

ABSTRACT
Aims/Introduction
Imeglimin is a novel oral hypoglycemic agent that improves blood glucose levels through multiple mechanisms of action including the enhancement of glucose‐stimulated insulin secretion (GSIS), however, the details of this mechanism have not been clarified. In the process of GSIS, activation of the transient receptor potential melastatin 2 (TRPM2) channel, a type of non‐selective cation channel (NSCCs) in β‐cells, promotes plasma membrane depolarization. The present study aimed to examine whether imeglimin potentiates GSIS via the TRPM2 channel in β‐cells.
Materials and Methods
Pancreatic islets were isolated by collagenase digestion from male wild‐type and TRPM2‐knockout (KO) mice. Insulin release and nicotinamide adenine dinucleotide (NAD+) production in islets were measured under static incubation. NSCC currents in mouse single β‐cells were measured by patch‐clamp experiments.
Results
Batch‐incubation studies showed that imeglimin enhanced GSIS at stimulatory 16.6 mM glucose, whereas it did not affect basal insulin levels at 2.8 mM glucose. Imeglimin increased the glucose‐induced production of NAD+, a precursor of cADPR, in islets and the insulinotropic effects of imeglimin were attenuated by a cADPR inhibitor 8‐Br‐cADPR. Furthermore, imeglimin increased NSCC current in β‐cells, and abolished this current in TRPM2‐KO mice. Imeglimin did not potentiate GSIS in the TRPM2‐KO islets, suggesting that imeglimin’s increase of NSCC currents through the TRPM2 channel is causally implicated in its insulin releasing effects.
Conclusions
Imeglimin may activate TRPM2 channels in β‐cells via the production of NAD+/cADPR, leading to the potentiation of GSIS. Developing approaches to stimulate cADPR‐TRPM2 signaling provides a potential therapeutic tool to treat type 2 diabetes.
Keywords: Cyclic ADP ribose, Imeglimin, Transient receptor potential melastatin 2
Imeglimin may activate TRPM2 channels in β‐cells via the production of NAD+/cADPR, leading to the potentiation of GSIS.

INTRODUCTION
Imeglimin, with the chemical name (6R)‐(+)‐4‐4‐dimethylamino‐2‐imino‐6‐methyl‐1,2,5,6‐tetrahydro‐1,3,5‐triazine hydrochloride, is the first member of the oral tetrahydrotriazine‐containing compounds, a glimin. It was originally developed to be used in combination therapies in addition to other drugs to improve insulin secretion and susceptibility in patients with type 2 diabetes mellitus 1 , 2 , 3 . Detailed studies have shown that this drug may have a positive effect on metabolism by improving glucose and lipid profiles in the context of diabetes 2 , 4 , 5 . Imeglimin is currently under clinical trials around the world and has been reported to improve both blood glucose and glycated hemoglobin (HbA1c) levels and to be safe 4 , 6 .
Evidence emerging from in vitro and in vivo studies suggests that imeglimin has a potent anti‐hyperglycemic effect and can normalize glucose homeostasis via several pathways including improving mitochondrial function 3 , maintaining β‐cell function, and increasing glucose‐induced insulin secretion 5 , 7 . Hallakou‐Bozec and colleagues have recently proposed that imeglimin stimulates insulin secretion by activating the salvage pathway in a dose‐dependent manner increasing the nicotinamide adenine dinucleotide (NAD+) pool; major metabolites of cyclic ADP‐ribose (cADPR), are also involved in the dose‐dependent mechanism 8 .
The transient receptor potential melastatin 2 (TRPM2) channel is one of the nonselective cation channels (NSCC) and is expressed in pancreatic β‐cells 9 . It has been long suggested that glucose‐induced insulin secretion in pancreatic β‐cells was assumed to be caused by the closure of ATP‐sensitive K+ (KATP) channels and thereby depolarization of the membrane 10 . Theoretically, the closure of KATP channels is an essential process for glucose‐induced insulin secretion, but it is not sufficient to induce a shift in membrane potential to a threshold level, because the membrane potential is determined by the overall balance of outward and inward currents 11 . During the initiation of insulin secretory stimulation in pancreatic β‐cells, glucose‐stimulated opening of NSCC results in increased background currents and increased Na+ and Ca2+ influx into the cell 11 . With respect to the regulation of the resting membrane potential, dual channel‐regulation by KATP channels and NSCCs is important, as this phenomenon works in conjunction with the increased membrane resistance due to the closure of KATP channels to promote plasma membrane depolarization 12 . We have demonstrated that the presence of TRPM2 plays an important role in β‐cell depolarization by GLP‐1 and glucose 13 , while adrenaline suppresses background currents, prolongs latency to depolarization, and reduces β‐cell glucose responsiveness 14 . It has been suggested that cADPR may be involved in the GSIS of imeglimin 8 due to its importance in the mechanism of insulin secretion via TRPM2 channels through its function as a TRPM2 promoter 15 . However, the relationship between imeglimin and the cADPR‐TRPM2 pathway and the involvement of imeglimin in phase 1 insulin secretion are unknown. In order to clarify these mechanisms, we conducted several experiments using male C57BL/6J mice and TRPM2‐knockout (KO) mice with reduced phase 1 insulin secretion for comparison 16 .
MATERIALS AND METHODS
Preparation of islets and single pancreatic β‐cell
Male C57BL/6J mice (CLEA Japan, Inc., Tokyo, Japan) and TRPM2‐KO mice were used according to the institutional guidelines and the Physiological Society of Japan’s animal husbandry guidelines for a 12 h light/dark cycle. TRPM2‐KO mice were kindly provided by Dr Y. Mori (Kyoto University) 17 . TRPM2‐KO mice were backcrossed with the C57BL/6J strain for at least nine generations. They were kept in an air‐conditioned room, and food and water were freely available. Islets of Langerhans were isolated by collagenase digestion from male C57BL/6J and TRPM2‐KO mice (aged 8–12 weeks) using a previously reported method 18 , 19 After anesthesia by intraperitoneal injection of pentobarbital (100 mg/kg), collagenase 1.05 mg/mL in Krebs‐Ringer bicarbonate buffer (HKRB) supplemented with HEPES containing 5 mM CaCl2 was injected directly into the common bile ducts. The composition of HKRB was 129 mM NaCl, 5 mM NaHCO3, 4.7 mM KCl, 1.2 mM KH2PO4, 2 mM CaCl2, 1.2 mM MgSO4, 10 mM HEPES, pH 7.4, NaOH. The pancreas was dissected and incubated in HKRB buffer for 15 min at 37°C. Size‐matched islets were harvested and used for insulin and dinucleotide release experiments in static incubation. In addition, electrophysiological experiments were performed using single β‐cells dispersed in Ca‐free HKRB 0.01% bovine serum albumin (fatty acid free; purchased from Sigma‐Aldrich). The HKRB was used to measure insulin secretion. All experimental protocols for animal experiments were conducted according to and approved by the Institutional Committee on Animal Care of Jichi Medical University. Every effort was made to minimize the suffering.
Measurement of insulin secretion and NAD+ production from mice islets
Each batch of 10 size‐matched islets was placed in HKRB with 2.8 mM glucose for stabilization for 30 min at 37°C, and then incubated in HKRB with 2.8 mM or 16.6 mM glucose with and without imeglimin (100 µM; Adooq Bioscience LLC, USA) for 10 min to evaluate the first phase of insulin secretion, and 60 min to evaluate the late phase of insulin secretion. Microtubes were centrifuged in a temperature of 4°C at 700 rpm for 15 s, and the supernatant of each microtube was collected and measured for insulin concentration using ELISA kits (Morinaga Institute of Biological Science, Yokohama, Japan). Other compounds were added to test culture batches at various concentrations, as described below. 2‐APB (10 μM; Cayman Chemical, Ann Arbor, MI, USA) was used as a TRP channel blocker; 8‐bromo‐cyclic adenosine diphosphate ribose (8‐Br cADPR; 100 µM; Santa Cruz Biotechnology, Inc. Dallas, Tx, USA) was used as a cADPR inhibitor. After 10 min incubation in HKRB with 2.8 or 16.6 mM glucose with imeglimin and 2‐APB, 8‐Br cADPR secreted insulin. The dinucleotide content was determined with 10 islets/microtube; islets were placed in HKRB with 16.6 mM glucose with or without imeglimin for 60 min. The supernatants were removed by centrifugation and the islets were stored at −80°C followed by lysis in PBS dodecyltrimethylammonium bromide solution; NAD+ was quantified by a luciferase assay provided in the NAD+/NADH Glo assay kit (#G9071, Promega, Madison, WI, USA).
Patch‐clamp experiments
Whole cell currents perforated were recorded using a pipette solution containing amphotericin B (200 mg/mL) dissolved in 0.1% DMSO. Membrane currents, which were recorded using an amplifier (Axopatch 200B; Axon, Foster, CA), were stored online on a computer with pCLAMP 10.2 software. When the series resistance was under 20 MΩ, the voltage clamp in perforated mode was considered appropriate. Patch pipettes were purchased from Narishige (Tokyo, Japan) and their resistances ranged from 3 to 5 MΩ when filled with a pipette solution containing 40 mmol/L K2SO4, 50 mmol/L KCl, 5 mmol/L MgCl2, 0.5 mmol/L EGTA, and 10 mmol/L HEPES at pH 7.2, KOH. To record the background current, the β‐cells were voltage‐clamped at a holding potential of −70 mV in the presence of 100 µmol/L tolbutamide, which is a sufficient concentration to specifically block the KATP channels; the residual current is a background current corresponding to NSCC conductance as reported previously 13 , 20 . Since glucose increases the TRPM2 current in a concentration‐dependent manner 13 , the membrane potentials were clamped at sub‐stimulatory concentrations of glucose (5.6 mM) in order to measure the actual effects of the drugs. Electrophysiological experiments were performed at 32–37°C. Since β‐cells have a wide variety of sizes and insulin secreting abilities, in this experiment, statistical processing was performed using current density (pA/pF) corrected by capacitance.
Statistical analysis
Data are presented as the mean ± standard error of the mean. Statistical analyses were performed by paired or unpaired Student’s t‐test with GraphPad Prism 7.0 software. Values of P < 0.05 were considered statistically significant.
RESULTS
Enhancement of GSIS by imeglimin
The effects of imeglimin on insulin release in mouse‐isolated islets were measured under basal (2.8 mM) and stimulatory (16.6 mM) glucose conditions. Insulin release from islets under batch‐incubation conditions was greater with 16.6 mM glucose than with 2.8 mM glucose. The early and late phases of glucose (16.6 mM)‐induced insulin release were both enhanced significantly by imeglimin (100 µM), whereas imeglimin did not affect basal insulin secretion at 2.8 mM glucose (Figure 1).
Figure 1.
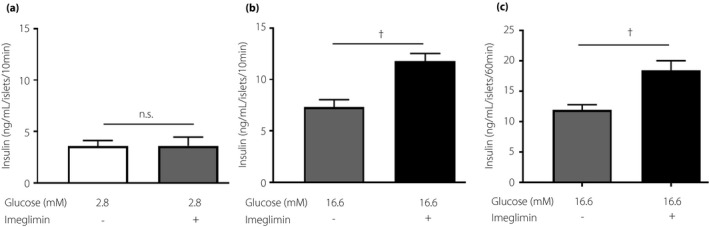
Imeglimin enhanced insulin secretion at supra threshold concentrations of glucose. Ten size‐matched mouse islets were placed in each batch and incubated for 10 min under (a) 2.8 mM glucose and (b) 16.6 mM glucose, and (c) for 60 min under 16.6 mM glucose. The number of data points was 4–7. n.s.; not significant. †P < 0.005, vs 16.6 mM glucose, by unpaired t‐test.
NAD+/cADPR‐mediated insulinotropic effects of imeglimin
We measured NAD+ production in mouse‐isolated islets. The production of NAD+ in the islets under static incubation was stimulated by 16.6 mM glucose compared with 2.8 mM glucose, but this difference was not statistically significant (Figure 2a). As NAD+ is a precursor of cADPR, the involvement of cADPR in the enhancing effects of imeglimin on the insulin release was assessed using 8‐Br‐cADPR (cADPR inhibitor). Glucose‐induced and imeglimin‐potentiated GSIS in isolated islets were significantly inhibited by 8‐Br‐cADPR. In the presence of 8‐Br‐cADPR, imeglimin did not significantly enhance GSIS (Figure 2b), indicating the NAD+/cADPR‐mediated effects of imeglimin.
Figure 2.
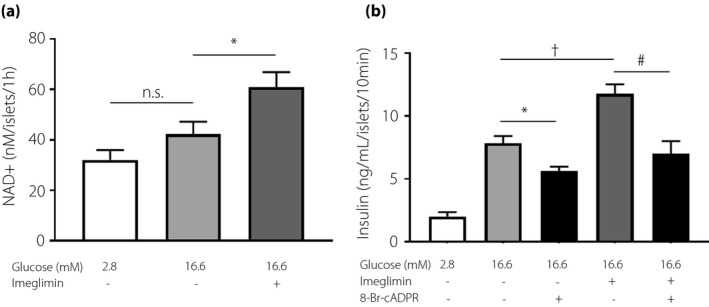
Insulin secretion effects by imeglimin related NAD+ and cADPR. Imeglimin increased NAD+ content and NAD+ biosynthesis in pancreatic islets of wild‐type mice. (a) Each batch containing 10 wild‐type mouse islets was incubated for 60 min in 2.8 or 16.6 mM of glucose, and imeglimin was added in 16.6 mM glucose. The number of data points was 5–6. Imeglimin‐induced insulin secretion was inhibited by cADPR inhibitors. (b) Each batch containing 10 wild‐type mouse islets was incubated for 10 min and insulin secretion was enhanced with 2.8 or 16.6 mM glucose. 100 μM Imeglimin or 100 μM 8‐Br‐cADPR was added. The number of data points was 5–10. n.s.; not significant. *P < 0.05, vs 16.6 mM glucose, †P < 0.005, vs 16.6 mM glucose, #P < 0.005, vs 16.6 mM glucose with 100 µM imeglimin, by unpaired‐t test.
Imeglimin potentiates GSIS via activation of TRPM2 channel
We examined whether the activation of the TRPM2 channel was involved in imeglimin‐induced enhancement of insulin release. The imeglimin‐potentiated insulin release was attenuated by a TRPM2 channel blocker 2‐APB (10 µM) (Figure 3a). In islets isolated from TRPM2‐KO mice, 16.6 mM glucose induced insulin release was significantly lower than in wild‐type mice, whereas basal insulin release at low glucose (2.8 mM) remained unchanged. Imeglimin did not enhance 16.6 mM glucose‐induced insulin release in the TRPM2‐KO islets (Figure 3b). To test the effects of imeglimin on the NSCC current, we voltage‐clamped the cells at −70 mV, which is close to the potassium equilibrium potential, and used tolbutamide to inhibit the KATP channel at 5.6 mM glucose.
Figure 3.
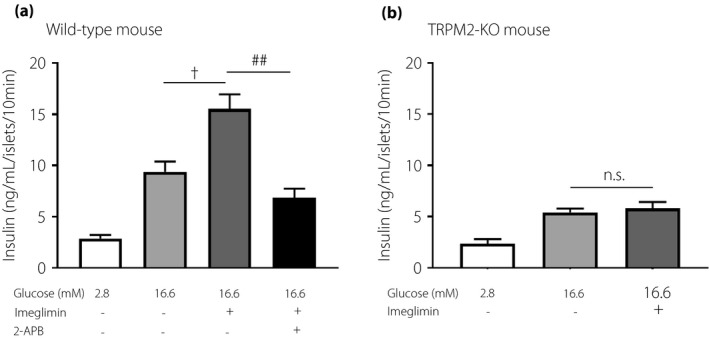
Imeglimin‐induced insulin secretion was inhibited by transient receptor potential (TRP) channel blockers and in TRPM2‐knock out mouse. (a) Each batch containing 10 wild‐type mouse islets was incubated for 10 min and insulin secretion was enhanced with 2.8 or 16.6 mM glucose. 100 μM imeglimin or 10 μM 2‐APB was added in 16.6 mM glucose. (b) Each batch containing 10 TRPM2 knockout mouse islets was incubated for 10 min and insulin secretion was enhanced with 2.8 or 16.6 mM glucose. 100 μM Imeglimin was added in 16.6 mM glucose. The number of data points was 5–11. n.s.; not significant. †P < 0.005 vs 16.6 mM glucose, ##P < 0.001 vs 16.6 mM glucose with 100 µM imeglimin by unpaired‐t test.
Imeglimin caused a significant increase of NSCC currents in β‐cells isolated from wild‐type mice, but not from TRPM‐KO mice (Figure 4a–d). In the absence of imeglimin stimulation, there was no change in the current in either the wild type or knockout mouse (Figure S1). These results suggest that the insulinotropic action of imeglimin is mediated by potentiating NSCC currents through TRPM2 channels.
Figure 4.
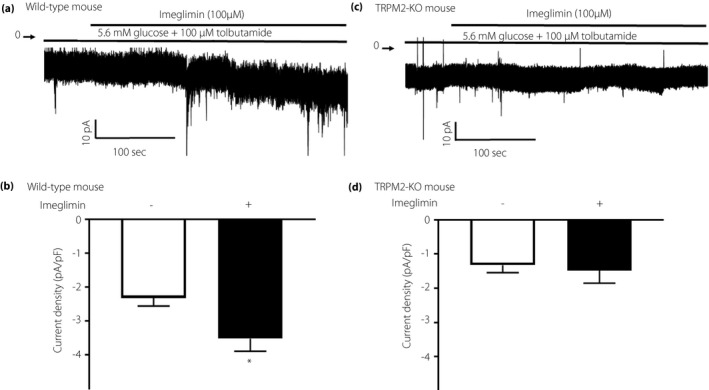
Nonselective cation channel (NSCC) current in pancreatic β‐cells are increased by imeglimin. Each mouse single β‐cell was voltage‐clamped at –70 mV in the presence or absence of 100 μM imeglimin, under the condition of 5.6 mM glucose and 100 μM tolbutamide throughout the experiments. (a) Inward NSCC current was increased by imeglimin in wild‐type β‐cell. (b) In β‐cells from TRPM2‐KO mice, imeglimin did not induce NSCC current. (c,d) The current density calculated from the averaged control current for 10 s just before the start of imeglimin stimulation is shown as a white column, and the current density calculated from the averaged peak current during 5 min of imeglimin stimulation is shown as a black column. The increase in NSCC current density induced by imeglimin was only seen in wild‐type mice. The number of data points was 5 β‐cells. *P < 0.05 by paired‐t test.
DISCUSSION
Imeglimin has been established to exert anti‐diabetic effects by improving mitochondrial function, inhibiting glucose production, and enhancing glucose‐dependent insulin secretion 21 . In this study, we confirmed the glucose‐dependent insulinotropic action of imeglimin in that the early and late phases of glucose (16.6 mM)‐induced insulin release from isolated islets were both enhanced significantly by imeglimin, whereas it did not affect basal insulin secretion at 2.8 mM glucose. While the details on the mechanism by which imeglimin exhibits GSIS were unavailable, we found that imeglimin increased NSCC current in β‐cells It is well known that background NSCC currents play an important role in pancreatic β‐cell insulin secretion 11 . Our group has previously reported the importance of background currents as well as the KATP pathway in insulin secretion of pancreatic β‐cells 13 . In addition, TRPC3, a member of the TRP family, contributes to GSIS via phospholipase C (PLC) and protein kinase C (PKC) 20 .
TRP proteins function not only as Ca2+ influx channels but also as anchor proteins in the plasma membrane 22 , 23 . The decrease in TRPM2 protein may also be due to the inability of pancreatic β‐cells to maintain EPAC and SUR1 (proteins of KATP channel), which are necessary for insulin secretion, in their normal positions, resulting in decreased insulin secretion. In the present study, imeglimin did not enhance GSIS in the TRPM2‐KO islets, suggesting that the NSCC currents through TRPM2 channels are implicated in imeglimin‐potentiated insulin secretion. Uchida et al. performed an oral glucose tolerance test on TRPM2‐KO mice and found that insulin secretion was significantly decreased 15 min after glucose loading compared with wild‐type mice, suggesting that TRPM2 channels contribute to phase 1 insulin secretion 15 . Since imeglimin showed GSIS at 10 min batch‐incubation, which was cancelled in TRPM2‐KO mice and by cADPR inhibitors, it is safe to assume that imeglimin enhances phase 1 insulin secretion via the cADPR‐TRPM2 pathway. Decreased phase 1 insulin secretion was thought to be the earliest detectable defect of β‐cell dysfunction 24 , and it is known to precede the onset of type 2 diabetes 25 . Since TRPM2‐KO mice have decreased phase 1 insulin secretion 15 , imeglimin, which improves phase 1 insulin secretion via TRPM2 channels, is expected to improve the pathogenesis of diabetes, a subject that warrants further investigation. In the present study, imeglimin was also effective on late phase insulin secretion. It has been reported that imeglimin enhances phase 2 insulin secretion. The fact that leucine and succinate, which are essential for the anaplerotic and oxidative pathways of mitochondrial metabolism, were found to enhance insulin secretion by imeglimin 3 suggests that mechanisms related to mitochondrial metabolism would be partly implicated in the late phase of the secretion. A relationship between mitochondrial metabolism and TRPM2 activity is interesting, and should be elucidated in future.
There are many mechanisms by which imeglimin improves blood glucose levels. Imeglimin enhances mitochondrial function by modulating the activity of complexes I and III, promoting the oxidation of mitochondrial fatty acids, and normalizing the phospholipid composition in the mitochondria of diabetic animals 4 . In addition, imeglimin decreases glycogenesis in the liver by inhibiting phosphoenolpyruvate carboxykinase (PEPCK) and glucose‐6‐phosphatase (G6Pase) 5 . Imeglimin has also been reported to stimulate pancreatic islets in diabetic rats and to activate NAD+‐dependent mechanisms and to salvage pathways in a dose‐dependent manner. The increase in NAD+ content by imeglimin had been shown to be due to an increase in NAD+ synthesis coming from nicotinamide via the salvage pathway and not to the de novo tryptophan pathway 8 . In the present study, we showed that imeglimin promoted NAD+ synthesis and that a cADPR inhibitor attenuated the insulinotropic response of imeglimin, suggesting that the mechanism is cADPR‐mediated.
cADPR was first isolated as a putative metabolite of NAD+ and it is known to be a second messenger of calcium (Ca2+) signaling 26 . It has been reported that a glucose‐induced increase in cADPR concentration was not observed in Ob/Ob mice, which are diabetic models 27 .
CD38, a glycoprotein found on the surface of many immune cells is active as a cADPR‐producing enzyme involved in glucose‐induced insulin release 28 . Transgenic mice overexpressing CD38 show enhanced glucose‐induced insulin release, whereas, conversely, CD38 knockout mice display a severe impairment in β‐cell function 29 . It has been shown that CD38 gene mutations are more common in non‐insulin‐dependent diabetic patients than in healthy subjects, and that administration of sera from patients with CD38 gene mutations attenuates insulin secretion 30 . The mechanism, as reported by Okamoto et al., is ATP‐mediated CD38 production inhibiting the hydrolysis of cADPR and increasing intracellular calcium, leading to insulin secretion 31 . Glucose‐stimulated ATP production has been shown to be reduced in Goto‐Kakizaki rats, a spontaneous model of diabetes and in patients with type 2 diabetes 32 , 33 .
In addition, cADPR is a potent opener of TRPM2 channels and has been reported to be involved in the insulin secretion of pancreatic β‐cells 16 . Recent investigations have demonstrated that there is a binding pocket where cADPR can dock to human TRPM2 channels and that cADPR directly activates human TRPM2 channels 34 . There is accumulating knowledge on the linkage between cADPR and TRPM2 channels, and our results suggest that imeglimin manifests GSIS through the cADPR‐TRPM2 pathway.
There are several limitations to this study. The fact that cADPR promotes Ca2+ release is controversial 35 , we recognize that the role of cADPR in islets is still open to debate. In assessing the role of mediators in enhancing insulin secretion in pancreatic β‐cells, our experiments were limited by the inability to measure levels of cADPR in islets from this diabetic mouse model. In future studies, it will be important to compare the results with those of mice treated with imeglimin and to measure cADPR itself. It is also worthwhile to examine how the opening of TRPM2 channels is affected when CD38‐KO mice are treated with imeglimin. Evaluating the involvement of TRPM2 in NAD+/cADPR production using TRPM2‐KO mice is also interesting.
In this study, we reported that imeglimin enhances insulin secretion. In clinical practice, caution should be exercised when imeglimin is used in combination with sulfonylureas. One randomized controlled trial revealed that imeglimin significantly improved HbA1c compared with placebo 36 . However, no study has compared it with sulfonylureas. In the present study, we demonstrated that imeglimin contributes to enhanced insulin secretion by opening TRPM2. Although the mechanism of action is different, we have reported that GLP‐1 opens TRPM2 and enhances insulin secretion 13 . Since there are reports that incretin‐related drugs increase the risk of hypoglycemia when used in combination with sulfonylureas 37 , it is of concern that imeglimin, which is also mediated by the TRPM2 pathway, may also increase the risk of hypoglycemia when used in combination with sulfonylureas. We need to be careful if imeglimin is used simultaneously with sulfonylureas in clinical situations. High‐dose sulfonylureas and imeglimin may induce more frequent hypoglycemic episodes.
In conclusion, our results suggest that the TRPM2 channel‐mediated mechanism plays an important role in imeglimin‐induced glucose‐dependent insulin secretion. The results of this study provide new insights into the first and late phase of insulin secretion of imeglimin, and the discovery of this pathway may lead to future signal analysis and new targets for diabetes treatment and the prevention of diabetes development.
DISCLOSURE
The authors declare having no conflicts of interest.
Approval of the research protocol: All experimental protocols for animal experiments were conducted according to and approved by the Institutional Committee on Animal Care of Jichi Medical University (20094‐01).
Informed consent: N/A.
Approval date of registry and the registration no. of the study: N/A.
Animal studies: All animal experiments were conducted following the national guidelines and the relevant national laws on the protection of animals.
Supporting information
Figure S1 | Nonselective cation channel (NSCC) current in pancreatic β‐cells did not differ between wild and TRPM2 knockout mice. (a) Wild and (b) TRPM2 knockout mouse Each mouse single β‐cells were voltage‐clamped at –70 mV, under the condition of 5.6 mM glucose and 100 μM tolbutamide throughout the experiments. (c,d) The current density calculated from the average current for 10 s immediately after the start of recording is shown as a white column (0 min), and the current density calculated from the averaged peak current during 3–5 min is shown as a black column (3–5 min). The number of data points was 5. *P < 0.05 by paired‐t test.
ACKNOWLEDGMENTS
This work was supported by a Grant‐in‐Aid for Young Scientist (JSPS KAKENHI Grant Number 19K18012), Scientific Research (C) (JSPS KAKENHI Grant Number 18K08525) and Scientific Research (C) (JSPS KAKENHI Grant Number 18K08524) to H.Y., M.Y. and K.D., and JMU Start‐Up Grant for Young Investigators to S.F.
J Diabetes Investig 2022; 13: 34–41
Contributor Information
Masashi Yoshida, Email: myoshida-md@jichi.ac.jp.
Katsuya Dezaki, Email: dezaki@jichi.ac.jp, Email: dezaki.katsuya@isu.ac.jp.
REFERENCES
- 1. Pirags V, Lebovitz H, Fouqueray P. Imeglimin, a novel glimin oral antidiabetic, exhibits a good efficacy and safety profile in type 2 diabetic patients. Diabetes Obes Metab 2012; 14: 852–858. [DOI] [PubMed] [Google Scholar]
- 2. Fouqueray P, Pirags V, Diamant M, et al. The efficacy and safety of imeglimin as add‐on therapy in patients with type 2 diabetes inadequately controlled with sitagliptin monotherapy. Diabetes Care 2014; 37: 1924–1930. [DOI] [PubMed] [Google Scholar]
- 3. Perry RJ, Cardone RL, Petersen MC, et al. Imeglimin lowers glucose primarily by amplifying glucose‐stimulated insulin secretion in high‐fat‐fed rodents. Am J Physiol Endocrinol Metab 2016; 311: E461–470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Vial G, Chauvin M‐A, Bendridi N, et al. Imeglimin normalizes glucose tolerance and insulin sensitivity and improves mitochondrial function in liver of a high‐fat, high‐sucrose diet mice model. Diabetes 2015; 64: 2254–2264. [DOI] [PubMed] [Google Scholar]
- 5. Fouqueray P, Leverve X, Fontaine E, et al. Imeglimin ‐ a new oral anti‐diabetic that targets the three key defects of type 2 diabetes. Diabetes Metab J 2011; 2:126. [Google Scholar]
- 6. Dubourg J, Ueki K, Grouin JM, et al. Efficacy and safety of imeglimin in Japanese patients with type 2 diabetes mellitus: a 24‐week, randomized, double‐blind, placebo‐controlled, dose‐ranging phase 2b trial. Diabetes Obes Metab 2021; 23:800–810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Vuylsteke V, Chastain LM, Maggu GA, et al. Imeglimin: a potential new multi‐target drug for type 2 diabetes. Drugs in R&D 2015; 15: 227–232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Hallakou‐Bozec S, Kergoat M, Fouqueray P, et al. Imeglimin amplifies glucose‐stimulated insulin release from diabetic islets via a distinct mechanism of action. PLoS One 2021; 16: e0241651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Uchida K, Tominaga M. The role of thermosensitive TRP (transient receptor potential) channels in insulin secretion. Endocrine J 2011; 58: 1021–1028. [DOI] [PubMed] [Google Scholar]
- 10. Ashcroft FM, Rorsman P. Diabetes mellitus and the β cell: the last ten years. Cell 2012; 148: 1160–1171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Kakei M, Yoshida M, Dezaki K, et al. Glucose and GTP‐binding protein‐coupled receptor cooperatively regulate transient receptor potential‐channels to stimulate insulin secretion [Review]. Endocrine J 2016; 63: 867–876. [DOI] [PubMed] [Google Scholar]
- 12. Leech CA, Habener JF. A role for Ca2+‐sensitive nonselective cation channels in regulating the membrane potential of pancreatic beta‐cells. Diabetes 1998; 47: 1066–1073. [DOI] [PubMed] [Google Scholar]
- 13. Yosida M, Dezaki K, Uchida K, et al. Involvement of cAMP/EPAC/TRPM2 activation in glucose‐ and incretin‐induced insulin secretion. Diabetes 2014; 63: 3394–3403. [DOI] [PubMed] [Google Scholar]
- 14. Ito K, Dezaki K, Yoshida M, et al. Endogenous α2A‐adrenoceptor‐operated sympathoadrenergic tones attenuate insulin secretion via cAMP/TRPM2 signaling. Diabetes 2017; 66: 699–709. [DOI] [PubMed] [Google Scholar]
- 15. Uchida K, Dezaki K, Damdindorj B, et al. Lack of TRPM2 impaired insulin secretion and glucose metabolisms in mice. Diabetes 2011; 60: 119–126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Togashi K, Hara Y, Tominaga T, et al. TRPM2 activation by cyclic ADP‐ribose at body temperature is involved in insulin secretion. EMBO J 2006; 25: 1804–1815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Yamamoto S, Shimizu S, Kiyonaka S, et al. TRPM2‐mediated Ca2+influx induces chemokine production in monocytes that aggravates inflammatory neutrophil infiltration. Nat Med 2008; 14: 738–747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Nakazaki M, Kakei M, Koriyama N, et al. Involvement of ATP‐sensitive K+ channels in free radical‐mediated inhibition of insulin secretion in rat pancreatic beta‐cells. Diabetes 1995; 44: 878–883. [DOI] [PubMed] [Google Scholar]
- 19. Dezaki K, Damdindorj B, Sone H, et al. Ghrelin attenuates cAMP‐PKA signaling to evoke insulinostatic cascade in islet β‐cells. Diabetes 2011; 60: 2315–2324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Yamada H, Yoshida M, Ito K, et al. Potentiation of glucose‐stimulated insulin secretion by the GPR40‐PLC‐TRPC pathway in pancreatic beta‐cells. Sci Rep 2016; 6: 25912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Yaribeygi H, Maleki M, Sathyapalan T, et al. Molecular mechanisms by which imeglimin improves glucose homeostasis. J Diabetes Res 2020; 2020: 8768954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Li HS, Montell C. TRP and the PDZ protein, INAD, form the core complex required for retention of the signalplex in Drosophila photoreceptor cells. J Cell Biol 2000; 150: 1411–1422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Tsunoda S, Sun Y, Suzuki E, et al. Independent anchoring and assembly mechanisms of INAD signaling complexes in Drosophila photoreceptors. J Neurosci 2001; 21: 150–158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Cerasi E, Luft R. "What is inherited–what is added” hypothesis for the pathogenesis of diabetes mellitus. Diabetes 1967; 16: 615–627. [DOI] [PubMed] [Google Scholar]
- 25. Kendall DM, Cuddihy RM, Bergenstal RM. Clinical application of incretin‐based therapy: therapeutic potential, patient selection and clinical use. Eur J Intern Med 2009; 20: S329–339. [DOI] [PubMed] [Google Scholar]
- 26. Guse AH. Regulation of calcium signaling by the second messenger cyclic adenosine diphosphoribose (cADPR). Curr Mol Med 2004; 4: 239–248. [DOI] [PubMed] [Google Scholar]
- 27. Takasawa S, Akiyama T, Nata K, et al. Cyclic ADP‐ribose and inositol 1,4,5‐trisphosphate as alternate second messengers for intracellular Ca2+ mobilization in normal and diabetic beta‐cells. J Biol Chem 1998; 273: 2497–2500. [DOI] [PubMed] [Google Scholar]
- 28. Guse AH. Biochemistry, biology, and pharmacology of cyclic adenosine diphosphoribose (cADPR). Curr Med Chem 2004; 11: 847–855. [DOI] [PubMed] [Google Scholar]
- 29. Antonelli A, Ferrannini E. CD38 autoimmunity: recent advances and relevance to human diabetes. J Endocrinol Investig 2004; 27: 695–707. [DOI] [PubMed] [Google Scholar]
- 30. Ikehata F, Satoh J, Nata K, et al. Autoantibodies against CD38 (ADP‐ribosyl cyclase/cyclic ADP‐ribose hydrolase) that impair glucose‐induced insulin secretion in noninsulin‐ dependent diabetes patients. J Clin Investig 1998; 102: 395–401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Tohgo A, Munakata H, Takasawa S, et al. Lysine 129 of CD38 (ADP‐ribosyl cyclase/cyclic ADP‐ribose hydrolase) participates in the binding of ATP to inhibit the cyclic ADP‐ribose hydrolase. J Biol Chem 1997; 272: 3879–3882. [DOI] [PubMed] [Google Scholar]
- 32. Mukai E, Fujimoto S, Sato H, et al. Exendin‐4 suppresses SRC activation and reactive oxygen species production in diabetic Goto‐Kakizaki rat islets in an Epac‐dependent manner. Diabetes 2011; 60: 218–226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Anello M, Lupi R, Spampinato D, et al. Functional and morphological alterations of mitochondria in pancreatic beta cells from type 2 diabetic patients. Diabetologia 2005; 48: 282–289. [DOI] [PubMed] [Google Scholar]
- 34. Yu P, Liu Z, Yu X, et al. Direct gating of the TRPM2 channel by cADPR via specific interactions with the ADPR binding pocket. Cell Rep 2019; 27: e3684. [DOI] [PubMed] [Google Scholar]
- 35. Islam MS, Berggren PO. Cyclic ADP‐ribose and the pancreatic beta cell: where do we stand? Diabetologia 1997; 40: 1480–1484. [DOI] [PubMed] [Google Scholar]
- 36. Dubourg J, Fouqueray P, Thang C, et al. Efficacy and safety of imeglimin monotherapy versus placebo in Japanese patients with type 2 diabetes (TIMES 1): a double‐blind, randomized, placebo‐controlled, parallel‐group, multicenter phase 3 trial. Diabetes Care 2021; 44: 952–959. [DOI] [PubMed] [Google Scholar]
- 37. Salvo F, Moore N, Arnaud M, et al. Addition of dipeptidyl peptidase‐4 inhibitors to sulphonylureas and risk of hypoglycaemia: systematic review and meta‐analysis. BMJ 2016; 353: i2231. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1 | Nonselective cation channel (NSCC) current in pancreatic β‐cells did not differ between wild and TRPM2 knockout mice. (a) Wild and (b) TRPM2 knockout mouse Each mouse single β‐cells were voltage‐clamped at –70 mV, under the condition of 5.6 mM glucose and 100 μM tolbutamide throughout the experiments. (c,d) The current density calculated from the average current for 10 s immediately after the start of recording is shown as a white column (0 min), and the current density calculated from the averaged peak current during 3–5 min is shown as a black column (3–5 min). The number of data points was 5. *P < 0.05 by paired‐t test.