

Abstract
Cellular autophagy is a protective mechanism where cells degrade damaged organelles to maintain intracellular homeostasis. Apoptosis, on the other hand, is considered as programmed cell death. Interestingly, autophagy inhibits apoptosis by degrading apoptosis regulators. In hypertension, an imbalance of autophagy and apoptosis regulators can lead to renal injury and dysfunction. Previously, we have reported that toll-like receptor 4 (TLR4) mutant mice are protective against renal damage, in part, due to reduced oxidative stress and inflammation. However, the detailed mechanism remained elusive. In this study, we tested the hypothesis of whether TLR4 mutation reduces Ang-II-induced renal injury by inciting autophagy and suppressing apoptosis in the hypertensive kidney. Male mice with normal TLR4 expression (TLR4N, C3H/HeOuJ) and mutant TLR4 (TLR4M, C3H/HeJLps-d) aged 10–12 weeks were infused with Ang-II (1000 ng/kg/d) for 4 weeks to create hypertension. Saline infused appropriate control were used. Blood pressure was increased along with increased TLR4 expression in TLR4N mice receiving Ang-II compared to TLR4N control. Autophagy was downregulated, and apoptosis was upregulated in TLR4N mice treated with Ang-II. Also, kidney injury markers plasma lipocalin-2 (LCN2) and kidney injury molecule 1 (KIM-1) were upregulated in TLR4N mice treated with Ang-II. Besides, increased nuclear translocation and activity of NF-κB were measured in Ang-II-treated TLR4N mice. TLR4M mice remained protected against all these insults in hypertension. Together, these results suggest that Ang-II-induced TLR4 activation suppresses autophagy, induces apoptosis and kidney injury through in part by activating NF-κB signaling, and TLR4 mutation protects the kidney from Ang-II-induced hypertensive injury.
Keywords: TLR4, autophagy, apoptosis, hypertension, kidney, injury
1. Introduction
Hypertension is the second main cause of kidney injury globally leading to end-stage renal disease [1]. Exaggerated cellular response and alterations of vascular structure, including various cytokines released from immune cells, matrix degradation, and fibrosis, are thought to be the leading contributing factors in the development and progression of organ damage in hypertension [2].
Toll-like receptors (TLRs) are an integral part of the innate immune system, and angiotensin II (Ang-II) is known for the development and progression of hypertension involving the innate immune system [3, 4] that include induction of TLR4 expression in the kidney cells [5]. It is also reported that Ang-II induces oxidative stress and inflammatory response, which are the key factors that lead to renal injury and fibrosis [6, 7], that were mitigated by TLR4 blunting [8]. Corroborating with these findings, we have previously reported that TLR4 deficient (mutant) mice are protected from Ang-II-induced hypertensive renal damage due to reduced oxidative and inflammatory insults [4]. Others have shown that TLR4 mutant mice are resistant to folic acid-induced renal injury and disease progression [9]. However, the molecular etiology of how TLR4 mutation protects the kidney from Ang-II-induced injury is still incompletely understood.
Autophagy is an intracellular degradation process that is involved in hypertension-induced kidney injury [10, 11]. Clinical data reports altered autophagy in hypertension-induced kidney injury [12]. Further, TLRs signaling, including TLR4, is associated with autophagy changes in various diseases [13–18]. Several studies demonstrated that autophagy plays a protective role in TLR4-dependent signaling cascades in diseases, such as acute kidney injury [19, 20] and diabetic nephropathy [21]. However, the precise role of TLR4-mediated autophagy in hypertensive kidney damage remains elusive.
Apoptosis is a form of programmed cell death that is vital for normal cell turnover, development, and function of the immune system. Uncontrolled apoptosis is an indication of many diseases, including degenerative disease, ischemic damage, and acute organ injury [22]. NF-κB (nuclear factor kappa-light-chain-enhancer of activated B cells) is a ubiquitously expressed transcription factor that mediates number of genes involved in several biological processes, including cell death and survival [23]. TLRs are capable of activating the NF-κB pathway leading to the progression of kidney disease [24–26]. Further, TLR4/NF-κB signaling is implicated in apoptosis of tubular cells in drug-induced acute kidney injury (AKI) [27]. However, whether TLR4-mediated NF-κB signaling plays a role in apoptotic cell signaling in the hypertensive kidney and whether TLR4 mutation protects the kidney from injury is unknown.
The present study was undertaken to determine whether TLR4 has a detrimental role in Ang-II-induced hypertensive kidney injury and whether mutation of TLR4 protects against kidney by modulating cellular death signaling.
2. Materials and methods:
2.1. Animals
All animal protocols were performed in accordance with the institutional animal care guidelines and conform to the Guide for the Care and Use of Laboratory Animals published by the U.S. National Institutes of Health (NIH Publication, 2011). This study was approved by the Institutional Animal Care and Use Committee (IACUC) of the University Of Louisville School Of Medicine. C3H/HeJ (Tlr4Lps-d, Stock no.: 000659) and C3H/HeOuJ (Stock no.:000635) mice aged 10–12 weeks were purchased from Jackson Laboratory (Bar Harbor, ME). The C3H/HeJ mice are endotoxin resistant due to a spontaneous mutation at the lipopolysaccharide response locus in the toll-like receptor 4 gene, Tlr4Lps-d, and we termed these mice as TLR4M hereafter. Whereas the C3H/HeOuJ is a substrain of C3H/HeJ, has normal TLR4, and we termed these mice as TLR4N hereafter. The animals were fed standard chow and regular water ad libitum.
2.2. Hypertension induction
Hypertension was induced through angiotensin II (Ang-II) treatment as described previously with modifications [28]. Briefly, under intraperitoneal tribromoethanol anesthesia and sterile conditions, a dorsal midline incision was made in the lower thorax, and a subcutaneous pocket was created in the right flank area. Alzet mini-osmotic pumps (model: 1004, delivery: 0.11 μL/h) loaded with saline or Ang-II were inserted to deliver Ang-II at 1000 ng/kg/min for a period of 4 weeks. Wound was closed with 5–0 nylon (Aros Surgical, Newport Beach, CA).
2.3. Blood pressure measurement
Blood pressure (BP) was measured by the tail-cuff method at 0, 1-, 2-, 3-, and 4-weeks using the Coda™ non-invasive BP system (Kent Scientific Corporation, Torrington, CT) as described before and by following manufacturer’s instructions [29]. Mice were acclimated with BP holder one week before commencement of BP measurement. All recordings were done between 10–11 AM at ambient room temperature, lighting, and absence of noise. Briefly, the mice were placed in the holder and the tail passed through the occlusion and volume pressure cuffs. Three cycles of BP were recorded, each with 20 readings. The data was exported (excel) and systolic and mean BP from all ‘regular’ and ‘accepted’ cycles were averaged and included in respective groups. The mean ± SEM for the groups were used to plot graphs in excel.
2.4. Antibodies and reagents
Antibodies against NF-κB (p-65) (Cat# 8242S), p-NF-κB (p65) (Cat# 3033S), IκBα (Cat# 9242S), p62 (Cat# 5114S), Atg3 (Cat# 3415P), Atg7 (Cat# 2631P), Atg12 (Cat# 4180P), and cleaved caspase 3 (Cat# 9664S), and 7 (Cat# 8438S) were purchased from Cell Signaling Technology (Danvers, MA). Lipocalin-2 (Cat# AF1857) was from R&D Systems. Bcl-2 (Cat# MA5-11757), Bax (Cat# MA5-14003), and fluorescently tagged secondary antibodies, anti-rabbit (Alexa Fluor 488, Cat# A-11008 and Alexa Fluor 594, Cat# A-11012), anti-mouse (Alexa Fluor 488, Cat# A-11001 and Alexa Fluor 594, Cat# A-11005) were purchased from Invitrogen (Carlsbad, CA). LC3-I/II (Cat# ab58610), KIM-1/TIM-1 (Cat# ab47635) were purchased from Abcam (Cambridge, MA), and Beclin 1 (Cat# NB500-249), TLR4 (Cat# NB100-56566) was from Novus Biologicals (Centennial, CO). GAPDH (Cat# sc-365062 HRP), Tubulin (Cat# sc-5286 HRP) and HRP conjugated secondary antibodies, anti-rabbit (Cat# sc-2357), anti-mouse (Cat# sc-516102) were from Santa Cruz Biotechnology (Dallas, TX). We followed the manufacturer’s recommendation for all antibody dilutions. All other chemicals used in activity assays were purchased from Sigma Aldrich (St. Louis, MO).
2.5. NF-κB (p65) transcription factor activity measurement
A semi-quantitative colorimetric NF-κB (p65) transcription factor activity assay was performed using a kit (Abcam, ab133112) with isolated nuclear fraction of kidney samples following the manufacturer’s guidelines. Nuclear fraction isolation was performed by Epiquik nuclear extraction kit (Epigentek, OP-0002-1).
2.6. Immunoblot analysis
Kidney homogenates were separated by SDS-PAGE and transferred to Polyvinylidene difluoride (PVDF) membrane. The membranes were incubated with appropriate primary antibodies overnight and corresponding secondary antibodies for 2 h at room temperature. The immunoreactive bands were developed with chemiluminescence and visualized using ChemiDoc MP System (BioRad, Hercules, CA). Band intensities were quantified using ImageJ software (https://imagej.nih.gov/ij/).
2.7. Immunostaining
Frozen 5 μm kidney sections were air-dried for 10 min and fixed with 4% paraformaldehyde for 20 min. Following blocking for 45 min at room temperature, sections were incubated with primary antibodies at 4°C overnight. Immune labeling was done with appropriate Alexa Fluor 488, AF 594, AF 647 conjugated secondary antibodies for 90 min at room temperature. Images were captured by Olympus FluoView1000 (B&B Microscope Ltd., PA). Mean fluorescent intensity was quantified using ImageJ software as described before [30, 31] and presented as bar graphs. For each mouse, three sections and ten regions of interest (ROI) in each section representing different parts of the kidney were evaluated. The mean values were exported to excel file to plot graph.
2.8. TUNEL assay
The terminal deoxynucleotidyl transferase-mediated dUTP-biotin nick end labeling (TUNEL) assay was performed to detect cell apoptosis in the kidney using in situ cell death detection kit, Fluorescein following the manufacturer’s instructions (Roche, Sigma Aldrich) as described previously [32]. Kidney tissue sections were fixed in fixation solution for 20 min at room temperature, washed twice with PBS, and then permeabilized with permeabilization solution for 5 min on ice, followed by washing twice with PBS. The sections were incubated in 50 μl TUNEL reaction mixture for 60 min at room temperature, washed thrice with PBS, and after mounting with coverslips, confocal microscopic analyses were performed.
2.9. Lipocalin-2 ELISA assay
The lipocalin-2 (LCN2) or NGAL mouse simple-step ELISA kit (ab199083, Abcam, Cambridge, UK) was used to determine plasma LCN2 levels according to the manufacturer’s instruction.
2.10. Statistical analysis
Data are expressed as mean ± SEM of the number of experiments or animals/group as stated in the figure legends. More precisely, n=5 for TLR4N and TLR4M controls (saline only), and n=6 for TLR4N+Ang-II and TLR4M+Ang-II. Following tests for normality and homogeneity, the significant differences between the groups were determined by one-way ANOVA. Individual comparisons between the groups were made by using Bonferroni’s multiple comparison tests using SPSS (Chicago, IL) software. A P-value of < 0.05 was accepted as significant.
3. Results
3.1. TLR4 mutant mice are markedly resistant to Ang-II on hypertension
In the TLR4N and TLR4M mice that received saline, there was no difference in systolic and mean blood pressure (BP) in four weeks of the experiment (Fig. 1A–B). In TLR4N mice, Ang-II increased systolic and mean BP within a week following Ang-II infusion and continued to increase until the end-point (Fig. 1A–B). In contrast, TLR4M mice demonstrated blunted response to Ang-II hypertension compared to TLR4N+Ang-II for the same period. The systolic and mean BP in TLR4M mice increased after the second week of Ang-II infusion and continued until four weeks; however, the increase was significantly lower compared to the TLR4N mice with Ang-II (Fig. 1A–B).
Fig. 1.
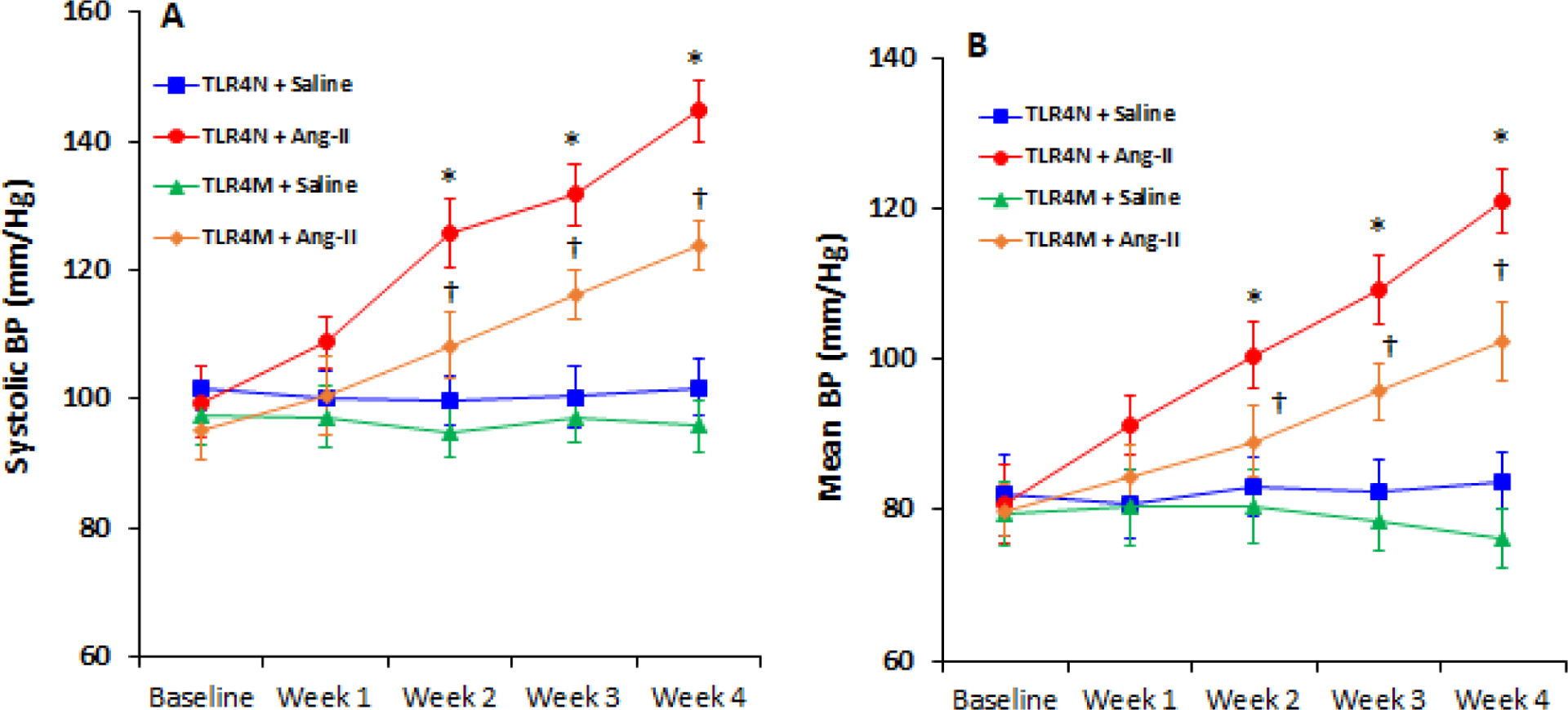
TLR4 mutant mice exhibited resistance to Ang-II induced hypertension. Time course blood pressure (BP) was measured following Ang-II and saline infusion through osmotic mini-pump. (A) systolic and (B) mean BP in TLR4N and TLR4M mice with or without Ang-II. Data presented as mean ± SEM, n = 5/saline (control) groups, and n=6/Ang-II treated groups: *P < 0.05 vs. TLR4N+saline (control), and †P < 0.05 vs. TLR4N+Ang-II.
3.2. Ang-II induced TLR4 in TLR4N mice, and TLR4 mutation maintained higher levels of ATGs expression in the kidney independent of hypertension
First, we determined whether Ang-II hypertension influences TLR4 expression in TLR4N and TLR4M mice infused with Ang-II. The results showed increased TLR4 expression in TLR4N mice treated with Ang-II compared to the TLR4N control group (Fig. 2A). In the TLR4M control group (saline), the expression of TLR4 showed non-significant decrease compared to TLR4N control (Fig. 2A). Interestingly, the expression of TLR4 significantly decreased in TLR4M mice treated with Ang-II compared to TLR4N with Ang-II (Fig. 2A).
Fig. 2.
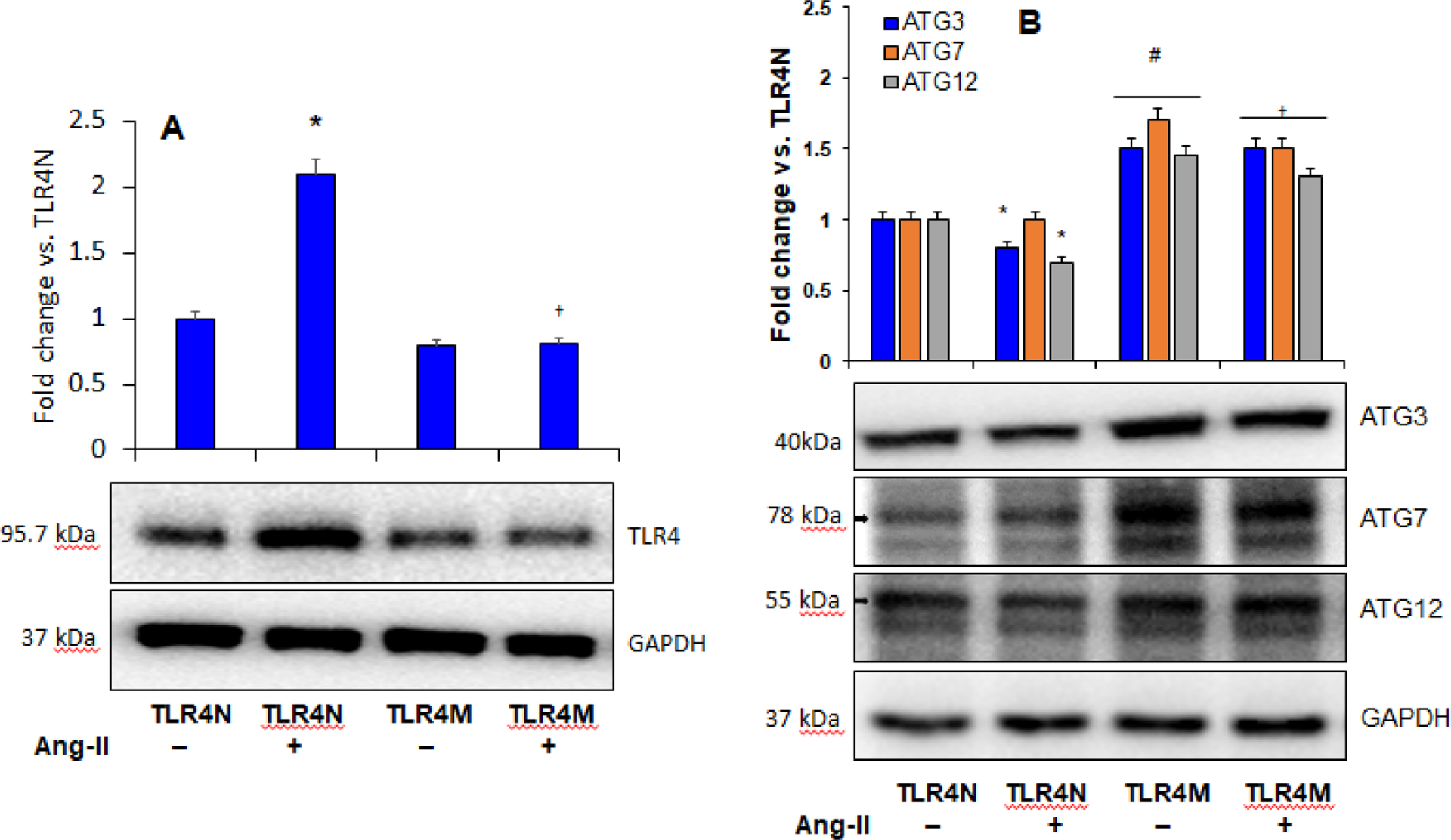
A) Ang-II increased TLR4 expression in TLR4N mice. Representative Western blot image of TLR4 protein expression in the kidney. Bar graph shows fold change of protein expression. Data presented as mean ± SEM, n=5/saline (control) groups, and n=6/Ang-II treated groups: *P < 0.05 vs. TLR4N+saline (control), and †P<0.05 vs. TLR4N+Ang-II. B) TLR4 mutation protected Ang-II-mediated inhibition of autophagy in the kidney. Representative Western blot images of autophagy marker proteins in the kidney. Bar graph represents fold change of protein expression. Data presented as mean ± SEM, n=5/saline (control) groups, and n=6/Ang-II treated groups: * and #P< 0.05 vs. TLR4N+saline (control), and †P< 0.05 vs. TLR4N+Ang-II of respective ATGs.
Autophagy process depends on the regulation of autophagy-related genes (ATGs). Autophagy is involved in regulating a variety of cellular functions [33], and TLR4 is known to regulate autophagy [34]. However, a relationship between TLR4 and autophagy in hypertensive kidney injury and repair is not definitely known. To determine whether TLR4 regulates autophagy in hypertensive renal injury, we measured protein expression of autophagy markers by Western blotting. Our results indicated that the expression of autophagy markers, particularly ATG3 and -12, was decreased in Ang-II hypertensive TLR4N kidney compared to the TLR4N control group, while ATG7 remained unaltered (Fig. 2B). The expressions of ATG3, -7, and -12 in the kidney of control TLR4M mice was significantly increased compared to TLR4N control (Fig. 2B). The expressions of these ATGs in TLR4M mice with Ang-II were significantly higher compared to TLR4N with or without Ang-II. Interestingly, in the TLR4M kidney with Ang-II, while ATG3 remained unaltered, the expressions of ATG7 and -12 were slightly but significantly decreased compared to TLR4M without Ang-II treatment (Fig. 2B).
3.3. TLR4 mutation increased autophagy in the kidney by modulating p62, Beclin 1, and LC3-I/II expression
While an increase in p62 indicates suppressed autophagic flux [35], Beclin 1 acts at the initiation stage of autophagy to stimulate the formation of autophagosome, and LC3-I-to-II conversion determines autophagic activity [36]. Therefore, we measured the expression of p62, Beclin 1, and LC3-I/II protein ratio through immunoblotting of kidney tissue to determine autophagic status in all groups. Our results indicated constitutive expressions of p62, Beclin 1, and LC3-I/II in TLR4N control kidneys (Fig. 3). The expression of p62 was increased, and Beclin 1 and LC3-I/II were decreased in TLR4N kidney treated with Ang-II compared to TLR4N control (Fig. 3). Interestingly, the expressions of p62 significantly decreased, and Beclin 1 and LC3-I/II were significantly increased in TLR4M control mice compared to TLR4N control (Fig. 3). Furthermore, while the expression of LC3-I/I and Beclin 1 remained high, the expression of p62 also increased in TLR4M mice receiving Ang-II compared to TLR4M control (Fig. 3). On the other hand, the expression of p62 decreased significantly, and Beclin 1 and LC3-I/II were increased significantly in TR4M mice that received Ang-II compared to TLR4N with Ang II (Fig. 3).
Fig. 3.
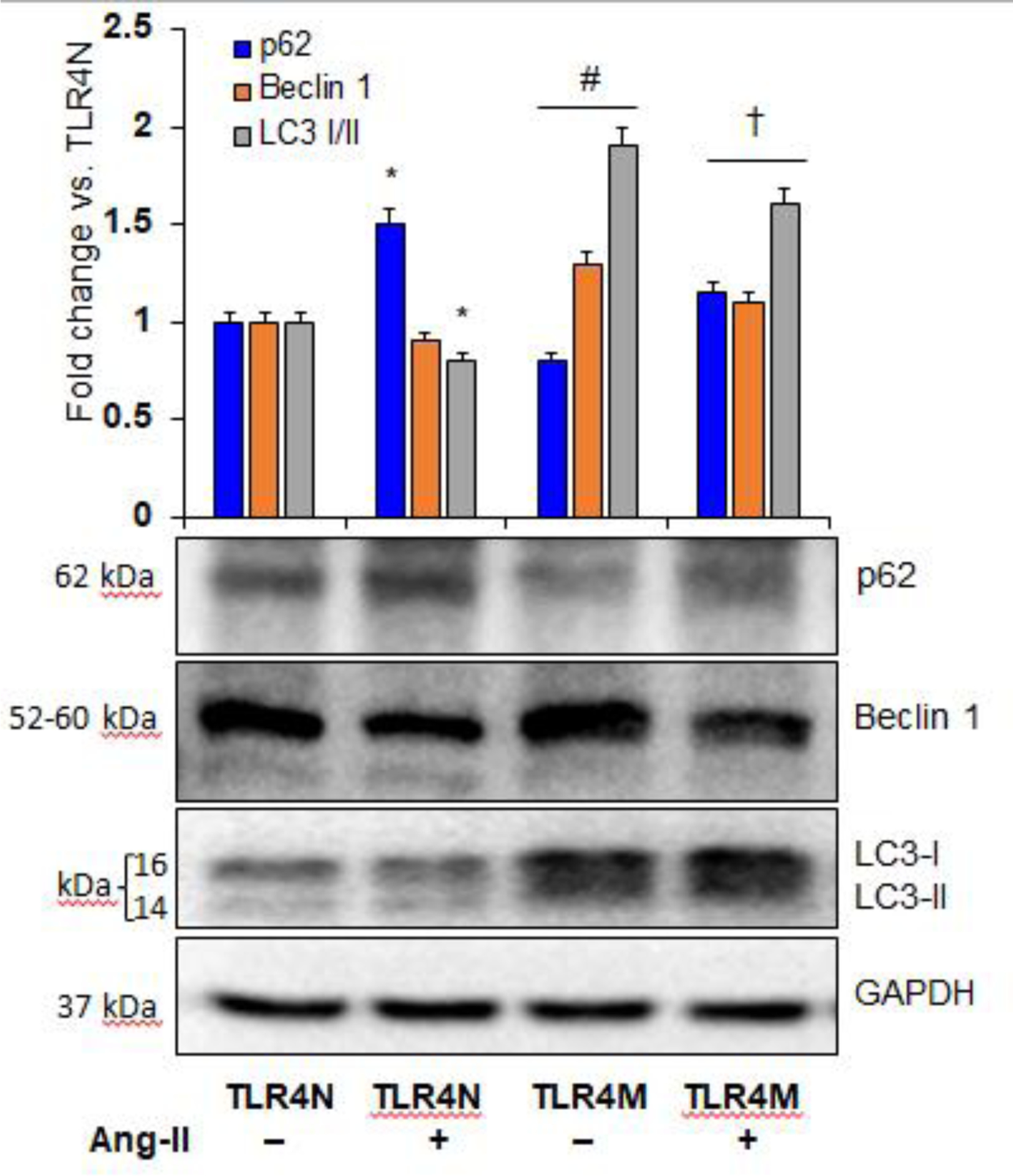
Ang II-mediated autophagy inhibition in TLR4N kidney was restored in TLR4M kidney. Representative Western blot images of autophagy marker proteins p62, Beclin 1, and LC3- I/II expression in the kidney. Respective bar graphs represent fold change of protein expression. Data presented as mean ± SEM, n=5/saline (control) groups, and n=6/Ang-II treated groups: * and #P< 0.05 vs. TLR4N+saline (control), and †P<0.05 vs. TLR4N+Ang-II of respective proteins.
3.4. TLR4 mutation normalized Bcl-2 and Bax expression in hypertension
Both Bcl-2 and Bax play important roles in apoptotic cell death in pathologic conditions [37, 38]. However, their roles in hypertensive kidney damage and whether TLR4 has a regulatory role is incompletely understood. Therefore, we measured Bcl-2 and Bax protein to determine their involvement in hypertensive kidney damage. Results indicated that the expression of Bcl-2 decreased while Bax was increased in TLR4N with Ang-II compared to TLR4N control (Fig. 4A). In the TLR4M mice without Ang-II, both Bcl-2 and Bax expressions did not change significantly compared to TLR4N control (Fig. 4A). Similarly, in TLR4M with Ang-II, the expression of these two molecules remained unchanged and were comparable to TLR4N and TLR4M without Ang-II (Fig. 4A).
Fig. 4.
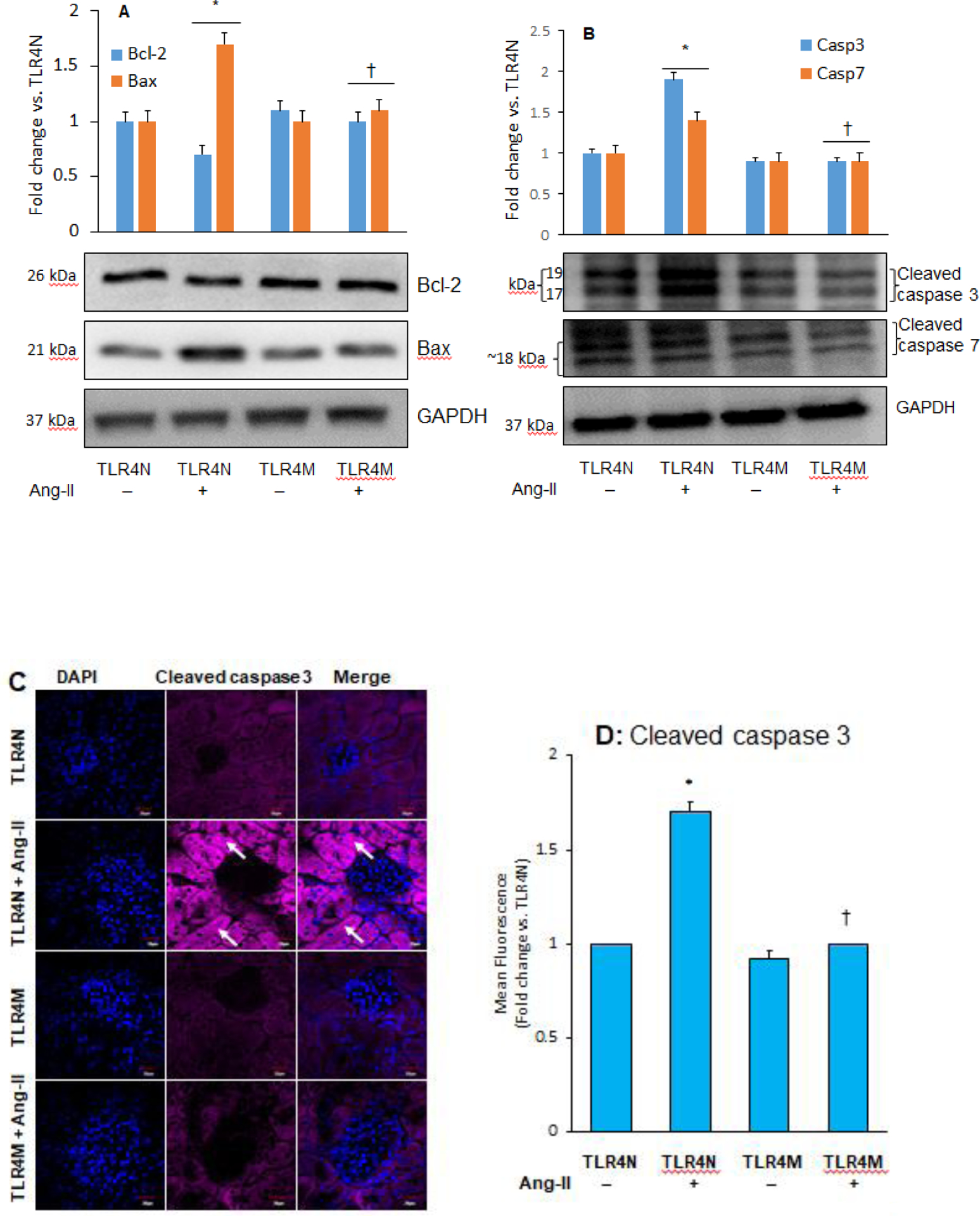
TLR4 mutation decreased Ang-II-induced apoptosis in the kidney. Representative Western blot images for apoptosis marker proteins (A) Bcl-2 and Bax, and (B) cleaved caspase 3 and 7 in the kidney. Bar graphs represents fold change of protein expression, Data presented as mean ± SEM, n=5/saline (control) groups, and n=6/Ang-II treated groups: *P<0.05 vs. TLR4N+saline (control), and †P< 0.05 vs. TLR4N+Ang-II of respective proteins. C) Increased cleaved caspase 3 expression in hypertensive kidney was normalized in TLR4 mutant mice. Arrows indicate cleaved caspase 3 expression in renal tubules. Representative images from random fields (5 fields/slide/animal), Magnification 60X. Scale bar 20 μM. (D) Bar graph represents fold change of cleaved caspase 3 expression in the immunostained sections. Data presented as mean ± SEM, n=5/saline (control) groups, and 6/Ang-II treated groups: *P<0.05 vs. TLR4N+saline (control), and †P<0.05 vs. TLR4N+Ang-II.
3.5. TLR4 mutation inhibited caspase 3 and 7 expressions in hypertension
Caspase 3 and 7 have been widely recognized as cell apoptosis markers [39], and therefore, we measured these molecules through immunoblotting. Results indicated that similar to Bax protein (Fig. 4A), the expression of cleaved caspase 3 and 7 increased significantly in TLR4N mice that received Ang-II compared to TLR4N control (Fig. 4B). In TLRM control mice, the expression of these caspases remained at the basal levels, which were comparable to TLR4N controls (Fig. 4B). Interestingly, in TLRM mice with Ang-II, their expression significantly decreased compared to TLR4N with Ang-II and was comparable to TLR4N controls (Fig. 4B). Further immunostaining results of cleaved caspase 3 revealed similar results of caspase 3 immunoblotting in all groups (Fig. 4C–D).
3.6. TLR4 mutation blunted Ang-II-induced apoptosis in hypertensive kidney
Apoptosis determines the turnover of entire cells, and loss and gain of apoptosis influence numerous pathological processes [40, 41]. To understand the effect of TLR4 mutation on apoptosis in the hypertensive kidney, we estimated apoptotic cell death in response to Ang-II treatment in TLR4N and TLR4M mice kidneys. For that reason, we performed a TUNEL assay, and the results indicated that the apoptotic cells were either non-detectable or at the minimum number in the TLR4N control group (Fig. 5A–B). The number of apoptotic cells was robustly and significantly increased in TLR4N mice that received Ang-II treatment compared to TLR4N control (Fig. 5A–B). In TLR4M control mice, the apoptotic cells were at the minimum numbers, which were also comparable to TLR4N control littermates. Interestingly, in the TLR4M mice that received Ang-II treatment, the number of apoptotic cells was significantly increased compared to TLR4N control or TLR4M control. The number of apoptotic cells in TLR4M mice with Ang-II was, however, significantly lowered compared to TLR4N with Ang-II (Fig. 5A–B).
Fig. 5.
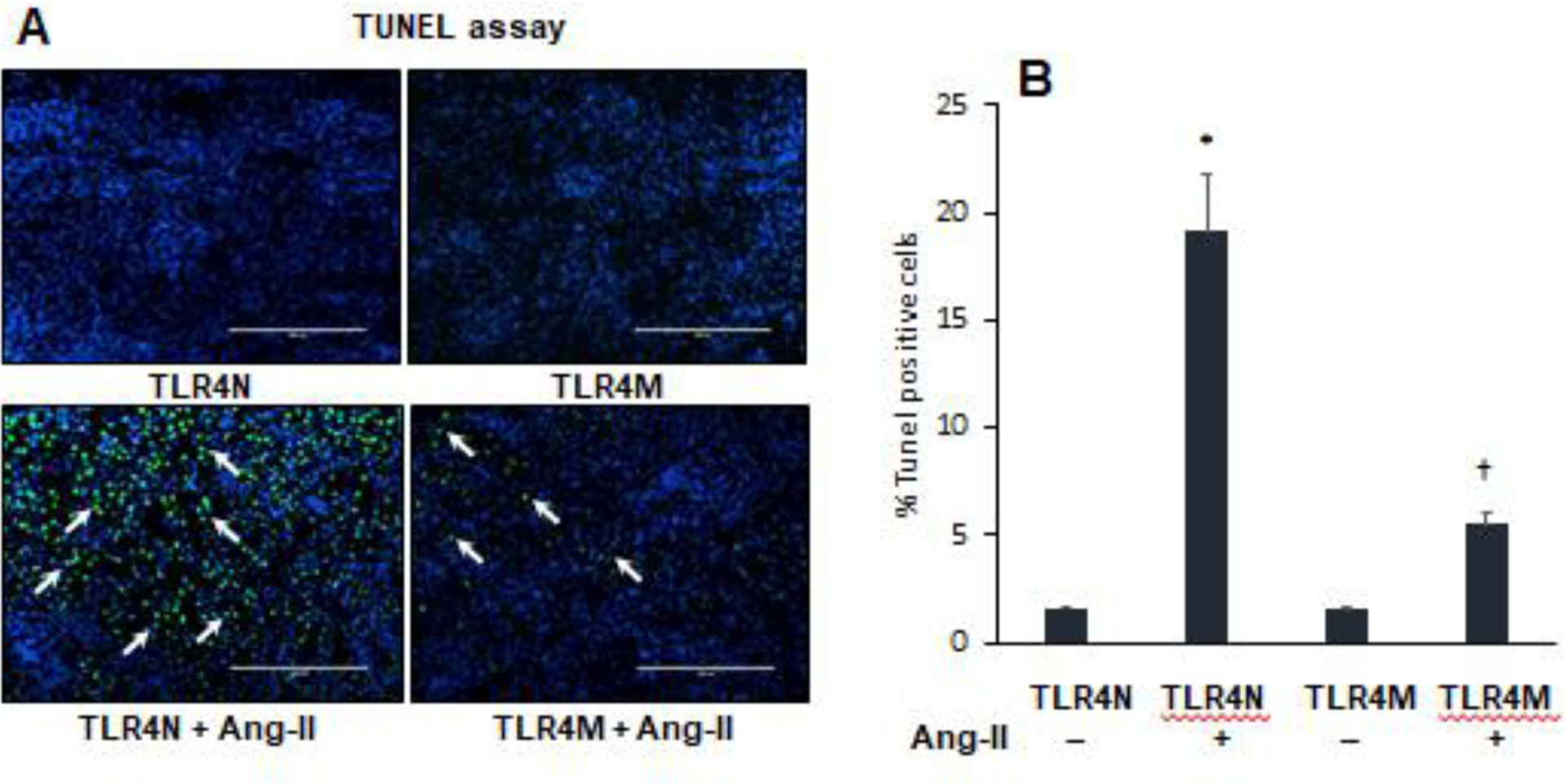
TLR4 mutant mice were protected from apoptotic cells death in hypertensive kidney. (A) Bright green cells are TUNEL positive cells (arrow heads) in the kidney section. Representative images from random fields (5 fields/slide/animal). Magnification 20X. Scale bar 200 μM. (B) Bar graph indicates percent change of number of dead cells. Data presented as mean ± SEM, n=5/saline (control) groups, and n=6/Ang-II treated groups: *P<0.05 vs. TLR4N+saline (control), and †P<0.05 vs. TLR4N+Ang-II.
3.7. TLR4 mutation attenuated Ang-II-induced activation of NF-κβ signaling pathway in hypertensive kidney
A major transcription factor that regulates innate and adaptive immune response genes is NF-κB, and TLR4 targets the NF-κB gene. Interestingly, a regulatory protein IkBα inhibits the activity of dimeric NF-kappa-B/REL complexes by trapping REL dimers in the cytoplasm through masking off their nuclear localization signals [42]. First, to determine whether Ang-II-induced NF-κB activation is TLR4 dependent or not, we measured levels of IkBα and NF-κB (p65) in the kidney tissue from Ang-II treated and control mice, TLR4N and TLR4M, by immunoblotting. Results suggested that IkBα was decreased and NF-κB (p65) was increased significantly in the whole tissue of TLR4N+Ang-II kidney compared to TLR4N control (Fig. 6A). Furthermore, the expression of IkBα was upregulated, and NF-κB (p65) was downregulated in TLR4M control mice compared to TLR4N control (Fig. 6A). Interestingly, the expression of these molecules was normalized in TLR4M kidney with Ang-II and was comparable to TLR4N control (Fig. 6A).
Fig. 6.
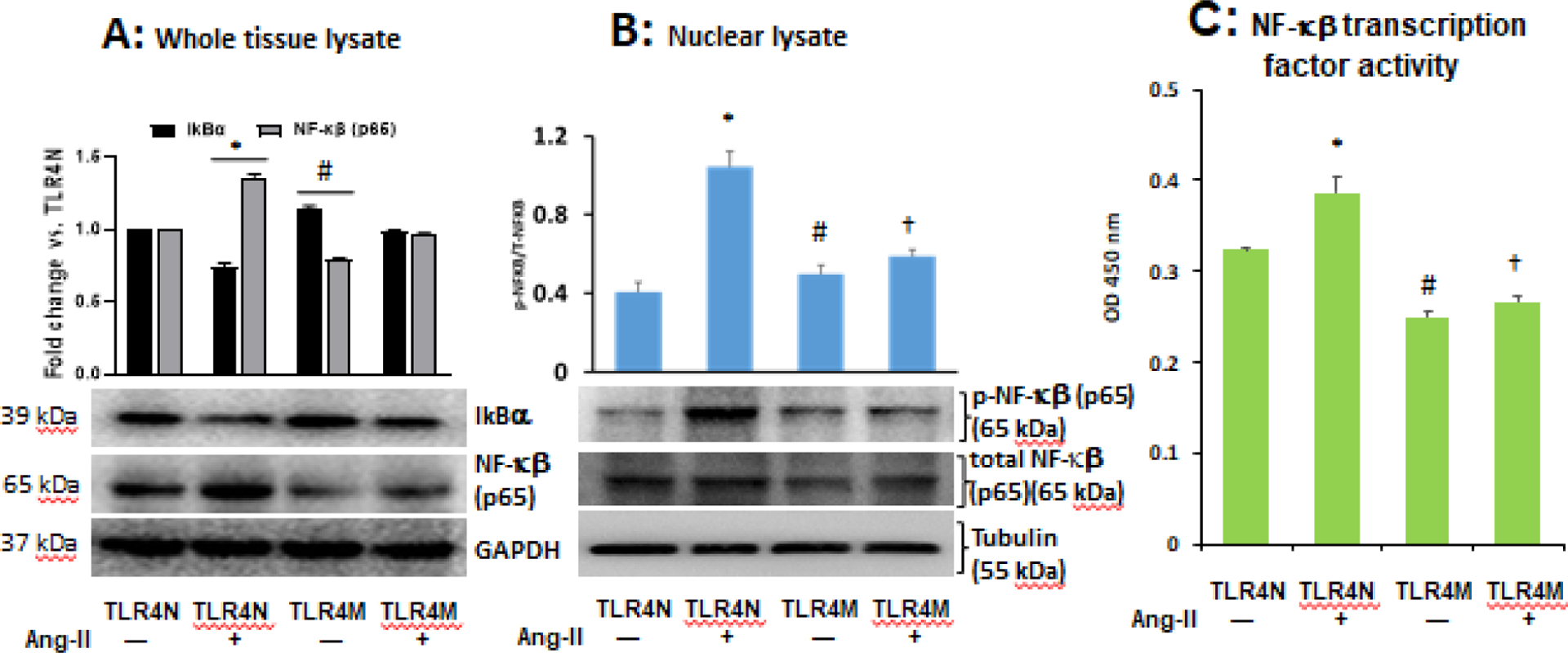
Ang-II-induced diminished IkBα and upregulated NF-κB (p65) expression in the TLR4N kidney was normalized in TLR4M kidney. (A) Representatives immunoblot images of IkBα and NF-κB (p65). GAPDH used as loading control. (B) TLR4 mutation prevented nuclear translocation of Ang-II-induced p-NF-κB (p65) in the kidney tissue. Tubulin was used as loading control; p-NF-κB p65, phospho-NF-κB (p65). C) TLR4 mutation prevented Ang-II-induced NF-κB transcription factor activity in kidney tissue. All bar graphs represent fold change compared to TLR4N+saline (control). Data presented as mean ± SEM, n=5/saline (control) groups, and n=6/Ang-II treated groups: * and #P<0.05 vs. TLR4N+saline (control), and †P<0.05 vs. TLR4N+Ang-II.
The NF-κB complex remains in an inactive state in the cytoplasm. Upon activation, it translocates to the nucleus through several intermediate ubiquitination and degradation steps, where it binds to the target genes [43]. To determine whether Ang-II induces nuclear translocation of NF-κB (p65) and TLR4 mutation inhibits translocation, we measured phosphorylated NF-κB (p65) in the nuclear fractions of kidney tissue from all groups. Our results indicated that Ang-II treatment robustly increased the nuclear fraction of phospho-NF-κB (p65) in TLR4N mice, and this increase was significant compared to TLR4N control (Fig. 6B). Although the nuclear fraction of phospho-NF-κB (p65) in TLR4M control mice was significantly higher compared to TLR4N control, it was not as robust as TLR4N+Ang-II (Fig. 6B). Interestingly, the phospho-NF-κB p65 in nuclear fraction did not increase significantly in TLR4M mice treated with Ang-II compared to non-treated TLR4M mice (Fig. 6B). Nevertheless, the nuclear fraction of phospho-NF-κB in TLR4M+Ang-II kidney was significantly lower compared to the fraction measured in TLR4N+Ang-II kidney (Fig. 6B).
We also measured NF-κB transcription factor activity, and the results indicated that the activity was increased in TLR4N mice treated with Ang-II compared to TLR4N control mice (Fig. 6C). Interestingly, the activity decreased significantly in TLR4M control mice compared to TLR4N control. The activity did not differ further in TLR4M mice treated with Ang-II and remained at comparable levels with TLR4M control mice (Fig. 6C); however, the activity was significantly lowered in TLR4M+Ang-II kidney compared to TLR4N+Ang-II (Fig. 6C)
3.8. TLR4 mutation protected the kidney from Ang-II-induced injury
Kidney injury molecule-1 (KIM-1) is a reliable biomarker of kidney damage and is highly expressed in proximal tubular cells following injury [44]. In addition, Lipocalin-2 (LCN2), also known as neutrophil gelatinase-associated lipocalin (NGAL), is an inflammatory biomarker of several disease processes, including kidney damage [45]. To demonstrate whether Ang-II hypertension-induced kidney damage and injury in TLR4N mice and whether TLR4M mice are protected from injury, we measured KIM-1 in the kidney section by immunostaining (Fig. 7A) and LCN2 in the plasma by ELISA (Fig. 7B). Our results indicated that KIM-1 and LCN2 were upregulated in the kidney proximal tubules and in blood plasma, respectively, in TLR4N mice treated with Ang-II compared to TLR4N control mice (Fig. 7A–B). The expression of KIM-1 in TLR4N and TLR4M mice without Ang-II treatment remained at the basal levels and were comparable between the groups (Fig. 7A). Similar comparable results were also observed for plasma LCN2 levels in TLR4N and TLR4M control mice (Fig. 7B). Interestingly, TLR4 mutation blunted increased expression of KIM-1 and LCN2 in Ang-II treated TLR4M mice, which otherwise were observed increased expression in TLR4N mice treated with Ang-II (Fig. 7A–B). Corroborating to KIM-1 immuno-expression in the kidney sections and LCN2 plasma levels, the immunoblotting results of kidney tissue extract indicated similar findings (Fig. 7C).
Fig. 7.
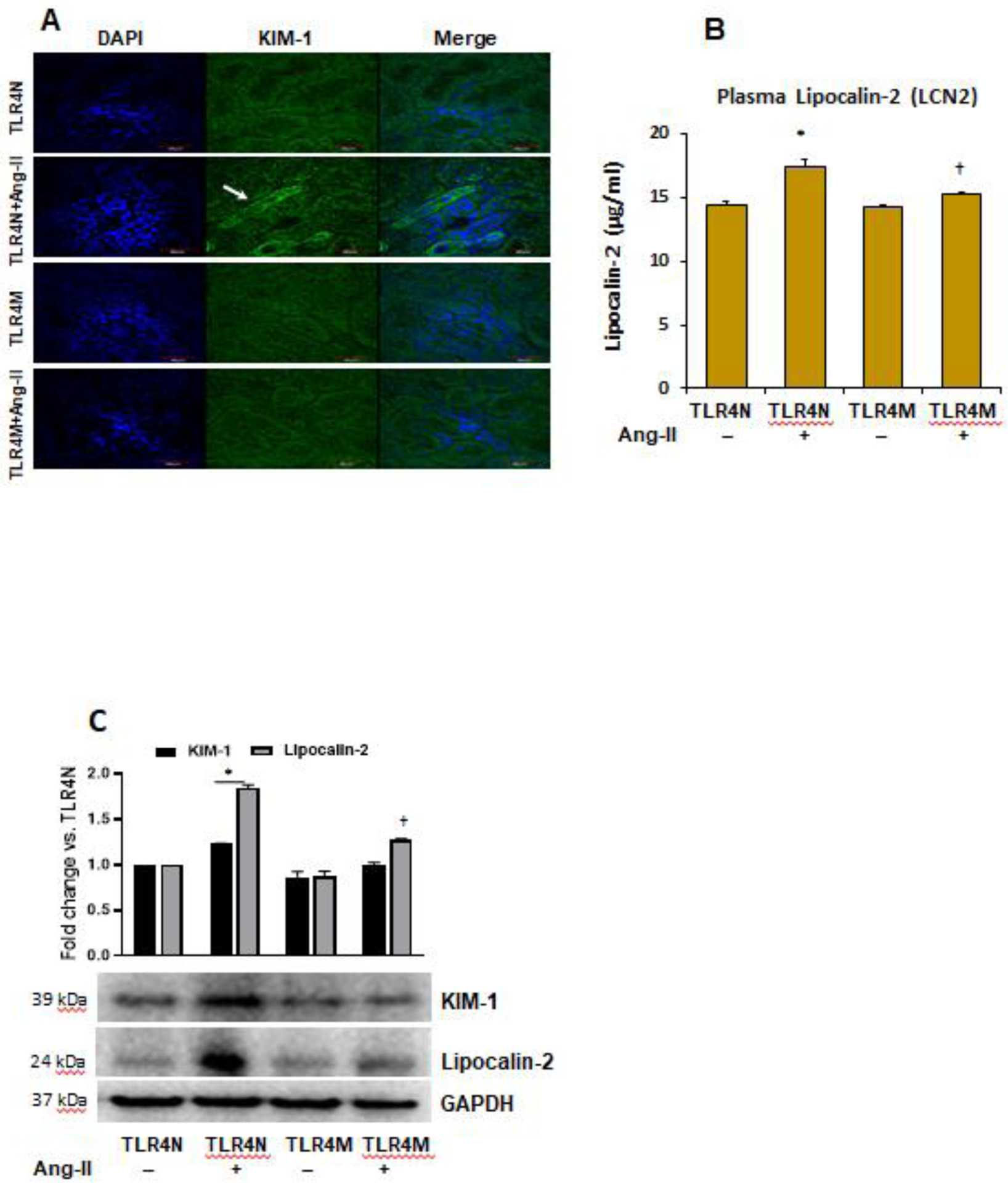
TLR4 mutation protected kidney from injury in Ang-II hypertension. (A) Kidney injury molecule-1 (KIM-1) was detected by immunostaining. Representative images from random fields (5 fields/slide/animal). Magnification 60X. Scale bar 40 μM. (B) plasma Lipocalin-2 (LCN2) was measured by ELISA. Data represent mean ± SEM, n=5/saline (control) groups, and n=6/Ang-II treated groups: *P<0.05 vs. TLR4N+saline (control), and †P<0.05 vs. TLR4N+Ang-II. (C) Western blot images of kidney tissue KIM-1 and Lipocalin-2 expression. GAPDH was used as loading control. Bar graphs represent fold change of protein expression. Data presented as mean ± SEM, n=5/saline (control) groups, and n=6/Ang-II treated groups: *P<0.05 vs. TLR4N+saline (control), and †P< 0.05 vs. TLR4N+Ang-II of respective proteins.
4. Discussion
Our overall results indicate that in response to hypertensive stress, autophagy was downregulated, and apoptosis was upregulated, leading to renal injury in TLR4N mice treated with Ang-II compared to TLR4N control mice (Fig. 8). These were due to, in part, increased nuclear translocation and activity of NF-κB resulting from inhibition of a regulatory protein, IkBα. Which, otherwise, when normally expressed, inhibits the activity of dimeric NF-κB/REL complexes by trapping REL dimers in the cytoplasm through masking off their nuclear localization signals. Our results also indicate that TLR4M blunted the renal injury in hypertension by preserving the normal expression of IkBα and preventing the increased activity of NF-κB and its nuclear translocation. Together, these results suggest a potential mechanistic pathway of renal injury and reveal that TLR4 could be considered as a target molecule to prevent kidney damage in hypertension.
Fig. 8.
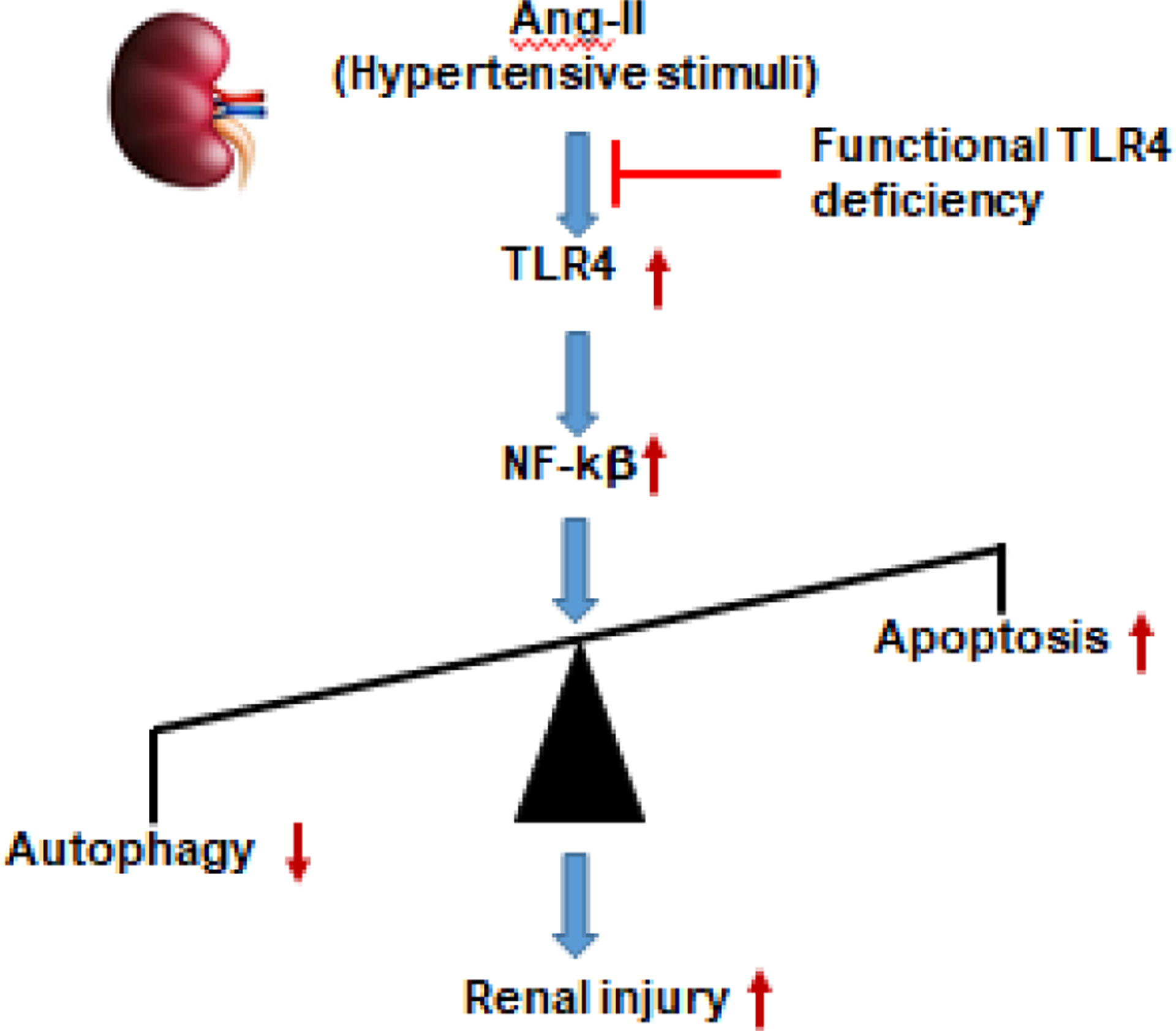
Schematic of findings. Ang-II-induced activation of TLR4 upregulates NF-κB in the kidney leading to impaired homeostasis of autophagy and apoptosis resulting in renal injury. Mutation of TLR4 protects Ang-II-induced renal injury by inducing autophagy and inhibiting apoptosis in the kidney during hypertension.
Kidney injury and dysfunction remain a constant challenge to successfully treat end-stage renal disease (ESRD), including hemodialysis patients with hypertension [46, 47]. Several plasma or urinary biomarkers are recognized as the early indicators of kidney injury. Of these, lipocalin-2 (LCN2), also known as neutrophil gelatinase-associated lipocalin (NGAL), and kidney injury molecule-1 (KIM-1) are the two most widely used biomarkers to detect AKI in clinical conditions [48, 49]. While LCN2 is released by a variety of cell types into the bloodstream and is recognized as a biomarker of several disease conditions, including kidney damage [45], KIM-1 is highly upregulated in proximal tubular cells following kidney injury [44]. Besides, in patients with essential and renovascular hypertension, these two biomarkers were found to be elevated [50]. Interestingly, increased KIM-1 and LCN2 expression in the kidney were reported to be associated with upregulated expression of TLRs, including TLR4 in myocardial infarcted diabetic rats [51], in a genetic model of spliced X-box binding protein 1, which is a key component of the endoplasmic reticulum stress-activated pathways [52], as well as in several AKI and CKD mouse models [53]. It is also reported that Ang-II triggers TLR4 activation in the kidney [3]. Corroborating with these previous reports, our results also indicate that these kidney injury biomarkers, i.e., KIM-1 and LCN2, were elevated (Fig. 7A–C) along with upregulated TLR4 expression (Fig. 2A) in hypertensive TLR4N mice (Fig. 1A–B) compared to their TLR4N control littermates. Importantly, the expression of these molecules was blunted in TLR4M mice treated with Ang-II compared to TLR4N mice with Ang-II, suggesting that TLR4M mice are protected from hypertensive kidney injury (Fig. 7A–C).
Among several other modulators, autophagy plays a crucial role in kidney injury during stress conditions [54]. It is a multi-step dynamic and occurs in a highly conserved lysosomal degradation pathway, which plays a critical role in maintaining cellular homeostasis in all major cell types in the kidney, including mesangial, podocytes, and renal tubular cells [55]. Precisely, it plays dual roles in modulating cellular survival and death [54]. A growing number of evidence indicate that abnormal autophagy regulation can induce renal damage in disease conditions, including AKI and CKD [56]. In response to hypertensive stress, autophagy has been reported to be activated in mesangial cells to induce apoptosis [57]. In addition, depletion in autophagy in proximal tubular cells resulted in severe tubular injury and renal dysfunction [58–60]. However, the detailed mechanism by which hypertension induces autophagy and regulates kidney injury is incompletely understood. Moreover, although it is well established that TLR4 is a sensor for autophagy [13], the causal relationship between TLR4, autophagy, and hypertension kidney injury is not clear. Furthermore, whether TLR4 mutation protects the kidney from hypertensive injury is not well documented. To address these knowledge gaps, we sought to delineate the molecular mechanism of how Ang-II-induced TLR4 activation affects the autophagy process in the kidney, leading to injury.
The core of the autophagy process largely depends on the regulation and activities of approximately 20 autophagy-related genes (ATGs) [61]. Homologs of ATG8 in the mammalian system are called microtubule-associated protein 1 light chain 3 (LC3), and the processed unlipidated LC3 are LC3-I and -II, and these are the most commonly used autophagy markers [61]. In addition, ATG3 and ATG7 form a stable covalent intracellular complex with LC3, a process crucial for functional autophagy [62]. Besides, the formation of autophagosome involves a ubiquitin-like conjugation system in which ATG12 is covalently bound to ATG5 [63], which is essential for LC3 lipidation, a regulatory machinery for autophagy [64]. We determined whether Ang-II hypertension diminishes autophagy in the kidney and whether TLR4 mutation protects the kidney from Ang-II threat by upregulating autophagy markers. Our results revealed that Ang-II treatment downregulated ATG3 and ATG12, but not the ATG7 in TLR4N mice compared to TLR4N control (Fig. 2B). These three ATGs were upregulated in TLR4M control mice compared to TLR4N, suggesting that TLR4M induces autophagy as a pro-defense mechanism against possible stress. This was further evidenced with sustained upregulation of ATG3, -7, and -12 in the kidney of TLR4M mice that received Ang-II (Fig. 2B). We also documented similar results for LC3-I/II (Fig. 3), further confirming that TLR4M is a defensive autophagy tool against Ang-II-induced hypertensive injury in the kidney.
Alongside ATGs, Beclin 1 is described as the mammalian homolog of ATG6 [65], which is part of a lipid complex that coordinates the cytoprotective function of autophagy [66]. Also, p62 is a multifunctional stress-inducible scaffold protein involved in a variety of cellular processes, including autophagy [67]. Degradation of p62 is another widely used marker to monitor autophagic activity because p62 directly binds to LC3 and is selectively degraded by autophagy [68]. Our results revealed that Ang-II treatment downregulated Beclin 1, and upregulated autophagy substrate p62 in the hypertensive kidney with normal functional TLR4 (TLR4N) compared to TLR4N control mice (Fig. 3). In contrast, TLR4M mice with dysfunctional TLR4 were protected from Ang-II-induced upregulation of autophagy marker Beclin 1 and downregulation of p62 in the kidney. These results suggest that the TLR4M kidney is protected against hypertensive stress through a mechanism that promotes autophagic induction.
While autophagy is a process by which dysfunctional sub-cellular organelles are destroyed and new ones are rebuilt to replace them, apoptosis is a programmed cell death through a series of protease pathways that the body uses to rid of damaged cells beyond repair. In general, autophagy blocks the induction of apoptosis, and apoptosis-associated caspase activation shuts off the autophagic process [41]. Bcl-2 family proteins are key regulators of apoptosis cell death, and Bax belongs to this family protein. These apoptosis-associated proteins, including Bax and cleaved caspase-3, were shown to be upregulated, and Bcl-2 downregulated in AKI kidneys [69]. Of note, Bcl-2 functions as anti-apoptotic, whereas Bax and cleaved caspase-3 are pro-apoptotic. In vivo and in vitro research also reported upregulated caspase -7 in agonist-activated renal tubular epithelial cells as well as in a model of unilateral ureteral obstruction (UUO) kidney [70]. Corroborating with these previous reports, our results also suggest that hypertensive injury following Ang-II treatment increased apoptosis in the kidney of TLR4N mice compared to TLR4N control as measured by downregulated expression of Bcl-2, and upregulated expression of Bax, and caspases-3 and -7 (Figs. 4A–D). TLR4N and TLR4M mice without Ang-II showed a basal and comparable level of expression of these apoptosis-associated proteins in the kidney. Interestingly, in the kidney of TLR4M mice treated with Ang-II, the expression of these apoptosis-associated proteins remained unchanged compared to TLR4M control and are comparable to TLR4N control as well (Fig. 4A–D). The TUNEL assay, in which fragmented DNAs were detected that occur in the last phase of apoptosis, further confirmed the above results across the groups (Fig. 5A–B).
Several studies have demonstrated TLR4 upregulation following Ang-II infusion in the kidney, heart, and brain, leading to organ injury [3, 71–73]. In the kidney, Xu et al. showed Ang-II induced renal inflammation and fibrosis was dependent on the formation of myeloid differentiation 2 (MD2)/TLR4 complex and subsequent recruitment of adaptor protein, MyD88, that resulted in ERK and NF-κB signaling [71]. In separate studies, Singh et al. demonstrated Ang-II induced hypertension is dependent on TLR4-TRIF signaling [74] whereas, MyD88 suppresses proinflammatory cytokine production in the kidney [75]. The studies above suggest the important and diverse mechanistic role of TLR4 in angiotensin-induced hypertension and renal inflammation. In this process, the activation of NF-κB is central to a variety of stress responses that, in addition to inflammation, include autophagy and apoptosis [76–78].
A link between NF-κB signaling and apoptosis was previously shown to be involved in kidney injury. For example, Marko et al. reported IRI-induced NF-κB activation in renal tubular epithelial and in interstitial cells followed by nuclear localization of phosphorylated NF-κB subunit p65 that led to apoptosis [79]. We have previously reported that functional TLR4 deficient, i.e., TLR4 mutant mice are protected from Ang-II-induced increased oxidative stress and inflammatory kidney injury due to improved antioxidant defense mechanisms [4]. However, whether Ang-II-induced kidney injury was a direct effect of NF-κB activation, which is one of the important target genes of TLR4, was not known. Reports showed that Ang-II-mediated NF-κB activation and cardiac remodeling were prevented by TLR4 knockdown [73]. Also, Ang-II promoted NF-κB mediated injury in kidney mesangial cells [80]. Furthermore, it is known that the degradation of IkB induces NF-κB activation [81, 82]. Corroborating with these earlier findings, our present report demonstrated that Ang-II decreased IkBα and increased NF-κB (p65) in TLR4N kidney tissue compared to TLR4N control (Fig. 6A). In addition, nuclear translocation of phosphorylated NF-κB (p65) was observed in the kidney of TLR4N mice treated with Ang-II, which was significantly decreased in TLR4M mice treated with Ang-II (Fig. 6B). NF-κB activity in these mice also corroborated with its phosphorylation and nuclear translocation levels (Fig. 6C). Together, these results suggest that TLR4M mice are protected from hypertension-induced increased apoptosis signaling in the kidney through reduced induction, nuclear translocation, and decreased activity of NF-κB (Fig. 6A–C). It also confirmed that autophagy is a defensive mechanism in TLR4M mice against Ang-II threat, which otherwise induces apoptosis leading to kidney injury (Fig. 8).
In summary, our study demonstrates that Ang-II-induced hypertension diminishes autophagy and induces apoptosis in TLR4N mice leading to renal injury. TLR4 mutant (TLR4M) mice are protected from Ang-II-induced renal insult through normalization of both autophagy and apoptosis. Further, we show that increased NF-κB activation, nuclear translocation, and activity played a major role in autophagy and apoptotic imbalance in hypertensive TLR4N kidneys. Finally, TLR4M inhibited NF-κB nuclear translocation and activity that protected the kidney from hypertensive injury.
Acknowledgments
This work was supported in part by the National Institutes of Health Grants, DK104653 and DK116591 (to U.S.), and American Heart Association Scientist Development Grant, 15SDG25840013 (to S.P.).
Abbreviations:
- Ang-II
Angiotensin II
- ATG
autophagy-related gene
- Bax
Bcl-2-associated X protein
- Bcl-2
B-cell lymphoma 2
- Beclin 1
a mammalian ortholog of the yeast ATG6
- Casp
caspase
- CKD
chronic kidney disease
- DAPI
4′,6-diamidino-2-phenylindole
- GAPDH
glyceraldehyde 3-phosphate dehydrogenase
- IκBα
a regulatory protein that inhibits NF-κB
- KIM-1
kidney injury molecule-1
- LC3- I/II
microtubule-associated protein light chain 3 I/II
- LCN2
Lipocalin-2
- NF-κB
nuclear factor kappa B
- p62
ubiquitin-binding protein p62 or sequestosome-1
- TLR4
toll-like receptor 4
- TUNEL
terminal deoxynucleotidyl transferase dUTP nick end labeling
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Disclosure of Potential Conflicts of Interest
The authors declare no conflicts of interest, financial or otherwise.
CRediT authorship contribution statement
SM, SP, and US conceived the idea and designed the experiments. SM, SP and SKJ performed the experiments. SM analyzed the data, prepared the figures, and drafted the manuscript; VRJ contributed to the discussion. US interpreted the results and edited the draft. All authors have approved the final version of the manuscript.
References
- [1].Hill NR, Fatoba ST, Oke JL, Hirst JA, O’Callaghan CA, Lasserson DS, Hobbs FD, Global Prevalence of Chronic Kidney Disease - A Systematic Review and Meta-Analysis, PLoS One 11(7) (2016) e0158765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Rai A, Narisawa M, Li P, Piao L, Li Y, Yang G, Cheng XW, Adaptive immune disorders in hypertension and heart failure: focusing on T-cell subset activation and clinical implications, J Hypertens 38(10) (2020) 1878–1889. [DOI] [PubMed] [Google Scholar]
- [3].Biancardi VC, Bomfim GF, Reis WL, Al-Gassimi S, Nunes KP, The interplay between Angiotensin II, TLR4 and hypertension, Pharmacol Res 120 (2017) 88–96. [DOI] [PubMed] [Google Scholar]
- [4].Pushpakumar S, Ren L, Kundu S, Gamon A, Tyagi SC, Sen U, Toll-like Receptor 4 Deficiency Reduces Oxidative Stress and Macrophage Mediated Inflammation in Hypertensive Kidney, Sci Rep 7(1) (2017) 6349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Bondeva T, Roger T, Wolf G, Differential regulation of Toll-like receptor 4 gene expression in renal cells by angiotensin II: dependency on AP1 and PU.1 transcriptional sites, Am J Nephrol 27(3) (2007) 308–14. [DOI] [PubMed] [Google Scholar]
- [6].Zhang Y, Peng W, Ao X, Dai H, Yuan L, Huang X, Zhou Q, TAK-242, a Toll-Like Receptor 4 Antagonist, Protects against Aldosterone-Induced Cardiac and Renal Injury, PLoS One 10(11) (2015) e0142456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Pulskens WP, Rampanelli E, Teske GJ, Butter LM, Claessen N, Luirink IK, van der Poll T, Florquin S, Leemans JC, TLR4 promotes fibrosis but attenuates tubular damage in progressive renal injury, J Am Soc Nephrol 21(8) (2010) 1299–308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Nair AR, Ebenezer PJ, Saini Y, Francis J, Angiotensin II-induced hypertensive renal inflammation is mediated through HMGB1-TLR4 signaling in rat tubulo-epithelial cells, Exp Cell Res 335(2) (2015) 238–47. [DOI] [PubMed] [Google Scholar]
- [9].Souza AC, Tsuji T, Baranova IN, Bocharov AV, Wilkins KJ, Street JM, Alvarez-Prats A, Hu X, Eggerman T, Yuen PS, Star RA, TLR4 mutant mice are protected from renal fibrosis and chronic kidney disease progression, Physiological reports 3(9) (2015) e12558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Chen K, Zhou X, Sun Z, Haplodeficiency of Klotho Gene Causes Arterial Stiffening via Upregulation of Scleraxis Expression and Induction of Autophagy, Hypertension 66(5) (2015) 1006–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Bensaada I, Robin B, Perez J, Salemkour Y, Chipont A, Camus M, Lemoine M, Guyonnet L, Lazareth H, Letavernier E, Henique C, Tharaux PL, Lenoir O, Calpastatin prevents Angiotensin II-mediated podocyte injury through maintenance of autophagy, Kidney international 100(1) (2021) 90–106. [DOI] [PubMed] [Google Scholar]
- [12].Chrysanthopoulou A, Gkaliagkousi E, Lazaridis A, Arelaki S, Pateinakis P, Ntinopoulou M, Mitsios A, Antoniadou C, Argyriou C, Georgiadis GS, Papadopoulos V, Giatromanolaki A, Ritis K, Skendros P, Angiotensin II triggers release of neutrophil extracellular traps, linking thromboinflammation with essential hypertension, JCI Insight 6(18) (2021) e148668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Xu Y, Jagannath C, Liu XD, Sharafkhaneh A, Kolodziejska KE, Eissa NT, Toll-like receptor 4 is a sensor for autophagy associated with innate immunity, Immunity 27(1) (2007) 135–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Bertin S, Pierrefite-Carle V, Autophagy and toll-like receptors: a new link in cancer cells, Autophagy 4(8) (2008) 1086–9. [DOI] [PubMed] [Google Scholar]
- [15].Delgado MA, Elmaoued RA, Davis AS, Kyei G, Deretic V, Toll-like receptors control autophagy, EMBO J 27(7) (2008) 1110–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Deretic V, Delgado M, Vergne I, Master S, De Haro S, Ponpuak M, Singh S, Autophagy in immunity against mycobacterium tuberculosis: a model system to dissect immunological roles of autophagy, Current topics in microbiology and immunology 335 (2009) 169–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Lee HM, Shin DM, Yuk JM, Shi G, Choi DK, Lee SH, Huang SM, Kim JM, Kim CD, Lee JH, Jo EK, Autophagy negatively regulates keratinocyte inflammatory responses via scaffolding protein p62/SQSTM1, J Immunol 186(2) (2011) 1248–58. [DOI] [PubMed] [Google Scholar]
- [18].Fujita K, Srinivasula SM, TLR4-mediated autophagy in macrophages is a p62-dependent type of selective autophagy of aggresome-like induced structures (ALIS), Autophagy 7(5) (2011) 552–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Andrade-Silva M, Cenedeze MA, Perandini LA, Felizardo RJF, Watanabe IKM, Agudelo JSH, Castoldi A, Goncalves GM, Origassa CST, Semedo P, Hiyane MI, Foresto-Neto O, Malheiros D, Reis MA, Fujihara CK, Zatz R, Pacheco-Silva A, Camara NOS, de Almeida DC, TLR2 and TLR4 play opposite role in autophagy associated with cisplatin-induced acute kidney injury, Clin Sci (Lond) 132(16) (2018) 1725–1739. [DOI] [PubMed] [Google Scholar]
- [20].Leventhal JS, Ni J, Osmond M, Lee K, Gusella GL, Salem F, Ross MJ, Autophagy Limits Endotoxemic Acute Kidney Injury and Alters Renal Tubular Epithelial Cell Cytokine Expression, PLoS One 11(3) (2016) e0150001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Lin M, Yiu WH, Wu HJ, Chan LY, Leung JC, Au WS, Chan KW, Lai KN, Tang SC, Toll-like receptor 4 promotes tubular inflammation in diabetic nephropathy, Journal of the American Society of Nephrology : JASN 23(1) (2012) 86–102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Havasi A, Borkan SC, Apoptosis and acute kidney injury, Kidney International 80(1) (2011) 29–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Albensi BC, What Is Nuclear Factor Kappa B (NF-kappaB) Doing in and to the Mitochondrion?, Frontiers in cell and developmental biology 7 (2019) 154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Anders HJ, Of Inflammasomes and Alarmins: IL-1beta and IL-1alpha in Kidney Disease, J Am Soc Nephrol 27(9) (2016) 2564–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Xia H, Bao W, Shi S, Innate Immune Activity in Glomerular Podocytes, Frontiers in immunology 8 (2017) 122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Knauf F, Brewer JR, Flavell RA, Immunity, microbiota and kidney disease, Nature reviews. Nephrology 15(5) (2019) 263–274. [DOI] [PubMed] [Google Scholar]
- [27].Bai J, Zhao J, Cui D, Wang F, Song Y, Cheng L, Gao K, Wang J, Li L, Li S, Jia Y, Wen A, Protective effect of hydroxysafflor yellow A against acute kidney injury via the TLR4/NF-kappaB signaling pathway, Sci Rep 8(1) (2018) 9173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Lu H, Howatt DA, Balakrishnan A, Moorleghen JJ, Rateri DL, Cassis LA, Daugherty A, Subcutaneous Angiotensin II Infusion using Osmotic Pumps Induces Aortic Aneurysms in Mice, J Vis Exp 103 (2015) e53191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Pushpakumar SB, Kundu S, Metreveli N, Tyagi SC, Sen U, Matrix Metalloproteinase Inhibition Mitigates Renovascular Remodeling in Salt-Sensitive Hypertension, Physiological reports 1(3) (2013) e00063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Jensen EC, Quantitative analysis of histological staining and fluorescence using ImageJ, Anat Rec (Hoboken) 296(3) (2013) 378–81. [DOI] [PubMed] [Google Scholar]
- [31].Hartig SM, Basic image analysis and manipulation in ImageJ, Curr Protoc Mol Biol Chapter 14 (2013) Unit14 15. [DOI] [PubMed] [Google Scholar]
- [32].Majumder S, Ren L, Pushpakumar S, Sen U, Hydrogen sulphide mitigates homocysteine-induced apoptosis and matrix remodelling in mesangial cells through Akt/FOXO1 signalling cascade, Cell Signal 61 (2019) 66–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Jing K, Lim K, Why is autophagy important in human diseases?, Exp Mol Med 44(2) (2012) 69–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Lee JW, Nam H, Kim LE, Jeon Y, Min H, Ha S, Lee Y, Kim SY, Lee SJ, Kim EK, Yu SW, TLR4 (toll-like receptor 4) activation suppresses autophagy through inhibition of FOXO3 and impairs phagocytic capacity of microglia, Autophagy 15(5) (2019) 753–770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Brown CN, Atwood D, Pokhrel D, Holditch SJ, Altmann C, Skrypnyk NI, Bourne J, Klawitter J, Blaine J, Faubel S, Thorburn A, Edelstein CL, Surgical procedures suppress autophagic flux in the kidney, Cell Death Dis 12(3) (2021) 248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Li P, Shi M, Maique J, Shaffer J, Yan S, Moe OW, Hu MC, Beclin 1/Bcl-2 complex-dependent autophagy activity modulates renal susceptibility to ischemia-reperfusion injury and mediates renoprotection by Klotho, Am J Physiol Renal Physiol 318(3) (2020) F772–F792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Song XF, Ren H, Andreasen A, Thomsen JS, Zhai XY, Expression of Bcl-2 and Bax in mouse renal tubules during kidney development, PLoS One 7(2) (2012) e32771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Borkan SC, The Role of BCL-2 Family Members in Acute Kidney Injury, Semin Nephrol 36(3) (2016) 237–50. [DOI] [PubMed] [Google Scholar]
- [39].Nguemo F, Nembo EN, Kamga Kapchoup MV, Enzmann F, Hescheler J, QuinoMit Q10-Fluid attenuates hydrogen peroxide-induced irregular beating in mouse pluripotent stem cell-derived cardiomyocytes, Biomed Pharmacother 142 (2021) 112089. [DOI] [PubMed] [Google Scholar]
- [40].Yu N, Yang L, Ling L, Liu Y, Yu Y, Wu Q, Gu Y, Niu J, Curcumin attenuates angiotensin II-induced podocyte injury and apoptosis by inhibiting endoplasmic reticulum stress, FEBS Open Bio 10(10) (2020) 1957–1966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Marino G, Niso-Santano M, Baehrecke EH, Kroemer G, Self-consumption: the interplay of autophagy and apoptosis, Nature reviews. Molecular cell biology 15(2) (2014) 81–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Zhang Q, Lenardo MJ, Baltimore D, 30 Years of NF-kappaB: A Blossoming of Relevance to Human Pathobiology, Cell 168(1–2) (2017) 37–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Bagaev AV, Garaeva AY, Lebedeva ES, Pichugin AV, Ataullakhanov RI, Ataullakhanov FI, Elevated pre-activation basal level of nuclear NF-kappaB in native macrophages accelerates LPS-induced translocation of cytosolic NF-kappaB into the cell nucleus, Sci Rep 9(1) (2019) 4563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Sabbisetti VS, Waikar SS, Antoine DJ, Smiles A, Wang C, Ravisankar A, Ito K, Sharma S, Ramadesikan S, Lee M, Briskin R, De Jager PL, Ngo TT, Radlinski M, Dear JW, Park KB, Betensky R, Krolewski AS, Bonventre JV, Blood kidney injury molecule-1 is a biomarker of acute and chronic kidney injury and predicts progression to ESRD in type I diabetes, J Am Soc Nephrol 25(10) (2014) 2177–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Moschen AR, Adolph TE, Gerner RR, Wieser V, Tilg H, Lipocalin-2: A Master Mediator of Intestinal and Metabolic Inflammation, Trends in endocrinology and metabolism: TEM 28(5) (2017) 388–397. [DOI] [PubMed] [Google Scholar]
- [46].Van Buren PN, Evaluation and Treatment of Hypertension in End-Stage Renal Disease Patients on Hemodialysis, Current cardiology reports 18(12) (2016) 125. [DOI] [PubMed] [Google Scholar]
- [47].Hannan M, Ansari S, Meza N, Anderson AH, Srivastava A, Waikar S, Charleston J, Weir MR, Taliercio J, Horwitz E, Saunders MR, Wolfrum K, Feldman HI, Lash JP, Ricardo AC, Investigators CS, Chronic I Renal Insufficiency Cohort Study, Risk Factors for CKD Progression: Overview of Findings from the CRIC Study, Clinical journal of the American Society of Nephrology : CJASN 16(4) (2021) 648–659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Sheanon NM, Mottl AK, Dagostino R, Suerken C, Afkarian M, Dabelea D, Imperatore G, Marcovina SM, Pettitt DJ, Saydah S, Dolan LM, Urinary NGAL and KIM-1 Are Significantly Elevated in Young Adults (YA) with Type 1 (T1D) and Type 2 (T2D) Diabetes, Diabetes 67 (2018) Supplement 1. [Google Scholar]
- [49].Lei L, Li LP, Zeng Z, Mu JX, Yang X, Zhou C, Wang ZL, Zhang H, Value of urinary KIM-1 and NGAL combined with serum Cys C for predicting acute kidney injury secondary to decompensated cirrhosis, Sci Rep 8(1) (2018) 7962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [50].Eirin A, Gloviczki ML, Tang H, Rule AD, Woollard JR, Lerman A, Textor SC, Lerman LO, Chronic renovascular hypertension is associated with elevated levels of neutrophil gelatinase-associated lipocalin, Nephrology, dialysis, transplantation : official publication of the European Dialysis and Transplant Association - European Renal Association 27(11) (2012) 4153–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [51].Ohno K, Kuno A, Murase H, Muratsubaki S, Miki T, Tanno M, Yano T, Ishikawa S, Yamashita T, Miura T, Diabetes increases the susceptibility to acute kidney injury after myocardial infarction through augmented activation of renal Toll-like receptors in rats, American Journal of Physiology-Heart and Circulatory Physiology 313(6) (2017) H1130–H1142. [DOI] [PubMed] [Google Scholar]
- [52].Ferre S, Deng Y, Huen SC, Lu CY, Scherer PE, Igarashi P, Moe OW, Renal tubular cell spliced X-box binding protein 1 (Xbp1s) has a unique role in sepsis-induced acute kidney injury and inflammation, Kidney Int 96(6) (2019) 1359–1373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [53].Sharifian R, Okamura DM, Denisenko O, Zager RA, Johnson A, Gharib SA, Bomsztyk K, Distinct patterns of transcriptional and epigenetic alterations characterize acute and chronic kidney injury, Sci Rep 8(1) (2018) 17870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [54].Sureshbabu A, Ryter SW, Choi ME, Oxidative stress and autophagy: Crucial modulators of kidney injury, Redox biology 4 (2015) 208–214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [55].Tang C, Livingston MJ, Liu Z, Dong Z, Autophagy in kidney homeostasis and disease, Nature reviews. Nephrology 16(9) (2020) 489–508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [56].Lin TA, Wu VC, Wang CY, Autophagy in Chronic Kidney Diseases, Cells 8(1) (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [57].Berman S, Abu Hamad R, Efrati S, Mesangial cells are responsible for orchestrating the renal podocytes injury in the context of malignant hypertension, Nephrology (Carlton) 18(4) (2013) 292–8. [DOI] [PubMed] [Google Scholar]
- [58].Jiang M, Wei Q, Dong G, Komatsu M, Su Y, Dong Z, Autophagy in proximal tubules protects against acute kidney injury, Kidney Int 82(12) (2012) 1271–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [59].Suzuki C, Tanida I, Oliva Trejo JA, Kakuta S, Uchiyama Y, Autophagy Deficiency in Renal Proximal Tubular Cells Leads to an Increase in Cellular Injury and Apoptosis under Normal Fed Conditions, Int J Mol Sci 21(1) (2019) 155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [60].Kimura T, Takabatake Y, Takahashi A, Kaimori JY, Matsui I, Namba T, Kitamura H, Niimura F, Matsusaka T, Soga T, Rakugi H, Isaka Y, Autophagy protects the proximal tubule from degeneration and acute ischemic injury, J Am Soc Nephrol 22(5) (2011) 902–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [61].Mizushima N, The ATG conjugation systems in autophagy, Curr Opin Cell Biol 63 (2020) 1–10. [DOI] [PubMed] [Google Scholar]
- [62].Frudd K, Burgoyne T, Burgoyne JR, Oxidation of Atg3 and Atg7 mediates inhibition of autophagy, Nat Commun 9(1) (2018) 95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [63].Pang Y, Yamamoto H, Sakamoto H, Oku M, Mutungi JK, Sahani MH, Kurikawa Y, Kita K, Noda NN, Sakai Y, Jia H, Mizushima N, Evolution from covalent conjugation to non-covalent interaction in the ubiquitin-like ATG12 system, Nat Struct Mol Biol 26(4) (2019) 289–296. [DOI] [PubMed] [Google Scholar]
- [64].Brier LW, Ge L, Stjepanovic G, Thelen AM, Hurley JH, Schekman R, Regulation of LC3 lipidation by the autophagy-specific class III phosphatidylinositol-3 kinase complex, Molecular Biology of the Cell 30(9) (2019) 1098–1107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [65].Menon MB, Dhamija S, Beclin 1 Phosphorylation - at the Center of Autophagy Regulation, Front Cell Dev Biol 6 (2018) 137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [66].Cao Y, Klionsky DJ, Physiological functions of Atg6/Beclin 1: a unique autophagy-related protein, Cell Research 17(10) (2007) 839–849. [DOI] [PubMed] [Google Scholar]
- [67].Sanchez-Martin P, Komatsu M, p62/SQSTM1 - steering the cell through health and disease, J Cell Sci 131(21) (2018) jcs222836. [DOI] [PubMed] [Google Scholar]
- [68].Pankiv S, Clausen TH, Lamark T, Brech A, Bruun JA, Outzen H, Overvatn A, Bjorkoy G, Johansen T, p62/SQSTM1 binds directly to Atg8/LC3 to facilitate degradation of ubiquitinated protein aggregates by autophagy, J Biol Chem 282(33) (2007) 24131–45. [DOI] [PubMed] [Google Scholar]
- [69].Yu XW, Xu M, Meng X, Li SM, Liu QQ, Bai M, You R, Huang SM, Yang L, Zhang Y, Jia ZJ, Zhang AH, Nuclear receptor PXR targets AKR1B7 to protect mitochondrial metabolism and renal function in AKI, Sci Transl Med 12(543) (2020) eaay7591. [DOI] [PubMed] [Google Scholar]
- [70].Ke Y, Tang H, Ye C, Lei CT, Wang YM, Su H, Zhang C, Jiang HJ, Role and Association of Inflammatory and Apoptotic Caspases in Renal Tubulointerstitial Fibrosis, Kidney Blood Press R 41(5) (2016) 643–653. [DOI] [PubMed] [Google Scholar]
- [71].Xu Z, Li W, Han J, Zou C, Huang W, Yu W, Shan X, Lum H, Li X, Liang G, Angiotensin II induces kidney inflammatory injury and fibrosis through binding to myeloid differentiation protein-2 (MD2), Scientific Reports 7 (2017) 44911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [72].Dange RB, Agarwal D, Masson GS, Vila J, Wilson B, Nair A, Francis J, Central blockade of TLR4 improves cardiac function and attenuates myocardial inflammation in angiotensin II-induced hypertension, Cardiovasc Res 103(1) (2014) 17–27. [DOI] [PubMed] [Google Scholar]
- [73].Han J, Ye S, Zou C, Chen T, Wang J, Li J, Jiang L, Xu J, Huang W, Wang Y, Liang G, Angiotensin II Causes Biphasic STAT3 Activation Through TLR4 to Initiate Cardiac Remodeling, Hypertension 72(6) (2018) 1301–1311. [DOI] [PubMed] [Google Scholar]
- [74].Singh MV, Cicha MZ, Nunez S, Meyerholz DK, Chapleau MW, Abboud FM, Angiotensin II-induced hypertension and cardiac hypertrophy are differentially mediated by TLR3- and TLR4-dependent pathways, American journal of physiology. Heart and circulatory physiology 316(5) (2019) H1027–H1038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [75].Singh MV, Cicha MZ, Meyerholz DK, Chapleau MW, Abboud FM, Dual Activation of TRIF and MyD88 Adaptor Proteins by Angiotensin II Evokes Opposing Effects on Pressure, Cardiac Hypertrophy, and Inflammatory Gene Expression, Hypertension 66(3) (2015) 647–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [76].Nandy A, Lin L, Velentzas PD, Wu LP, Baehrecke EH, Silverman N, The NF-kappaB Factor Relish Regulates Atg1 Expression and Controls Autophagy, Cell Rep 25(8) (2018) 2110–2120 e3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [77].Verzella D, Pescatore A, Capece D, Vecchiotti D, Ursini MV, Franzoso G, Alesse E, Zazzeroni F, Life, death, and autophagy in cancer: NF-kappaB turns up everywhere, Cell Death Dis 11(3) (2020) 210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [78].Yu XW, Meng X, Xu M, Zhang XJ, Zhang Y, Ding GX, Huang SM, Zhang AH, Jia ZJ, Celastrol ameliorates cisplatin nephrotoxicity by inhibiting NF-kappa B and improving mitochondrial function, Ebiomedicine 36 (2018) 266–280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [79].Marko L, Vigolo E, Hinze C, Park JK, Roel G, Balogh A, Choi M, Wubken A, Cording J, Blasig IE, Luft FC, Scheidereit C, Schmidt-Ott KM, Schmidt-Ullrich R, Muller DN, Tubular Epithelial NF-kappa B Activity Regulates Ischemic AKI, Journal of the American Society of Nephrology 27(9) (2016) 2658–2669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [80].Alique M, Sanchez-Lopez E, Rayego-Mateos S, Egido J, Ortiz A, Ruiz-Ortega M, Angiotensin II, via angiotensin receptor type 1/nuclear factor-kappaB activation, causes a synergistic effect on interleukin-1-beta-induced inflammatory responses in cultured mesangial cells, Journal of the renin-angiotensin-aldosterone system : JRAAS 16(1) (2015) 23–32. [DOI] [PubMed] [Google Scholar]
- [81].Zhang G, Ghosh S, Toll-like receptor-mediated NF-kappaB activation: a phylogenetically conserved paradigm in innate immunity, J Clin Invest 107(1) (2001) 13–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [82].Yoo JY, Cha DR, Kim B, An EJ, Lee SR, Cha JJ, Kang YS, Ghee JY, Han JY, Bae YS, LPS-Induced Acute Kidney Injury Is Mediated by Nox4-SH3YL1, Cell Rep 33(3) (2020) 108245. [DOI] [PubMed] [Google Scholar]