

Summary
CD8+ T cells responding to chronic infection adapt an altered differentiation program that provides some restrain on pathogen replication yet limits immunopathology. This adaptation is imprinted in stem-like cells and propagated to their progeny. Understanding the molecular control of CD8+ T cell differentiation in chronic infection has important therapeutic implications. Here, we found that the chemokine receptor CXCR3 was highly expressed on viral-specific stem-like CD8+ T cells and that one of its ligands, CXCL10, regulated the persistence and heterogeneity of responding CD8+ T cells in spleens of mice chronically infected with lymphocytic choriomeningitis virus. CXCL10 was produced by inflammatory monocytes and fibroblasts of the splenic red pulp where it granted stem-like cells access to signals promoting differentiation and limited their exposure to pro-survival niches in the white pulp. Consequently, functional CD8+ T cell responses were greater in Cxcl10−/− mice and were associated with a lower viral set point.
Keywords: CD8 T cell differentiation, CXCR3, CXCL10, LCMV, chronic viral infection, chemokine
eTOC Blurb
The chemokine CXCL10 limits formation of memory CD8+ T cells during acute infection but its role in CD8+ T cell differentiation during chronic infection is unknown. Ozga et al. reveal that CXCL10 expression in the splenic red pulp of chronically infected mice promotes exposure of virus-specific stem-like CD8+ T cells to differentiation signals and limits access to pro-survival signals.
Graphical Abstract
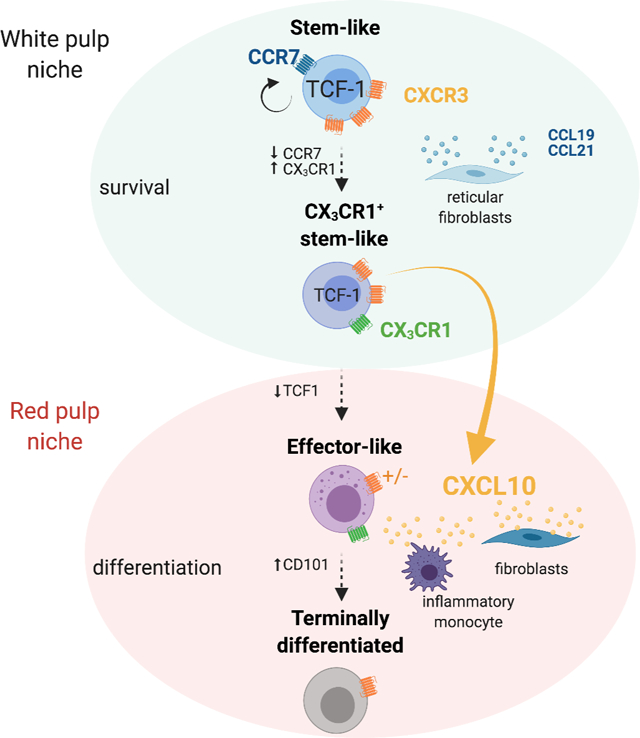
Introduction
Functional CD8+ T cell responses are indispensable for the control of viral infections and rejection of tumors. During chronic infection and cancer, the persistence of antigen induces a differentiation program in CD8+ T cells that restrains their effector function thus limiting immunopathology and autoreactivity. This adaptation of T cell responses to antigen persistence is known as T cell exhaustion. T cells responding to chronic infection highly express the inhibitory receptor programmed cell death 1 (PD1) (Barber et al., 2006) and the transcription factor TOX (Alfei et al., 2019; Khan et al., 2019; Scott et al., 2019). Nevertheless, these T cells are heterogeneous, and three functionally distinct subsets have been defined based on the expression of the T cell specific transcription factor 1 (TCF1) and the chemokine receptor CX3CR1 (Hudson et al., 2019; Zander et al., 2019). TCF1+CX3CR1− T cells self-renew and propagate a stably imprinted exhaustion program to their progeny (Utzschneider et al., 2020), and are referred to as stem-like T cells (Tstem-l) or progenitors of exhausted T cells (Blank et al., 2019). TCF1−CX3CR1+ cells, referred to as effector-like (Teff-l) or transitory T cells are a direct progeny of Tstem-l, have increased cytotoxic capacity and are essential for viral control (Paley et al., 2012). TCF1−CX3CR1− cells, referred to as terminally differentiated (Tterm) or terminally exhausted (Blank et al., 2019), have impaired cytokine production and express multiple inhibitory receptors, such as mucin domain containing-3 (TIM3) and natural killer cell receptor 2B4 (2B4), and have impaired proliferative potential and survival (Wherry et al., 2003; Zajac et al., 1998). Tterm can be identified by the expression of glycoprotein CD101 (Hudson et al., 2019).
Tstem-l are critical for the proliferative burst observed following anti-PD1 or anti-PDL1 therapy (He et al., 2016; Im et al., 2016; Utzschneider et al., 2016). Only a small number of PD1 responsive CD8+ T cells exist in patients with cancer or chronic infection (Huang et al., 2017), and their abundance can erode over time (Jansen et al., 2019). The frequency of TCF1+ T cells in tumor tissues correlates with the responsiveness to cancer immunotherapy (Miller et al., 2019; Sade-Feldman et al., 2018; Siddiqui et al., 2019), and in blood with the ability to control HIV infection (Rutishauser et al., 2021). Therefore, understanding the cellular and molecular mechanisms that regulate terminal differentiation and durability of Tstem-l cells could have important therapeutic implications for treating chronic infections and cancer.
Chemokines orchestrate migratory patterns and positioning of immune cells and therefore are critical for the generation and modulation of CD8+ T cell responses. Tstem-l and more differentiated T cells show very distinct chemokine receptor expression. Tstem-l express more CCR7, CXCR3, and CXCR5 and less CXCR6 and CX3CR1 compared to more differentiated T cells (Im et al., 2016; Zander et al., 2019). CXCR3, a chemokine receptor for the interferon-inducible chemokines - CXCL9, CXCL10, and CXCL11 - has been shown to regulate CD8+ T cell fate decisions during acute infection where CXCR3 expression favors the generation of effector CD8+ T cells (Teff) over memory CD8+ T cells (Tmem) (Hu et al., 2011; Kurachi et al., 2011). However, the role of CXCR3 and its ligands in the control of T cell differentiation during chronic infection remains undefined.
Here we sought to understand if the CXCR3 chemokine system regulates CD8+ T cell heterogeneity and differentiation during a chronic infection. To address this question, we used the clone 13 (Cl13) strain of lymphocytic choriomeningitis virus (LCMV) that induces chronic infection. We observed that in the absence of CXCL10, LCMV-specific CD8+ T cells show improved persistence within the spleen accompanied by major changes in T cell subset composition. Cxcl10−/− mice had an increase in the numbers and frequency of a previously unrecognized CX3CR1+ Tstem-l subset as well as in Teff-l at the expense of Tterm. Spatially restricted CXCL10 expression limited exposure of Tstem-l to pro-survival signals in the white pulp but granted these cells access to signals driving differentiation in the red pulp. Consequently, in Cxcl10−/− mice, enhanced persistence of LCMV-specific CD8+ T cells and a decrease in their terminal differentiation correlated with improved viral control. Moreover, the inhibition of CXCL10 enhanced the proliferation of PD1+ CD8+ T cells and frequency of Teff-l cells in chronically infected mice treated with anti-PDL1. These observations support the role of CXCL10 in the regulation of T cell heterogeneity during chronic infections.
Results
CXCR3 controls the magnitude and heterogeneity of the LCMV-specific CD8+ T cell response in a cell intrinsic manner.
Infection of mice with LCMV Armstrong (Arm) results in virus clearance by ~day (d) 8 post-infection (pi), whereas infection with LCMV Cl13 results in viremia that can last for several weeks. The two strains differ by only three amino acids, preserving CD8+ T cell response towards all known CD8+ T cell epitopes, including GP33-41 and GP276-286 (Wherry et al., 2003). To determine if CXCR3 participates in the differentiation of CD8+ T cells during chronic infection, we first compared the kinetics of CXCR3 expression on splenic GP33-specific cells during both infections. Percentages of GP33-specific cells that were CXCR3 positive were comparable between these two infections, with ~90% of cells being positive on d8 and ~75% on d16 and d24 (Figure 1A). Of note, GP33-specific cells isolated from d16 or d24 Cl13-infected mice showed reduced CXCR3 expression compared to GP33-specific cells isolated from Arm-infected mice (Figure 1B, C).
Figure 1. CXCR3 controls the magnitude and heterogeneity of the LCMV-specific CD8+ T cell response in cell intrinsic manner.
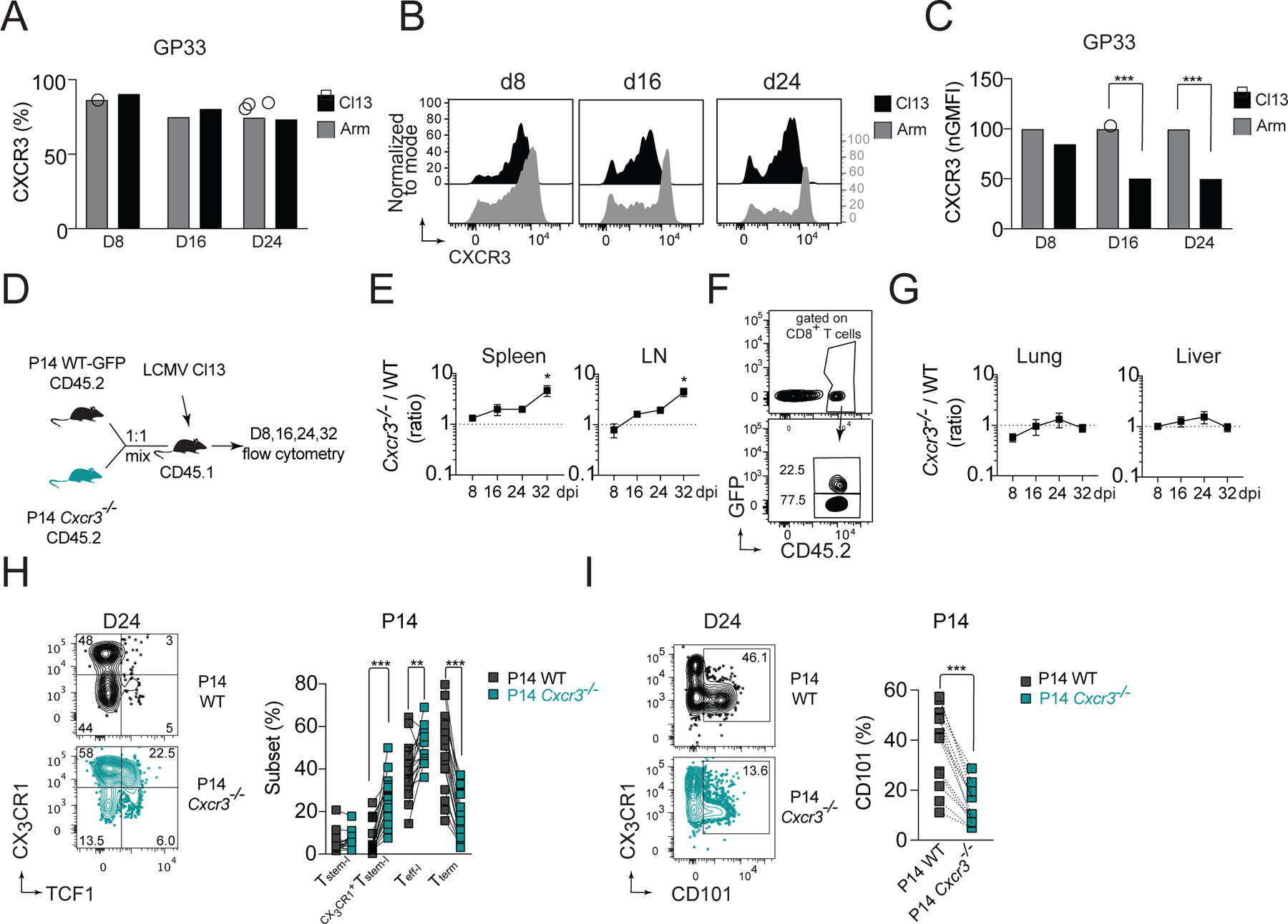
(A-C) Kinetics of CXCR3 expression on splenic GP33-specific cells isolated from C57BL/6 WT mice during acute (Arm) and chronic (Cl13) infection. Percentage of CXCR3+ cells among GP33-specific cells (A); flow cytometry histograms of CXCR3 expression on GP33-specific cells (B), and amount of CXCR3 expression on GP33-specific cells (normalized to Arm) (C). (D-I) CD45.1 mice received an adoptive transfer of 1:1 mixture of CD45.2 positive GFP+ WT P14 and Cxcr3−/− P14 cells followed by infection with Cl13. (D) Experimental scheme. (E-G) The ratio of Cxcr3−/−/WT P14 cells recovered from spleen and LNs (E) with exemplary gating of transferred cells on d24 (F), or the ratio of Cxcr3−/−/WT P14 cells recovered from parenchyma of liver and lung (G) pi. with Cl13. (H-I) Frequency of T cell subsets based on the expression of TCF1 and CX3CR1 among WT and Cxcr3−/− P14 cells (H) and frequency of CD101+ P14 cells (I) in spleens on d24 pi. with Cl13. Data are from at least two independent experiments (n=3–4 per time point). Data in A, C and H were analyzed with an ordinary one-way Anova test with Sidak’s multiple comparison test; data in E and G with Dunnett’s multiple comparison test; and data in I with paired Student’s t-test; *, P < 0.05; **, P < 0.01; ***, P < 0.001. See also Figure S1.
The fact that CXCR3 was downregulated on GP33-specific cells during Cl13 infection suggested ligand-induced activation and internalization (Sauty et al., 2001), prompting us to ask if CXCR3 regulates LCMV-specific CD8+ T cell responses to chronic infection. To address this question, we co-transferred wild-type (WT) and Cxcr3−/− GP33-specific P14 TCR-transgenic (tg) cells (P14) into WT mice that were subsequently infected with Cl13 (Figure 1D), which allowed for a direct comparison of WT and Cxcr3−/− P14 cells in the same environment. The initial recovery of WT and Cxcr3−/− P14 cells from spleen and lymph nodes (LNs) was comparable, suggesting similar early expansion of P14 cells. However, at later stages of Cl13 infection, Cxcr3−/− P14 cells markedly outnumbered WT P14 cells (Figure 1E, F). This was only observed in secondary lymphoid organs (SLO), as the ratio of intraparenchymal Cxcr3−/− and WT P14 cells in the liver and lung were close to 1 at all dpi. (Figure 1G). These Cxcr3−/− and WT P14 cells had higher expression of PD1 and TOX than P14 cells recovered from acute infection, suggesting that CXCR3 does not interfere with establishment of the exhaustion program in responding P14 cells (Figure S1A). To understand whether CXCR3 affects the heterogeneity of responding P14 cells, we analyzed the frequency of functional T cell subsets identified by expression of TCF1 and CX3CR1. On d24, the frequency of Tstem-l among Cxcr3−/− P14 cells was comparable with WT P14 cells (Figure 1H). However, the frequency of TCF1+CX3CR1+ cells among Cxcr3−/− P14 cells was markedly increased compared to the frequency of this subset among WT P14 cells (27% vs 5%) (Figure 1H). The frequency of Teff-l among Cxcr3−/− P14 cells was also slightly increased (51% vs 40%), whereas the frequency of Tterm was markedly reduced compared to WT P14 cells (16% vs 49%), which was associated with lower percentage of cells expressing CD101 among Cxcr3−/− P14 cells (Figure 1I). A similar pattern was observed when total numbers of functional subsets were compared (Figure S1C). TCF1+CX3CR1+ cells barely formed during chronic infection but were abundant among memory cells following acute infection (Figure S1B). The increase in TCF1+CX3CR1+ cells in the absence of CXCR3 signaling correlated with improved persistence of P14 cells specifically within SLO (Figure 1E, F), which are known to support Tstem-l maintenance and quiescence (Im et al., 2020). Given that the TCF1+CX3CR1+ subset seemed to be restricted to SLO and still expressed TCF1, a key transcription factor of Tstem-l, we referred to this subset as CX3CR1+Tstem-l. Thus, these findings demonstrate that CXCR3 signaling on T cells promotes the formation of CD101+Tterm, but limits formation of CX3CR1+Tstem-l. Moreover, in the setting of antigen persistence, the absence of CXCR3 signaling improves the persistence of P14 cells specifically within SLO.
CXCR3 deficiency results in increased CX3CR1+Tstem-l and Teff-l LCMV-specific CD8+ T cell responses.
To understand if the CXCR3 chemokine system affects the heterogeneity of endogenous LCMV-specific T cells, we infected WT and Cxcr3−/− mice with Cl13 and analyzed GP33- and GP276-specific responses within spleens on d24 pi. (Figure 2A). The magnitude of these LCMV-specific responses was enhanced in Cxcr3−/− mice compared to WT mice. Cxcr3−/− mice had an increased percentage of GP33-specific cells (Figure 2B) and a trend toward an increase in the percentage of GP276-specific cells among CD8+ T cells (Figure 2B). The increase in frequency of these LCMV-specific cells together with an increase in total numbers of CD8+ T cells in Cxcr3−/− mice (Figure S1D) resulted in a marked ~6-fold increase in the total numbers of GP33- and GP276-specific cells compared to WT mice (Figure 2C). Similar to Cxcr3−/− P14 cells, these endogenous LCMV-specific cells in Cxcr3−/− mice had a ~3-fold increase in CX3CR1+Tstem-l and ~3-fold decrease in the frequency of Tterm compared to LCMV-specific cells in WT mice (Figure 2D, E). Furthermore, the total number of CX3CR1+Tstem-l and Teff-l was markedly increased in Cxcr3−/− mice compared to WT mice (Figure 2F). These changes correlated with reduced expression of markers of terminal differentiation, such as CD101 (Figure 2G), Tim3 (Figure 2H), and (Figure 2I), on LCMV-specific cells from Cxcr3−/− mice compared to WT mice. Together, these data demonstrate that CXCR3 deficiency permits improved persistence of T cells during chronic infection and results in an increased frequency of CX3CR1+Tstem-l and Teff-l at the expense of Tterm.
Figure 2. CXCR3 deficiency results in increased CX3CR1+Tstem-l and Teff-l LCMV-specific CD8+ T cell responses.
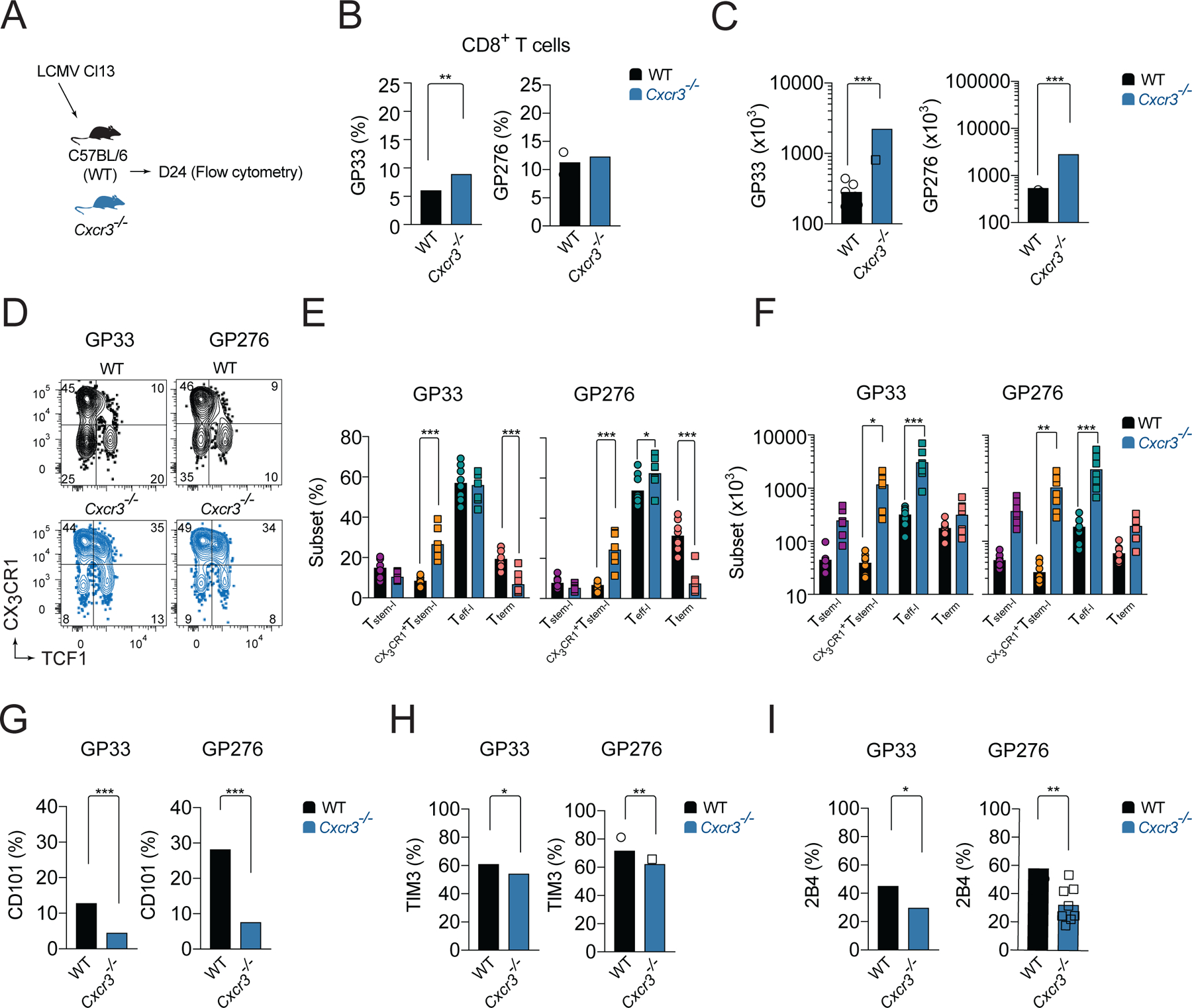
(A-I) WT or Cxcr3−/− mice were infected with Cl13 and the phenotype of splenic LCMV-specific responses was analyzed on d24 pi. (A) Experimental scheme. (B-C) Percentage (B) and total numbers (C) of GP33- and GP276-specific cells. (D-F) Frequency and total number of T cell subsets based on the expression of TCF1 and CX3CR1 among LCMV-specific cells. Flow cytometry representative graphs (D), quantification (E) and total numbers (F). (G-I) Frequency of CD101 (G), TIM3 (H), and 2B4 (I) expressing cells among LCMV-specific cells. Data are from at least two independent experiments (n=3–4 per time point). Data in B and G-I were analyzed with unpaired Student’s t-test; data in C with Mann-Whitney test; and data in E, F with an ordinary one-way Anova test with Sidak’s multiple comparison test; *, P < 0.05; **, P < 0.01; ***, P < 0.001. See also Figure S1.
CXCL10 controls the magnitude and heterogeneity of LCMV-specific CD8+ T cell response.
To further characterize how the CXCR3 chemokine system controls T cell heterogeneity during chronic infection, we compared the kinetics of CXCR3 ligand expression in the spleens of Cl13- and Arm-infected mice. In C57BL/6 mice, there are only two functional CXCR3 ligands - CXCL9 and CXCL10 - as the third CXCR3 ligand, CXCL11, is a null mutant in this strain (Sierro et al., 2007). Cxcl9 and Cxcl10 mRNA expression was induced in spleens of Cl13-infected mice (Figure 3A). Early Cxcl9 mRNA expression was comparable in both infections. However, on d25 Cxcl9 mRNA expression remained upregulated in spleens of Cl13-infected mice but decreased in Arm-infected mice (Figure 3A). In contrast, Cxcl10 mRNA expression was higher in Cl13-infected mice at all time-points analyzed compared to Arm-infected mice (Figure 3A). Expression of CXCL9 and CXCL10 is induced by IFNγ, whereas CXCL10 can also be induced by IFNα/β (Groom and Luster, 2011). We could detect increased Ifng and Ifnb1 mRNA expression in Cl13-infected mice compared to uninfected controls (Figure S1E). Moreover, on d8 and d16 pi. Ifnb1 mRNA expression was higher in Cl13- than in Arm-infected mice (Figure S1E). To determine the role of type I and type II IFNs for induction of CXCR3 ligands, WT, Ifnar−/− and Ifngr1−/− mice were infected with Cl13. The expression of Cxcl9 was markedly reduced in Ifngr1−/− mice at all dpi, confirming the importance of IFNγ for Cxcl9 expression (Figure 3B). The early expression of Cxcl10 was also IFNγ-dependent (Figure 3B). However, as the disease progressed, the role of type I IFN signaling in Cxcl10 expression become more pronounced (Figure 3B).
Figure 3. CXCL10 controls the magnitude and heterogeneity of LCMV-specific CD8+ T cell response.
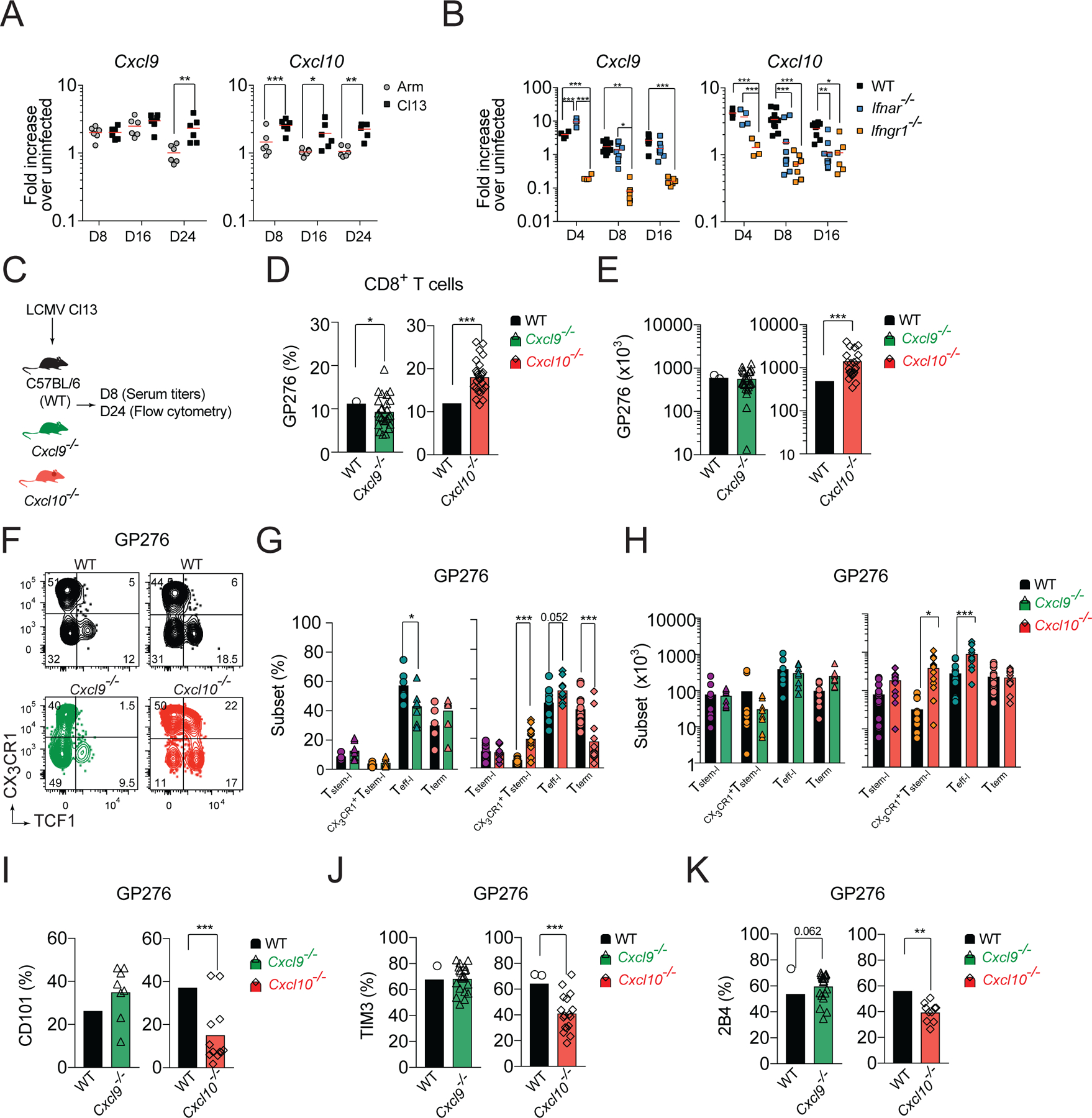
(A) Fold increase in Cxcl9 and Cxcl10 mRNA expression in the spleen of WT mice on the indicated days pi. with Cl13 or Arm relative to uninfected. (B) Fold increase in Cxcl9 and Cxcl10 mRNA expression in the spleen of WT, Ifnar−/−, and Ifnγr−/− mice on the indicated days pi. with Cl13 relative to uninfected. (C-J) WT, Cxcl9−/−, or Cxcl10−/− mice were infected with Cl13 and phenotype of splenic GP276-specific cells was analyzed on d24 pi. (C) Experimental scheme. (D-E) Percentage (D) and total numbers (E) of GP276-specific cells. (F-H) Flow cytometry representative graphs (F), quantification (G) and total numbers (H) of T cell subsets based on the expression of TCF1 and CX3CR1 among GP276-specific cells. (I-K) Frequency of CD101 (I); TIM3 (J); and 2B4 (K) positive cells among GP276-specific cells. Data are from at least two independent experiments (n=3–4 per time point). Data in A, B and G, H were analyzed with an ordinary one-way Anova with Sidak’s multiple comparison test; and data in D-E and I-K with unpaired Student’s t-test; *, P < 0.05; **, P < 0.01; ***, P < 0.001. See also Figure S1.
Type I IFN signaling limits the expansion and persistence of Tstem-l and promotes their terminal differentiation (Wu et al., 2016). Therefore, we hypothesized that CXCL10 might be involved in regulation of T cell differentiation. To test this hypothesis, we infected Cxcl9−/−, Cxcl10−/−, and WT mice with Cl13 and analyzed the phenotype of LCMV-specific cells on d24 pi. (Figure 3C). On d24 pi., similar to Cxcr3−/− mice, the percentage and total numbers of GP276-specific cells were markedly increased in Cxcl10−/− mice compared to WT mice (Figure 3D, E). In contrast, the frequency of GP276-specific cells was slightly decreased in Cxcl9−/− mice, but their total number was comparable to WT mice (Figure 3D, E). In Cxcl10−/− mice, analysis of GP276-specific cell heterogeneity showed a 3.7-fold increase in the percentage of CX3CR1+Tstem-l and a trend towards an increase in Teff-l and a corresponding decrease in Tterm cells compared to WT mice (Figure 3F, G). Moreover, the total number of CX3CR1+Tstem-l and Teff-l were increased in Cxcl10−/− mice compared to WT mice, whereas the numbers of Tstem-l and Tterm were comparable (Figure 3H). In contrast, in Cxcl9−/− mice, analysis of GP276-specific cell heterogeneity revealed a mild reduction in the frequency of Teff-l but no significant differences in other subsets (Figure 3F, G). Moreover, the total number of different T cells was comparable between Cxcl9−/− and WT mice (Figure 3H). The decrease in the frequency of Tterm in Cxcl10−/− mice correlated with reduced expression of markers of terminal differentiation, such as CD101 (Figure 3I), TIM3 (Figure 3J), and 2B4 (Figure 3K), among GP276-specific cells. In contrast, GP276-specific cells from Cxcl9−/− mice showed comparable expression of CD101, TIM3, and a trend towards increased expression of 2B4 compared to WT mice (Figure 3I–K). Analysis of GP33-specific T cells yielded similar results (Figure S1F–J). These data demonstrate that indeed IFNα/β- and IFNγ-inducible CXCL10, but not IFNγ-dependent-CXCL9, regulates CXCR3-dependent T cell subset composition during chronic infection.
CX3CR1+Tstem-l cells have a distinct transcriptional signature
Cxcr3−/− or Cxcl10−/− mice showed ~2-fold increase in the frequency of CX3CR1+Tstem-l and a smaller increase in Teff-l (Figure 2E, 3G). CX3CR1+Tstem-l are barely detectable during chronic infection in WT mice and have not been identified by others (Hudson et al., 2019; Zander et al., 2019). To understand why CXCR3 and CXCL10 signaling predominantly affects CX3CR1+Tstem-l, we better defined their differentiation stage. To this end, we analyzed the pattern of gene expression in the four T cell subsets from d24 Cl13-infected WT mice. Subsets were sorted based on CX3CR1 and Ly108 whose expression correlates with TCF1 expression (Utzschneider et al., 2016). The transcriptional signature of CX3CR1+Tstem-l was very different from the signature of Tterm and less so from the signature of Tstem-l or Teff-l (Figure 4A, B, S2A–C, Table S1). Compared to Tterm, CX3CR1+Tstem-l had much lower amounts of transcripts for inhibitory molecules (Cd244, Cd160, Ctla4, Pdcd1, Tigit), and a very distinct expression pattern of genes involved in cytokine, TCR and type I IFN signaling, and regulation of apoptosis (Figure 4B, S2A–C). Genes expressed the highest in Tstem-l included genes involved in T cell stemness (Tcf7, Il7r, Sell, Nfkbia), chemokine signaling (Ccr7, Xcl1, Cxcl10), and co-stimulation (Cd28, Icos). These genes were expressed at intermediate amounts in CX3CR1+Tstem-l and at lower amounts in Teff-l (Figure 4B, Table S1). Similarly, genes upregulated during differentiation into Teff-l, such as genes encoding effector molecules (Gzma, Gzmb, Prf, Fasl), transcription factors (Tbx21, Prdm1), natural killer receptors (Klra8, Klrc1), chemokines (Ccl3, Ccl4, Ccl5) and chemokine receptors (Cx3cr1, Cxcr6), were also upregulated in CX3CR1+Tstem-l, but to a lesser extent (Figure 4B, Table S1). Thus, these data suggest that CX3CR1+Tstem-l are developmentally intermediate between Tstem-l and Teff-l.
Figure 4. CX3CR1+Tstem-l have a distinct transcriptional signature.
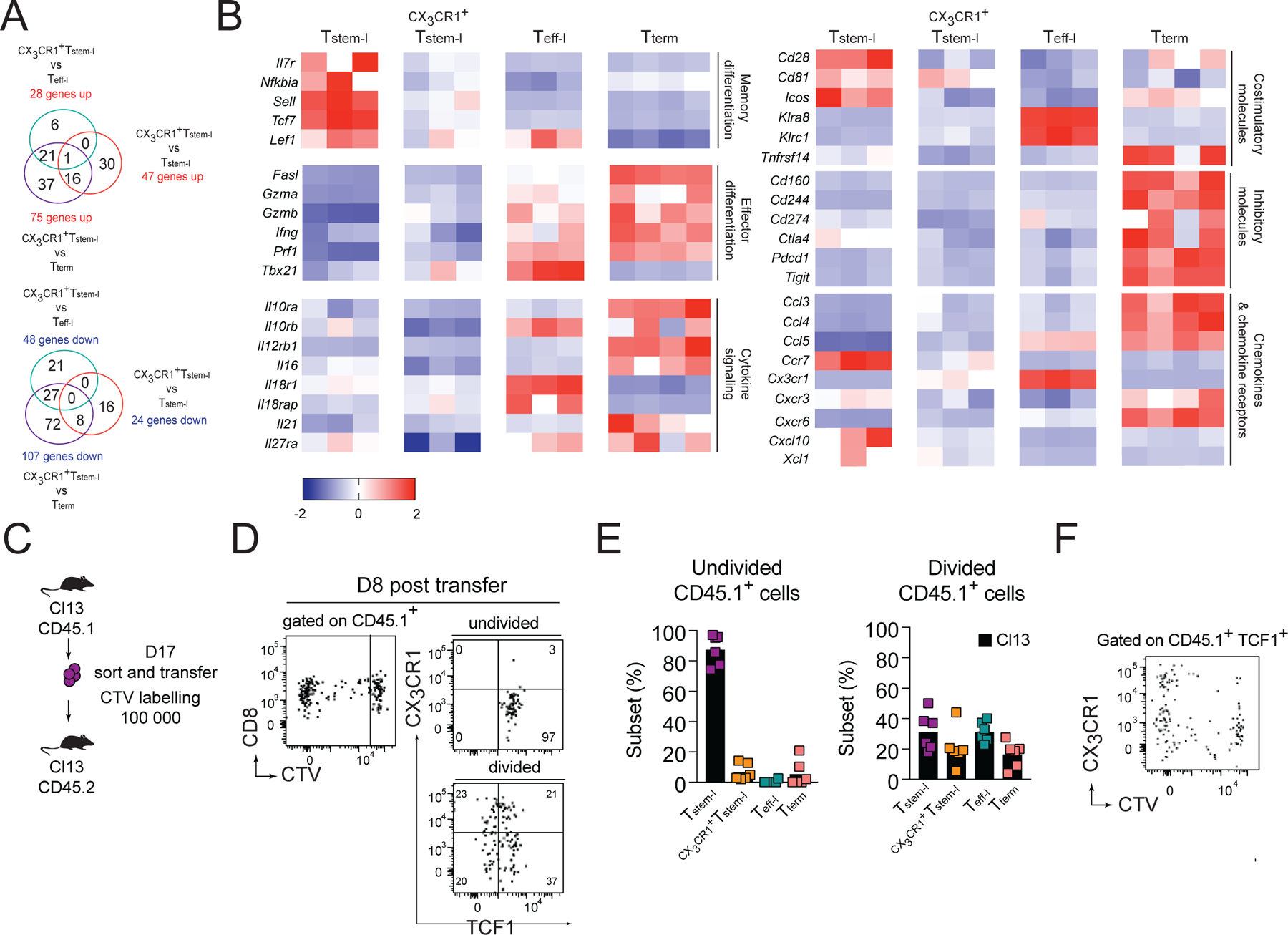
(A-B) Ly108, CX3CR1 and CD101 were used to distinguish and sort four T cell subsets from WT mice for analysis of differentially expressed genes using NanoString. (A) Number of differentially expressed genes between CX3CR1+Tstem-l and other T cell subsets. (B) Heatmap representing relative expression of selected differentially expressed genes between all subsets. (C-F) Congenically marked CX3CR1−Ly108+CD44+PD1+ CD8+ T cells (Tstem-l) were sorted from d17 Cl13-infected mice, labelled with cell tracker violet (CTV) and transferred into infection matched recipients. (C) Experimental scheme. (D) Proliferation and differentiation of Tstem-l in the spleen on d8 post-transfer. (E) Frequency of distinct subsets among undivided and divided cells. (F) Representative flow cytometry plot showing CX3CR1 and CTV expression on transferred cells that remained TCF1 positive. Data are from at least two independent experiments (n=3–4 per time point). Data in B were analyzed with the use of nSolver and multiple comparison test with Benjamini-Yekutieli False Discovery Rate method; *, P < 0.05; **, P < 0.01; ***, P < 0.001. See also Figure S2.
Finally, to address if Tstem-l can give rise to CX3CR1+Tstem-l, we sorted Tstem-l (PD1+CX3CR1−Ly108+ CD8+ T cells; Figure S2D) from d16 Cl13-infected mice and labeled them with cell tracker violet (CTV) prior to transfer into infection matched recipient mice (Figure 4C). On d8 post-transfer, undivided cells were predominantly TCF1+ and CX3CR1− (Figure 4D, E). In contrast, divided cells contained ~21% of CX3CR1+TCF1+ cells, suggesting that Tstem-l can generate CX3CR1+Tstem-l (Figure 4D, E). Upregulation of CX3CR1 was most prominent among TCF1+ cells that had undergone several rounds of division (Figure 4F). These results demonstrate that CX3CR1 upregulation is coupled to cell division, and that CX3CR1 expression identifies more differentiated TCF1+ T cells whose abundance is markedly increased in Cxcr3−/− and Cxcl10−/− mice. The accumulation of CX3CR1+Tstem-l in Cxcr3−/− or Cxcl10−/− mice may result from either accelerated differentiation of Tstem-l and/or decelerated further differentiation of CX3CR1+Tstem-l into Teff-l.
Cxcr3−/− stem-like subsets have decreased TCF1 downregulation and proliferation and improved survival
CXCR3 was most highly expressed on the Tstem-l subset of GP276-specific cells (Figure 5A). Therefore, we hypothesized that the CXCR3 chemokine system might affect division of Tstem-l. To address this question, we sorted Tstem-l from d17 Cl13-infected CD45.2 or CD45.2 Cxcr3−/− mice and transferred them into infection matched CD45.1 WT recipient. On d8 post-transfer, cells were analyzed by flow cytometry for cell numbers, proliferation, and differentiation (Figure 5B, C). The recovery of transferred Cxcr3−/− cells was comparable with WT cells (Figure 5D), even though Cxcr3−/− cells showed a slight reduction in the frequency of divided cells (Figure 5E). Cxcr3−/− cells that divided showed an increase in the frequency of Tstem-l (~2.5 fold) and a decrease in the frequency of Teff-l (~2-fold) compared to divided WT cells (Figure 5F, C). Thus, these data suggest that CXCR3 favors the proliferation of Tstem-l and downregulation of TCF1 in proliferating cells. The fact that reduced division of Cxcr3−/− Tstem-l was not accompanied by a reduction in the total number of recovered cells implied improved survival of Tstem-l in the absence of CXCR3 signaling. These findings were corroborated in P14 cell adoptive transfer experiments. On d24 pi., a smaller percentage of TCF1+ Cxcr3−/− P14 cells incorporated a modified thymidine analog (EdU) compared to TCF1+ WT P14 cells, suggesting a decreased proliferation rate of Cxcr3−/− Tstem-l cells (Figure S3A); no differences were detected between TCF1− WT and TCF1− Cxcr3−/− P14 cells (Figure S3A). The decreased proliferation rate was not accompanied by reduced recovery of Cxcr3−/− P14 cells. Consistent with improved persistence, Cxcr3−/− P14 cells expressed higher amounts of the anti-apoptotic protein BCL2 compared to WT P14 cells (Figure S3B). Thus, decreased susceptibility of Cxcr3−/− P14 cells to apoptosis might, in part, contribute to their improved persistence. In sum, it is likely that as Tstem-l differentiate into CX3CR1+Tstem-l and downregulate CCR7 (Figure 4B), they become more responsive to CXCR3-dependent signaling, which likely facilitates their exposure to factors promoting proliferation and TCF1 downregulation, but restricts their access to pro-survival signals.
Figure 5. CXCR3 deficiency results in improved survival and decreased TCF1 downregulation of TCF1+ cells and decreased terminal differentiation of Teff-l.
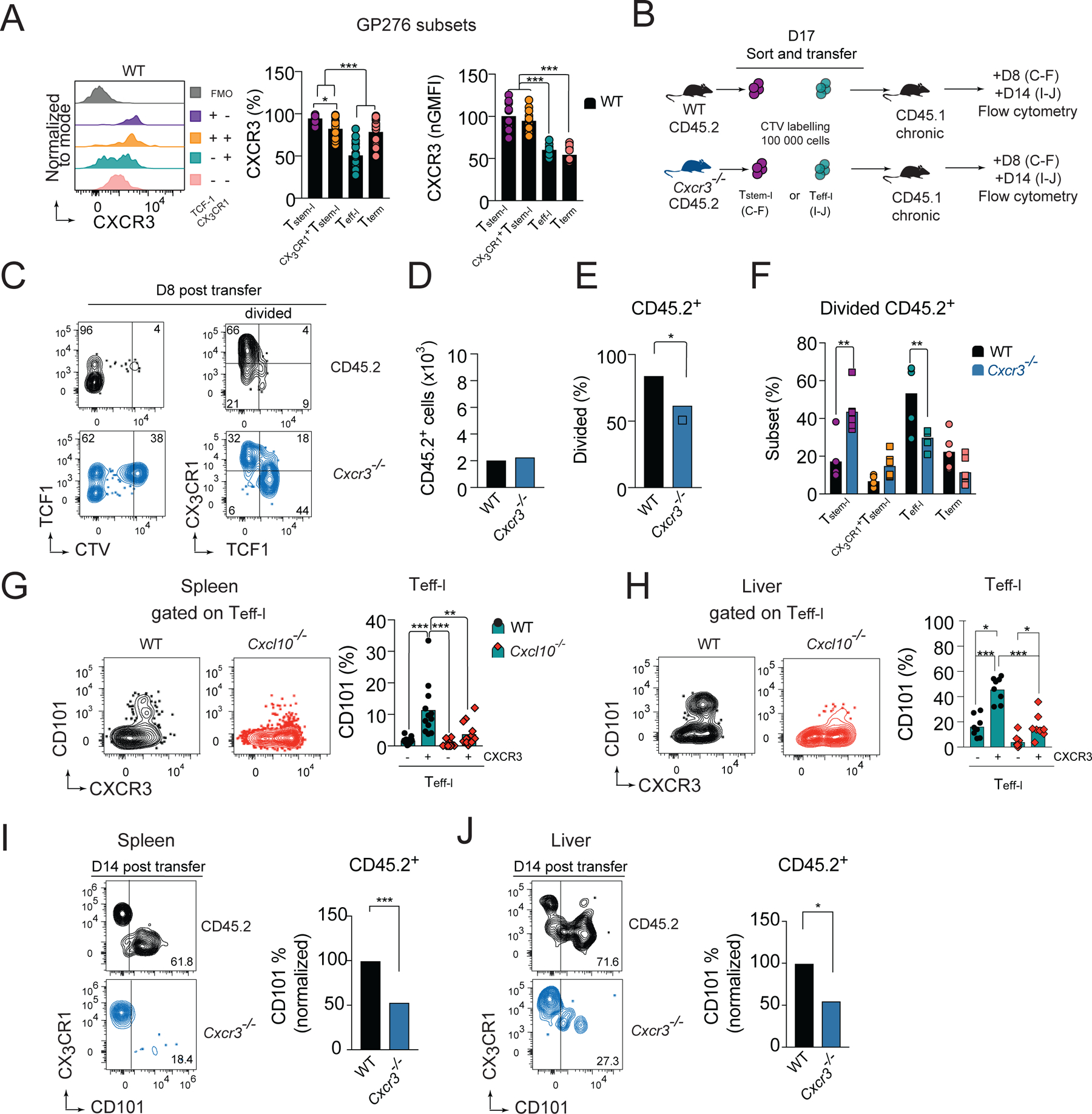
(A) CXCR3 expression normalized to expression on Tstem-l and the frequency of CXCR3+ cells on distinct subsets of GP276-specific cells on d24 of Cl13 infection in WT mice. (B-F) Congenically marked cell tracker violet (CTV)-labelled CX3CR1−Ly108+CD44+PD1+ CD8+ T cells (Tstem-l) from chronically infected CD45.2 WT, or CD45.2 Cxcr3−/− mice were adoptively transferred into infection matched CD45.1 recipient. (B) Experimental scheme. (C) Flow cytometry plots of recovered cells. (D-F) The total number of recovered cells (D), percentage of divided cells (E), and phenotype of divided cells (F) among adoptively transferred WT and Cxcr3−/− cells on d8 post-transfer. (G-H) Flow cytometry plots and quantification of CD101 expression on CXCR3− and CXCR3+ Teff-l on d24 pi. with Cl13 in WT and Cxcl10−/− mice in spleen (G) and liver (H). (B, I-J) Congenically marked CX3CR1+Ly108−PD1+ CD8+ T cells (Teff-l) from chronically infected CD45.2 WT or CD45.2 Cxcr3−/− mice were adoptively transferred into infection matched WT CD45.1 recipient that were treated with 150μg of CD4 depleting mAb one day before and after infection to induce stable viremia. (B) Experimental scheme. (I-J) Flow cytometry plots and frequency of CD101+ cells among transferred WT and Cxcr3−/− cells recovered from spleen (I) and liver (J) on d14 post-transfer. Data are from at least two independent experiments (n=3–4 per time point). Data in A,G and H were analyzed with an ordinary one-way Anova test with Tukey’s multiple comparison test; data in D-E, I-J with unpaired Student’s t-test; and data in F with an ordinary one-way Anova test Sidak’s multiple comparison test; *, P < 0.05; **, P < 0.01; ***, P < 0.001. See also Figure S3.
CXCR3 deficiency results in decreased terminal differentiation of Teff-l
The lower efficiency of TCF1 downregulation in dividing Cxcr3−/− Tstem-l would be predicted to translate into a decrease in the frequency of Teff-l. On the contrary, Cxcr3−/− mice have an increase in Teff-l and a decrease in Tterm. We therefore hypothesized that CXCR3 might promote terminal differentiation of Teff-l. CXCR3 expression on Teff-l were lower compared to other T cell subsets (Figure 5A) and there was a clear CXCR3 negative population among Teff-l (Figure 5A). Therefore, we hypothesized that downregulation of CXCR3 enables these cells to delay terminal differentiation before CXCR3 cell surface expression is restored by de novo expression of CXCR3 (Meiser et al., 2008). Indeed, CXCR3+ Teff-l show enhanced upregulation of CD101 compared to CXCR3− Teff-l in spleen (Figure 5G) and liver (Figure 5H). Moreover, in these organs, the upregulation of CD101 occurred in a CXCL10-dependent manner (Figure 5G, H). Finally, we sorted Teff-l (PD1+CX3CR1+Ly108− CD8+ T cells) from d17 Cl13-infected CD45.2 or CD45.2 Cxcr3−/− mice and transferred them to infection matched CD45.1 WT recipients. On d14 post-transfer, cells were analyzed for signs of terminal differentiation (Figure 5B). WT cells recovered from the spleen (Figure 5I) and liver (Figure 5J) showed enhanced upregulation of CD101 compared to Cxcr3−/− cells. Thus, these data suggest that expression of CXCR3 on Teff-l accelerates their terminal differentiation in a CXCL10-dependent manner.
CXCL10 and CXCR3 control the exposure of CD8+ T cells to differentiation signals
We hypothesized that CXCL10 and CXCR3 controls the quantity and quality of pro-survival and inflammatory signals that T cells receive within SLO, affecting their differentiation and persistence. To address these questions, we analyzed the gene expression profile of the four T cell subsets from d24 Cl13-infected WT and Cxcl10−/− mice. Pairwise comparison revealed that these sorted cell subsets had a very similar transcriptional signature. There were no statistically significant differences in the expression of analyzed genes (516 targets) (Figure S3C, Table S2). These data suggest that the CXCL10 chemokine axis regulates transitions between T cell subsets rather than their functionality. CXCL10 promoted the formation of Tterm, which have increased expression of genes involved in TCR and type I IFN signaling, and factors promoting apoptosis (Figure S2A–C). Therefore, we hypothesized that CXCR3 and CXCL10 regulate exposure of T cells to antigen and type I IFNs, which are known to contribute to the terminal differentiation of T cells. To address these questions, we co-transferred WT and Cxcr3−/− P14 cells into WT recipients, followed by infection with Cl13. On d24 or d16 pi, transferred cells were analyzed for the expression of IRF4, CD69 and IRF1, genes known to respond to TCR stimulation and type I IFN signaling. IRF4 is a TCR-signaling responsive transcription factor which, together with BATF and NFAT, binds to the promoter region of the Tcf1 gene to repress its expression during chronic infection (Man et al., 2017). We detected a modest reduction in IRF4 expression among Cxcr3−/− P14 cells on d16 pi (Figure S3D). Moreover, on d24 pi, Cxcr3−/− P14 cells showed decreased expression of CD69 and IRF1, factors that are known to be upregulated by type I IFN stimulation (Figure S3E, F) (Huang et al., 2019; Shiow et al., 2006). Thus, these data suggest that CXCL10 and CXCR3 promote exposure of dividing T cells to antigen and type I IFN, favoring TCF1 downregulation and terminal differentiation.
CXCL10 high producers are predominantly detected outside of T cell zones
To gain better insight into signals that differentiating cells might receive within distinct splenic regions, we utilized REX3 tg mice that express RFP and BFP under the control of the CXCL9 and CXCL10 promoter, respectively (Groom et al., 2012). TCF1+ T cells localize within naïve T cell zones due to high expression of CCR7 and responsiveness to the CCR7 T cell zone ligands, CCL21 and CCL19. In contrast, activated TCF1− T cell subsets downregulate CCR7 expression and are primarily found within red pulp (Im et al., 2016). To define splenic architecture, spleen sections were stained with monoclonal antibodies (mAb) against B220 (white pulp) or F4/80 (red pulp) (Figure 6A). On d16, CXCL10 single producers were predominant within bridging channels and red pulp (Figure 6A). In contrast, CXCL9 single and double producers were more abundant within the T cell zone (white dotted circle) (Figure 6A). Accordingly, TCF1+ CD8+ T cells were predominantly localized in the proximity of CXCL9 single and double producers (Figure 6B). Although some CD101+CD8+ T cells were detected within the T cell zone, they were more abundant in the proximity of CXCL10+ cells in peripheral areas of the spleen (Figure 6B). These data suggest that CXCR3 on dividing Tstem-l might predominantly regulate their interactions with CXCL9 single and double producers. Only when these cells differentiate and downregulate CCR7, CXCR3 might regulate their migration towards red pulp and their interactions with CXCL10 single producers.
Figure 6. CXCR3 ligand expression distinguishes stimulatory and inhibitory myeloid cell subsets.
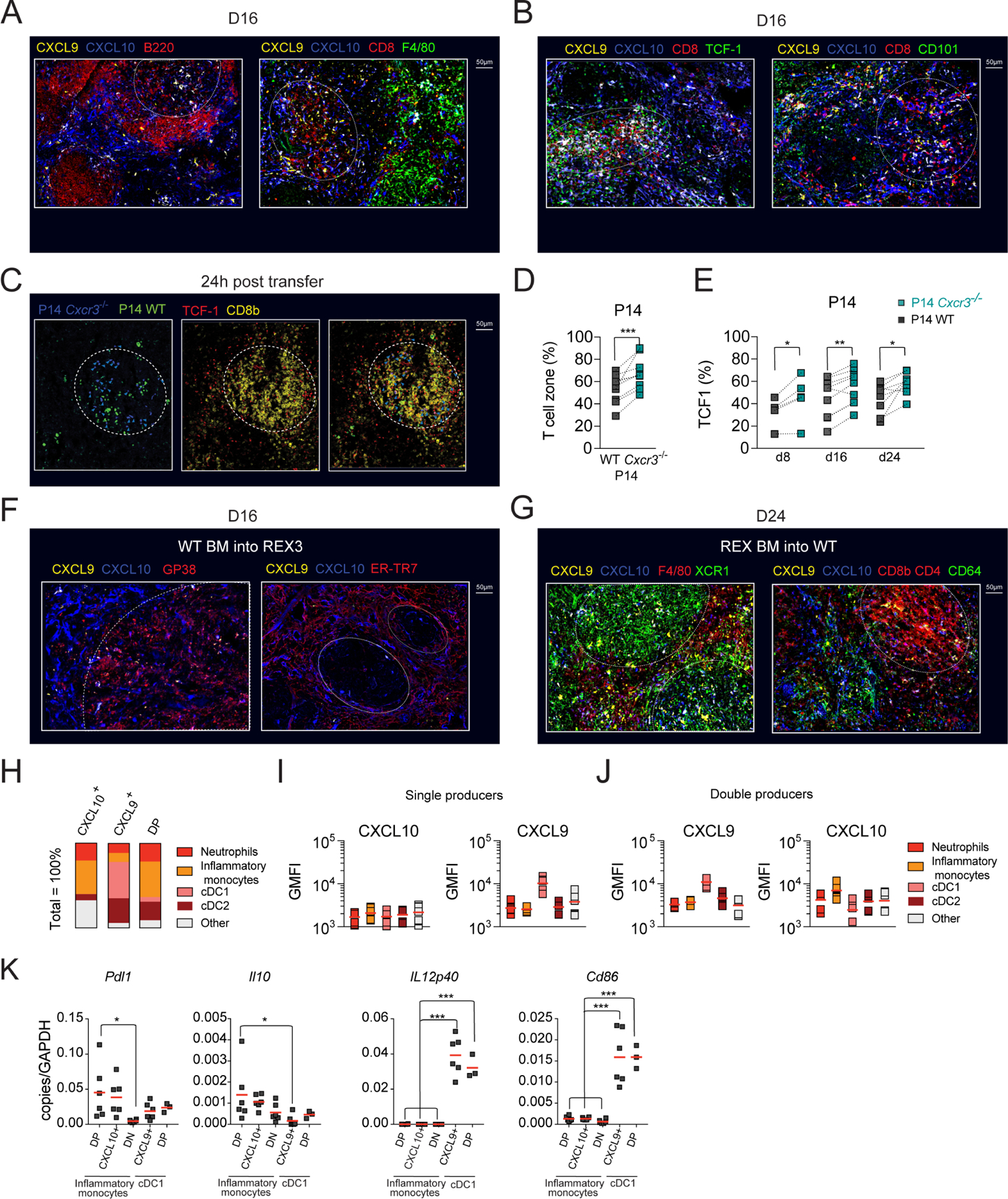
(A-B) Pattern of CXCL10 and CXCL9 expression in Cl13-infected spleens. REX3 (CXCL9 and CXCL10 dual fluorescent reporter) mice were infected with Cl13 and on d16 pi. spleens were harvested for immunofluorescence. (A) B cell follicles (B220, red, left panel) or red pulp macrophages (F4/80, green, right panel) in infected spleens with respect to CXCL10 (blue), CXCL9 (yellow), and CXCL10 and CXCL9 double producers (white). (B) Localization of TCF1+ CD8+ T cells (left panel) and CD101+ CD8+ T cells (right panel) in infected spleens with respect to CXCL10 (blue) and CXCL9 (yellow). (C-E) WT GFP+ P14 and Cxcr3−/− P14 cells in 1:1 ratio were cultured in vitro in conditions favoring TCF1+ phenotype (Figure S4A–C) followed by transfer into d8, d16, d24 Cl13-infected CD45.1 mice and analyzed by immunofluorescence (24h post-transfer) and flow cytometry analysis (d4 post-transfer). (C) Localization of in vitro transferred cells in infected spleens. In vitro generated TCF1+ P14 WT (green asterisk) and TCF1+ Cxcr3−/− P14 cells (blue arrow) in infected spleens 24h post-transfer (left panel). Identification of T cell zones defined as TCF1 (red) and CD8b (yellow) dense regions (middle panel, white dashed circle). Localization of in vitro transferred cells with respect to naïve T cell zones (TCF1+ CD8b+, white dashed circle) (right panel). (D) Percentage of transferred cells that localized within TCF1+ CD8b+ dense areas (white dashed circle). (E) Percentage of P14 cells that express TCF1 in infected spleens on d4 post-transfer into d8, d16 and d24 Cl13-infected mice. (F-G) Localization and identity of CXCL10 and CXCL9 expressing stromal (F) or hematopoietic (G) cells in Cl13-infected spleens of WT BM into REX3 (F) or REX3 BM into WT (G) chimeras. On d24 when stroma compartment recovered from Cl13-induced destruction (F) or d16 when the reporter expression in hematopoietic compartment was highest (G), spleens were harvested and processed for immunofluorescence. (F) Gp38+ reticular fibroblasts (red, left panel) or ER-TR7+ red pulp fibroblasts (red, right panel) in infected spleens with respect to CXCL10 (blue) and CXCL9 (yellow). (G) XCR1+ (green), F4/80+ cells (red, left panel), CD8 and CD4+ (red), and CD64+ cells (green, right panel) in infected spleens with respect to CXCL10 (blue) and CXCL9 (yellow) (H-J) REX3 mice were infected with Cl13 and on d16 pi., the phenotype of CXCL10+ and CXCL9+ single positive (SP) and CXCL9+CXCL10+ double positive (DP) were analyzed by flow cytometry according to the gating strategy in Figure S5A and tabulated (H). (I-J) Expression of CXCL10 and CXCL9 among single producer (I) and double producer (J) myeloid cell subsets (GMFI calculated for positive cells). (K) Distinct myeloid cell subsets were sorted from d15 Cl13-infected mice and Pdl1, Il10, Cd86, and IL12p40 mRNA expression was determined by RT-qPCR. Images are representative of two independent experiments with n=1–2 per time point. Data are from at least two independent experiments (n=3–4 per time point). White dashed circles mark T cell zones, Data in D and E were analyzed with paired t-test; and data in K with an ordinary one-way Anova with Dunnett’s multiple comparison test; *, P < 0.05; **, P < 0.01; ***, P < 0.001. See also Figures S4 and S5.
CXCR3 affects the localization of LCMV-specific CD8+ T cells within infected spleens
To determine if the absence of CXCR3 indeed can affect localization of TCF1+ cells within infected spleens, we compared the localization of in vitro generated WT or Cxcr3−/− TCF1+ P14 cells after transfer into d8, d16, or d24 Cl13-infected CD45.1 mice (Figure S4A). Cultured P14 cells were 80–90% TCF1-positive and 70–90% of WT P14 cells expressed CXCR3 (Figure S4B, C). Comparable numbers of Cxcr3−/− and WT P14 cells were detected within spleens by immunofluorescence 24h post-transfer (Figure 6C, S4D, E). Although similar percentages of WT and Cxcr3−/− P14 cells localized within the white pulp (data not shown), an increased frequency of transferred Cxcr3−/− TCF1+ P14 cells was detected within T cell zones of the white pulp (Figure 6D). The enhanced localization of Cxcr3−/− TCF1+ cells within these regions early post-transfer correlated with improved persistence of Cxcr3−/− TCF1+ P14 cells on d4 after transfer into d16 and d24 infected mice (Figure S4F). Moreover, a higher percentage of P14 Cxcr3−/− cells remained TCF1 positive (Figure 6E). Thus, these data demonstrate that CXCR3 expression controls positioning of T cells within niches of infected spleens and their exposure to pro-survival and differentiation signals.
CXCR3 ligand expression distinguishes stimulatory and inhibitory myeloid cell subsets
Next, we defined the identity of CXCR3-ligand producing cells in distinct splenic locations. To distinguish stromal versus hematopoietic contributions, we generated BM chimeras between WT and REX3 mice. Splenic sections from Cl13-infected chimeric mice (WT into REX3) were stained with mAbs distinguishing ER-TR7+ red pulp fibroblasts from gp38+ white pulp fibroblastic reticular cells. A minor population of gp38+ stromal cells was CXCL9 positive (Figure 6F). In contrast, a much greater proportion of red pulp fibroblasts produced CXCL10 (Figure 6F), or, less frequently, CXCL9 and CXCL10 (Figure S4G). CXCL10+ cells formed a characteristic ring at the border between the white and the red pulp (Figure 6F). CXCL9+ cells located within the T cell zone were predominantly of hematopoietic origin (Figure S4G) and were identified as XCR1+ conventional dendritic cells type 1 (cDC1) by immunofluorescence (REX3 into WT) (Figure 6G) and flow cytometry analysis (Figure 6H, S5A). As high as 40–60% of cDC1 cells were CXCL9+ at all stages of Cl13 infection (Figure S5C) and were the predominant population that contributed to CXCL9 production (Figure 6H; S5E). cDC1 had the highest CXCL9 MFI among all myeloid cells (Figure 6I, S5C). T cells and CD64+ and F4/80+ myeloid cells contributed to CXCL10-production (Figure 6G). We focused our analysis on the myeloid compartment, given that myeloid cells were a predominant source of CXCL10 in the red pulp where terminal differentiation occurs (Figure 6G). Moreover, Cl13 induced a more pronounced production of CXCR3 ligands within the myeloid lineage than Arm infection (Figure S5F–H). On d16 and d24 pi., the percentage of CXCL10 single producers were consistently higher during Cl13 compared to Arm infection (Figure S5F–H). CXCL10 single producers included predominantly CD11b+Ly6G−F4/80+CD64+ cells (inflammatory monocytes) and, to a lesser extent, CD11b+Ly6G+ (neutrophils) and CD11c+XCR1− cells (cDC2) (Figure 6H; S5B). At all stages of Cl13 infection, ~30% of inflammatory monocytes and 18–30% of cDC2 were CXCL10+ (Figure S5E). Other subsets did not show sustained CXCL10 production (Figure S5E). CXCL9 and CXCL10 double producers were predominantly inflammatory monocytes and cDC2 (Figure 6H; S5E). Although the cDC1 subset showed a small initial contribution, by d24 pi., they constituted ~20% of the double producing cells (Figure 6H, S5E). These three subsets differ in their pattern of expression of CXCL9 and CXCL10. Inflammatory monocytes showed the highest expression of CXCL10, followed by cDC2 and cDC1 (Figure 6J; S5D), while CXCL9 expression was highest among cDC1 and much lower among inflammatory monocytes and cDC2 (Figure 6J; S5D). Thus, different myeloid cells show unique patterns of CXCR3 ligand expression. Chronic infection induces sustained accumulation of CD11b+ myeloid cells with an immunosuppressive phenotype (Norris et al., 2013) while cDC1 are essential to sustain T cell cytotoxic and effector functions during chronic infection (Argilaguet et al., 2019). We, therefore, hypothesized that myeloid subsets that show differential CXCR3 ligand expression differ in their ability to stimulate T cells. To this end, we compared the expression of IFN-regulated inhibitory signals (Il10 and Pdl1) and stimulatory signals (Il12p40 and Cd86) in myeloid subsets that were sorted based on differential pattern of CXCL9 and CXCL10 expression, which was confirmed by RT-qPCR (Figure S5I). We chose two subsets that were characterized by high CXCL10 (CXCL10hi) and low CXCL9 expression: CXCL10 single producing (SP) and double-producing (DP) inflammatory monocytes (Figure 6H–J); two subsets that were characterized by high CXCL9 expression (CXCL9hi) and low CXCL10: CXCL9 single producing and double-producing cDC1 (Figure 6H–J). We also sorted double negative (DN) inflammatory monocytes as a control. There was increased expression of Il10 and Pdl1 in CXCL10hi inflammatory monocytes (Figure 6K). In contrast, CXCL9hi cDC1 showed high expression of Il12p40 and Cd86 (Figure 6K). We confirmed increased expression of CD86 on the surface of CXCL9hi cDC1 and PDL1 on the surface of CXCL10hi inflammatory monocytes by flow cytometry (Figure S5J–M). On d24 of Cl13 infection, a greater percentage of CXCL9hi cDC1 expressed CD86 compared to double negative cDC1 and inflammatory monocytes and CD86 expression was highest on double producing cDC1 (Figure S5J, K). In contrast, a greater percentage of CXCL10hi inflammatory monocytes expressed PDL1 compared to double negative inflammatory monocytes and cDC1 (Figure S5L, M). Moreover, PDL1 expression was highest on CXCL10hi inflammatory monocytes (Figure S5L, M). These data suggest that CXCL10 and CXCL9 expression might reflect the inflammatory milieu to which these respective myeloid cells are exposed and their capacity to provide stimulatory or inhibitory signals for differentiating T cells. Thus, it is likely that within the T cell zone, T cells are predominantly exposed to differentiation signals in the presence of co-stimulation, and only when they downregulate CCR7 and migrate out of the T cell zone in a CXCR3-dependent manner they become exposed to signals known to inhibit T cell function (Brooks et al., 2006; Ejrnaes et al., 2006).
CXCL10 deficiency is associated with lower viral set point
Viral replication in chronic viral infections reaches a set-point determined by an equilibrium with the residual function of the antiviral T cell response (Wherry and Kurachi, 2015). On d8 pi., Cxcr3−/−, Cxcl9−/− and Cxcl10−/− mice showed comparable serum viral titers as WT controls, suggesting similar early viral containment (Figure S6A). To determine if enhanced persistence of functional T cell subsets correlated with better viral control, we assessed virus titers in serum, brain and liver of WT and gene-ablated mice on d24 pi. Cxcl9−/− mice that do not show expansion of LCMV-specific responses had similar viral titers as WT controls (Figure 7A–C). Despite the parallels in CD8+ T cell subset dynamics, viral replication in Cxcr3−/− and Cxcl10−/− mice was not identical. Cxcr3−/− mice showed comparable viral titers in serum (Figure 7A) and increased viral burden in the brain compared to WT controls (Figure 7B). In contrast, Cxcl10−/− mice had reduced viral titers in serum (75% decrease), brain (68% decrease), and liver (65% decrease) compared to WT mice (Figure 7A–C). The improved viral control in Cxcl10−/− mice correlated with increased effector functions of GP276-specific cells in the spleen (Figure 7D). The percentage of IFNγ+TNFα+ producers was increased ~2.2-fold among GP276-specific cells in Cxcl10−/− mice but this was not seen in Cxcr3−/− mice (Figure 7D). In contrast, in Cxcl9−/− mice, the frequency of IFNγ+TNFα+ producers among GP276-specific cells was decreased compared to WT controls (Figure 7D). Thus, these data demonstrate that in the absence of CXCL10 the increase in effector cytokine producing GP276-specific cells correlates with improved viral control reflected in a lower viral set point.
Figure 7. CXCL10 deficiency is associated with lower viral set point and improved response to anti-PDL1.
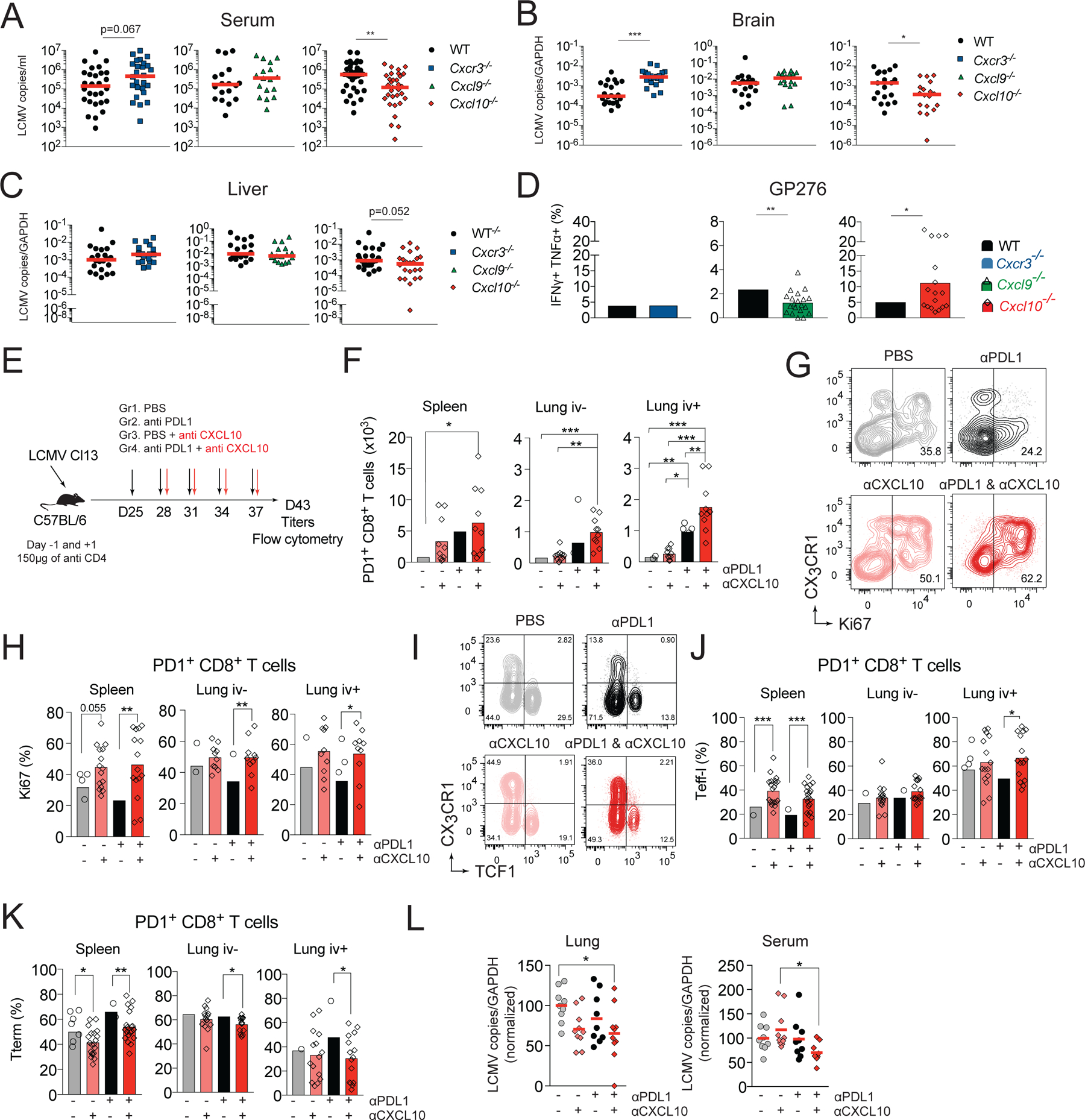
(A-D) Paired groups of WT and Cxcr3−/−, Cxcl9−/−, or Cxcl10−/− mice were infected with Cl13 and on day 24 pi. mice were analyzed for viral titer in serum (A), brain (B) and liver (C). (D) Splenocytes from d24 infected mice were restimulated with GP276 peptide and analyzed for cytokine production by flow cytometry. Percentage of GP276-specific cells that are IFNγ+TNFα+. (E-L) WT mice were infected with Cl13 and treated with 150μg of anti-CD4 i.p. to induce stable viremia. Beginning on day 24, mice were treated with PBS, anti-CXCL10, anti-PDL1, or anti-PDL1 and anti-CXCL10 i.p. every third day for 2 weeks. Six days after the last treatment, mice were sacrificed, and organs processed for flow cytometry and viral titers. (E) Experimental scheme. (F) Total number of PD1+ CD8+ T cells in spleen and lung vascular and parenchymal compartments. (G-H) Frequency of Ki67+ cells among PD1+ CD8+ T cells in spleen and lungs. Representative flow cytometry plots (G) and quantification (H). (I-K) Representative flow cytometry plots (I) and quantification of the frequency of Teff-l (J) or Tterm (K) in spleen and lungs. (L) Viral titers in lung and serum (normalized to the no treatment group). Data are from at least two independent experiments (n=3–4 per time point). Data in A-C were analyzed with Mann-Whitney test, the red line marks median, data in D with unpaired Student’s t-test; data in F-K with Anova with Sidak’s multiple comparison test; and data in L with Tukey’s multiple comparison test *, P < 0.05; **, P < 0.01; ***, P < 0.001. See also Figure S6.
CXCL10 neutralization enhances the CD8+ T cell proliferative response to anti-PDL1
PDL1 blockade temporarily increases the numbers and functionality of Teff-l (Hudson et al., 2019). Nevertheless, in the case of incomplete antigen clearance, Teff-l ultimately progress towards Tterm (Pauken et al., 2016). CXCR3 and CXCL10 facilitate terminal differentiation of T cells. Therefore, we determined if CXCL10 blockade during anti-PDL1 treatment had therapeutic potential. To this end, WT mice were infected with Cl13 and treated with blocking mAbs to PDL1 and CXCL10 (Figure 7E). Mice treated with anti-PDL1 treatment alone had increased numbers of PD1+ CD8+ T cells only in the lung vasculature compared to control treated mice. In contrast, mice treated with combination therapy showed increased numbers of PD1+ T cells not only in the lung vasculature, but also in the lung parenchyma and spleen, suggesting enhanced T cell responses in mice treated with combination therapy (Figure 7F). These changes were accompanied by an increase in the frequency of Ki67+ cells in several organs, including the spleen, lung (Figure 7G, H), liver and blood (Figure S6B), suggesting enhanced proliferation of PD1+ T cells in mice that received combination therapy. Treatment with anti-CXCL10 alone resulted in a mild increase in Ki67+ cells in the spleen and a more marked increase in the liver compared to untreated mice. Finally, combination treatment increased the frequency of Teff-l at the expense of Tterm in several organs, including the spleen, lung (Figure 7I–K), liver and blood (Figure S6C, D) compared to anti-PDL1 treatment alone. These changes correlated with improved viral control in the lungs and serum of mice that received combination therapy (Figure 7L). No improvement in viral control was observed in liver (Figure S6E), suggesting organ specific differences in the kinetics or quality of the response to the combination therapy.
Discussion
In this study, we identified a role for CXCL10 in the control of LCMV-specific CD8+ T cell heterogeneity and persistence during chronic LCMV infection. CXCL10 acted at two distinct stages of T cell differentiation: 1) it limited survival of Tstem-l, promoting their proliferation and TCF1 downregulation; and 2) it facilitated terminal differentiation of Teff-l into CD101+Tterm. Thus, CXCL10 played an important role in balancing fates of differentiating stem-like cells, which had an impact on viral control as the absence of CXCL10 signaling resulted in a lower viral set point.
During chronic infection, depletion of CX3CR1+ cells in the diphtheria toxin model results in increased viral titers, which has been explained by the loss of Teff-l (Hudson et al., 2019). Therefore, it is likely that the increase in numbers of CX3CR1+Tstem-l and Teff-l contributes to improved viral control in Cxcl10−/− mice. Cxcr3−/− mice did not show improved viral control, suggesting that LCMV-specific cells that expanded in Cxcr3−/− mice have lower functionality compared to phenotypically similar populations of T cells that expanded in Cxcl10−/− mice. This might be due to the role CXCR3 and its other ligand CXCL9 play in the acquisition of effector functions in differentiating Tstem-l. In addition, lack of CXCR3 might impair the ability of T cells to locate and eliminate virally infected cells in non-lymphoid tissues (Hickman et al., 2015), and impair the function of other CXCR3+ immune cells involved in differentiation and functionality of CD8+ T cells (Groom et al., 2012). Thus, these findings suggest that targeting individual chemokine ligands might be a better strategy to manipulate CD8+ T cell responses during chronic infection rather than targeting the chemokine receptor itself.
Our immunofluorescence data suggest that the absence of CXCR3 on Tstem-l promotes their localization within T cell zones. This can be explained by 1) reduced responsiveness of Cxcr3−/− Tstem-l to CXCL10 signals within red pulp and/or 2) enhanced responsiveness to ligands expressed within T cell zone, e.g., CCL19 and CCL21. Therefore, the balance of CXCR3 and CCR7 signaling might control timing of exposure of differentiating Tstem-l to pro-survival signals located within T cell zone, such as IL-7. Our data suggest that when Tstem-l differentiate into CX3CR1+Tstem-l they downregulate CCR7 expression, which might explain why the frequency and number of CX3CR1+Tstem-l was more affected by the absence of CXCR3 signaling compared to Tstem-l. It is also likely that as cells differentiate and upregulate other chemokine receptors, such as CXCR6 and CX3CR1, the absence of CXCR3 signaling promotes their increased responsiveness to these other chemokine cues likely permitting their exposure to critical pro-survival signals (Di Pilato et al., 2021). Indeed, activated Teff-l downregulate CXCR3, which likely delays their terminal differentiation before de novo expression of CXCR3 licenses their responsiveness to CXCL10 produced by inflammatory monocytes in niches that promote terminal differentiation.
During chronic infection, T cell responses must be fine-tuned to prevent immunopathology while simultaneously ensuring viral control. Our data suggest that CXCR3 and its ligands might serve as a decision point to either promote the acquisition of effector functions (through CXCL9-dependent signaling) or to drive terminal differentiation (through CXCL10-dependent signaling). Thus, blocking CXCL10 might represent an attractive strategy to control viral-specific CD8+ T cell subset composition and to establish a lower viral set point. Previous studies show that the spatially restricted and myeloid cell-specific character of CXCL9 and CXCL10 expression in LNs correlates with a non-redundant role of CXCL10 in driving Teff formation during Arm infection (Duckworth et al., 2021). Similarly, the divergent roles of CXCR3 ligands during Cl13 infection might, in part, be explained by the specific expression pattern of the two ligands within the myeloid compartment and the spatially restricted character of their expression. High CXCL9 expression was found in cDC1 that were predominantly localized within T cell zones. These cells are necessary for the formation of efficient CD8+ T cell responses during chronic infection (Argilaguet et al., 2019), tumorigenesis (Broz et al., 2014) and response to anti-PD1 therapy (Chow et al., 2019). We also detected high expression of costimulatory signals, such as IL12p40 and CD86 in this cell subset, suggesting that CXCL9 might control Tstem-l interactions with cDC1 and promote acquisitions of effector functions. In contrast, CXCL10 signals were abundant within red pulp of Cl13-infected spleens and were highly expressed by red pulp fibroblasts and inflammatory monocytes. Inflammatory monocytes also expressed inhibitory molecules, such as IL10 and PDL1. Chronic infection induces sustained expansion of CD11b+CD11clo/− myeloid cells, and their depletion enhances effector T cell responses (Norris et al., 2013). Moreover, blockade of IL10 and PDL1 during chronic infection improves T cell effector function and results in reduced viral titers (Brooks et al., 2006; Ejrnaes et al., 2006). We hypothesize that stroma-derived CXCL10 plays a dominant role in positioning differentiating T cells within the red pulp of the spleen. In addition, CXCL10 produced by inflammatory monocytes likely controls the quality of their interactions with Teff-l, promoting T cell terminal differentiation.
Overall, these data identify an important role of the CXCL10-CXCR3 axis in the control of T cell heterogeneity during chronic infection. Chronic CXCL10 signaling reduces survival of stem-like cells and favors differentiation of T cells, resulting in impaired persistence of T cells over-time and diminished frequency of CX3CR1+Tstem-l and Teff-l. Further work is required to better understand the impact of chronic CXCL10 signaling on T cell terminal differentiation in various tissues and during checkpoint inhibition to define the optimal CXCL10 neutralization regimens to fine tune T cell fates.
Limitations of Study
It remains to be defined to what extent the increase in CX3CR1+ stem-like cells occur due to their improved survival versus their slower rate of TCF1 downregulation and differentiation. Likewise, it remains to be determined to what extent the decreased upregulation of CD101 on Cxcr3−/− effector-like cells occurs due to their reduced exposure to differentiating signals versus their improved exposure to pro-survival signals. Moreover, this study did not address whether CXCL10 derived from inflammatory monocytes is necessary for T cell terminal differentiation and to what extent this mechanism operates in peripheral tissues and within tumors.
Star Methods
Resource availability
Lead Contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Andrew Luster (aluster@mgh.harvard.edu).
Materials Availability
This study did not generate new unique reagents.
Data and Code Availability
NanoString data generated during this study, including genes counts pre-and post-normalization, as well as the raw nCounter data files (RCC files), have been deposited in the Gene Expression Omnibus (GEO) and are publicly available as of the date of publication. Accession number is listed in the key resources table. Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
Key Resources Table.
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| CXCL10 neutralizing mAb (IF11) | (Khan et al., 2000) | N/A |
| PDL1 blocking mAb (10F.9G2) | BioXcell | Cat# BE0101, RRID:AB_10949073 |
| CD4 depleting mAb (GK1.5) | BioXcell | Cat# BE003-1, RRID:AB_1107592 |
| CD8a (BUV395 conjugate) (53-6.7) | BD Bioscience | Cat# 563786, RRID:AB_2732919 |
| CD244.2 (Brilliant Violet 605™ conjugate) (2B4) | BD Bioscience | Cat# 740345, RRID:AB_2740078 |
| CD45.2 (Brilliant Violet 785™ conjugate) (104) | BD Bioscience | Cat# 563686, RRID:AB_2738375 |
| CD45.2 (Brilliant Violet 711™ conjugate) (104) | BD Bioscience | Cat# 563685, RRID:AB_2738374 |
| rat IgG2b (Brilliant Violet 711™ conjugate) (G15-337) | BD Bioscience | Cat# 743222, RRID:AB_2741358 |
| CD45.1 (Brilliant Violet BV421™ conjugate) (A20) | BD Bioscience | Cat# 563983, RRID:AB_2738523 |
| CD45.2 (Brilliant Violet BV421™ conjugate) (104) | BD Bioscience | Cat# 562895, RRID:AB_2737873 |
| Thy 1.1 (BUV395 conjugate) (53-2.1) | BD Bioscience | Cat# 740261, RRID:AB_2721773 |
| CD19 (BUV395 conjugate) (1D3/CD19) | BD Bioscience | Cat# 563557, RRID:AB_2722495 |
| NK1.1 (BUV395 conjugate) (PK136) | BD Bioscience | Cat# 564144, RRID:AB_2738618 |
| Ly-108 (BUV395 conjugate) (13G3) | BD Bioscience | Cat# 745730, RRID:AB_2743205 |
| Ly6G (Brilliant Violet 785™ conjugate) (1A8) | BD Bioscience | Cat# 740953, RRID:AB_2740578 |
| CXCR3 (Brilliant Violet BV421™ conjugate) (CXCR3-173) | BD Bioscience | Cat# 562937, RRID:AB_2687551 |
| Streptavidin (Brilliant Violet BV711™) | BD Biosciences | Cat# 563262, RRID:AB_2869478 |
| CD8a (Brilliant Violet 711™ conjugate) (53-6.7) | BD Biosciences | Cat# 563046, RRID:AB_2737972 |
| CD3 (Alexa Fluor® 700 conjugate) | BioLegend | Cat# 100216, RRID:AB_493697 |
| CD4 (Alexa 647 conjugate) (RM405) | BioLegend | Cat# 100530, RRID:AB_389325 |
| TIM3 (Brilliant Violet 421™ conjugate) (RMT3-23) | Biolegend | Cat# 119723, RRID:AB_2616908 |
| PD-1 (PE conjugate) (RMPI-30) | Biolegend | Cat# 109104, RRID:AB_313421 |
| PD-1 (PE/Cy7 conjugate) (RMP1-30) | BioLegend | Cat# 109110, RRID:AB_572017 |
| CX3CR1 (APC conjugate) (SA011F110) | BioLegend | Cat# 149008, RRID:AB_2564492 |
| CX3CR1 (Brilliant Violet BV605™ conjugate) (SA011F110) | BioLegend | Cat# 149027, RRID:AB_2565937 |
| CD8b (Alexa Fluor 488 conjugate) (YTS156.7.7) | BioLegend | Cat# 126628, RRID:AB_2800619 |
| CD8b (PE conjugate) (YTS156.7.7) | BioLegend | Cat# 126608, RRID:AB_961298 |
| BCL-2 (PE/Cy7) (BCL/10C4) | BioLegend | Cat# 633512, RRID:AB_2565247 |
| BCL-2 (PE) (BCL/10C4) | BioLegend | Cat# 633508, RRID:AB_2290367 |
| CD11c (Brilliant Violet BV605™ conjugate)(N418) | BioLegend | Cat# 117334, RRID:AB_2562415 |
| CD11b (Alexa Fluor™ 700 conjugate)(M1/70) | BioLegend | Cat# 101222, RRID:AB_493705 |
| XCR1 (APC conjugate) (ZET) | BioLegend | Cat# 148206, RRID:AB_2563932 |
| PODOPLANIN/GP38 (APC) (8.1.1) | BioLegend | Cat#127409, RRID:AB_10612940 |
| CD64 (PE/Cyanine7 conjugate) (X54-5/7.1) | BioLegend | Cat# 139313, RRID:AB_2563903 |
| F4/80 (Alexa488 conjugate) (BM8) | BioLegend | Cat# 123120, RRID:AB_893479 |
| F4/80 (APC conjugate) (BM8) | BioLegend | Cat# 123116, RRID:AB_893481 |
| F4/80 (Brilliant Violet BV711™ conjugate) (BM8) | BioLegend | Cat# 123147, RRID:AB_2564588 |
| Ly108 (APC conjugate) (330-AJ) | BioLegend | Cat# 134610, RRID:AB_2728155 |
| Ly108 (Pacific Blue™ conjugate) (330-AJ) | BioLegend | Cat# 134608, RRID:AB_2188093 |
| Ly6G (PerCP/Cyanine5.5 conjugate) (1A8) | BioLegend | Cat# 127616, RRID:AB_1877271 |
| Ly6C (Brilliant Violet 785™ conjugate) (HK1.4) | BioLegend | Cat# 128041, RRID:AB_2565852 |
| CD69 (Brilliant Violet 421™ conjugate) (H1.2F3) | BioLegend | Cat# 104545, RRID:AB_2686969 |
| TruStain fcX™ (anti-mouse CD16/32) (93) | BioLegend | Cat# 101320, RRID:AB_1574975 |
| CD45 (purified) (30-F11) | BioLegend | Cat# 103102, RRID:AB_312967 |
| TNF alpha (APC conjugate) (MP6-XT22) | BioLegend | Cat# 506308, RRID:AB_315429 |
| IFN-gamma (Brilliant Violet 421™ conjugate) (XMG1.2) | BioLegend | Cat# 505830, RRID:AB_2563105 |
| CD44 (Brilliant Violet 785™) (IM7) | BioLegend | Cat# 103059, RRID:AB_2571953 |
| CXCR3 (FITC conjugate) (CXCR3-173) | BioLegend | Cat# 126536, RRID:AB_2566565 |
| CXCR3 (PerCP/Cyanine5.5) (CXCR3-173) | BioLegend | Cat# 126514, RRID:AB_1186015 |
| T-bet (PE/Cy7 conjugate) (4B10) | BioLegend | Cat# 644824, RRID:AB_2561761 |
| CD45.2 (PE conjugate) (104) | BioLegend | Cat# 109808, RRID:AB_313445 |
| TCF1/TCF7 Rabbit mAb (Pacific BlueTM conjugate) (clone C63D9) | Cell Signaling Technology | Cat# 9066 RRID: AB_2797696 |
| TCF1/TCF7 Rabbit mAb (PE Conjugate) (clone C63D9) | Cell Signaling Technology | Cat# 14456S, RRID:AB_2798483 |
| TCF1/TCF7 Rabbit mAb (Alexa Fluor 488 Conjugate) (clone C63D9) | Cell Signaling Technology | Cat# 6444S, RRID:AB_2797627 |
| TCF1/TCF7 Rabbit mAb (Alexa Fluor 647® Conjugate) (Clone C63D9) | Cell Signaling Technology | Cat# 6709S, RRID:AB_2797631 |
| TCF1/TCF7 Rabbit mAb (purified) (Clone C63D9) | Cell Signaling Technology | Cat# 2203S, RRID:AB_2199302 |
| CD44 (FITC conjugate) (5035-41.1 D] | GeneTex | Cat# GTX74574, RRID:AB_378218 |
| Normal goat serum | Jackson ImmunoResearch | Cat# 005-000-121, RRID:AB_2336990 |
| XCR1 (VioBright FITC conjugate) (REA707) | Miltenyi Biotec | Cat# 130-111-378, RRID:AB_2654280 |
| IRF-4 (PE, REAfinityTM) (REA201) | Miltenyi Biotec | Cat# 130-100-917, RRID:AB_2652515 |
| H-2D(b) LCMV gp 33-41 KAVYNFATC Alexa 488 | NIH Tetramer Core Facility | N/A |
| H-2D(b) LCMV GP 33-41 KAVYNFATC (APC conjugate) | NIH Tetramer Core Facility | N/A |
| H-2D(b) LCMV GP 276-286 SGVENPGGYCL (APC conjugate) | NIH Tetramer Core Facility | N/A |
| CCR2 (APC conjugate) (475301) | R and D Systems | Cat# FAB5538A, RRID:AB_10645617 |
| CD86 (FITC conjugate) (GL1) | Thermo Fisher Scientific | Cat# 11-0862-82, RRID:AB_465148 |
| CD64 (purified) (27) | Thermo Fisher Scinetific | Cat# MA5-29706, RRID:AB_2785530 |
| CD101 (purified) (polyclonal) | Thermo Fisher Scientific | Cat# 26047-1-AP, RRID:AB_2880350 |
| CD101 (PE/Cy7 conjugate) (Moushi 101) | Thermo Fisher Scientific | Cat# 25-1011-82, RRID:AB_2573378 |
| CD274 (Alexa Fluor 488) (MIH5) | Thermo Fisher Scientific | Cat# 53-5982-82, RRID:AB_2811871 |
| PD-1 (FITC conjugate) (RMP1-30) | Thermo Fisher Scientific | Cat# 11-9981-82, RRID:AB_465467 |
| TNF alpha (FITC conjugate) (MP6-XT22) | Thermo Fisher Scientific | Cat# 11-7321-41, RRID:AB_10670212 |
| GFP Polyclonal Antibody, (Alexa Fluor 488 conjugate) | Thermo Fisher Scientific | Cat# A-21311, RRID:AB_221477 |
| IRF1 (purified) (SR44-08) | Thermo Fisher Scientific | Cat# MA5-31996, RRID:AB_2809290 |
| ER-TR7 (purified) (ER-TR7) | Thermo Fisher Scientific | Cat# MA1-40076, RRID:AB_1074409 |
| Anti-rat IgG (H+L) Highly Cross-Adsorbed (Alexa Fluor 488 conjugate) | Thermo Fisher Scientific | Cat# A11006, RRID:AB_141373 |
| Anti-rat IgG (H+L) Highly Cross-Adsorbed (Alexa Fluor 647 conjugate) | Thermo Fisher Scientific | Cat#A21247, RRID:AB_141778 |
| Anti-rabbit IgG (H+L) Highly Cross-Adsorbed (Alexa Fluor 488 conjugate) | Thermo Fisher Scientific | Cat# MA5-29706, RRID:AB |
| anti-rabbit IgG (H+L) Highly Cross-Adsorbed (Alexa Fluor 647 conjugate) | Thermo Fisher Scientific | Cat# A-21245, RRID:AB_2535813 |
| CD127 (biotin conjugate) (A7R34) | Thermo Fisher Scientific | Cat# 13-1271-82, RRID:AB_466588 |
| Bacterial and Virus strain | ||
| LCMV Armstrong (Arm) | kind gift of J. Wherry | Grew up in house |
| LCMV Cl-13 (Cl-13) | kind gift of J. Wherry | Grew up in house |
| Chemicals, Peptides, and Recombinant Proteins | ||
| LCMV gp33 – 41, GP33, (KAVYNFATM) | AnaSpec | Cat# AS-61296 |
| LCMV gp276 – 286, GP276, (SGVENPGGYCL) | AnaSpec | Cat# AS-62539 |
| BD GolgiStop™ (Containing Monensin) | BD Bioscience | Cat# 554274 |
| BD Golgi Plug™ (Containing Brefeldin A) | BD Bioscience | Cat# 555029 |
| 16% Paraformaldehyde Aqueous Solution | Electron Microscopy Sciences | Cat# 50-980-487 |
| Percoll | GE Healthcare | Cat# 17-0891-01 |
| Recombinant murine IL-12 protein | Peprotech | Cat# 210-12P40H |
| Recombinant murine IL-2 protein | Peprotech | Cat# 212-12 |
| Recombinant mouse CXCL10 protein | Peprotech | Cat# 250-16 |
| Liberase TM Research Grade | Sigma Aldrich | Cat# 5401119001 |
| DNAse I, grade II, from bovine pancreas | Sigma Aldrich | Cat# 10104159001 |
| TRIzol Reagent | Thermo Fisher Scientific | Cat# 15596018 |
| MultiScribe Reverse Transcriptase | Thermo Fisher Scientific | Cat# 4311235 |
| GeneAMP dNTP mix with dTTP | Thermo Fisher Scientific | Cat# N8080260 |
| MgCl2 Solution 25 mM | Thermo Fisher Scientific | Cat# 4486224 |
| 10x PCR Buffer II | Thermo Fisher Scientific | Cat# 4486220 |
| Oligio d(T)16 | Thermo Fisher Scientific | Cat# 100023441 |
| Random Hexamers 50 mM | Thermo Fisher Scientific | Cat# 100026484 |
| RNase Inhibitor | Thermo Fisher Scientific | Cat# 100021540 |
| Fixable Viability Dye eFluor 780 | Thermo Fisher Scientific | Cat# 65-0865-14 |
| Glycogen RNA grade | ThermoFisher Scientific | Cat# R0551 |
| ProLong Diamond Antifade Mountant | ThermoFisher Scientific | Cat# P36961 |
| OCT Compound | Tissue-Tek | Cat# 4583 |
| Critical Commercial Assays | ||
| nCounter® Mouse Immunology Panel (CodeSet Only) | NanoString | Cat# XT-CSO-MIM1-12 |
| QIAMP Viral RNA Mini kit | Qiagen | Cat# 52906 |
| QuantiFast SYBR Green RT-qPCR Kit | Qiagen | Cat# 204154 |
| FastStart Essential DNA Green Master | Roche | Cat# 25595200 |
| EasySep™ Mouse Naïve CD8+ T Cell Isolation Kit | Stem Cell Technologies | Cat# 19858 |
| FOXP3/Transcription Factor staining Kit | Thermo Fisher Scientific | Cat# 00-5523-00 |
| DNA-free™ DNA Removal Kit | Thermo Fisher Scientific | Cat# AM1906 |
| Click-iT™ Plus EdU Pacific Blue™ Flow Cytometry Assay Kit | Thermo Fisher Scientific | Cat# C10636 |
| Live/dead fixable viability dye eFluorTM 780 | Thermo Fisher Scientific | Cat# 65-0865-18 |
| Image-iT FX Signal Enhancer | Cell Signaling Technology | Cat# 11932S |
| BD Cytofix/Cytoperm | BD Bioscience | Cat# 554714 |
| CellTrace™ Violet Cell Proliferation Kit | Thermo Fisher Scientific | Cat# C34571 |
| Experimental Models: Organisms/Strains | ||
| Mouse: B6.129S7-Ifngr1tm1A9t/J | The Jackson Laboratory | Cat# 3288 |
| Mouse: C57BL/6-Tg(CAG-EGFP)131Osb/LeySopJ | The Jackson Laboratory | Cat# 006567 |
| Mouse: Cxcl10−/− | (Dufour et al., 2002) | N/A |
| Mouse: Cxcl9−/− | (Park et al., 2002) | N/A |
| Mouse: Cxcr3−/− | (Hancock et al., 2000) | N/A |
| Mouse: ifnar−/− | Müller et al., 1994 | Cat# N/A |
| Mouse: REX3 | Groom et al., 2012 | Cat# N/A |
| Mouse: WT: B6.SJL-Ptprca Pepcb/BoyJ | The Jackson Laboratory | Cat# 002014 |
| Mouse: WT: C57BL6 NCI | Charles River Laboratory | Cat# 027 |
| Mouse: WT: C57BL6/J | The Jackson Laboratory | Cat# 664 |
| Mouse: WT: CD45.1 | Charles River Lab - NCI Grantee Pricing’ | Cat# 564 |
| Oligonucleotides | ||
| Il10 | IDT technologies | N/A |
| forward; 5’ – GCT CTT ACT GAC TGG CAT GAG -3’ | ||
| reverse 5’- CGC AGC TCT AGG AGC ATG TG -3’ | ||
| Il12p40 | IDT technologies | N/A |
| forward 5’-TGG TTT GCC ATC GTT TTG CTG -3’ | ||
| reverse 5’- ACA GGT GAG GTT CAC TGT TTC T -3’ | ||
| Cd86 | IDT technologies | N/A |
| forward 5’- TGT TTC CGT GGA GAC GCA AG -3’ | ||
| reverse 5’ -TTG AGC CTT TGT AAA TGG GCA -3’ | ||
| Cxcl10 | IDT technologies | N/A |
| forward; 5’-GCCGTCATTTTCTGCCTCA-3’ | ||
| reverse 5’-CGTCCTTGCGAGAGGGATC-3’ | ||
| Cxcl9 | IDT technologies | N/A |
| forward; 5’-AATGCACGATGCTCCTGCA-3’ | ||
| reverse 5’-AGGTCTTTGAGGGATTTGTAGTGG-3’ | ||
| Pdl1 | IDT technologies | N/A |
| forward; 5’-GCT CCA AAG GAC TTG TAC GTG -3’ | ||
| reverse 5’- TGA TCT GAA GGG CAG CAT TTC -3’ | ||
| Gp | IDT technologies | N/A |
| forward; 5’-CATTCACCTGGACTTTGTCAGACTC-3’ | ||
| reverse GCAACTGCTGTGTTCCCGAAAC | ||
| Gapdh | IDT technologies | N/A |
| forward; 5’-GGCAAATTCAACGGCACAGT-3’ | ||
| reverse 5’-AGATGGTGATGGGCTTCCC-3’ | ||
| Np | IDT technologies | N/A |
| forward; 5’-CAGAAATGTTGATGCTGGACTGC-3’ | ||
| reverse 5’-CAGACCTTGGCTTGCTTTACACAG-3’ | ||
| Mm_Ifng_2_SG cc Assay | Qiagen | Cat# QT02423428 |
| Mm_Ifnb1_1_SG QuantiTect Primer Assay | Qiagen | Cat# QT00249662 |
| Software | ||
| BD FACS Diva v8 | BD Biosciences | https://www.bdbiosciences.com/us/instruments/clinical/software/flow-cytometry-acquisition/bd-facsdiva-software/m/333333/overview; RRID: SCR_001456 |
| Biorender | Biorender | http://biorender.com;RRID:SCR_018361 |
| Fiji | Fiji | http://fiji.sc; RRID:SCR_002285 |
| GraphPad Prism Version 8 | GraphPad | https://www.graphpad.com; RRID: SCR_002798 |
| nSolver Analysis Software 4.0 | nSolver Analysis Software | http://www.nanostring.com/products/nSolver; RRID:SCR_003420 |
| Light Cycler 96 | Roche | http://www.roche-applied-science.com/shop/products/absolute-quantification-with-the-lightcycler-carousel-based-system; RRID:SCR_012155 |
| FlowJo v10 | Tree Star | https://www.flowjo.com/solutions/flowjo/downloads; RRID: SCR_008520 |
| Zen Black 2012 v8 | Zeiss | https://www.zeiss.com/microscopy/us/products/microscope-software/zen.html; RRID: SCR_013672 |
| Recombinant DNA | ||
| pCITE-GP plasmid | J. de la Torre | N/A |
Experimental model and subject details
Mice
C57BL/6 CD45.2 and CD45.1 mice (experiments in Figures 4C–F; 5B–F; 5I–J) were purchased from Charles River Laboratory. C57BL6/J and B6.SJL-Ptrprc<a>Pepc<b>/BoyJ (CD45.1) mice were purchased from Jackson laboratory (experiments in Figures 1D–I; 6C–E; S1A–C; S3A–B; S3D–F; S4A–F). P14 tg CD45.1 mice were provided by A. Sharpe (Harvard Medical School, Taconic B6.Cg-Tcratm1Mom Tg(TcrLCMV)327Sdz backcrossed 10 generations to Jackson C57BL/6J; (LaFleur et al., 2019)). P14 CD45.2 WT GFP mice were generated by crossing of C57BL/6-Tg(-CAG-EGFP)131Osb/LeySobJ with P14 CD45.1 mice. Cxcr3−/− CD45.2 P14 mice were generated by crossing P14 mice with Cxcr3−/− mice. Cxcr3−/− CD45.2 P14 GFP mice were generated by crossing P14 Cxcr3−/− mice with C57BL/6-Tg(-CAG-EGFP)131Osb/LeySobJ from Jackson laboratory. All mice, including REX3 tg (Groom et al., 2012), Ifnar1−/− (Muller et al., 1994), Ifnγr−/− (Jackson Laboratory), Cxcr3−/− (Hancock et al., 2000), Cxcl9−/− (Park et al., 2002), and Cxcl10−/− (Dufour et al., 2002) were on a C57BL/6 background and were housed under specific-pathogen-free conditions of animal facility of MGH Charlestown Navy Yard. Ifnar1−/− and Ifnγr−/− mice crossed into the REX3 background were used for experiments in Figure 3B. Female mice were used between 6– 15 weeks of age. All animal work has been approved by the Massachusetts General Hospital Subcommittee on Research and Animal Care.
Virus infection
LCMV strains were propagated from stocks obtained from the laboratory of J. Wherry (University of Pennsylvania), and titers were measured as described (Ahmed et al., 1984). Mice were infected with 2× 105 plaque-forming units (PFU) of LCMV Armstrong (Arm) or with 4× 106 PFU of LCMV Cl13. LCMV Arm was injected intraperitoneally (i.p.), whereas LCMV Cl13 was injected intravenously (i.v.) to facilitate establishment of persistent infection (Dangi et al., 2020). For experiments in Figure 5I–J, Figure 7E–L, and Figure S6B–E mice were treated with 150μg of anti-CD4 (GK1.5) i.p. one day before and after infection with Cl13 to induce stable viremia (Barber et al., 2006; Wherry et al., 2007).
Antibody treatments
Mice were treated with 200μg of anti-PDL1 mAb (clone 10F.9G2) i.p. on day 24 post Cl13 infection and then each third day for total of two weeks (5 doses). One day after the second, third, fourth and fifth dose of anti-PDL1 mice received 200μg of anti-CXCL10 blocking mAb (clone IF11) i.p. (Khan et al., 2000).
P14 cells adoptive transfer model
For adoptive transfer experiments, naïve CD8+ T cells were isolated from spleens and peripheral lymph nodes of GFP+ CD45.2 P14 or Cxcr3−/− CD45.2 P14 mice with a negative selection kit (EasySep mouse naive CD8+ T cell Isolation Kit, STEMCELL Technologies) according to the manufacturer’s protocol. CD8+ T cells purity was typically > 95%. Three hundred cells of each genotype were mixed in a 1:1 ratio and transferred i.v. into CD45.1 C57BL6/J mice from Jackson Laboratory. Two hours post-transfer of P14 cells, mice were infected with 2× 105 PFU of Arm or with 4× 106 PFU of LCMV Cl13.
Method details
Tissue preparation
If staining of lymphocytes in blood was necessary, a few drops of blood were collected by submandibular bleeding into tubes containing 10–20μl of EDTA. After euthanizing mice, lymph nodes and spleens were removed and homogenized using 70-μm cell strainers. Spleens and blood were treated with red blood cell lysing solution (Sigma-Aldrich) for 3min at room temperature. Cell numbers were counted with trypan blue using Kova counting chambers. Livers and lungs were harvested, cut in small pieces, and incubated with 100μg/ml of liberase TM (Sigma Aldrich) and 50μg/ml of DNAse solution (Roche) for 30min at 37°C. After 30min, organs were homogenized using 70-μm cell strainers and lymphocytes were isolated using Percoll (Fisher Scientific) gradient. Total cell numbers were counted with trypan blue staining using Kova counting chambers.
Bone marrow isolation
Femurs, tibias and humerus bones were isolated from hind and front legs of donor mice. In sterile conditions, the endings of the bones were cut off, and bones were placed in a 0.5 ml microcentrifuge tube. Tubes were centrifuged at >10,000g for 20sec, the efficacy of BM recovery was assessed by visual inspection of bones. The pellet was resuspended in PBS, cells were counted, and 3–5 × 106 cells were injected i.v. into recipient mice.
Generation of bone marrow chimeras
To generate bone marrow chimeric recipient mice, C57BL/6 CD45.1 WT mice or REX3 mice were irradiated with a lethal dose of 10 Grey. After ~2 hours, mice received i.v. injection of 3–5 × 106 BM donor cells (CD45.1 or REX3). The following chimeras were generated: CD45.1 BM into REX3 recipient and REX3 BM into CD45.1 recipient. After minimum of 8 weeks, mice which showed reconstitution of the hematopoietic compartment with donor BM higher than 80% (REX3 BM into CD45.1) or 90% (CD45.1 into REX3) were used for experiments.
Intravascular staining of lymphocytes
To distinguish cells located within vasculature vs. parenchyma, ketamine/xylazine anesthetized mice received i.v. injection of 3μg of purified anti CD45 Ab (30-F11) and were sacrificed after 3min by cervical dislocation.
Antibodies and flow cytometry
Single-cell suspensions obtained from murine spleens, lymph nodes, livers, or lungs were stained for flow cytometry. Fc receptors were blocked with purified TruStain FcX™ anti-mouse CD16/32 mAb (1:100) in FACS buffer (PBS with 1% FCS and 0.05% NaN3) in the presence of live/dead fixable viability dye eFluorTM 780 (1:1000, ThermoFisher Scientific) for 10 min at room temperature. For experiments in which mice received an i.v. injection of purified anti-CD45 (30-F11), the Fc receptor blocking mix also contained an anti-rat IgG2b mAb (G15–337; 1:400). Cell were then washed and stained with fluorochrome-conjugated mAbs against CD8 (53–6.7), CD3 (17A2), CXCR3 (CXCR3–173), TIM3 (RMT3–23), CD244.2 (2B4), CD101 (Moushi101), CX3CR1 (SA011F11), CD69 (H1.2F3), CD45.2 (104), CD45.1 (A20), CD44 (IM7), Ly108 (330-AJ), PD1 (RPMI-30).Tetramers specific for GP33 and GP276 were obtained from the NIH tetramer facility. For analysis of myeloid compartment, cells were stained with fluorochrome-conjugated mAbs against CD19 (1D3/CD19), NK1.1 (PK136), Thy1.2 (53–2.1), CD11b (M1/70), CD11c (N418), Ly6G (1A8), Ly6C (HK1.4), F4/80 (BM8), XCR1 (ZET), CD64 (X54–5/7.1), BCL-2 (BCL/10C4), CD86 (GL1), PD-L1 (MIH5). All Ab stainings were performed at room temperature (RT) for 30min. If no intracellular staining was needed, stained cells were fixed with 2% paraformaldehyde (PFA, Electron Microscopy Sciences) at RT for 20min. For intracellular detection of transcription factors cells were fixed and permeabilized with Foxp3 staining buffers (ThermoFisher Scientific) at RT for 20min followed by staining with intracellular mAbs against BCL2 (BCL/10C4), T-bet (4B10), TCF1 (C63D9), IRF4 (REA201), IRF1 (SR44–08), anti-GFP polyclonal Ab in FOXP3 permeabilization buffer for 30–60min at RT. For the IRF1 Ab, primary staining was followed by the secondary staining with fluorochrome-conjugated anti-rabbit Ab (20min, RT). Samples were analyzed using the Fortessa X-20 flow cytometer (BD) and FlowJo software (Tree Star).
Intracellular cytokine staining
For analysis of cytokine production, 2× 106 splenocytes were cultured in the presence of GP276 peptide (0.2μg/ml), brefeldin A (BD GolgiPlug™) and monensin (BD GolgiStop™) for 5 h at 37°C. Post incubation, cells were stained for extracellular molecules, including CD8 and CD3, as described above. Following extracellular staining, cells were fixed with 2% PFA and stained for intracellular cytokines using the Cytofix/Cytoperm kit (BD/Pharmingen) according to the manufacturer’s instruction. Following fluorochrome-conjugated mAbs were used for intracellular cytokine detection anti-IFNγ (XMG1.2), and anti-TNFα (MP6-XT22).
Edu staining
To measure the rate of new DNA synthesis, mice received i.p. injection of 1mg/kg of EdU (5-ethynyl-2’-deoxyuridine, a nucleoside analog of thymidine) 24h before euthanasia. Then single cell suspension from spleen was stained as described in antibodies and flow cytometry section. After the intracellular staining, cells were resuspended in Click-iT permeabilization and wash reagent and incubated at RT for 15min. Followed by 30min incubation with fluorescent dye picolyl azide prepared as described in manufacturer’s protocol (Click-iT Plus EdU Flow Cytometry Assay Kits, ThermoFisher Scientific). Samples were then analyzed using Fortessa X-20 flow cytometer (BD) and FlowJo software (Tree Star).
Cell Sorting experiments
Cell sorting of Tstem-l (PD1+ CD44+ Ly108+ CX3CR1− CD8+ T cells) or Teff-l (PD1+ Ly108− CX3CR1+ CD101− CD8+ T cells) from spleens of WT CD45.2, CD45.1, or Cxcr3−/− mice or of distinct myeloid cell subsets from the spleen of REX3 mice was performed on FACSAria Fusion Cell Sorter, BSL2+, with 70μ nozzle and a 4°C circulating cool-down system. Post-sorting CD8+ T cells were stained with CellTraceTM violet cell proliferation kit (ThermoFisher Scientific) according to the manufacturer’s protocol. Stained cells were then washed and injected i.v. into recipient mice (~105 cells). Sorted myeloid cell subsets were used for RNA isolation and qPCR.
NanoString analysis of gene expression
Four T cell subsets were sorted from spleens of WT or Cxcl10−/− mice on FACSAria Fusion Cell Sorter, BSL2+, with 70μ nozzle and a 4°C circulating cool-down system. The sorting was based on following markers: stem-like T cells (PD1+Ly108+CX3CR1−CD101− CD8+ T cells); CX3CR1+ stem-like T cells (PD1+Ly108+CX3CR1+CD101− CD8+ T cells); effector-like T cells (PD1+Ly108−CX3CR1+CD101− CD8+ T cells); and terminally differentiated T cells (PD1+Ly108−CX3CR1+CD101+ CD8+ T cells). Post sorting cells were spun down and lysed using TRIZOL reagent (ThermoFisher Scientific). Total RNA was chloroform-extracted, precipitated overnight at −20°C with isopropanol in the presence of 3μg of RNA grade glycogen (ThermoFisher Scientific), washed two times with 75% ethanol, and dissolved in water. 50ng of RNA was used for downstream preparation according to manufacturer’s recommendation. Briefly, RNA was incubated at 65°C with Reporter Code Set, Hybridization Buffer and Capture ProbeSet for 17 hours (nCounter ® Mouse Immunology Panel gene expression CodeSet). Once the hybridization reaction was terminated, samples were injected into loading ports on the cartridge and sealed. The cartridge was loaded into nCounter SPRINT Profiler and processed for digital count of transcript abundance. Data were processed and analyzed with nSolver 4.0. In brief, data were controlled for any imaging and binding quality control issues, positive control linearity and limit of detection parameters. The counts for each sample were normalized based on housekeeping gene expression. Analysis of differentially expressed genes was performed, accounting for multiple comparison corrections with Benjamini-Yekutieli False Discovery Rate method. The relative expression of selected differentially expressed genes between all subsets was represented as a heatmap as the standard deviation from the mean.
RNA isolation and Reverse Transcription (RT) - Quantitative (q) PCR
Livers, kidneys or brains were frozen at dry ice and stored at −80°C until further processed. Tissues or sorted myeloid cell subsets were lysed using TRIzol™ reagent (ThermoFisher Scientific). Total RNA was chloroform-extracted, precipitated with isopropanol, washed two times with 75% ethanol, and dissolved in water. To remove genomic DNA, RNA samples were treated with rDNase I according to the manufacturer’s protocol (DNA-free kit, Life Technologies). RNA was reverse transcribed using multiscribe reverse transcriptase (Applied Biosystems). Reverse transcription mix contained random hexamers and oligoDTs or reverse primer for GP33 and oligoDTs. qPCR was performed using the LightCycler 96 System (Roche) and FastStart Essential DNA Green Master kit (Roche). Specific target sequences were amplified with the use of self-designed primers for Gapdh (forward; 5’-GGCAAATTCAACGGCACAGT-3’and reverse 5’-AGATGGTGATGGGCTTCCC-3’), Cxcl9 (forward; 5’-AATGCACGATGCTCCTGCA-3’ and reverse 5’-AGGTCTTTGAGGGATTTGTAGTGG-3’); Cxcl10 (forward; 5’-GCCGTCATTTTCTGCCTCA-3’ and reverse 5’-CGTCCTTGCGAGAGGGATC-3’) GP (forward; 5’-CATTCACCTGGACTTTGTCAGACTC-3’ and reverse GCAACTGCTGTGTTCCCGAAAC), IL12p40 (forward 5’-TGG TTT GCC ATC GTT TTG CTG −3’ and reverse 5’- ACA GGT GAG GTT CAC TGT TTC T −3’), CD86 (5’- TGT TTC CGT GGA GAC GCA AG −3’ and reverse 5’ -TTG AGC CTT TGT AAA TGG GCA −3’), NP (forward; 5’-CAGAAATGTTGATGCTGGACTGC-3’ and reverse 5’-CAGACCTTGGCTTGCTTTACACAG-3’), PD-L1 (forward; 5’-GCT CCA AAG GAC TTG TAC GTG −3’ and reverse 5’- TGA TCT GAA GGG CAG CAT TTC −3’), IL-10 (forward; 5’ – GCT CTT ACT GAC TGG CAT GAG −3’ and reverse 5’- CGC AGC TCT AGG AGC ATG TG −3’). All primers were purchased from Integrated DNA Technologies. For analysis of Ifng and Ifnb1 expression commercially available QuantiTect primers were used (Qiagen). GAPDH was used as a reference gene. Known amounts of the linearized pCITE-GP plasmid (kind gift of J.de la Torre) were used to create a standard curve necessary for the quantification of GP RNA expression (McCausland and Crotty, 2008). For experiments in Figures 3A, 3B and Figure S1F, RNA expression was calculated as a fold change over uninfected spleen using the delta-delta Ct method. For figures 6K, 7B–C, 7L, S5I, and S6E RNA expression was normalized to GAPDH expression.
RT-qPCR analysis of serum viral titers
Mice were bled by submandibular bleeding and the blood was allowed to coagulate for a minimum of 30min, followed by centrifugation (15min, 20 000 g). The supernatants were then collected and frozen for RT-qPCR analysis of viral titers. Viral RNA was isolated from twenty microliters of serum using the QIAmp Viral RNA Mini kit (Qiagen). RT-qPCR was performed using QuantiFast SYBR Green RT-qPCR Kit (Qiagen) and primers for GP (forward; 5’-CATTCACCTGGACTTTGTCAGACTC-3’ and reverse GCAACTGCTGTGTTCCCGAAAC) or NP (forward; 5’-CAGAAATGTTGATGCTGGACTGC-3’ and reverse 5’-CAGACCTTGGCTTGCTTTACACAG-3’). Known amounts of the linearized pCITE-GP plasmid (kind gift of J.de la Torre) were used to create a standard curve necessary for the quantification of GP RNA expression (McCausland and Crotty, 2008)
In vitro TCF1+ cell culture
In vitro TCF1+ cell culture was based on the protocol published in (Di Pilato et al., 2021). In brief, splenocytes from CD45.2 P14 GFP mice and CD45.2 Cxcr3−/− P14 mice were cultured at concentration of 107/ml in the presence of 1μg/ml of GP33–41 peptide (KAVYNFATM, AnaSpec). Alternatively, CD45.1 P14 mice and GFP+ CD45.2 Cxcr3−/− P14 cells were used for culturing. Twenty-four hours later, cells were harvested counted and re-plated at concentration of 106/ml with low amounts of rIL-2 (5ng/mL, Peprotech). Cells were cultured for 4 days, with a daily exchange of medium supplemented with fresh rIL-2. In some experiments, on day 3 of culture, 200ng/ml of murine recombinant CXCL10 (Peprotech) was added to the culture. On day 4, cells were controlled for the percentage of TCF1 and CXCR3 expression. For some experiments, cultured cells were i.v. transferred into Cl13-infected CD45.1 or CD45.2 C57BL6/J recipient mice to control their differentiation and localization within spleens.
Immunofluorescence
Unfortunately, due to the low abundance of endogenously activated TCF1+ P14 cells at later stages of chronic infection, it was difficult to perform a meaningful analysis of their distribution. Therefore, we compared the localization of in vitro generated WT or Cxcr3−/− TCF1+ P14 cells after transfer into d8, d16, or d24 Cl13-infected CD45.1 recipient. To examine the localization of adoptively transferred in vitro cultured P14 cells or expression of CXCR3 ligands, the spleens or REX3 infected mice (d8,16,24 of Cl13 infection) and spleens of d24 Cl13-infected mice that received P14 cells (24h post-transfer of P14 cells) were harvested and fixed with BD cytofix cytoperm buffer (BD Bioscience) diluted in PBS (1:3) for 2h at 4 °C. Fixed tissues were then immersed in 30% sucrose solution and left until tissue sinks. After an exchange of 30% sucrose, tissues were embedded in OCT-Compound (Tissue-Tek) and frozen on dry ice. 8–12μm thick sections were cut using a cryostat. Sections were stored at −20°C until processed. The slides were surrounded by DAKO-Pen (Vector Laboratories), permeabilized in 0.3% Triton X-100 for 30min, then incubated for 30min at RT with Image-iT FX Signal Enhancer (Cell Signaling Technology) followed by blocking with 10% normal goat serum for 1h at RT. Sections were stained with primary mAbs at 4°C overnight. Staining with primary mAbs was followed by washing and staining with secondary fluorochrome-conjugated Abs for 1h at RT. Primary and secondary Abs listed below were used: purified rabbit anti-TCF1 (clone C63D9; Cell Signaling Technology), Alexa647 conjugated anti-B220 (clone RA3–6B2; BD Bioscience), Alexa647 or PE conjugated anti-CD8b (clone YTS156.7.7; Biolegend), Alexa647 conjugated anti-CD4 (clone RM405, Biolegend), APC or Alexa488 conjugated anti-F4/80 (clone BM8; Biolegend), VioBright FITC conjugated anti-XCR1 (clone REA707; Miltenyi Biotec) purified rabbit anti-CD64 (clone 27; Thermo Fisher Scientific), purified rabbit anti-CD101 (polyclonal; Thermo Fisher Scientific), purified rat anti-red pulp fibroblasts (clone ER-TR7; Thermo Fisher Scientific), APC conjugated anti-podoplanin (clone 8.1.1, Biolegend), Alexa647 or Alexa488 goat anti-rabbit, and Alexa 647 or Alexa488 goat anti-rat. After washing with PBS, the slides were cured with Prolong Gold mounting medium and coverslipped for 24h at RT. Images were taken using an LSM 710 confocal laser scanning microscope from Zeiss using a 20x air objective. The T cell zone (white dashes circle) was defined based on the localization of red pulp macrophages, B cell follicles, TCF1 staining or white and red pulp stromal cells. The characteristic lower intensity of autofluorescence within white pulp splenic regions further guided the demarcation of distinct splenic regions.
Statistics
Data were analyzed using Prism 7 (GraphPad Software) using unpaired or paired t-test, or Mann-Whitney test, or one-way Anova with Dunnett’s, Tukey’s, or Sidak’s multicomparison post-test. P-values < 0.05 were considered significant.
Supplementary Material
HIGHLIGHTS.
Inflammatory monocytes and fibroblasts of the splenic red pulp produce CXCL10.
CXCL10 limits exposure of stem-like CD8+ T cells to pro-survival signals.
CXCL10 promotes access of virus-specific CD8+ T cells to differentiation signals.
Cxcl10−/− mice chronically infected with LCMV have a lower viral set point.
Acknowledgments
We thank J. Wherry and his lab members (M. Kurachi, S. Ngiow, and R. Staupe) for LCMV Arm and Cl13 viral stocks and valuable advice; A. Sharpe and her lab members (K. Pauken, and M. LaFleur) for sharing P14 mice; J. Farber for Cxcl9−/− mice and C. Gerard for the Cxcr3−/− mice; J. de la Torre for sharing pCITE-GP plasmid and protocols. We also thank W. Hudson for critical comments on the manuscript.
This work was supported by grants from the National Institutes of Health (R01 CA204028) and the Cancer Research Institute to A.D.L; the Swiss National Science Foundation Early Postdoctoral Mobility Award (P3BEP3_165406) and the American Association Cancer Research Basic Science Fellowship Program (18–40-01-OZGA) to A.J.O
Footnotes
Declaration of interests
T.R.M is a founder, shareholder, and member of the advisory board of Monopteros Therapeutics, Inc.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Ahmed R, Salmi A, Butler LD, Chiller JM, and Oldstone MB 1984. Selection of genetic variants of lymphocytic choriomeningitis virus in spleens of persistently infected mice. Role in suppression of cytotoxic T lymphocyte response and viral persistence. J Exp Med 160:521–540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alfei F, Kanev K, Hofmann M, Wu M, Ghoneim HE, Roelli P, Utzschneider DT, von Hoesslin M, Cullen JG, Fan Y, Eisenberg V, Wohlleber D, Steiger K, Merkler D, Delorenzi M, Knolle PA, Cohen CJ, Thimme R, Youngblood B, and Zehn D 2019. TOX reinforces the phenotype and longevity of exhausted T cells in chronic viral infection. Nature 571:265–269. [DOI] [PubMed] [Google Scholar]
- Argilaguet J, Pedragosa M, Esteve-Codina A, Riera G, Vidal E, Peligero-Cruz C, Casella V, Andreu D, Kaisho T, Bocharov G, Ludewig B, Heath S, and Meyerhans A 2019. Systems analysis reveals complex biological processes during virus infection fate decisions. Genome Res 29:907–919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barber DL, Wherry EJ, Masopust D, Zhu B, Allison JP, Sharpe AH, Freeman GJ, and Ahmed R 2006. Restoring function in exhausted CD8 T cells during chronic viral infection. Nature 439:682–687. [DOI] [PubMed] [Google Scholar]
- Blank CU, Haining WN, Held W, Hogan PG, Kallies A, Lugli E, Lynn RC, Philip M, Rao A, Restifo NP, Schietinger A, Schumacher TN, Schwartzberg PL, Sharpe AH, Speiser DE, Wherry EJ, Youngblood BA, and Zehn D 2019. Defining ‘T cell exhaustion’. Nat Rev Immunol 19:665–674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brooks DG, Trifilo MJ, Edelmann KH, Teyton L, McGavern DB, and Oldstone MB 2006. Interleukin-10 determines viral clearance or persistence in vivo. Nat Med 12:1301–1309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Broz ML, Binnewies M, Boldajipour B, Nelson AE, Pollack JL, Erle DJ, Barczak A, Rosenblum MD, Daud A, Barber DL, Amigorena S, Van’t Veer LJ, Sperling AI, Wolf DM, and Krummel MF 2014. Dissecting the tumor myeloid compartment reveals rare activating antigen-presenting cells critical for T cell immunity. Cancer Cell 26:638–652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chow MT, Ozga AJ, Servis RL, Frederick DT, Lo JA, Fisher DE, Freeman GJ, Boland GM, and Luster AD 2019. Intratumoral activity of the CXCR3 chemokine system Is required for the efficacy of anti-PD-1 therapy. Immunity 50:1498–1512 e1495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dangi T, Chung YR, Palacio N, and Penaloza-MacMaster P 2020. Interrogating Adaptive Immunity Using LCMV. Curr Protoc Immunol 130:e99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Di Pilato M, Kfuri-Rubens R, Pruessmann J, Ozga A, Messemaker M, Cadilha B, Sivakumar R, Cianciaruso C, Warner R, Marangoni F, Carrizosa E, Lesch S, Billingsley J, Perez-Ramos D, Zavala F, Rheinbay E, Lustrer A, Gerner M, Kobold S, Pittet M, and Mempel T 2021. CXCR6 Positions Cytotoxic T cells to Receive Critical Survival Signals in the Tumor Microenvironment. Cell (in press) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duckworth BC, Lafouresse F, Wimmer VC, Broomfield BJ, Dalit L, Alexandre YO, Sheikh AA, Qin RZ, Alvarado C, Mielke LA, Pellegrini M, Mueller SN, Boudier T, Rogers KL, and Groom JR 2021. Effector and stem-like memory cell fates are imprinted in distinct lymph node niches directed by CXCR3 ligands. Nat Immunol 22:434–448. [DOI] [PubMed] [Google Scholar]
- Dufour JH, Dziejman M, Liu MT, Leung JH, Lane TE, and Luster AD 2002. IFN-gamma-inducible protein 10 (IP-10; CXCL10)-deficient mice reveal a role for IP-10 in effector T cell generation and trafficking. J Immunol 168:3195–3204. [DOI] [PubMed] [Google Scholar]
- Ejrnaes M, Filippi CM, Martinic MM, Ling EM, Togher LM, Crotty S, and von Herrath MG 2006. Resolution of a chronic viral infection after interleukin-10 receptor blockade. J Exp Med 203:2461–2472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Groom JR, and Luster AD 2011. CXCR3 ligands: redundant, collaborative and antagonistic functions. Immunol Cell Biol 89:207–215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Groom JR, Richmond J, Murooka TT, Sorensen EW, Sung JH, Bankert K, von Andrian UH, Moon JJ, Mempel TR, and Luster AD 2012. CXCR3 chemokine receptor-ligand interactions in the lymph node optimize CD4+ T helper 1 cell differentiation. Immunity 37:1091–1103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hancock WW, Lu B, Gao W, Csizmadia V, Faia K, King JA, Smiley ST, Ling M, Gerard NP, and Gerard C 2000. Requirement of the chemokine receptor CXCR3 for acute allograft rejection. J Exp Med 192:1515–1520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- He R, Hou S, Liu C, Zhang A, Bai Q, Han M, Yang Y, Wei G, Shen T, Yang X, Xu L, Chen X, Hao Y, Wang P, Zhu C, Ou J, Liang H, Ni T, Zhang X, Zhou X, Deng K, Chen Y, Luo Y, Xu J, Qi H, Wu Y, and Ye L 2016. Follicular CXCR5- expressing CD8(+) T cells curtail chronic viral infection. Nature 537:412–428. [DOI] [PubMed] [Google Scholar]
- Hickman HD, Reynoso GV, Ngudiankama BF, Cush SS, Gibbs J, Bennink JR, and Yewdell JW 2015. CXCR3 chemokine receptor enables local CD8(+) T cell migration for the destruction of virus-infected cells. Immunity 42:524–537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu JK, Kagari T, Clingan JM, and Matloubian M 2011. Expression of chemokine receptor CXCR3 on T cells affects the balance between effector and memory CD8 T-cell generation. Proc Natl Acad Sci U S A 108:E118–127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang Z, Zak J, Pratumchai I, Shaabani N, Vartabedian VF, Nguyen N, Wu T, Xiao C, and Teijaro JR 2019. IL-27 promotes the expansion of self-renewing CD8(+) T cells in persistent viral infection. J Exp Med 216:1791–1808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hudson WH, Gensheimer J, Hashimoto M, Wieland A, Valanparambil RM, Li P, Lin JX, Konieczny BT, Im SJ, Freeman GJ, Leonard WJ, Kissick HT, and Ahmed R 2019. Proliferating transitory T Cells with an effector-like transcriptional signature emerge from PD-1(+) stem-like CD8(+) T cells during chronic infection. Immunity 51:1043–1058 e1044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Im SJ, Hashimoto M, Gerner MY, Lee J, Kissick HT, Burger MC, Shan Q, Hale JS, Lee J, Nasti TH, Sharpe AH, Freeman GJ, Germain RN, Nakaya HI, Xue HH, and Ahmed R 2016. Defining CD8+ T cells that provide the proliferative burst after PD-1 therapy. Nature 537:417–421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Im SJ, Konieczny BT, Hudson WH, Masopust D, and Ahmed R 2020. PD-1+ stemlike CD8 T cells are resident in lymphoid tissues during persistent LCMV infection. Proc Natl Acad Sci U S A 117:4292–4299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jansen CS, Prokhnevska N, Master VA, Sanda MG, Carlisle JW, Bilen MA, Cardenas M, Wilkinson S, Lake R, Sowalsky AG, Valanparambil RM, Hudson WH, McGuire D, Melnick K, Khan AI, Kim K, Chang YM, Kim A, Filson CP, Alemozaffar M, Osunkoya AO, Mullane P, Ellis C, Akondy R, Im SJ, Kamphorst AO, Reyes A, Liu Y, and Kissick H 2019. An intra-tumoral niche maintains and differentiates stem-like CD8 T cells. Nature 576:465–470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khan IA, MacLean JA, Lee FS, Casciotti L, DeHaan E, Schwartzman JD, and Luster AD 2000. IP-10 Is Critical for Effector T Cell Trafficking and Host Survival in Toxoplasma gondii Infection. Immunity 12:483–494. [DOI] [PubMed] [Google Scholar]
- Khan O, Giles JR, McDonald S, Manne S, Ngiow SF, Patel KP, Werner MT, Huang AC, Alexander KA, Wu JE, Attanasio J, Yan P, George SM, Bengsch B, Staupe RP, Donahue G, Xu W, Amaravadi RK, Xu X, Karakousis GC, Mitchell TC, Schuchter LM, Kaye J, Berger SL, and Wherry EJ 2019. TOX transcriptionally and epigenetically programs CD8(+) T cell exhaustion. Nature 571:211–218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kurachi M, Kurachi J, Suenaga F, Tsukui T, Abe J, Ueha S, Tomura M, Sugihara K, Takamura S, Kakimi K, and Matsushima K 2011. Chemokine receptor CXCR3 facilitates CD8(+) T cell differentiation into short-lived effector cells leading to memory degeneration. J Exp Med 208:1605–1620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LaFleur MW, Nguyen TH, Coxe MA, Miller BC, Yates KB, Gillis JE, Sen DR, Gaudiano EF, Al Abosy R, Freeman GJ, Haining WN, and Sharpe AH 2019. PTPN2 regulates the generation of exhausted CD8(+) T cell subpopulations and restrains tumor immunity. Nat Immunol 20:1335–1347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Man K, Gabriel SS, Liao Y, Gloury R, Preston S, Henstridge DC, Pellegrini M, Zehn D, Berberich-Siebelt F, Febbraio MA, Shi W, and Kallies A 2017. Transcription factor IRF4 promotes CD8(+) T cell exhaustion and limits the development of memory-like T cells during chronic infection. Immunity 47:1129–1141 e1125. [DOI] [PubMed] [Google Scholar]
- McCausland MM, and Crotty S 2008. Quantitative PCR technique for detecting lymphocytic choriomeningitis virus in vivo. J Virol Methods 147:167–176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meiser A, Mueller A, Wise EL, McDonagh EM, Petit SJ, Saran N, Clark PC, Williams TJ, and Pease JE 2008. The chemokine receptor CXCR3 is degraded following internalization and is replenished at the cell surface by de novo synthesis of receptor. J Immunol 180:6713–6724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller BC, Sen DR, Al Abosy R, Bi K, Virkud YV, LaFleur MW, Yates KB, Lako A, Felt K, Naik GS, Manos M, Gjini E, Kuchroo JR, Ishizuka JJ, Collier JL, Griffin GK, Maleri S, Comstock DE, Weiss SA, Brown FD, Panda A, Zimmer MD, Manguso RT, Hodi FS, Rodig SJ, Sharpe AH, and Haining WN 2019. Subsets of exhausted CD8(+) T cells differentially mediate tumor control and respond to checkpoint blockade. Nat Immunol 20:326–336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muller U, Steinhoff U, Reis LF, Hemmi S, Pavlovic J, Zinkernagel RM, and Aguet M 1994. Functional role of type I and type II interferons in antiviral defense. Science 264:1918–1921. [DOI] [PubMed] [Google Scholar]
- Norris BA, Uebelhoer LS, Nakaya HI, Price AA, Grakoui A, and Pulendran B 2013. Chronic but not acute virus infection induces sustained expansion of myeloid suppressor cell numbers that inhibit viral-specific T cell immunity. Immunity 38:309–321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paley MA, Kroy DC, Odorizzi PM, Johnnidis JB, Dolfi DV, Barnett BE, Bikoff EK, Robertson EJ, Lauer GM, Reiner SL, and Wherry EJ 2012. Progenitor and terminal subsets of CD8+ T cells cooperate to contain chronic viral infection. Science 338:1220–1225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park MK, Amichay D, Love P, Wick E, Liao F, Grinberg A, Rabin RL, Zhang HH, Gebeyehu S, Wright TM, Iwasaki A, Weng Y, DeMartino JA, Elkins KL, and Farber JM 2002. The CXC chemokine murine monokine induced by IFN-gamma (CXC chemokine ligand 9) is made by APCs, targets lymphocytes including activated B cells, and supports antibody responses to a bacterial pathogen in vivo. J Immunol 169:1433–1443. [DOI] [PubMed] [Google Scholar]
- Rutishauser RL, Deguit CDT, Hiatt J, Blaeschke F, Roth TL, Wang L, Raymond KA, Starke CE, Mudd JC, Chen W, Smullin C, Matus-Nicodemos R, Hoh R, Krone M, Hecht FM, Pilcher CD, Martin JN, Koup RA, Douek DC, Brenchley JM, Sekaly RP, Pillai SK, Marson A, Deeks SG, McCune JM, and Hunt PW 2021. TCF-1 regulates HIV-specific CD8+ T cell expansion capacity. JCI Insight 6: [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sade-Feldman M, Yizhak K, Bjorgaard SL, Ray JP, de Boer CG, Jenkins RW, Lieb DJ, Chen JH, Frederick DT, Barzily-Rokni M, Freeman SS, Reuben A, Hoover PJ, Villani AC, Ivanova E, Portell A, Lizotte PH, Aref AR, Eliane JP, Hammond MR, Vitzthum H, Blackmon SM, Li B, Gopalakrishnan V, Reddy SM, Cooper ZA, Paweletz CP, Barbie DA, Stemmer-Rachamimov A, Flaherty KT, Wargo JA, Boland GM, Sullivan RJ, Getz G, and Hacohen N 2018. Defining T Cell states associated with response to checkpoint immunotherapy in melanoma. Cell 175:998–1013 e1020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sauty A, Colvin RA, Wagner L, Rochat S, Spertini F, and Luster AD 2001. CXCR3 internalization following T cell-endothelial cell contact: preferential role of IFN-inducible T cell alpha chemoattractant (CXCL11). J Immunol 167:7084–7093. [DOI] [PubMed] [Google Scholar]
- Scott AC, Dundar F, Zumbo P, Chandran SS, Klebanoff CA, Shakiba M, Trivedi P, Menocal L, Appleby H, Camara S, Zamarin D, Walther T, Snyder A, Femia MR, Comen EA, Wen HY, Hellmann MD, Anandasabapathy N, Liu Y, Altorki NK, Lauer P, Levy O, Glickman MS, Kaye J, Betel D, Philip M, and Schietinger A 2019. TOX is a critical regulator of tumour-specific T cell differentiation. Nature 571:270–274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shiow LR, Rosen DB, Brdickova N, Xu Y, An J, Lanier LL, Cyster JG, and Matloubian M 2006. CD69 acts downstream of interferon-alpha/beta to inhibit S1P1 and lymphocyte egress from lymphoid organs. Nature 440:540–544. [DOI] [PubMed] [Google Scholar]
- Siddiqui I, Schaeuble K, Chennupati V, Fuertes Marraco SA, Calderon-Copete S, Pais Ferreira D, Carmona SJ, Scarpellino L, Gfeller D, Pradervand S, Luther SA, Speiser DE, and Held W 2019. Intratumoral Tcf1(+)PD-1(+)CD8(+) T Cells with Stem-like Properties Promote Tumor Control in Response to Vaccination and Checkpoint Blockade Immunotherapy. Immunity 50:195–211 e110. [DOI] [PubMed] [Google Scholar]
- Sierro F, Biben C, Martinez-Munoz L, Mellado M, Ransohoff RM, Li M, Woehl B, Leung H, Groom J, Batten M, Harvey RP, Martinez AC, Mackay CR, and Mackay F 2007. Disrupted cardiac development but normal hematopoiesis in mice deficient in the second CXCL12/SDF-1 receptor, CXCR7. Proc Natl Acad Sci U S A 104:14759–14764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Utzschneider DT, Charmoy M, Chennupati V, Pousse L, Ferreira DP, Calderon-Copete S, Danilo M, Alfei F, Hofmann M, Wieland D, Pradervand S, Thimme R, Zehn D, and Held W 2016. T cell factor 1-expressing memory-like CD8(+) T cells sustain the immune response to chronic viral infections. Immunity 45:415–427. [DOI] [PubMed] [Google Scholar]
- Utzschneider DT, Gabriel SS, Chisanga D, Gloury R, Gubser PM, Vasanthakumar A, Shi W, and Kallies A 2020. Early precursor T cells establish and propagate T cell exhaustion in chronic infection. Nat Immunol 21:1256–1266. [DOI] [PubMed] [Google Scholar]
- Wherry EJ, Blattman JN, Murali-Krishna K, van der Most R, and Ahmed R 2003. Viral persistence alters CD8 T-cell immunodominance and tissue distribution and results in distinct stages of functional impairment. J Virol 77:4911–4927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wherry EJ, Ha SJ, Kaech SM, Haining WN, Sarkar S, Kalia V, Subramaniam S, Blattman JN, Barber DL, and Ahmed R 2007. Molecular signature of CD8+ T cell exhaustion during chronic viral infection. Immunity 27:670–684. [DOI] [PubMed] [Google Scholar]
- Wherry EJ, and Kurachi M 2015. Molecular and cellular insights into T cell exhaustion. Nat Rev Immunol 15:486–499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu T, Ji Y, Moseman EA, Xu HC, Manglani M, Kirby M, Anderson SM, Handon R, Kenyon E, Elkahloun A, Wu W, Lang PA, Gattinoni L, McGavern DB, and Schwartzberg PL 2016. The TCF1-Bcl6 axis counteracts type I interferon to repress exhaustion and maintain T cell stemness. Sci Immunol 1: [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zajac AJ, Blattman JN, Murali-Krishna K, Sourdive DJ, Suresh M, Altman JD, and Ahmed R 1998. Viral immune evasion due to persistence of activated T cells without effector function. J Exp Med 188:2205–2213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zander R, Schauder D, Xin G, Nguyen C, Wu X, Zajac A, and Cui W 2019. CD4(+) T Cell Help Is Required for the Formation of a Cytolytic CD8(+) T Cell Subset that Protects against Chronic Infection and Cancer. Immunity 51:1028–1042 e1024. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
NanoString data generated during this study, including genes counts pre-and post-normalization, as well as the raw nCounter data files (RCC files), have been deposited in the Gene Expression Omnibus (GEO) and are publicly available as of the date of publication. Accession number is listed in the key resources table. Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
Key Resources Table.
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| CXCL10 neutralizing mAb (IF11) | (Khan et al., 2000) | N/A |
| PDL1 blocking mAb (10F.9G2) | BioXcell | Cat# BE0101, RRID:AB_10949073 |
| CD4 depleting mAb (GK1.5) | BioXcell | Cat# BE003-1, RRID:AB_1107592 |
| CD8a (BUV395 conjugate) (53-6.7) | BD Bioscience | Cat# 563786, RRID:AB_2732919 |
| CD244.2 (Brilliant Violet 605™ conjugate) (2B4) | BD Bioscience | Cat# 740345, RRID:AB_2740078 |
| CD45.2 (Brilliant Violet 785™ conjugate) (104) | BD Bioscience | Cat# 563686, RRID:AB_2738375 |
| CD45.2 (Brilliant Violet 711™ conjugate) (104) | BD Bioscience | Cat# 563685, RRID:AB_2738374 |
| rat IgG2b (Brilliant Violet 711™ conjugate) (G15-337) | BD Bioscience | Cat# 743222, RRID:AB_2741358 |
| CD45.1 (Brilliant Violet BV421™ conjugate) (A20) | BD Bioscience | Cat# 563983, RRID:AB_2738523 |
| CD45.2 (Brilliant Violet BV421™ conjugate) (104) | BD Bioscience | Cat# 562895, RRID:AB_2737873 |
| Thy 1.1 (BUV395 conjugate) (53-2.1) | BD Bioscience | Cat# 740261, RRID:AB_2721773 |
| CD19 (BUV395 conjugate) (1D3/CD19) | BD Bioscience | Cat# 563557, RRID:AB_2722495 |
| NK1.1 (BUV395 conjugate) (PK136) | BD Bioscience | Cat# 564144, RRID:AB_2738618 |
| Ly-108 (BUV395 conjugate) (13G3) | BD Bioscience | Cat# 745730, RRID:AB_2743205 |
| Ly6G (Brilliant Violet 785™ conjugate) (1A8) | BD Bioscience | Cat# 740953, RRID:AB_2740578 |
| CXCR3 (Brilliant Violet BV421™ conjugate) (CXCR3-173) | BD Bioscience | Cat# 562937, RRID:AB_2687551 |
| Streptavidin (Brilliant Violet BV711™) | BD Biosciences | Cat# 563262, RRID:AB_2869478 |
| CD8a (Brilliant Violet 711™ conjugate) (53-6.7) | BD Biosciences | Cat# 563046, RRID:AB_2737972 |
| CD3 (Alexa Fluor® 700 conjugate) | BioLegend | Cat# 100216, RRID:AB_493697 |
| CD4 (Alexa 647 conjugate) (RM405) | BioLegend | Cat# 100530, RRID:AB_389325 |
| TIM3 (Brilliant Violet 421™ conjugate) (RMT3-23) | Biolegend | Cat# 119723, RRID:AB_2616908 |
| PD-1 (PE conjugate) (RMPI-30) | Biolegend | Cat# 109104, RRID:AB_313421 |
| PD-1 (PE/Cy7 conjugate) (RMP1-30) | BioLegend | Cat# 109110, RRID:AB_572017 |
| CX3CR1 (APC conjugate) (SA011F110) | BioLegend | Cat# 149008, RRID:AB_2564492 |
| CX3CR1 (Brilliant Violet BV605™ conjugate) (SA011F110) | BioLegend | Cat# 149027, RRID:AB_2565937 |
| CD8b (Alexa Fluor 488 conjugate) (YTS156.7.7) | BioLegend | Cat# 126628, RRID:AB_2800619 |
| CD8b (PE conjugate) (YTS156.7.7) | BioLegend | Cat# 126608, RRID:AB_961298 |
| BCL-2 (PE/Cy7) (BCL/10C4) | BioLegend | Cat# 633512, RRID:AB_2565247 |
| BCL-2 (PE) (BCL/10C4) | BioLegend | Cat# 633508, RRID:AB_2290367 |
| CD11c (Brilliant Violet BV605™ conjugate)(N418) | BioLegend | Cat# 117334, RRID:AB_2562415 |
| CD11b (Alexa Fluor™ 700 conjugate)(M1/70) | BioLegend | Cat# 101222, RRID:AB_493705 |
| XCR1 (APC conjugate) (ZET) | BioLegend | Cat# 148206, RRID:AB_2563932 |
| PODOPLANIN/GP38 (APC) (8.1.1) | BioLegend | Cat#127409, RRID:AB_10612940 |
| CD64 (PE/Cyanine7 conjugate) (X54-5/7.1) | BioLegend | Cat# 139313, RRID:AB_2563903 |
| F4/80 (Alexa488 conjugate) (BM8) | BioLegend | Cat# 123120, RRID:AB_893479 |
| F4/80 (APC conjugate) (BM8) | BioLegend | Cat# 123116, RRID:AB_893481 |
| F4/80 (Brilliant Violet BV711™ conjugate) (BM8) | BioLegend | Cat# 123147, RRID:AB_2564588 |
| Ly108 (APC conjugate) (330-AJ) | BioLegend | Cat# 134610, RRID:AB_2728155 |
| Ly108 (Pacific Blue™ conjugate) (330-AJ) | BioLegend | Cat# 134608, RRID:AB_2188093 |
| Ly6G (PerCP/Cyanine5.5 conjugate) (1A8) | BioLegend | Cat# 127616, RRID:AB_1877271 |
| Ly6C (Brilliant Violet 785™ conjugate) (HK1.4) | BioLegend | Cat# 128041, RRID:AB_2565852 |
| CD69 (Brilliant Violet 421™ conjugate) (H1.2F3) | BioLegend | Cat# 104545, RRID:AB_2686969 |
| TruStain fcX™ (anti-mouse CD16/32) (93) | BioLegend | Cat# 101320, RRID:AB_1574975 |
| CD45 (purified) (30-F11) | BioLegend | Cat# 103102, RRID:AB_312967 |
| TNF alpha (APC conjugate) (MP6-XT22) | BioLegend | Cat# 506308, RRID:AB_315429 |
| IFN-gamma (Brilliant Violet 421™ conjugate) (XMG1.2) | BioLegend | Cat# 505830, RRID:AB_2563105 |
| CD44 (Brilliant Violet 785™) (IM7) | BioLegend | Cat# 103059, RRID:AB_2571953 |
| CXCR3 (FITC conjugate) (CXCR3-173) | BioLegend | Cat# 126536, RRID:AB_2566565 |
| CXCR3 (PerCP/Cyanine5.5) (CXCR3-173) | BioLegend | Cat# 126514, RRID:AB_1186015 |
| T-bet (PE/Cy7 conjugate) (4B10) | BioLegend | Cat# 644824, RRID:AB_2561761 |
| CD45.2 (PE conjugate) (104) | BioLegend | Cat# 109808, RRID:AB_313445 |
| TCF1/TCF7 Rabbit mAb (Pacific BlueTM conjugate) (clone C63D9) | Cell Signaling Technology | Cat# 9066 RRID: AB_2797696 |
| TCF1/TCF7 Rabbit mAb (PE Conjugate) (clone C63D9) | Cell Signaling Technology | Cat# 14456S, RRID:AB_2798483 |
| TCF1/TCF7 Rabbit mAb (Alexa Fluor 488 Conjugate) (clone C63D9) | Cell Signaling Technology | Cat# 6444S, RRID:AB_2797627 |
| TCF1/TCF7 Rabbit mAb (Alexa Fluor 647® Conjugate) (Clone C63D9) | Cell Signaling Technology | Cat# 6709S, RRID:AB_2797631 |
| TCF1/TCF7 Rabbit mAb (purified) (Clone C63D9) | Cell Signaling Technology | Cat# 2203S, RRID:AB_2199302 |
| CD44 (FITC conjugate) (5035-41.1 D] | GeneTex | Cat# GTX74574, RRID:AB_378218 |
| Normal goat serum | Jackson ImmunoResearch | Cat# 005-000-121, RRID:AB_2336990 |
| XCR1 (VioBright FITC conjugate) (REA707) | Miltenyi Biotec | Cat# 130-111-378, RRID:AB_2654280 |
| IRF-4 (PE, REAfinityTM) (REA201) | Miltenyi Biotec | Cat# 130-100-917, RRID:AB_2652515 |
| H-2D(b) LCMV gp 33-41 KAVYNFATC Alexa 488 | NIH Tetramer Core Facility | N/A |
| H-2D(b) LCMV GP 33-41 KAVYNFATC (APC conjugate) | NIH Tetramer Core Facility | N/A |
| H-2D(b) LCMV GP 276-286 SGVENPGGYCL (APC conjugate) | NIH Tetramer Core Facility | N/A |
| CCR2 (APC conjugate) (475301) | R and D Systems | Cat# FAB5538A, RRID:AB_10645617 |
| CD86 (FITC conjugate) (GL1) | Thermo Fisher Scientific | Cat# 11-0862-82, RRID:AB_465148 |
| CD64 (purified) (27) | Thermo Fisher Scinetific | Cat# MA5-29706, RRID:AB_2785530 |
| CD101 (purified) (polyclonal) | Thermo Fisher Scientific | Cat# 26047-1-AP, RRID:AB_2880350 |
| CD101 (PE/Cy7 conjugate) (Moushi 101) | Thermo Fisher Scientific | Cat# 25-1011-82, RRID:AB_2573378 |
| CD274 (Alexa Fluor 488) (MIH5) | Thermo Fisher Scientific | Cat# 53-5982-82, RRID:AB_2811871 |
| PD-1 (FITC conjugate) (RMP1-30) | Thermo Fisher Scientific | Cat# 11-9981-82, RRID:AB_465467 |
| TNF alpha (FITC conjugate) (MP6-XT22) | Thermo Fisher Scientific | Cat# 11-7321-41, RRID:AB_10670212 |
| GFP Polyclonal Antibody, (Alexa Fluor 488 conjugate) | Thermo Fisher Scientific | Cat# A-21311, RRID:AB_221477 |
| IRF1 (purified) (SR44-08) | Thermo Fisher Scientific | Cat# MA5-31996, RRID:AB_2809290 |
| ER-TR7 (purified) (ER-TR7) | Thermo Fisher Scientific | Cat# MA1-40076, RRID:AB_1074409 |
| Anti-rat IgG (H+L) Highly Cross-Adsorbed (Alexa Fluor 488 conjugate) | Thermo Fisher Scientific | Cat# A11006, RRID:AB_141373 |
| Anti-rat IgG (H+L) Highly Cross-Adsorbed (Alexa Fluor 647 conjugate) | Thermo Fisher Scientific | Cat#A21247, RRID:AB_141778 |
| Anti-rabbit IgG (H+L) Highly Cross-Adsorbed (Alexa Fluor 488 conjugate) | Thermo Fisher Scientific | Cat# MA5-29706, RRID:AB |
| anti-rabbit IgG (H+L) Highly Cross-Adsorbed (Alexa Fluor 647 conjugate) | Thermo Fisher Scientific | Cat# A-21245, RRID:AB_2535813 |
| CD127 (biotin conjugate) (A7R34) | Thermo Fisher Scientific | Cat# 13-1271-82, RRID:AB_466588 |
| Bacterial and Virus strain | ||
| LCMV Armstrong (Arm) | kind gift of J. Wherry | Grew up in house |
| LCMV Cl-13 (Cl-13) | kind gift of J. Wherry | Grew up in house |
| Chemicals, Peptides, and Recombinant Proteins | ||
| LCMV gp33 – 41, GP33, (KAVYNFATM) | AnaSpec | Cat# AS-61296 |
| LCMV gp276 – 286, GP276, (SGVENPGGYCL) | AnaSpec | Cat# AS-62539 |
| BD GolgiStop™ (Containing Monensin) | BD Bioscience | Cat# 554274 |
| BD Golgi Plug™ (Containing Brefeldin A) | BD Bioscience | Cat# 555029 |
| 16% Paraformaldehyde Aqueous Solution | Electron Microscopy Sciences | Cat# 50-980-487 |
| Percoll | GE Healthcare | Cat# 17-0891-01 |
| Recombinant murine IL-12 protein | Peprotech | Cat# 210-12P40H |
| Recombinant murine IL-2 protein | Peprotech | Cat# 212-12 |
| Recombinant mouse CXCL10 protein | Peprotech | Cat# 250-16 |
| Liberase TM Research Grade | Sigma Aldrich | Cat# 5401119001 |
| DNAse I, grade II, from bovine pancreas | Sigma Aldrich | Cat# 10104159001 |
| TRIzol Reagent | Thermo Fisher Scientific | Cat# 15596018 |
| MultiScribe Reverse Transcriptase | Thermo Fisher Scientific | Cat# 4311235 |
| GeneAMP dNTP mix with dTTP | Thermo Fisher Scientific | Cat# N8080260 |
| MgCl2 Solution 25 mM | Thermo Fisher Scientific | Cat# 4486224 |
| 10x PCR Buffer II | Thermo Fisher Scientific | Cat# 4486220 |
| Oligio d(T)16 | Thermo Fisher Scientific | Cat# 100023441 |
| Random Hexamers 50 mM | Thermo Fisher Scientific | Cat# 100026484 |
| RNase Inhibitor | Thermo Fisher Scientific | Cat# 100021540 |
| Fixable Viability Dye eFluor 780 | Thermo Fisher Scientific | Cat# 65-0865-14 |
| Glycogen RNA grade | ThermoFisher Scientific | Cat# R0551 |
| ProLong Diamond Antifade Mountant | ThermoFisher Scientific | Cat# P36961 |
| OCT Compound | Tissue-Tek | Cat# 4583 |
| Critical Commercial Assays | ||
| nCounter® Mouse Immunology Panel (CodeSet Only) | NanoString | Cat# XT-CSO-MIM1-12 |
| QIAMP Viral RNA Mini kit | Qiagen | Cat# 52906 |
| QuantiFast SYBR Green RT-qPCR Kit | Qiagen | Cat# 204154 |
| FastStart Essential DNA Green Master | Roche | Cat# 25595200 |
| EasySep™ Mouse Naïve CD8+ T Cell Isolation Kit | Stem Cell Technologies | Cat# 19858 |
| FOXP3/Transcription Factor staining Kit | Thermo Fisher Scientific | Cat# 00-5523-00 |
| DNA-free™ DNA Removal Kit | Thermo Fisher Scientific | Cat# AM1906 |
| Click-iT™ Plus EdU Pacific Blue™ Flow Cytometry Assay Kit | Thermo Fisher Scientific | Cat# C10636 |
| Live/dead fixable viability dye eFluorTM 780 | Thermo Fisher Scientific | Cat# 65-0865-18 |
| Image-iT FX Signal Enhancer | Cell Signaling Technology | Cat# 11932S |
| BD Cytofix/Cytoperm | BD Bioscience | Cat# 554714 |
| CellTrace™ Violet Cell Proliferation Kit | Thermo Fisher Scientific | Cat# C34571 |
| Experimental Models: Organisms/Strains | ||
| Mouse: B6.129S7-Ifngr1tm1A9t/J | The Jackson Laboratory | Cat# 3288 |
| Mouse: C57BL/6-Tg(CAG-EGFP)131Osb/LeySopJ | The Jackson Laboratory | Cat# 006567 |
| Mouse: Cxcl10−/− | (Dufour et al., 2002) | N/A |
| Mouse: Cxcl9−/− | (Park et al., 2002) | N/A |
| Mouse: Cxcr3−/− | (Hancock et al., 2000) | N/A |
| Mouse: ifnar−/− | Müller et al., 1994 | Cat# N/A |
| Mouse: REX3 | Groom et al., 2012 | Cat# N/A |
| Mouse: WT: B6.SJL-Ptprca Pepcb/BoyJ | The Jackson Laboratory | Cat# 002014 |
| Mouse: WT: C57BL6 NCI | Charles River Laboratory | Cat# 027 |
| Mouse: WT: C57BL6/J | The Jackson Laboratory | Cat# 664 |
| Mouse: WT: CD45.1 | Charles River Lab - NCI Grantee Pricing’ | Cat# 564 |
| Oligonucleotides | ||
| Il10 | IDT technologies | N/A |
| forward; 5’ – GCT CTT ACT GAC TGG CAT GAG -3’ | ||
| reverse 5’- CGC AGC TCT AGG AGC ATG TG -3’ | ||
| Il12p40 | IDT technologies | N/A |
| forward 5’-TGG TTT GCC ATC GTT TTG CTG -3’ | ||
| reverse 5’- ACA GGT GAG GTT CAC TGT TTC T -3’ | ||
| Cd86 | IDT technologies | N/A |
| forward 5’- TGT TTC CGT GGA GAC GCA AG -3’ | ||
| reverse 5’ -TTG AGC CTT TGT AAA TGG GCA -3’ | ||
| Cxcl10 | IDT technologies | N/A |
| forward; 5’-GCCGTCATTTTCTGCCTCA-3’ | ||
| reverse 5’-CGTCCTTGCGAGAGGGATC-3’ | ||
| Cxcl9 | IDT technologies | N/A |
| forward; 5’-AATGCACGATGCTCCTGCA-3’ | ||
| reverse 5’-AGGTCTTTGAGGGATTTGTAGTGG-3’ | ||
| Pdl1 | IDT technologies | N/A |
| forward; 5’-GCT CCA AAG GAC TTG TAC GTG -3’ | ||
| reverse 5’- TGA TCT GAA GGG CAG CAT TTC -3’ | ||
| Gp | IDT technologies | N/A |
| forward; 5’-CATTCACCTGGACTTTGTCAGACTC-3’ | ||
| reverse GCAACTGCTGTGTTCCCGAAAC | ||
| Gapdh | IDT technologies | N/A |
| forward; 5’-GGCAAATTCAACGGCACAGT-3’ | ||
| reverse 5’-AGATGGTGATGGGCTTCCC-3’ | ||
| Np | IDT technologies | N/A |
| forward; 5’-CAGAAATGTTGATGCTGGACTGC-3’ | ||
| reverse 5’-CAGACCTTGGCTTGCTTTACACAG-3’ | ||
| Mm_Ifng_2_SG cc Assay | Qiagen | Cat# QT02423428 |
| Mm_Ifnb1_1_SG QuantiTect Primer Assay | Qiagen | Cat# QT00249662 |
| Software | ||
| BD FACS Diva v8 | BD Biosciences | https://www.bdbiosciences.com/us/instruments/clinical/software/flow-cytometry-acquisition/bd-facsdiva-software/m/333333/overview; RRID: SCR_001456 |
| Biorender | Biorender | http://biorender.com;RRID:SCR_018361 |
| Fiji | Fiji | http://fiji.sc; RRID:SCR_002285 |
| GraphPad Prism Version 8 | GraphPad | https://www.graphpad.com; RRID: SCR_002798 |
| nSolver Analysis Software 4.0 | nSolver Analysis Software | http://www.nanostring.com/products/nSolver; RRID:SCR_003420 |
| Light Cycler 96 | Roche | http://www.roche-applied-science.com/shop/products/absolute-quantification-with-the-lightcycler-carousel-based-system; RRID:SCR_012155 |
| FlowJo v10 | Tree Star | https://www.flowjo.com/solutions/flowjo/downloads; RRID: SCR_008520 |
| Zen Black 2012 v8 | Zeiss | https://www.zeiss.com/microscopy/us/products/microscope-software/zen.html; RRID: SCR_013672 |
| Recombinant DNA | ||
| pCITE-GP plasmid | J. de la Torre | N/A |