

Abstract
Tuberculosis consistently causes more deaths worldwide annually than any other single pathogen, making new effective vaccines an urgent priority for global public health. Amongst potential adjuvants, STING-activating cyclic-di-nucleotides (CDNs) uniquely stimulate a cytosolic sensing pathway activated only by pathogens. Recently, we demonstrated that a CDN-adjuvanted protein subunit vaccine robustly protects against tuberculosis infection in mice. Here we delineate the mechanistic basis underlying the efficacy of CDN vaccines for tuberculosis. CDN vaccines elicit CD4 T cells that home to lung parenchyma and penetrate into macrophage lesions in the lung. Although CDNs, like other mucosal vaccines, generate B cell-containing lymphoid structures in the lungs, protection is independent of B cells. Mucosal vaccination with a CDN vaccine induces Th1, Th17 and Th1-Th17 cells, and protection is dependent upon both IL-17 and IFN-γ. scRNA-seq experiments reveal that vaccination enhances a metabolic state in Th17 cells reflective of activated effector function and implicate expression of Tnfsf8 (CD153) in vaccine induced protection. Finally, we demonstrate that simply eliciting Th17 cells via mucosal vaccination with any adjuvant is not sufficient for protection. A vaccine adjuvanted with deacylated monophosphoryl lipid A (MPLA) failed to protect against TB infection when delivered mucosally, despite eliciting Th17 cells, highlighting the unique promise of CDNs as adjuvants for TB vaccines.
Introduction
Tuberculosis (TB) causes more deaths worldwide than any other single infectious agent (1). The existing live-attenuated vaccine against TB, M. bovis Bacillus Calmette-Guérin (BCG), is widely administered in many countries with endemic TB and is effective against severe forms of childhood disease (1). However, efficacy wanes over time and BCG provides minimal protection against adult pulmonary TB (2–4). There is a clear need for an improved vaccine to combat this global health threat. One of the barriers to the development of effective vaccines for TB is the lack of reliable immune correlates of protection. Immunity to TB has long been considered to be dependent on the production of IFN-γ by CD4 T cells (5), as defects in IFN-γ signaling are linked to susceptibility to mycobacterial infection (6–8). Thus, the development of new TB vaccines to date has focused on eliciting IFN-γ-producing CD4 T cells and polyfunctional CD4 T cells that produce IFN-γ, TNF-α, and IL-2 (9, 10). A highly anticipated clinical trial of the BCG booster protein subunit vaccine MVA85A showed successful induction of M. tuberculosis-specific polyfunctional CD4 T cells in infants (11). However, this immune response did not translate into significant protection against TB infection or disease (11). More recently, the protein subunit vaccine candidate M72/AS01E induced an IFN-γ-producing T cell response and demonstrated 50% efficacy in preventing the transition from latent to active disease over a 3-year period (12). This protein subunit vaccine contains AS01E, an adjuvant consisting of the TLR4 agonist 3-deacylated monophosphoryl lipid A (MPLA) and saponin. Why two vaccines that both elicit IFN-γ-producing CD4 T cells bore different results is unclear, however, these results suggest that alternate biomarkers may more clearly predict vaccine efficacy.
Animal studies have suggested the existence of IFN-γ-independent mechanisms of CD4 T cell-mediated control of M. tuberculosis infection (6, 13–15). However, the basis of this control has remained elusive (16). IL-17-producing Th17 cells do not contribute to control of primary TB infection in mice, possibly because they are not robustly elicited during primary infection. However, intranasal administration of BCG (17, 18) or protein subunit vaccines (18, 19) elicits IL-17-producing memory T cells that correlate with enhanced protection against M. tuberculosis challenge in rodent models. Th17 cells are a heterogenous effector subset of CD4 T cells that can range in function from inflammatory to regulatory and can even transdifferentiate into Th1 cells (20). Although TB vaccine-elicited memory Th17s can persist in lung-draining lymph nodes and acquire Th1 characteristics after challenge (21), it is unclear which Th17 subtype is most important for vaccine-elicited control of TB, or how this subtype enhances control of M. tuberculosis infection. Furthermore, it is unclear whether different vaccines administered intranasally will uniformly elicit Th17-dependent protection, or whether the specific adjuvant or antigen used in the vaccine is paramount. It is also unclear whether mucosal vaccination results in IL-17-independent impacts on vaccine efficacy.
In order to exert control of infection, T cells must both differentiate into a protective effector state and adopt an effector phenotype that allows them to migrate to the site of infected macrophages in the lung. In mouse models, lung parenchyma-homing CXCR3+ CD153+ KLRG1− CD4 T cells mediate superior protection against M. tuberculosis compared to vasculature-associated KLRG1+ CD4 T cells that do not migrate into the lung tissue (22, 23). Furthermore, T cells that are blocked from entering the granuloma core are unable to provide as effective protection as T cells that penetrate into the central core of infected macrophages (24). However, it is unclear how to rationally tailor a vaccine that will elicit CD4 T cell homing to granuloma cores.
We recently demonstrated that an experimental protein subunit vaccine formulated with STING-activating cyclic-di-nucleotide (CDN) adjuvants is highly efficacious in mice, particularly when delivered by a mucosal route (25). Here we find that protective efficacy correlates with penetration of T cells into lung lesions and robust IL-17 production by CD4 T cells. We further show that type I IFN, IL-17, and IFN-γ are all required for complete intranasal CDN-adjuvanted vaccine-induced protection. While intranasal vaccine delivery elicits IFN-γ-producing CD4 T cells, vaccine-mediated protection is partially IFN-γ-independent and does not correlate with increased Th1 responses. Intranasal vaccination induces T cell transcriptional heterogeneity, particularly within the Il17a-expressing subset, and a large number of CD4 T cells co-expressing Il17a and Ifng, hereafter referred to as Th1-Th17 cells. To define additional functional characteristics of protective CD4 T cells, we performed single cell RNA-sequencing (scRNA-seq). Parenchyma-homing CXCR3+ T cells have higher levels of expression of markers of memory and activation and are also enriched for expression of Tnfsf8 (CD153), a protein previously shown to enhance the efficacy of CD4 T cells against M. tuberculosis in the mouse model (22). Relative to Th17 cells, Th1 cells express markers of TCR signaling and exhaustion as well as type I IFN signaling. Cxcr3-expressing T cells and Il17a-expressing T cells both display transcriptional signatures of HIF-1α activation. Importantly, we find that the efficacy of CDN vaccines delivered via the intranasal route is not merely a function of the route of delivery, as an experimental vaccine adjuvanted with MPLA delivered via intranasal administration does not induce significant protection. Finally, we also show that, although we and others have found that intranasal vaccination results in B cell aggregates in the lungs of infected mice, B cells are dispensable for vaccine-induced protection. These results demonstrate that CDN-adjuvanted protein subunit vaccines generate a multifaceted CD4 T cell response that leads to enhanced protection against TB disease.
Materials and Methods
Ethics Statement.
All procedures involving the use of mice were approved by the University of California, Berkeley Institutional Animal Care and Use Committee (protocol 2015-09-7979). All protocols conform to federal regulations, the National Research Council Guide for the Care and Use of Laboratory Animals, and the Public Health Service Policy on Humane Care and Use of Laboratory Animals.
Vaccine Reagents.
ML-RR-cGAMP was synthesized at Aduro Biotech as described previously (26). Monophosphoryl lipid A (MPLA) was purchased from InvivoGen (San Diego, CA). AddaVax (InvivoGen) was used for the subcutaneous formulation of antigen and adjuvant as directed by the manufacturer. 5Ag fusion protein was provided by Aeras, H1 fusion protein was provided by Aeras and Statens Serum Institut, and peptide pools were provided by the NIH BEI Resources Repository.
Mice.
C57BL/6J (#000664), Ighmtm1Cgn (muMt−, # 002288), Il17aCre (Il17−/−, #016879), Ifng−/− (#002287) and Stat6−/− (#005977) mice were obtained from The Jackson Laboratory (Bar Harbor, ME) and bred in-house. Ifnar1−/− (Ifnar−/−, Jackson Laboratory strain #028288) mice were obtained from the Vance lab (University of California, Berkeley). Mice were sex- and age-matched for all experiments.
Bacterial Culture.
M. tuberculosis strain Erdman and M. bovis BCG (Pasteur) were grown in Middlebrook 7H9 liquid medium supplemented with 10% albumin-dextrose-saline (M. tuberculosis) or 10% oleic acid, albumin, dextrose, catalase (OADC) (BCG), 0.4% glycerol, and 0.05% Tween 80 or on solid 7H10 agar plates supplemented with 10% Middlebrook OADC (BD Biosciences) and 0.4% glycerol. Frozen stocks of M. tuberculosis and BCG were made from single cultures and used for all experiments.
Vaccinations.
5 μg ML-RR-cGAMP (CDN) or 5 μg MPLA and 3 μg fusion antigen protein (5Ag or H1) were formulated in PBS for intranasal (i.n.) delivery or in 2% AddaVax in PBS for subcutaneous (s.c.) delivery. Mice were vaccinated three times at 4-week intervals with 20 μL i.n. or with 100 μL s.c. at the base of the tail (50 μL on each flank). BCG-vaccinated mice were injected once with 2.5 × 105 CFU/mouse in 100 μL of PBS plus 0.05% Tween 80 s.c. in the scruff of the neck.
Challenge Experiments with M. tuberculosis.
Twelve weeks after the initial vaccine injection, mice were infected via the aerosol route with M. tuberculosis strain Erdman. Aerosol infection was done using a nebulizer and full-body inhalation exposure system (Glas-Col, Terre Haute, IN). A total of 9 mL of culture diluted in sterile water was loaded into the nebulizer calibrated to deliver ~100 bacteria per mouse as measured by CFU in the lungs 1 day following infection (data not shown).
Pre-challenge ICS Assay.
Heparinized blood lymphocytes were isolated 9 weeks post priming, processed as previously described (25), stained with Live/Dead stain (Thermo Fisher Scientific L34970), Fc receptor block, and antibodies specific for CXCR3, major histocompatibility complex (MHC) class II, and TCRγ/δ (BioLegend 101319, 126522, 107606, and 118124, respectively), and CD4, CD8, CD90.2, CD153, KLRG1 and Ly6G (BD Biosciences 564933, 563898, 561616, 740542, 740279 and 551460, respectively), and fixed/permeabilized with BD Cytofix/Cytoperm Fixation/Permeabilization Solution Kit (Thermo Fisher, 554714) before staining with antibodies specific for IFN-γ (eBioscience 12-73111-81) and IL-17 (BioLegend 506904). Data were collected using a BD LSR Fortessa flow cytometer and analyzed using FlowJo Software (Tree Star, Ashland, OR).
Post Challenge Cell Preparation.
For bacterial enumeration, the largest lung lobe was homogenized in PBS plus 0.05% Tween 80, and serial dilutions were plated on 7H10 plates. CFU were counted 21 days after plating. The second largest lung lobe was fixed in 10% formalin for histological analysis or used for single cell RNA-sequencing. The remaining lung lobes were harvested into complete RPMI, dissociated, and strained through a 40 μm strainer. Cells were restimulated with no peptide or Ag85B peptide pools (2 μg/mL) with Protein Transport Inhibitor Cocktail (eBioscience 00-4980-93) for 5 hours at 37°C. Cells were washed and stained with antibodies used for pre-challenge ICS, or with the following for myeloid cell staining: live/dead (eBioscience 65-0865-14), and antibodies specific for CD4 (BD 564933), CD3, CD8a, CD11b, CD11c, CD45, B220, Ly6C, and Ly6G (Biolegend 100205, 100750, 101257, 117317, 103127, 103211, 128011, and 127628, respectively). Data were collected and analyzed as outlined above. Data presented are from Ag85B-stimulated samples for cytokine staining, and unstimulated samples for all other flow cytometric analyses.
Histological and immunohistochemical analysis.
Lung lobes were fixed in 10% formalin at room temperature for at least 24 hours. Histology was performed by HistoWiz Inc. (histowiz.com) using a Standard Operating Procedure and fully automated workflow. Samples were processed, embedded in paraffin, and sectioned at 4 μm. Antibodies used were rat monoclonal anti-CD45R/B220 (Biolegend 103229), rat monoclonal anti-CD3 primary antibody (Abcam), and rabbit anti-rat secondary antibody (Vector). BOND polymer Refine Detection (Leica Biosystems) was used according to manufacturers’ protocol. After staining, sections were dehydrated and film coverslipped using a TissueTek-Prisma and Coverslipper (Sakura). Whole slide scanning (40x) was performed on an Aperio AT2 (Leica Biosystems). Slide images are available online at https://app.histowiz.com/shared_orders/6d2c7816-b292-42c4-9ee2-71ffe032957e/slides/. Tissue sections were evaluated by a veterinary anatomic pathologist at the UC Davis Comparative Pathology Laboratory. Hemotoxylin and eosin (H&E) images were analyzed using Olympus cellSens software. Semiquantitative scoring was used to evaluate the degree of inflammation and the presence and severity of an array of inflammatory features. Lymphoid nodules are defined as discrete, nodular or ovoid, homogenous aggregates of lymphocytes comprising an area > 1000 μm2; lymphoid follicles exhibit germinal center development as well as the previously stated features; granulomas are defined as discrete, nodular aggregates of macrophages that efface pulmonary parenchyma, occasionally exhibit circumferential arrangement, and contain multinucleate giant cells. Immunohistochemical and immunofluorescent staining was analyzed using QuPath software (27). In brief, classifiers were trained to detect regions of inflammation, cytoplasmic CD3 immunoreactivity, and cytoplasmic B220 immunoreactivity by manual annotation of a subset of images. Classifiers were applied to either the total area of a lung section or to manually annotated regions of interest (ROIs) within a lung section. A single measurement classifier was generated for detection of cytoplasmic CD3 immunoreactivity within lesion. The classifier was applied to three equal-area ROI in each section of lung. For detection of cytoplasmic B220 immunoreactivity, classifiers were applied to the total sectional area of each lung section and the percentage of B220+ immune cells was determined. Lymphoid nodules were defined as discrete, nodular or ovoid, homogenous aggregates of lymphocytes with an area > 10000 μm2. Granulomas were defined as discrete, nodular aggregates of macrophages that efface pulmonary parenchyma, occasionally exhibit circumferential arrangement, and contain multinucleate giant cells.
Single Cell RNA Sequencing.
The second largest lung lobes were pooled from three mock or i.n. H1/ML-RR-cGAMP-vaccinated mice, tissue dissociated in RPMI containing liberase and DNase I in gentleMACS C tubes using the gentleMACS dissociator and strained through a 70 μm filter for sorting on a Sony SH-800 sorter using a 100 μm nozzle. Sorted CD4 T cells were washed in RPMI and counted. 5000–6000 CD4 T cells per sample were used for single cell RNA sequencing (scRNA-seq) according to the 10x Genomics protocol. Briefly, single cell suspensions were partitioned into Gel Beads in emulsion (GEMs) using the 3’ Chromium Next GEM single Cell v3.1 system. 10x barcoded primers were used to reverse transcribe poly-adenylated mRNA to produce barcoded, full-length cDNA. Purified DNA was amplified, fragmented, and amplified again to attach Illumina adapter sequences by the Functional Genomics Laboratory at UC Berkeley. Libraries were sequenced on an Illumina NovaSeq SP 100SR and demultiplexed by the Vincent J. Coates Genomics Sequencing Laboratory at UC Berkeley. Reads were aligned to the mouse transcriptome mm10 with the 10x Cell Ranger analysis pipeline (28), using the Savio computational cluster at UC Berkeley. After filtering, barcode counting, and UMI counting, the Scanpy toolkit (29) was used to preprocess the data. The single cell variational inference (scVI) (30) approach was utilized to project the data from both vaccinated and vaccine-naïve mice into a shared lower-dimensional latent space. Uniform manifold approximation and projection (UMAP) and Leiden clustering were then applied on the k-Nearest-Neighbor graph of the latent space for visualization and clustering of the data. Leiden clusters that expressed high levels of myeloid, B cell, or CD8 T cell markers were excluded from the data before utilizing the scVI model a second time. The filtered data was annotated using signature genes from CD4 T cell subset in cellxgene (https://github.com/chanzuckerberg/cellxgene), a single-cell data exploration tool. Pathway analysis was performed using Ingenuity Pathway Analysis (IPA). Metabolic flux analysis was performed using Compass v 0.9.9.5 (31). All data has been deposited in GEO (GSE164590).
Scanpy was used to visualize data and test differential expression of genes between samples and CD4 T cell subsets. The single-cell ANnotation using Variational Inference (scANVI) variant of scVI was used to assign annotations to unassigned cells based on high-confidence seed labels annotated on cellxgene (32). High-confidence seed cells were identified using a high threshold of expression as cutoffs. In the scANVI analysis, Tbx21 and Rorc were also used to define classical Th1 and Th17, respectively.
Statistical Analysis.
Data are presented as mean values, and error bars represent SD. Symbols represent individual animals. The number of samples and statistical tests used are denoted in the legend of the corresponding ure for each experiment. Analysis of statistical significance was performed using GraphPad Prism 8 (GraphPad, La Jolla, CA), and p < 0.05 was considered significant.
Results
We previously found that mucosal administration of a protein subunit vaccine with CDN adjuvants elicits significant protection against M. tuberculosis in the mouse model of infection (26). We therefore set out to define the mechanisms underlying the efficacy of CDN adjuvants for TB vaccines. For antigen we used either 5Ag—a five antigen fusion of M. tuberculosis proteins that includes the immunodominant proteins Ag85B and ESAT-6 and three proteins predicted to be expressed in bacterial latency—or H1 antigen, a fusion of Ag85b-ESAT6 alone, as 5Ag and H1 were functionally equivalent in their ability to elicit immune cells and for protection out to 4 weeks post challenge (25). Protein antigen was formulated with the CDN ML-RR-cGAMP (33).
ML-RR-cGAMP-adjuvanted protein subunit vaccines elicit an expanded population of CD4 T cells in the lungs of mice prior to challenge with M. tuberculosis.
Previously, we demonstrated that CDN vaccines elicit robust protection at 4 weeks post challenge (25). We next set out to characterize vaccination elicited changes in the cellular immune response that could be detected prior to challenge with M. tuberculosis. Mice were vaccinated by the intranasal route (i.n.) with H1/ML-RR-cGAMP three times at 4-week intervals, and immune cells residing within the lungs were evaluated 4 weeks after the third vaccination. Only CD4 T cell and alveolar macrophage populations changed substantially with vaccination, with both cell types constituting a larger percentage of lung CD45+ cells in vaccinated mice (Figure 1a). Previously, we found that vaccination with H1/ML-RR-cGAMP via the intranasal route resulted in increased numbers of IL-17- and IFN-γ/IL-17-producing T cells in the lungs at the peak of the immune response at 4 weeks post challenge, as well as an increase in CXCR3+KLRG1− cells, which are known to home to lung parenchyma. We next sought to determine whether these CD4 T cell subsets are present in the lungs after vaccination but prior to infection with M. tuberculosis. We found that vaccination with H1/ML-RR-cGAMP resulted in increased percentages of IFN-γ+, IL-17+, and IFN-γ+/IL-17+ cells in the lungs (Figure 1b, 1c, 1d). Furthermore, vaccination resulted in a dramatic increase in the fraction of all CD4 T cells expressing lung parenchyma-homing marker CXCR3, and a decreased fraction expressing terminal differentiation and pulmonary vasculature retention marker KLRG1 (Figure 1e, 1f). Thus, intranasal vaccination with H1/ML-RR-cGAMP changes the characteristics of the resident CD4 T cell population existing in the lungs prior to challenge with M. tuberculosis.
Figure 1. Intranasal immunization with H1/CDN enhances CD4 T cell responses in the lungs of vaccinated mice both pre and post-challenge.
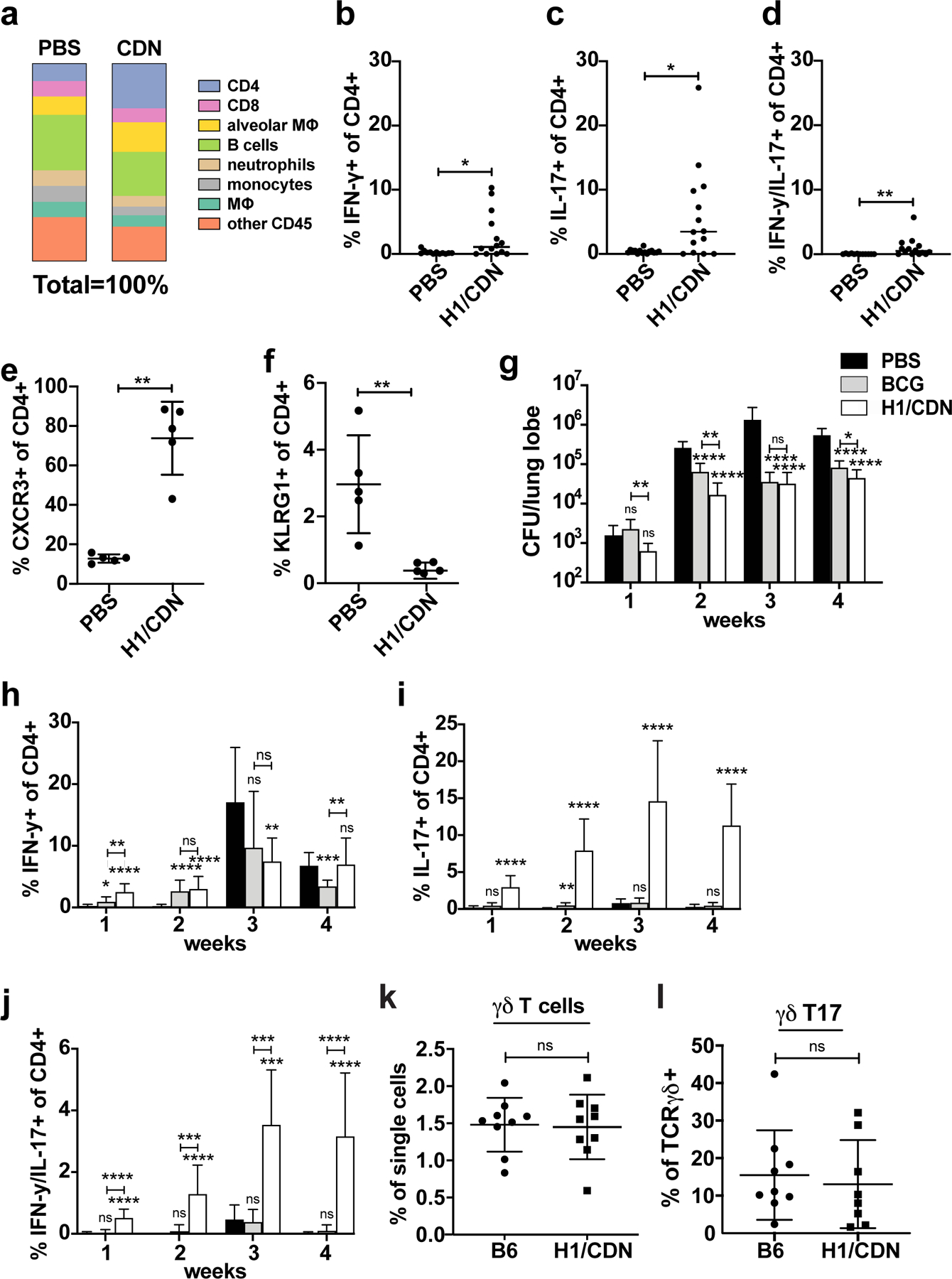
Mice were vaccinated with intranasal M. tuberculosis H1 antigen and ML-RR-cGAMP cyclic-di-nucleotide adjuvant (H1/CDN) 12 weeks before challenge, then boosted twice at 4-week intervals after priming, before M. tuberculosis aerosol challenge with 50–100 CFU. Mice were mock primed and boosted with PBS as a control. (a) Cellular percentages were enumerated in the lungs using flow cytometry in vaccinated and mock vaccinated mice on day 0 prior to challenge with M. tuberculosis. (b) Intracellular cytokine staining (ICS) for percentage of lung CD4 T cells that produce IFN-γ (Th1), (c) IL-17 (Th17), (d) both IFN-γ and IL-17 (Th1-Th17) after restimulation ex vivo with recombinant Ag85b peptide pool on day 0 prior to challenge with M. tuberculosis. Percentage of CD4 T cells that express (e) CXCR3 (parenchymal T cells) or (f) KLRG1 (vascular T cells) on day 0 prior to challenge with M. tuberculosis. Mice were vaccinated with s.c. BCG or i.n. H1/CDN as above and (g) lung bacterial burden was enumerated at 1 – 4 weeks post M. tuberculosis challenge. (h) Intracellular cytokine staining (ICS) for percentage of lung CD4 T cells that produce IFN-γ (Th1) (i) IL-17 (Th17), or (j) both IFN-γ and IL-17 (Th1-Th17) after restimulation ex vivo with recombinant Ag85b peptide pool at indicated timepoints post challenge. (k) Enumeration of TCRɣδ and (l) IL-17-producing TCRɣδ T cells at 4 weeks post challenge. Data are expressed as mean (± SD) of eight to ten mice per group from two independent experiments; in (b-f; k, l) each animal is represented by a point. Mann-Whitney t test p values calculated in comparison to PBS vaccinated controls except where indicated; *p ≤ 0.05, **p ≤ 0.01, ***p ≤ 0.001, ****p ≤ 0.0001. Significance is relative to PBS control unless otherwise indicated. All experiments were performed a minimum of two times; for a-d two pooled experiments are shown; otherwise a representative experiment is shown.
ML-RR-cGAMP-adjuvanted protein subunit vaccines elicit a rapid influx of T cells and control of infection upon M. tuberculosis challenge.
We next set out to determine how early the i.n. H1/ML-RR-cGAMP vaccine is able to establish control in the lungs. For analysis of vaccine kinetics, we used the control of s.c. administration of BCG, the standard route of administration, which was previously shown to accelerate the arrival of Th1 cells into the lungs post challenge with M. tuberculosis (34). Vaccination with H1/ML-RR-cGAMP resulted in increased protection when compared with BCG at 1- and 2-weeks post challenge, demonstrating that mucosal CDN-adjuvanted vaccination induces immunity earlier than BCG vaccination (Fig. 1g). We next set out to determine whether vaccination accelerates the influx of CD4 T cells into the lung. Th1 frequency dramatically increased in mock (PBS) vaccinated mice between 2–3 weeks post challenge (Fig. 1h), eclipsing that of the vaccinated mice and coinciding with a larger bacterial burden (Fig. 1g). We also observed an influx of antigen-specific IL-17-producing T cells into the lungs at 1 week post challenge that increased steadily until 3 weeks post challenge (Fig. 1i). Neither mock nor BCG-vaccinated mice showed appreciable Th17 responses (Fig. 1i). A smaller population of CD4 T cells produced both IFN-γ and IL-17, predominantly in H1/ML-RR-cGAMP-vaccinated mice (Fig. 1j). While several sources have suggested that TCRγ/δ T cells are the main source of IL-17 in M. tuberculosis-infected mouse lungs during primary infection (35), we observed no difference in frequencies of TCRγ/δ+ T cells or in the subset of IL-17+ TCRγ/δ+ T cells in mock and H1/ML-RR-cGAMP-vaccinated mice after infection (Fig. 1k, 1l). Thus, the evidence points to a role for vaccine-induced conventional IL-17-producing CD4 T cells in early and sustained mucosal vaccine efficacy.
Lymphocytes home to infected tissue in ML-RR-cGAMP-adjuvanted vaccinated mice.
Proper localization of T cells in infected lungs is crucial for effective T cell-mediated immunity (36). CXCR3hi CD4 T cells can localize to the infected mouse lung parenchyma and mediate superior bacterial resistance compared to KLRG1hi T cells, which remain trapped in the vasculature (23, 37). In agreement with what we previously reported, i.n. vaccination with H1/ML-RR-cGAMP induced higher frequencies of CXCR3+ and lower frequencies of KLRG1+ CD4 T cells in infected mouse lungs than s.c. BCG vaccination or mock vaccination (Fig. 2a, 2b). Conversely, BCG vaccination induced a higher percentage of KLRG1+ cells than i.n. H1/ML-RR-cGAMP vaccination (Fig. 2b). In vaccine-naïve infected mice, significant numbers of KLRG1+ T cells appeared in the lungs at 3–4 weeks post M. tuberculosis challenge (Fig. 2b). We next sought to determine whether i.n. vaccination changed T cell localization within infected lung tissue at 4 weeks post challenge. Consistent with previous reports, we found that in vaccine-naïve infected mice, very few CD3+ T cells were observed in lung lesions (Fig. 2c). However, i.n. vaccination effectively enhanced infiltration of CD3+ T cells into the lesion (Fig. 2c, e) without increasing overall lung inflammation (Fig. S1a). Indeed, overall levels of inflammation did not correlate with protection (Fig. S1a–g).
Figure 2. Intranasal immunization with H1/CDN promotes T cell influx into macrophage aggregates in lungs.
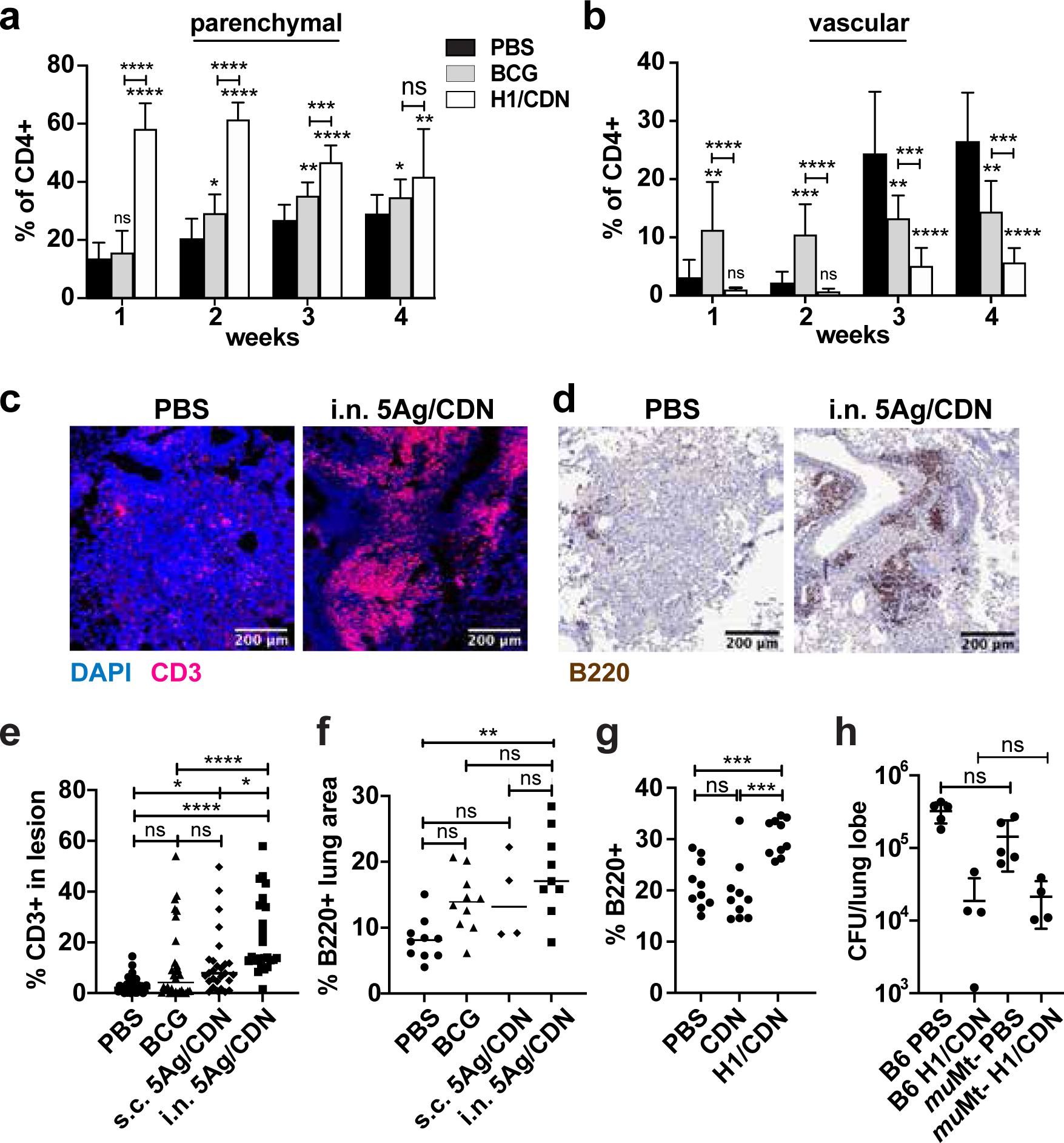
Mice were immunized as described and analyzed at indicated timepoints. (a) Surface staining for percentage of parenchymal CXCR3+ KLRG1− lung CD4 T cells from mock and i.n. H1/ML-RR-cGAMP (i.n. H1/CDN) vaccinated mice. (b) Surface staining for percentage of vascular CXCR3− KLRG1+ lung CD4 T cells. Mice were immunized as described and analyzed at 4 weeks post challenge with M. tuberculosis. (c) Representative immunofluorescent staining of formalin-fixed, paraffin-embedded lung sections from mock and 5Ag/ML-RR-cGAMP-vaccinated mice for T cell marker CD3 and nuclear DAPI stain. (d) Representative immunohistochemical staining of lung sections for B cell marker B220. (e) Quantification of CD3+ immune cells out of total cells in lung lesions. (f) Quantification of B220+ lung area. (g) Percentage of B220+ cells in the lungs at 4 weeks post challenge analyzed by flow cytometry. (h) WT and muMT− mice were immunized with H1/CDN and M. tuberculosis CFU in the lungs was enumerated at 4 weeks post challenge. Data are expressed as mean (± SD) of eight to ten mice per group from two independent experiments; each animal is shown as an individual point. (a, b, g, h) Mann-Whitney t test p values; (e, f) Kruskal Wallis test followed by Dunn multiple comparison posthoc p-values; *p ≤ 0.05, **p ≤ 0.01, ***p ≤ 0.001, ****p ≤ 0.0001. Significance is relative to PBS control unless otherwise indicated. All experiments were performed a minimum of twice; a-f a representative experiment is shown, for g-h pooled data from 2 experiments is shown.
B cells are dispensable for H1/ML-RR-cGAMP-elicited protection against M. tuberculosis infection.
Although previous reports associated the efficacy of mucosal TB protein subunit vaccines with the formation of B cell follicles in vaccinated murine lungs (38, 39), we did not observe B cell follicles with germinal center organization in any of the mice at 4 weeks post challenge (Fig. S1b). However, i.n. vaccinated mouse lungs were more likely to contain lymphocyte aggregates without germinal centers (lymphoid nodules, Fig. S1c), B220+ staining in discrete cellular aggregates was increased in i.n. vaccinated mouse lungs (Fig. 2d, f), and overall B cell numbers were increased in vaccinated mice (Fig. 2g), suggesting that mucosal vaccination may boost the B cell response. To determine whether B cells contribute to vaccine-elicited protection in the mouse model, wild type and Ighmtm1Cgn B cell-deficient mice (muMt−) were immunized with H1/ML-RR-cGAMP and efficacy of protection evaluated at 4 weeks post challenge with M. tuberculosis. No differences in bacterial burden were observed when comparing infection in unimmunized mice, or in mice immunized with the CDN vaccine (Fig. 2h). Thus, B cells are not required for the efficacy of mucosally delivered H1/ML-RR-cGAMP.
IL-17 is required for full efficacy of ML-RR-cGAMP-adjuvanted protein subunit vaccines.
To determine whether IL-17 is necessary for protective efficacy, we vaccinated wild type and Il17–/– mice and enumerated bacterial burdens in the lungs 4 weeks post challenge with M. tuberculosis. Bacterial burden was unaffected by the loss of IL-17 in mice that were mock vaccinated, vaccinated s.c. with 5Ag/ML-RR-cGAMP, or vaccinated s.c. with BCG (Fig. 3a). However, we observed a significant loss of protection in Il17–/– mice that were vaccinated with i.n. 5Ag/ML-RR-cGAMP. While IL-17 has been reported to impact the development and recruitment of vaccine-induced Th1 cells (40), Th1 cell production was unaffected in Il17–/– mice at this timepoint (Fig. 3b). These data suggest that the efficacy of mucosal CDN vaccines is partially IL-17-dependent.
Figure 3. IL-17, Type I IFN, and IFN-γ all contribute to vaccine-elicited control of infection with M. tuberculosis.
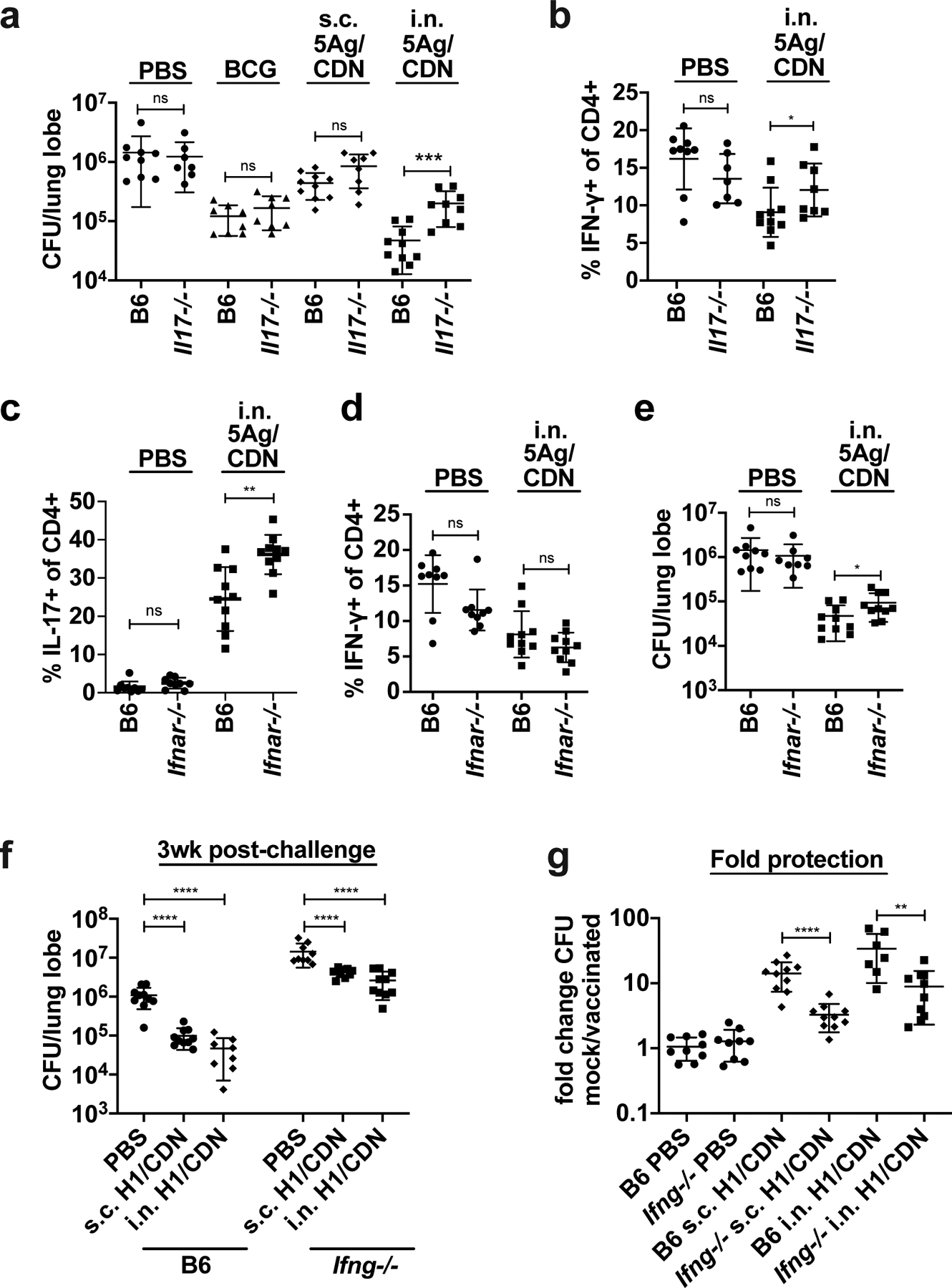
Wild-type C57BL/6 (B6) and Il17−/− mice were vaccinated once with subcutaneous M. bovis BCG 12 weeks before M. tuberculosis aerosol challenge or primed with either subcutaneous or intranasal M. tuberculosis 5Ag antigen and ML-RR-cGAMP cyclic-di-nucleotide adjuvant (i.n. 5Ag/CDN) 12 weeks before challenge, followed by two boosts. (a) At 4 weeks post challenge, mouse lungs were harvested and analyzed for bacterial burden. (b) ICS analysis of ex vivo restimulated lung leukocytes for IFN-γ. (c) Wild-type C57BL/6 (B6) and Ifnar−/− mice were vaccinated as described and CD4 T cells expressing IL-17 or (d) IFN-γ were evaluated by ICS and flow cytometry at 4 weeks post challenge. (e) Bacterial burdens in the lungs were enumerated at 4 weeks post challenge. Wild-type C57BL/6 (B6) and Ifng−/− mice were vaccinated as described and (f) bacterial burden was enumerated at 4 weeks post challenge. (g) data is reiterated in (f) expressed as fold change. Data are expressed as mean (± SD) of eight to ten mice per group from two independent experiments pooled with value from each individual animal displayed. Mann-Whitney t test p values; *p ≤ 0.05, **p ≤ 0.01, ***p ≤ 0.001.
Type I interferon and IFN-γ contribute to ML-RR-cGAMP-adjuvanted mucosal vaccine efficacy.
STING agonists strongly induce type I IFN expression through IRF3-dependent signaling (41). As type I IFN responses can negatively regulate the development of Th17 cells (43) and type I IFN can play a detrimental role in primary M. tuberculosis infection immunity it is somewhat surprising that STING agonists elicit robust Th17-dependent protective immunity (42–44). We tested the efficacy of i.n. 5Ag/ML-RR-cGAMP vaccination in IFNα/β receptor-deficient mice (Ifnar−/−). As expected, the Th17 response was more robust in the lungs of Ifnar−/− mice at 4 weeks post challenge (Fig. 3c), while the Th1 response was unaffected (Fig. 3d). However, despite an increased M. tuberculosis-specific Th17 response, we observed a small but statistically significant loss of control in vaccinated Ifnar−/− mice (Fig. 3e). The increased Th17 response in Ifnar−/− mice thus cannot compensate for the loss of type I IFN in vaccine-elicited protective immunity.
We next sought to determine whether the classical Th1 response plays a significant role in protection. We vaccinated mice lacking IFN-γ (Ifng–/–) with H1/ML-RR-cGAMP subcutaneously or intranasally and harvested lungs at 3 weeks post challenge to assess vaccine efficacy. Loss of IFN-γ did not affect the robust Th17 response in i.n. vaccinated mice prior to challenge (Fig. S2a) or at the peak of the T cell response at 3 weeks post challenge (Fig. S2b). While all Ifng–/– mice were more susceptible to M. tuberculosis infection, the subcutaneous and mucosal vaccines both provided significant protection compared to mock vaccination (Fig. 3f, g). While this demonstrates that both routes of inoculation elicit IFN-γ-independent mechanisms of control, the magnitude of protection was significantly reduced in Ifng–/– mice (Fig. 3f), indicating that IFN-γ does play some role in vaccine efficacy, in conjunction with IFN-γ-independent mechanisms.
Single cell analysis reveals shifts in vaccine-naïve and vaccinated CD4 T cell compartment transcriptomes.
To gain a more detailed understanding of the characteristics of the CD4 T cell response during M. tuberculosis infection, we performed scRNA-seq (47) on CD4 T cells sorted from vaccine-naïve and i.n. H1/ML-RR-cGAMP-vaccinated infected mouse lungs at the peak of the T cell response, 4 weeks post infection. Vaccination provided potent protection at this timepoint (Fig.1g, S3a). The filtered dataset, analyzed by single cell variational inference (scVI), consisted almost exclusively of CD4 T cells (Fig. S3b). Using signature transcription factor, cytokine, and cell surface marker expression, we were able to define naïve, Th1, Th2, Th17, Treg and Tfh cells in both mock vaccinated and vaccinated mice (Fig. 4a, S3c). In addition, we found that the Th1 subset contained the majority of proliferating cells (Mki67+), as well as the majority of cells expressing markers of cytotoxic potential (Gzma, Gzmb, Pfr1) (Fig. S3c).
Figure 4. scRNA-seq analysis of CD4 T cells reveals vaccine-elicited differences in CD4 T cells subsets and shifts in Th17 metabolism.
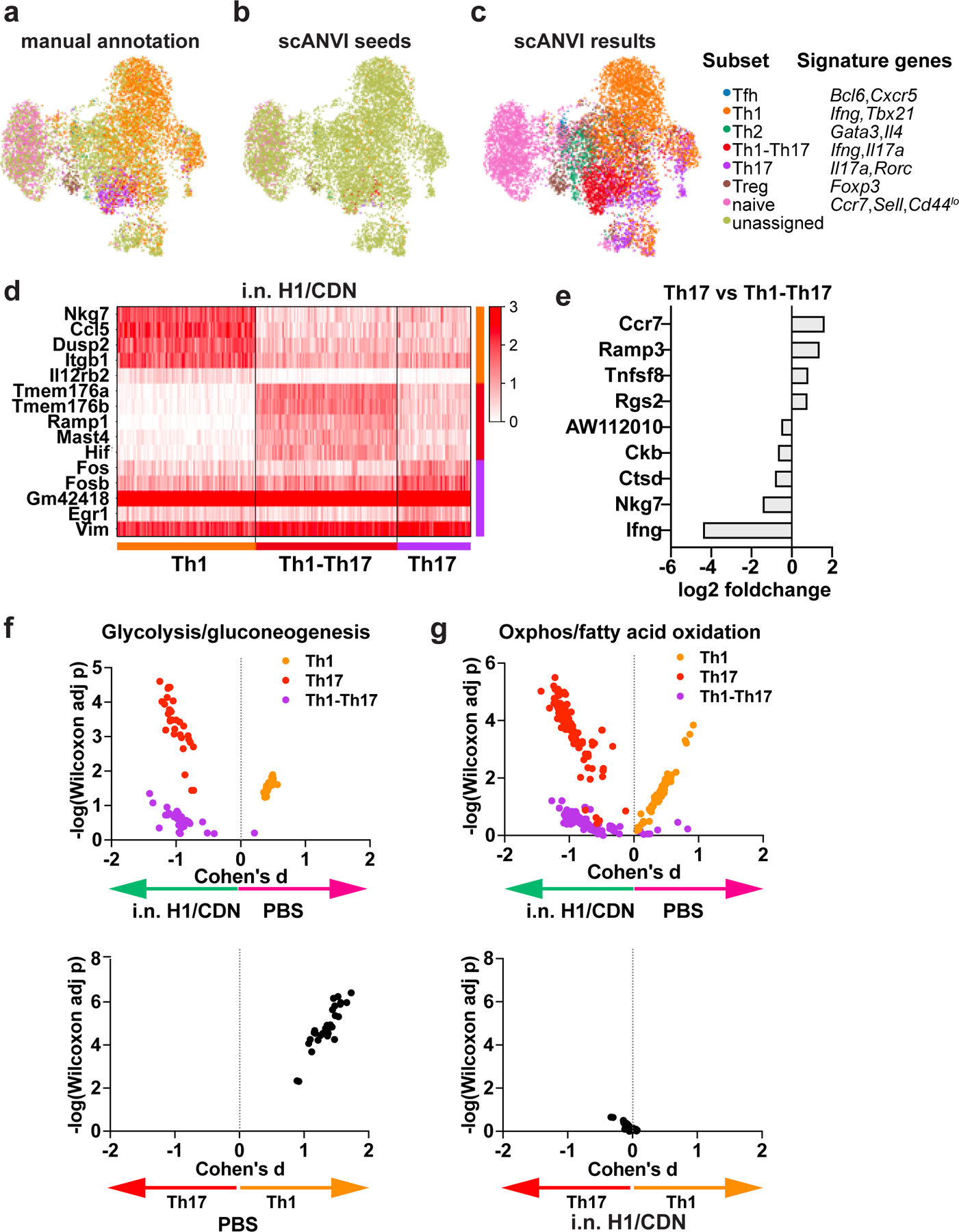
Subsets of CD4 T cells were (a) manually annotated based on signature expression of lineage-specific transcription factor, cytokine, and cell surface markers or (b, c) annotated using scANVI. (d) Heatmap depicting genes that were highly expressed in Th1, Th1-Th17, and Th17 cells. (e) Genes that were differentially expressed in Th17 vs Th1-Th17 cells. (f) Compass analysis of glycolysis in Th1, Th17, and Th1-Th17 subsets. (g) Compass analysis of oxidative phosphorylation in Th1, Th17, and Th1-Th17 subsets.
Only 56% of CD4 T cells were assigned to a particular helper T cell subset using signature gene expression. This effect may be due to the limitations of scRNA-seq (45), which can lead to difficulty distinguishing inherent CD4 T cell compartment heterogeneity (46). We aimed to overcome these issues by using the single-cell Annotation using Variational Inference (scANVI) deep generative model, which leverages high confidence seed labels based on signature marker genes to assign labels to unannotated cells (Fig 4b). We selected seed cells based on high expression of the gene signatures used in manual annotation (Fig. S3c), and also added lineage-specific transcription factor signature genes (Tbx21 for Th1 and Rorc for Th17) to increase confidence in denoting these classical Th subsets. No cells of the nonclassical Th1-Th17 subset co-expressed Tbx21 and Rorc; thus, scANVI seed labels for Th1-Th17 only required Ifng and Il17a expression. scANVI successfully annotated all previously unassigned cells and delineated similar clusters as manual annotation with signature genes, performing well at classifying cell types (Fig. 4c) and assigning the majority of CD4 T cells to a known subset. These results show the promise and utility of recently developed tools in single cell transcriptomics and probabilistic models to study the heterogeneity of CD4 T cells in the context of vaccination and infection.
To determine whether functionality differed between the CD4 T cell compartments, we first identified genes upregulated in mock vaccinated mouse CD4 T cells. Ifng, Klrg1, and Cx3cr1 expression was increased in CD4 T cells from vaccine-naïve infected lungs compared to vaccinated infected lungs (Fig. S3d), as expected. Interestingly, Nkg7, a regulator of cytotoxic granule exocytosis (47), was highly expressed in the bulk CD4 population of vaccine-naïve infected lungs (Fig. S4a, b), as were cytotoxic markers Gzma, Gzmb, and to a lesser-extent, Prf1 (Fig. S3c, S4a). Mucosal vaccination led to upregulation of genes associated with the Rorc-expressing Th17 lineage, such as Tmem176a, Tmem176b (51), and JunB (48–50) (Fig. S3d). The naïve and central memory T cell marker Il7r (51) was highly expressed in the i.n. vaccinated CD4 T cell compartment (Fig. S3d, S4c), while Ccr7 and Sell expression were unchanged (Fig. S3c), indicating that vaccination did not alter the naïve T cell population. Increased Rbpj and Cd44 expression in vaccinated mice (Fig. S4c,d) may have contributed to survival of effector and memory T cells (50, 52) and memory Th1 development (53). Other activation marker genes such as Il2ra, Cd69, and Ctla4, also had increased expression within the bulk CD4 population (Fig. S4c). Comparison of Th1 cells in vaccinated and vaccine-naïve mice demonstrated that these subsets are broadly similar, with Th1 cells in both subsets expressing markers of memory and activation relative to T cells from naïve infected animals (Fig. S4b, S4d).
We next sought to further delineate the transcriptional phenotypes of Th1, Th17 and Th1-Th17 cells in vaccinated mice. We therefore compared the top differentially expressed genes in scANVI-predicted Th1, Th17, and Th1-Th17 subsets. Th1 cells were substantially different from both Th17 and Th1-Th17 cells at the transcriptional level, with hundreds of genes differentially expressed. The top differentially expressed genes in Th1 cells included Nkg7, which mediates both the cytotoxic activity of NK and CD8 T cells as well as CD4 function in infection with Leishmania donovani; the chemokine Ccl5, as well as Dusp2, and Il12rb2, genes implicated in Th1 differentiation (54, 55). The top differentially expressed genes in Th1-Th17 cells included cation channels selectively expressed on Th17 cells (Tmem176a, Tmem176b) as well as regulators of lymphangiogenesis (Ramp1) and T cell activation (Fos, Fosb, Egr1) (Fig. 4d) (48, 56, 57). Ingenuity Pathway Analysis comparing the transcriptional profile of cells expressing Il17a (Th17) or Ifng (Th1) revealed that Th1 cells were enriched for phosphoinositide signaling and TCR signaling pathways. In contrast, Th17 cells exhibited a transcriptional profile indicative of HIF-1α activation (Table 1), a transcription factor known to promote both aerobic glycolysis and Th17 differentiation, as well as cellular responses to hypoxia (58). Interestingly, we found very few transcriptional differences between Th1 and Th1-Th17 cells. Th17 cells expressed moderately higher levels of Ccr7, a marker of memory T cells, Ramp3, and Tnfsf8, a protein previously associated with CD4 T cell-based control of M. tuberculosis infection in mice. Th1-Th17 cells preferentially expressed the lncRNA AW112010, known to suppress IL-10 and promote inflammatory cytokine production by T cells, as well as Ckb, Ctsd, Nkg7 and Ifng (Fig. 4e).
Table 1. Ingenuity Canonical Pathways that are differentially represented in Th1 and Th17 cells from vaccinated mice.
A negative z-score indicates that the pathway is overrepresented in Th17 cells, whereas a positive z-score indicates that the pathway is more highly represented in Th1 cells.
| Ingenuity Canonical Pathways | −log(p-value) | z-score |
|---|---|---|
| HIF1a± Signaling | 2.43E+00 | −2.121 |
| 14-3-3-mediated Signaling | 1.73E+00 | −2 |
| IL-17 Signaling | 1.26E+00 | −2 |
| IL-23 Signaling Pathway | 2.12E+00 | −1.89 |
| Th17 Activation Pathway | 8.47E+00 | −1.667 |
| ILK Signaling | 3.09E+00 | −1.667 |
| Acute Phase Response Signaling | 2.56E+00 | −1.633 |
| Inhibition of ARE-Mediated mRNA Degradation Pathway | 1.07E+00 | −1.633 |
| HOTAIR Regulatory Pathway | 1.35E+00 | −1.342 |
| NRF2-mediated Oxidative Stress Response | 1.33E+00 | −1.342 |
| Estrogen Receptor Signaling | 1.18E+00 | −1.342 |
| Th2 Pathway | 4.49E−01 | −1.342 |
| HMGB1 Signaling | 4.37E+00 | −1.134 |
| Aryl Hydrocarbon Receptor Signaling | 3.71E+00 | −1.134 |
| IL-6 Signaling | 2.50E+00 | −1.134 |
| IGF-1 Signaling | 5.46E+00 | −1 |
| Sphingosine-1-phosphate Signaling | 2.08E+00 | −1 |
| Insulin Receptor Signaling | 1.27E+00 | −1 |
| PPAR Signaling | 1.05E+00 | −1 |
| CCR5 Signaling in Macrophages | 5.24E+00 | 1 |
| CD28 Signaling in T Helper Cells | 1.41E+00 | 1 |
| cAMP-mediated signaling | 1.32E+00 | 1 |
| VDR/RXR Activation | 5.29E−01 | 1 |
| TREM1 Signaling | 3.45E+00 | 1.342 |
| ERK/MAPK Signaling | 2.63E+00 | 1.342 |
| D-myo-inositol-5-phosphate Metabolism | 2.35E+00 | 1.342 |
| Superpathway of Inositol Phosphate Compounds | 2.06E+00 | 1.342 |
| Regulation of IL-2 Expression in T Lymphocytes | 1.65E+00 | 1.342 |
| Natural Killer Cell Signaling | 9.79E−01 | 1.414 |
| D-myo-inositol (1,4,5,6)-Tetrakisphosphate Biosynthesis | 3.15E+00 | 1.667 |
| D-myo-inositol (3,4,5,6)-tetrakisphosphate Biosynthesis | 1.71E+00 | 2 |
| 3-phosphoinositide Degradation | 1.71E+00 | 2 |
| 3-phosphoinositide Biosynthesis | 1.58E+00 | 2 |
| T Cell Receptor Signaling | 1.45E+00 | 2 |
Naïve T cells rely primarily on oxidative metabolism, however, engagement of the TCR results in a dramatic upregulation of glycolysis in activated effector cells. Although resting memory cells revert to a more oxidative metabolism, upon restimulation, activated memory cells more efficiently engage in glycolysis to proliferate and engage in effector functions. Using Compass, an algorithm for characterizing the metabolic state of cells based on scRNA-seq data, we evaluated the glycolytic state of Th1, Th17, and Th1-Th17 cells in vaccine-naïve and vaccinated mice. Both Th17 and Th1-Th17 exhibited a transcriptional state indicative of higher glycolysis in vaccinated mice (Fig. 4f). Surprisingly, however, we found that Th1 cells were slightly less glycolytic in vaccinated mice when compared with mock vaccinated infected mice (Fig. 4f). By examining each subset within an infection condition, we found that in vaccine-naïve infected mice, Th1 cells are likely to be far more glycolytic than Th17 cells (Fig. 4f). However, this difference was not evident in vaccinated mice (Fig. 4g). Thus, we hypothesize that vaccination significantly boosts glycolytic metabolism in Th17 cells, whereas Th1 cells are maximally glycolytic in vaccine-naïve infected mice.
CXCR3+ cells confer protection against M. tuberculosis via enhanced localization to lung tissues relative to KLRG1+ cells that remain trapped in the vasculature. We next asked whether differential gene expression in these CD4 subsets might provide insights into the differentiation or function of these cells. Cells that express Cxcr3 more highly express keratin (Krt83), which may influence cellular stiffness and invasiveness, restraint of Th2 differentiation (Rbpj), insulin responsiveness (Tbc1d4), memory and activation (Il7r, Ctla4), and polyamine synthesis (Odc1) (Fig. 5a) (59–61). As our scRNA-seq analysis repeatedly associated Tnfsf8 expression with protective T cells subsets, we used flow cytometry to characterize expression of the protein associated with this gene on CD4 T cells in vaccinated and vaccine-naïve mice. We found an increase in the percentage of CD4 T cells that express CD153 (the protein encoded by Tnfsf8) both prior to challenge and at 4 weeks post challenge in vaccinated mice (Fig. 5b, 5c). Ingenuity Pathway Analysis suggested that Cxcr3-expressing parenchyma-homing cells express Th17 cell signatures, including Rorc, and IL-23, IL-6, and HIF-1α signaling pathways (Fig. 5d).
Figure 5. Vaccination enhances expression of Tnfsf8 (CD153) in parenchyma-homing T cells and in the CD4 T cell compartment.
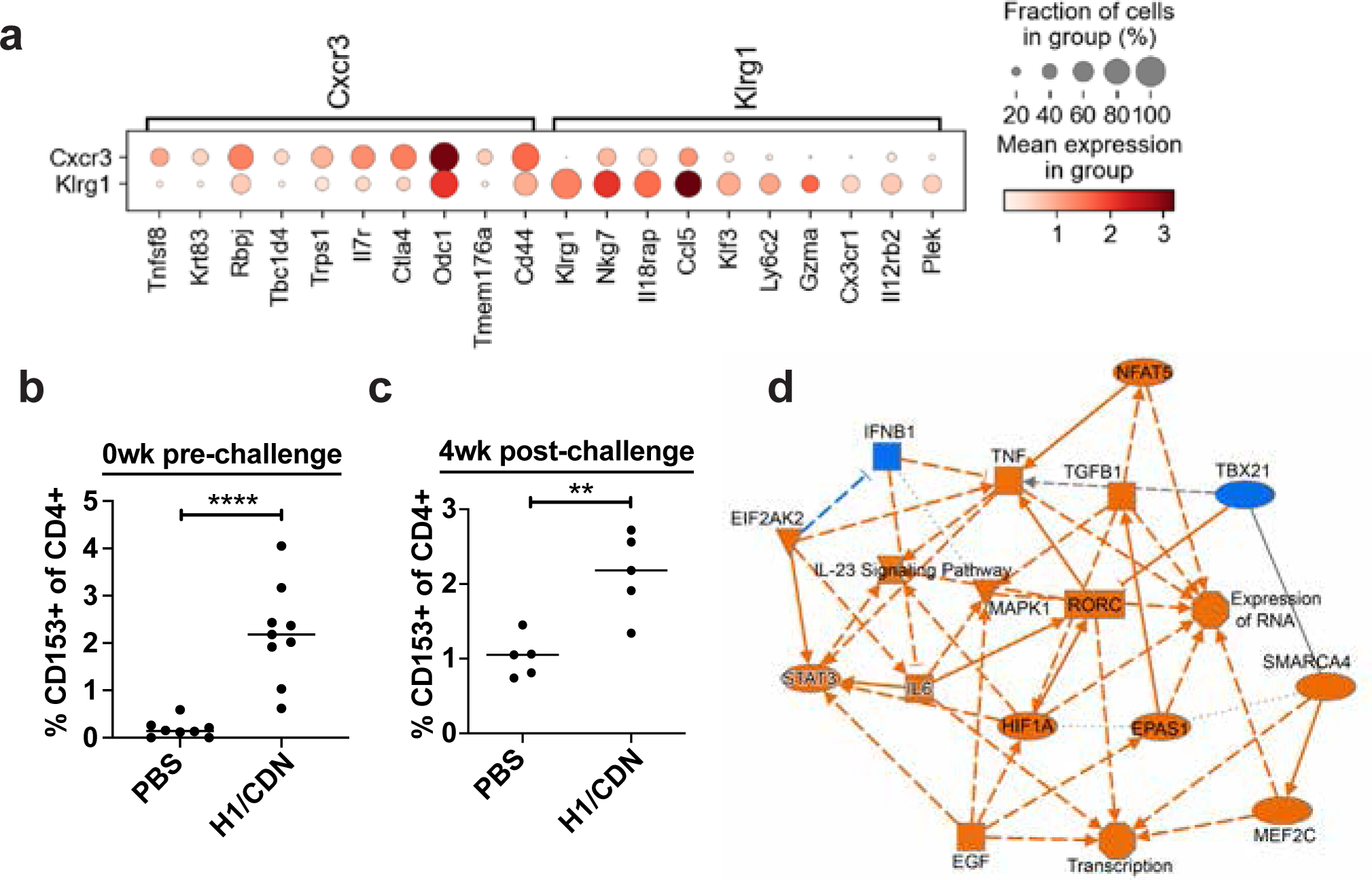
Mice were vaccinated three times at 4 week intervals with intranasal H1/CDN. (a) scRNA-seq results from all CXCR3+KLRG1− vs. CXCR3−KLRG1+ CD4 T cells were compared. Top genes that were differentially expressed in these subtypes. (b) Flow cytometry analysis for CD153 expression on CD4 T cells at day 0 prior to challenge or (c) at 4 weeks post challenge. (d) Network analysis of scRNA-seq results comparing CXCR3+KLRG1− and CXCR3−KLRG1+. Orange nodes are overrepresented in CXCR3+ cells; blue nodes are overrepresented in KLRG1+ cells.
Protection elicited by i.n. H1/ML-RR-cGAMP vaccination is not solely a function of route.
We next sought to disentangle the contributions of the specific CDN adjuvant from the route of vaccination in the efficacy of i.n. H1/ML-RR-cGAMP vaccination. Having previously demonstrated that i.n. is superior to s.c. delivery when CDN-adjuvanted vaccines are compared head-to-head (25), we next compared H1/ML-RR-cGAMP with an H1 vaccine adjuvanted with MPLA, one of the major immunostimulatory molecules in the successful vaccine M72/AS01E (12). Both vaccines were formulated by mixing the adjuvant with H1 in PBS and were delivered to mice intranasally three times at 4-week intervals before challenge. At 3 weeks pre-challenge, we found a significantly larger population of both Th1 and Th1-Th17 T cells in H1/ML-RR-cGAMP-vaccinated mice; IL-17+/IFN-γ− Th17 T cells were equivalent (Fig. 6a, b, c). In these experiments, we were unable to observe enough antigen specific T cells on day 0 prior to challenge to enable analysis. However, we found that although both vaccines elicited equivalent increases in total CD4 T cells in the lungs (Fig. 6d), the CDN-adjuvanted vaccine elicited more CXCR3+ parenchyma-homing T cells (Fig. 6e). Similarly, at 4 weeks after challenge we observed no differences in the percentages of Th1, Th17 or Th1-Th17 cells in the lungs of mice vaccinated with CDN- or MPLA-adjuvanted vaccines (Fig. 6f, g, h). However, the CDN-adjuvanted vaccine elicited more CXCR3+ parenchyma-homing T cells than the MPLA-adjuvanted vaccine (Fig. 6i). Despite an apparent similarity in the ability of both vaccines to elicit CD4 T cell responses, the MPLA-adjuvanted vaccine failed to protect mice from infection (Fig. 6j). These results highlight the unique efficacy of the CDN vaccine for tuberculosis. Furthermore, these results suggest that simply eliciting Th17 cells via mucosal vaccination is not sufficient for protection; T cell homing, and perhaps other unidentified factors also play a major role in vaccine efficacy for tuberculosis.
Figure 6. Vaccination with H1/CDN by the intranasal route elicits more robust protection than vaccination with H1/MPLA.
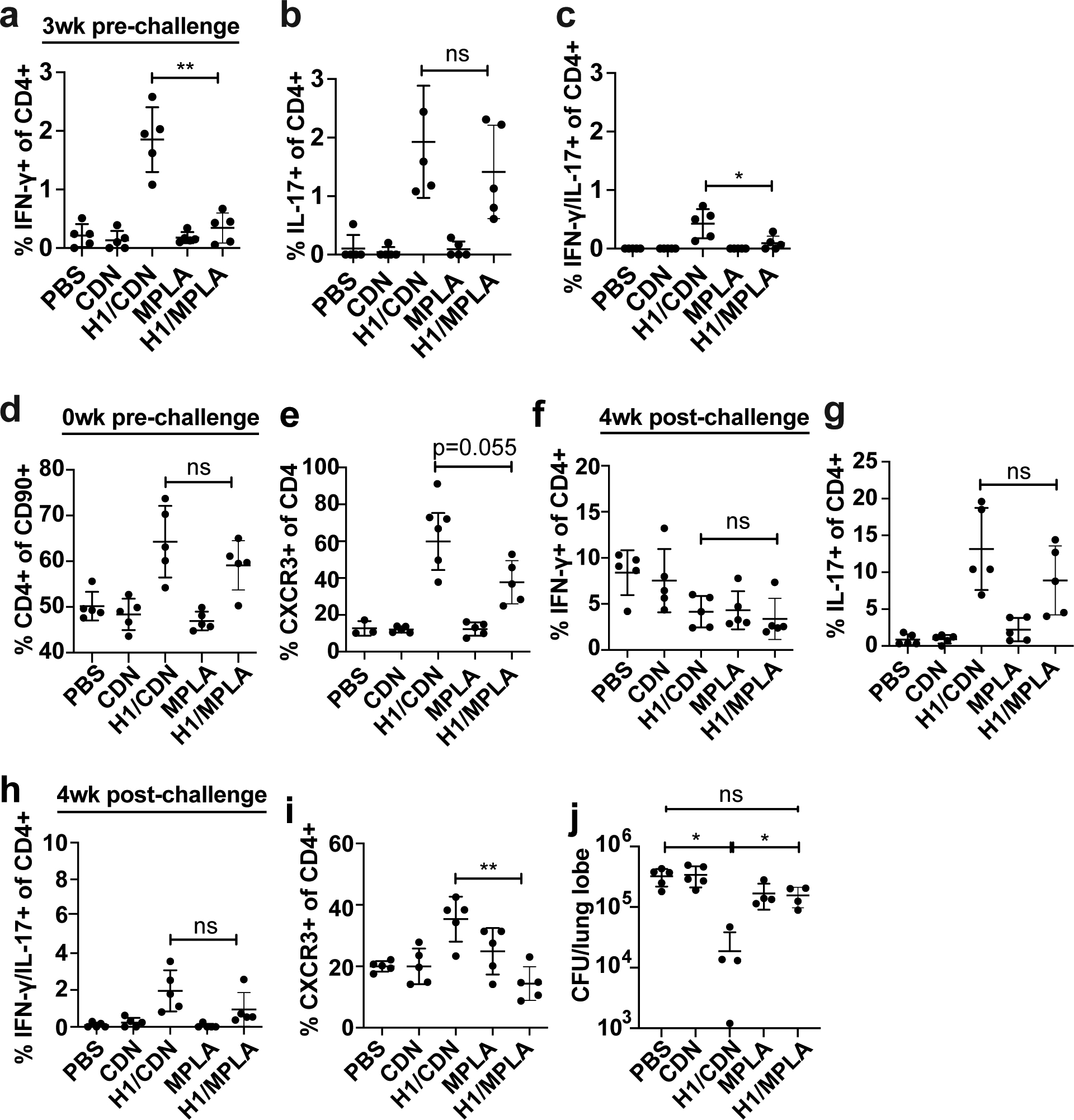
Mice were vaccinated three times at 4 week intervals with either intranasal H1/CDN or H1/MPLA. (a) Intracellular cytokine staining (ICS) for percentage of blood CD4 T cells that produce IFN-γ, (b) IL-17, or (c) IFN-γ and IL-17 at 3 weeks prior to challenge. (d) Flow cytometry analysis for CD4 expression on T cells or (e) CXCR3 expression on CD4 T cells at day 0 prior to challenge. (f) Intracellular cytokine staining (ICS) for percentage of lung CD4 T cells that produce IFN-γ, (g) IL-17, or (h) IFN-γ and IL-17 at 4 weeks after challenge with M. tuberculosis. (i) Flow cytometry analysis for CXCR3 expression on T cells at 4 weeks post challenge. (j) CFU from mice vaccinated with CDN or MPLA-adjuvanted vaccines at 4 weeks post challenge. Data are expressed as mean (± SD) of five mice per group with value from each individual animal displayed. Each experiment was performed a minimum of twice and a representative experiment is shown. Mann-Whitney t test p values; *p ≤ 0.05, **p ≤ 0.01, ***p ≤ 0.001.
Discussion
The recent success of the phase IIb clinical trial of the M72/AS01E protein subunit vaccine suggests that improved adjuvants and protein antigens may contribute to a vaccine that confers complete protection against TB infection. Importantly, protein subunit vaccines have potential for safe use in HIV-infected or otherwise immunocompromised individuals. Our results provide mechanistic insight into a highly efficacious CDN-adjuvanted TB protein subunit vaccine. We found that intranasal vaccine-induced protection is evident 1 week post M. tuberculosis challenge in mouse lungs, correlates with a robust Th17 and Th1-Th17 response, and is dependent on IL-17 for full efficacy. Furthermore, the CDN-adjuvanted vaccine is able to elicit T cells that home not only into the lung parenchyma, but also into macrophage-containing granuloma-like lesions. Using scRNA-seq, we found that both Th17 and Th1-Th17 cells in vaccinated mice have higher levels of expression of glycolytic genes than Th17 cells in vaccine-naïve infected mice, suggesting a more effector activated state, whereas Th1 cells, in contrast, appear to be less glycolytic in vaccinated mice, suggesting that CDN adjuvanted vaccines suppress (or limit) Th1 effector function activation while enhancing Th17 and Th1-Th17 effector functdions. CXCR3+ parenchyma-homing T cells express markers of memory and activation more highly than KLRG1+ vascular-homing T cells and have transcriptional signatures of STAT-3 and HIF-1α activation. Finally, CD4 T cells from vaccinated mice are associated with higher expression of CD153, which has been previously shown to promote T cell-based control of M. tuberculosis infection. Interestingly, although the MPLA adjuvant also elicits Th1, Th17, and Th1-Th17 cells, this vaccine is unable to confer significant protection in the mouse model compared with the CDN adjuvant. T cells in MPLA-vaccinated mice have diminished expression of Cxcr3 relative to CDN-immunized mice. Thus, simply eliciting Th17 cells using immunization via a mucosal route is not sufficient for vaccine-elicited protection; localization and likely other as of yet unidentified factors also play a major role.
Taken together, our data strongly suggest that vaccine-elicited protection is predominantly reliant upon CD4 T cells. Our analysis of immune cells present in the lungs after vaccination but prior to challenge demonstrates that other major cell types are not strongly elicited by mucosal vaccination with a CDN-adjuvanted vaccine, as CD8 T cells, B cells, and innate immune cells are not significantly elicited by vaccination. Although we and others have observed that control of infection in vaccinated mice is correlated with the presence of B cells aggregates in the lungs at 4 weeks post infection, we demonstrated that B cells are dispensable for CDN-adjuvanted vaccine elicited protection.
While it is clear that IFN-γ from CD4 T cells is essential for host defense against M. tuberculosis, our understanding of IFN-γ-independent mechanisms of control has been limited. Although Ifng–/– mice are extremely susceptible to M. tuberculosis, both subcutaneous and intranasal administration of a CDN-adjuvanted vaccine induced protection that was only partially dependent on IFN-γ. It is surprising that Th1 cells in vaccinated mice have a transcriptional signature indicative of a diminished metabolic state relative to Th1 cells in vaccine-naïve mice. Furthermore, the opposite is observed both with Th17 and Th1-Th17 cells; these populations in vaccinated mice have transcriptional profiles indicative of higher rates of glycolysis than the corresponding populations in vaccine-naïve mice. It is possible that one reason that Th1-Th17 cells are emerging as a strong correlate of protective immunity is that these cells can produce two effector cytokines and maintain a more differentiated effector state than traditional Th1 cells.
It is becoming more evident that proper positioning of T cells is as important as effector cytokine production for protection in M. tuberculosis infection. In macaques, defective intralesional positioning limits the ability of T cells to interact with and promote M. tuberculosis killing in myeloid cells (62). While CXCR3 has primarily been described as a marker of protective Th1 parenchyma-homing cells during M. tuberculosis infection (24), its role in tissue-homing of pathogen-specific T cells in the context of vaccination has not been well-studied. Early and sustained lung parenchymal localization of protective CXCR3+ CD4 T cells may mediate our mucosal vaccine’s protection. As IL-17 is primarily known to recruit neutrophils to the site of infection, it is of great interest to determine if and how this cytokine might induce and maintain this protective memory T cell response. Although a robust Th1 response was seen in mock vaccinated mouse lungs at 3 weeks post infection, CD4 T cells in these mice expressed markers of terminal differentiation—Klrg1 and Cx3cr1—and failed to infiltrate into lesions. Cytotoxic genes such as Nkg7, Gzma, and Gzmb were also expressed in a subset of these cells. Terminally differentiated Th1s are not associated with protection against TB (23, 63), and blood signature cytotoxic gene expression is anticorrelated with mouse susceptibility and progression from latent to active TB disease in humans (64). Further investigation is necessary to determine if cytotoxic CD4 T cells can mediate resistance to TB.
In contrast to the terminally differentiated Th1 marker expression seen in mock vaccinated mice, mucosal vaccination elicited higher expression of central memory T cell marker Il7r and activation/memory marker Cd44 in the total CD4 and Th1 populations. Interestingly, i.n. vaccination also led to increased expression of Tnfsf8, which has been shown to be essential for IFN-γ-independent control of TB in a mouse model (22) and implicated as a mediator of protection against human TB (65). We thus find potential markers of both IFN-γ-dependent and IFN-γ-independent CD4 T cell memory function. These markers may also suggest mechanisms for protection: signaling through these molecules may contribute to enhanced activation, survival, function, and memory development of CD4 T cells during infection. It will be crucial to evaluate whether these transcriptional changes result in memory responses that can provide enduring protection, past the point at which BCG-mediated immunity wanes.
While we had previously found that Th17 cells were induced by i.n. vaccination (25), our latest findings reveal a surprisingly heterogeneous Th17 compartment, with 20–30% of Il17a-expressing cells in vaccinated mice also expressing Ifng. While CD4 T cell plasticity has been well documented in different contexts in vivo, it is uncertain whether this works to the host’s advantage. In fact, Th1-Th17 presence correlates with TB disease severity in humans (66), and numerous studies have identified pathogenic IFN-γ+ Th17s as a driver of disease in mouse models of autoimmunity (20). Furthermore, a previous study using adoptively transferred Th17s reported that the ability to coproduce IFN-γ limits Th17 protective capacity during M. tuberculosis infection in mice (38, 39). In our model, it is currently uncertain whether co-producing cells differ in protective capacity to classical Th1s or Th17s, and what cell types respond to IL-17 to mediate protection. Interestingly, our scRNA-seq data revealed significant metabolic differences within specific subsets when comparing vaccinated and mock vaccinated infected mice, with Th1s adopting a metabolic signature more indicative of effector function in naïve mice, and Th17 and Th1-Th17 cells exhibiting a more active glycolytic mechanism in vaccinated mice.
A recent mass cytometry study showed that the Th17 lineage maintains plasticity after in vitro differentiation (67). Our scANVI analysis classified a surprisingly large proportion of both unassigned and Il17a-expressing “Th17” cells as Th1-Th17. This finding may indicate that Th1-Th17 cells are part of a larger pool of heterogeneous helper T cells, whose transcriptomes and effector functions may shift dynamically in response to inflammatory signals. Future fate-mapping studies could reveal whether CD4 T cell expression of IFN-γ and IL-17 changes over the course of vaccination, infection, and memory recall. While the plasticity in CD4 T cell phenotypes and function may be a hurdle in finding reliable correlates of vaccine correlates protection, true delineation of subtypes may be impossible, and it may be time to embrace heterogeneity in CD4 T cell-mediated protection for better TB vaccine design.
Our studies add to the growing body of literature proving that mucosal vaccination leads to the development of Th17 and Th1-Th17 compartments. This heterogeneity distinguishes mucosal vaccination from the terminally differentiated Th1-dominated response elicited by systemic delivery of a vaccine. While intranasal delivery of the BCG vaccine also induces Th17 responses and protection, there remains a need to find a vaccine with an increased safety profile (68). CDN-adjuvanted vaccines are safe and effective when administered twice as a booster to BCG and when delivered three times as a sole vaccine (25), inducing a mixed IFN-γ- and IL-17-dependent response and protection that can outperform that of BCG. Importantly, however, our data suggests that simply eliciting these subsets via mucosal delivery of a vaccine is not sufficient to confer protection. We found that administering a vaccine adjuvanted with MPLA via the intranasal route conferred no protection against infection, despite eliciting similar numbers of Th1, Th17, and Th1-Th17 cells at the peak of the immune response prior to challenge. We did find that MPLA induced a lower proportion of CXCR3+ parenchyma-homing T cells when compared with CDN-vaccinated mice, however it is not clear whether this accounts for the difference in efficacy of the two vaccine adjuvants. Future studies will reveal additional determinants that mediate protective efficacy of CDN-adjuvanted vaccines against M. tuberculosis. The studies presented here thus highlight the potentially profound impact of T cell diversity and lineage plasticity on antibacterial immune defense. Our findings suggest multiple avenues for improvement and optimism in designing improved vaccines against infectious respiratory diseases.
Supplementary Material
Key Points.
Mucosal CDN vaccine elicits IL-17 and IFN-γ dependent protection against TB
T cells in CDN immunized mice traffic to granulomatous lesions
Mucosal MPLA adjuvanted vaccine does not protect despite eliciting IL-17
Acknowledgments
We thank Russell Vance for the kind gift of Ifnar1−/− mice. We thank Sagar Rawal for technical assistance, Kiran Magee and Lily McCann for assistance with mouse colony maintenance, Bianca Blackshire and Kiran Magee for assistance with in vitro experiments, and Dmitri Kotov for advice and assistance with scRNA-seq experiments. We thank Aduro Biotech for the gift of cyclic-di-nucleotides. We thank Rasmus Skaarup Mortensen and Ida Rosenkrands from the Statens Serum Institut for H1 antigen. We thank members of the Stanley and Cox labs for helpful discussions.
This work was supported by NIH grant T32 GM 7232-40 and NSF Graduate Research Fellowship DGE-1752814 (to E.V.D. and R.J.), and NIH 1R01AI113270-01A1 to SAS.
Footnotes
Disclosures
E.V.D and S.A.S declare that parts of this work are related to a United States patent application titled “Intranasal delivery of a cyclic-di-nucleotide adjuvanted vaccine for tuberculosis” (United States Patent Application 20200338182).
References
- 1.Geneva. 2018. Global tuberculosis report 2018,. World Health Organization. [Google Scholar]
- 2.Colditz GA 1994. The efficacy of bcg vaccine in the prevention of tuberculosis. Pediatr. Infect. Dis. J 13: 761. [Google Scholar]
- 3.Fine PE 1995. Variation in protection by BCG: implications of and for heterologous immunity. Lancet 346: 1339–1345. [DOI] [PubMed] [Google Scholar]
- 4.Mangtani P, Abubakar I, Ariti C, Beynon R, Pimpin L, Fine PEM, Rodrigues LC, Smith PG, Lipman M, Whiting PF, and Sterne JA. 2014. Protection by BCG vaccine against tuberculosis: a systematic review of randomized controlled trials. Clin. Infect. Dis 58: 470–480. [DOI] [PubMed] [Google Scholar]
- 5.Cooper AM 2009. Cell-mediated immune responses in tuberculosis. Annu. Rev. Immunol 27: 393–422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Caruso AM, Serbina N, Klein E, Triebold K, Bloom BR, and Flynn JL. Mice deficient in CD4 T cells have only transiently diminished levels of IFN-gamma, yet succumb to tuberculosis. J. Immunol 162: 5407–5416. [PubMed] [Google Scholar]
- 7.Flynn JL, Chan J, Triebold KJ, Dalton DK, Stewart TA, and Bloom BR. 1993. An essential role for interferon gamma in resistance to Mycobacterium tuberculosis infection. J. Exp. Med 178: 2249–2254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Green AM, Difazio R, and Flynn JL. 2013. IFN-γ from CD4 T cells is essential for host survival and enhances CD8 T cell function during Mycobacterium tuberculosis infection. J. Immunol 190: 270–277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Lewinsohn DA, Lewinsohn DM, and Scriba TJ. 2017. Polyfunctional CD4+ T cells as targets for tuberculosis vaccination. Front. Immunol 8: 1262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Rodo MJ, Rozot V, Nemes E, Dintwe O, Hatherill M, Little F, and Scriba TJ. 2019. A comparison of antigen-specific T cell responses induced by six novel tuberculosis vaccine candidates. PLoS Pathog. 15: e1007643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Tameris MD, Hatherill M, Landry BS, Scriba TJ, Snowden MA, Lockhart S, Shea JE, McClain JB, Hussey GD, Hanekom WA, Mahomed H, McShane H, and MVA85A 020 Trial Study Team. 2013. Safety and efficacy of MVA85A, a new tuberculosis vaccine, in infants previously vaccinated with BCG: a randomised, placebo-controlled phase 2b trial. Lancet 381: 1021–1028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Tait DR, Hatherill M, Van Der Meeren O, Ginsberg AM, Van Brakel E, Salaun B, Scriba TJ, Akite EJ, Ayles HM, Bollaerts A, Demoitié M-A, Diacon A, Evans TG, Gillard P, Hellström E, Innes JC, Lempicki M, Malahleha M, Martinson N, Mesia Vela D, Muyoyeta M, Nduba V, Pascal TG, Tameris M, Thienemann F, Wilkinson RJ, and Roman F. 2019. Final analysis of a trial of M72/AS01E vaccine to prevent tuberculosis. N. Engl. J. Med 381: 2429–2439. [DOI] [PubMed] [Google Scholar]
- 13.Cowley SC, and Elkins KL. 2003. CD4+ T cells mediate IFN-gamma-independent control of Mycobacterium tuberculosis infection both in vitro and in vivo. J. Immunol 171: 4689–4699. [DOI] [PubMed] [Google Scholar]
- 14.Mittrücker H-W, Steinhoff U, Köhler A, Krause M, Lazar D, Mex P, Miekley D, and Kaufmann SHE. 2007. Poor correlation between BCG vaccination-induced T cell responses and protection against tuberculosis. Proc. Natl. Acad. Sci. USA 104: 12434–12439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Gallegos AM, Pamer EG, and Glickman MS. 2008. Delayed protection by ESAT-6-specific effector CD4+ T cells after airborne M. tuberculosis infection. J. Exp. Med 205: 2359–2368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Nunes-Alves C, Booty MG, Carpenter SM, Jayaraman P, Rothchild AC, and Behar SM. 2014. In search of a new paradigm for protective immunity to TB. Nat. Rev. Microbiol 12: 289–299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Aguilo JI, Alonso H, Uranga S, Marinova D, Arbués A, de Martino A, Anel A, Monzon M, Badiola J, Pardo J, Brosch R, and Martin C. 2013. ESX-1-induced apoptosis is involved in cell-to-cell spread of Mycobacterium tuberculosis. Cell Microbiol 15: 1994–2005. [DOI] [PubMed] [Google Scholar]
- 18.Perdomo C, Zedler U, Kühl AA, Lozza L, Saikali P, Sander LE, Vogelzang A, Kaufmann SHE, and Kupz A. 2016. Mucosal BCG Vaccination Induces Protective Lung-Resident Memory T Cell Populations against Tuberculosis. MBio 7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Gopal R, Rangel-Moreno J, Slight S, Lin Y, Nawar HF, Fallert Junecko BA, Reinhart TA, Kolls J, Randall TD, Connell TD, and Khader SA. 2013. Interleukin-17-dependent CXCL13 mediates mucosal vaccine-induced immunity against tuberculosis. Mucosal Immunol. 6: 972–984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Bystrom J, Clanchy FIL, Taher TE, Al-Bogami M, Ong VH, Abraham DJ, Williams RO, and Mageed RA. 2019. Functional and phenotypic heterogeneity of Th17 cells in health and disease. Eur. J. Clin. Invest 49: e13032. [DOI] [PubMed] [Google Scholar]
- 21.Lindenstrøm T, Woodworth J, Dietrich J, Aagaard C, Andersen P, and Agger EM. 2012. Vaccine-induced th17 cells are maintained long-term postvaccination as a distinct and phenotypically stable memory subset. Infect. Immun 80: 3533–3544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Sallin MA, Kauffman KD, Riou C, Du Bruyn E, Foreman TW, Sakai S, Hoft SG, Myers TG, Gardina PJ, Sher A, Moore R, Wilder-Kofie T, Moore IN, Sette A, Lindestam Arlehamn CS, Wilkinson RJ, and Barber DL. 2018. Host resistance to pulmonary Mycobacterium tuberculosis infection requires CD153 expression. Nat. Microbiol 3: 1198–1205. [DOI] [PubMed] [Google Scholar]
- 23.Sakai S, Kauffman KD, Schenkel JM, McBerry CC, Mayer-Barber KD, Masopust D, and Barber DL. 2014. Cutting edge: control of Mycobacterium tuberculosis infection by a subset of lung parenchyma-homing CD4 T cells. J. Immunol 192: 2965–2969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Gern BH, Adams KN, Plumlee CR, Stoltzfus CR, Shehata L, Busman-Sahay K, Hansen SG, Axthelm MK, Picker LJ, Estes JD, Urdahl KB, and Gerner MY. 2019. TGFβ restricts T cell function and bacterial control within the tuberculous granuloma. BioRxiv. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Van Dis E, Sogi KM, Rae CS, Sivick KE, Surh NH, Leong ML, Kanne DB, Metchette K, Leong JJ, Bruml JR, Chen V, Heydari K, Cadieux N, Evans T, McWhirter SM, Dubensky TW, Portnoy DA, and Stanley SA. 2018. STING-Activating Adjuvants Elicit a Th17 Immune Response and Protect against Mycobacterium tuberculosis Infection. Cell Rep. 23: 1435–1447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Gaffney BL, Veliath E, Zhao J, and Jones RA. 2010. One-flask syntheses of c-di-GMP and the [Rp,Rp] and [Rp,Sp] thiophosphate analogues. Org. Lett 12: 3269–3271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Bankhead P, Loughrey MB, Fernández JA, Dombrowski Y, McArt DG, Dunne PD, McQuaid S, Gray RT, Murray LJ, Coleman HG, James JA, Salto-Tellez M, and Hamilton PW. 2017. QuPath: Open source software for digital pathology image analysis. Sci. Rep 7: 16878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Zheng GXY, Terry JM, Belgrader P, Ryvkin P, Bent ZW, Wilson R, Ziraldo SB, Wheeler TD, McDermott GP, Zhu J, Gregory MT, Shuga J, Montesclaros L, Underwood JG, Masquelier DA, Nishimura SY, Schnall-Levin M, Wyatt PW, Hindson CM, Bharadwaj R, Wong A, Ness KD, Beppu LW, Deeg HJ, McFarland C, Loeb KR, Valente WJ, Ericson NG, Stevens EA, Radich JP, Mikkelsen TS, Hindson BJ, and Bielas JH. 2017. Massively parallel digital transcriptional profiling of single cells. Nat. Commun 8: 14049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Wolf FA, Angerer P, and Theis FJ. 2018. SCANPY: large-scale single-cell gene expression data analysis. Genome Biol. 19: 15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Lopez R, Regier J, Cole MB, Jordan MI, and Yosef N. 2018. Deep generative modeling for single-cell transcriptomics. Nat. Methods 15: 1053–1058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Wagner A, Wang C, Fessler J, DeTomaso D, Avila-Pacheco J, Kaminski J, Zaghouani S, Christian E, Thakore P, Schellhaass B, Akama-Garren E, Pierce K, Singh V, Ron-Harel N, Douglas VP, Bod L, Schnell A, Puleston D, Sobel RA, Haigis M, Pearce EL, Soleimani M, Clish C, Regev A, Kuchroo VK, and Yosef N. 2021. Metabolic modeling of single Th17 cells reveals regulators of autoimmunity. Cell 184: 4168–4185.e21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Xu C, Lopez R, Mehlman E, Regier J, Jordan MI, and Yosef N. 2021. Probabilistic harmonization and annotation of single-cell transcriptomics data with deep generative models. Mol. Syst. Biol 17: e9620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Corrales L, Glickman LH, McWhirter SM, Kanne DB, Sivick KE, Katibah GE, Woo S-R, Lemmens E, Banda T, Leong JJ, Metchette K, Dubensky TW, and Gajewski TF. 2015. Direct activation of STING in the tumor microenvironment leads to potent and systemic tumor regression and immunity. Cell Rep. 11: 1018–1030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Goter-Robinson C, Derrick SC, Yang AL, Jeon BY, and Morris SL. 2006. Protection against an aerogenic Mycobacterium tuberculosis infection in BCG-immunized and DNA-vaccinated mice is associated with early type I cytokine responses. Vaccine 24: 3522–3529. [DOI] [PubMed] [Google Scholar]
- 35.Lockhart E, Green AM, and Flynn JL. 2006. IL-17 production is dominated by gammadelta T cells rather than CD4 T cells during Mycobacterium tuberculosis infection. J. Immunol 177: 4662–4669. [DOI] [PubMed] [Google Scholar]
- 36.Urdahl KB 2014. Understanding and overcoming the barriers to T cell-mediated immunity against tuberculosis. Semin. Immunol 26: 578–587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Lindenstrøm T, Moguche A, Damborg M, Agger EM, Urdahl K, and Andersen P. 2018. T cells primed by live mycobacteria versus a tuberculosis subunit vaccine exhibit distinct functional properties. EBioMedicine 27: 27–39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Gopal R, Monin L, Slight S, Uche U, Blanchard E, Fallert Junecko BA, Ramos-Payan R, Stallings CL, Reinhart TA, Kolls JK, Kaushal D, Nagarajan U, Rangel-Moreno J, and Khader SA. 2014. Unexpected role for IL-17 in protective immunity against hypervirulent Mycobacterium tuberculosis HN878 infection. PLoS Pathog. 10: e1004099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Monin L, Griffiths KL, Slight S, Lin Y, Rangel-Moreno J, and Khader SA. 2015. Immune requirements for protective Th17 recall responses to Mycobacterium tuberculosis challenge. Mucosal Immunol. 8: 1099–1109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Khader SA, Bell GK, Pearl JE, Fountain JJ, Rangel-Moreno J, Cilley GE, Shen F, Eaton SM, Gaffen SL, Swain SL, Locksley RM, Haynes L, Randall TD, and Cooper AM. 2007. IL-23 and IL-17 in the establishment of protective pulmonary CD4+ T cell responses after vaccination and during Mycobacterium tuberculosis challenge. Nat. Immunol 8: 369–377. [DOI] [PubMed] [Google Scholar]
- 41.McWhirter SM, Barbalat R, Monroe KM, Fontana MF, Hyodo M, Joncker NT, Ishii KJ, Akira S, Colonna M, Chen ZJ, Fitzgerald KA, Hayakawa Y, and Vance RE. 2009. A host type I interferon response is induced by cytosolic sensing of the bacterial second messenger cyclic-di-GMP. J. Exp. Med 206: 1899–1911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Stanley SA, Johndrow JE, Manzanillo P, and Cox JS. 2007. The Type I IFN response to infection with Mycobacterium tuberculosis requires ESX-1-mediated secretion and contributes to pathogenesis. J. Immunol 178: 3143–3152. [DOI] [PubMed] [Google Scholar]
- 43.Donovan ML, Schultz TE, Duke TJ, and Blumenthal A. 2017. Type I interferons in the pathogenesis of tuberculosis: molecular drivers and immunological consequences. Front. Immunol 8: 1633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Ji DX, Yamashiro LH, Chen KJ, Mukaida N, Kramnik I, Darwin KH, and Vance RE. 2019. Type I interferon-driven susceptibility to Mycobacterium tuberculosis is mediated by IL-1Ra. Nat. Microbiol 4: 2128–2135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Papalexi E, and Satija R. 2018. Single-cell RNA sequencing to explore immune cell heterogeneity. Nat. Rev. Immunol 18: 35–45. [DOI] [PubMed] [Google Scholar]
- 46.Zemmour D, Kiner E, and Benoist C. 2020. CD4+ teff cell heterogeneity: the perspective from single-cell transcriptomics. Curr. Opin. Immunol 63: 61–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Ng SS, De Labastida Rivera F, Yan J, Corvino D, Das I, Zhang P, Kuns R, Chauhan SB, Hou J, Li X-Y, Frame TCM, McEnroe BA, Moore E, Na J, Engel JA, Soon MSF, Singh B, Kueh AJ, Herold MJ, Montes de Oca M, Singh SS, Bunn PT, Aguilera AR, Casey M, Braun M, Ghazanfari N, Wani S, Wang Y, Amante FH, Edwards CL, Haque A, Dougall WC, Singh OP, Baxter AG, Teng MWL, Loukas A, Daly NL, Cloonan N, Degli-Esposti MA, Uzonna J, Heath WR, Bald T, Tey S-K, Nakamura K, Hill GR, Kumar R, Sundar S, Smyth MJ, and Engwerda CR. 2020. The NK cell granule protein NKG7 regulates cytotoxic granule exocytosis and inflammation. Nat. Immunol 21: 1205–1218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Drujont L, Lemoine A, Moreau A, Bienvenu G, Lancien M, Cens T, Guillot F, Bériou G, Bouchet-Delbos L, Fehling HJ, Chiffoleau E, Nicot AB, Charnet P, Martin JC, Josien R, Cuturi MC, and Louvet C. 2016. RORγt+ cells selectively express redundant cation channels linked to the Golgi apparatus. Sci. Rep 6: 23682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Carr TM, Wheaton JD, Houtz GM, and Ciofani M. 2017. JunB promotes Th17 cell identity and restrains alternative CD4+ T-cell programs during inflammation. Nat. Commun 8: 301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Maekawa Y, Ishifune C, Tsukumo S, Hozumi K, Yagita H, and Yasutomo K. 2015. Notch controls the survival of memory CD4+ T cells by regulating glucose uptake. Nat. Med 21: 55–61. [DOI] [PubMed] [Google Scholar]
- 51.Surh CD, and Sprent J. 2008. Homeostasis of naive and memory T cells. Immunity 29: 848–862. [DOI] [PubMed] [Google Scholar]
- 52.Helbig C, Gentek R, Backer RA, de Souza Y, Derks IAM, Eldering E, Wagner K, Jankovic D, Gridley T, Moerland PD, Flavell RA, and Amsen D. 2012. Notch controls the magnitude of T helper cell responses by promoting cellular longevity. Proc. Natl. Acad. Sci. USA 109: 9041–9046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Baaten BJG, Li C-R, Deiro MF, Lin MM, Linton PJ, and Bradley LM. 2010. CD44 regulates survival and memory development in Th1 cells. Immunity 32: 104–115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Lang R, Hammer M, and Mages J. 2006. DUSP meet immunology: dual specificity MAPK phosphatases in control of the inflammatory response. J. Immunol 177: 7497–7504. [DOI] [PubMed] [Google Scholar]
- 55.Zhu J, Yamane H, and Paul WE. 2010. Differentiation of effector CD4 T cell populations (*). Annu. Rev. Immunol 28: 445–489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Yukawa M, Jagannathan S, Vallabh S, Kartashov AV, Chen X, Weirauch MT, and Barski A. 2020. AP-1 activity induced by co-stimulation is required for chromatin opening during T cell activation. J. Exp. Med 217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Cogswell PC, Mayo MW, and Baldwin AS. 1997. Involvement of Egr-1/RelA synergy in distinguishing T cell activation from tumor necrosis factor-alpha-induced NF-kappa B1 transcription. J. Exp. Med 185: 491–497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Shi LZ, Wang R, Huang G, Vogel P, Neale G, Green DR, and Chi H. 2011. HIF1alpha-dependent glycolytic pathway orchestrates a metabolic checkpoint for the differentiation of TH17 and Treg cells. J. Exp. Med 208: 1367–1376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Seltmann K, Fritsch AW, Käs JA, and Magin TM. 2013. Keratins significantly contribute to cell stiffness and impact invasive behavior. Proc. Natl. Acad. Sci. USA 110: 18507–18512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Delacher M, Schmidl C, Herzig Y, Breloer M, Hartmann W, Brunk F, Kägebein D, Träger U, Hofer A-C, Bittner S, Weichenhan D, Imbusch CD, Hotz-Wagenblatt A, Hielscher T, Breiling A, Federico G, Gröne H-J, Schmid RM, Rehli M, Abramson J, and Feuerer M. 2019. Rbpj expression in regulatory T cells is critical for restraining TH2 responses. Nat. Commun 10: 1621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Peck GR, Chavez JA, Roach WG, Budnik BA, Lane WS, Karlsson HKR, Zierath JR, and Lienhard GE. 2009. Insulin-stimulated phosphorylation of the Rab GTPase-activating protein TBC1D1 regulates GLUT4 translocation. J. Biol. Chem 284: 30016–30023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Kauffman KD, Sallin MA, Sakai S, Kamenyeva O, Kabat J, Weiner D, Sutphin M, Schimel D, Via L, Barry CE, Wilder-Kofie T, Moore I, Moore R, and Barber DL. 2018. Defective positioning in granulomas but not lung-homing limits CD4 T-cell interactions with Mycobacterium tuberculosis-infected macrophages in rhesus macaques. Mucosal Immunol. 11: 462–473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Sallin MA, Sakai S, Kauffman KD, Young HA, Zhu J, and Barber DL. 2017. Th1 Differentiation Drives the Accumulation of Intravascular, Non-protective CD4 T Cells during Tuberculosis. Cell Rep. 18: 3091–3104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Moreira-Teixeira L, Tabone O, Graham CM, Singhania A, Stavropoulos E, Redford PS, Chakravarty P, Priestnall SL, Suarez-Bonnet A, Herbert E, Mayer-Barber KD, Sher A, Fonseca KL, Sousa J, Cá B, Verma R, Haldar P, Saraiva M, and O’Garra A. 2020. Mouse transcriptome reveals potential signatures of protection and pathogenesis in human tuberculosis. Nat. Immunol 21: 464–476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Du Bruyn E, Ruzive S, Lindestam Arlehamn CS, Sette A, Sher A, Barber DL, Wilkinson RJ, and Riou C. 2021. Mycobacterium tuberculosis-specific CD4 T cells expressing CD153 inversely associate with bacterial load and disease severity in human tuberculosis. Mucosal Immunol. 14: 491–499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Jurado JO, Pasquinelli V, Alvarez IB, Peña D, Rovetta AI, Tateosian NL, Romeo HE, Musella RM, Palmero D, Chuluyán HE, and García VE. 2012. IL-17 and IFN-γ expression in lymphocytes from patients with active tuberculosis correlates with the severity of the disease. J. Leukoc. Biol 91: 991–1002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Tortola L, Jacobs A, Pohlmeier L, Obermair F-J, Ampenberger F, Bodenmiller B, and Kopf M. 2020. High-Dimensional T Helper Cell Profiling Reveals a Broad Diversity of Stably Committed Effector States and Uncovers Interlineage Relationships. Immunity 53: 597–613.e6. [DOI] [PubMed] [Google Scholar]
- 68.Aguilo N, Alvarez-Arguedas S, Uranga S, Marinova D, Monzón M, Badiola J, and Martin C. 2016. Pulmonary but Not Subcutaneous Delivery of BCG Vaccine Confers Protection to Tuberculosis-Susceptible Mice by an Interleukin 17-Dependent Mechanism. J. Infect. Dis 213: 831–839. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.