

Abstract
Aims/Introduction
Diabetic nephropathy (DN) is among the leading causes of end‐stage renal disease worldwide. DN pathogenesis remains largely unknown. Weighted gene co‐expression network analysis is a powerful bioinformatic tool for identifying key genes in diseases.
Materials and Methods
The datasets GSE30122, GSE104948, GSE37463 and GSE47185 containing 23 DN and 23 normal glomeruli samples were obtained from the National Center for Biotechnology Information Gene Expression Omnibus database. After data pre‐processing, weighted gene co‐expression network analysis was carried out to cluster significant modules. Then, Gene Set Enrichment Analysis‐based Gene Ontology analysis and visualization of network were carried out to screen the key genes in the most significant modules. The connectivity map analysis was carried out to find the significant chemical compounds. Finally, some key genes were validated in in vivo and in vitro experiments.
Results
A total of 454 upregulated and 392 downregulated genes were identified. A total of 16 modules were clustered, and the most significant modules (green, red and yellow modules) were determined. The green module was associated with extracellular matrix organization, the red module was associated with immunity reaction and the yellow module was associated with kidney development. We found several key genes in these three modules separately, and part of them were validated in vivo and in vitro successfully. We found the top 15 chemical compounds that could perturb the overall expression of key genes in DN.
Conclusion
Weighted gene co‐expression network analysis was applied to DN expression profiling in combination with connectivity map analysis. Several novel key genes and chemical compounds were screened out, providing new molecular targets for DN.
Keywords: Connectivity map analysis, Diabetic nephropathy, Weighted gene co‐expression network analysis
Weighted gene co‐expression network analysis was applied to diabetic nephropathy expression profiling in combination with connectivity map analysis. Several novel key genes and chemical compounds were screened out, providing new molecular targets for diabetic nephropathy.

INTRODUCTION
Diabetic nephropathy (DN) is among the leading causes of end‐stage renal disease worldwide 1 , 2 , 3 . The classical pathological characteristics of DN include excessive organization and accumulation of extracellular matrix (ECM) that finally deteriorates into nodular glomerulosclerosis 4 , 5 . Stimulated by advanced glycation end‐products, Smad1 can upregulate the expression of Col4a1/a2, the main component of ECM, and then cause mesangial area expansion; meanwhile, the upstream bone morphogenetic protein 4 could regulate Smad1, thereby showing that the bone morphogenetic protein 4/Smad1 signaling pathway plays an important role in DN 6 , 7 , 8 . Furthermore, the Notch1/Jagged1 and Wnt/β‐catenin signaling pathways reportedly regulate the organization of ECM in DN 9 , 10 , 11 , 12 , 13 .
The immune system also plays a key role in DN. The innate immune and complement systems are involved in DN development, whereas cell‐mediated immunity in DN requires further investigation 14 , 15 , 16 . Previous studies have shown that excessively activated T cells are related to proteinuria in DN, but the specific mechanism remains unclear 17 . Subsequent studies suggested that it might be related to the increased infiltration of activated CD3+, CD4+ and CD8+ T cells in the renal interstitium. Meanwhile, the interferon‐γ and tumor necrosis factor‐α produced by CD3+ T cells increased, which could lead to inflammatory responses in DN 18 , 19 . Relevant studies showed that DN could be alleviated by inhibiting the activation of T cells 20 , 21 .
However, information on the molecular mechanism remains incomplete, and these previous studies insufficiently provided global evidence on the molecular characteristics of DN. Thus, a new and systematic research strategy is required. Weighted gene co‐expression network analysis (WGCNA), a new and powerful bioinformatics analysis method, can construct scale‐free gene co‐expression networks and identify functionally similar modules by analyzing gene expression profiles and calculating the gene‐weighted correlation. Furthermore, a relationship network and significant modules can be constructed. Thus, WGCNA is widely implemented in medical research on topics such as clear cell renal cell carcinoma, hepatocellular carcinoma, Schmid‐type metaphyseal chondrodysplasia and others, thereby leading to research progress 22 , 23 , 24 , 25 .
In the present study, after the integration of different batches of gene expression profiling microarray data, we used WGCNA to construct a scale‐free network and identified significant modules in DN. Then, we used Gene Set Enrichment Analysis (GSEA) on significant modules to screen key Gene Ontology (GO) terms. We also visualized and analyzed the networks of the key GO terms to identify key genes. Next, we carried out connectivity map analysis based on the key genes to determine the chemical compounds perturbing the overall expression of the key genes in DN. Finally, we constructed the animal and cell models of DN to verify some key genes obtained by WGCNA to provide new key molecular targets and data resources for DN pathogenesis research.
MATERIALS AND METHODS
Searching and downloading of microarray data
Microarray datasets of DN were downloaded from the National Center for Biotechnology Information Gene Expression Omnibus (GEO) database (http://www.nibi.nih.gov/geo/) with the following search strategies: (i) keyword ‘diabetic nephropathy’; (ii) expression profiling of Homo sapiens; and (iii) glomeruli tissue. Four datasets, namely, GSE30122, GSE104948, GSE37463 and GSE47185, were obtained. By comparing the samples information, we deleted some duplicate samples in DN and normal samples. Thus, a total of 23 DN samples and 23 normal glomeruli samples were used in the present study. However, the clinical information is not available in the GEO database. All the raw data were downloaded for further analysis.
Data pre‐processing
Raw data were imported into an R Affy package (version 1.66.0, Harvard School of Public Health, Boston, MA, USA) using the robust multichip analysis algorithm for background correction, the quantile method for normalization and log2 transformation. The three microarray datasets are from different batches based on the Affymetrix platform. Thus, to merge the datasets and remove the batch effect, we implemented the ComBat method by using the R sva package (version 3.28.2, Department of Biostatistics, Bloomberg School of Public Health, Johns Hopkins University, Baltimore, ML, USA) 26 .
Identification of differentially expressed genes
We used the linear fit method, Bayesian analysis and t‐test algorithm in the R package limma (version 3.36.5, Bioinformatics Division, The Walter and Eliza Hall Institute of Medical Research, Parkville, Victoria, Australia.) to identify the differentially expressed genes (DEGs) between DN and the normal controls. The values of adjusted P < 0.05 and |log2FC|≥0.585 (fold change) were set as the cut‐off criteria.
Selection of genes for WGCNA
Before genes were selected for WGCNA, we identified and removed the outlier samples. A hierarchical cluster analysis was carried out in the R. Regarding gene selection, the genetic variance in the DN samples was calculated. The top 30% of genes with the largest variance were selected for WGCNA.
Identification of soft‐thresholding power (β)
Soft‐thresholding power (β) should be determined to identify a network that satisfies a scale‐free topology. We implemented the pickSoftThreshold function of WGCNA package (version 1.64.1, Department of Human Genetics and Department of Biostatistics, University of California, Los Angeles, CA, USA) to calculate the scale‐free topology fit index and mean connectivity of each power. If the value of the scale‐free topology fit index was > 0.9 for low powers (<30), and the mean connectivity was as large as possible, then the soft‐thresholding power (β) was determined.
Construction of weighted gene co‐expression networks
One‐step function blockwiseModules were used to construct the WGCNA and identify the modules. To identify the modules, the related parameters were set as follows: minModuleSize was 30, and mergeCutHeight was 0.25. To further evaluate the interaction of all modules, eigengene adjacency was calculated by the flashClust function. A heatmap was operated to visualize the correlations of each module.
Identification of important modules
Several modules were obtained by WGCNA. Combined differential expression analysis was carried out to identify the module with a high correlation with DN. First, the average values of the |log2(FC)| of genes were calculated in each module. Then, the average values of ‐Log (adjusted P‐value) of genes were calculated. Only the module whose value was highest in both calculations was considered the most significant to DN.
GSEA and visualization of DEGs in selected modules
We imported the most significant modules into R and clusterProfiler package (version 3.5, Institute of Life and Health Engineering, Key Laboratory of Functional Protein Research of Guangdong Higher Education Institutes, Jinan University, Guangzhou, People's Republic of China) using predefined gene sets from the Molecular Signatures Database (MSigDB v5.0) to carry out GO enrichment analysis, including biological process (BP), cellular component (CC) and molecular function gene sets. To identify the key genes in each module, the relationships among genes were visualized.
Connectivity map analysis
To connect the disease–gene–drug, we carried out the connectivity map analysis using a gene expression profile database comprising cultured human cells treated with bioactive small molecules. First, we established the gene query signature of DN derived from the aforementioned analysis results. Then, the pattern‐matching algorithms were implemented to score each reference profile data with the query signature for the direction and strength of enrichment through the online tool https://clue.io/cmap 27 .
Cell culture
The rat glomerular mesangial cells were cultured in Dulbecco’s modified Eagle’s medium containing 5.6 mmol/L glucose. The medium was supplemented with 5% fetal bovine serum, and 1% penicillin and streptomycin. The culture was grown in a 5% CO2 humidified atmosphere at 37°C. The cells were passaged at 80% confluence. The confluent cells were grown in serum‐free Dulbecco’s modified Eagle’s medium for 24 h before the experiments. The mesangial cells were then cultured with normal glucose (5.6 mmol/L glucose), high glucose (30 mmol/L glucose) or high osmolarity (osmotic control; 5.6 mmol/L glucose + 24.4 mmol/L mannitol) for 24, 48 and 72 h. Conditionally immortalized murine podocyte cells were cultured at 33°C in RPMI medium 1,640 supplemented with 10% fetal bovine serum and 10 U/mL mouse recombinant interferon‐γ. To induce differentiation, podocytes were grown at 37°C in the absence of interferon‐γ for 14 days before the experiments. Podocytes were then maintained in normal glucose (5.6 mmol/L), passaged at 80% confluence. The cells were then cultured with normal glucose (5.6 mmol/L glucose), high glucose (30 mmol/L glucose) or high osmolarity (osmotic control; 5.6 mmol/L glucose + 24.4 mmol/L mannitol) for 24, 48 and 72 h, respectively.
Establishment of DN rat models
Specific pathogen‐free male Sprague–Dawley rats (200–250 g and 8 weeks old) were obtained from the animal center of the Sichuan University, Chengdu, Sichuan, China, and fed on standard food and water. All animal experiments were carried out in the Experimental Animal Facility of West China Hospital, Sichuan University. The animals were randomly distributed into two groups, namely the control and DN groups (n = 6). After a week of adaptive feeding and fasting, 1% streptozotocin (STZ) solution was administered by intraperitoneal injection at a dose of 65 mg/kg, whereas the control group was injected with the same amount of citrate buffer. After 3 days, the rats’ blood glucose levels were measured. A blood glucose level of at least 16.7 mmol/L for 3 consecutive days represented the development of diabetes mellitus. Rats of the control and DN groups were killed at 0 and 20 weeks. The right kidney was immediately removed, fixed by 4% paraformaldehyde, embedded by paraffin and made into serial sections.
All animal procedures were approved by the Institutional Animal Care and Use Committee of Sichuan University (approval number: 2018012A; date: 9 October 2018).
Quantification by real‐time quantitative polymerase chain
Total ribonucleic acid (RNA) was extracted from mesangial cells and podocytes by Trizol (Biotek, Winooski, VT, USA). Reverse transcription of total RNA was operated by the Reverse Transcription Kit (Thermo Scientific, Waltham, MA, USA). Then, the SYBR Premix Ex Taq (Takara Bio, Kusatsu, Japan) was used in a Bio‐Rad CFX96 Real PCR System (Bio‐Rad, Hercules, CA, USA.) to detect messenger RBA (mRNA) expressions. The primers used for quantitative polymerase chain reaction amplification are listed in Table S1. Finally, the fold changes of genes were calculated by the method 2–ΔΔCt.
Immunohistochemistry
The paraffin‐embedded slides of kidney tissue were deparaffinized and dehydrated. After eliminating endogenous peroxidase activity, antigen retrieval and blocking non‐specific antigen, the slides were incubated with primary antibody (PYCARD1:100; LUM 1:100; COMP 1:100) overnight at 4°C. Then, a biotin‐labeled secondary antibody working solution was added, and the slides were incubated. Next, the horseradish peroxidase‐labeled streptavidin was dropped to the slices, and they were incubated before staining with 3,3′‐diaminobenzidine and counterstaining with hematoxylin. After conventional dewatering and neutral balsam mounting, photographs were blindly taken at random fields under a fluorescence microscope (AX10 imager A2/AX10 Cam HRC, Carl Zeiss AG, Oberkochen, Germany). The slides were digitized and analyzed by Image Pro Plus 6.0 (Media Cybernetics, Inc., Rockville, MD, USA).
Statistical analysis
Bioinformatic analysis was carried out in R platform (version 3.5.1). The results of validation experiments are presented as the mean ± standard error of the mean. One‐way anova was used for comparison among groups, and Student–Newman–Keuls test was used for multiple comparison by SPSS software (SPSS, Chicago, IL, USA). P < 0.05 was considered statistically significant. Data visualization was carried out by GraphPad Prism 6.0 (GraphPad, San Diego, CA, USA).
RESULTS
Evaluation and preparation of data
Four microarray datasets, namely, GSE30122, GSE104948, GSE37463 and GSE47185, were hit and downloaded from the GEO, then the duplicate samples were excluded. After background correction, normalization and log2 transformation, batch effect removal was carried out to eliminate systematic and technical differences between different platforms and datasets (Figure 1a,b).
Figure 1.
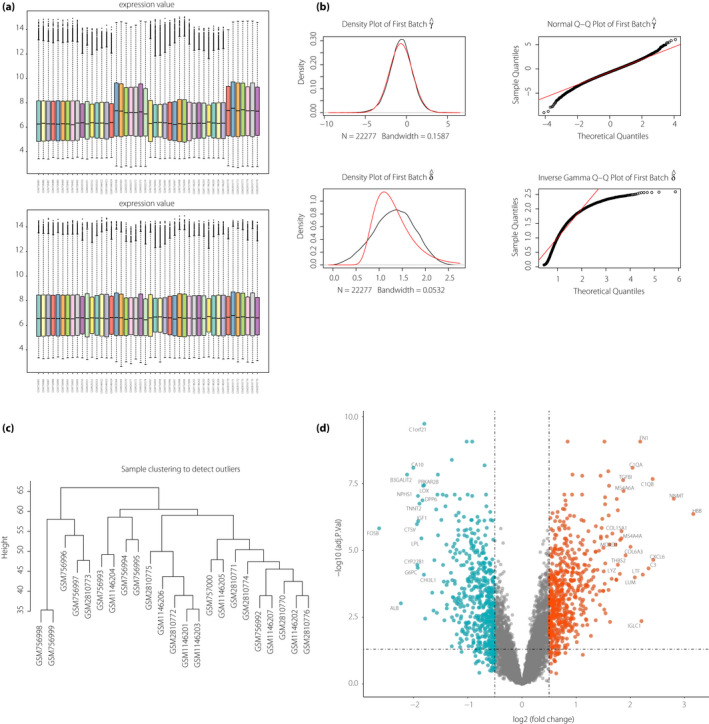
Evaluation and preparation of data. (a) Overall expression levels of all samples in the microarray after normalization. (b) The batch removal of the samples. The first line shows the distribution of the data after batch removal. The second line shows the distribution of the original data. (c) Clustering dendrogram of 23 diabetic nephropathy samples used to detect outliers. The vertical axis represents the cut height. The branches of the dendrogram represent the samples that are clustered together. (d) Volcano plot of differentially expressed genes. Differentially expressed genes in diabetic nephropathy were compared with those in normal samples. Green, red and gray plots represent downregulated, upregulated and non‐significant genes, respectively. The horizontal axis represents the log2(FC), whereas the vertical axis represents the ‐Log10 (adjusted P‐value).
The dendrogram of DN samples was used to identify and discard the outlier samples in DN. As shown in Figure 1c, all 23 DN samples were clustered and reserved for the subsequent analysis. Finally, 23 DN samples and 23 normal samples were included.
After calculating the variance of genes in the DN samples, 3,731 genes obtained from 23 DN samples were operated in the WGCNA by the one‐step function.
Identification of DEGs was carried out and visualized by a volcano plot (Figure 1d). A total of 454 upregulated genes and 392 downregulated genes were included. The number of upregulated genes (red plots) presented a pattern similar to that of downregulated genes (green plots). The distribution of genes was continuous. The hemoglobin subunit beta, nicotinamide N‐methyltransferase, C‐X‐C motif chemokine ligand 6, complement C1q B chain and complement C3 were the top five upregulated genes, whereas AP‐1 transcription factor subunit, albumin, beta‐1,3‐galactosyltransferase 2, carbonic anhydrase 10 and cathepsin V were the top five downregulated genes. Furthermore, the total DEGs in DN are shown in Table S2. After data pre‐processing, the quality of our data was suitable for the next WGCNA.
Construction of gene co‐expression networks and identification of modules
Before WGCNA, we implemented the pickSoftThreshold function to raise the soft‐thresholding powers for the calculation of the scale‐free topology fit index and mean connectivity in WGCNA. As shown in Figure 2a, when the power was 8, the scale‐free topology fit index was nearest 0.9, thereby showing that the networks were scale‐free. Thus, the power 8 was selected as the soft‐thresholding value.
Figure 2.
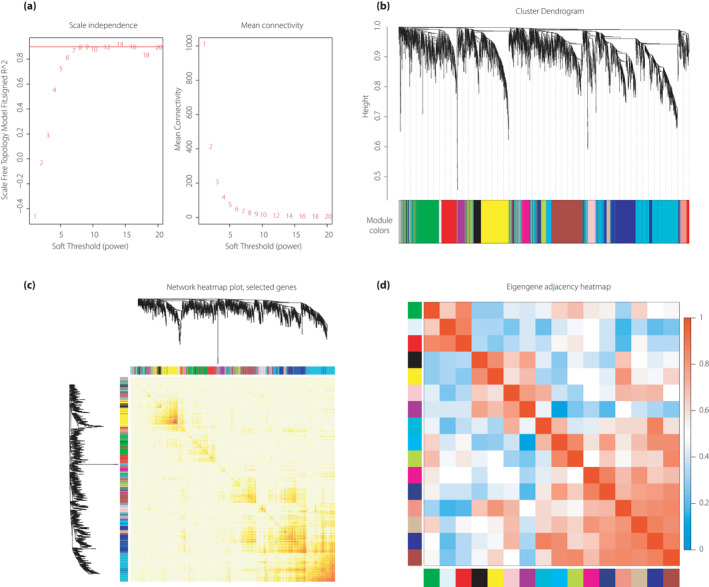
Construction of gene co‐expression networks and identification of modules by weighted gene co‐expression network analysis. (a) Analysis of different soft‐thresholding power network topology for constructing the scale‐free network. The left panel shows the scale‐free topology model fit, signed R 2 (y‐axis) as a function of the soft‐thresholding power. The red line shows that the value of the y‐axis is 0.9. The right panel represents the mean connectivity (y‐axis) as a function of the soft‐thresholding power. (b) Cluster dendrogram of genes in diabetic nephropathy samples with dissimilarity based on topological overlap. The different color row below the dendrogram represents module membership clustered by the dynamic tree cut method. (c) Network heatmap plot. Branches in the hierarchical clustering dendrograms correspond to each module on the left and top of the dendrograms. In this heatmap, the progressively more saturated yellow and red colors indicate the high co‐expression interconnectedness. (d) Heatmap plot of the adjacencies of modules. The colors of columns and row squares represent the adjacency of corresponded modules. Red represents high adjacency, whereas blue represents low adjacency.
After hierarchical clustering tree analysis based on co‐expression relationships, 16 modules were identified and labeled with different colors (Figure 2b). The gray module was particularly reserved for genes that can no longer be clustered into specific modules and that feature low intramodular connectivity.
The network heatmap plot (Figure 2c) shows the topological overlap matrix among all genes in the modules with each module showing independent validation to each other. To further evaluate the interaction of all modules, eigengene adjacency was calculated (Figure 2d). Based on co‐expression relationships, the modules were distinguished well among one another. Finally, the WGCNA network data were obtained and prepared for the subsequent analysis.
Selection of significant module
In the identified modules, the average values of |log2(FC)| and ‐Log (adjusted P‐value) of genes were calculated. Higher average values of the |log2(FC)| of genes in the module implied higher differential gene expression level, indicating that the modules were more significant and functional in the disease. As shown in Figure 3a, the values of the green, red and yellow modules were the highest among the 16 modules. A higher value of ‐Log (adjusted P‐value) presented a higher statistical significance. As shown in Figure 3b, the values of the green, red and yellow modules also remained the highest. Therefore, considering these results, the green, red and yellow modules were the most significant and valuable for further analysis.
Figure 3.
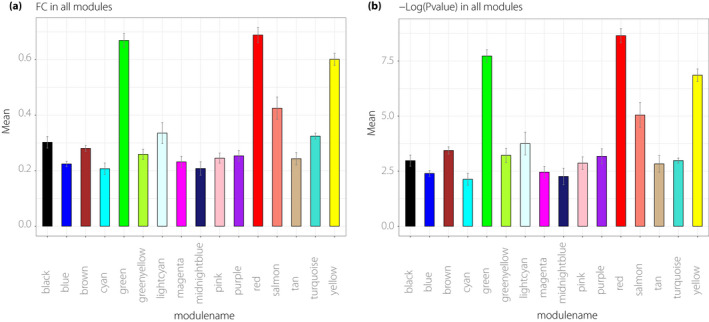
Selection of significant module. (a) The mean of the |Log2(FC)| of genes in each module. The y‐axis represents the mean of |Log2(FC)|, whereas the x‐axis represents the different modules. (b) The mean of the ‐Log (adjusted P‐value) of genes in each module. The y‐axis represents the mean of the ‐Log (adjusted P‐value), whereas the x‐axis represents the different modules. FC, fold change.
GSEA analysis and visualization of significant modules
To analyze the related functions of these three modules, GO enrichment based on the GSEA analysis was carried out.
The results of the green module are shown in Figure 4a. Most GO terms were related to ECM organization. Especially in BP, ECM organization (GO: 0030198), extracellular structure organization (GO: 0043082) and collagen catabolic process (GO: 0030574) were ranked at the top three of the GO terms. Figure 4b shows the chord diagram of the BP in the green module to identify the key genes. The upregulated genes FN1 (fibronectin 1), LUM (lumican), THBS2 (thrombospondin 2), TGFBI (transforming growth factor beta induced) and COMP (cartilage oligomeric matrix protein), and the downregulated genes CYR61 (cysteine rich angiogenic inducer 61), FGB (fibrinogen beta chain) and CRISPLD2 (cysteine rich secretory protein LCCL domain containing 2) were the key genes in the green module based on the weighted correlations.
Figure 4.
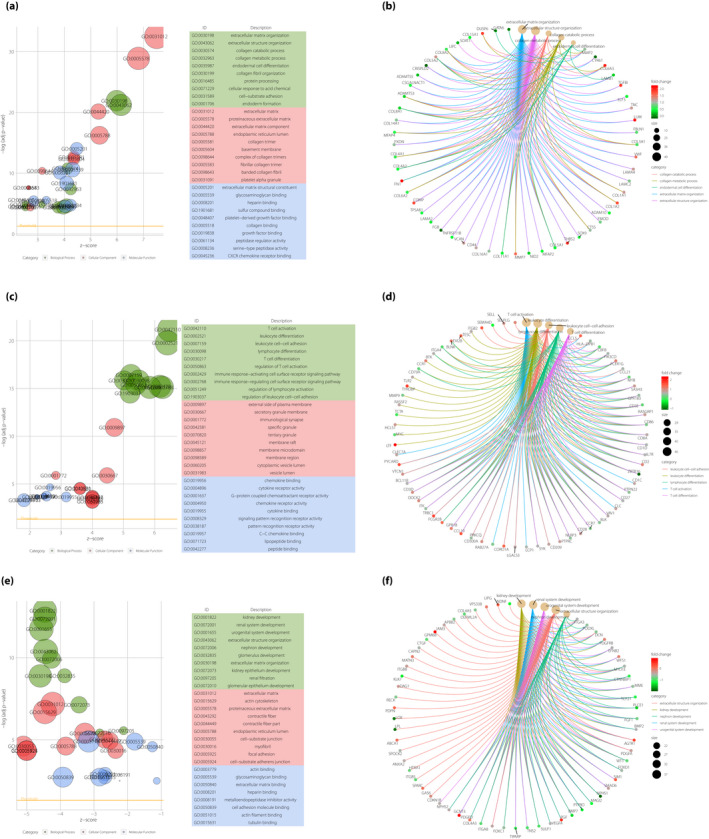
Gene Ontology (GO) enrichment analysis and network visualization of green, red and yellow modules. (a) The diagram of the GO enrichment based on the Gene Set Enrichment Analysis (GSEA) analysis in the green module. In the left part, the horizontal axis represents the z‐score of GO terms, whereas the vertical axis represents the term of the ‐Log (adjusted P‐value). The green nodes represent the biological process (BP) GO term. The red nodes represent the cellular component (CC) GO term. The blue nodes represent the molecular function (MF) GO term. The top 10 most significant GO terms are ranked in the right part. (b) The chord diagram of the weighted gene co‐expression network analysis (WGCNA) network in the green module. The node represents the genes of top five GO term clusters in the green module. The edges of genes represent genes’ weighted correlation in the sub‐network. The depth of color represents the fold change values of the gene. (c) The diagram of GO enrichment based on the GSEA analysis in the red module. In the left part, the horizontal axis represents the z‐score of GO terms, whereas the vertical axis represents the term of the ‐Log (adjusted P‐value). The green nodes represent the BP GO term. The red nodes represent the CC GO term. The blue nodes represent MF GO term. The top 10 most significant GO terms are ranked in the right part. (d) The chord diagram of the WGCNA network in the red module. The node represents the genes of the top five GO term clusters in the green module. The edges of genes represent genes’ weighted correlation in the sub‐network. The depth of color represents the fold change values of the gene. (e) The diagram of the GO enrichment based on the GSEA analysis in the yellow module. In the left part, the horizontal axis represents the z‐score of GO terms, whereas the vertical axis represents the term of the ‐Log (adjusted P‐value). The green nodes represent the BP GO term. The red nodes represent the CC GO term. The blue nodes represent the MF GO term. The top 10 most significant GO terms are ranked in the right part. (f) The chord diagram of the WGCNA network in the yellow module. The node represents the genes of the top five GO term clusters in the green module. The edges of genes represent the genes’ weighted correlation in the sub‐network. The depth of color represents the fold change values of the gene.
The results of the red module are shown in Figure 4c. Most GO terms were related to immunity and inflammation. Especially in BP, T‐cell activation (GO:0042110) was ranked at the top of the GO terms. Figure 4d shows the chord diagram of the BP in the red module. The upregulated genes TRBC1 (T‐cell receptor beta constant 1), PYCARD (PYD and CARD domain containing), CCL19 (C‐C motif chemokine ligand 19), CCL5 (C‐C motif chemokine ligand 19) and CORO1A (coronin 1A), and the downregulated gene ZBTB16 (zinc finger and BTB domain containing 16) were the key genes in the red module based on the weighted correlations.
The results of the yellow module are shown in Figure 4e. Most GO terms were related to kidney development. Especially in BP, kidney development (GO:0001822), renal system development (GO:0072001) and urogenital system development (GO:0001655) were ranked at the top three of the GO terms. Figure 4f shows the chord diagram of the BP in the green module to identify the key genes. The upregulated genes GPM6B (glycoprotein M6B), GCNT3 (glucosaminyl [N‐acetyl] transferase 3, mucin type), PGF (placental growth factor), AGTR1 (angiotensin II receptor type 1) and SIM1 (single‐minded family BHLH transcription factor 1), and the downregulated genes LOX (lysyl oxidase), LPL (lipoprotein lipase), NPHS1 (nephrin), PLCE1 (phospholipase C epsilon 1) and bone morphogenetic protein 4 were the key genes in the green module based on the weighted correlations.
Results of the connectivity map
We established the DN gene query signature based on the aforementioned results. The top 10 upregulated genes were FN1, LUM, THBS2, TGFBI, COMP, PYCARD, CCL19, CCL5, CORO1A and FCGR2B (Fc fragment of immunoglobulin G receptor IIb). The downregulated genes were CYR61, ELF3 (E74 like ETS transcription factor 3), FGB, CRISPLD2, GATA6 (GATA‐binding protein 6), IGFBP1 (insulin‐like growth factor‐binding protein 1), ZBTB16, SEMA4D (semaphoring 4D), ENPEP (glutamyl aminopeptidase) and GHR (growth hormone receptor). We carried out the connectivity MAP analysis. The signature file was imported into the online tool of connectivity MAP. The results are shown in Table 1, which shows that the scores of these top 15 chemical compounds, including triclabendazole, altizide, triamcinolone, QL‐XII‐47 and memantine, were negative. These findings showed that the overall perturbation of DN by these chemical compounds was opposite to those of the aforementioned genes. Thus, these compounds or their analogs might potentially play antagonistic roles in DN.
Table 1.
Top 15 chemical compounds in connectivity map
| Name | Score | Description |
|---|---|---|
| Triclabendazole | −98.41 | Microtubule inhibitor |
| Altizide | −94.08 | Thiazide diuretic |
| Triamcinolone | −92.74 | Glucocorticoid receptor agonist |
| QL‐XII‐47 | −91.6 | BTK inhibitor |
| Memantine | −90.21 | Glutamate receptor antagonist |
| Etamivan | −90.07 | Respiratory stimulant |
| Fluocinolone | −89.82 | Glucocorticoid receptor agonist |
| Dexketoprofen | −88.99 | Cyclooxygenase inhibitor |
| Niridazole | −88.53 | Phosphofructokinase inhibitor |
| Triciribine | −88.08 | AKT inhibitor |
| Beclometasone | −87.47 | Glucocorticoid receptor agonist |
| Azacitidine | −87.07 | DNA methyltransferase inhibitor |
| Ketoprofen | −87.02 | Protein synthesis inhibitor |
| PD‐169316 | −86.01 | p38 MAPK inhibitor |
| Cytochalasin‐D | −85.97 | Actin polymerization inhibitor |
AKT, serine threonine kinase; BTK, Bruton's tyrosine kinase; DNA, deoxyribonucleic acid; MAPK, mitogen‐activated protein kinase.
Gene expression validation in vitro
The green module is closely related to ECM organization, whereas the red module is associated with T cell activation. The representative genes we selected were the most obviously changed in these two modules.
We focused on these 13 key genes that all played pivotal roles in DN, as follows: the upregulated genes Fn1, Lum, Thbs2, Tgfbi and Comp, and the downregulated genes Fgb and Crispld2 in the green module; and the upregulated genes Trbc1, Pycard, Ccl19, Ccl5 and Corp1a, and the downregulated gene Zbtb16 in the red module. We validated the expressions of these 13 hub genes in the DN model in vitro (Figure 5).
Figure 5.
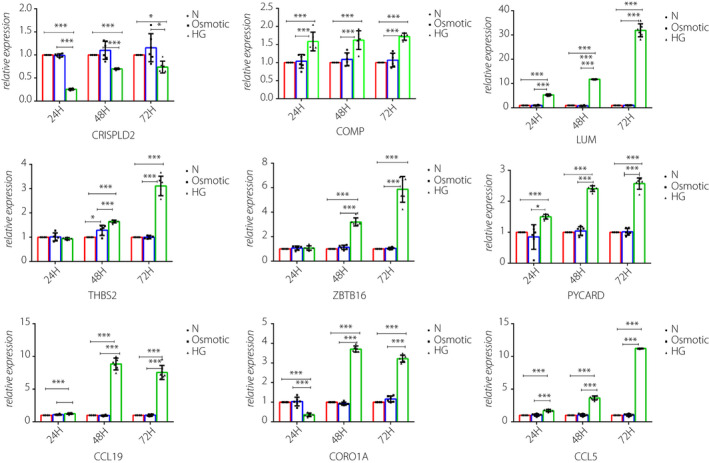
In vitro validation for novel key genes in green and red modules. The messenger ribonucleic acid expressions of CRISPLD2, COMP and LUM in the rat glomerular mesangial cells, THBS2, PYCARD, CCL19, CORO1A, CCL5 and ZBTB16 in the murine podocyte cells were measured by quantitative real‐time polymerase chain reaction under high‐glucose stimulation, osmotic control and normalized with glyceraldehyde 3‐phosphate dehydrogenase. All data are represented as the mean ± standard deviation (n = 6). *P < 0.05 versus control; ***P < 0.001 versus Control.
Quantitative real‐time polymerase chain reaction showed that the mRNA levels of COMP, LUM, THBS2, ZBTB16, PYCARD, CCL19, CORO1A and CCL5 were upregulated, and that of CRISPLD2 was downregulated after they were stimulated by high glucose. The changes in these expression levels were consistent with the bioinformatic analysis results of all genes except for ZBTB16. Meanwhile, the osmotic pressure did not affect the level of gene expression. The expressions of Fn1, Tgfbi, Fgb and Trbc1 changed, but the difference was not statistically significant.
Gene expression validation in vivo
In the present study, we verified the expressions of the proteins encoded by COMP, LUM and PYCARD genes at 0 and 20 weeks in DN rats.
Figure 6a shows the immunohistochemical staining of PYCARD in the glomeruli. The staining of the glomerular mesangial area deepened. The IOD/area at 0 and 20 weeks were 0.171629 ± 0.0082202 and 0.223739 ± 0.009221, respectively, and the P‐value was <0.001, Figure 6b shows the immunohistochemical staining of COMP in the glomeruli. The figure shows that the glomerular mesangial area had deepened staining. The IOD/area at 0 and 20 weeks were 0.1468 ± 0.002612 and 0.26029 ± 0.00511, respectively, and the P‐value was <0.001.
Figure 6.
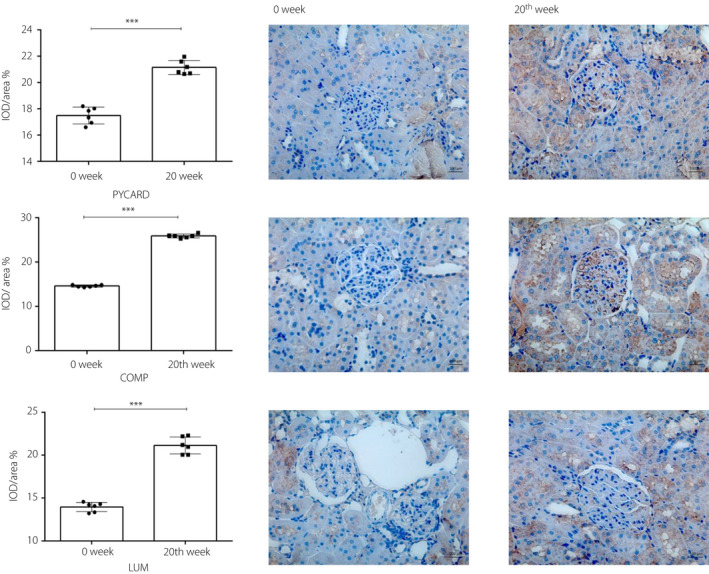
In vivo validation for key genes in green and red modules. (a) The immunohistochemical staining of PYD and CARD domain containing (PYCARD) at 0 and 20 weeks in diabetic nephropathy rats (scale, 100 μm, magnification: ×400). All data are represented as the mean ± standard deviation (n = 6). ***Significant changes were observed in the week 20 compared with those in week 0 (P < 0.001). (b) The immunohistochemical staining of cartilage oligomeric matrix protein (COMP) in weeks 0 and 2 in diabetic nephropathy rats (scale, 100 μm, magnification: ×400). All data are represented as the mean ± standard deviation (n = 6). ***Significant changes were observed in week 20 compared with those in week 0 (P < 0.001). (c) The immunohistochemical staining of lumican (LUM) in weeks 0 and 20 in diabetic nephropathy rats (scale, 100 μm, magnification: ×400). All data are represented as the mean ± standard deviation (n = 6). ***Significant changes were observed in week 20 compared with those in week 0 (P < 0.001).
Figure 6c shows the immunohistochemical staining of LUM in the glomeruli. The figure shows that the staining of the glomerular mesangial area deepened. The IOD/area at 0 and 20 weeks were 0.1387 ± 0.00673 and 0.2205 ± 0.01882, respectively, and the P‐value was <0.001.
DISCUSSION
DN is the leading cause of end‐stage renal disease worldwide. The pathogenesis of DN is not well known. ECM proliferation, immune system and inflammatory response play key roles in DN development and progression. In the present study, we carried out a bioinformatics analysis on DN. We obtained the transcriptome data of DN from the GEO database. After pre‐processing the raw data, we constructed the WGCNA network based on the gene expression patterns. Through GO enrichment analysis by GSEA, we found some of the most significant gene sets related to various aspects of DN, including ECM organization, T‐cell activation and kidney development.
In the green module, the most significant gene set is related to ECM organization. The expressions of LUM, THBS2 and COMP were significantly upregulated under the stimulation of high glucose, and these genes were reportedly associated with the formation of ECM in other diseases 28 , 29 , 30 , 31 . Thus, we concluded that these genes play important roles in DN by promoting ECM organization. Under the stimulation of high glucose, the expression of CRISPLD2 was downregulated. This gene is novel and not well known. Considering the results of bioinformatics analysis and verification, we hypothesized that this gene could ameliorate DN by inhibiting ECM organization.
In the red module, the most significant gene set is related to T‐cell activation. The expressions of TRBC1, PYCARD, CCL19, CCL5 and CORO1A were upregulated over time under the stimulation of high glucose in the early stages of DN, and these findings were consistent with the results of bioinformatics analysis. Thus, T‐cell activation might be regulated by these genes. Then, the activated T cells act as the initiating factor that results in the occurrence of immune and inflammatory responses. With DN development, excessive amounts of T cells are continuously activated, further intensifying the responses and aggravating the disease. However, the expression of ZBTB16 was inconsistent with the results of the bioinformatics analysis. ZBTB16 is reportedly associated with metabolic syndromes 32 . Furthermore, ZBTB16 could regulate the maturation of innate lymphoid cells (ILCs) through T helper cells; the ILCs play important roles in renal inflammation and fibrosis in DN 33 , 34 , 35 , 36 , 37 . We hypothesized that in the early stage of DN, ILCs play a vital role along with upregulated ZBTB16. With DN development, the function of ILCs declines, and ZBTB16 expression is downregulated.
Through the analysis of the connectivity map database, we found some of the most significant chemical compounds. These compounds have opposite effects on the gene expression of DN, indicating their potential interference effects on DN. For example, the microtubule inhibitor, triclabendazole, could inhibit the upregulated genes and activate the downregulated genes, thereby possibly correcting the overall gene expression levels to achieve a balanced state in DN.
The p38 mitogen‐activated protein kinase (MAPK) signaling pathway plays a key role in DN, leading to albuminuria and glomerular mesangial expansion, and affecting DN prognosis 38 , 39 , 40 . Therefore, the inhibition of p38 MAPK signaling pathway can effectively ameliorate DN 41 , 42 , 43 . PD‐169316 is a novel inhibitor of the p38 MAPK pathway in DN. Therefore, PD‐169316 and its related derivatives could possibly be used to treat DN, because they inhibit the p38 MAPK pathway.
Autophagy in kidney has protective effects that could influence the outcome of DN 44 , 45 , 46 . Microtubules consist of microtubule‐associated proteins and are reportedly involved in autophagy 47 , 48 . We also found triclabendazole, a microtubule inhibitor. Thus, we believe that triclabendazole and its derivatives can ameliorate DN by repairing the injured autophagy in the kidney.
Serine threonine kinase (Akt), a multifunctional serine threonine kinase, is involved in various signal pathways. It could participate in the occurrence and development of DN through Akt‐related signal pathways. Akt‐related inhibitors could significantly ameliorate the clinical manifestation of DN 49 , 50 , 51 , 52 . Triciribine is a novel Akt inhibitor in kidney diseases. We hypothesized that triciribine and its derivatives are drugs that can potentially ameliorate DN by inhibiting Akt‐related signal pathways.
Other compounds, such as triamcinolone, fluocinolone and beclometasone, are glucocorticoid receptor agonists that can regulate glucose and lipid metabolism, and immune and inflammation response, and can be used as an intervention for DN in the early stage.
The present study featured various advantages. First, we used WGCNA, a powerful and prevalent method to cluster significant genes. Second, we implemented the connectivity map analysis in DN for the first time to identify disease‐related compounds. Third, some key genes were validated successively in vitro and in vivo. However, the study suffers from certain limitations. Gene expression levels at different DN stages were not obtained. In addition, the clinical information of data from GEO was unavailable. Thus, the relationship between gene expression values and clinical features has not been determined. In the in vivo experiments, it would be more vigorous to evaluate the mRNA expressions of the genes of interest using isolated glomeruli. Additionally, using STZ‐induced diabetic rats with insulin treatment as controls to eliminate the possibility for toxic effects of STZ would be more convincing.
This study presents the first attempt to use WGCNA and connectivity map analysis in DN. Several novel key genes and important compounds closely associated with DN were screened. The present study provided a new idea for future research on DN. Furthermore, it provided a large number of data resources, novel targets and research clues in DN.
Supplementary data
The supplementary data showed the detailed information of DEGs in the present study and the primers for quantitative polymerase chain reaction in green and red modules.
ACKNOWLEDGMENTS
This work was supported by the National Natural Science Foundation of China (grant no. 81270805), the Science and Technology Department of Sichuan Province (grant no. 2018SZ0378), and Chengdu Science and Technology Bureau Grant (grant no. 2019‐YF09‐00090‐SN).
DISCLOSURE
The authors declare no conflict of interest.
Supporting information
Table S1 | Primers for quantitative polymerase chain reaction in green and red modules
Table S2 | Detailed information of differentially expressed genes in the represent study
J Diabetes Investig 2022; 13: 112–124.
References
- 1. Zheng Y, Ley SH, Hu FB. Global aetiology and epidemiology of type 2 diabetes mellitus and its complications. Nature reviews. Endocrinology 2018; 14: 88–98. [DOI] [PubMed] [Google Scholar]
- 2. Umanath K, Lewis JB. Update on diabetic nephropathy: core curriculum 2018. Am J Kidney Dis 2018; 71: 884–895. [DOI] [PubMed] [Google Scholar]
- 3. NCD Risk Factor Collaboration (NCD‐RisC) . Worldwide trends in diabetes since 1980: a pooled analysis of 751 population‐based studies with 4.4 million participants. Lancet (London, England), 2016; 387: 1513–1530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Najafian B, Fogo AB, Lusco MA, et al. AJKD Atlas of renal pathology: diabetic nephropathy. Am J Kidney Dis 2015; 66: E37–E38. [DOI] [PubMed] [Google Scholar]
- 5. Qi C, Mao X, Zhang Z, et al. Classification and differential diagnosis of diabetic nephropathy. J Diabetes Res 2017; 2017: 8637138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Chen C, Lin J, Li L, et al. The role of the BMP4/Smad1 signaling pathway in mesangial cell proliferation: a possible mechanism of diabetic nephropathy. Life Sci 2019; 220: 106–116. [DOI] [PubMed] [Google Scholar]
- 7. Matsubara T, Araki M, Abe H, et al. Bone morphogenetic protein 4 and Smad1 mediate extracellular matrix production in the development of diabetic nephropathy. Diabetes 2015; 64: 2978–2990. [DOI] [PubMed] [Google Scholar]
- 8. Tominaga T, Abe H, Ueda O, et al. Activation of bone morphogenetic protein 4 signaling leads to glomerulosclerosis that mimics diabetic nephropathy. J Biol Chem 2011; 286: 20109–20116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Sirin Y, Susztak K. Notch in the kidney: development and disease. J Pathol 2012; 226: 394–403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Bonegio R, Susztak K. Notch signaling in diabetic nephropathy. Exp Cell Res 2012; 318: 986–992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Sweetwyne MT, Gruenwald A, Niranjan T, et al. Notch1 and Notch2 in podocytes play differential roles during diabetic nephropathy development. Diabetes 2015; 64: 4099–4111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Guo Q, Zhong W, Duan A, et al. Protective or deleterious role of Wnt/beta‐catenin signaling in diabetic nephropathy: an unresolved issue. Pharmacol Res 2019; 144: 151–157. [DOI] [PubMed] [Google Scholar]
- 13. He W, Dai C, Li Y, et al. Wnt/beta‐catenin signaling promotes renal interstitial fibrosis. J Am Soc Nephrol 2009; 20: 765–776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Wada J, Makino H. Innate immunity in diabetes and diabetic nephropathy. Nat Rev Nephrol 2016; 12: 13–26. [DOI] [PubMed] [Google Scholar]
- 15. Flyvbjerg A. The role of the complement system in diabetic nephropathy. Nat Rev Nephrol 2017; 13: 311–318. [DOI] [PubMed] [Google Scholar]
- 16. Zheng Z, Zheng F. Immune cells and inflammation in diabetic nephropathy. J Diabetes Res 2016; 2016: 1841690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Bending JJ, Lobo‐Yeo A, Vergani D, et al. Proteinuria and activated T‐lymphocytes in diabetic nephropathy. Diabetes 1988; 37: 507–511. [DOI] [PubMed] [Google Scholar]
- 18. Moon J‐Y, Jeong K‐H, Lee T‐W, et al. Aberrant recruitment and activation of T cells in diabetic nephropathy. Am J Nephrol 2012; 35: 164–174. [DOI] [PubMed] [Google Scholar]
- 19. Lampropoulou IT, Stangou Μ, Sarafidis P, et al. TNF‐α pathway and T‐cell immunity are activated early during the development of diabetic nephropathy in Type II Diabetes Mellitus. Clin Immunol 2020; 215: 108423. [DOI] [PubMed] [Google Scholar]
- 20. Zhang F, Wang C, Wen X, et al. Mesenchymal stem cells alleviate rat diabetic nephropathy by suppressing CD103 DCs‐mediated CD8 T cell responses. J Cell Mol Med 2020; 24: 5817–5831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Herrera M, Söderberg M, Sabirsh A, et al. Inhibition of T‐cell activation by the CTLA4‐Fc Abatacept is sufficient to ameliorate proteinuric kidney disease. Am J Physiol Renal Physiol 2017; 312: F748–F759. [DOI] [PubMed] [Google Scholar]
- 22. Langfelder P, Horvath S. WGCNA: an R package for weighted correlation network analysis. BMC Bioinformatics 2008; 9: 559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Chen L, Yuan L, Wang Y, et al. Co‐expression network analysis identified FCER1G in association with progression and prognosis in human clear cell renal cell carcinoma. Int J Biol Sci 2017; 13: 1361–1372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Yin LI, Cai Z, Zhu B, et al. Identification of key pathways and genes in the dynamic progression of HCC based on WGCNA. Genes (Basel) 2018; 9: 92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Wang B, He L, Miao W, et al. Identification of key genes associated with Schmid‐type metaphyseal chondrodysplasia based on microarray data. Int J Mol Med 2017; 39: 1428–1436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Gautier L, Cope L, Bolstad BM, et al. affy–analysis of Affymetrix GeneChip data at the probe level. Bioinformatics (Oxford, England) 2004; 20: 307–315. [DOI] [PubMed] [Google Scholar]
- 27. Subramanian A, Narayan R, Corsello SM, et al. A next generation connectivity map: L1000 platform and the first 1,000,000 profiles. Cell 2017; 171: 1437–1452 e17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Boguslawska J, Kedzierska H, Poplawski P, et al. Expression of genes involved in cellular adhesion and extracellular matrix remodeling correlates with poor survival of patients with renal cancer. J Urol 2016; 195: 1892–1902. [DOI] [PubMed] [Google Scholar]
- 29. Posey KL, Coustry F, Hecht JT. Cartilage oligomeric matrix protein: COMPopathies and beyond. Matrix Biol 2018; 71–72: 161–173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Halper J, Kjaer M. Basic components of connective tissues and extracellular matrix: elastin, fibrillin, fibulins, fibrinogen, fibronectin, laminin, tenascins and thrombospondins. Adv Exp Med Biol 2014; 802: 31–47. [DOI] [PubMed] [Google Scholar]
- 31. Dupuis LE, Berger MG, Feldman S, et al. Lumican deficiency results in cardiomyocyte hypertrophy with altered collagen assembly. J Mol Cell Cardiol 2015; 84: 70–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Šeda O, Šedová L, Včelák J, et al. ZBTB16 and metabolic syndrome: a network perspective. Physiol Res 2017; 66(Suppl 3): S357–S365. [DOI] [PubMed] [Google Scholar]
- 33. Shih H‐Y, Sciumè G, Mikami Y, et al. Developmental acquisition of regulomes underlies innate lymphoid cell functionality. Cell 2016; 165: 1120–1133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Savage AK, Constantinides MG, Han J, et al. The transcription factor PLZF directs the effector program of the NKT cell lineage. Immunity 2008; 29: 391–403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Kovalovsky D, Uche OU, Eladad S, et al. The BTB‐zinc finger transcriptional regulator PLZF controls the development of invariant natural killer T cell effector functions. Nat Immunol 2008; 9: 1055–1064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Lu P, Ji X, Wan J, et al. Activity of group 2 innate lymphoid cells is associated with chronic inflammation and dysregulated metabolic homoeostasis in type 2 diabetic nephropathy. Scand J Immunol 2018; 87: 99–107. [DOI] [PubMed] [Google Scholar]
- 37. Liu C, Qin L, Ding J, et al. Group 2 innate lymphoid cells participate in renal fibrosis in diabetic kidney disease partly via TGF‐1 signal pathway. J Diabetes Res 2019; 2019: 8512028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Chang P‐C, Chen T‐H, Chang C‐J, et al. Advanced glycosylation end products induce inducible nitric oxide synthase (iNOS) expression via a p38 MAPK‐dependent pathway. Kidney Int 2004; 65: 1664–1675. [DOI] [PubMed] [Google Scholar]
- 39. Ma FY, Tesch GH, Nikolic‐Paterson DJ. ASK1/p38 signaling in renal tubular epithelial cells promotes renal fibrosis in the mouse obstructed kidney. Am J Physiol Renal Physiol 2014; 307: F1263–F1273. [DOI] [PubMed] [Google Scholar]
- 40. Komers R, Lindsley JN, Oyama TT, et al. Renal p38 MAP kinase activity in experimental diabetes. Lab Investig 2007; 87: 548–558. [DOI] [PubMed] [Google Scholar]
- 41. Tesch GH, Ma FY, Han Y, et al. ASK1 inhibitor halts progression of diabetic nephropathy in Nos3‐deficient mice. Diabetes 2015; 64: 3903–3913. [DOI] [PubMed] [Google Scholar]
- 42. Wang S, Zhou Y, Zhang Y, et al. Roscovitine attenuates renal interstitial fibrosis in diabetic mice through the TGF‐β1/p38 MAPK pathway. Biomed Pharmacother 2019; 115: 108895. [DOI] [PubMed] [Google Scholar]
- 43. Peng L, Li J, Xu Y, et al. The protective effect of beraprost sodium on diabetic nephropathy by inhibiting inflammation and p38 MAPK signaling pathway in high‐fat diet/streptozotocin‐induced diabetic rats. Int J Endocrinol 2016; 2016: 1690474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Tagawa A, Yasuda M, Kume S, et al. Impaired podocyte autophagy exacerbates proteinuria in diabetic nephropathy. Diabetes 2016; 65: 755–767. [DOI] [PubMed] [Google Scholar]
- 45. Huang C, Zhang Y, Kelly DJ, et al. Thioredoxin interacting protein (TXNIP) regulates tubular autophagy and mitophagy in diabetic nephropathy through the mTOR signaling pathway. Sci Rep 2016; 6: 29196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Lenoir O, Jasiek M, Hénique C, et al. Endothelial cell and podocyte autophagy synergistically protect from diabetes‐induced glomerulosclerosis. Autophagy 2015; 11: 1130–1145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Polletta L, Vernucci E, Carnevale I, et al. SIRT5 regulation of ammonia‐induced autophagy and mitophagy. Autophagy 2015; 11: 253–270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Wang S, Livingston MJ, Su Y, et al. Reciprocal regulation of cilia and autophagy via the MTOR and proteasome pathways. Autophagy 2015; 11: 607–616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Yan R, Wang Y, Shi M, et al. Regulation of PTEN/AKT/FAK pathways by PPARγ impacts on fibrosis in diabetic nephropathy. J Cell Biochem 2019; 120: 6998–7014. [DOI] [PubMed] [Google Scholar]
- 50. Zhang YH, Wang B, Guo F, et al. Involvement of the TGFβ1‐ ILK‐Akt signaling pathway in the effects of hesperidin in type 2 diabetic nephropathy. Biomed Pharmacother 2018; 105: 766–772. [DOI] [PubMed] [Google Scholar]
- 51. Wu W, Hu W, Han WB, et al. Inhibition of Akt/mTOR/p70S6K signaling activity with Huangkui capsule alleviates the early glomerular pathological changes in diabetic nephropathy. Front Pharmacol 2018; 9: 443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Huang S, Xu Y, Ge X, et al. Long noncoding RNA NEAT1 accelerates the proliferation and fibrosis in diabetic nephropathy through activating Akt/mTOR signaling pathway. J Cell Physiol 2019; 234: 11200–11207. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1 | Primers for quantitative polymerase chain reaction in green and red modules
Table S2 | Detailed information of differentially expressed genes in the represent study