

Abstract
People living with human immunodeficiency virus (PLWH) have significantly increased risk for cardiovascular disease in part due to inflammation and immune dysregulation. Clonal hematopoiesis of indeterminate potential (CHIP), the age-related acquisition and expansion of hematopoietic stem cells due to leukemogenic driver mutations, increases risk for both hematologic malignancy and coronary artery disease (CAD). Since increased inflammation is hypothesized to be both a cause and consequence of CHIP, we hypothesized that PLWH have a greater prevalence of CHIP. We searched for CHIP in multi-ethnic cases from the Swiss HIV Cohort Study (SHCS, n = 600) and controls from the Atherosclerosis Risk in the Communities study (ARIC, n = 8111) from blood DNA-derived exome sequences. We observed that HIV is associated with a twofold increase in CHIP prevalence, both in the whole study population and in a subset of 230 cases and 1002 matched controls selected by propensity matching to control for demographic imbalances (SHCS 7%, ARIC 3%, p = 0.005). We also observed that ASXL1 is the most commonly mutated CHIP-associated gene in PLWH. Our results suggest that CHIP may contribute to the excess cardiovascular risk observed in PLWH.
Subject terms: Immunology, Infectious diseases, HIV infections, Population genetics, Genomics
Introduction
As current treatments have rendered human immunodeficiency virus (HIV) a chronic condition, coronary artery disease has emerged as a major source of morbidity in people living with human immunodeficiency virus (PLWH). Inflammation and immune dysregulation likely accelerate CAD risk among PLWH1. Recently, ‘clonal hematopoiesis of indeterminate potential’ (CHIP), the age-related acquisition and expansion of leukemogenic mutations (primarily in DNMT3A, TET2, ASXL1, JAK2) in white blood cells, was found to increase risk for both hematologic malignancy2,3 and CAD4,5 among asymptomatic individuals in the general population. The proatherogenic mechanisms for CHIP included heightened inflammation4,6. Given converging proposed mechanisms promoting CAD risk and increased hematologic malignancy risk among PLWH, we tested the hypothesis that HIV-infected individuals have heightened prevalence of CHIP.
Methods
We identified CHIP in a multi-ethnic sample of 600 PLWH from the Swiss HIV Cohort Study (SHCS), aged 21–83. The SHCS is a multicenter, prospective observational study for interdisciplinary HIV research7. Established in 1988, the SHCS currently comprises more than 20,000 PLWH with median 51 years of age. Samples of 600 patients, used for exome sequencing, were chosen randomly in terms of gender (genetic sex, gender at birth), age, category of transmission, as well as HIV management and control8.
We utilized a set of 8111 individuals with available exome sequences from the Atherosclerotic Risk in the Community study (ARIC), aged 45–84 years, as population controls9. The ARIC study is a prospective longitudinal investigation of the development of atherosclerosis and its clinical sequelae which enrolled 15,792 individuals aged 45 to 64 years at baseline10. At study enrollment (1987–1989), the participants were selected by probability sampling from four United States communities: Forsyth County, North Carolina; Jackson, Mississippi; the northwestern suburbs of Minneapolis, Minnesota; and Washington County, Maryland.
Exome capture kits used in the compared cohorts were different (SHCS: xGen Exome Research Panel v 1.0, Sureselect All Exon V5 and TruSeq DNA Exome, ARIC: HGSC VCRome 2.1 design (42 Mb, NimbleGen)), which is the major limitation of the study. To minimise these differences we performed a set of statistical analysis to normalize the coverage among cohorts (see below the adjusting for the depth of sequencing and table S4).
CHIP was called in both exome sequenced cohorts using an identical and previously described pipeline4,11. Briefly, short read sequence data were aligned to the hg19 reference genome using the BWA-mem algorithm and processed with the Genome Analysis Toolkit MuTect2 tool to detect somatic variants12. To identify individuals with CHIP, we used a pre-specified list of variants in 74 genes known to be recurrent drivers of myeloid malignancies (Table S3) and variant filtration process (> = 20 reads in total, > = 3 Alt reads including one on a forward and one on a reverse strand, VAF limit > 2%) which filter in the biologically relevant cases13.
As CHIP prevalence depends strongly on age, we performed a 1:5 case/control propensity matching on age, sex and self-reported ethnicity using nearest neighbor matching14 as implemented by the MatchIt package version 3.0.2 in R. Next, we used univariate Fisher’s exact test and multivariable logistic regression to test the association between HIV status and CHIP prevalence. Multivariable models were adjusted for age, sex, self-reported ethnicity, and smoking status.
To take into account potential difference in the depth of sequencing of the CHIP-associated genes between the matched cohorts, we used a backward stepwise multiple logistic model, describing CHIP status (0/1) as a function of cohort (0/1) and coverage of the four most common CHIP-associated genes (DNMT3A, TET2, ASXL1 and JAK2). Analyses were performed in R version 3.6. A threshold of p < 0.05 was considered statistically significant.
The Swiss HIV Cohort Study was approved by the local ethical committees of the participating centres: Ethikkommission beider Basel ("Die Ethikkommission beider Basel hat die Dokumente zur Studie zustimmend zur Kenntnis genommen und genehmigt."); Kantonale Ethikkommission Bern (21/88); Comité départemental d'éthique des spécialités médicales et de médecine communautaire et de premier recours, Hôpitaux Universitaires de Genève (01–142); Commission cantonale d'éthique de la recherche sur l'être humain, Canton de Vaud (131/01); Comitato etico cantonale, Repubblica e Cantone Ticino (CE 813); Ethikkommission des Kantons St. Gallen (EKSG 12/003); Kantonale Ethikkommission Zürich (KEK-ZH-NR: EK-793), and written informed consent was obtained from all participants. Secondary analysis of the SHCS data in this manuscript was covered by the original approvals mentioned above, and secondary analysis of ARIC was approved by the Mass General Brigham Institutional Review Board.
All methods were performed in accordance with the relevant guidelines and regulations.
Results
First, we compared the prevalence of CHIP across the entire SHCS PLWH cohort (N = 600) and ARIC cohort (N = 8111) (Fig. 1). SHCS PLWH and ARIC participants had mean (SD) age 44 (11) and 57 (6) years (p = 1.8 × 10–167), were 25% and 56% female (p = 1.9 × 10−46), and were 95% and 74% of European ancestry (p = 5.2 × 10−36) respectively. With adjustment for age, age2, sex and self-reported ethnicity, we observed a significant association between HIV case status and CHIP (OR: 1.77, 95% CI: 1.33–2.21, p = 0.02).
Figure 1.
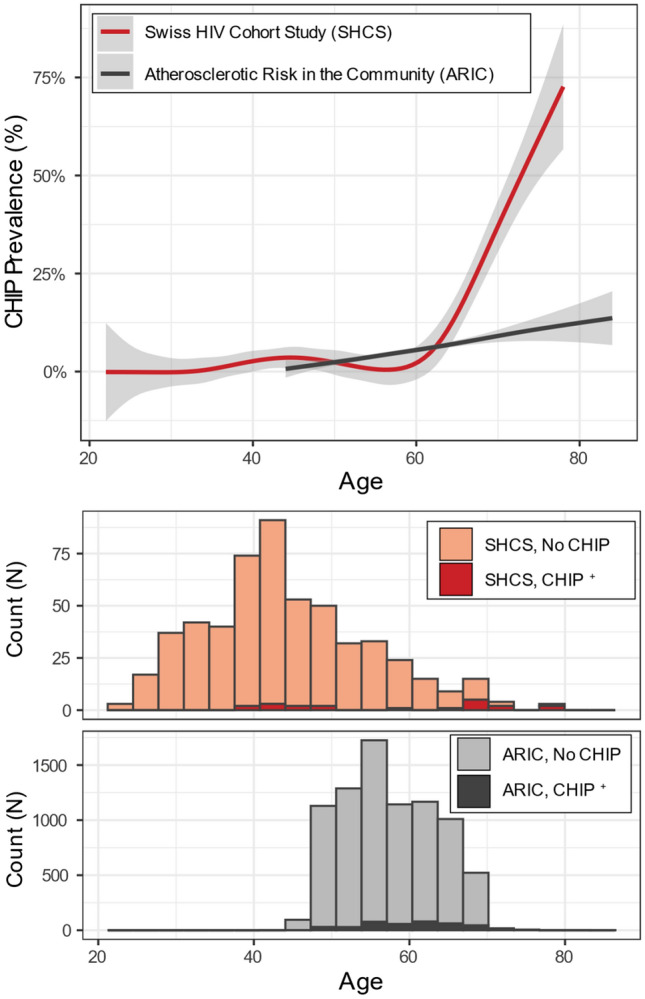
CHIP prevalence in Swiss HIV Cohort Study and Atherosclerotic Risk in the Community Study. Upper panel: fraction of cohort observed to have CHIP over time fit with a general additive model spline. 95% confidence interval displayed as shaded area. Lower panel: Count of number of individuals with and without CHIP binned by age of time of blood sampling across the entire sequenced cohort. Complete datasets (SHSC N = 600, ARIC N = 8111).
Second, given the overall demographic imbalances, to confirm an excess of CHIP amongst SHCS under all else equal, we used a propensity matching strategy to match the two cohorts by age, gender, self-reported ethnicity and smoking status (ever-smoker or not). Propensity matching analyses yielded a set of 230 (out of 600) PLWH cases and 1002 (out of 8111) ARIC population controls. Neither age nor sex differed significantly between the matched cohorts (Table 1) and the standardized mean difference across age, sex and self-reported ethnicity were all less than 0.1 indicative of adequate matching. In this subset, CHIP was detected in 7% of exomes from PLWH, but only 3% of the controls (Table 1, univariate p = 0.005; multivariable p = 0.004). Of note, the statistical association strengthened despite a significantly decreased sample size, demonstrating the robustness of our matching approach.
Table 1.
Demographics and CHIP association in matched samples.
| n | HIV + Individuals (SHCS) | Population Controls (ARIC) | p-value |
|---|---|---|---|
| 230 | 1002 | ||
| Age at blood draw, mean (st. dev.) | 54.2 (7.4) | 55.0 (6.8) | 0.12 |
| Female, N (%) | 44 (19%) | 240 (24%) | 0.086 |
| Ever smoker, N (%) | 143 (62%) | 651 (65%) | 0.408 |
| Diabetes mellitus, N (%) | 18 (8%) | 80 (8%) | 0.936 |
| Black, N (%) | 7 (3%) | 80 (8%) | 0.017 |
| CHIP carrier, N (%) | 16 (7%) | 30 (3%) | 0.005 |
P-value derived from Fisher's exact test for counts and t-test for continuous variables.
Third, we tested if sequencing coverage differs between the matched sub cohorts. Comparing the average coverage of the four most common CHIP genes (DNMT3A, TET2, ASXL1, JAK2) between the matched SHCS and ARIC sub cohorts we observed higher coverage in SHCS (median coverages are 66 and 47 reads per nucleotide for SHCS and ARIC correspondingly, p < 2 * 10−16 Mann–Whitney U test). Increased sequence coverage in SHCS can facilitate CHIP discovery in SHCS and to take into account the differences in the coverage we performed a multivariable logistic regression analysis with the included depth of sequencing. Inclusion into the models the total coverage of the four most common CHIP genes (DNMT3A, TET2, ASXL1, JAK2) as well as individual coverage of those genes demonstrated non-significant effect of the coverage (models 2 and 3 in table S4), confirming the robustness of our main finding—an excess of CHIP amongst PLWH (model 1 in table S4).
The limited sample size precluded inference on the association of HIV status with specific CHIP driver genes, however we observed differences in the genes most likely to carry CHIP mutations between PLWH (table S1) and population controls (table S2). The most common CHIP gene in the SHCS was ASXL1 (13 out of 27 CHIP mutations, 48%) followed by TET2 (8 out of 27 CHIP mutations, 30%) and DNMT3A (5 out of 27 CHIP mutations, 19%). Overall this distribution was inverted from the control cohort where CHIP mutations were more frequent in DNMT3A (14 out of 28 CHIP mutations, 50%), followed by TET2 (5 out of 28 CHIP mutations, 18%) and ASXL1 (5 out of 28 CHIP mutations, 18%). In total, 22 PLWH had a single CHIP mutation, while one individual had 2 mutations and one individual had 3 mutations, while in the ARIC cohort all CHIP carriers had a single CHIP mutation (tables S1 and S2). Additionally, we compared VAFs between matched ARIC and SHCS CHIP carriers (tables S1 and S2) and observed a trend of increased VAF in ARIC (28 CHIP mutations in ARIC and 14 CHIP mutations in SHCS: 12 patients among which one patient has 3 CHIP variants, p-value = 0.026, Mann–Whitney U test).
Within the full PLWH cohort (N = 600) we considered additional phenotypes, which might be a cause or consequence of CHIP. First, we observed a trend toward an increase in CAD among CHIP carriers (Fisher’s exact test OR: 2.99, p = 0.068) and increased cases of diabetes among CHIP carriers (Fisher’s exact test OR: 3.76, p = 0.037) (see the patient-specific information in Table S1). Second, we observed that duration of antiretroviral therapy (ART) was twice as long in CHIP carriers versus non-carriers (ART mean [st. dev.] 2675 [1850] days vs. 1322 [1454] days in carriers vs. non-carriers, respectively; p = 0.0004, Mann–Whitney U test). This association was directionally concordant after adjusting for patient age in multiple logistic regression (p = 0.066). It is important to note that although ART duration positively correlated with the total duration of HIV infection (Spearman’s rho = 0.58, p = 2.0 × 10−54), the total duration of HIV infection was not associated with CHIP (p = 0.452; paired Mann–Whitney U test on matched CHIP carriers and non-carriers, p = 0.22).
Discussion
Here, we report that HIV infection is associated with increased prevalence of CHIP. In the present samples, we identify a more than twofold enrichment of CHIP among PLWH versus controls after careful adjustment for known factors predisposing to CHIP (age, smoking status, ethnicity, gender). Although PLWH and controls originate from different cohorts, identical bioinformatics pipelines were used for the identification of CHIP-associated variants, and the statistical matching of cohorts and multiple logistic models controlling for the gene coverage (see Methods) assure robustness of our results. Of note, a very similar twofold excess of CHIP among PLWH has been described recently in an independent study from Australia15. Altogether, we demonstrate that HIV infection is the second strongest factor, after age, associated with increased CHIP prevalence among PLWH.
Our finding is based on cross‐sectional design and thus we cannot infer causality. However, assuming that CHIP is highly unlikely to be a risk factor for HIV acquisition, we focus on potential mechanisms that could promote CHIP development among PLWH. HIV infection or, more generally, HIV-related factors may promote CHIP development either through an increased rate of occurrence of CHIP-associated mutations and/or increased rate of clonal expansion of these somatic variants.
An increased somatic mutational rate in PLWH up to date has been shown only for the mitochondrial genome, which is particularly sensitive to some antiretroviral therapies16–19. However, taking into account several potential factors such as: a mutagenic effect of the virus, DNA-replication errors associated with an increased turnover rate of hematopoietic stem cells, mutagenic effects of antiretroviral therapy and other HIV-specific confounders (such as tobacco smoke, the effect of which has been controlled in the current study) we can not rule out an increased rate of somatic mutagenesis in the nuclear genome of PLWH.
A potentially higher rate of occurrence of somatic mutations alone is unlikely to provide a comprehensive explanation of increased CHIP prevalence among PLWH. A recent study, performing accurate detection of rare (with variant allele frequency higher than or equals to 0.0003) CHIP-associated mutations, demonstrated that such CHIP variants are nearly universal in healthy individuals by the age of 50 and often stable longitudinally20, showing that for the majority of people, decades elapse between the acquisition of a CHIP-associated mutation and CHIP itself. Thus, an understanding of the rate of clonal expansion of initially rare CHIP-associated variants is of great importance to shed light on the excess of CHIP among PLWH. A recent model proposed that many of the CHIP-associated mutations increase cell fitness, ensuring their proliferation with age21. Thus, HIV infection may modify the fitness landscape of CHIP-associated mutations, accelerating their clonal expansion and thus providing a fertile substrate for CHIP development. Various HIV-related mechanisms may be responsible for this, including induced immunodeficiency, increased prevalence of tobacco smoking and other comorbid conditions, as well as chronic immune activation from antigenic stimulation. Indeed, it has been recently shown that mutations in both the most common CHIP-associated genes DNMT3A22 and TET223 are getting selective advantage in case of chronic infection. According to our results, ART could additionally induce CHIP development, however an elucidation of the mechanisms as well as relative contribution of different HIV-specific factors to CHIP risk requires future studies.
The relationship we identify between HIV and CHIP may be a mechanistic basis of shared phenotypes. For example, recent study showed that HIV infection leads to a greater risk of myelodysplastic syndrome (MDS), a downstream consequence of CHIP and precursor to myeloid malignancy24. Furthermore, similar to the gene distribution in MDS, we find a greater relative prevalence of ASXL1 mutations among PLWH compared to controls. Of note, while cigarette smoking selects for ASXL1 clonal hematopoiesis25, our cohort of PLWH still had an increased prevalence of ASXL1 mutations compared to the control cohort despite being matched for smoking status. Another shared phenotype is an increased risk for cardiovascular disease. We propose that CHIP may be one mechanism that elevates risk for CAD among PLWH and further studies are required to evaluate this hypothesis.
PLWH have accelerated biologic aging. CHIP detection may represent a new opportunity for identification of at-risk patients with particular relevance for HIV medicine. Conversely, PLWH may provide a rich source of information to understand mechanisms of clonal expansion of different CHIP-associated variants under long-term low-grade inflammation.
Supplementary Information
Acknowledgements
SHCS data are gathered by the Five Swiss University Hospitals, two Cantonal Hospitals, 15 affiliated hospitals and 36 private physicians (listed in http://www.shcs.ch/180-health-care-providers). The authors acknowledge the effort and commitment of SHCS participants, investigators, study nurses, laboratory personnel, and administrative assistance by the SHCS coordination and data center.
Author contributions
J.F. and P.N. designed the study. A.B., K.P. and C.T. performed main statistical analyses. All authors prepared the manuscript.
Funding
This study has been financed within the framework of the Swiss HIV Cohort Study, supported by the Swiss National Science Foundation (Grant #177499), by SHCS project #860 and by the SHCS research foundation. This work was also supported by the Swiss National Science Foundation Grant #175603 to JF. AGB is supported by a Burroughs Wellcome Foundation career award for medical scientists and a grant from the National Institute of Health Common Fund (DP5 OD029586). SG is supported by P30 DK040561 and U01HL123336. PN is supported by a Hassenfeld Scholar Award from the Massachusetts General Hospital, and grants from the National Heart, Lung, and Blood Institute (R01HL1427, R01HL148565, R01HL148050, and R01HL151283) and Fondation Leducq (TNE-18CVD04). Dr. Libby receives funding support from the National Heart, Lung, and Blood Institute (1R01HL134892), the American Heart Association (18CSA34080399), the RRM Charitable Fund, and the Simard Fund. The Atherosclerosis Risk in Communities study has been funded in whole or in part with Federal funds from the National Heart, Lung, and Blood Institute, National Institutes of Health, Department of Health and Human Services (Contract Numbers HHSN268201700001I, HHSN268201700002I, HHSN268201700003I, HHSN268201700004I and HHSN268201700005I). The authors thank the staff and participants of the ARIC study for their important contributions. Funding support for “Building on GWAS for NHLBI-diseases: the U.S. CHARGE consortium” was provided by the NIH through the American Recovery and Reinvestment Act of 2009 (ARRA) (5RC2HL102419). Sequencing was carried out at the Baylor College of Medicine Human Genome Sequencing Center (U54 HG003273 and R01HL086694).
Data availability
CHIP-associated genetic variant callsets and associated participant level phenotype data used in this study are available to qualified investigators by application to the SHCS and ARIC.
Competing interests
Dr. Libby is an unpaid consultant to, or involved in clinical trials for Amgen, AstraZeneca, Baim Institute, Beren Therapeutics, Esperion, Therapeutics, Genentech, Kancera, Kowa Pharmaceuticals, Medimmune, Merck, Norvo Nordisk, Merck, Novartis, Pfizer, Sanofi-Regeneron. Dr. Libby is a member of scientific advisory board for Amgen, Corvidia Therapeutics, DalCor Pharmaceuticals, Kowa Pharmaceuticals, Olatec Therapeutics, Medimmune, Novartis, and XBiotech, Inc. Dr. Libby’s laboratory has received research funding in the last 2 years from Novartis. Dr. Libby is on the Board of Directors of XBiotech, Inc. Dr. Libby has a financial interest in Xbiotech, a company developing therapeutic human antibodies. Dr. Libby's interests were reviewed and are managed by Brigham and Women's Hospital and Partners HealthCare in accordance with their conflict of interest policies. Dr Natarajan reported investigator-initiated grants from Amgen, Apple, AstraZeneca, Boston Scientific, and Novartis, personal fees from Apple, AstraZeneca, Blackstone Life Sciences, Foresite Labs, Novartis, Roche / Genentech, and TenSixteen Bio, is a scientific advisory board member of Esperion Therapeutics, geneXwell, and TenSixteen Bio, shareholder of geneXwell and TenSixteen Bio, and spousal employment at Vertex, all unrelated to the present work. None of the authors other than those already mentioned share any competing interests.
Footnotes
The original online version of this Article was revised: The Competing interests section in the original version of this Article was incomplete. It now reads: "Dr. Libby is an unpaid consultant to, or involved in clinical trials for Amgen, AstraZeneca, Baim Institute, Beren Therapeutics, Esperion, Therapeutics, Genentech, Kancera, Kowa Pharmaceuticals, Medimmune, Merck, Norvo Nordisk, Merck, Novartis, Pfizer, Sanofi-Regeneron. Dr. Libby is a member of scientific advisory board for Amgen, Corvidia Therapeutics, DalCor Pharmaceuticals, Kowa Pharmaceuticals, Olatec Therapeutics, Medimmune, Novartis, and XBiotech, Inc. Dr. Libby’s laboratory has received research funding in the last 2 years from Novartis. Dr. Libby is on the Board of Directors of XBiotech, Inc. Dr. Libby has a financial interest in Xbiotech, a company developing therapeutic human antibodies. Dr. Libby's interests were reviewed and are managed by Brigham and Women's Hospital and Partners HealthCare in accordance with their conflict of interest policies. Dr Natarajan reported investigator-initiated grants from Amgen, Apple, AstraZeneca, Boston Scientific, and Novartis, personal fees from Apple, AstraZeneca, Blackstone Life Sciences, Foresite Labs, Novartis, Roche / Genentech, and TenSixteen Bio, is a scientific advisory board member of Esperion Therapeutics, geneXwell, and TenSixteen Bio, shareholder of geneXwell and TenSixteen Bio, and spousal employment at Vertex, all unrelated to the present work. None of the authors other than those already mentioned share any competing interests."
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
These authors contributed equally: Alexander G. Bick, Konstantin Popadin, Pradeep Natarajan and Jacques Fellay.
A list of authors and their affiliations appears at the end of the paper.
Change history
7/8/2022
A Correction to this paper has been published: 10.1038/s41598-022-16151-0
Contributor Information
Pradeep Natarajan, Email: pnatarajan@mgh.harvard.edu.
Jacques Fellay, Email: jacques.fellay@epfl.ch.
the Swiss HIV Cohort Study:
I. Abela, K. Aebi-Popp, A. Anagnostopoulos, M. Battegay, E. Bernasconi, D. L. Braun, H. C. Bucher, A. Calmy, M. Cavassini, A. Ciuffi, G. Dollenmaier, M. Egger, L. Elzi, J. Fehr, J. Fellay, H. Furrer, C. A. Fux, H. F. Günthard, A. Hachfeld, D. Haerry, B. Hasse, H. H. Hirsch, M. Hoffmann, I. Hösli, M. Huber, C. R. Kahlert, L. Kaiser, O. Keiser, T. Klimkait, R. D. Kouyos, H. Kovari, K. Kusejko, G. Martinetti, B. Martinez de Tejada, C. Marzolini, K. J. Metzner, N. Müller, J. Nemeth, D. Nicca, P. Paioni, G. Pantaleo, M. Perreau, A. Rauch, P. Schmid, R. Speck, M. Stöckle, P. Tarr, A. Trkola, G. Wandeler, and S. Yerly
Supplementary Information
The online version contains supplementary material available at 10.1038/s41598-021-04308-2.
References
- 1.Zanni MV, Schouten J, Grinspoon SK, Reiss P. Risk of coronary heart disease in patients with HIV infection. Nat. Rev. Cardiol. 2014;11:728–741. doi: 10.1038/nrcardio.2014.167. [DOI] [PubMed] [Google Scholar]
- 2.Jaiswal S, et al. Age-related clonal hematopoiesis associated with adverse outcomes. N. Engl. J. Med. 2014;371:2488–2498. doi: 10.1056/NEJMoa1408617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Genovese G, et al. Clonal hematopoiesis and blood-cancer risk inferred from blood DNA sequence. N. Engl. J. Med. 2014;371:2477–2487. doi: 10.1056/NEJMoa1409405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Jaiswal S, et al. Clonal hematopoiesis and risk of atherosclerotic cardiovascular disease. N. Engl. J. Med. 2017;377:111–121. doi: 10.1056/NEJMoa1701719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bick AG, et al. Genetic interleukin 6 signaling deficiency attenuates cardiovascular risk in clonal hematopoiesis. Circulation. 2020;141:124–131. doi: 10.1161/CIRCULATIONAHA.119.044362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Fuster JJ, et al. Clonal hematopoiesis associated with TET2 deficiency accelerates atherosclerosis development in mice. Science. 2017;355:842–847. doi: 10.1126/science.aag1381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Swiss HIV Cohort Study et al. Cohort profile: The Swiss HIV Cohort study. Int. J. Epidemiol.39, 1179–1189 (2010). [DOI] [PubMed]
- 8.McLaren PJ, et al. Evaluating the impact of functional genetic variation on HIV-1 control. J. Infect. Dis. 2017;216:1063–1069. doi: 10.1093/infdis/jix470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Li AH, et al. Analysis of loss-of-function variants and 20 risk factor phenotypes in 8,554 individuals identifies loci influencing chronic disease. Nat. Genet. 2015;47:640–642. doi: 10.1038/ng.3270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.The Atherosclerosis Risk in Communities (ARIC) Study: design and objectives. The ARIC investigators. Am. J. Epidemiol.129, 687–702 (1989). [PubMed]
- 11.Bick AG, et al. Inherited causes of clonal haematopoiesis in 97,691 whole genomes. Nature. 2020;586:763–768. doi: 10.1038/s41586-020-2819-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Benjamin, D. et al. Calling somatic SNVs and indels with Mutect2. bioRxiv (2019). 10.1101/861054.
- 13.Steensma DP, et al. Clonal hematopoiesis of indeterminate potential and its distinction from myelodysplastic syndromes. Blood. 2015;126:9–16. doi: 10.1182/blood-2015-03-631747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ho DE, Imai K, King G, Stuart EA. Matching as nonparametric preprocessing for reducing model dependence in parametric causal inference. Polit. Anal. 2007;15:199–236. doi: 10.1093/pan/mpl013. [DOI] [Google Scholar]
- 15.Dharan NJ, et al. HIV is associated with an increased risk of age-related clonal hematopoiesis among older adults. Nat. Med. 2021;27:1006–1011. doi: 10.1038/s41591-021-01357-y. [DOI] [PubMed] [Google Scholar]
- 16.Li M, et al. High frequency of mitochondrial DNA mutations in HIV-infected treatment-experienced individuals. HIV Med. 2017;18:45. doi: 10.1111/hiv.12390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Payne BAI, et al. Mitochondrial aging is accelerated by anti-retroviral therapy through the clonal expansion of mtDNA mutations. Nat. Genet. 2011;43:806. doi: 10.1038/ng.863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ziada, A. S. et al. Mitochondrial DNA somatic mutation burden and heteroplasmy are associated with chronological age, smoking, and HIV infection. Aging Cell18, e13018 (2019). [DOI] [PMC free article] [PubMed]
- 19.Gardner K, Hall PA, Chinnery PF, Payne BAI. HIV treatment and associated mitochondrial pathology: Review of 25 years of in vitro, animal, and human studies. Toxicol. Pathol. 2014;42:811–822. doi: 10.1177/0192623313503519. [DOI] [PubMed] [Google Scholar]
- 20.Young AL, Challen GA, Birmann BM, Druley TE. Clonal haematopoiesis harbouring AML-associated mutations is ubiquitous in healthy adults. Nat. Commun. 2016;7:12484. doi: 10.1038/ncomms12484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Watson CJ, et al. The evolutionary dynamics and fitness landscape of clonal hematopoiesis. Science. 2020;367:1449–1454. doi: 10.1126/science.aay9333. [DOI] [PubMed] [Google Scholar]
- 22.Hormaechea-Agulla D, et al. Chronic infection drives Dnmt3a-loss-of-function clonal hematopoiesis via IFNγ signaling. Cell Stem Cell. 2021 doi: 10.1016/j.stem.2021.03.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Abegunde SO, Buckstein R, Wells RA, Rauh MJ. An inflammatory environment containing TNFα favors Tet2-mutant clonal hematopoiesis. Exp. Hematol. 2018;59:60–65. doi: 10.1016/j.exphem.2017.11.002. [DOI] [PubMed] [Google Scholar]
- 24.Kaner JD, et al. HIV portends a poor prognosis in myelodysplastic syndromes. Leuk. Lymphoma. 2019;60:3529–3535. doi: 10.1080/10428194.2019.1633631. [DOI] [PubMed] [Google Scholar]
- 25.Dawoud AAZ, Tapper WJ, Cross NCP. Clonal myelopoiesis in the UK Biobank cohort: ASXL1 mutations are strongly associated with smoking. Leukemia. 2020;34:2660–2672. doi: 10.1038/s41375-020-0896-8. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
CHIP-associated genetic variant callsets and associated participant level phenotype data used in this study are available to qualified investigators by application to the SHCS and ARIC.