how collateral flow is increased during large vessel occlusion, and ways in which it is impaired during hypertension, could lead to therapeutics that target pial collaterals to improve stroke outcome.
Summary – We found several important targets of vasodilation for pial collaterals that could be important for increasing collateral flow during large vessel occlusion, including during hypertension.
Acknowledgements
We want to thank Dr. Gerry Herrera from the department of Pharmacology at the University of Vermont, for this tremendous help and support in setting up the experimental system.
Funding
This work was generously supported by the National Institutes of Health, National Institute of Neurological Disorders and Stroke grant 2R01 NS093289, and National Center for Research Resources grant No. 1S10OD025030-01, the Cardiovascular Research Institute of Vermont, the Totman Medical Research Trust, and the American Heart Association grant No. 19PRE34430175/Zhaojin Li/2019-2020.
Non-standard Abbreviations and Acronyms:
- Ang II
Angiotensin II
- ACE
Angiotensin converting enzyme
- IK channel
intermediate-conductance calcium-activated potassium channel
- LMA
leptomeningeal anastomoses
- L-NAME
L-nitro-arginine methyl ester
- MCAO
middle cerebral artery occlusion
- NOX
NADPH oxidase
- PAI-1
plasminogen activator inhibitor-1
- tPA
tissue plasminogen activator
- TRPV4
transient receptor potential vanilloid type 4
- SHR
spontaneous hypertensive rats
- SK channel
small-conductance calcium-activated potassium channel
Footnotes
Declaration of conflicting interests
The authors declared no conflict of interest.
References
- 1.Benjamin EJ, Muntner P, Alonso A, et al. Heart disease and stroke statistics—2019 update: a report from the American Heart Association. Circulation 2019;139:e56–e528. [DOI] [PubMed] [Google Scholar]
- 2.Baron J-C. Protecting the ischaemic penumbra as an adjunct to thrombectomy for acute stroke. Nat Rev Neurol 2018;14:325–337. [DOI] [PubMed] [Google Scholar]
- 3.Astrup J, Siesjö BK, Symon L. Thresholds in cerebral ischemia-the ischemic penumbra. Stroke 1981;12:723–725. [DOI] [PubMed] [Google Scholar]
- 4.Chuang Y-M, Chan L, Lai Y-J, et al. Configuration of the circle of Willis is associated with less symptomatic intracerebral hemorrhage in ischemic stroke patients treated with intravenous thrombolysis. J Crit Care 2013;28:166–172. [DOI] [PubMed] [Google Scholar]
- 5.Kucinski T, Koch C, Eckert B, et al. Collateral circulation is an independent radiological predictor of outcome after thrombolysis in acute ischaemic stroke. Neuroradiol 2003;45:11–18. [DOI] [PubMed] [Google Scholar]
- 6.Miteff F, Levi CR, Bateman GA, et al. The independent predictive utility of computed tomography angiographic collateral status in acute ischaemic stroke. Brain 2009;132:2231–2238. [DOI] [PubMed] [Google Scholar]
- 7.Chan S-L, Sweet JG, Bishop N, et al. Pial collateral reactivity during hypertension and aging: understanding the function of collaterals for stroke therapy. Stroke 2016;47:1618–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Beard DJ, McLeod DD, Logan CL, et al. Intracranial pressure elevation reduces flow through collateral vessels and the penetrating arterioles they supply. A possible explanation for ‘collateral failure’and infarct expansion after ischemic stroke. J Cereb Blood Flow Metab 2015;35:861–872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Chalothorn D, Faber JE. Formation and maturation of the native cerebral collateral circulation. J Mol Cell Cardiol 2010;49:251–259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Resnick N, Yahav H, Shay-Salit A, et al. Fluid shear stress and the vascular endothelium: for better and for worse. Prog Biophys Mol Biol 2003;81:177–199. [DOI] [PubMed] [Google Scholar]
- 11.Nam D, Ni C-W, Rezvan A, et al. Partial carotid ligation is a model of acutely induced disturbed flow, leading to rapid endothelial dysfunction and atherosclerosis. Am J Physiol 2009;297:H1535–H1543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Barakat AI, Lieu DK. Differential responsiveness of vascular endothelial cells to different types of fluid mechanical shear stress. Cell Biochem Biophys 2003;38:323–343. [DOI] [PubMed] [Google Scholar]
- 13.Mendoza SA, Fang J, Gutterman DD, et al. TRPV4-mediated endothelial Ca2+ influx and vasodilation in response to shear stress. Am J Physiol 2010;298:H466–H476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hannah RM, Dunn KM, Bonev AD, et al. Endothelial SKCa and IKCa channels regulate brain parenchymal arteriolar diameter and cortical cerebral blood flow. J Cereb Blood Flow Metab 2011;31:1175–1186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Wu S, Moore TM, Brough GH, et al. Cyclic nucleotide-gated channels mediate membrane depolarization following activation of store-operated calcium entry in endothelial cells. J Biol Chem 2000;275:18887–18896. [DOI] [PubMed] [Google Scholar]
- 16.Cipolla MJ, Chan S-L. Impact of Acute and Chronic Hypertension on Changes in Pial Collateral Tone In Vivo During Transient Ischemia. Hypertension 2020;76:1019–10126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.McCabe C, Gallagher L, Gsell W, et al. Differences in the evolution of the ischemic penumbra in stroke-prone spontaneously hypertensive and Wistar-Kyoto rats. Stroke 2009;40:3864–3868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Lind L Endothelium-dependent vasodilation in hypertension: a review. Blood Press 2000;9:4–15. [DOI] [PubMed] [Google Scholar]
- 19.Antony I, Lerebours G, Nitenberg A. Angiotensin-converting enzyme inhibition restores flow-dependent and cold pressor test–induced dilations in coronary arteries of hypertensive patients. Circulation 1996;94:3115–3122. [DOI] [PubMed] [Google Scholar]
- 20.Krenz GS, Dawson CA. Flow and pressure distributions in vascular networks consisting of distensible vessels. Am J Physiol 2003;284:H2192–H2203. [DOI] [PubMed] [Google Scholar]
- 21.Bryan RM Jr, Marrelli SP, Steenberg ML, et al. Effects of luminal shear stress on cerebral arteries and arterioles. Am J Physiol 2001;280:H2011–H2022. [DOI] [PubMed] [Google Scholar]
- 22.Katritsis D, Kaiktsis L, Chaniotis A, Pantos J, Efstathopoulos EP, Marmarelis V. Wall shear stress: theoretical considerations and methods of measurement. Prog Cardiovasc Dis. 2007. Mar-Apr;49(5):307–29. doi: 10.1016/j.pcad.2006.11.001. [DOI] [PubMed] [Google Scholar]
- 23.Bader M. Tissue renin-angiotensin-aldosterone systems: Targets for pharmacological therapy. Ann Review Pharml Tox 2010;50:439–465. [DOI] [PubMed] [Google Scholar]
- 24.Takeda K, Ichiki T, Tokunou T, et al. Critical role of Rho-kinase and MEK/ERK pathways for angiotensin II–induced plasminogen activator inhibitor type-1 gene expression. Aterio Thromb Vasc Biol 2001;21:868–873. [DOI] [PubMed] [Google Scholar]
- 25.Cui M, Qin G, Yu K, et al. Targeting the small-and intermediate-conductance Ca2+-activated potassium channels: The drug-binding pocket at the channel/calmodulin interface. Neurosig 2014;22:65–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Cai H, Harrison DG. Endothelial dysfunction in cardiovascular diseases: the role of oxidant stress. Circ Res 2000;87:840–844. [DOI] [PubMed] [Google Scholar]
- 27.Gaw A, Bevan J. Flow-induced relaxation of the rabbit middle cerebral artery is composed of both endothelium-dependent and-independent components. Stroke 1993;24:105–109. [DOI] [PubMed] [Google Scholar]
- 28.Wellman GC, Bevan JA. Barium inhibits the endothelium-dependent component of flow but not acetylcholine-induced relaxation in isolated rabbit cerebral arteries. J Pharm Exper Thera 1995;274:47–53. [PubMed] [Google Scholar]
- 29.Sonkusare SK, Bonev AD, Ledoux J, et al. Elementary Ca2+ signals through endothelial TRPV4 channels regulate vascular function. Science 2012;336:597–601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Hartmannsgruber V, Heyken W-T, Kacik M, et al. Arterial response to shear stress critically depends on endothelial TRPV4 expression. PloS One 2007;2:e827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Marrelli SP, O’Neil RG, Brown RC, et al. PLA2 and TRPV4 channels regulate endothelial calcium in cerebral arteries. Am J Physiol 2007;292:H1390–H1397. [DOI] [PubMed] [Google Scholar]
- 32.Sobey CG, Heistad DD, Faraci FM. Mechanisms of bradykinin-induced cerebral vasodilatation in rats: evidence that reactive oxygen species activate K+ channels. Stroke 1997;28:2290–2295. [DOI] [PubMed] [Google Scholar]
- 33.Muller JM, Chilian WM, Davis MJ. Integrin signaling transduces shear stress-dependent vasodilation of coronary arterioles. Circ Res 1997;80:320–326. [DOI] [PubMed] [Google Scholar]
- 34.Madden JA, Christman NJ. Integrin signaling, free radicals, and tyrosine kinase mediate flow constriction in isolated cerebral arteries. Am J Physiol 1999;277:H2264–H2271. [DOI] [PubMed] [Google Scholar]
- 35.Bryan RM Jr, Steenberg ML, Marrelli SP. Role of Endothelium in Shear Stress–Induced Constrictions in Rat Middle Cerebral Artery. Stroke 2001;32:1394–1400. [DOI] [PubMed] [Google Scholar]
- 36.Ngai AC, Winn HR. Modulation of cerebral arteriolar diameter by intraluminal flow and pressure. Circ Res 1995;77:832–840. [DOI] [PubMed] [Google Scholar]
- 37.Shimoda LA, Norins NA, Jeutter DC, et al. Flow-induced responses in piglet isolated cerebral arteries. Ped Res 1996;39:574–583. [DOI] [PubMed] [Google Scholar]
- 38.Koller A, Huang A. Impaired nitric oxide-mediated flow-induced dilation in arterioles of spontaneously hypertensive rats. Circ Res 1994;74:416–421. [DOI] [PubMed] [Google Scholar]
- 39.Paniagua OA, Bryant MB, Panza JA. Role of endothelial nitric oxide in shear stress–induced vasodilation of human microvasculature: diminished activity in hypertensive and hypercholesterolemic patients. Circulation 2001;103:1752–1758. [DOI] [PubMed] [Google Scholar]
- 40.Garcia V, Park EJ, Siragusa M, et al. Unbiased proteomics identifies plasminogen activator inhibitor-1 as a negative regulator of endothelial nitric oxide synthase. Proc Nat Acad Sci 2020;117:9497–9507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Boe AE, Eren M, Murphy SB, et al. Plasminogen activator inhibitor-1 antagonist TM5441 attenuates Nω-nitro-l-arginine methyl ester–induced hypertension and vascular senescence. Circulation 2013;128:2318–2324. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.