

Abstract
Purpose:
To evaluate the mechanisms of how therapeutic upregulation of the transcription factor, CCAAT/enhancer-binding protein alpha (C/EBPα), prevents tumor progression in patients with advanced hepatocellular carcinoma (HCC) and in different mouse tumor models.
Experimental Design:
We conducted a phase I trial in 36 patients with HCC (NCT02716012) who received sorafenib as part of their standard care, and were given therapeutic C/EBPα small activating RNA (saRNA; MTL-CEBPA) as either neoadjuvant or adjuvant treatment. In the preclinical setting, the effects of MTL-CEBPA were assessed in several mouse models, including BNL-1ME liver cancer, Lewis lung carcinoma (LLC), and colon adenocarcinoma (MC38).
Results:
MTL-CEBPA treatment caused radiologic regression of tumors in 26.7% of HCC patients with an underlying viral etiology with 3 complete responders. MTL-CEBPA treatment in those patients caused a marked decrease in peripheral blood monocytic myeloid-derived suppressor cell (M-MDSC) numbers and an overall reduction in the numbers of protumoral M2 tumor-associated macrophages (TAM). Gene and protein analysis of patient leukocytes following treatment showed CEBPA activation affected regulation of factors involved in immune-suppressive activity. To corroborate this observation, treatment of all the mouse tumor models with MTL-CEBPA led to a reversal in the suppressive activity of M-MDSCs and TAMs, but not polymorphonuclear MDSCs (PMN-MDSC). The antitumor effects of MTL-CEBPA in these tumor models showed dependency on T cells. This was accentuated when MTL-CEBPA was combined with checkpoint inhibitors or with PMN-MDSC–targeted immunotherapy.
Conclusions:
This report demonstrates that therapeutic upregulation of the transcription factor C/EBPα causes inactivation of immune-suppressive myeloid cells with potent antitumor responses across different tumor models and in cancer patients. MTL-CEBPA is currently being investigated in combination with pembrolizumab in a phase I/Ib multicenter clinical study (NCT04105335).
Translational Relevance.
In this paper, we demonstrate preclinical data across a range of tumor types that establish the mechanism of action of MTL-CEBPA in modulating myeloid cells. The safety of combination treatment with sorafenib is confirmed in a phase Ib clinical trial that demonstrates a strong efficacy signal in a subgroup of patients with hepatocellular carcinoma of viral etiology who are tyrosine kinase inhibitor naïve. These data provide evidence for combination treatment with immunotherapies across other primary tumor types.
Introduction
Myeloid-derived suppressor cells (MDSC), which are composed of monocytic (M-MDSC) and polymorphonuclear (PMN-MDSC), and tumor-associated macrophages (TAM) play an important role in immune suppression and tumor progression (1) and are closely associated with negative clinical outcome in cancer (2). Transcriptional factors regulating the function of myeloid cells represent an attractive targeting opportunity with broad effects on the function of these cells. The transcription factor CCAAT/enhancer-binding protein alpha (C/EBPα) is involved in differentiation of myeloid cells as well as in proliferation, metabolism, and immunity (3, 4). Deregulation of C/EBPα has been reported in several solid tumors, including liver, breast, and lung (5). This is in contrast to C/EBPβ, which is upregulated in MDSCs and involved in their suppressive activity (6); C/EBPα was found to be downregulated in MDSCs and, furthermore, C/EBPα knockout mice displayed greater MDSC tumor infiltration (7). Upregulation of C/EBPα inhibits tumor growth in rodent liver cancer models; however, it is still unclear if this is mediated directly on tumor cells (8, 9). We have developed a first-in-class small activating RNA (saRNA) therapeutic (MTL-CEBPA) comprising of a SMARTICLES liposomal nanoparticle encapsulating CEBPA-51, a 2′O-Me RNA oligonucleotide duplex designed to specifically target and upregulate transcription of the CEBPA gene (10). The compound demonstrated safety in a phase I clinical trial (11, 12). However, the mechanism of the antitumor effects of MTL-CEBPA, as well as its possible effects in cancer patients, remains unclear.
Sorafenib, a multikinase inhibitor, has been the first-line systemic treatment for hepatocellular carcinoma (HCC) for many years. Despite this, the overall survival benefit of sorafenib in previously untreated patients with preserved liver function, good performance status, and advanced disease, although statistically significant, remains disappointing (10.7 vs. 7.9 months; ref. 13). Recently, the immune-checkpoint inhibitor pembrolizumab, when combined with bevacizumab, a VEGF-specific monoclonal antibody, showed a one-year progression-free survival rate increase from 54.6% to 67.2% when compared with sorafenib in a phase III clinical trial (14). This illustrates the need to search for novel combination therapeutics for patients with unresectable HCC. Because sorafenib is associated with increased MDSC infiltration, and is an identified mechanism for acquired resistance, we used this as a rationale for combining MTL-CEBPA with sorafenib. In this study, we evaluated the effect of MTL-CEBPA on the function of myeloid cells in patients with advanced HCC. We performed mechanistic studies in mouse tumor models and identified the mechanism of antitumor effect of MTL-CEBPA.
Materials and Methods
Patients in phase Ib clinical trial
Study design and participants
This study was conducted in accordance with the Declaration of Helsinki.
We report an international multicenter, noncomparative, open-label, phase Ib study in patients with advanced HCC to evaluate the safety and tolerability of MTL-CEBPA once weekly at 90 mg/m2 or 130 mg/m2 in combination with sorafenib 400 mg twice daily administered to HCC patients either concomitantly or sequentially, in cohorts either tyrosine kinase inhibitor (TKI) naïve or resistant. This study was conducted at 10 tertiary centers and university hospitals in three countries (Singapore, Taiwan, and the United Kingdom). Clinical trial information: NCT02716012.
Eligible patients were at least 16 years old with histologically confirmed advanced HCC with cirrhosis, or resulting from nonalcoholic steatohepatitis (NASH), with or without cirrhosis, and unsuitable for liver surgery and/or refractory to radiotherapy, ablation, and other therapies. Patients were required to have a Child–Pugh score of B8 or less and Eastern Cooperative Oncology Group (ECOG) performance status of 0–1. Full inclusion and exclusion criteria are described in Supplementary Appendix SA. All patients provided written informed consent, and the study protocol and amendments were approved by the relevant regulatory authority and each site's institutional review board or independent ethics committee.
MTL-CEBPA was administered by intravenous infusion over 60 minutes. MTL-CEBPA dosing was preceded by corticosteroid and anti-histamine administration to minimize the risk of infusion reactions. Two doses of MTL-CEBPA were explored. Patient dosing was based on body surface area calculation on day 1 of each cycle. The recommended starting dose for sorafenib was 400 mg twice daily. For relevant algorithms for sorafenib dose modification, interruption, or stoppage, please see study protocol in Supplementary Material.
Each treatment cycle was 28 days and continued until disease progression. Seven days elapsed between the first dose of the first participant and the first dose of the subsequent participants in each dose cohort. Patients off treatment were followed up for survival every three months.
In the combination cohorts, MTL-CEBPA 90 or 130 mg/m2 was administered once weekly on days 1, 8, and 15; sorafenib was initiated on day 1 of cycle 1 and continued for the duration of each cycle. In the sequential cohorts, MTL-CEBPA was administered at 130 mg/m2 once weekly on days 1, 8, and 15 for the first two cycles only and discontinued thereafter; sorafenib was initiated on day 1 of cycle 3 and continued for the complete duration of each cycle.
Safety
The dose-limiting toxicities (DLT) were determined on the basis of the incidence and severity of adverse events (AE) occurring in the first cycle (28 days). Patients were treated until disease progression or unacceptable toxicity. A Safety Review Committee was convened to oversee safety, scientific integrity, and validity of the study. Safety and tolerability of MTL-CEBPA was evaluated in terms of frequency of AEs graded according to toxicity criteria (NCI Common Terminology Criteria for Adverse Events, CTCAE v 4.03).
Endpoints
The primary endpoint was the objective response rate. Tumor response was evaluated using CT or MRI every two cycles using the Response Evaluation Criteria in Solid Tumors v1.1 by investigator assessment.
The secondary endpoints were to evaluate the safety and tolerability of coadministering/sequentially administering MTL-CEBPA and sorafenib (frequency of AEs graded according to toxicity criteria) and to characterize the pharmacokinetic (PK) parameters of MTL-CEBPA during these treatments (Cmax, Tmax, and AUC).
Additional outcome measures included determining the antitumor activity of MTL-CEBPA in combination with sorafenib as assessed by complete response rate and progression-free survival (PFS), and overall survival (OS).
Pharmacokinetics
Plasma samples for the analysis of CEBPA-51 were collected over the first dosing interval and for 72 hours after administration of the second dose. Due to the rapid degradation and elimination of free CEBPA-51 in plasma, the measured concentration of CEBPA-51 reflects the concentration of CEBPA-51 encapsulated in MTL-CEBPA nanoparticles. A fluorescently labeled peptide nucleic acid (PNA)-probe, designed against the guide strand of CEBPA-51, was used to extract the single-stranded parent compound. RNA species are quantitated using anion-exchange HPLC and fluorescence detection. Plasma CEBPA-51 is expressed as μg/mL of double-stranded RNA, and the lower limit of quantitation is 0.001 μg/mL.
MDSC measurement from patient blood
Whole blood (8 mL) was collected from trial subjects in Streck CytoChex vacutainers at D0 (pretreatment), 24 hours after first infusion (D1) and 7 days after infusion (D7) of MTL-CEBPA.
The blood was processed within 30 minutes of collection. Briefly, following red blood lysis, the isolated peripheral blood mononuclear cells (PMBC) were adjusted to a concentration of 20 × 106 cells/mL with cold FACS staining buffer. Cell suspension (50 μL), which was equivalent to 1 × 106 cells, was used per FACS staining in 50 μL antibody cocktail with Brilliant Stain buffer comprising of (CD10, CD66b−; CD16; CD14, CD15, CD11b, LOX-1, HLA-DR, CD38, and DRAQ7). Together with the appropriate FMO controls and compensation bead set up, all mixtures were performed at 4°C in the dark for 15 minutes. All samples were washed with PBS and fixed BD Cytofix. Samples were resuspended in 500 μL of cold FACS staining buffer and transferred through 30-μm cell strainer into round bottom tubes ready for FACS analysis. All samples were analyzed with a BD LSRFortessa. All analyses were based on 60,000 events captured from 1 × 106 cells.
Animal experiments
Mouse experiments were approved by the Institutional Animal Care and Use Committee of The Wistar Institute. C57BL/6 mice (female, 6–8 weeks old) and NOD-SCID mice (female, 6–8 weeks old) were purchased from Charles River. B6.Cg-Thy1a/Cy Tg(TcraTcrb)8Rest/J (PMEL, female, 6–8 weeks old) mice were purchased from the Jackson Laboratory.
Cell lines
LLC lung carcinoma cell line was obtained from ATCC, and MC 38 colon carcinoma cell line was obtained from I. Turkova, University of Pittsburgh, Pittsburgh, PA. Murine BNL 1ME A.7R.1 cell line (BNL; American Type Culture Collection, Manassas, VA) were derived from BALB/c mice. BNL cells (3 × 105) in 100 mL HBSS were injected into livers of BALB/c mice to generate orthotopic tumors. All cells were cultured in DMEM (Corning Incorporated) supplemented with 10% FBS (Atlanta Biologicals, Inc.) and 1% antibiotics (penicillin–streptomycin, Thermo Fisher Scientific Inc.) at 37°C, 5% CO2. The cells were harvested using 0.25% Trypsin (Thermo Fisher Scientific Inc.), suspended in DPBS, and then subcutaneously injected to the mice at 5 × 105 per mouse. After tumors were established, the mice were randomized into groups based on their tumor sizes and used for the studies. The tumor diameters (width and length) were measured using digital calipers and used for the calculation of tumor area (width × length).
Reagents and treatment
MTL-CEBPA, a liposomal nanoparticle encapsulating a saRNA for CEBPA (CEBPA-51), and its control liposomal nanoparticle with a nonspecific oligonucleotide (NOV-FLUC) encapsulating siFLUC were supplied by MiNA Therapeutics Ltd. MTL-CEBPA or NOV-FLUC was intravenously injected into the tumor-bearing mice twice per week at 3 mg/kg. For the T-cell depletion study, intraperitoneal injection with 100 μg of anti-mouse CD8a antibody (Bio X Cell, BE0004-1) or rat IgG2a isotype control antibody (Bio X Cell) was started 2 days before tumor injection and repeated twice a week for 2 weeks. Anti-mouse CTLA-4 antibody (Bio X Cell, BE0164) or mouse IgG2b isotype control antibody was intraperitoneally injected into the tumor-bearing mice: 100 μg per mouse on days 10, 17, and 24. Celecoxib, a selective cox2 inhibitor (Selleck Chemicals), was suspended in 0.5% methylcellulose and orally treated at 50 mg/kg to the tumor-bearing mice every day. Lipofermata was dissolved in DMSO and diluted in 30% (v/v) Kolliphor, and then subcutaneously injected into the tumor-bearing mice at 2 mg/kg twice a day.
Transfection studies
LLC and MC38 cells were seeded into 24-well plates at a density of 40,000 cells per well. We used saRNA for CEBPA (CEBPA-51) and control siFLUC, which are synthesized at MiNA Therapeutics for in vitro studies. CEBPA-51 or siFLUC was added to the cells at a final concentration of 10 nmol/L with lipofectamine 2000, following the manufacturer's instructions (Life Technologies). The treatment was repeated 24 hours later, and the cells were harvested at the 72-hour time point and used for MTS assay and RNA extraction for qRT-PCR. The transfected cells were seeded into 96-well plates from 625 to 10,000 cells per well, and cell proliferation was detected by CellTiter 96 AQueous One Solution Cell Proliferation Assay (MTS assay, Promega).
Isolation of cells
Single-cell suspensions were prepared from spleens and bone marrow from femur and tibia and followed by red blood cell removal using ammonium chloride lysis buffer. Tumor tissues, lungs, and livers were processed to obtain single-cell suspensions using the Mouse Tumor Dissociation Kit according to the manufacturer's recommendation (Miltenyi), and followed by red blood cell removal.
T-cell suppression assay
PMN-MDSCs (Ly6G+) were purified from spleens and tumors. Isolated cells were subsequently incubated with biotinylated Ly6G antibody and streptavidin microbeads (Miltenyi). M-MDSCs (CD45+CD11b+Ly6ChiLy6G−) and macrophages (CD45+CD11b+F4/80+Ly6C−) were sorted using FACSAria (BD Biosciences). PMN-MDSCs, M-MDSCs, or macrophages were plated in U-bottom 96-well plates (3 replicates) in RPMI supplemented with 10% FBS, penicillin–streptomycin, and 0.05 mmol/L 2-mercaptoethanol, and cocultured at different ratios with splenocytes from PMEL mice in the presence of 0.1 μg/mL of murine gp100 peptide (EGSRNQDWL, AnaSpec, Inc.). After 48 hours, the cells were incubated with 3H-thymidine (1 μCi/well; GE Healthcare) for 16 hours. Proliferation was measured using the TopCount NXT instrument (PerkinElmer).
Flow cytometry
Monoclonal antibodies specific to the mouse cell-surface markers CD45, CD11b, CD11c, Ly6G, Ly6C, F4/80, I-Ab, and CD16/32 (Fc block) were purchased from BD Bioscience. Cells were incubated with Fc block for 10 minutes and stained with fluorochrome-conjugated antibodies for 15 minutes at 4°C. The cells were run on LSR II flow cytometer (BD Bioscience) and analyzed with FlowJo (FlowJo, LLC.).
qRT-PCR
RNA was extracted using the Quick-RNA MicroPrep Kit (Zymo Research) according to the manufacturer's instruction. Single-stranded cDNA was synthesized from RNA samples using the High-Capacity cDNA reverse transcription kit (Applied Biosystems). RT-PCR was performed using Power SYBR Green PCR Master Mix (Applied Biosystems) in 96-well plates, and then read using QuantStudio 5 Real-Time PCR System (Applied Biosystems). Amplifications were carried out with the primers described in the table. Bioinformatically validated primer set for mouse CEBPA (QuantiTect Primer Assays) was purchased from Qiagen.
β-actin: 5′-ATGGAGGGGAATACAGCCC-3′; 5′-TTCTTTGCAGCTCCTTCGTT-3′
Arg1: 5′-GCTGTCTTCCCAAGAGTTGGG-3′; 5′-ATGGAAGAGACCTTCAGCTAC-3′
Nos2: 5′-AACGGAGAACGTTGGATTTG-3′; 5′-CAGCACAAGGGGTTTTCTTC-3′
Ptgs2: 5′-CCAGCACTTCACCCATCAGTT-3′; 5′-ACCCAGGTCCTCGCTTATGA-3′
Ptges: 5′-GCACACTGCTGGTCATCAAG-3′; 5′-ACGTTTCAGCGCATCCTC-3′
Cy3 uptake and trace
MTL-CEBPA labeled with Cy3 dye was intravenously injected to the LLC tumor–bearing mice at 3 mg/kg. Peripheral blood, spleen, lung, bone marrow, liver, and tumor were taken from the mice 4 hours after injection or before injection (0 hours). Single cells from the tissues were analyzed using a flow cytometry.
HalioDx brightplex technology: sequential multiplex IHC
H&E staining was performed on 4-μm-thick formalin-fixed paraffin-embedded (FFPE) tissue sections for a preliminary tissue evaluation. Slides were scanned with the NanoZoomer-XR (Hamamatsu) to generate digital images (20×). A pathologist identified the tumor area and provided qualitative assessment. The multiplex IHC panels used were part of Immunoscore Suppressor Cells family (MDSC/Macrophages Brightplex; Neutrophils Brightplex; MacrophagesBrightplex). They were carried out on 4 FFPE biopsies to identify and quantify myeloid cell subsets in a patient's tumor microenvironment. The 4-μm-thick unstained sections from the pre- and posttreated patients were stained in a Leica Bond RX autostainer (Leica Biosystems). Slides were deparaffinized and rehydrated in the autostainer according to the manufacturer's instructions. Antigen retrieval was performed with Bond Epitope Retrieval Solution #2 (Leica Biosystems), equivalent to EDTA pH 9.0, for detection of all biomarkers. Successive stainings were performed on the same FFPE slide. The primary antibodies used for the different Brightplex panels (Supplementary Table S4) were anti-CD68 (Abcam, catalog #ab213363), anti-CD163 (Bio-Rad, catalog #MCA1853), anti-LOX1 (Merck Millipore, catalog #MABS186), anti-CD11B (Cell Signaling Technology, catalog #49420S), anti-CD15 (BD Biosciences, catalog #555400), anti-CD14 (Cell Marque, catalog #114R-15), anti-S100A9 (Origene, catalog #UM800066), anti-IL10 (RandD Systems, catalog #MAB92101), anti-TNFα (Proteintech, catalog #60291-1-Ig), anti-MPO (Abcam, catalog #ab93665), anti-IDO (Thermo Fisher, catalog #14-9750-82), anti-CD64 (Abcam, catalog #ab140779), anti-CD163 (Abcam, catalog #ab182422), anti-CD86 (Cell Signaling Technology, catalog #91882S), and anti-CD206 (R&D Systems, catalog #MAB25341). Antibodies were diluted in Emerald antibody diluent (ESBE Scientifique; catalog #CMQ-936B09). The primary antibodies were detected using MACH 2 rabbit HRP polymer (Biocare, RHRP520L) or MACH 4 Universal HRP polymer as secondary antibody (Biocare, M4U534L). The labeling was visualized using aminoethyl carbazole (AEC Peroxidase Substrate Kit, Biocare, catalog #SK-4200; ImmPACT AMEC Red, Vector Lab, catalog #SK-4285) and hematoxylin counterstaining. A human tonsil specimen was used as control for all immune biomarker's detection using qualitative acceptance criteria [specificity, staining location (nucleus/membrane), cell type, and lack of background or unspecific staining]. After each individual staining, coverslipping was performed automatically by the workstation CTM6 with aqueous mounting (VectaMount AQ, VECTOR Laboratories, catalog #H-5501). The slides were digitalized in a NanoZoomer-XR scanner (Hamamatsu; ×20) and a visual quality control carried out. Between each staining cycle of the sequential multiplex, the labeling was eliminated by incubating the samples in ethanol, and the antibody complexes were denatured using a denaturing buffer.
HalioDx brightplex technology: digital pathology analysis
Each biopsy was analyzed using the HalioDx Digital Pathology Platform. Images obtained following sequential multiplex IHC workflow were aligned with Brightplex-fuse (HalioDx software). A pseudo-color image containing the information for the expression of all biomarkers was created. The latter was analyzed by HALO software (Indica Labs) for the identification of tumor areas using annotation tools. Next, positively stained cells were detected and quantified in the selected regions of interest using HALO software (Indica Labs).
Phenotypes of myeloid cells were visually verified according to expected staining and quantified with Brightplex MultiplexR (HalioDx software). The final data were expressed as the myeloid cell density (cells/m2) in the analyzed tumor regions.
HalioDx brightplex technology: bioinformatics analysis
The descriptive analysis of the data was performed using heatmaps and unsupervised clustering. The heatmaps of immune cell populations were based on the log2 fold change between pre- and posttreatment. The phenotypes or patient clustering associated with each heatmap was performed by using Euclidean distance analysis. The data analysis was carried out with the R software (v 3.6.1, https://www.R-project.org/). The heatmaps were achieved with the ComplexeHeatmap and FactoMineR packages, respectively.
RNA-seq
RNA sequencing (RNA-seq) raw sequencing reads were aligned using bowtie 2 (15) algorithm against mm10 human genome version and RSEM v1.2.12 software (16) was used to estimate read counts and RPKM values using gene information from Ensemble transcriptome version. Raw counts were used to estimate significance of differential expression difference between two experimental groups using DESeq2 (17). Genes that passed nominal P < 0.05 threshold were subject to enrichment analysis using QIAGEN's Ingenuity Pathway Analysis software (IPA, QIAGEN; www.qiagen.com/ingenuity) using the “Canonical pathways” and “Upstream Regulators” options. Select pathways and regulators that passed P < 0.05 threshold and were significantly predicted to be activated or inhibited based on activation state absolute Z-score of at least 2 were reported.
NanoString analysis
Blood (6 mL) was collected in EDTA vacutainers (BD) and captured in a LeukoLOCK filter system (Ambion) modified for use for the OUTREACH study. Briefly, the filter captured white blood cells (WBC) from whole blood, whereas all remaining blood components were flushed out. The filter content was then preserved with RNALater solution and stored at −80°C for total RNA extraction. Total RNA was then isolated from the captured WBC by using a modified TRIzol extraction method. The captured RNA was then analyzed for concentration (Nanodrop) and RNA integrity (Qbit) before proceeding to NanoString analysis or real-time quantitative PCR analysis using the Quantitect reverse transcription (Qiagen) kit.
NanoString RCC files were imported into nSolver 4.0.70 Analysis Software (NanoString Technologies Inc). The quality of the data was checked using the default QC parameters of the nSolver, that is, positive control limit of detection was required to be less than or equal to 2 standard deviations above the mean of the negative controls. All samples were found to be of analysis-ready quality. Positive control and codeset content normalization procedures were run using nSolver for QC purposes to detect samples whose normalization factors were outside of the recommended ranges. Both normalization factors were computed using the geometric mean of either positive controls or housekeeping genes, and the accepted ranges of these factors were 0.3–3 and 0.1–10, respectively. No QC flags for codeset normalization were raised, and thus raw counts of all samples were exported for further analysis using R, v. 3.5.1 (18).
As an additional QC step before normalization and differential gene-expression analysis, R package NanoStringDiff, v. 1.12.0, was used for checking that the expressions of positive controls were linearly related to their concentrations and that the expressions of housekeeping genes had relatively low variation across samples. For data normalization, top 6 housekeeping genes with lowest variation across the samples were chosen. R package RUVSeq, v. 1.16.1, was first used for estimating factors of unwanted variation using housekeeping gene counts. These factors were then included in the DESEq2 model for differential gene-expression analysis using the raw counts. Principal component analysis (PCA) was performed for the data after variance stabilizing transformation using the varianceStabilizingTransformation function of DESeq2, v. 1.22.2. In addition, removeBatchEffect function of the limma R package, v. 3.38.3, was used for removing any effects that might be due to differences in the amount of input RNA by incorporating the factors of unwanted variation that were estimated above based on the housekeeping genes as covariates in the function. PCA was done using basic R functions and custom plotting scripts utilizing ggplot2 package, v. 3.1.1 (19).
Differentially expressed genes (DEG) were analyzed using DESeq2, v. 1.22.2 (17), contrasting day 1 samples and day 0 samples and adjusting for patient as well as the NanoString cartridge. Before the analysis, lowly expressed genes were filtered out, including only those with at least 10 counts in total across all samples. Statistical significance of the DEGs was assessed using a Wald test, and the obtained P values were adjusted for multiple testing using the Benjamini–Hochberg procedure (20). Statistically significant differential gene expression was regarded if adjusted P value was below 0.05. The obtained DEG lists were further filtered for biological significance to include only genes with at least 1.5-fold upregulation or downregulation (absolute log2 fold change > 0.585) between the day 1 and day 0 sample groups.
Protein analysis
Proteomics experiments were performed using mass spectrometry essentially as reported (21, 22).
Briefly, LeukoLOCK-captured WBCs were lysed in urea lysis buffer (8M urea, 10 mmol/L Na3VO4, 50 mmol/L NaF, 100 mmol/L β-glycerol phosphate, and 25 mmol/L Na2H2P2O7) and proteins reduced and alkylated by sequential addition of 1 mmol/L DTT and 5 mmol/L iodoacetamide. Immobilized trypsin was then added to digest proteins into peptides. After overnight incubation with trypsin, peptides were desalted by solid phase extraction (SPE) using OASIS HLB columns (Waters) in a vacuum manifold following manufacturer's guidelines with the exception that the elution buffer contained 1M glycolic acid. Peptides were enriched from the resulting peptide mixture using TiO2 chromatography essentially as described (23) with the modifications (24). TiO2 chromatographic media were added to the SPE-eluted peptides and incubated 5 minutes with rotation. The TiO2 media were then packed in empty spin-tips and washed three times with 1M glycolic acid, 5% TFA. Peptides were eluted with 5% NH4OH and dried in a vacuum concentrator. Dried peptide extracts were dissolved in 0.1% TFA and analyzed by nanoflow LC-MS/MS in an LTQ-orbitrap as described before (21, 22). Gradient elution was from 2% to 35% buffer B in 90 minutes with buffer A being used to balance the mobile phase (buffer A was 0.1% formic acid in water and B was 0.1% formic acid in acetonitrile). MS/MS was acquired in multistage acquisition mode. MS raw files were converted into Mascot Generic Format using Mascot Distiller (version 1.2) and searched against the SwissProt database (2013.03 version) restricted to human entries using the Mascot search engine (version 2.3; ref. 25). Allowed mass windows were 10 ppm and 600 mmu for parent and fragment mass to charge values, respectively. Variable modifications included in searches were oxidation of methionine, pyro-glu (N-term), and phosphorylation of serine, threonine, and tyrosine. Results were filtered to include those with a potential for false discovery rate less than 1% by comparing with searches against decoy databases. Quantification was performed by obtaining peak areas of extracted ion chromatographs (XICs) for the first three isotopes of each peptide ion using Pescal (26, 27). Mass and retention time windows of XICs were 7 ppm and 1.5 minutes, respectively (26, 27).
NanoString RCC files were imported into nSolver 4.0.70 Analysis Software (NanoString Technologies Inc). Quality of the data was checked using the default QC parameters of the nSolver, that is, positive control limit of detection was required to be less than or equal to 2 standard deviations above the mean of the negative controls. All samples were found to be of analysis-ready quality. Positive control and codeset content normalization procedures were run using nSolver for QC purposes to detect samples whose normalization factors were outside of the recommended ranges. Both normalization factors were computed using the geometric mean of either positive controls or housekeeping genes, and the accepted ranges of these factors were 0.3–3 and 0.1–10, respectively. No QC flags for codeset normalization were raised, and thus raw counts of all samples were exported for further analysis using R, v. 3.5.1.
As an additional QC step before normalization and differential gene-expression analysis, R package NanoStringDiff, v. 1.12.0, was used for checking that the expressions of positive controls were linearly related to their concentrations and that the expressions of housekeeping genes had relatively low variation across samples. For data normalization, top 6 housekeeping genes with lowest variation across the samples were chosen. R package RUVSeq, v. 1.16.1, was first used for estimating factors of unwanted variation using housekeeping gene counts. These factors were then included in the DESEq2 model for differential gene-expression analysis using the raw counts.
PCAs were performed and the results were visualized using ggplot2 (19). The analysis was performed using all samples and therein both using all available measurement data and only the measurements of the top 574 proteins and top with the greatest variance in expression. For this analysis, the provided normalized data were log2 transformed. In these analyses, the top 574 proteins with greatest variance in expression were used using voom (28) logcpm transformed normalized values. Pearson correlation coefficients were calculated pairwise for all samples, using the log2 transformed data of all available measurements. Proteins with P < 0.05 and absolute log2 fold change > 1 were considered as significantly differentially expressed. Adjusted P values were also calculated by correcting for multiple testing using the Benjamini–Hochberg method (20). Differentially expressed proteins were compared between D1 and D0 samples. Statistical testing between sample groups was performed with limma using the voom logcpm transformed data. The information of sample pairedness was included into the experimental design in limma analysis. Proteins with P < 0.05 and absolute log2 fold change > 1 were considered as significantly differentially expressed. Adjusted P values were also calculated by correcting for multiple testing using the Benjamini–Hochberg method (20).
Statistical analysis
After test for normal distribution of data, statistical analyses were performed using two-tailed Student t test and GraphPad Prism 8.2. software (GraphPad Software Inc.). One-way ANOVA test with correction for multiple comparisons (Kruskal–Wallis or Tukey tests) was used in experiments with more than two groups. In other cases, an unpaired two-tailed Student t test was used. Significance was determined at P of 0.05. Estimation of variation within each group of data was performed, and variance was similar between groups that were compared. Animal experiments were not blinded. Tumor growth was evaluated using two-way ANOVA test with Bonferroni correction for multiple comparisons.
Results
MTL-CEBPA treatment in patients with advanced HCC shows antitumor responses
We previously reported the effects of MTL-CEBPA in a phase Ia clinical trial on 38 patients with advanced-stage liver cancer (11). A recommended phase II dose was identified (130 mg/m2 once weekly) where MTL-CEBPA was found to be safe, well tolerated, and demonstrated pharmacodynamic activity with evidence of target engagement. This prompted the initiation of a phase Ib dose escalation and cohort expansion study of MTL-CEBPA in combination with sorafenib in patients with advanced HCC. In this international multicenter, noncomparative, open-label, phase Ib study (NCT02716012), we evaluated safety, tolerability, and preliminary assessment of the activity of MTL-CEBPA 90–130 mg/m2 once weekly in combination with sorafenib 400 mg twice daily administered to HCC patients either concomitantly or sequentially, in cohorts of either TKI-naïve or -resistant patients. Eligible patients were at least 16 years old with histologically confirmed advanced HCC with cirrhosis, or resulting from NASH, with or without cirrhosis, and unsuitable for liver surgery and/or refractory to radiotherapy, ablation, and other therapies. Patients were required to have a Child–Pugh score of B8 or less and ECOG performance status of 0–1. Between November 2018 and January 2020, 36 patients were enrolled in the phase Ib trial. Twenty-two patients were enrolled in the coadministration cohorts receiving MTL-CEBPA and sorafenib concomitantly during both treatment cycle 1 and cycle 2. Fourteen patients were enrolled into the sequential cohorts receiving MTL-CEBPA alone for 2 cycles followed by sorafenib alone in cycle 3 (Supplementary Fig. S1; Supplementary Table S1). The safety profile was acceptable (Supplementary Table S2) with no dose-limiting toxicities observed.
Of the 36 patients with advanced HCC enrolled in the study, 15 were not previously treated with TKI and had established viral etiology of the disease (Table 1). Four patients in this group demonstrated objective response (OR) to the treatment with MTL-CEBPA in combination with sorafenib (26.7%). Remarkably, 3 patients had developed complete response (Fig. 1A and Table 1). The responses were durable with complete eradication of target lesions at month 12 when compared with pretreatment (Fig. 1B), and a complete radiologic response of lung metastases (Fig. 1C). Notably, only 1 patient out of 11 with HCC of nonviral etiology had OR (Table 1). These results compared favorably with contemporary OR rate in HCC patients treated with sorafenib (7%–11.9%; ref. 14).
Table 1.
Best objective response.
| Best objective response | TKI-naïve viral HCC, n = 15 | TKI-naïve nonviral HCC, n = 11 | TKI-experienced viral HCC, n = 6 | TKI-experienced nonviral HCC, n = 4 |
|---|---|---|---|---|
| Total evaluable (%) | 15 (100) | 11 (100) | 6 (100) | 4 (100) |
| Objective response (%) | 4 (26.7) | 1 (9.1) | 0 | 0 |
| Complete response | 2 (13.3) | 0 | 0 | 0 |
| Partial response | 2 (13.3) | 1 (9.1) | 0 | 0 |
| Stable disease (%) | 7 (46.7) | 7 (63.6) | 6 (100) | 4 (100) |
| Progressive disease (%) | 4 (26.7) | 3 (27.3) | 0 | 0 |
Figure 1.
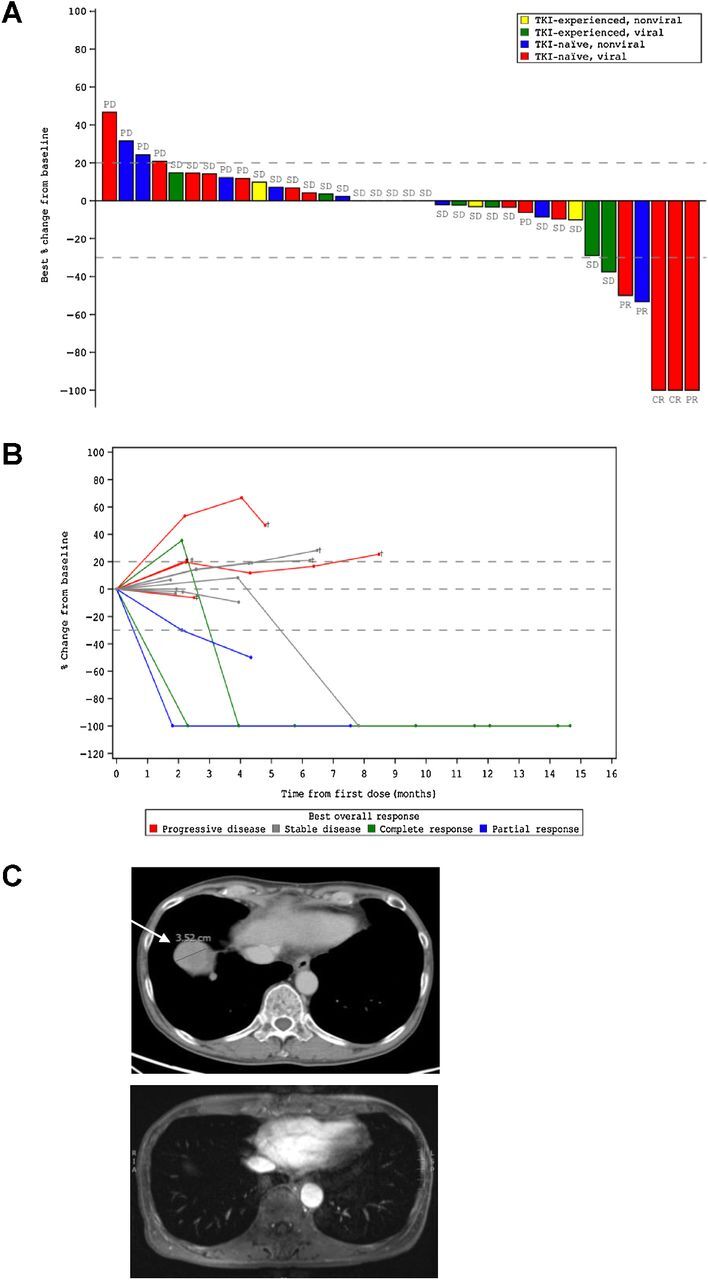
Clinical activity of MTL-CEBPA in advanced HCC patients treated in combination with sorafenib. A, Waterfall plot of patients in phase Ib study showing best percentage (%) change from baseline, with identification of groups that had previously been treated with TKI and those that had HCC of viral etiology. B, Durable responses of patients previously naïve to TKI with HCC of viral etiology. Spider plot in phase Ib patients who had not previously been treated with TKI and had HCC of viral etiology, showing tumor response for target lesions. C, Complete radiologic response of lung metastases following treatment with MTL-CEBPA and sorafenib. Cross-sectional imaging of a patient with baseline imaging on top from June 12, 2018, showing right lung metastases and on bottom from December 31, 2018, showing complete resolution of lung metastases. This patient maintains a complete radiologic response to both liver and lung metastases on last surveillance imaging on March 13, 2020.
The effect of MTL-CEBPA therapy on myeloid cells in cancer patients
We assessed the effect of MTL-CEBPA on gene and protein expression changes specifically in mononuclear cells. Because our preliminary studies demonstrated that MTL-CEBPA was rapidly taken up by myeloid cells, we evaluated cells isolated from blood before (day 0) and 24 hours (day 1) after the initial dose of MTL-CEBPA. No additional therapy was administrated during that time. Mononuclear cells were collected from 7 patients. Gene-expression profile was evaluated by NanoString analysis using the human PanCaner IO 360 panel (29). Genes with > 1.4-fold up- or downregulation with FDR < 5% are shown in Fig. 2A. We observed marked decrease in the expression of NFKB1, MAPK8, ILF3, CCR3, IDO1, CLEC4C, ISG15, CCL4, C2 encoding complement C2, CYBB that encodes NADPH oxidase, LTF encoding lactoferrin, CEACAM8 encoding CD66b, and ITGB1 encoding integrin β1 protein. These genes are known to be implicated in immune-suppressive activity of MDSCs (Fig. 2A). In parallel, mononuclear cells were collected from 6 patients, and the total protein content was extracted and solubilized for whole proteome mass spectrometry analysis. Five hundred and seventy-four differentially enriched proteins (DEP; P < 0.05) were identified from the paired day 1 vs. day 0 samples. Five hundred and thirty-one proteins showed more than 2-fold upregulation, and 43 proteins showed more than 2-fold downregulation. Marked downregulation of different subunits of NF-kB, complement C2, LAMP1, and TGFβ was seen (Fig. 2B). These paralleled the changes also measured from the gene-expression profile (Fig. 2A). Genes and proteins associated with monocyte and neutrophil function including TNFR, TLR4, TLR5, TLR2, integrins, neutrophil cytosolic factor 4, CD14, MHC class I, neutrophil elastase, ICAM3, and MAPK14 (p38α) were upregulated (Fig. 2B). From this pattern of factors changing, we proposed that MTL-CEBPA caused downregulation of proteins associated with suppressive activity of myeloid cells while promoting activation of classic monocytes and granulocytes.
Figure 2.
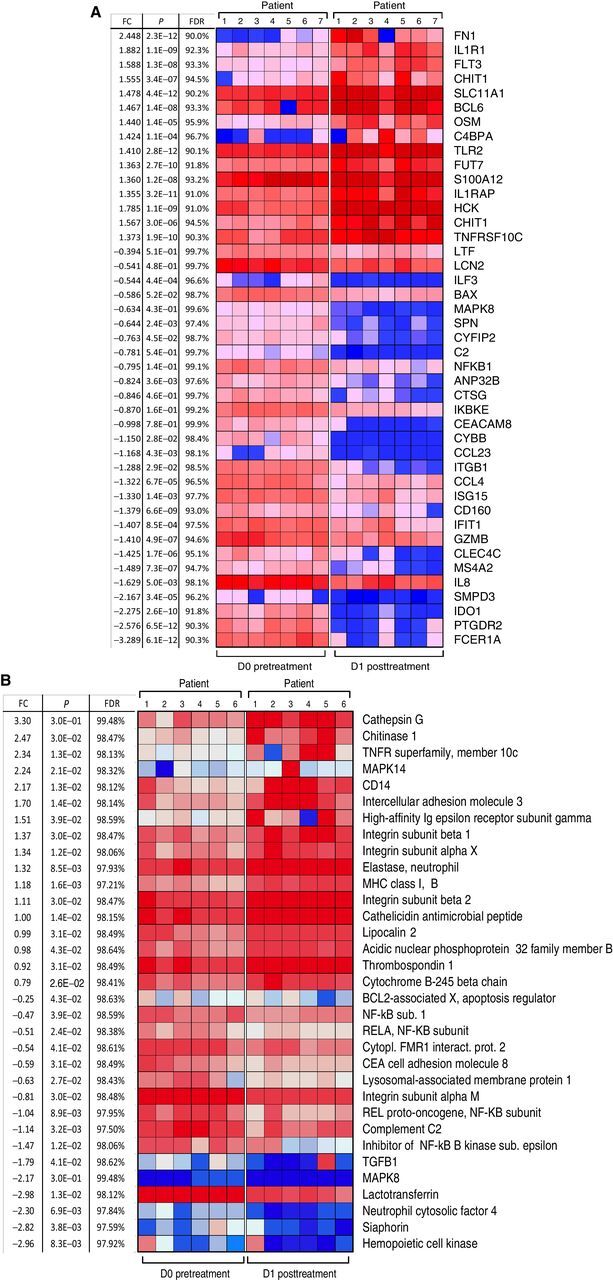
Effect of MTL-CEBPA treatment of patients with HCC on gene and protein expression in myeloid cells. A, Gene-expression profile was evaluated by NanoString using the human PanCaner IO 360 panel. Heat map of gene-expression upregulated (+1> log2 fold change and above) or downregulated (−1< log2 fold change and below) with a false discovery rate (FDR) of <5% is shown. B, Protein expression profile was evaluated by mass spectroscopy. Proteins with P < 0.05 and absolute log2 fold change > 1 were considered as significantly differentially expressed. Adjusted P values were calculated by correcting for an FDR of <5%.
To expand on these observations, we performed independent gene-expression analysis of total leukocyte population using qRT-PCR from 12 patients treated only with MTL-CEBPA. As expected, MTL-CEBPA upregulated expression of CEBPA. It was associated with upregulation of TLR5, IL18R1, IL18AP, and MAPK14. At the same time, marked decrease of CSF1, OLR1 encoding LOX-1, IL8, and TNFA was found (Fig. 3A). This observation therefore supported the result of our unbiased gene-expression analysis (Fig. 2A) of the mononuclear cells. Downregulation of these genes suggested that MTL-CEBPA treatment affected the presence of M-MDSCs or PMN-MDSCs in blood. Therefore, we evaluated these cells by flow cytometry in patients treated with MTL-CEBPA. M-MDSCs (CD66b− CD14+ HLA-DR−/loCD15− CD11b+CD38+) were dramatically reduced 24 hours after MTL-CEBPA injection and further decreased 7 days after the treatment. A decrease in PMN-MDSCs (CD66b+CD14−CD15+CD11b+ LOX1+) was also evident, albeit at a lesser extent and with a slight rebound observed after 7 days (Fig. 3B). In contrast, no significant changes in the circulating populations of monocytes and neutrophils were observed in those patients (Fig. 3C). Thus, we proposed that an increase of CEBPA transcription factor expression in monocytes driven by MTL-CEBPA caused rapid downregulation of genes and proteins involved in MDSC suppressive activity.
Figure 3.
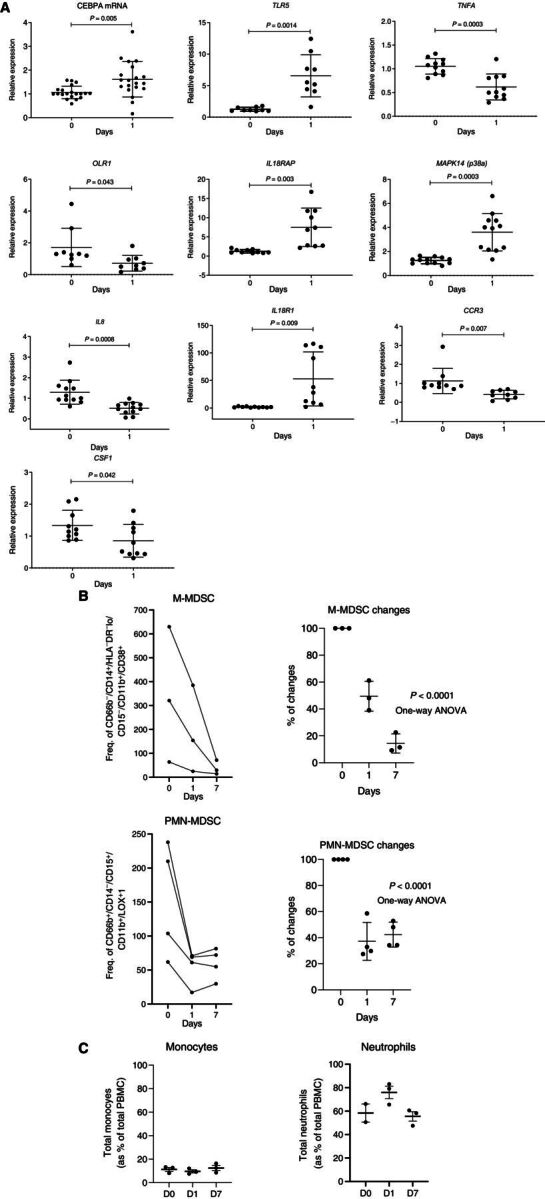
Changes in gene expression in total leukocytes in patients treated with MTL-CEBPA. A, Expression of indicated genes in leukocytes from 12 patients. Gene expression was evaluated by qRT-PCR. Individual results, mean, and standard deviation are shown. P values are calculated using two-sided Student t test. B, the presence of M-MDSCs (CD66b− CD14+ HLA-DR−/loCD15− CD11b+CD38+) and PMN-MDSCs (CD66b+CD14−CD15+CD11b+ LOX1+) among mononuclear cells was analyzed by flow cytometry and represented as frequency of gated cell population at 60K event per 1 × 106 cells (n = 3). C, Total circulating population of monocytes and neutrophils in the same patients was measured as a percentage of total PBMC.
We explored the effect of therapy on the presence of 8 major subpopulations of TAMs including the M1- and M2-type polarization using sequential multiplex, brightplex IHC in liver biopsies of representative patients who showed complete response, stable disease (SD), and progressive disease (PD; Fig. 4A). A heatmap of the different macrophage populations was established based on the log2 fold change of cell density in the liver biopsies obtained before commencement of MTL-CEBPA treatment (pretreatment) and compared with end-of-study biopsies (posttreatment). We observed a strong downregulation of M2-polarized TAMs [where reduced staining of CD68, CD163, and CD64 was quantified from the posttreatment biopsy of the complete response (CR) patient (Fig. 4B)], whereas in patients with SD and PD, the decrease in these cells after MTL-CEBPA treatment was less prominent (Fig. 4B). Overall, data from the biopsy staining and from the gene-expression analysis suggested a possible shift from M2-type to M1-type polarized TAMs after treatment with MTL-CEBPA and sorafenib. Because sorafenib alone was not part of the therapeutic regimen in this trial, we cannot at this stage exclude the contribution of sorafenib on the observed changes in myeloid cells within the tumors.
Figure 4.

The effects of MTL-CEBPA treatment on TAMs in patients with HCC. A, Representative images of a CR patient's biopsies with a complete loss of protumoral M2 macrophages (blue squares: CD68+ CD163+ CD64− cells) are shown in bottom (CR posttreatment) panel when compared with top (CR pretreatment) panel. White squares represent the pan-macrophage population expressing CD68+. Pseudo-color image: created by virtual slides alignment and imported in Halo software for biomarker analysis. B, A heat map of macrophage subsets was set up based on log2 fold change between pre- and posttreatment (cell densities) samples of 3 HCC patients including the CR as shown in A, stable disease (SD), and progressive disease (PD). The macrophage populations analyzed were: pan-macrophage (CD68+ cells), antitumoral M1 macrophages (CD68+CD64+CD163−CD206− cells), activated M1 macrophages (CD68+CD64+CD163−CD206− cells), protumoral M2 macrophages subset 1 (CD68+CD163+CD64− cells), protumoral M2 macrophages subset 2 (CD68+CD206+CD64− cells), protumoral M2 macrophages subset 3 (CD68+CD163+CD206+CD64− cells), and activated M2 subsets characterized by IL10 production.
MTL-CEBPA abrogates the immune-suppressive activity of M-MDSCs and macrophages in mouse tumor models
To better understand the contribution of MTL-CEBPA with sorafenib or checkpoint inhibitors on the tumor responses seen from the clinical study, we used different mouse tumor models. First, we asked if the antitumor effects of MTL-CEBPA alone were observed in an orthotopic BNL model of HCC, where tumor cells were injected into livers of BALB/c mice. After establishment of tumor nodules, we observed marked reduction in tumor growth following MTL-CEBPA treatment (Fig. 5A). This was associated with an increase in the presence of T cells (CD3+CD4+ and CD3+CD8+) but not NK cells (CD3−CD49b+) in the spleens when compared with the control group (Fig. 5B). Antitumor effect of MTL-CEBPA was similar to that of sorafenib alone. A combination of sorafenib with MTL-CEBPA demonstrated increased antitumor effects; however, this did not reach statistical significance (Fig. 5C).
Figure 5.

The effect of MTL-CEBPA on tumor growth in mouse tumor models. MTL-CEBPA and control NOV-FLUC were intravenously injected to the tumor-bearing mice at 3 mg/kg twice a week from day 5. A, Kinetic of tumor growth in mice bearing BNL HCC cell line (n = 5). P value was calculated using two-way ANOVA test. B, The presence of the indicated cell population in spleens of NBL tumor–bearing mice presented as percentages (%). C, Tumor volume in NBL tumor–bearing mice treated with MTL-CEBPA and sorafenib after 10 days of treatment. Mean and standard deviation are shown. n = 6 for PBS control- and sorafenib-treated groups, n = 10 for the MTL-CEBPA–treated group, n = 8 for the combination group. P values were calculated in one-way ANOVA test with corrections for multiple comparisons. D, Kinetics of LLC tumor growth (n = 5 per group). P value was calculated using two-way ANOVA test. E, Kinetics of LLC tumor growth in the mice depleted of CD8 T cells and treated with MTL-CEBPA (n = 5 per group). Mean and standard deviation are shown. P values were calculated using two-way ANOVA. F, Kinetics of tumor growth in NOD-SCID mice (n = 4 and 5 per group).
To expand on these observation to models other than HCC, we used a Lewis lung carcinoma (LLC) model. Treatment with MTL-CEBPA resulted in modest but significant delay in tumor progression when compared with mice treated with a control oligonucleotide (NOV-FLUC; Fig. 5D). The antitumor activity of MTL-CEBPA was mediated by CD8+ T cells, since the depletion of these cells with anti-CD8 antibodies abrogated the antitumor response (Fig. 5E). MTL-CEBPA treatment failed to control tumor growth in immune deficient SCID-NOD mice (Fig. 5F). Furthermore, in vitro transfection of tumor cells with CEBPA-saRNA (CEBPA-51) did not affect their survival and proliferation (Supplementary Fig. S2). Taken together, these results indicated that the antitumor effect of MTL-CEBPA was mediated by the immune system rather than by a direct effect on tumor cells.
We next evaluated the blood distribution profile of MTL-CEBPA after intravenous injection of Cy3-labeled compound in LLC-C57BL/6 mice. Time-course experiments indicated that maximum uptake was observed 4 hours after intravenous administration. Cy3-labeled MTL-CEBPA was readily detectable in myeloid cells (Supplementary Fig. S3A), but was also picked up by a substantial (>10%) proportion of macrophages and dendritic cells (DC) in spleen, tumor, lung, and liver tissues. About 10% of monocytes/M-MDSCs and only very few PMN/PMN-MDSCs were positive for Cy3-labeled MTL-CEBPA (Supplementary Fig. S3A and S3B). The liver was the exception where monocytes/M-MDSCs, PMN/PMN-MDSCs, DCs, and macrophages all showed an equal distribution of about 10% of positively stained cells across all the subpopulation when measured by FACS analysis (Supplementary Fig. S3B). Expression of Cebpa was measured by qPCR in M-MDSCs, PMN-MDSCs, and TAMs dissociated from the tumor nodules. Although the expression levels were low, we observed increased Cebpa levels across all the cell populations in the MTL-CEBPA–treated group. Only M-MDSCs showed a significant increase consistent with the data on MTL-CEBPA uptake. Tumor M-MDSCs and to a lesser extent TAMs also demonstrated upregulation of Cebpa expression. In PMN-MDSCs, these changes were less prominent (Supplementary Fig. S3C).
Although MTL-CEBPA treatment of the LLC tumor–bearing mice did not appear to affect the presence of the major population of myeloid cells within the spleen and tumors (Supplementary Fig. S4), MTL-CEBPA treatment caused a significant reversal in the suppressive activity of M-MDSCs and TAMs. Consistent with the absence of Cy3-MTL-CEBPA internalization within PMN-MDSCs, MTL-CEBPA treatment did not affect the T-cell suppressive activity of PMN-MDSCs (Fig. 6A). Similarly, in spleens the T-cell suppressive activity of M-MDSCs was abrogated following MTL-CEBPA treatment, whereas no effect on PMN-MDSC activity was observed (Fig. 6B).
Figure 6.

Effect of MTL-CEBPA treatment on immune-suppressive function of myeloid cells. A, Suppression of T-cell proliferation by M-MDSCs, macrophages, and PMN-MDSCs isolated from the tumors of the LLC tumor–bearing mice treated with NOV-FLUC or MTL-CEBPA for 2 weeks (n = 4). Mean and standard deviation are shown. P values were calculated using two-sided Student t test. B, Suppression of T-cell proliferation by M-MDSCs and PMN-MDSCs isolated from the spleens of the LLC tumor–bearing mice treated with NOV-FLUC or MTL-CEBPA for 2 weeks (n = 4). Mean and standard deviation are shown. P values were calculated using a two-sided Student t test. C and D, TAMs and PMN-MDSCs were isolated from the tumors of LLC tumor–bearing mice treated with NOV-FLUC or MTL-CEBPA for 2 weeks and used for RNA-seq analysis. C, Pathways predicted to be inhibited (z-score < −2) in TAMs in MTL-CEBPA as compared with NOV-FLUC–treated groups. D, Regulators predicted to be activated (z-score > 2) or inhibited (z-score < −2) in TAMs in MTL-CEBPA as compared with NOV-FLUC–treated group.
To confirm that MTL-CEBPA exploits an immune modulating axis via M-MDSCs and TAMs for its tumor suppressive activity, we next investigated the effects of MTL-CEBPA in a colon adenocarcinoma (MC38) tumor-bearing mouse model. We showed a modest but significant reduction in tumor progression (Supplementary Fig. S5A) and a significant reversal in T-cell suppression by M-MDSCs and TAMS upon MTL-CEBPA treatment (Supplementary Fig. S5C).
Similar to our observations with the LLC cell lines, an in vitro cell proliferation assay with MC38, when transfected with CEBPA-saRNA (CEBPA-51), failed to show a direct antitumor effect (Supplementary Fig. S5B).
MTL-CEBPA treatment controls regulators of macrophages by increasing expression of cebpa
To better understand mechanistically how MTL-CEBPA exerts its immune-modulating properties, TAMs (where suppressive activity was abrogated) and PMN-MDSCs (where suppressive activity was not changed) were sorted by flow cytometry from tumors of vehicle control (NOV-FLUC) and MTL-CEBPA treated LLC tumor–bearing mice. The transcriptome of these cells was evaluated using RNA-seq. Three hundred genes were differentially expressed (P < 0.05) in TAMs (150 genes were upregulated and 153 downregulated), whereas only 100 genes showed changes in PMN-MDSCs. No overlapping gene changes between TAMs and PMN-MDSCs were observed (Supplementary Fig. S6). The top-scoring changes in gene expression showed MTL-CEBPA caused downregulation of pathways associated with integrin signaling and extravasation in TAMs (Fig. 6C). A marked decrease in the expression of genes regulated by NF-κB, type I interferon, IL1β, and STAT4 was observed (Fig. 6D). No statistically significant changes in pathways were observed in the PMN-MDSC population.
Next, we focused on the analysis of genes known to be directly involved in the suppressive activity of myeloid cells. MTL-CEBPA caused substantial decrease in the expression of Arg1 and Nos2 in TAMs and M-MDSCs, whereas expression of genes involved in PGE2 production (Ptges and Ptgs2) were increased (Supplementary Fig. S7A and S7B). No changes in Arg1 expression were observed in PMN-MDSCs. However, expression of Ptges and Ptgs2 was increased (Supplementary Fig. S7C) suggesting that MTL-CEBPA showed evidence of affecting gene expression in PMN-MDSCs albeit at a lesser extent when compared with M-MDSCs and TAMs. Importantly, upregulation of C/EBPα caused increase in the expression of genes responsible for synthesis of PGE2, a potent immune-suppressive mediator (30).
Therapeutic effect of MTL-CEBPA
Our data demonstrated that MTL-CEBPA caused significant regulatory changes in factors affecting myeloid function where increase in Cebpa expression reduced the immune-suppressive activity of M-MDSCs and TAMs. However, we also identified upregulation of Ptges, which is directly involved in PGE2 synthesis known for its immune-suppressive activity. We hypothesized that upregulation of Ptges could potentially limit the effect of MTL-CEBPA. Our data also demonstrated that MTL-CEBPA treatment did not affect the function of PMN-MDSCs. We set out to evaluate the effects of combining MTL-CEBPA with checkpoint inhibitors (anti–PD-1 or anti–CTLA-4), with an inhibitor of PGE2 (celocoxib) and with an inhibitor of PMN-MDSCs (lipofermata). We first used the MC38 colon carcinoma model as it is known to respond to PD-1 antibody treatment (31). As expected, MTL-CEBPA monotherapy had little antitumor effects, whereas treatment with PD-1 antibody alone had modest but significant antitumor activity (Fig. 7A). A combination of MTL-CEBPA with PD-1 antibody, however, showed marked abrogation of tumor progression (Fig. 7A). Next, we tested LLC tumor–bearing mice that poorly responded to the checkpoint inhibitor anti–CTLA-4. A weak antitumor effect of either MTL-CEBPA or anti–CTLA-4 alone was only slightly enhanced by a combination of these compounds (Fig. 7B). Because MTL-CEBPA caused upregulation of genes involved in PGE2 synthesis, we used celecoxib, an inhibitor of PGE2 synthesis. A combination of MTL-CEBPA and celecoxib failed to show significant antitumor effects. However, when MTL-CEBPA and celecoxib were combined with anti–CTLA-4, we observed a complete suppression of tumor progression (Fig. 7C). Recently, an inhibitor of fatty acid transport protein 2 (FATP2), lipofermata, was shown to selectively inhibit the immune-suppressive activity of PMN-MDSCs but not M-MDSCs or TAMs (32). A combination of MTL-CEBPA with lipofermata caused marked inhibition of tumor progression in LLC tumor–bearing mice (Fig. 7D). Thus, our data support the beneficial effect of simultaneously targeting both major groups of myeloid cells (M-MDSCs and PMN-MDSCs), or targeting the escape mechanism of myeloid cells through immune checkpoint inhibition, after upregulation of C/EBPα with MTL-CEBPA.
Figure 7.

Therapeutic activity of MTL-CEBPA in combination with checkpoint inhibitors. A, MC38 tumor–bearing mice were treated with MTL-CEBPA or NOV-FLUC control at 5 mg/kg from day 4 (twice a week). Anti–PD-1 antibody was intraperitoneally injected to the mice twice a week at 10 mg/kg. n = 5 per group. Mean and SEM are shown. P values were calculated using two-way ANOVA test. B, LLC tumor–bearing mice were treated with MTL-CEBPA or NOV-FLUC control at 3 mg/kg from day 3 (twice a week). Anti–CTLA-4 antibody was intraperitoneally injected to the mice on days 10, 17, and 24 (100 μg/mouse). Celecoxib was orally given to the mice at 50 mg/kg from day 3 (daily). Mean and SEM (n = 4) are shown. P values were calculated using two-way ANOVA test. C, LLC tumor–bearing mice were treated with MTL-CEBPA or NOV-FLUC (3 mg/kg from day 3, twice a week) in combination with lipofermata (2 mg/kg, twice per day from day 3, subcutaneously). In each experiment, P values were calculated in two-way ANOVA.
Discussion
This study describes our unexpected finding that C/EPBα upregulation in myeloid cells elicits a potent inhibition in the suppressive activity of M-MDSCs and TAMs. The C/EBP family of transcription factors are generally characterized as regulators of several cellular processes including cell differentiation, proliferation, and tumorigenesis. Although C/EBPβ is known for its ability to enhance the suppressive function of MDSCs (6) and protumoral polarization of M2 macrophages (33), the role of C/EBPα in regulating immune-suppressive myeloid cells is less well characterized. Our study revealed that the liposomal formulation MTL-CEBPA was largely taken up by M-MDSCs and TAMs, subsequent to which its CEBPA-saRNA payload (CEBPA-51) enhanced expression of C/EBPα. Here, we observed C/EBPα induced downregulation of major genes implicated in the suppressive activity of M-MDSCs and TAMs. However, given the diverse regulatory role of C/EBPα in different cell types and our observation of small changes occurring in PMN-MDSCs, we did not dismiss the notion that this subpopulation of MDSCs escaped the effects of MTL-CEBPA. Within the hematopoietic system, C/EBPα is predominantly expressed in myeloblast progenitors and granulocytes. Monocytes however have lower endogenous levels of C/EBPα. Reports have shown that ectopic expression of C/EBPα in bipotential myeloid cells induces granulopoiesis while blocking monocyte differentiation (34). In contrast, loss of C/EBPα results in an absence of granulocytes (35). Therefore, it is possible that upregulation of C/EBPα preferentially affects cells where its endogenous levels are very low (i.e., in mononuclear cells). This selective effect of MTL-CEBPA was further supported when we combined MTL-CEBPA with the PMN-MDSC inhibitor lipofermata (32). Of significance, we demonstrated that targeting both arms of myeloid cells (mononuclear and polymorphonuclear) resulted in tumor suppression even without combination with standard immunotherapy. We observed that MTL-CEBPA treatment caused a decrease in factors involved in immune suppression (Arg1, Nos2) while concomitantly increasing factors involved in PGE2 synthesis (Ptges and Ptgs2). Because PGE2 has been shown to be a potent immune-suppressive factor produced by myeloid cells (36), we postulated that if we abrogated the synthesis of PGE2 synthesis, we would observe a more pronounced effect of MTL-CEBPA on its suppressive activity of myeloid cells. The addition of celecoxib, a COX2 inhibitor in combination with MTL-CEBPA, confirmed this by significantly reducing tumor progression. This supports the biological role of PGE2 in the dynamics of macrophage immunometabolism, and also suggests a potential therapeutic opportunity.
In previous studies, we demonstrated that treatment of HCC tumor–bearing mice with sorafenib resulted in decreased presence of regulatory T cells and MDSCs, and a substantial regression of tumor growth (37). However, it was not clear if the effect of therapy was the result of changes in tumor burden. It is reported that an accumulation of TAMs and neutrophils (possibly PMN-MDSCs) is responsible for the progression of resistance to sorafenib (38). Although sorafenib is still maintained as standard-of-care treatment for nonresectable HCC, there is an urgent need to find alternative therapy. Recent examples of this include the immune-checkpoint inhibitor pembrolizumab in combination with bevacizumab, a monoclonal antibody targeting VEGF, that increased OS at one year from 54.6% to 67.2% compared with sorafenib in a phase III clinical trial (14). This provides both a rationale for combining MTL-CEBPA with sorafenib and an explanation for the clinical response we observed in TKI-naïve patients. Because accumulation of MDSCs in patients with HCC is associated with negative clinical outcomes (39), then, the specific targeting of these cells would be a valuable therapeutic strategy.
The disparity in observing better treatment response in patients with viral hepatitis–associated HCC when compared with much weaker response in patients with NASH-associated HCC shows anecdotal, but interesting correlation of immune responsiveness in patients to treatment outcome. As a way of validating this observation, we showed that by depleting mice of T cells, we lost the antitumor effects of MTL-CEBPA despite its continued activity in myeloid cells. We therefore hypothesized that the better treatment responses seen in virally associated HCC are likely due to a more potent immune response when compared with NASH-associated HCC. This hypothesis is further supported by a recent study that demonstrated that the immune response of T cells to tumor-associated antigen in NASH-associated HCC patients was substantially weaker than the responses in patients with the virally associated HCC (40). This hypothesis requires further confirmation. We hypothesize that patients with any solid tumor will be suitable for combination treatment with MTL-CEBPA. MTL-CEBPA is currently being used in a large international multicenter study in combination with pembrolizumab (a PD-1 inhibitor) in adult patients with advanced solid tumors (NCT04105335).
Authors' Disclosures
D. Sarker reports non-financial support from MiNA Therapeutics during the conduct of the study. D. Sarker also reports personal fees and non-financial support from Ipsen and Eisai; personal fees from Bayer, AstraZeneca, Surface Oncology, MSD, Sirtex, and AAA; grants from UCB; and non-financial support from Medivir outside the submitted work. V. Reebye reports grants from MiNA Therapeutics Ltd during the conduct of the study, as well as other support from MiNA Therapeutics Ltd outside the submitted work. M.H. Sodergren reports grants from MiNA Therapeutics during the conduct of the study, as well as personal fees from Verb Robotics and EMMAC Life Sciences outside the submitted work. E. Sanseviero is currently a full-time employee of AstraZeneca. N. Raulf reports other support from MiNA Therapeutics Ltd during the conduct of the study, as well as other support from MiNA Therapeutics Ltd outside the submitted work. T. Meyer reports personal fees from Eisai, Ipsen, Roche, AstraZeneca, and Bayer outside the submitted work. R. Plummer reports other support from MiNA Therapeutics during the conduct of the study; R. Plummer also reports personal fees from Pierre Faber, Bayer, Novartis, Biosceptre, BMS, Cybrexa, Ellipses, CV6 Therapeutics, Astex Pharmaceuticals, Medivir, GammaDelta Therapeutics, Sanofi Aventis, and AstraZeneca, as well as other support from MSD outside the submitted work. D.J. Pinato reports personal fees from MiNA Therapeutics during the conduct of the study, as well as grants and personal fees from BMS, MSD, Eisai, Roche, Ipsen, AstraZeneca, DaVolterra, and Bayer outside the submitted work. R. Sharma reports grants from AAA Pharmaceuticals, Bayer, Astex Pharmaceuticals, Incyte Pharmaceuticals, and Boston Scientific, as well as other support from Esai, Sirtex, and Shingoi outside the submitted work. D. Palmer reports grants and personal fees from BMS, Sirtex, and Bayer, as well as personal fees from Eisai outside the submitted work. Y.-T. Ma reports personal fees from Ipsen, Roche, AstraZeneca, Eisai, Bayer, and Faron outside the submitted work. J. Evans reports other support from MiNA Therapeutics during the conduct of the study. J. Evans also reports other support from AstraZeneca, Bayer, Adaptimmune, Basilea, Starpharma, CytomX, Bicycle Therapeutics, Codiak, Boehringer, Verastem, Sierra, and Medivir; personal fees and other support from Eisai, MSD, Bristol-Myers Squibb, and Roche/Genentech; and personal fees from Pierre Fabre outside the submitted work. R. Habib reports personal fees from MiNA (Holdings) Limited during the conduct of the study. A. Martirosyan is a HalioDx employee and HalioDx is a service provider for MiNA Therapeutics. N. Elasri is a HalioDx employee and HalioDx is a service provider for MiNA Therapeutics. A. Reynaud is a HalioDx employee and HalioDx is a service provider for MiNA Therapeutics. M. Cobbold is an employee of AstraZeneca. N.A. Habib reports grants, personal fees, and other support from MiNA Therapeutics Ltd during the conduct of the study. D.I. Gabrilovich reports grants from NIH and MiNA Therapeutics during the conduct of the study. No disclosures were reported by the other authors.
Supplementary Material
Supplementary Figure 1, Supplementary Figure 2, Supplementary Figure 3, Supplementary Figure 4, Supplementary Figure 5, Supplementary Figure 6, Supplementary Figure 7
Demographic and baseline characteristics
Most frequently reported drug related adverse event
Phenotype of cells used for the analysis of tumour tissues
Acknowledgments
This work was supported by Experimental Cancer Medicine Centres, NIHR Biomedical Research Centres, and NHS Trust Tissue Banks at Imperial College London, Newcastle University, Kings College London, University of Cambridge, University of Liverpool, University of Birmingham, University of Glasgow, and University College London. We thank Drs. DiRusso and Black (University of Nebraska) for providing lipofermata for our research of MDSC. This work was partially supported by Wistar Cancer Center Support NIH grant P50 CA168536. Funding for preclinical work was provided by MiNA Therapeutics.
The costs of publication of this article were defrayed in part by the payment of page charges. This article must therefore be hereby marked advertisement in accordance with 18 U.S.C. Section 1734 solely to indicate this fact.
Footnotes
Note: Supplementary data for this article are available at Clinical Cancer Research Online (http://clincancerres.aacrjournals.org/).
Authors' Contributions
A. Hashimoto: Data curation, formal analysis, validation, investigation, visualization, methodology, writing–original draft, writing–review and editing. D. Sarker: Clinical. V. Reebye: Data curation, formal analysis, validation, investigation, visualization, methodology, writing–original draft, writing–review and editing. S. Jarvis: Data curation, software, formal analysis, visualization. M.H. Sodergren: Formal analysis, writing–review and editing, clinical. A. Kossenkov: Writing–review and editing. E. Sanseviero: Investigation. N. Raulf: Data curation, formal analysis, visualization. J. Vasara: Data curation. P. Andrikakou: Data curation. T. Meyer: Clinical. K.-W. Huang: Data curation, methodology, clinical. R. Plummer: Clinical. C.E. Chee: Clinical. D. Spalding: Clinical. M. Pai: Clinical. S. Khan: Clinical. D.J. Pinato: Clinical. R. Sharma: Clinical. B. Basu: Clinical. D. Palmer: Clinical. Y.-T. Ma: Clinical. J. Evans: Clinical. R. Habib: Resources, funding acquisition. A. Martirosyan: Formal analysis, visualization. N. Elasri: Formal analysis, visualization. A. Reynaud: Formal analysis, visualization. J.J. Rossi: Resources. M. Cobbold: Writing–review and editing. N.A. Habib: Conceptualization, data curation, formal analysis, supervision, investigation, methodology, writing–original draft, project administration, writing–review and editing. D.I. Gabrilovich: Conceptualization, data curation, formal analysis, supervision, validation, investigation, visualization, methodology, writing–original draft, writing–review and editing.
References
- 1. Binnewies M, Roberts EW, Kersten K, Chan V, Fearon DF, Merad M, et al. Understanding the tumor immune microenvironment (TIME) for effective therapy. Nat Med 2018;24:541–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Veglia F, Perego M, Gabrilovich D. Myeloid-derived suppressor cells coming of age. Nat Immunol 2018;19:108–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Newman JR, Keating AE. Comprehensive identification of human bZIP interactions with coiled-coil arrays. Science 2003;300:2097–101. [DOI] [PubMed] [Google Scholar]
- 4. Avellino R, Delwel R. Expression and regulation of C/EBPalpha in normal myelopoiesis and in malignant transformation. Blood 2017;129:2083–91. [DOI] [PubMed] [Google Scholar]
- 5. Lourenco AR, Coffer PJ. A tumor suppressor role for C/EBPalpha in solid tumors: more than fat and blood. Oncogene 2017;36:5221–30. [DOI] [PubMed] [Google Scholar]
- 6. Marigo I, Bosio E, Solito S, Mesa C, Fernandez A, Dolcetti L, et al. Tumor-induced tolerance and immune suppression depend on the C/EBPbeta transcription factor. Immunity 2010;32:790–802. [DOI] [PubMed] [Google Scholar]
- 7. Mackert JR, Qu P, Min Y, Johnson PF, Yang L, Lin PC. Dual negative roles of C/EBPalpha in the expansion and protumor functions of MDSCs. Sci Rep 2017;7:14048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Reebye V, Huang KW, Lin V, Jarvis S, Cutilas P, Dorman S, et al. Gene activation of CEBPA using saRNA: preclinical studies of the first in human saRNA drug candidate for liver cancer. Oncogene 2018;37:3216–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Huan H, Wen X, Chen X, Wu L, Liu W, Habib NA, et al. C/EBPalpha short-activating RNA suppresses metastasis of hepatocellular carcinoma through inhibiting EGFR/beta-catenin signaling mediated EMT. PLoS One 2016;11:e0153117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Voutila J, Reebye V, Roberts TC, Protopapa P, Andrikakou P, Blakey DC, et al. Development and mechanism of small activating RNA targeting CEBPA, a novel therapeutic in clinical trials for liver cancer. Mol Ther 2017;25:2705–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Sarker D, Plummer R, Meyer T, Sodergren MH, Basu B, Chee CE, et al. MTL-CEBPA, a small activating RNA therapeutic upregulating C/EBP-alpha, in patients with advanced liver cancer: a first-in-human, multicenter, open-label, phase I trial. Clin Cancer Res 2020;26:3936–46. [DOI] [PubMed] [Google Scholar]
- 12. Sarker D, Sodergren M, Plummer ER, Basu B, Meyer T, Huang K-W, et al. Phase Ib dose escalation and cohort expansion study of the novel myeloid differentiating agent MTL-CEBPA in combination with sorafenib in patients with advanced hepatocellular carcinoma (HCC). J Clin Oncol 2020;38:4601. [Google Scholar]
- 13. Llovet JM, Ricci S, Mazzaferro V, Hilgard P, Gane E, Blanc JF, et al. Sorafenib in advanced hepatocellular carcinoma. N Engl J Med 2008;359:378–90. [DOI] [PubMed] [Google Scholar]
- 14. Finn RS, Qin S, Ikeda M, Galle PR, Ducreux M, Kim TY, et al. Atezolizumab plus bevacizumab in unresectable hepatocellular carcinoma. N Engl J Med 2020;382:1894–905. [DOI] [PubMed] [Google Scholar]
- 15. Langmead B, Salzberg SL. Fast gapped-read alignment with Bowtie 2. Nat Methods 2012;9:357–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Li B, Dewey CN. RSEM: accurate transcript quantification from RNA-seq data with or without a reference genome. BMC Bioinformatics 2011;12:323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Love MI, Huber W, Anders S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol 2014;15:550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Team RC. R: A language and environment for statistical computing. R Foundation for Statistical Computing, Vienna, Austria; 2013. Available from: http://www.R-project.org/. [Google Scholar]
- 19. Wickham H. ggplot2: Elegant graphics for data analysis. New York: Springer-Verlag; 2016. [Google Scholar]
- 20. Benjamini Y, Hochberg Y. Controlling the false discovery rate: a practical and powerful approach to multiple testing. J R Stat Soc Series B Stat Methodol 1995;B, 57:289–300. [Google Scholar]
- 21. Rajeeve V, Vendrell I, Wilkes E, Torbett N, Cutillas PR. Cross-species proteomics reveals specific modulation of signaling in cancer and stromal cells by phosphoinositide 3-kinase (PI3K) inhibitors. Mol Cell Proteomics 2014;13:1457–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Casado P, Rodriguez-Prados JC, Cosulich SC, Guichard S, Vanhaesebroeck B, Joel S, et al. Kinase-substrate enrichment analysis provides insights into the heterogeneity of signaling pathway activation in leukemia cells. Sci Signal 2013;6:rs6. [DOI] [PubMed] [Google Scholar]
- 23. Larsen MR, Thingholm TE, Jensen ON, Roepstorff P, Jørgensen TJ. Highly selective enrichment of phosphorylated peptides from peptide mixtures using titanium dioxide microcolumns. Mol Cell Proteomics 2005;4:873–86. [DOI] [PubMed] [Google Scholar]
- 24. Montoya A, Beltran L, Casado P, Rodriguez-Prados JC, Cutillas PR. Characterization of a TiO(2) enrichment method for label-free quantitative phosphoproteomics. Methods (San Diego, Calif) 2011;54:370–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Perkins DN, Pappin DJ, Creasy DM, Cottrell JS. Probability-based protein identification by searching sequence databases using mass spectrometry data. Electrophoresis 1999;20:3551–67. [DOI] [PubMed] [Google Scholar]
- 26. Casado P, Cutillas PR. A self-validating quantitative mass spectrometry method for assessing the accuracy of high-content phosphoproteomic experiments. Mol Cell Proteomics 2011;10:M110 003079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Cutillas PR, Vanhaesebroeck B. Quantitative profile of five murine core proteomes using label-free functional proteomics. Mol Cell Proteomics 2007;6:1560–73. [DOI] [PubMed] [Google Scholar]
- 28. Law CW, Chen Y, Shi W, Smyth GK. Voom: precision weights unlock linear model analysis tools for RNA-seq read counts. Genome biol 2014;15:R29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Warren S, Danaher P, Mashadi-Hossein A, Skewis L, Wallden B, Ferree S, et al. Development of gene expression-based biomarkers on the ncounter((R)) platform for immuno-oncology applications. Methods Mol Biol 2020;2055:273–300. [DOI] [PubMed] [Google Scholar]
- 30. Zelenay S, van der Veen AG, Bottcher JP, Snelgrove KJ, Rogers N, Acton SE, et al. Cyclooxygenase-dependent tumor growth through evasion of immunity. Cell 2015;162:1257–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Efremova M, Rieder D, Klepsch V, Charoentong P, Finotello F, Hackl H, et al. Targeting immune checkpoints potentiates immunoediting and changes the dynamics of tumor evolution. Nat Commun 2018;9:32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Veglia F, Tyurin VA, Blasi M, De Leo A, Kossenkov AV, Donthireddy L, et al. Fatty acid transport protein 2 reprograms neutrophils in cancer. Nature 2019;569:73–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Ruffell D, Mourkioti F, Gambardella A, Kirstetter P, Lopez RG, Rosenthal N, et al. A CREB-C/EBPbeta cascade induces M2 macrophage-specific gene expression and promotes muscle injury repair. Proc Natl Acad Sci U S A 2009;106:17475–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Radomska HS, Huettner CS, Zhang P, Cheng T, Scadden DT, Tenen DG. CCAAT/enhancer binding protein alpha is a regulatory switch sufficient for induction of granulocytic development from bipotential myeloid progenitors. Mol Cell Biol 1998;18:4301–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Zhang DE, Zhang P, Wang ND, Hetherington CJ, Darlington GJ, Tenen DG. Absence of granulocyte colony-stimulating factor signaling and neutrophil development in CCAAT enhancer binding protein alpha-deficient mice. Proc Natl Acad Sci U S A 1997;94:569–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Prima V, Kaliberova LN, Kaliberov S, Curiel DT, Kusmartsev S. COX2/mPGES1/PGE2 pathway regulates PD-L1 expression in tumor-associated macrophages and myeloid-derived suppressor cells. Proc Natl Acad Sci U S A 2017;114:1117–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Cao M, Xu Y, Youn JI, Cabrera R, Zhang X, Gabrilovich D, et al. Kinase inhibitor sorafenib modulates immunosuppressive cell populations in a murine liver cancer model. Lab Invest 2011;91:598–608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Zhou SL, Zhou ZJ, Hu ZQ, Huang XW, Wang Z, Chen EB, et al. Tumor-associated neutrophils recruit macrophages and T-regulatory cells to promote progression of hepatocellular carcinoma and resistance to sorafenib. Gastroenterology 2016;150:1646–58e17. [DOI] [PubMed] [Google Scholar]
- 39. Sachdeva M, Arora SK. Prognostic role of immune cells in hepatocellular carcinoma. EXCLI J 2020;19:718–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Inada Y, Mizukoshi E, Seike T, Tamai T, Iida N, Kitahara M, et al. Characteristics of immune response to tumor-associated antigens and immune cell profile in patients with hepatocellular carcinoma. Hepatology 2019;69:653–65. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Figure 1, Supplementary Figure 2, Supplementary Figure 3, Supplementary Figure 4, Supplementary Figure 5, Supplementary Figure 6, Supplementary Figure 7
Demographic and baseline characteristics
Most frequently reported drug related adverse event
Phenotype of cells used for the analysis of tumour tissues