

Abstract
Aberrant promoter DNA hypermethylation is a typical characteristic of cancer and it is often seen in malignancies. Recent studies showed that regulatory cis-elements found up-stream of many tumor suppressor gene promoter CpG island (CGI) attract DNA methyltransferases (DNMT) that hypermethylates and silence the genes. As epigenetic alterations are potentially reversible, they make attractive targets for therapeutic intervention. The currently used decitabine (DAC) and azacitidine (AZA) are DNMT inhibitors that follow the passive demethylation pathway. However, they lead to genome-wide demethylation of CpGs in cells, which makes difficult to use it for causal effect analysis and treatment of specific epimutations. Demethylation through specific demethylase enzymes is thus critical for epigenetic resetting of silenced genes and modified chromatins. Yet DNA-binding factors likely play a major role to guide the candidate demethylase enzymes upon its fusion. Before the advent of clustered regulatory interspaced short palindromic repeats (CRISPR), both zinc finger proteins (ZNFs) and transcription activator-like effector protein (TALEs) were used as binding platforms for ten-eleven translocation (TET) enzymes and both systems were able to induce transcription at targeted loci in an in vitro as well as in vivo model. Consequently, the development of site-specific and active demethylation molecular trackers becomes more than hypothetical to makes a big difference in the treatment of cancer in the future. This review is thus to recap the novel albeit distinct studies on the potential use of site-specific demethylation for the development of epigenetic based cancer therapy.
Keywords: Cancer, DNA, demethylase, epigenetics, promoter, methylase, tumor suppressor
Introduction
Genetic mutation and epigenetic alteration are the two important perigenetic changes that cause cancer. Epigenetic alteration due to DNA methylation, histone modifications and related altered chromatin accessibility that regulate patterns of gene expression are the commonly studied epigenetic events involved in the pathogenesis of cancer.1,2 Such epigenetic alteration or epimutation affects the expression of a gene by modifying the DNA or changing the structure of chromatin without altering the native nucleotide sequence. The epimutation study on mouse becomes more clear evidence that alteration in the epigenetic pathway alone can cause cancer. 3 Contrary to the global hypomethylation in cancer, local hypermethylation of CpG islands in the promoter region that silence tumor suppressor genes emerged as a major epigenetic pathway driving the hallmark of the neoplastic epigenome for the development and progression of cancer. 4
Disease-linked DNA hypermethylation mainly at promoters, enhancers, gene bodies and other site that regulate chromatin conformation not only drive oncogenesis of cancer but also disease like atherosclerosis, autoimmune, neurological diseases and other age-associated diseases. 5 Remarkably, silencing of tumor suppressor genes by promoter hypermethylation is a key mechanism to facilitate cancer progression in many malignancies. While promoter hypermethylation can occur at later stages of the carcinogenesis process, constitutional methylation of key tumor suppressors may be an initiating event whereby cancer is started. 6
Epigenetic dysregulation is often notably linked to cancer. Mutation of genes associated in the cell cycle control, DNA repair, regulatory genes and tumor suppressor genes also often leads to alteration in epigenetic regulation. As the early diagnosis and preclinical pathology of cancer often unnoticeable under normal circumstances, tracing the immediate cause of carcinogenesis is difficult, the commonly used cytotoxic and alternative chemotherapy have variable prognostic outcomes. Once cancer reached a clinically detectable stage, the chance of accumulating more mutation and epimutation will increase. This allows them to evade the commonly used therapy and recur aggressively. Current work in translational research seeks to identify epigenetic regulators whose aberrant activity contributes to oncogenesis thereby developing drugs that inhibit the aberrant activity of these regulators. 7 Such studies on cancer treatment drive further studies of dissecting the potentially targetable of epigenetic factors in cancer cells and opens the high demands for the development of better therapeutic approaches.
The high-throughput analysis of cancer epigenomes and genomes indicated an epimutation can be revealed in cancer genes where no mutation yet to be seen.8-11 DNA methylation is one of the most plentiful epigenetic alterations that reliably affect the eukaryotic DNA molecules. An enzyme of the DNMT family catalyzes the methyl group addition to 5-cytosine and producing 5-methylcytosine (5mC). Such methylations often occur at the CpG islands located at the gene promoters and regulatory regions. 12 CpG sites that are outside of CpG islands tend to be highly methylated in normal cells, whereas CpGs in promoter CpG islands are typically unmethylated. However, in cancer cells the promoter CpG islands of important tumor suppressor genes and DNA repair genes are found to be methylated (Table 1). The presence of regulatory cis-elements found up-stream of the tumor suppressor genes were also reported to dictate the methylation status of the promoter CGI. 3 Since epigenetic changes are potentially reversible, they make attractive targets for therapeutic intervention. 13 Hence this review is to give insightful information on the potential application of site-specific demethylation for the development of Epigenetic based cancer therapy.
Table 1.
Lists of genes with aberrantly hypermethylated promoter in cancer.
| Gene | Function | Impact on carcinogenesis |
|---|---|---|
| p53 | Tumor suppressor | Uncontrolled cell cycle progression 14 |
| p16 | Cyclin dependent kinase inhibitor | Sensitivity to antigrowth signal 15 |
| p15 | Tumor suppressor | Uncontrolled cell cycle 16 |
| VHL | Suppression of metastasis | Tissue invasion and metastasis 17 |
| RB | Regulation of cell cycle | Uncontrolled replication 18 |
| VEGF-2 | Angiogenesis | Uncontrolled angiogenesis 19 |
| CDH1 | Cellular adhesion | Invasiveness and metastasis 20 |
| BRCA1 | Tumor suppressor | Altered DNA damage repair 21 |
| PAX6 | Transcription factor | Oncogene that facilitates cell growth 22 |
| MLH1 | Tumor suppressor | Altered DNA mismatch repair 23 |
| MGMT | DNA repair enzyme | Rescues tumor cells from apoptosis 24 |
DNA Methylation as a Hallmark of Cancer
Recent epigenomic studies revealed that nearly all tumor types harbor abnormally hypermethylated promoter CGIs. Such alterations in epigenetic modifications in cancer regulate various cellular responses, including cell proliferation, apoptosis, invasion, and senescence. 2 Through DNA methylation, epigenetics plays an important role in tumorigenesis. In mammalian non-cancer cells as the genome-wide CpGs are hypomethylated, gene silencing is usually modulated by repressive chromatin marks. In cancer cells it is often modulated by methylation of promoter CpG islands. In this regard it has been proved that epimutation that leads to hypermethylation of tumor suppressor gene promotor subsequently induced tumor development and reduced mice survival. 3 This aspect of epigenetics presents reversible effects on gene silencing via epigenetic enzymes and related proteins. DNA methyltransferases (DNMTs) along with methyl-CpG binding domain proteins (MBDs) is responsible for the transfer of methyl groups to specific DNA residue (Figure 1).
Figure 1.

Repression of tumor suppressor gene by hypermethylation of its promoter. DNMT1, DNMT3A and DNMT3B are DNA methyltransferase enzymes that methylate cytosine residues of promotor CpGs. The figure indicates the change in the putative tumor suppressor gene methylation pattern comparing the normal cell with hypomethylation (expression ON) and cancer cells with hypermethylation (expression OFF).
Aberrant DNA methylation is thought to serve as a hallmark in cancer development by inactivating or repressing gene transcription. 25 In normal tissues, gene promoters, especially key tumor suppressor genes, are unmethylated but hypermethylated in cancer. 13 p16Ink4a (p16) is a tumor suppressor gene that regulates the retinoblastoma (Rb) protein ability to control the cell cycle at the G1 stage. 26 The inhibitory cyline dependent kinase 4a (INK4a) locus is normally involved both in the pRb and p53 pathways which are an important regulator of cell cycle.26,27 p16, p15, and PAX6 are also often aberrantly methylated in bladder cancer and show enhanced methylation in cell culture 22 and p16 found to gain de novo methylation in –20% of different primary neoplasms. 28 Beyond its cell cycle regulatory and tumor-suppressive effect, p16 also plays an important role in other cellular events like cellular differentiation, cell quiescence, cell senescence and the aging process. 29 This makes p16 a key biomolecule involved in different cellular pathways. Inactivation of these pathways occurs in most human tumors and methylation of p16 promoter CGI is often underlies epigenetic incidences in human cancer. 15
Promotor of tumor suppressor genes like p53, RB and BRAC1 which are well known key players for the hallmark of cancer has been also reported to be methylated in different cancers.18,30-32 Methylation of promoters of VHL genes which is responsible for tissue invasion and metastasis 17 and promoter of VEGF-2 gene which is responsible for sustained angiogenesis of cancer has been reported. 19 Hypermethylation at gene body leads to activation of oncogenes (DLX1, POU3F3), by increasing its transcriptional activity. 33 Signals from the microenvironment, especially those from transforming growth factor-β, has been reported to induce targeted de novo epigenetic alterations of cancer-related genes. 1 On top of cancer driving role, aberrant methylations were also found to trigger drug resistance of cancer cells. This has been observed with hypermethylation of thymidine kinase 34 and O-6-methylguanine DNA methyltransferase (MGMT).35-37 Some of the several genes associated with the development and progressions cancer due to aberrant promoter DNA methylation are detailed in Table 1.
The epigenetic events that contribute to oncogenesis often have cross-talks with other perigenetic causes of cancer. For instance, a mutation that happens as an early cytogenetic event leads to the aberrant activity of epigenetic regulators. 7 In fact next to p53, p16 was the most frequently mutated gene in the majority of tumors. Such mutations often involve comparable epigenetic alterations. In this regard, DNA methyltransferase inhibitors have been approved as first-generation epigenetic inhibitors for cancer therapy.38,39 On the other hand knowing the status of DNA methylation as with DNA epigenetic mark, 5-methylcytosine (m5C) can be used as an efficient and reliable way to diagnose tumor and monitor its pathological processes. 40 Hence, it is amicable to note that the reversible nature of DNA methylation also makes demethylase enzymes as appealing drug targets for epigenetic cancer therapy (Figure 2).
Figure 2.

DNA methylation and demethylation mechanisms. The methylation of Cytosine (C) nuclotide (the ring highlighted red at the top) leads to transcriptional repression. While the reverse reaction of passive and active demethylation (the dark green highlighted rings) step-wisely removes a methyl group from carbon number 5 of the cytosine residue.
General Mechanisms of DNA Demethylation
Epigenetic alteration can be best addressed focusing on the methylation status of biomolecules (DNA, RNA and Protein). DNA methylation, histone methylation and miRNA mediated DNMT expression control can be addressed by harnessing the demethylation mechanisms. DNA methylation is a reversible process in which DNMTs enzymes methylate the cytosine residue in the CpG of target gene promoter thereby cause transcriptional repression. In this regard the 5-methyl cytosine (5-mC) shown in Figure 2 can be demethylated through numerous overlapping pathways of passive and active demethylation mechanisms. 41
The passive DNA demethylation takes place in the absence of methylation of newly synthesized DNA strands by DNMT1 during several replication rounds. 42 It has been identified that active 5-mC demethylation pathway commenced by the proteins of ten-eleven translocation family enzymes (TET1, TET2 and TET3) that oxidizes 5-mC in to 5-hydroxy-methyl cytosine (5-hmC). 43 This became a crucial step that allows numerous alternative and/or overlapping ways to remake normal cytosine residue either autonomously or in a DNA replication-dependent manner thereby causing final removal of the methyl mark.25,44-49
Active DNA demethylation can occur via direct removal of a methyl group independently of DNA replication. The underlying various mechanisms include enzymatic exclusion of the methyl group, base excision repair, deamination followed by mismatch repair, nucleotide excision repair and oxidative demethylation. 50 5-mC is oxidized to 5-hmC which is then oxidized to 5-formylcytosine (5-fC) then 5-carboxylcytosine (5-caC) which is the final oxidized derivative of 5-methylcytosine (5mC). 46 Each of these oxidation steps are catalyzed by TET family of enzymes. Both 5-fC and 5-caC can be converted to unmodified cytosine by thymine DNA glycosylase (TDG) or by base excision repair (BER). 51 The overall cascade of TET1-TDG-BER-dependent active DNA demethylation is described in Figure 2 below.
Recitation of the demethylation phenomenon, studies indicated that demethylation of tumor suppressor gene promoter can be a key target for future epigenetic therapy of cancer.1,52-54 In this regard, the study on the hypermethylation status of the p16 CpG island in colon cancer cells showed, it was effectively reversed using DNMT inhibitor 5-aza-2’-deoxycytidine (DAC) in which demethylation suppressed the growth of colon cancer cells and also induced apoptosis. 52 The two food and drug authority (FDA) approved conventional drugs (DAC and AZA) are passive hypomethylators and these chemotherapeutic approach causes genome-wide demethylation of promoters but lacks sequence-specificity to the promoter of interest. Due to their pleitropic effects, it has been difficult to confirm the mechanism of action of such DNA methyltransferase inhibitors. Nevertheless, the pre-clinical data showed that reactivation of silenced promoter through selective DNA demethylation may have significant therapeutic alternatives. 55
Epigenetic effects can also control miRNA expression. Restoration of important miRNA which down-regulates DNMT induces a global hypomethylated state in cancer cells. It also induces concomitant re-expression of tumor suppressor genes whose expression is silenced in cancer by promoter hypermethylation. 56 Over-all, the involvement of DNA methylation event in the well-characterized epigenetic regulatory pathways makes demethylation as a fabulous point to design therapeutic strategy.
Site-Specific DNA Demethylation as Epigenetic Therapy
Cancer therapy is as complex as it’s intertwined etiological diversity. Prevention and treatment of cancer thus remain a challenging and researchable area of study. Abnormal promoter hypermethylation is a stamp of many cancers. But the inherent reversibility of such epigenetic alterations makes them viable therapeutic targets. 13 For instance, the hypermethylation status of the p16 promoter associated 5’CpG Island in cancer cells was efficiently reversed using the DNMT inhibitor. 52 However such treatments cause global demethylation with some unforeseen side effects. The involvement of aberrant hyper-methylation of many tumor suppressor gene promoters and associated CpG sites in most cancer,57-60 makes them providential candidates and it is likely to hypothesize that site-specific demethylation of cancer-associated gene promoter might have a tremendous impact in future therapy of cancer. Hence developing a strategy to target cancer-associated site-specific CpG promoter demethylation has paramount importance.
Site-specific demethylation modalities
Applying the currently advanced biotechnological tools it is possible to produce the candidate demethylating enzyme fused with the engineered construct of DNA binding proteins that able to sequence-specifically localize the methylated promoter CpG of interest. Such proteins include the rel-homology domain (RHD) and zinc finger domains (ZFD) that bound sequences flanking a specific CpG site. 41 Other transcriptional activator components can also be used for the activation of the target promoter as well as site-specific delivery. The demethylase enzyme cloned in fusion with such DNA site-specific tracker can be transduced and the co-expression will enable nuclear localization as the transcription factor has special structures to recognize DNA. 55 Studies also indicate that, DNA-binding factors like hormone receptors can cause DNA demethylation at specific promoter or enhancer area thereby allowing the binding and localization of fused pioneer factors.61-64
The molecular tool through which DNA demethylase targeted to the tumor suppressor gene promoter CpG sites can be made by fusing human TET1 demethylase enzyme to the recombinant transcription activator-like effector (TALE) repeat that has targeted DNA-binding specificities. 54 The modified TALE repeat makes an attractive platform to guide TET1 activity because monomeric proteins that bind to nearly any target DNA sequence of interest can be enormously produced by assembling individual repeat domains with known nucleotide specificities. 65 Cloning of demethylase enzyme with DNA site-specific tracker can also be constructed and transduced. 55
Concomitant to the TALE-TET fusion module, the well-known DNA-binding proteins utilized in targeted editing were the eukaryotic ZNFs and represented the beginning of a new era in genomic and epigenomic manipulation. 66 ZNFs are transcription factors comprising protein motifs or fingers that recognize and bind three DNA nucleotides. Different ZNF modules are used in combination, based on their respective affinities for a particular three-base sequence, to target specific genomic regions. ZNF DNA binding domains are therefore commonly fused with a nuclease or other effector protein, to mediate a site-specific genetic or epigenetic response.66-69 Both ZNFs and TALEs were used as binding podiums for TET enzymes and both systems were able to induce transcription at targeted loci.54,70 Here the Site-specific DNA Demethylation using programmable TALE-TETs fusion is shown as representative modalities (Figure 3).
Figure 3.

Site-specific DNA demethylation using programmable TALE-TETs fusion. In this module, the fusion can be made using the specific-site DNA binding TALE at N-terminus and full-length or catalytic domain of TET1 at C-terminus. The amino acids for the specific TALE can be computationally predicted for its binding to the putative tumor suppressor gene promoter target sequences. 71
Another molecular tool currently used for epigenome editing is a CRISPR module. CRISPR can be used for site-specific Demethylation harnessing its specific DNA-binding ability by deactivating its Caspase 9 (dCas9) function. Here, for demethylation of 5-mC marks, the TET hydroxylase catalytic domain fused to dCas9 (Figure 4). So far numerous demethylation studies have been published using CRISPR-dCas9 systems.53,72-75 Generally, each system utilizes the CRISPR-dCas9-TET1 fusion protein paired with a programmable 20 nucleotide sgRNA guide homologous to the target locus. The first study using a transient and lentiviral-based dCas9-TET1 system showed selective targeting of the BRCA1 promoter to induce robust gene expression. 53 This was followed by modification made on sgRNA by inserting bacteriophage MS2 RNA elements into the conventional sgRNA, thereby making MS2-fused TET-CD proteins. 72 Further modification on the length of SunTag linker improved the efficiency of antibody-fused TET1 recruitment and in vitro and in vivo demethylation. 76 With this dCas9-TET1 fusion system, it has been possible to make site-specific demethylation that led to an increase in the expression of target genes.72,73,77 The dCas9-based systems can also achieve targeting of multiple genomic loci simultaneously by the delivery of several guide RNAs (gRNAs), offering far greater multiplexing simplicity compared to the delivery of multiple different ZFN or TALE DNA binding proteins. 78 Simplified modality of CRISPR-dCas9-TET1-CD-based site-specific demethylation is indicated in Figure 4 below.
Figure 4.
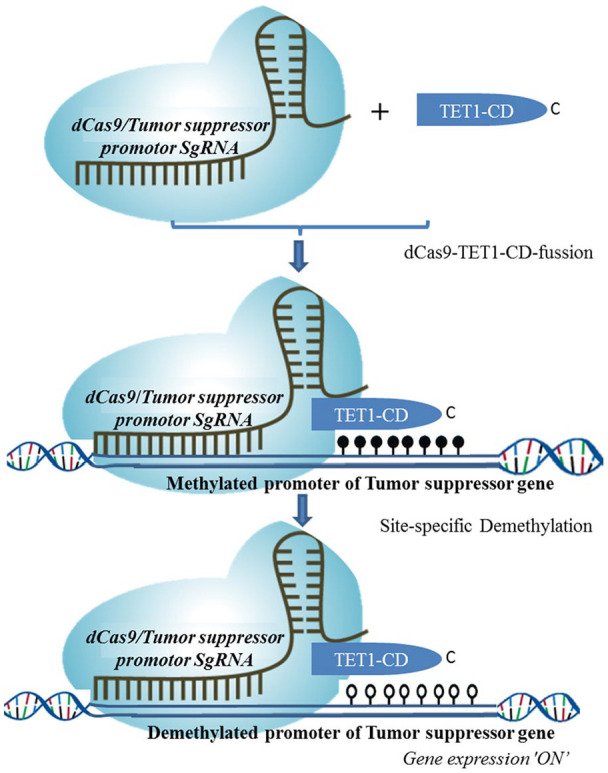
CRISPR-dCas9-TET-CD-based site-specific demethylation. The dCas9 fused to the catalytic domain (CD) of TET1 enzyme (indicated dark blue) and the single guide RNA (sgRNA) targeting the putative hypermethylated promoter of the tumor suppressor gene is used in this molecular tracker module.
It is important to note that there is also newly emerging dCas9 system in which the DNMT1 is recruited by an R2 loop, preventing DNA methylation maintenance during replication. It avoids the potential side effects of exogenous TET protein expression while conserving better targeting accuracy. 75 Concomitant to the above mentioned CRISPR-dCas9-based promoter targeted demethylation strategies have been mainly validated in vitro for specific gene reactivation, this reprogrammable tool is also useful to demethylate other part of the gene to induce reprogramming in fibroblasts, 77 as well as pluripotent stem cells for sustainable reactivation in a human-mouse chimeric model. 79 Numerous modification of dCas9 system including the use of thymidine DNA glycosylase (TDG) 80 and ROS1 5mC DNA glycosylase (ROS1CD) 74 instead of TET were made and found to methylate targeted promoters and increase the expression of target gene. Such site-specific demethylation system however requires further in vitro optimization and adoption in to in vivo research model to be used in the near future.
Potential application of Site-specify molecular trackers
To develop therapeutic modalities using epigenomic engineering techniques, in vitro test can be conducted on a well-characterized cancer cell line with hypermethylation of tumor suppressor gene to be used as a target. TALE-TET1 fusions, ZNF-TDG, or CRISPR-dCas9-TET-CD-based modality or another demethylase based modalities can be used for in vivo study of cancer epigenetic therapeutic trials although these systems are still relatively new and still requires further optimization. On top of its use for demethylation study for cancer, such specific DNA targeted modalities can be used to study other diseases including Alzheimer’s, Parkinson’s, and Huntington’s diseases that are caused by epigenetic alteration. 81 It is not only to remove the aberrant promoter methylation (to reactivate the silenced genes) but also it is possible to track the methylation phenomenon using DNMTs to methylate and silence the over-activation of oncogenic genes or genes involved in oncogenic signaling pathway in cancer. 82
The current technical developments of molecular tools help us not only to understand the oncogenesis of cancer with epigenetic cause but also to evaluate the responses of targeted epigenetic therapy. For instance methylation status of the targeted a given tumor suppressor gene 83 and the overall efficacy of the site-specific epigenetic therapy can be evaluated using laboratory technique like pyrosequencing, assay for transposase-accessible chromatin using sequencing (ATAC-Seq) and quantitative polymerase chain reaction, 84 Chromatin Immunoprecipitation sequencing, 85 CRISPR86,87 and DNA methylation analysis are worth to mention. 88 Sideway different routes of drug delivery and its efficacy compared to prevailing therapeutic agents can be validated.
Epigenetic alterations can also be used as potential predictive biomarkers for the detection of cancer and associated diseases and to design corresponding targeted therapy. There are also many diseases caused epimutation (aberrant DNA hypermethylation or hypomethylation) that could be reverted using the above epigenomic engineering techniques. As these tools use the biological system and do not directly affect the DNA sequence mammalian genomes, it is likely to be safer and efficient than conventionally used chemotherapy for such cancers. On top of this, it encourages the development of alternative therapeutic ventures like with that of nanotechnology and screening of small-molecule inhibitors that have translational value to develop targeted epigenetic therapy in the near future.
Limitations associated with these strategies
Multiple tumor-suppressor genes are silenced by DNA methylation in cancer cells, and it has been reported that simultaneous re-activation of these multiple genes than only one specific gene is important for therapeutic efficacy. 89 By target gene-specific demethylation strategy, simultaneous re-activation of multiple genes seems to be difficult. Therefore, the potential strategies to overcome this disadvantage should be studies.
Another key factor that often hinders the development of therapy for cancer and related disease is the technical complexity in understanding the disease itself and to design targeted therapy. On top of tumor suppressor gene hypermethylation, some cancer may harbor other epigenetic alteration during its course of progression. Hence efficacies of these site-specific demethylation tools still possess a variety of limitations in efficacy, implementation, and targeting specificity. 78
Conclusion
Cancer is a complex disease mostly associated with mutation and alteration in the epigenetic pathway. The widespread presence of an epigenetic abnormality in the human cancer genome suggests the need for persistent efforts of developing strategies that encompass epigenetic aberrations. Aberrant methylation of tumor suppressor genes can be best addressed focusing on the methylation status of DNA and its subsequent effects. Site-specific demethylation for reactivation of epigenetically silenced genes might provide a useful strategy to further explore biological mechanisms and to develop targeted epimutation therapy for cancer. The above discussed site-specific demethylation modalities for tumor suppressor genes can be used as an exemplary molecular tool to chase and develop a novel and potential target for epigenetic therapy of cancer and related diseases in the near future.
Acknowledgments
The author acknowledges Hawassa University for providing internet facility and reading materials used to prepare this manuscript.
Footnotes
Funding: The author(s) received no financial support for the research, authorship, and/or publication of this article.
Declaration of conflicting interests: The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Authors’ Contributions: The first author drafted the manuscript, written, and made proofreading. The author designed illustrated figures and properly sited for the statement taken from the open-access journal used in this manuscript.
Data Availability Statement: The corresponding author can also be reached for any data enquiry.
Authors’ Information (optional): Dr. Sultan Abda Neja pursed DVM and MvSc in Microbiology at Addis Ababa University, Ethiopia, MSc in Biotechnology from Asia University, Taiwan, and PhD in Biochemistry from the National University of Singapore. He has published more than 15 research papers in reputable international journals available online. He participated in conferences including an International Congress on Hematologic Malignancies 2017 in New York. His PhD research work was on the oncogenesis of human cancer. He has 3-year of academia and 6 years of research experience. He is currently working as Assistant Professor at Hawassa University, Ethiopia.
ORCID iD: Sultan Abda Neja  https://orcid.org/0000-0003-3972-0016
https://orcid.org/0000-0003-3972-0016
References
- 1. Khin SS, Kitazawa R, Kondo T, et al. Epigenetic alteration by DNA promoter hypermethylation of genes related to Transforming Growth Factor-beta (TGF-beta) signaling in cancer. Cancers (Basel). 2011;3:982-993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Cheng Y, He C, Wang M, et al. Targeting epigenetic regulators for cancer therapy: mechanisms and advances in clinical trials. Signal Transduct Target Ther. 2019;4:62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Yu DH, Waterland RA, Zhang P, et al. Targeted p16(Ink4a) epimutation causes tumorigenesis and reduces survival in mice. J Clin Invest. 2014;124:3708-3712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Issa JP. CpG island methylator phenotype in cancer. Nat Rev Cancer. 2004;4:988-993. [DOI] [PubMed] [Google Scholar]
- 5. Ehrlich M. DNA hypermethylation in disease: mechanisms and clinical relevance. Epigenetics. 2019;14:1141-1163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Lonning PE, Eikesdal HP, Loes IM, Knappskog S. Constitutional mosaic epimutations – a hidden cause of cancer? Cell Stress. 2019;3:118-135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Wee S, Dhanak D, Li H, et al. Targeting epigenetic regulators for cancer therapy. Ann NY Acad Sci. 2014;1309:30-36. [DOI] [PubMed] [Google Scholar]
- 8. Cancer Genome Atlas Network. Comprehensive molecular portraits of human breast tumours. Nature. 2012;490:61-70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Cancer Genome Atlas Network. Comprehensive molecular characterization of human colon and rectal cancer. Nature. 2012;487:330-337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Cancer Genome Atlas Research Network. Integrated genomic analyses of ovarian carcinoma. Nature. 2011;474:609-615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Cancer Genome Atlas Research Network. Comprehensive genomic characterization of squamous cell lung cancers. Nature. 2012;489:519-525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Law JA, Jacobsen SE. Establishing, maintaining and modifying DNA methylation patterns in plants and animals. Nat Rev Genet. 2010;11:204-220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Baylin SB, Jones PA. A decade of exploring the cancer epigenome – biological and translational implications. Nat Rev Cancer. 2011;11:726-734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Kang JH, Kim SJ, Noh DY, et al. Methylation in the p53 promoter is a supplementary route to breast carcinogenesis: correlation between CpG methylation in the p53 promoter and the mutation of the p53 gene in the progression from ductal carcinoma in situ to invasive ductal carcinoma. Lab Invest. 2001;81:573-579. [DOI] [PubMed] [Google Scholar]
- 15. Herman JG, Merlo A, Mao L, et al. Inactivation of the CDKN2/p16/MTS1 gene is frequently associated with aberrant DNA methylation in all common human cancers. Cancer Res. 1995;55:4525-4530. [PubMed] [Google Scholar]
- 16. Krajnovic M, Jovanovic MP, Mihaljevic B, et al. Hypermethylation of p15 gene in diffuse – large B-cell lymphoma: association with less aggressiveness of the disease. Clin Transl Sci. 2014;7:384-390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Herman JG, Latif F, Weng Y, et al. Silencing of the VHL tumor-suppressor gene by DNA methylation in renal carcinoma. Proc Natl Acad Sci USA. 1994;91:9700-9704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Greger V, Passarge E, Hopping W, Messmer E, Horsthemke B. Epigenetic changes may contribute to the formation and spontaneous regression of retinoblastoma. Hum Genet. 1989;83:155-158. [DOI] [PubMed] [Google Scholar]
- 19. Cooper MP, Keaney JF., Jr. Epigenetic control of angiogenesis via DNA methylation. Circulation. 2011;123:2916-2918. [DOI] [PubMed] [Google Scholar]
- 20. Caldeira JR, Prando EC, Quevedo FC, Neto FA, Rainho CA, Rogatto SR. CDH1 promoter hypermethylation and E-cadherin protein expression in infiltrating breast cancer. BMC Cancer. 2006;6:48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Esteller M, Silva JM, Dominguez G, et al. Promoter hypermethylation and BRCA1 inactivation in sporadic breast and ovarian tumors. J Natl Cancer Inst. 2000;92:564-569. [DOI] [PubMed] [Google Scholar]
- 22. Markl ID, Cheng J, Liang G, Shibata D, Laird PW, Jones PA. Global and gene-specific epigenetic patterns in human bladder cancer genomes are relatively stable in vivo and in vitro over time. Cancer Res. 2001;61:5875-5884. [PubMed] [Google Scholar]
- 23. Li X, Yao X, Wang Y, et al. MLH1 promoter methylation frequency in colorectal cancer patients and related clinicopathological and molecular features. PLoS One. 2013;8:e59064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Nagasaka T, Goel A, Notohara K, et al. Methylation pattern of the O6-methylguanine-DNA methyltransferase gene in colon during progressive colorectal tumorigenesis. Int J Cancer. 2008;122:2429-2436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Ito S, D’Alessio AC, Taranova OV, Hong K, Sowers LC, Zhang Y. Role of Tet proteins in 5mC to 5hmC conversion, ES-cell self-renewal and inner cell mass specification. Nature. 2010;466:1129-1133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Sharpless NE, DePinho RA. The INK4A/ARF locus and its two gene products. Curr Opin Genet Dev. 1999;9:22-30. [DOI] [PubMed] [Google Scholar]
- 27. Serrano M. The INK4a/ARF locus in murine tumorigenesis. Carcinogenesis. 2000;21:865-869. [DOI] [PubMed] [Google Scholar]
- 28. Merlo A, Herman JG, Mao L, et al. 5’ CpG island methylation is associated with transcriptional silencing of the tumour suppressor p16/CDKN2/MTS1 in human cancers. Nat Med. 1995;1:686-692. [DOI] [PubMed] [Google Scholar]
- 29. Agarwal P, Sandey M, DeInnocentes P, Bird RC. Tumor suppressor gene p16/INK4A/CDKN2A-dependent regulation into and out of the cell cycle in a spontaneous canine model of breast cancer. J Cell Biochem. 2013;114:1355-1363. [DOI] [PubMed] [Google Scholar]
- 30. Rideout WM, 3rd, Coetzee GA, Olumi AF, Jones PA. 5-Methylcytosine as an endogenous mutagen in the human LDL receptor and p53 genes. Science. 1990;249:1288-1290. [DOI] [PubMed] [Google Scholar]
- 31. Kleihues P, Schauble B, zur Hausen A, Esteve J, Ohgaki H. Tumors associated with p53 germline mutations: a synopsis of 91 families. Am J Pathol. 1997;150:1-13. [PMC free article] [PubMed] [Google Scholar]
- 32. Caputo S, Benboudjema L, Sinilnikova O, et al. Description and analysis of genetic variants in French hereditary breast and ovarian cancer families recorded in the UMD-BRCA1/BRCA2 databases. Nucleic Acids Res. 2012;40:D992-1002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Su J, Huang YH, Cui X, et al. Homeobox oncogene activation by pan-cancer DNA hypermethylation. Genome Biol. 2018;19:108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Nyce J, Leonard S, Canupp D, Schulz S, Wong S. Epigenetic mechanisms of drug resistance: drug-induced DNA hypermethylation and drug resistance. Proc Natl Acad Sci USA. 1993;90:2960-2964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Esteller M, Garcia-Foncillas J, Andion E, et al. Inactivation of the DNA-repair gene MGMT and the clinical response of gliomas to alkylating agents. N Engl J Med. 2000;343:1350-1354. [DOI] [PubMed] [Google Scholar]
- 36. Hegi ME, Diserens AC, Gorlia T, et al. MGMT gene silencing and benefit from temozolomide in glioblastoma. N Engl J Med. 2005;352:997-1003. [DOI] [PubMed] [Google Scholar]
- 37. Esteller M, Gaidano G, Goodman SN, et al. Hypermethylation of the DNA repair gene O(6)-methylguanine DNA methyltransferase and survival of patients with diffuse large B-cell lymphoma. J Natl Cancer Inst. 2002;94:26-32. [DOI] [PubMed] [Google Scholar]
- 38. Pechalrieu D, Etievant C, Arimondo PB. DNA methyltransferase inhibitors in cancer: from pharmacology to translational studies. Biochem Pharmacol. 2017;129:1-13. [DOI] [PubMed] [Google Scholar]
- 39. Gnyszka A, Jastrzebski Z, Flis S. DNA methyltransferase inhibitors and their emerging role in epigenetic therapy of cancer. Anticancer Res. 2013;33:2989-2996. [PubMed] [Google Scholar]
- 40. Barciszewska AM, Giel-Pietraszuk M, Perrigue PM, Naskret-Barciszewska M. Total DNA methylation changes reflect random oxidative DNA damage in gliomas. Cells. 2019;8:1065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Hackett JA, Zylicz JJ, Surani MA. Parallel mechanisms of epigenetic reprogramming in the germline. Trends Genet. 2012;28:164-174. [DOI] [PubMed] [Google Scholar]
- 42. Kohli RM, Zhang Y. TET enzymes, TDG and the dynamics of DNA demethylation. Nature. 2013;502:472-479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Wu H, Zhang Y. Mechanisms and functions of Tet protein-mediated 5-methylcytosine oxidation. Genes Dev. 2011;25:2436-2452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Tahiliani M, Koh KP, Shen Y, et al. Conversion of 5-methylcytosine to 5-hydroxymethylcytosine in mammalian DNA by MLL partner TET1. Science. 2009;324:930-935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Tu ZQ, Xue HY, Chen W, Cao LF, Zhang WQ. Identification of potential peripheral blood diagnostic biomarkers for patients with juvenile idiopathic arthritis by bioinformatics analysis. Rheumatol Int. 2017;37:423-434. [DOI] [PubMed] [Google Scholar]
- 46. Ito S, Shen L, Dai Q, et al. Tet proteins can convert 5-methylcytosine to 5-formylcytosine and 5-carboxylcytosine. Science. 2011;333:1300-1303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. He YF, Li BZ, Li Z, et al. Tet-mediated formation of 5-carboxylcytosine and its excision by TDG in mammalian DNA. Science. 2011;333:1303-1307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Xu Y, Wu F, Tan L, et al. Genome-wide regulation of 5hmC, 5mC, and gene expression by Tet1 hydroxylase in mouse embryonic stem cells. Mol Cell. 2011;42:451-464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Williams K, Christensen J, Pedersen MT, et al. TET1 and hydroxymethylcytosine in transcription and DNA methylation fidelity. Nature. 2011;473:343-348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Wu SC, Zhang Y. Active DNA demethylation: many roads lead to Rome. Nat Rev Mol Cell Biol. 2010;11:607-620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Zhang L, Lu X, Lu J, et al. Thymine DNA glycosylase specifically recognizes 5-carboxylcytosine-modified DNA. Nat Chem Biol. 2012;8:328-330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Christman JK. 5-Azacytidine and 5-aza-2’-deoxycytidine as inhibitors of DNA methylation: mechanistic studies and their implications for cancer therapy. Oncogene. 2002;21:5483-5495. [DOI] [PubMed] [Google Scholar]
- 53. Choudhury SR, Cui Y, Lubecka K, Stefanska B, Irudayaraj J. CRISPR-dCas9 mediated TET1 targeting for selective DNA demethylation at BRCA1 promoter. Oncotarget. 2016;7:46545-46556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Maeder ML, Angstman JF, Richardson ME, et al. Targeted DNA demethylation and activation of endogenous genes using programmable TALE-TET1 fusion proteins. Nat Biotechnol. 2013;31:1137-1142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Gregory DJ, Mikhaylova L, Fedulov AV. Selective DNA demethylation by fusion of TDG with a sequence-specific DNA-binding domain. Epigenetics. 2012;7:344-349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Fabbri M, Garzon R, Cimmino A, et al. MicroRNA-29 family reverts aberrant methylation in lung cancer by targeting DNA methyltransferases 3A and 3B. Proc Natl Acad Sci USA. 2007;104:15805-15810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Esteller M. Aberrant DNA methylation as a cancer-inducing mechanism. Annu Rev Pharmacol Toxicol. 2005;45:629-656. [DOI] [PubMed] [Google Scholar]
- 58. Wu L, Shen Y, Peng X, et al. Aberrant promoter methylation of cancer-related genes in human breast cancer. Oncol Lett. 2016;12:5145-5155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Ng JM, Yu J. Promoter hypermethylation of tumour suppressor genes as potential biomarkers in colorectal cancer. Int J Mol Sci. 2015;16:2472-2496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Esteller M. CpG island hypermethylation and tumor suppressor genes: a booming present, a brighter future. Oncogene. 2002;21:5427-5440. [DOI] [PubMed] [Google Scholar]
- 61. Stadler MB, Murr R, Burger L, et al. DNA-binding factors shape the mouse methylome at distal regulatory regions. Nature. 2011;480:490-495. [DOI] [PubMed] [Google Scholar]
- 62. Serandour AA, Avner S, Percevault F, et al. Epigenetic switch involved in activation of pioneer factor FOXA1-dependent enhancers. Genome Res. 2011;21:555-565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Kim MS, Kondo T, Takada I, et al. DNA demethylation in hormone-induced transcriptional derepression. Nature. 2009;461:1007-1012. [DOI] [PubMed] [Google Scholar]
- 64. Metivier R, Gallais R, Tiffoche C, et al. Cyclical DNA methylation of a transcriptionally active promoter. Nature. 2008;452:45-50. [DOI] [PubMed] [Google Scholar]
- 65. Joung JK, Sander JD. TALENs: a widely applicable technology for targeted genome editing. Nat Rev Mol Cell Biol. 2013;14:49-55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Adli M. The CRISPR tool kit for genome editing and beyond. Nat Commun. 2018;9:1911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Urnov FD, Rebar EJ, Holmes MC, Zhang HS, Gregory PD. Genome editing with engineered zinc finger nucleases. Nat Rev Genet. 2010;11:636-646. [DOI] [PubMed] [Google Scholar]
- 68. Chatterjee A, Eccles MR. DNA methylation and epigenomics: new technologies and emerging concepts. Genome Biol. 2015;16:103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Grimmer MR, Stolzenburg S, Ford E, Lister R, Blancafort P, Farnham PJ. Analysis of an artificial zinc finger epigenetic modulator: widespread binding but limited regulation. Nucleic Acids Res. 2014;42:10856-10868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Chen H, Kazemier HG, de Groote ML, Ruiters MH, Xu GL, Rots MG. Induced DNA demethylation by targeting ten-eleven translocation 2 to the human ICAM-1 promoter. Nucleic Acids Res. 2014;42:1563-1574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Erkes A, Mucke S, Reschke M, Boch J, Grau J. PrediTALE: a novel model learned from quantitative data allows for new perspectives on TALE targeting. PLoS Comput Biol. 2019;15:e1007206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Xu X, Tao Y, Gao X, et al. A CRISPR-based approach for targeted DNA demethylation. Cell Discov. 2016;2:16009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Morita S, Noguchi H, Horii T, et al. Targeted DNA demethylation in vivo using dCas9-peptide repeat and scFv-TET1 catalytic domain fusions. Nat Biotechnol. 2016;34:1060-1065. [DOI] [PubMed] [Google Scholar]
- 74. Parrilla-Doblas JT, Ariza RR, Roldan-Arjona T. Targeted DNA demethylation in human cells by fusion of a plant 5-methylcytosine DNA glycosylase to a sequence-specific DNA binding domain. Epigenetics. 2017;12:296-303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Lu A, Wang J, Sun W, et al. Reprogrammable CRISPR/dCas9-based recruitment of DNMT1 for site-specific DNA demethylation and gene regulation. Cell Discov. 2019;5:22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Lei Y, Zhang X, Su J, et al. Targeted DNA methylation in vivo using an engineered dCas9-MQ1 fusion protein. Nat Commun. 2017;8:16026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Liu XS, Wu H, Ji X, et al. Editing DNA methylation in the mammalian genome. Cell. 2016;167:233-247.e17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Pflueger C, Swain T, Lister R. Harnessing targeted DNA methylation and demethylation using dCas9. Essays Biochem. 2019;63:813-825. [DOI] [PubMed] [Google Scholar]
- 79. Liu XS, Wu H, Krzisch M, et al. Rescue of fragile X syndrome neurons by DNA methylation editing of the FMR1 gene. Cell. 2018;172:979-992.e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Gregory DJ, Zhang Y, Kobzik L, Fedulov AV. Specific transcriptional enhancement of inducible nitric oxide synthase by targeted promoter demethylation. Epigenetics. 2013;8:1205-1212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Jakovcevski M, Akbarian S. Epigenetic mechanisms in neurological disease. Nat Med. 2012;18:1194-1204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Rodger EJ, Chatterjee A, Morison IM. 5-hydroxymethylcytosine: a potential therapeutic target in cancer. Epigenomics. 2014;6:503-514. [DOI] [PubMed] [Google Scholar]
- 83. Ehrlich M, Gama-Sosa MA, Huang LH, et al. Amount and distribution of 5-methylcytosine in human DNA from different types of tissues of cells. Nucleic Acids Res. 1982;10:2709-2721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Chatterjee A, Rodger EJ, Morison IM, Eccles MR, Stockwell PA. Tools and strategies for analysis of genome-wide and gene-specific DNA methylation patterns. Methods Mol Biol. 2017;1537:249-277. [DOI] [PubMed] [Google Scholar]
- 85. O’Geen H, Henry IM, Bhakta MS, Meckler JF, Segal DJ. A genome-wide analysis of Cas9 binding specificity using ChIP-seq and targeted sequence capture. Nucleic Acids Res. 2015;43:3389-3404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Polstein LR, Perez-Pinera P, Kocak DD, et al. Genome-wide specificity of DNA binding, gene regulation, and chromatin remodeling by TALE- and CRISPR/Cas9-based transcriptional activators. Genome Res. 2015;25:1158-1169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87. Perez-Pinera P, Kocak DD, Vockley CM, et al. RNA-guided gene activation by CRISPR-Cas9-based transcription factors. Nat Methods. 2013;10:973-976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88. Patterson K, Molloy L, Qu W, Clark S. DNA methylation: bisulphite modification and analysis. J Vis Exp. 2011;56:3170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89. Tsai HC, Li H, Van Neste L, et al. Transient low doses of DNA-demethylating agents exert durable antitumor effects on hematological and epithelial tumor cells. Cancer Cell. 2012;21:430-446. [DOI] [PMC free article] [PubMed] [Google Scholar]