

Abstract
Chimeric antigen receptor T-cell (CAR-T) therapy is associated with significant toxicities secondary to immune activation including a rare but increasingly recognized severe toxicity resembling hemophagocytic lymphohistiocytosis (carHLH). We report the development of carHLH in 14.8% of pediatric and young adults treated with CD19-specific CAR-T therapy with carHLH occurring most commonly in those with high disease burden. Diagnosis and treatment of carHLH required a high index of suspicion and included multidrug immunomodulation with variable response to therapies. Compared to patients without carHLH, patients with carHLH had both reduced response to CAR-T therapy (P-value = 0.018) and overall survival (P-value = <0.0001).
Keywords: Chimeric Antigen Receptor T-cell Therapy, Refractory Acute B-cell Lymphoblastic Leukemia, Hemophagocytic Lymphohistiocytosis, Cytokine Release Syndrome
Chimeric antigen receptor T-cell (CAR-T) therapy is associated with a significant risk of toxicity secondary to immune system activation and inflammation. The most well described toxicities are cytokine release syndrome (CRS) and neurotoxicity (1). Additionally, a toxicity resembling hemophagocytic lymphohistiocytosis (hereafter carHLH) has been described as a second inflammatory wave after initial improvement in CRS signs and symptoms (2–7). While reported as a rare toxicity, Shah et al recently described carHLH in one third of patients in a pediatric cohort treated with CD22-CAR T-cells (2, 5, 6, 8–10). While diagnostic criteria for carHLH have varied, the most commonly employed include hyperferritinemia, multiorgan dysfunction (liver, renal, and/or pulmonary toxicity) and/or evidence of hemophagocytosis on bone marrow evaluation (5, 6, 10). Given variability in diagnostic criteria, it is difficult to compare experiences across cohorts. Furthermore, carHLH risk factors, monitoring and treatment strategies, as well as outcomes including CAR-T cell efficacy and associated morbidity and mortality, are not well known.
Methods
Herein, we describe carHLH in a pediatric and adolescent/young adult cohort with relapsed and/or refractory CD19-positive acute lymphoblastic leukemia (ALL), treated with lymphodepletion and CD19-CAR T-cells. This cohort includes 27 patients treated over a 2-year period (8/2018–8/2020). Patients received either the commercially available product tisagenlecleucel (Kymriah, Novartis; n=12) or an institutional product as part of an ongoing Phase I/II clinical trial (SJCAR19; NCT03573700; n=15). Clinical outcomes of the Phase I portion of SJCAR19 have been previously reported in abstract form, and highlight the tolerability and expected side effect profile of this therapy(11). Both products utilize a lentiviral vector encoding the CD19-specific single chain variable fragment (scFv) FMC63 and a 41BBζ signaling domain. IRB approval was obtained; all records were retrospectively reviewed. Pre-treatment leukemia disease burden was evaluated using bone marrow in all patients, in conjunction with lumbar puncture (cerebrospinal fluid [CSF]) and positron emission tomography (PET) in a subset of patients. Disease evaluations were performed after receipt of any bridging therapy and prior to start of CAR-T therapy. On the day of CAR-T infusion, a subset of patients also had peripheral blood minimal residual disease (MRD) testing. CRS was graded using the ASTCT consensus grading criteria (1). Intervention for CRS was guided by institutional treatment algorithms, including tocilizumab with or without steroids for ≥ grade 3 CRS. However, some patients received tocilizumab at lower grades based on clinical status and/or concern for high-risk of symptom progression. Presence of carHLH was determined in real-time using the clinical criteria of recurrent high-grade fever, precipitous rise in ferritin, and new/worsening organ dysfunction. For this work, the Shah criteria were retrospectively applied to define date of carHLH onset (6). Change in end-organ function was calculated as the difference between minimum and maximum lab values; peak organ dysfunction was defined as the maximum lab value in the 30 days following CAR-T infusion. Of note, the maximum ferritin value able to be reported is >100,000 ng/mL; these values were categorized as 100,001 for this analysis. Continuous variables were compared using Kruskal-Wallis test and categorical variables were compared using the Fisher’s Chi square test (SASv9.4, Cary, NC). Statistical significance was defined as P-value ≤ 0.05. Survival time was defined from date of infusion to date of death or the date of last contact. Survival distributions were estimated using the Kaplan Meier method. Comparison of the survival distribution between groups was performed using the log-rank exact test (StatXact v9.4).
Results
Among the 27 patients, 4 (14.8%) had CRS and carHLH, 11 (40%) had CRS alone and 12 (44%) had no CRS. Among these three subgroups, there was no statistical difference between baseline characteristics (Table 1). Clinical diagnosis of carHLH occurred at a median of 10 days (range: 7–12) after CAR-T infusion; using the Shah criteria, the median onset of carHLH was 11.5 days (range: 8–20) after infusion. There was evidence of higher pre-treatment ALL burden in carHLH patients, including bone marrow (P-value = 0.002) and pre-infusion peripheral blood (P-value = 0.011). Furthermore, patients with carHLH had increased evidence of extramedullary disease. This included detectable disease in the CSF (P-value = 0.0004) and a trend towards increased PET-avidity concerning for extramedullary disease (Table 1). Active infection 14 days prior to infusion and in the 30 days after CAR-T infusion was not significantly associated with development of carHLH (Table 1). Additionally, there was no evidence of increased active infection at the time of CRS development in the carHLH group compared to the CRS alone group. Two patients (50%) had active infection at time of carHLH diagnosis.
Table 1.
Patient Characteristics and Laboratory Data
| All N=27 | carHLH and CRS N=4 | CRS alone N=11 | No CRS N=12 | P-Value | |
|---|---|---|---|---|---|
|
| |||||
| Age (Median; years) | 10.4(1.78–23.6) | 12.6(6.18–20.43) | 7.8(1.78–15.4) | 14.4(3.33–23.6) | 0.051 |
|
| |||||
| Sex N (%) | |||||
| Male | 15 (56.3) | 3 (75) | 5 (45,5) | 7 (58.3) | 0.67 |
| Female | 12 (43.8) | 1 (25) | 6 (54.5) | 5 (41.7) | |
|
| |||||
| Disease Burden^ N(%) | |||||
| PET-avidity | |||||
| Yes | 6 (22.3) | 3 (75) | 1 (9.1) | 2 (16.7) | 0.3 |
| No | 10 (37) | 1 (25) | 5 (45.5) | 4 (33.3) | |
| Not evaluated | 11 (40.7) | 0 (0) | 5 (45.5) | 6 (50) | |
| CNS Status | |||||
| CNS1 | 23 (87.5) | 0 | 11 (100) | 12 (100) | 0.0004* |
| CNS2/3 | 3 (9.4) | 3(75.0) | 0 (0) | 0 (0) | |
| Not evaluated | 1 (3.1) | 1 (25.0) | 0 (0) | 0 (0) | |
| Bone Marrow & | |||||
| <50% | 19 (70.4) | 1 (25) | 6 (54.5) | 12 (100) | 0.002* |
| ≥50% | 8 (29.6) | 3 (75) | 5 (45.5) | 0 | |
| Median (range) | 6.18 (0–100) | 71 (24.6–95.4) | 32.3 (0–100) | 0.92 (0–43.06) | 0.06 |
| Peripheral Blood # & | |||||
| <25% | 12 (44.4) | 0 (0) | 6 (54.5) | 6 (50) | 0.011* |
| ≥25% | 2 (7.4) | 2(50) | 0 (0) | 0 (0) | |
| Median (range) | 0.4 (0–56.2) | 43 (29.9–56.2) | 0.2 (0.05–2.7) | 0.4 (0–0.47) | 0.087 |
| Not evaluated | 13 (50) | 2 (50) | 5 (45.5) | 6 (50) | |
|
| |||||
| Infection** N(%) | |||||
| 14 days prior to infusion | |||||
| Yes | 4 (14.8) | 0 (0) | 2 (18.2) | 2 (16.7) | >0.999 |
| No | 23 (85.2) | 4 (100) | 9 (81.8) | 10 (83.3) | |
| 30 days after infusion | |||||
| Yes | 11(40.7) | 3 (75) | 5 (45.5) | 3 (25) | 0.255 |
| No | 16 (59.3) | 1(25) | 6 (54.5) | 9 (75) | |
| Active infection at CRS diagnosis | |||||
| Yes | 5 (33.3) | 1 (25) | 4 (36.4) | NA | >0.999 |
| No | 10 (66.7) | 3 (75) | 7 (63.6) | NA | |
|
| |||||
| CRS (Max Grade; N(%) | |||||
| 0 | 12 (44.4) | 0 (0) | 0 (0) | NA | 0.09 |
| 1 | 8 (29.6) | 1 (25) | 7 (63.6) | ||
| 2 | 2 (7.4) | 0 (0) | 2 (18.2) | ||
| 3 | 3 (11.1) | 1 (25) | 2 (18.2) | ||
| 4 | 2 (7.4) | 2 (50) | 0 (0) | ||
|
| |||||
| Disease Response N(%) | |||||
| CR | 19 (70.4) | 0 (0) | 10 (90.9) | 9 (75) | 0.018* |
| NR | 7 (25.9) | 3 (75) | 1 (9.1) | 3 (25) | |
| Not Evaluable | 1 (3.7) | 1 (25) | 0 (0) | 0 (0) | |
|
| |||||
| Survival %(CI) | |||||
| 1 month | 75 (12.8–96.1) | 100 | 100 | <0.0001* | |
| 2 months | 25 (0.89–66.5) | 90.9 (53.9–98.8) | 100 | ||
| 6 months | 0 | 91 (53.9–98.8) | 91.7 (63.2–99.1) | ||
| 12 months | 0 | 79.5 (36.1–94.4) | 92 (63.2–99.1) | ||
|
| |||||
| Laboratory Data [median value (range)] | |||||
|
| |||||
| Ferritin (ng/mL) | |||||
| Peak | 1711 (42– >100,000) | >100,000 (max) | 1767 (95–86211) | 1056 (42–6314) | 0.0032* |
| Change | 775 (0–98927) | 98,000 (95667–98927) | 1611 (73–84302) | 275 (0–3525) | 0.0015* |
|
| |||||
| CRP (mg/dL) | |||||
| Baseline | 0.5 (0.05–21.2) | 7.05 (6.1–21.2) | 1 (0.05–12.1) | 0.3 (0.05–1.9) | 0.0056* |
| Peak | 0.51(0.18–3.56) | 2.13(0.34–3.56) | 0.44(0.18–2.38) | 0.58(0.34–1.18) | 0.05 |
| Change | 0.22(0.09–2.97) | 1.82(0.18–2.97) | 0.18(0.09–2.08) | 0.24(0.11–0.4) | 0.035* |
|
| |||||
| Creatinine (mg/dL) | |||||
| Peak | 0.51(0.18–3.56) | 2.13(0.34–3.56) | 0.44(0.18–2.38) | 0.58(0.34–1.18) | 0.05 |
| Change | 0.22(0.09–2.97) | 1.82(0.18–2.97) | 0.18(0.09–2.08) | 0.24(0.11–0.4) | 0.035* |
|
| |||||
| ALT (U/L) | |||||
| Peak | 81(8–2128) | 934(77–2128) | 98(24–584) | 41(8–582) | 0.07 |
| Change | 70(2–2118) | 925(66–2118) | 82(17–572) | 28.5(2–566) | 0.06 |
|
| |||||
| Bilirubin (mg/dL) | |||||
| Peak | 0.7(0.15–8) | 3.35(1.3–8) | 0.6(0.15–1.2) | 0.55(0.2–2.5) | 0.011* |
| Change | 0.5(0–7.6) | 3.13(1–7.6) | 0.45(0–0.8) | 0.33(0.05–1.9) | 0.01* |
|
| |||||
| Fibrinogen (mg/dL) | |||||
| Minimum | 183(53–426) | 116(53–168) | 186(88–426) | 327(257–372) | 0.03* |
| Change | 64(0–506) | 298(64–506) | 67(0–291) | 0 | 0.02* |
| No data | 12 | 0 | 3 | 9 | |
|
| |||||
| INR | |||||
| Peak | 1.24(0.91–2.2) | 1.59(1.22–2.2) | 1.25(0.97–1.73) | 0.93(0.91–1.05) | 0.04* |
| Change | 0.2(0–0.74) | 0.45(0.11–0.74) | 0.21(0–0.54) | 0 (0–0.24) | 0.03* |
Cytokine Release Syndrome (CRS); CAR-mediated Hemophagocytic Lymphohistiocytosis (carHLH); Central Nervous System (CNS); Positron Emission Topography (PET); Minimal Residual Disease (MRD); Complete Response (CR; <5% blasts in bone marrow); No Response (NR; persistent disease post-CAR T-cell therapy at same magnitude as pre-CAR T-cell [MRD-positive pre/post or >5% blasts); C-reactive protein (CRP); Alanine Aminotransferase (ALT); International Normalized Ratio (INR); “Change” refers to the median change in minimum to peak level
statistically significant
unless otherwise noted: disease burden was assessed within 2-weeks of start of CAR T-cell therapy and after completion of any bridging therapy
Disease burden determined using flow-based technique for minimal residual disease (MRD) except for 2 patients without available MRD, and percent morphologic blasts was used for categorization
obtained on day of CAR T-cell infusion using MRD
Includes any viral, fungal or bacterial infections; Statistical significance between groups was compared using exact Chi-square test for categorical variables and the Kruskal-Wallis test for continuous variables. Missing data were excluded from the p-value.
Median time to development of carHLH was significantly longer than CRS (11.5 vs. 5 days; P-value = 0.012). Among patients with high-grade CRS (grades 3–4), 60% developed carHLH. Notably, all carHLH cases developed after CRS onset, during a phase of initial laboratory and clinical improvement (Figure 1A–D). Patients with carHLH had the most severe peak end-organ function (Table 1) and were more likely to require ICU admission (75% vs. 18.2%; P-value = 0.003). Baseline CRP was higher in the carHLH group compared to those with CRS alone or no CRS (P-value = 0.0056; Table 1). After CAR-T treatment, the carHLH cohort had a higher maximum CRP, although timing of maximum CRP did not correlate with carHLH onset (Table 1; Figure 1A–D). Peak ferritin and change in ferritin were significantly different between groups, with hyperferritinemia markedly more severe in carHLH patients (Table 1). Uniquely, the rate of rise of ferritin (change in ferritin/24 hours) was highest just prior to onset of carHLH compared to onset of CRS (carHLH: median 29167 ng/mL over 24 hours [range: 18161–30340] vs. CRS alone: median 1363 ng/mL over 24 hours [range: 0–37061]; P-value 0.026; Supplemental Figure 1). No bone marrow biopsies were performed for evaluation of hemophagocytosis.
Figure 1. Clinical course of patients with carHLH after CAR T-cell therapy.
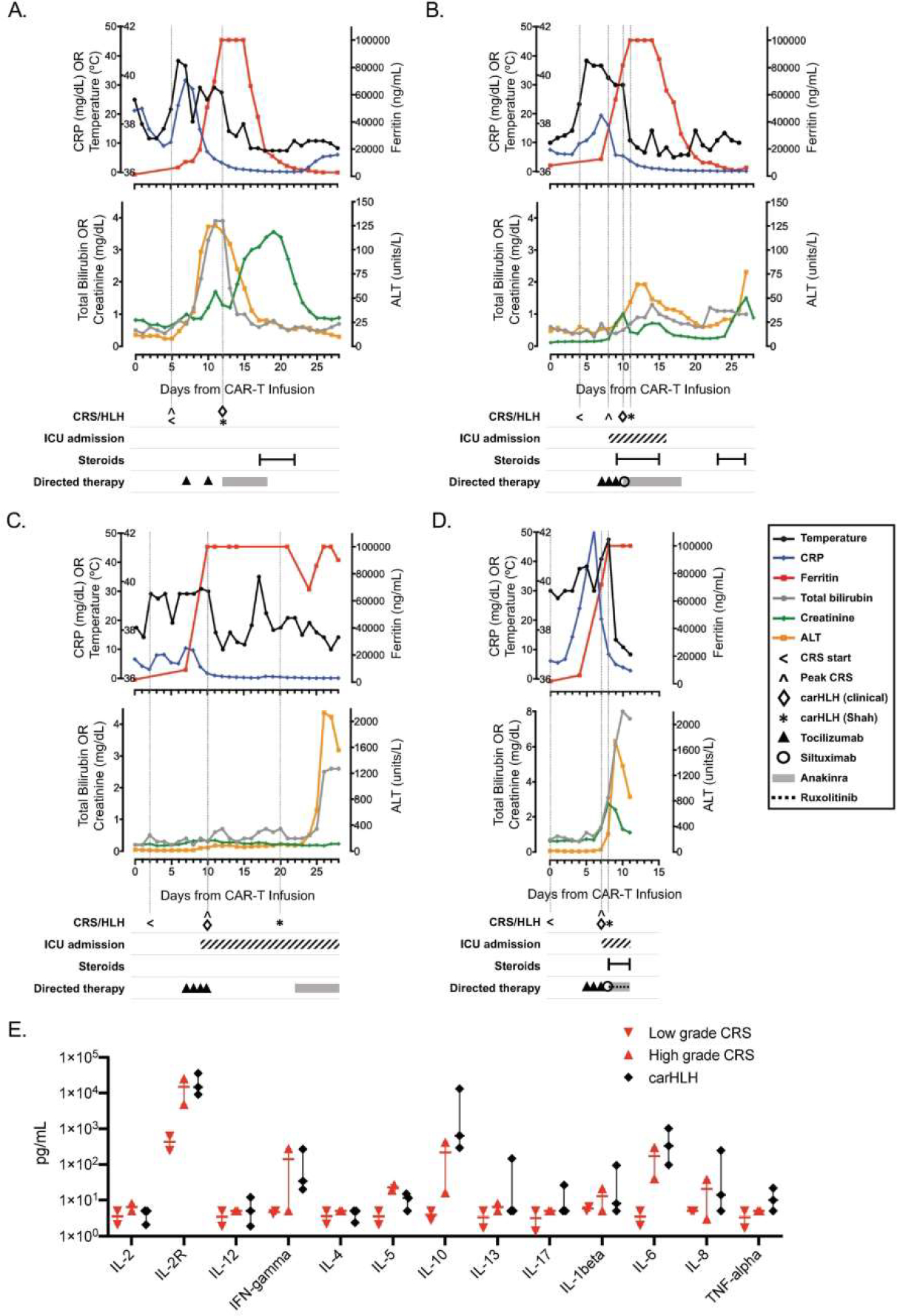
Graphical representation of clinical course of patients (n=4) who developed carHLH after receipt of CD19-CAR T-cell therapy, with each panel (A-D) representing a unique patient/infusion. Temperature and laboratory markers of inflammation and organ function are displayed temporally from CAR T-cell infusion until 30-days post-infusion (or until death, whichever was sooner), with corresponding timing of ICU admission (if applicable) and therapeutics administered for CRS and/or carHLH. Temperature (black), CRP (C-reactive protein; blue) and ferritin (red) are shown on top graphs for each patient. Total bilirubin (grey), creatinine (green) and ALT (alanine aminotransferase; yellow) are shown on the lower graphs for each patient (A-D). Vertical lines are shown to indicate start of CRS, day of peak CRS grade, and day of carHLH diagnosis (clinically and by Shah (6) criteria). Panel E depicts comparison of available serum cytokine levels between low grade CRS (red, downward triangle; n=2; Grade 1), high grade CRS (red, upward triangle n=2; Grade 3) and carHLH (n=3) patients. Bar represents median values with range. Cytokine analysis (ARUP Laboratories, Salt Lake City, UT) were obtained +/−3 days from carHLH/CRS diagnosis.
Cytokine profiles in our carHLH patients were similar to those previously described in both severe CRS and carHLH, including elevations in IFN-gamma, TNF-alpha, IL-6, IL-10, and IL-1beta (Figure 1E; (12, 13). All four carHLH patients had previously received CRS-directed therapy with multiple doses of tocilizumab and/or siltuximab (n=2; Figure 1A–D). Additionally, all carHLH patients received anakinra at time of clinical diagnosis of carHLH, with addition of steroids (n=3; 75%) and ruxolitinib (n=1; 25%) due to progressive symptoms. Three patients showed signs of response to this therapy, including stabilization of clinical course, improvement of fever, down-trending ferritin, and improving hepatitis (Figure 1A–C). Notably, the incidence of no leukemic response (NR) to CAR-T therapy was significantly higher in the carHLH cohort (75%) when compared to the CRS alone (9.1%) or no CRS cohorts (25%; P-value = 0.018; Table 1). Furthermore, overall survival was significantly reduced in carHLH patients (Table 1). Patients with carHLH all died, at a median of 44.5 days (range: 11–111) after CAR-T infusion (cause of death: overwhelming carHLH toxicity [n=1]; leukemic disease [n=3]). Of note, 5 of the 27 patients received a 2nd treatment course of lymphodepleting chemotherapy followed by CAR-T infusion for treatment of relapsed disease after initial remission. None of these patients developed carHLH. One patient developed CRS (grade 2); this patient had grade 1 CRS with 1st infusion.
Discussion
There are very few reports describing the incidence, treatment and outcome of patients who develop carHLH after CAR-T therapy. In our cohort, carHLH developed after 14.8% of CD19-CAR T-cell infusions and was significantly associated with decreased leukemic response and survival, despite aggressive and early intervention with immunomodulators. While reported response to CAR-T therapy in patients with carHLH has varied, Shah et al did not report a correlation between development of carHLH and disease response or CAR T-cell expansion (2, 3, 6). This highlights the variability of outcomes in these patients and the need to further evaluate the potential relationship between carHLH and leukemic response.
Similar to a previous report (3), all 4 patients with carHLH had down-trending CRP at the time of carHLH onset. However, these patients were more likely to have an elevated pre-infusion CRP possibly revealing increased baseline inflammation that could not readily be attributed to other factors such as infection. Importantly, we found that ferritin rate of rise was significantly increased prior to onset of carHLH (Supplemental Figure 1; >20,000 ng/mL in 24 hours), possibly indicating high risk for development of carHLH. Due to clinical concern for developing carHLH, 75% of our patients were treated prior to meeting Shah diagnostic criteria, again highlighting the necessity for increased clinical suspicion for evolution of carHLH.
The pathophysiologic mechanisms underlying of the development of carHLH remain unclear. It has been suggested that the risk to develop HLH-like manifestations may be higher in patients that receive a CAR-T product generated using a CD4/CD8 selected apheresis product (6). However, within our cohort, development of carHLH was equally distributed among patients receiving a product generated using T-cell selected starting material (SJCAR19; n=2) versus not (tisagenlecleucel; n=2). Other considerations include persistent, uncontrolled CAR-T expansion or re-expansion, which has been seen in a perforin deficient murine model (12). Additionally, there is evidence of increased circulating CAR-T cells on day 28 in those with carHLH compared to those with CRS in patients treated with CD22-CAR T-cells (12). In our cohort, CAR-T cell expansion was measured by qPCR for those patients treated on SJCAR19, with no difference in peak expansion noted between carHLH and non-carHLH patients (data not shown). Alternatively, NR to CAR-T therapy and resultant persistent leukemic burden could theoretically drive the development of carHLH, as seen in our group.
As reported by others, our carHLH patients all had clinical and laboratory improvement of CRS prior to development of carHLH (2–7), suggesting distinct processes. However, similarities in cytokine profiles during CRS versus carHLH may point to continued evolution of an underlying hyperinflammatory process (Figure 1E) (13). In contrast to primary HLH patients with a predominate adaptive immune response and cytokine profile, cytokine profiles of carHLH patients, including our cohort, show evidence of both innate and adaptive type responses including elevation of IL-1beta (Figure 1E)(12, 14). These findings support the use of steroids and cytokine-directed therapies, such as anakinra and/or specific JAK inhibitors with guarded consideration of emapalumab due to loss of anti-leukemic CAR activity in a preclinical model (2, 4, 6, 7, 15, 16). Further studies are needed to determine the efficacy and optimal dosing and timing of these agents, as well as any effects on CAR-T expansion, persistence, and anti-leukemic activity.
While early studies reported higher rates of CRS using varied grading scales, our experience aligns with recent reports using the ASTCT grading system. This includes data from the CIBMTR reporting ‘real-world’ experience of using tisagenlecleucel in a comparable patient population (n=255), with 54.9% of patients developing any grade CRS and only 16.1% of cases being high-grade (17). This may relate to the overall lower disease burden of patients prior to treatment with CAR-T therapy, with 37.2% of patients in morphologic CR prior to infusion, of which 46% were MRD-negative (17). Furthermore, a recent cohort of patients treated with CTL019 (n=70) also demonstrated most patients had lower pre-CAR disease burden (n=55), 80% of which had <5% marrow disease. CRS rates in this cohort (using the ASTCT grading system) were also similar to our cohort, with 33% of patients having no CRS and only 14.3% having high-grade CRS, most of which occurred in patients with high-burden disease (18).
In conclusion, in this pediatric cohort with relapsed/refractory ALL treated with CD19-CAR T-cell therapy, 14.8% of patients developed carHLH. While the data presented are limited due to the low number of patients and retrospective nature of the study, they nevertheless highlight the necessity for sensitive and specific diagnostic criteria for carHLH, as well as further investigations into the underlying pathophysiology, risk factors, and treatment of this severe toxicity of CAR-T therapy.
Supplementary Material
Acknowledgements
This work was supported by the National Cancer Institute grant P30 CA021765, American Society of Transplantation and Cellular Therapy (New Investigator Award to AT), Cookies for Kids Cancer (BT), and the American Lebanese Syrian Associated Charities. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
Disclosures
KN and MH receive research funding from Incyte. SG has patent applications in the fields of T-cell and/or gene therapy for cancer. He consults for Catamaran Bio, Nektar Therapeutics, TESSA Therapeutics, is on the Scientific Advisory Board of Tidal, and is a DSMB member of Immatics. BT has received financial support for presentations from Miltenyi Biotec. GM receives research funding from Astellas Inc. AS’s institution receives support for the conduct of industry sponsored trials from Vertex Pharmaceuticals, CRISPR Therapeutics and Novartis. AS has received consulting fee from Spotlight Therapeutics; neither are related to the work being presented.
References
- 1.Lee DW, Santomasso BD, Locke FL, Ghobadi A, Turtle CJ, Brudno JN, et al. ASTCT Consensus Grading for Cytokine Release Syndrome and Neurologic Toxicity Associated with Immune Effector Cells. Biol Blood Marrow Transplant. 2019;25(4):625–38. [DOI] [PubMed] [Google Scholar]
- 2.Ahmed S, furqan f, Strati P, Westin J, Fayad L, Hagemeister FB, et al. Haemophagocytic lymphohistiocytosis (HLH) in patients with large B-cell lymphoma treated with standard of care (SOC) axicabtagene ciloleucel (Axi-cel). Journal of Clinical Oncology. 2020;38(15_suppl):8057-. [Google Scholar]
- 3.Hashmi H, Bachmeier C, Chavez JC, Song J, Hussaini M, Krivenko G, et al. Haemophagocytic lymphohistiocytosis has variable time to onset following CD19 chimeric antigen receptor T cell therapy. Br J Haematol. 2019;187(2):e35–e8. [DOI] [PubMed] [Google Scholar]
- 4.Maus MV, Alexander S, Bishop MR, Brudno JN, Callahan C, Davila ML, et al. Society for Immunotherapy of Cancer (SITC) clinical practice guideline on immune effector cell-related adverse events. J Immunother Cancer. 2020;8(2). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Neelapu SS, Tummala S, Kebriaei P, Wierda W, Gutierrez C, Locke FL, et al. Chimeric antigen receptor T-cell therapy - assessment and management of toxicities. Nat Rev Clin Oncol. 2018;15(1):47–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Shah NN, Highfill SL, Shalabi H, Yates B, Jin J, Wolters PL, et al. CD4/CD8 T-Cell Selection Affects Chimeric Antigen Receptor (CAR) T-Cell Potency and Toxicity: Updated Results From a Phase I Anti-CD22 CAR T-Cell Trial. J Clin Oncol. 2020;38(17):1938–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Sheth VS, Gauthier J. Taming the beast: CRS and ICANS after CAR T-cell therapy for ALL. Bone Marrow Transplant. 2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Sandler RD, Tattersall RS, Schoemans H, Greco R, Badoglio M, Labopin M, et al. Diagnosis and Management of Secondary HLH/MAS Following HSCT and CAR-T Cell Therapy in Adults; A Review of the Literature and a Survey of Practice Within EBMT Centres on Behalf of the Autoimmune Diseases Working Party (ADWP) and Transplant Complications Working Party (TCWP). Front Immunol. 2020;11:524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Neelapu SS, Tummala S, Kebriaei P, Wierda W, Locke FL, Lin Y, et al. Toxicity management after chimeric antigen receptor T cell therapy: one size does not fit ‘ALL’. Nat Rev Clin Oncol. 2018;15(4):218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Shalabi H, Gust J, Taraseviciute A, Wolters PL, Leahy AB, Sandi C, et al. Beyond the storm - subacute toxicities and late effects in children receiving CAR T cells. Nat Rev Clin Oncol. 2021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Talleur AC, Madden RM, Qudeimat A, Mamcarz E, Sharma A, Srinivasan A, et al. Allogeneic Hematopoietic Cell Transplantation Is Critical to Maintain Remissions after CD19-CAR T-Cell Therapy for Pediatric ALL: A Single Center Experience. Blood. 2020;136(Supplement 1):39–40. [Google Scholar]
- 12.Ishii K, Pouzolles M, Chien CD, Erwin-Cohen RA, Kohler ME, Qin H, et al. Perforin-deficient CAR T cells recapitulate late-onset inflammatory toxicities observed in patients. J Clin Invest. 2020;130(10):5425–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Teachey DT, Lacey SF, Shaw PA, Melenhorst JJ, Maude SL, Frey N, et al. Identification of Predictive Biomarkers for Cytokine Release Syndrome after Chimeric Antigen Receptor T-cell Therapy for Acute Lymphoblastic Leukemia. Cancer Discov. 2016;6(6):664–79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Chaturvedi V, Marsh R, Lorenz AZ, Owsley E, Chaturvedi V, Nguyen T, et al. T cell activation profiles distinguish hemophagocytic lymphohistiocytosis and early sepsis. Blood 2020;ahead of print. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Strati P, Ahmed S, Kebriaei P, Nastoupil LJ, Claussen CM, Watson G, et al. Clinical efficacy of anakinra to mitigate CAR T-cell therapy-associated toxicity in large B-cell lymphoma. Blood Adv. 2020;4(13):3123–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Huarte E, O’Connor RS, Peel MT, Nunez-Cruz S, Leferovich J, Juvekar A, et al. Itacitinib (INCB039110), a JAK1 Inhibitor, Reduces Cytokines Associated with Cytokine Release Syndrome Induced by CAR T-cell Therapy. Clin Cancer Res. 2020;26(23):6299–309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Pasquini MC, Hu ZH, Curran K, Laetsch T, Locke F, Rouce R, et al. Real-world evidence of tisagenlecleucel for pediatric acute lymphoblastic leukemia and non-Hodgkin lymphoma. Blood Adv. 2020;4(21):5414–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kadauke S, Myers RM, Li Y, Aplenc R, Baniewicz D, Barrett DM, et al. Risk-Adapted Preemptive Tocilizumab to Prevent Severe Cytokine Release Syndrome After CTL019 for Pediatric B-Cell Acute Lymphoblastic Leukemia: A Prospective Clinical Trial. J Clin Oncol. 2021;39(8):920–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.