

Abstract
Background
The benefits of erythropoiesis‐stimulating agents (ESA) for dialysis patients have been demonstrated. However, it remains unclear whether the efficacy and safety of new, longer‐acting ESA given less frequently is equivalent to recombinant human erythropoietin (rHuEPO) preparations. This is an update of a review first published in 2002 and last updated in 2005.
Objectives
This review aimed to establish the optimal frequency of ESA administration in terms of effectiveness (correction of anaemia, and freedom from adverse events) and efficiency (optimal resource use) of different ESA dose regimens.
Search methods
We searched the Cochrane Renal Group's Specialised Register to 21 March 2013 through contact with the Trials' Search Co‐ordinator using search terms relevant to this review.
Selection criteria
We included randomised control trials (RCTs) comparing different frequencies of ESA administration in dialysis patients.
Data collection and analysis
Two authors independently assessed study eligibility, risk of bias and extracted data. Results were expressed as risk ratio (RR) or risk differences (RD) with 95% confidence intervals (CI) for dichotomous outcomes. For continuous outcomes the mean difference (MD) or standardised mean difference (SMD) with 95% confidence intervals (CI) was used. Statistical analyses were performed using the random‐effects model.
Main results
This review included 33 studies (5526 participants), 22 of which were added for this update. Risk of bias was generally high; only nine studies were assessed at low risk of bias for sequence generation and 14 studies for allocation concealment. Although only four studies were placebo‐controlled, all were considered to be at low risk of performance or detection bias because the primary outcome of haemoglobin level was a laboratory‐derived assessment and unlikely to be influenced by lack of blinding. We found that 16 studies were at low risk of attrition bias and five were at low risk of selection bias; only one study reporting sources of support was not funded by a pharmaceutical company.
We compared four different interventions: Continuous erythropoietin receptor agonists (CERA) versus other ESA (darbepoetin or rHuEPO); different frequencies of darbepoetin administration; darbepoetin versus rHuEPO; and different frequencies of rHuEPO administration.
There were no significant differences in maintaining final haemoglobin between CERA administered at two weekly intervals (4 studies, 1762 participants: MD 0.08 g/dL, 95% CI ‐0.04 to 0.21) or four weekly intervals (two studies, 1245 participants: MD ‐0.03 g/dL, 95% CI ‐0.17 to 0.12) compared with rHuEPO administered at two to three weekly intervals. In one study comparing CERA administered every two weeks with darbepoetin administered once/week, there was no significant difference in final haemoglobin (313 participants: MD 0.30 g/dL, 95% CI 0.05 to 0.55). In comparisons of once/week with once every two weeks darbepoetin (two studies, 356 participants: MD 0.04 g/dL, 95% CI ‐0.45 to 0.52) and once every two weeks with monthly darbepoetin (one study, 64 participants: MD 0.40 g/dL, 95% CI ‐0.37 to 1.17) there were no significant differences in final haemoglobin levels. There was marked heterogeneity among studies comparing weekly darbepoetin with once every two weeks and was possibly related to different administration protocols. Eight studies compared weekly darbepoetin with rHuEPO given two to three times/week; no statistical difference in final haemoglobin was demonstrated (6 studies, 1638 participants: MD 0.02 g/dL, 95% CI ‐0.09 to 0.12). Fourteen studies compared different frequencies of rHuEPO. No statistical difference was demonstrated in final haemoglobin (7 studies, 393 participants: SMD ‐0.17 g/dL, 95% CI ‐0.39 to 0.05). Adverse events did not differ significantly within comparisons; however, mortality and quality of life were poorly reported, particularly in earlier publications.
Authors' conclusions
Longer‐acting ESA (darbepoetin and CERA) administered at one to four week intervals are non‐inferior to rHuEPO given one to three times/week in terms of achieving haemoglobin targets without any significant differences in adverse events in haemodialysis patients. Additional RCTs are required to evaluate different frequencies of ESA in peritoneal and paediatric dialysis patients and to compare different longer‐acting ESA (such as darbepoetin compared with CERA).
Plain language summary
Frequency of administration of erythropoiesis‐stimulating agents for the anaemia of end‐stage kidney disease in dialysis patients
Anaemia (having too few red blood cells) is a major cause of tiredness and other problems often experienced by people on dialysis. Dialysis is treatment for kidney disease using an artificial kidney machine (haemodialysis) or by exchanging fluid through a tube in the abdomen (peritoneal dialysis).
Manufactured erythropoietin (a hormone that increases red blood cell production) improves anaemia and is often prescribed for people on dialysis. Several different forms of manufactured erythropoietin that can be given less often are now available.
We looked at evidence from 33 studies that involved over 5500 people that were published before March 2013 to find out if how often erythropoietin agents are given to people who are receiving dialysis can help to improve anaemia.
We found that the newer erythropoietin agents given less often (weekly to every four weeks) resulted in similar correction of anaemia compared with older agents (given two to three times per week) for people on haemodialysis. We did not find any significant differences in side effects between the newer and older agents, but not all studies reported information on side effects.
There was not enough information about use of these agents for children or people on peritoneal dialysis to determine if less frequent administration was effective for these groups of people.
This review updates information previously published in 2002 and 2005.
Summary of findings
Summary of findings for the main comparison. CERA versus other ESA for the anaemia of end‐stage kidney disease in dialysis patients.
| CERA versus other ESA for the anaemia of end‐stage kidney disease in dialysis patients | ||||||
| Patient or population: patients with the anaemia of end‐stage kidney disease undergoing dialysis Settings: tertiary centres Intervention: CERA versus other ESA | ||||||
| Outcomes | Illustrative comparative risks* (95% CI) | Relative effect (95% CI) | No of participants (studies) | Quality of the evidence (GRADE) | Comments | |
| Assumed risk | Corresponding risk | |||||
| Control | CERAversus other ESA | |||||
| Final Hb g/dL: CERA every 2 weeks versus rHuEPO | Mean final Hb g/dL: CERA every 2 weeks versus rHuEPO in the intervention groups was 0.08 higher (0.04 lower to 0.21 higher) | 1126 (4) | ⊕⊕⊕⊕ high | |||
| Final Hb g/dL: CERA every 4 week versus rHuEPO | Mean final Hb g/dL: CERA every 4 weeks versus rHuEPO in the intervention groups was 0.03 lower (0.17 lower to 0.12 higher) | 672 (2) | ⊕⊕⊕⊕ high | |||
| Final Hb g/dL: CERA every 2 week versus darbepoetin | Mean final Hb g/dL: CERA every 2 weeks versus darbepoetin in the intervention groups was 0.3 higher (0.05 to 0.55 higher) | 249 (1) | ⊕⊕⊕⊝ moderate¹ | |||
| All‐cause mortality: CERA every 2 weeks versus rHuEPO | Study population | RR 1.03 (0.67 to 1.57) | 1341 (4) | ⊕⊕⊕⊝ moderate² | ||
| 62 per 1000 | 64 per 1000 (41 to 97) | |||||
| Moderate | ||||||
| 61 per 1000 | 63 per 1000 (41 to 96) | |||||
| Transfusions: CERA every 2 weeks versus rHuEPO | Study population | RR 0.92 (0.64 to 1.32) | 1341 (4) | ⊕⊕⊕⊝ moderate² | ||
| 90 per 1000 | 83 per 1000 (58 to 119) | |||||
| Moderate | ||||||
| 88 per 1000 | 81 per 1000 (56 to 116) | |||||
| Numbers of adverse events due to hypertension: CERA every 2 weeks versus rHuEPO | Study population | RR 0.93 (0.69 to 1.26) | 1341 (4) | ⊕⊕⊕⊕ high | ||
| 151 per 1000 | 140 per 1000 (104 to 190) | |||||
| Moderate | ||||||
| 149 per 1000 | 139 per 1000 (103 to 188) | |||||
| Numbers of adverse events due to access thrombosis: CERA every 2 weeks versus rHuEPO | Study population | RR 0.96 (0.56 to 1.65) | 1341 (4) | ⊕⊕⊕⊝ moderate³ | ||
| 92 per 1000 | 88 per 1000 (52 to 152) | |||||
| Moderate | ||||||
| 85 per 1000 | 82 per 1000 (48 to 140) | |||||
| *The basis for the assumed risk (e.g. the median control group risk across studies) is provided in footnotes. The corresponding risk (and its 95% confidence interval) is based on the assumed risk in the comparison group and the relative effect of the intervention (and its 95% CI). CI: Confidence interval; RR: Risk ratio | ||||||
| GRADE Working Group grades of evidence High quality: Further research is very unlikely to change our confidence in the estimate of effect Moderate quality: Further research is likely to have an important impact on our confidence in the estimate of effect and may change the estimate Low quality: Further research is very likely to have an important impact on our confidence in the estimate of effect and is likely to change the estimate Very low quality: We are very uncertain about the estimate | ||||||
¹ One study, 249 participants ² Small numbers of events ³ Heterogeneity among studies
Summary of findings 2. Different frequencies of CERA for the anaemia of end‐stage kidney disease in dialysis patients.
| Different frequencies of CERA for the anaemia of end‐stage kidney disease in dialysis patients | ||||||
| Patient or population: patients with the anaemia of end‐stage kidney disease undergoing dialysis Settings: tertiary Intervention: different frequencies of CERA | ||||||
| Outcomes | Illustrative comparative risks* (95% CI) | Relative effect (95% CI) | No of participants (studies) | Quality of the evidence (GRADE) | Comments | |
| Assumed risk | Corresponding risk | |||||
| Control | Different frequencies of CERA | |||||
| Final Hb (g/dL) | Mean final Hb in the intervention groups was 0.11 lower (0.35 lower to 0.14 higher) | 675 (2) | ⊕⊕⊕⊝ moderate¹ | |||
| All‐cause mortality | Study population | RR 1.03 (0.6 to 1.78) | 822 (2) | ⊕⊕⊕⊕ high | ||
| 78 per 1000 | 80 per 1000 (47 to 139) | |||||
| Moderate | ||||||
| 77 per 1000 | 79 per 1000 (46 to 137) | |||||
| Transfusion | Study population | RR 1.11 (0.52 to 2.37) | 822 (2) | ⊕⊕⊕⊝ moderate¹ | ||
| 80 per 1000 | 89 per 1000 (42 to 190) | |||||
| Moderate | ||||||
| 79 per 1000 | 88 per 1000 (41 to 187) | |||||
| Numbers of adverse effects due to hypertension | Study population | RR 1.18 (0.83 to 1.67) | 822 (2) | ⊕⊕⊕⊕ high | ||
| 122 per 1000 | 144 per 1000 (101 to 203) | |||||
| Moderate | ||||||
| 123 per 1000 | 145 per 1000 (102 to 205) | |||||
| Numbers of adverse events due to access thrombosis | Study population | RR 0.96 (0.64 to 1.43) | 822 (2) | ⊕⊕⊕⊕ high | ||
| 105 per 1000 | 100 per 1000 (67 to 150) | |||||
| Moderate | ||||||
| 104 per 1000 | 100 per 1000 (67 to 149) | |||||
| *The basis for the assumed risk (e.g. the median control group risk across studies) is provided in footnotes. The corresponding risk (and its 95% confidence interval) is based on the assumed risk in the comparison group and the relative effect of the intervention (and its 95% CI). CI: Confidence interval; RR: Risk ratio | ||||||
| GRADE Working Group grades of evidence High quality: Further research is very unlikely to change our confidence in the estimate of effect Moderate quality: Further research is likely to have an important impact on our confidence in the estimate of effect and may change the estimate Low quality: Further research is very likely to have an important impact on our confidence in the estimate of effect and is likely to change the estimate Very low quality: We are very uncertain about the estimate | ||||||
¹ Heterogeneity among studies
Summary of findings 3. Darbepoetin versus rHuEPO for the anaemia of end‐stage kidney disease in dialysis patients.
| Darbepoetin versus rHuEPO for the anaemia of end‐stage kidney disease in dialysis patients | ||||||
| Patient or population: patients with the anaemia of end‐stage kidney disease in dialysis patients Settings: tertiary Intervention: darbepoetin versus rHuEPO | ||||||
| Outcomes | Illustrative comparative risks* (95% CI) | Relative effect (95% CI) | No of participants (studies) | Quality of the evidence (GRADE) | Comments | |
| Assumed risk | Corresponding risk | |||||
| Control | Darbepoetin versusrHuEPO | |||||
| Final/change in Hb (g/dL) | Mean final/change in Hb in the intervention groups was 0.02 higher (0.09 lower to 0.12 higher) | 1245 (6) | ⊕⊕⊕⊝ moderate¹ | |||
| Final/change in ESA dose | Mean final/change in ESA dose in the intervention groups was 12.27 lower (21.72 to 2.82 lower) | 757 (3) | ⊕⊕⊝⊝ low¹,² | |||
| All‐cause mortality | Study population | RR 1.29 (0.82 to 2.02) | 1596 (5) | ⊕⊕⊕⊝ moderate¹ | ||
| 55 per 1000 | 71 per 1000 (45 to 112) | |||||
| Moderate | ||||||
| 64 per 1000 | 83 per 1000 (52 to 129) | |||||
| Total treatment‐related adverse events | Study population | See comment | 570 (3) | Risks were calculated from pooled risk differences | ||
| 84 per 1000 | 91 per 1000 (54 to 134) | |||||
| Moderate | ||||||
| 17 per 1000 | 18 per 1000 (11 to 27) | |||||
| Hypertension | Study population | See comment | 1475 (4) | ⊕⊕⊝⊝ low¹,² | Risks were calculated from pooled risk differences | |
| 144 per 1000 | 137 per 1000 (84 to 194) | |||||
| Moderate | ||||||
| 70 per 1000 | 67 per 1000 (41 to 95) | |||||
| Access thrombosis/vascular complication | Study population | See comment | 1475 (4) | ⊕⊕⊕⊝ moderate¹ | Risks were calculated from pooled risk differences | |
| 95 per 1000 | 81 per 1000 (65 to 95) | |||||
| Moderate | ||||||
| 29 per 1000 | 25 per 1000 (20 to 29) | |||||
| Transfusion | Study population | See comment | 1069 (3) | ⊕⊕⊕⊝ moderate¹ | Risks were calculated from pooled risk differences | |
| 53 per 1000 | 29 per 1000 (3 to 53) | |||||
| Moderate | ||||||
| 50 per 1000 | 28 per 1000 (3 to 50) | |||||
| *The basis for the assumed risk (e.g. the median control group risk across studies) is provided in footnotes. The corresponding risk (and its 95% confidence interval) is based on the assumed risk in the comparison group and the relative effect of the intervention (and its 95% CI). CI: Confidence interval; RR: Risk ratio | ||||||
| GRADE Working Group grades of evidence High quality: Further research is very unlikely to change our confidence in the estimate of effect Moderate quality: Further research is likely to have an important impact on our confidence in the estimate of effect and may change the estimate Low quality: Further research is very likely to have an important impact on our confidence in the estimate of effect and is likely to change the estimate Very low quality: We are very uncertain about the estimate | ||||||
¹ High risk of bias for several domains in each study ² Significant heterogeneity between studies
Summary of findings 4. rHuEPO once/week versus rHuEPO 2 to 3 times/week for the anaemia of end‐stage kidney disease in dialysis patients.
| rHuEPO once/week versus with rHuEPO 2 to 3 times/week for the anaemia of end‐stage kidney disease in dialysis patients | ||||||
| Patient or population: patients with the anaemia of end‐stage kidney disease undergoing dialysis Settings: tertiary Intervention: rHuEPO once/week Comparison: rHuEPO 2 to 3 times/week | ||||||
| Outcomes | Illustrative comparative risks* (95% CI) | Relative effect (95% CI) | No of participants (studies) | Quality of the evidence (GRADE) | Comments | |
| Assumed risk | Corresponding risk | |||||
| RHuEPO 2 to 3 times/week | RHuEPO once/week | |||||
| Final/change in Hb | Mean final/change in Hb in the intervention groups was 0.17 SD lower (0.39 lower to 0.05 higher) | 363 (7) | ⊕⊕⊝⊝ low¹,² | SMD ‐0.17 (‐0.39 to 0.05) | ||
| Final/change in EPO dose | Mean final/change in EPO dose in the intervention groups was 8.47 higher (1.01 lower to 17.95 higher) | 217 (5) | ⊕⊕⊝⊝ low¹,² | |||
| Adverse effects: transfusions | Study population | See comment | 173 (1) | See comment | Risks were calculated from pooled risk differences | |
| 90 per 1000 | 71 per 1000 (‐10 to 150) | |||||
| Moderate | ||||||
| 90 per 1000 | 71 per 1000 (‐10 to 150) | |||||
| Adverse effects: hypertension | Study population | See comment | 175 (4) | ⊕⊕⊝⊝ low¹,² | Risks were calculated from pooled risk differences | |
| 265 per 1000 | 260 per 1000 (146 to 374) | |||||
| Moderate | ||||||
| 240 per 1000 | 235 per 1000 (132 to 338) | |||||
| Adverse effects: access problems | Study population | See comment | 173 (1) | See comment | Risks were calculated from pooled risk differences | |
| 34 per 1000 | 24 per 1000 (‐26 to 74) | |||||
| Moderate | ||||||
| 34 per 1000 | 24 per 1000 (‐27 to 74) | |||||
| *The basis for the assumed risk (e.g. the median control group risk across studies) is provided in footnotes. The corresponding risk (and its 95% confidence interval) is based on the assumed risk in the comparison group and the relative effect of the intervention (and its 95% CI). CI: Confidence interval; RR: Risk ratio | ||||||
| GRADE Working Group grades of evidence High quality: Further research is very unlikely to change our confidence in the estimate of effect Moderate quality: Further research is likely to have an important impact on our confidence in the estimate of effect and may change the estimate Low quality: Further research is very likely to have an important impact on our confidence in the estimate of effect and is likely to change the estimate Very low quality: We are very uncertain about the estimate | ||||||
¹ Significant risk of bias in several domains in all studies ² Small patient numbers
Background
Description of the condition
Anaemia is a condition very often encountered among people with chronic kidney disease (CKD). There is a direct relationship between anaemia severity and decline in kidney function (Koch 1991). Anaemia results in significant morbidity causing symptoms including lack of energy, breathlessness, dizziness, angina, poor appetite and decreased exercise tolerance (Canadian EPO 1990a; Lundin 1989). Decreased production of erythropoietin, a naturally occurring hormone mainly produced by the kidney, is the major cause of anaemia among people with CKD (Jensen 1994). Improvement in energy levels (Wolcott 1989), increased cardiac performance and ejection fraction (Pappas 2008), and normalisation of increased cardiac output and left ventricular mass (Cannella 1990) occurred with increasing haemoglobin levels. Before recombinant human erythropoietin (rHuEPO) became available, anaemia was treated by blood transfusion together with iron and folate supplements. Blood transfusions were used sparingly because they have associated risks of infection transmission and inducing cytotoxic antibodies that could jeopardise future kidney transplantation (Ward 1990). Cloning of the human gene for erythropoietin was achieved in 1983 (Lin 1985), and production of rHuEPO followed. The efficacy of rHuEPO treatment in dialysis patients was demonstrated by 1986 (Winearls 1986). National (CARI 2011) and international guidelines (KDIGO 2012) currently recommend target haemoglobin levels of 110 mg/L to 120 mg/L in people with CKD.
Description of the intervention
Administration of erythropoiesis‐stimulating agents (ESA) aims to replace endogenous erythropoietin production, which is reduced in CKD, and to raise haemoglobin levels to alleviate signs and symptoms of anaemia.
ESA products, such as epoetin‐α and epoetin‐β, have proven efficacy in treating anaemia in people with CKD (Eschbach 1987). However, because of the relatively short half‐life of these agents (six to eight hours when administered intravenously, and 19 to 24 hours when administered subcutaneously), they require administration at two to three weekly intervals in most people, although some stable haemodialysis patients with low dose requirements may require weekly administration only (Locatelli 2011). Such frequent administration may be inconvenient for both patients and healthcare workers. Following red cell aplasia, a complication predominantly associated with subcutaneous administration of epoetin‐α, intravenous administration was recommended (Ortho Biotech Jansen Cilag 2001).
There has been a general trend towards less frequent dosing regimens using new long‐acting ESA preparations that offer greater patient comfort and convenience. Darbepoetin was the first ESA with a prolonged half‐life to enter the market. It has five N‐linked carbohydrate chains, whereas rHuEPO has only three. Because of its increased sialic acid‐containing carbohydrate content, darbepoetin has a threefold longer terminal half‐life (25 hours intravenous and 48 hours subcutaneous) compared with rHuEPO. This enables once a week or once every two weeks administration in people with CKD (Macdougall 1999). More recently, the continuous erythropoietin receptor activator (CERA), methoxy polyethylene glycol‐epoetin‐β has been developed to ensure stable maintenance and correction of haemoglobin levels at less frequent administration in people with CKD. CERA differs from rHuEPO through the formation of a chemical bond between either the N‐terminal amino group or the ɛ‐amino group of any lysine present in erythropoietin and methoxy polyethylene glycol‐butanoic acid (Macdougall 2005). Despite lower receptor affinity, CERA induces a more prolonged response than epoetin‐α or epoetin‐β.
How the intervention might work
The primary cause of anaemia in CKD is the relative insufficiency of erythropoietin production associated with CKD. ESA accelerate erythropoiesis, increase iron utilisation and raise haemoglobin levels with clinical improvement in signs and symptoms of anaemia. ESA requirements are difficult to predict in individual patients, and may be increased in people with associated comorbidities including cardiovascular disease, diabetes and chronic inflammation. ESA requirements are generally lower in patients not receiving dialysis. ESA therapy aims to increase haemoglobin levels slowly at a rate of < 1 g/dL to 2 g/dL per month during the correction phase. This is done to avoid major side effects including hypertension, vascular access thrombosis and cardiovascular events, and then maintain stable haemoglobin.
A major issue in ESA use relates to the haemoglobin target to be achieved. Recent systematic reviews have suggested that aiming for haemoglobin levels similar to those seen in healthy adults is associated with a significantly higher risk of all‐cause mortality (Palmer 2010).
Why it is important to do this review
ESA are effective in correcting anaemia associated with kidney disease and increasing haemoglobin levels but choice of agent should include the drug's pharmacodynamics, pharmacokinetics, route and frequency of administration, adverse effects, availability and economic issues. The 2002 review (Cody 2002) and subsequent 2005 update (Cody 2005a) assessed different frequencies of rHuEPO. This update (2013) evaluated the benefits and adverse effects of the newer ESA, which are administered at less frequent intervals than rHuEPO.
This review assessed different frequencies of ESA in patients on dialysis. A review by (Cody 2005b), which assessed rHuEPO frequencies in non‐dialysis patients, is being updated to include longer‐acting ESA.
Objectives
This review aimed to establish optimal frequency of ESA administration in terms of:
effectiveness (correction of anaemia, and freedom from adverse events); and
efficiency (optimal resource use) of different ESA dose regimens.
Methods
Criteria for considering studies for this review
Types of studies
All randomised controlled trials (RCTs) or quasi‐RCTs (RCTs in which allocation to treatment was obtained by alternation, use of alternate medical records, date of birth or other predictable methods) comparing different frequencies of ESA administration in patients dialysed for end‐stage kidney disease (ESKD) were eligible for inclusion.
Types of participants
Haemodialysis or peritoneal dialysis patients (adults and children) with the anaemia of ESKD.
Types of interventions
Different frequencies and administration of ESA: rHuEPO, darbepoetin and continuous erythropoietin receptor agonists (CERA)
CERA versus other ESA (darbepoetin or rHuEPO)
Different frequencies of darbepoetin administration
Darbepoetin versus rHuEPO
Different frequencies of rHuEPO administration.
Types of outcome measures
Measures of correction of anaemia: values of haemoglobin/haematocrit or change in haemoglobin/haematocrit at the end of the study
Change in ESA requirements
All‐cause mortality; cardiovascular mortality
Measures of hypertension: systolic and diastolic blood pressure, numbers with hypertension
Other adverse events: numbers of blood transfusions; numbers discontinued due to adverse events (e.g. access problems)
Quality of life.
Search methods for identification of studies
Electronic searches
We searched the Cochrane Renal Group's Specialised Register to 21 March 2013 through contact with the Trials' Search Co‐ordinator using search terms relevant to this review. The Cochrane Renal Group’s Specialised Register contains studies identified from the following sources.
Quarterly searches of the Cochrane Central Register of Controlled Trials (CENTRAL)
Weekly searches of MEDLINE OVID SP
Handsearching of renal‐related journals and the proceedings of major renal conferences
Searching of the current year of EMBASE OVID SP
Weekly current awareness alerts for selected renal journals
Searches of the International Clinical Trials Register (ICTRP) Search Portal and ClinicalTrials.gov.
Studies contained in the Specialised Register are identified through search strategies for CENTRAL, MEDLINE, and EMBASE based on the scope of the Cochrane Renal Group. Details of these strategies, as well as a list of handsearched journals, conference proceedings and current awareness alerts, are available in the Specialised Register section of information about the Cochrane Renal Group.
See Appendix 1 for search terms used in strategies for this review.
Searching other resources
Reference lists of clinical practice guidelines, review articles and relevant studies.
Letters seeking information about unpublished or incomplete studies to investigators known to be involved in previous studies.
For search strategies used in the previous reviews please refer to Cody 2002 and Cody 2005a.
Data collection and analysis
Selection of studies
In the previous versions of this review (2002 and 2005) all electronically‐derived abstracts and study titles were assessed by a single author for subject relevance and methodological quality. All possible RCTs or quasi‐RCTs which were relevant were assigned specific topic keywords in Reference Manager, and the full published paper was obtained for full assessment.
In the current review (2014), study titles and abstracts were reviewed by two authors (DH, EH). Full text articles of studies considered relevant were obtained and reviewed for eligibility by both authors.
Data extraction and management
A data abstraction form was devised to record details of data elements such as outcome measures, participants and intervention from each included study for the 2002 and 2005 reviews. Only comparisons and outcomes which were prespecified in the protocol were included. For these reviews, data were abstracted by a single assessor and a sample was double checked.
For the current review, data extraction and assessment of risk of bias was performed by two authors (DH, EH) using standardised data extraction forms. Disagreements not resolved by discussion between authors could be referred to a third person. Studies reported in languages other than English were to be translated before data extraction, but no foreign language reports were identified. Where more than one report of a study was identified, data were extracted from all reports. Where there were discrepancies between reports, data from the primary source were used. Study authors were contacted for additional information about studies; however, no additional information was obtained.
Assessment of risk of bias in included studies
Hard copies of studies were independently assessed for methodological quality by two assessors for the 2002 and 2005 reviews. Quality assessments were made for allocation concealment, blinding, description of withdrawals and drop‐outs, numbers lost to follow‐up, and whether intention‐to‐treat (ITT) analysis was possible.
In this review, the following items were assessed using the risk of bias assessment tool (Higgins 2011) (see Appendix 2).
Was there adequate sequence generation (selection bias)?
Was allocation adequately concealed (selection bias)?
-
Was knowledge of the allocated interventions adequately prevented during the study (detection bias)?
Participants and personnel
Outcome assessors
Were incomplete outcome data adequately addressed (attrition bias)?
Are reports of the study free of suggestion of selective outcome reporting (reporting bias)?
Was the study apparently free of other problems that could put it at a risk of bias?
Measures of treatment effect
For dichotomous outcomes (all‐cause mortality, adverse effects), relative risk (RR) with 95% confidence intervals (CI) were calculated. For continuous outcomes (final haemoglobin or change in haemoglobin), mean difference (MD) with 95% CI were calculated. Either final haemoglobin or changes in haemoglobin were included in meta‐analyses. When both measures were provided, final haemoglobin was included in meta‐analyses. Standard mean difference (SMD) with 95% CI was used to combine different units of measurement which measured the same underlying concept (e.g. haematocrit and haemoglobin).
Unit of analysis issues
Data from cross‐over studies were to be included in meta‐analyses if separate data for the first study phase were available. However, no such data were available.
Dealing with missing data
We aimed to analyse available data in meta‐analyses using ITT data. However, where ITT data were only available graphically or not provided and additional information could not be obtained from the study authors, per‐protocol (PP) data were used in analyses.
Assessment of heterogeneity
Heterogeneity was analysed using Chi² on N‐1 degrees of freedom, with an alpha of 0.05 used for statistical significance and with the I² test (Higgins 2003). I² of 25%, 50% and 75% correspond to low, medium and high levels of heterogeneity respectively.
Assessment of reporting biases
The search strategy applied aimed to reduce publication bias caused by lack of publication of studies with negative results. We had planned to investigate for publication bias using funnel plots but there were too few studies on each comparison. Where there were multiple publications of the same study, all reports were reviewed to ensure that all details of methods and results were included.
Data synthesis
Data were combined using a random‐effects model for dichotomous and continuous data.
Subgroup analysis and investigation of heterogeneity
Included studies were divided into four groups based on the ESA comparisons. Subgroup analysis was planned based on dialysis modality (haemodialysis versus peritoneal dialysis), patient age (paediatric versus adult) and route of ESA administration (intravenous versus subcutaneous). However, there were few data on peritoneal dialysis patients, no data on paediatric patients and insufficient studies to evaluate route of administration.
Sensitivity analysis
Sensitivity analyses tested decisions where inclusion of a study may have altered the results of the meta‐analysis. In particular, sensitivity analysis was used to test decisions where ITT and PP data were included in the same analyses.
Results
Description of studies
Results of the search
In the first version of this review (Cody 2002), there were 38 studies identified and assessed for relevance. Of these, eight studies met the inclusion criteria (Canaud 1995; Frifelt 1996; Lago 1996; Lui 1991; Lui 1992; Miranda 1990; Paganini 1991; Weiss 2000). For the first update (Cody 2005a) of this review, three new studies were added (Brahm 1999; Leung 1995; Locatelli 2002) to provide a total of 11 studies that involved 719 participants.
For the 2014 update, 181 potentially relevant articles were identified. Of these, 22 studies satisfied the inclusion criteria (AMICUS Study 2007; BA16285 Study 2007; BA16286 Study 2007; Carrera 2003; Coyne 2000; Coyne 2006a; Hori 2004; Nagaya 2010; Kwan 2005; Lee 2008; Locatelli 2004; MAXIMA Study 2007; Mircescu 2006; Muirhead 1989; Murtagh 2000; Nissenson 2002; PROTOS Study 2007; RUBRA Study 2008; STRIATA Study 2008; Tessitore 2008; Vanrenterghem 2002; Yoon 2004). Two studies were identified from study registration databases with no results to date. We excluded 32 studies. In this update, 33 studies (92 reports) involving 5526 participants were included for analysis (Figure 1).
1.
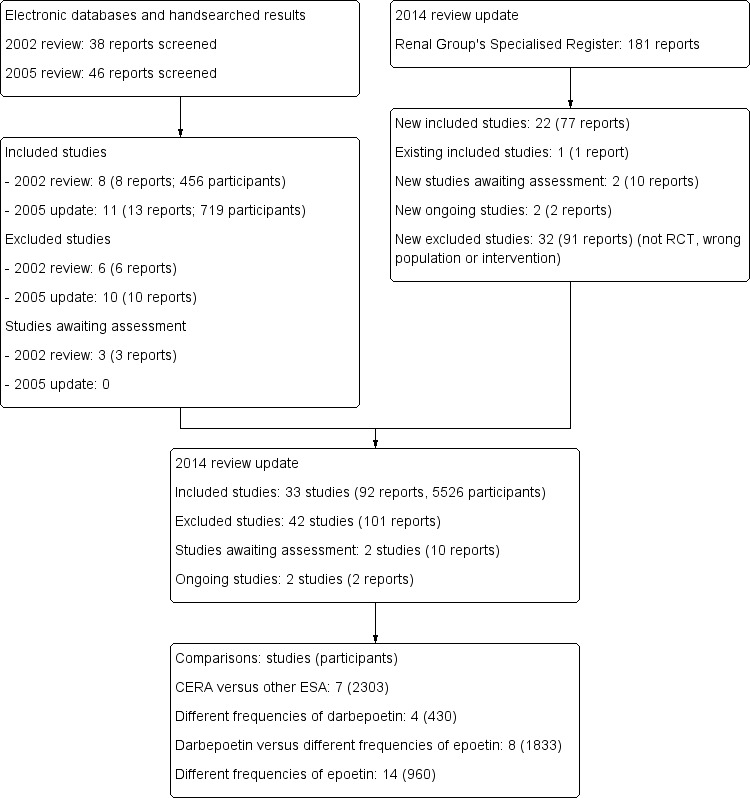
Flow diagram for study selection
CERA ‐ continuous erythropoietin receptor agonists; ESA ‐ erythropoiesis‐stimulating agents
Following external review, the literature search was updated to March 2013. Two additional studies (EMERALD 1 Study 2013; EMERALD 2 Study 2013) were identified that assessed peginesatide in patients with anaemia undergoing dialysis. These studies are listed in Studies awaiting classification and will be assessed for inclusion during the next update of this review.
Included studies
The 33 included studies were divided into four groups according to ESA comparisons. There were 20 studies available as full papers and 13 in abstract form only. Data from 10 studies could not be included in any meta‐analyses.
CERA versus other ESA
Seven studies (2303 participants) compared CERA to another ESA (AMICUS Study 2007; BA16285 Study 2007; BA16286 Study 2007; MAXIMA Study 2007; PROTOS Study 2007; RUBRA Study 2008; STRIATA Study 2008). Six studies (AMICUS Study 2007; BA16285 Study 2007; BA16286 Study 2007; MAXIMA Study 2007; PROTOS Study 2007; RUBRA Study 2008) compared CERA given two weekly with rHuEPO, with evaluation of efficacy at 24 to 36 weeks and assessment of safety up to 52 weeks. Two studies (MAXIMA Study 2007; PROTOS Study 2007) had third arms that compared CERA given four weekly with rHuEPO. Four studies (AMICUS Study 2007; MAXIMA Study 2007; PROTOS Study 2007; RUBRA Study 2008) recorded dosages of CERA and rHuEPO as median and interquartile range (IQR), and therefore, final ESA dosages could not be meta‐analysed. The STRIATA Study 2008 compared CERA given two weekly with darbepoetin given once weekly or once every two weeks. Since more than 80% of participants received darbepoetin weekly, and the results were not separated according to the frequency of darbepoetin use, data for darbepoetin‐treated patients included in the meta‐analyses included all darbepoetin‐treated patients. Three of these studies exclusively used intravenous administration, with one study using a combination of subcutaneous and intravenous administration, and one study using only subcutaneous administration
Two studies (BA16285 Study 2007; BA16286 Study 2007) provided the results of the ITT population graphically only so data could not be included in meta‐analyses. CERA and rHuEPO dosages were not recorded at the end of the evaluation period. In the STRIATA Study 2008 dosages of CERA and darbepoetin were recorded as medians with IQR and could not be meta‐analysed. Only 70 participants in these studies received peritoneal dialysis, and they were not analysed separately, so a distinct analysis could not be made.
Different frequencies of darbepoetin administration
Four studies (430 participants) compared differing frequencies of darbepoetin administration (Nagaya 2010; Kwan 2005; Locatelli 2004; Murtagh 2000). Murtagh 2000 reported results for all patients together so could not be meta‐analysed.
Darbepoetin once/week versus rHuEPO two to three times/week
Eight studies (1833 participants) compared darbepoetin with rHuEPO (Carrera 2003; Coyne 2000; Coyne 2006a; Hori 2004; Nissenson 2002; Tessitore 2008, Vanrenterghem 2002; Yoon 2004). Participant numbers in each group were not provided by Yoon 2004 so data from this study could not be meta‐analysed.
Different frequencies of rHuEPO administration
The remaining 14 studies (960 participants) evaluated different frequencies of rHuEPO administration.
Ten studies compared weekly dosing of rHuEPO with administration two to three times/week (Canaud 1995; Frifelt 1996; Lago 1996; Lee 2008; Locatelli 2002; Lui 1991; Lui 1992; Muirhead 1989; Paganini 1991; Weiss 2000)
Canaud 1995 included a third arm comparing rHuEPO daily versus weekly
Mircescu 2006 compared weekly dosing with doses given every second week
Leung 1995 compared twice weekly dosing with dosing three times weekly
Brahm 1999 compared four different frequencies (weekly, twice weekly, three times/week, and daily)
Miranda 1990 compared daily, twice weekly and three times weekly dosing.
Data from six studies could not be included in the meta‐analyses. Brahm 1999, Miranda 1990 and Muirhead 1989 were cross‐over studies and provided combined data for all patients. Frifelt 1996 provided data only as medians and IQR; Leung 1995 did not provide standard deviations; and Locatelli 2002 provided data as graphical presentations only.
Excluded studies
We excluded 42 studies (101 reports); these included 32 studies (91 reports) that were identified for the 2014 update.
Although 28 studies were RCTs, they investigated interventions that were not eligible for inclusion for this review (BA16260 Study 2006; Besarab 1998; Bhuiyan 2004; Brandt 1999; Brown 1988; Canadian EPO Study 1990; Chazot 2009; Dougherty 2004; Ifudu 1998; Kawanishi 2005; Kim 2009a; Macdougall 2003; Macdougall 2007a; Martin 2007; Moiz 2000; Muirhead 1992; Parfrey 2005; PATRONUS Study 2009; Pawlak 2007; Smith 2007; Provenzano 2006; Schmitt 2006; Smyth 2006; Spaia 1995; Spinowitz 2006; Stockenhuber 1990; Tolman 2005; Yalcinkaya 1997). Four were chiefly pharmacokinetic or safety studies (Allon 2002; Knebel 2008; Macdougall 2006; Raftery 2000); two included participants who were not on dialysis (Bennett 1991; Hirakata 2010); and five were not randomised studies (Castro 1994; Fan 1992; Ifudu 1998; Raftery 2000; Wang 2000). The frequency of administration of the intervention in the control group was unclear in Locatelli 2008.
Risk of bias in included studies
2.
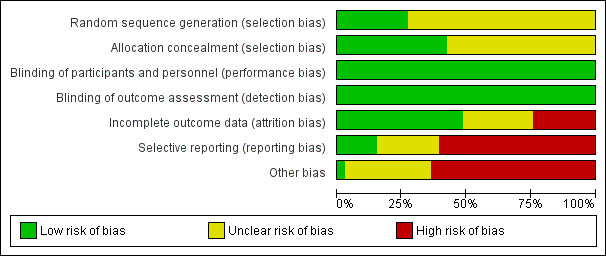
Risk of bias graph: review authors' judgements about each risk of bias item presented as percentages across all included studies
3.
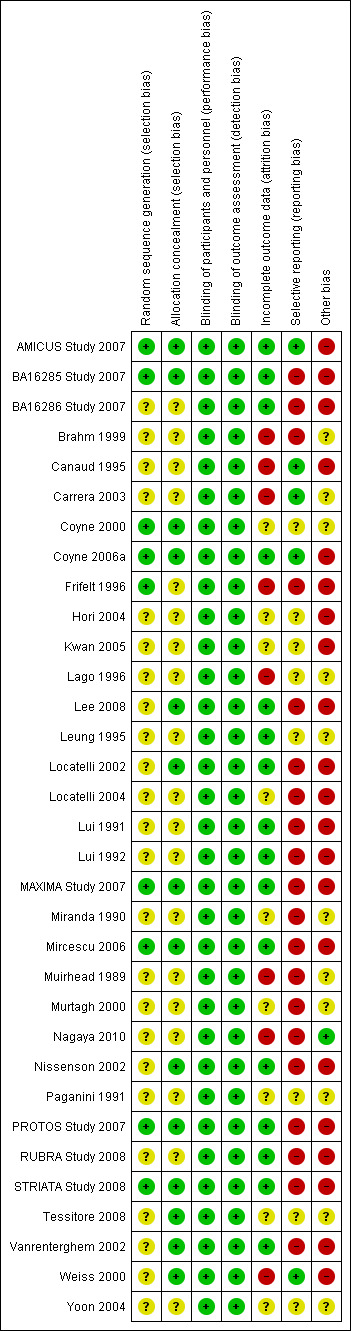
Risk of bias summary: review authors' judgements about each risk of bias item for each included study
Allocation
Nine studies reported sequence generation at low risk of bias (AMICUS Study 2007; BA16285 Study 2007; Coyne 2000; Coyne 2006a; Frifelt 1996; MAXIMA Study 2007; Mircescu 2006; PROTOS Study 2007; STRIATA Study 2008). Sequence generation methodology was unclear in all other included studies.
There were 14 studies that reported central randomisation methods at low risk of bias (AMICUS Study 2007; BA16285 Study 2007; Coyne 2000; Coyne 2006a; Lee 2008; Locatelli 2002; MAXIMA Study 2007; Mircescu 2006; Nissenson 2002; PROTOS Study 2007; STRIATA Study 2008; Tessitore 2008; Vanrenterghem 2002; Weiss 2000). Allocation concealment methodology was unclear in all other included studies.
Blinding
Four studies (Coyne 2006a; Hori 2004; Locatelli 2004; Nissenson 2002) were placebo‐controlled, and eight studies (AMICUS Study 2007; Canaud 1995; Lee 2008; Locatelli 2002; RUBRA Study 2008; STRIATA Study 2008; Vanrenterghem 2002; Weiss 2000) were described as open‐label. However, because the primary outcome (haemoglobin, haematocrit) in all studies was based on laboratory‐based assessment, and unlikely to be influenced by blinding, all included studies were considered to be at low risk of performance and detection biases.
Incomplete outcome data
Sixteen studies were considered to be at low risk of attrition bias (AMICUS Study 2007; BA16285 Study 2007; BA16286 Study 2007; Coyne 2006a; Lee 2008; Leung 1995; Locatelli 2002; Lui 1991; Lui 1992; MAXIMA Study 2007; Mircescu 2006; Nissenson 2002; PROTOS Study 2007; RUBRA Study 2008; STRIATA Study 2008; Vanrenterghem 2002). Eight studies were considered to be at high risk of attrition bias because between 19% and 41% of participants were lost to follow‐up or excluded from analysis (Brahm 1999; Canaud 1995; Carrera 2003; Frifelt 1996; Nagaya 2010; Lago 1996; Muirhead 1989; Weiss 2000). Nine studies were considered to be at unclear risk of attrition bias.
Selective reporting
Studies were considered to be at high risk of reporting bias if they did not provide data on final or change in haemoglobin or haematocrit levels, final ESA requirement, all‐cause mortality, and adverse effects (transfusions, hypertension, problems with haemodialysis access). Studies were also considered to be at high risk of bias if they provided results of ITT analyses in formats that could not be meta‐analysed or provided combined data for all periods in cross‐over studies.
Five studies (AMICUS Study 2007; Canaud 1995; Carrera 2003; Coyne 2006a; Weiss 2000) were considered to be at low risk of reporting bias. We assessed eight studies as unclear risk of bias (Coyne 2000; Hori 2004; Kwan 2005; Lago 1996; Leung 1995; Paganini 1991; Tessitore 2008; Yoon 2004), and 20 studies were considered to be at high risk of reporting bias.
Other potential sources of bias
Studies reporting any funding from pharmaceutical companies were considered to be at high risk of bias. There were 21 funded studies (AMICUS Study 2007; BA16285 Study 2007; BA16286 Study 2007; Canaud 1995; Coyne 2006a; Frifelt 1996; Hori 2004; Kwan 2005; Lee 2008; Locatelli 2002; Locatelli 2004; Lui 1991; Lui 1992; MAXIMA Study 2007; Mircescu 2006; Nissenson 2002; PROTOS Study 2007; RUBRA Study 2008; STRIATA Study 2008; Vanrenterghem 2002; Weiss 2000). Nagaya 2010 was considered to be at low risk of bias because it was funded by the Japan Dialysis Outcome Research Group. Risk of bias was unclear in the remaining 11 studies because no information on funding sources was provided.
Effects of interventions
See: Table 1; Table 2; Table 3; Table 4
Outcomes assessed included final or change in haemoglobin/haematocrit, final ESA dose, and adverse events including all‐cause mortality, hypertension, transfusion requirements and vascular access complications.
CERA every two or four weeks versus rHuEPO or darbepoetin
Meta‐analysis of four studies (AMICUS Study 2007; MAXIMA Study 2007; PROTOS Study 2007; RUBRA Study 2008) showed no significant difference in final haemoglobin between CERA given every two weeks and rHuEPO given two to three times/week (Analysis 1.1.1 (4 studies, 1126 participants) MD 0.08 g/dL, 95% CI ‐0.04 to 0.21; I² = 0%). No significant heterogeneity was demonstrated.
1.1. Analysis.
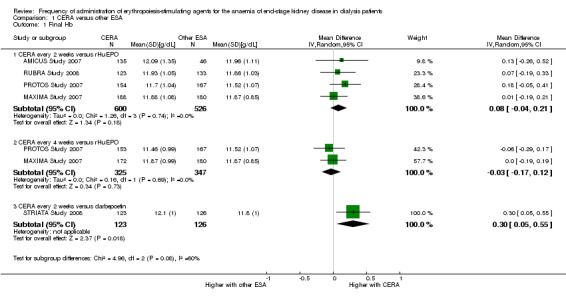
Comparison 1 CERA versus other ESA, Outcome 1 Final Hb.
In two studies (MAXIMA Study 2007; PROTOS Study 2007) a third group treated monthly with CERA was compared with rHuEPO given two to three times/week. There was no significant difference in final haemoglobin (Analysis 1.1.2 (2 studies, 672 participants): MD ‐0.03 g/dL, 95% CI ‐0.17 to 0.12; I² = 0%).
STRIATA Study 2008 compared CERA given every two weeks with weekly darbepoetin. This study reported haemoglobin was statistically significantly higher among participants who received CERA compared with those on darbepoetin, although this difference was unlikely to be of clinical significance (Analysis 1.1.3 (1 study, 249 participants): MD 0.30 g/dL, 95% CI 0.05 to 0.55).
Among the four studies that compared CERA given every two weeks with rHuEPO, and two studies comparing CERA given every four weeks with rHuEPO, there were no significant differences in all‐cause mortality (Analysis 1.2.1 (4 studies, 1341 participants): RR 1.03, 95% CI 0.67 to 1.57; I² = 0%); (Analysis 1.2.2 (2 studies, 827 participants): RR 1.15, 95% CI 0.70 to 1.89; I² = 5%), numbers of adverse events due to hypertension (Analysis 1.3.1 (4 studies, 1341 participants): RR 0.93, 95% CI 0.69 to 1.26; I² = 24%); (Analysis 1.3.2 (2 studies, 827 participants): RR 1.00, 95% CI 0.71 to 1.40; I² = 3%), numbers of patients requiring transfusions (Analysis 1.4.1 (4 studies, 1341 participants): RR 0.92, 95% CI 0.64 to 1.32; I² = 0%; Analysis 1.4.2 (2 studies, 827 participants): RR 1.01 95% CI 0.65 to 1.57; I² = 0%). Furthermore, there were no significant differences in or haemodialysis access thrombosis (Analysis 1.5.1 (4 studies, 1341 participants): RR 0.96 95% CI 0.56 to 1.65; I² = 50%; Analysis 1.5.2 (2 studies, 827 participants) RR 1.16, 95% CI 0.53 to 2.54; I² = 63%). For the outcomes of haemodialysis access thrombosis, there was significant heterogeneity in the analyses which was eliminated by removal of one study (PROTOS Study 2007), which had significantly fewer access events in the rHuEPO‐treated group compared with either CERA treated groups.
1.2. Analysis.
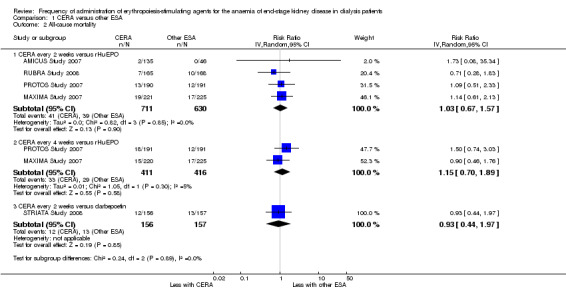
Comparison 1 CERA versus other ESA, Outcome 2 All‐cause mortality.
1.3. Analysis.
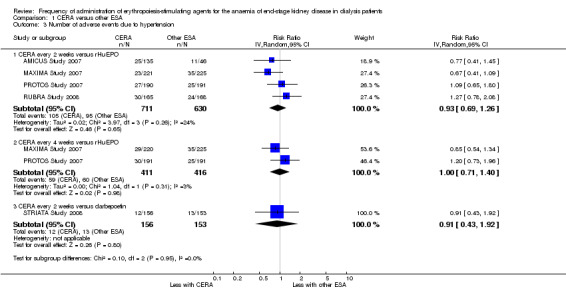
Comparison 1 CERA versus other ESA, Outcome 3 Number of adverse events due to hypertension.
1.4. Analysis.
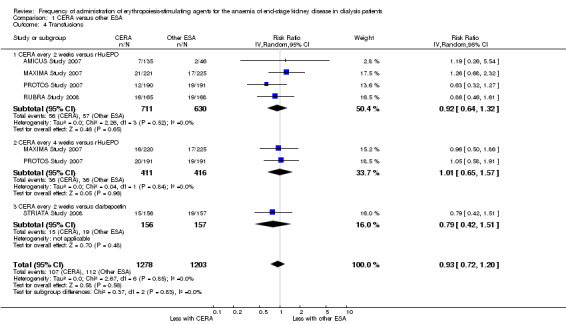
Comparison 1 CERA versus other ESA, Outcome 4 Transfusions.
1.5. Analysis.
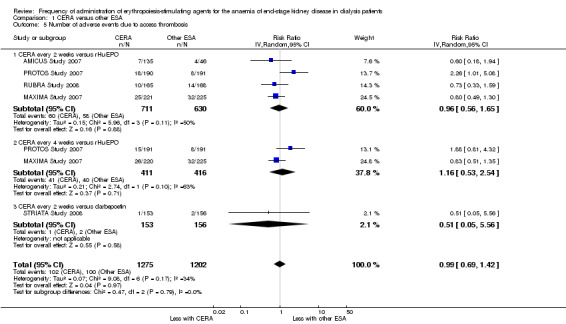
Comparison 1 CERA versus other ESA, Outcome 5 Number of adverse events due to access thrombosis.
One study comparing CERA with darbepoetin reported no significant differences in all‐cause mortality (Analysis 1.2.3 (1 study, 313 participants): RR 0.93, 95% CI 0.44 to 1.97), numbers of adverse events due to hypertension (Analysis 1.3.3 (1 study, 309 participants): RR 0.91, 95% CI 0.43 to 1.92), numbers of patients requiring transfusions (Analysis 1.4.3 (1 study, 313 participants): RR 0.79, 95% CI 0.42 to 1.51) or haemodialysis access thrombosis (Analysis 1.5.3 (1 study, 309 participants): RR 0.51, 95% CI 0.05 to 5.56). Only AMICUS Study 2007, an open‐label study, reported any quality of life assessment findings: CERA produced clinically significant improvements in 7/8 subscales of quality of life at the end of the assessment period compared with 2/8 subscales with rHuEPO. No studies reported specifically on cardiovascular mortality.
Different frequencies of CERA
In two studies with three arms, CERA given every four weeks could be compared with CERA given every two weeks. There was no significant difference in final haemoglobin (Analysis 2.1 (2 studies, 675 participants): MD ‐0.11 g/dL, 95% CI ‐0.35 to 0.14; I² = 62%), all‐cause mortality (Analysis 2.2 (2 studies, 822 participants): RR 1.03, 95% CI 0.60 to 1.78; I² = 24%), numbers of adverse events due to hypertension (Analysis 2.3 (2 studies, 822 participants): RR 1.18, 95% CI 0.83 to 1.67; I² = 0%), numbers of participants requiring transfusions (Analysis 2.4 (2 studies, 822 participants): RR 1.11, 95% CI 0.52 to 2.37; I² = 63%), or numbers of adverse events due to access thrombosis (Analysis 2.5 (2 studies, 822 participants): RR 0.96, 95% CI 0.64 to 1.43; I² = 0%). Although the I² analyses for final haemoglobin and transfusions were 62% and 63% respectively, the 95% CI overlapped and Chi² analyses did not indicate significant heterogeneity.
2.1. Analysis.
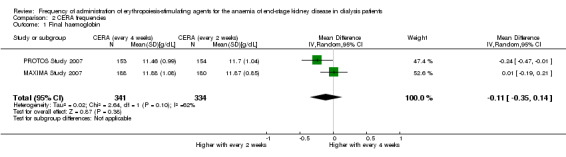
Comparison 2 CERA frequencies, Outcome 1 Final haemoglobin.
2.2. Analysis.
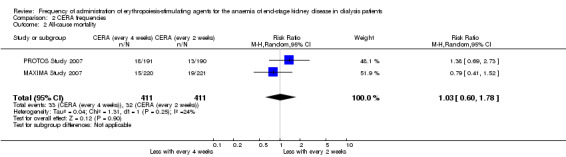
Comparison 2 CERA frequencies, Outcome 2 All‐cause mortality.
2.3. Analysis.
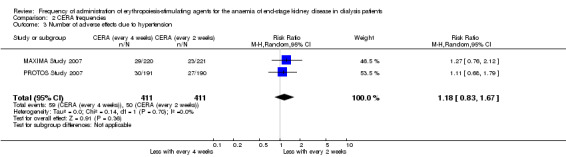
Comparison 2 CERA frequencies, Outcome 3 Number of adverse effects due to hypertension.
2.4. Analysis.
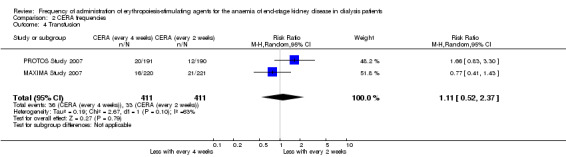
Comparison 2 CERA frequencies, Outcome 4 Transfusion.
2.5. Analysis.
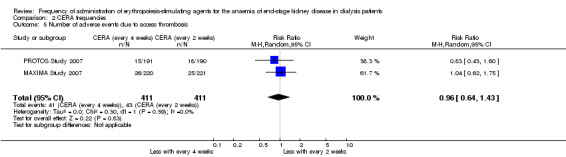
Comparison 2 CERA frequencies, Outcome 5 Number of adverse events due to access thrombosis.
Assessment or evaluation dosages of CERA, darbepoetin or rHuEPO were expressed as medians with IQR in four studies (AMICUS Study 2007; MAXIMA Study 2007; PROTOS Study 2007; RUBRA Study 2008) and therefore could not be meta‐analysed.
Only 70 of the included participants received peritoneal dialysis and these data were not presented separately from haemodialysis patients' data in study results. Likewise, different routes of administration were not separated in the study results.
Different frequencies of darbepoetin
Two studies (Locatelli 2004; Nagaya 2010) compared once/week with every two weeks dosing of darbepoetin. There were no significant differences in haemoglobin (Analysis 3.1 (2 studies, 252 participants): MD 0.04 g/dL, 95% CI ‐0.45 to 0.52; I² = 69%) or darbepoetin dose (Analysis 3.2 (2 studies, 252 participants): MD ‐8.03 µg/wk, 95% CI ‐21.64 to 5.59; I² = 77%) between treatment groups at the end of the evaluation period. However, there was significant heterogeneity in both analyses which could be related to different protocols used to determine the initial darbepoetin dose in patients who converted from once/week to every two weeks.
3.1. Analysis.
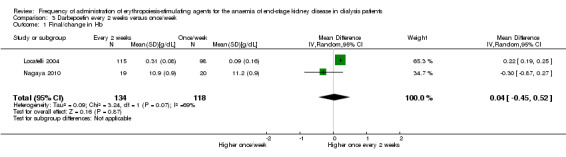
Comparison 3 Darbepoetin every 2 weeks versus once/week, Outcome 1 Final/change in Hb.
3.2. Analysis.
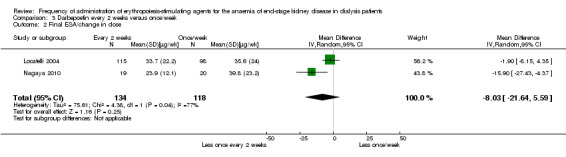
Comparison 3 Darbepoetin every 2 weeks versus once/week, Outcome 2 Final ESA/change in dose.
Locatelli 2004 reported no significant differences in all‐cause mortality (Analysis 3.3 (1 study, 306 participants) RR 1.11, 95% CI 0.46, 2.66), total treatment related adverse events (Analysis 3.4.1 (1 study, 306 participants): RR 3.50, 95% CI 0.74 to 16.58) and numbers who required transfusions (Analysis 3.4.2 (1 study, 206 participants): RR 0.60, 95% CI 0.22 to 1.61). Nagaya 2010 did not provide information on all‐cause mortality or transfusions, and neither study provided comparative information on hypertension and access thrombosis.
3.3. Analysis.

Comparison 3 Darbepoetin every 2 weeks versus once/week, Outcome 3 All‐cause‐mortality.
3.4. Analysis.
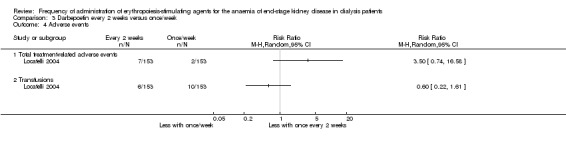
Comparison 3 Darbepoetin every 2 weeks versus once/week, Outcome 4 Adverse events.
Kwan 2005 reported no significant difference in final haemoglobin between darbepoetin once/month compared with every two weeks (Analysis 4.1 (1 study, 64 participants): MD 0.40 g/dL, 95% CI ‐0.37 to 1.17). The only adverse outcome reported was transfusion requirement; there was no significant difference reported between the two groups (Analysis 4.2.1 (1 study, 64 participants): RR 0.33, 95% CI 0.10 to 1.14).
4.1. Analysis.
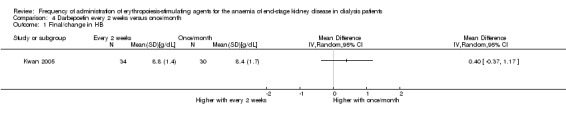
Comparison 4 Darbepoetin every 2 weeks versus once/month, Outcome 1 Final/change in HB.
4.2. Analysis.
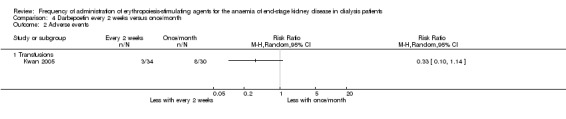
Comparison 4 Darbepoetin every 2 weeks versus once/month, Outcome 2 Adverse events.
Murtagh 2000 compared weekly with three times/week darbepoetin, and combined the results for both groups. It was reported that all patients achieved target haemoglobin. Murtagh 2000 reported that one graft thrombosis occurred, but did not specify the participant's treatment group.
Darbepoetin versus rHuEPO two to three times/week
Seven (Carrera 2003; Coyne 2000; Coyne 2006a; Hori 2004; Nissenson 2002; Tessitore 2008; Vanrenterghem 2002) of the eight studies that evaluated this comparison contributed data to one or more meta‐analyses.
There was no significant difference between final haemoglobin or change in haemoglobin at end of the evaluation period (Analysis 5.1 (6 studies, 1245 participants): MD 0.02 g/dL, 95% CI ‐0.09 to 0.12; I² = 0%). Hori 2004 reported the mean change in haemoglobin concentration from baseline only between groups and found no significant difference (0.07 g/dL, 95% CI ‐0.25 to 0.39).
5.1. Analysis.
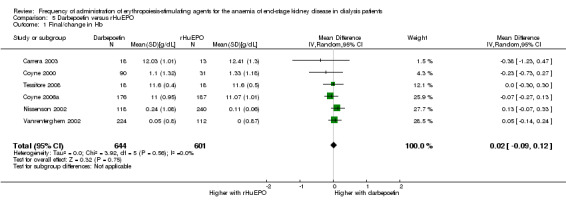
Comparison 5 Darbepoetin versus rHuEPO, Outcome 1 Final/change in Hb.
Three studies (Coyne 2006a; Nissenson 2002; Tessitore 2008) reported ESA dose at evaluation. To enable dose comparisons, rHuEPO doses were converted from units to μg using the formula: 200 U rHuEPO = 1 μg darbepoetin (Nissenson 2002). Compared with rHuEPO‐treated participants, people on darbepoetin received significantly lower weekly ESA doses (Analysis 5.2 (3 studies, 757 participants): MD ‐12.27 µg/wk, 95% CI ‐21.72 to ‐2.82; I² = 52%). All studies favoured darbepoetin although there was a moderate degree of heterogeneity in this analysis, which was eliminated by exclusion of Tessitore 2008 (I² = 0%).
5.2. Analysis.
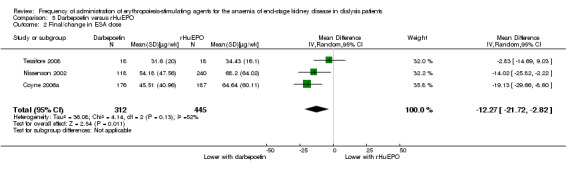
Comparison 5 Darbepoetin versus rHuEPO, Outcome 2 Final/change in ESA dose.
There were no significant differences in all‐cause mortality (Analysis 5.3 (5 studies, 1596 participants): RR 1.29, 95% CI 0.82 to 2.02; I² = 10%), hypertension (Analysis 5.4 (4 studies, 1475 participants): RR ‐0.01, 95% CI ‐0.06 to 0.05; I² = 68%), total treatment‐related adverse events (Analysis 5.6 (3 studies, 570 participants): RR ‐0.01, 95% CI ‐0.03 to 0.05; I² = 0%), or vascular access complications (Analysis 5.7 (4 studies, 1477 participants) RR ‐0.01, 95% CI‐0.03 to 0.00; I² = 0%). There was significant heterogeneity in the analysis of hypertension, which was eliminated in sensitivity analyses by excluding study data from Vanrenterghem 2002, which reported a significantly higher risk of hypertension among rHuEPO‐treated patients. Transfusion requirements were significantly increased among rHuEPO‐treated patients (Analysis 5.5 (3 studies, 1069 participants): RD ‐0.02, 95% CI ‐0.05 to ‐0.00; I² = 0%).
5.3. Analysis.
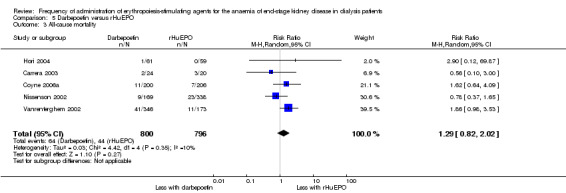
Comparison 5 Darbepoetin versus rHuEPO, Outcome 3 All‐cause mortality.
5.4. Analysis.
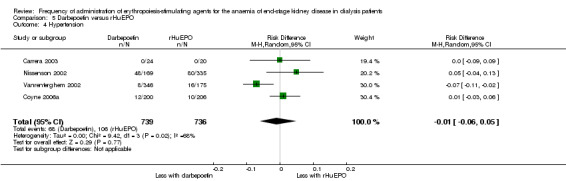
Comparison 5 Darbepoetin versus rHuEPO, Outcome 4 Hypertension.
5.6. Analysis.
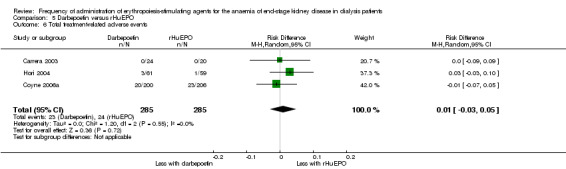
Comparison 5 Darbepoetin versus rHuEPO, Outcome 6 Total treatment‐related adverse events.
5.7. Analysis.
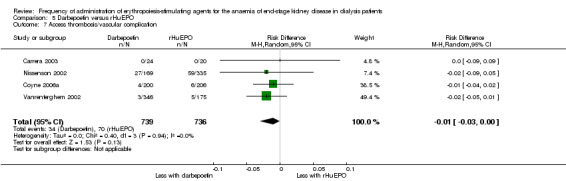
Comparison 5 Darbepoetin versus rHuEPO, Outcome 7 Access thrombosis/vascular complication.
5.5. Analysis.
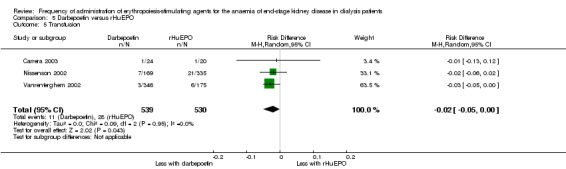
Comparison 5 Darbepoetin versus rHuEPO, Outcome 5 Transfusion.
Data from Yoon 2004 could not be included in meta‐analyses because numbers of participants in each treatment group were not provided. The reported difference in haemoglobin between treatments was ‐0.30 g/dL (95% CI ‐0.84 to 0.23), which was not significantly different.
Different frequencies of rHuEPO
Among the included studies, 14 compared different frequencies of rHuEPO administration.
Once/week versus two to three times/week
Of 10 studies that evaluated rHuEPO given weekly with rHuEPO given two to three times/week, seven could be included in meta‐analyses (Canaud 1995; Lago 1996; Lee 2008; Lui 1991; Lui 1992; Paganini 1991; Weiss 2000). There was no significant difference between final haemoglobin and haematocrit at the end of the evaluation period (Analysis 6.1 (7 studies, 363 participants): SMD ‐0.17, 95% CI ‐0.39 to 0.05; I² = 0%). Three studies reported no significant differences among frequencies but results could not be meta‐analysed because: results were provided as medians with IQR (Frifelt 1996), graphically (Locatelli 2002), or as combined data in a cross‐over study (Muirhead 1989).
6.1. Analysis.
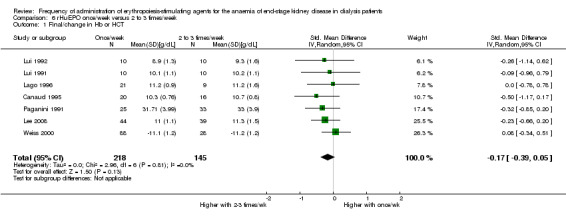
Comparison 6 rHuEPO once/week versus 2 to 3 times/week, Outcome 1 Final/change in Hb or HCT.
Five studies reported final or change in EPO dose (Canaud 1995; Lee 2008; Lui 1991; Lui 1992; Paganini 1991). There was no significant difference between weekly and two to three times/week (Analysis 6.2 (5 studies, 504 participants): MD 8.47 U/kg/wk, 95% CI ‐1.01 to 17.95; I² = 0%). There were no significant differences in hypertension (Analysis 6.3.1 (4 studies, 175 participants): RR ‐0.00, 95% CI ‐0.12 to 0.11: I² = 0%), transfusion requirements (Analysis 6.3.2 (1 study, 173 participants): RR ‐0.02, 95% CI ‐.010 to 0.06)) and haemodialysis access thrombosis (Analysis 6.3.3 (1 study, 173 participants): RR ‐0.01 95% CI ‐0.06 to 0.04).
6.2. Analysis.
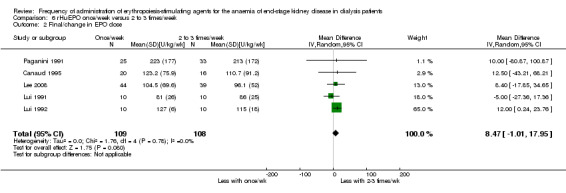
Comparison 6 rHuEPO once/week versus 2 to 3 times/week, Outcome 2 Final/change in EPO dose.
6.3. Analysis.
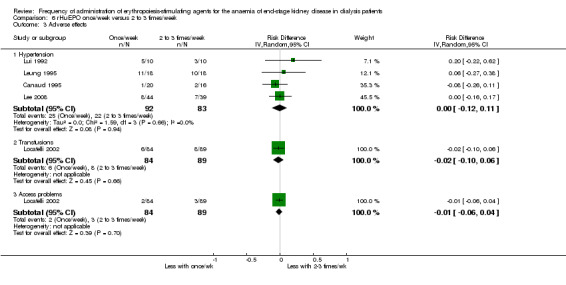
Comparison 6 rHuEPO once/week versus 2 to 3 times/week, Outcome 3 Adverse effects.
Once/week versus every two weeks
One study compared once weekly rHuEPO with rHuEPO given every two weeks (Mircescu 2006). This study reported no significant differences in haemoglobin (Analysis 7.1 (1 study, 203 participants): MD ‐0.03 g/dL, 95% CI ‐0.27 to 0.21) or final rHuEPO dose (Analysis 7.2 (1 study, 203 participants): MD 4.0 IU/kg/wk, 95% CI ‐2.05 to 10.05). Systolic blood pressure was significantly lower among patients treated with rHuEPO weekly compared with two weekly (Analysis 7.3 (1 study, 203 participants): MD ‐ 8.7 mm Hg, 95% CI ‐13.08 to ‐4.32), but there was no significant difference in diastolic blood pressure (Analysis 7.4 (1 study, 203 participants): MD 0.40 mm Hg, 95% CI ‐2.73 to 3.53). No vascular access complications were reported.
7.1. Analysis.
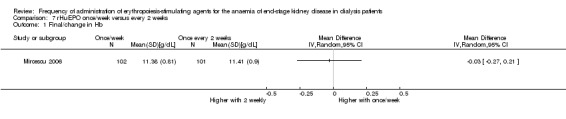
Comparison 7 rHuEPO once/week versus every 2 weeks, Outcome 1 Final/change in Hb.
7.2. Analysis.
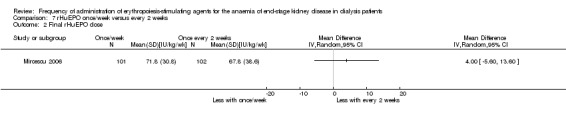
Comparison 7 rHuEPO once/week versus every 2 weeks, Outcome 2 Final rHuEPO dose.
7.3. Analysis.

Comparison 7 rHuEPO once/week versus every 2 weeks, Outcome 3 Systolic blood pressure.
7.4. Analysis.

Comparison 7 rHuEPO once/week versus every 2 weeks, Outcome 4 Diastolic blood pressure.
Daily versus weekly
Canaud 1995 enrolled a third group comparing daily rHuEPO with once weekly dosing. This study reported no significant differences in mean haemoglobin (Analysis 8.1 (1 study, 42 participants): MD ‐0.20 g/dL, 95% CI ‐0.65 to 0.25) or final rHuEPO dose (Analysis 8.2 (1 study, 42 participants) MD 34.60 U/kg/wk, 95% CI ‐6.34 to 75.54) at the end of the study.
8.1. Analysis.
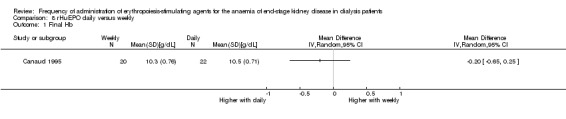
Comparison 8 rHuEPO daily versus weekly, Outcome 1 Final Hb.
8.2. Analysis.
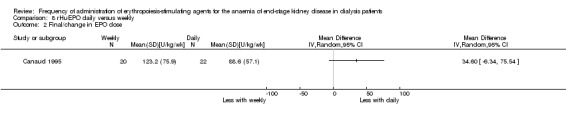
Comparison 8 rHuEPO daily versus weekly, Outcome 2 Final/change in EPO dose.
Other results
Brahm 1999 compared multiple different rHuEPO frequencies; Leung 1995 compared rHuEPO twice/week with three times/week; and Miranda 1990 reported on rHuEPO daily compared with twice/week. Brahm 1999 (a cross‐over study) and Miranda 1990 reported data for combined treatment groups only. Leung 1995 did not provide standard deviations. Brahm 1999 maintained stable haemoglobin levels and found no difference in numbers of rHuEPO injections required/week to achieve this outcome. All‐cause mortality and adverse effects were not reported. Leung 1995 reported that there were no significant differences in haemoglobin change, rHuEPO dose or numbers of antihypertensive drugs required between the twice and three times/week dosing regimen. Miranda 1990 reported satisfactory rises in haemoglobin in patients and no differences in required rHuEPO doses between treatment groups.
Discussion
Summary of main results
Previous versions of this review presented analyses based on 11 studies that assessed different frequencies of rHuEPO ‐ epoetin‐α or epoetin‐β. An additional 22 studies were included in this update, most of which evaluated the longer‐acting ESA darbepoetin and CERA. For ease of comparison, we divided the included studies into four groups.
CERA versus other ESA
Our findings demonstrated that CERA administered at reduced frequencies compared with rHuEPO provides comparable degrees of anaemia correction without new or increased numbers of adverse effects or increase in all‐cause mortality. In addition, no significant differences in efficacy or adverse effects were demonstrated using different frequencies of CERA. Two studies investigated intravenous ESA administration, one used subcutaneous administration and another used both routes. Interpretation of the meta‐analyses revealed no obvious variation in results according to route of administration, but this could not be formally tested in subgroup analyses.
Only AMICUS Study 2007, an open‐label study, reported on quality of life with improvement and determined greater improvement with CERA compared with rHuEPO.
MAXIMA Study 2007 and PROTOS Study 2007 compared administration of CERA at two weekly and four weekly intervals. No significant differences were found in final haemoglobin or reports of adverse events.
CERA administered intravenously every two weeks showed no significant difference in maintaining haemoglobin compared with darbepoetin administered at once/week. There were no significant differences between groups for all‐cause mortality and adverse events including hypertension, vascular access complications and transfusion requirements.
Different frequencies of darbepoetin
Two studies compared darbepoetin administered once weekly with every two weeks. There were no significant differences in final haemoglobin, all‐cause mortality and other reported adverse events between frequencies. There was significant heterogeneity between studies for the outcomes of haemoglobin and darbepoetin dosage, which may have resulted from different dosing schedules.
Darbepoetin versus rHuEPO
Six studies compared once/week darbepoetin with two to three times/week administration of rHuEPO, with no significant differences in final haemoglobin or change in haemoglobin, all‐cause mortality and adverse effects. ESA dose was significantly lower in darbepoetin‐treated patients but this outcome was only reported in three studies (757 participants).
Different frequencies of rHuEPO
No significant differences in final haemoglobin, haematocrit or rHuEPO dose were identified in 10 studies that compared rHuEPO once/week with two to three times/week. No studies reported all‐cause mortality, and there were limited data on other adverse effects. The other included studies assessed different frequencies of rHuEPO administration and identified no significant differences in final haemoglobin or rHuEPO dose with limited data on other outcomes.
Overall completeness and applicability of evidence
This review included only studies that evaluated ESA for people on dialysis. However, most participants were on haemodialysis. Lui 1991 only included peritoneal dialysis patients and evaluated rHuEPO. Other studies included few participants on peritoneal dialysis but did not separate data according to mode of dialysis. Furthermore, we did not identify any studies in children on dialysis. An open‐label, non‐inferiority study that compared darbepoetin‐α and rHuEPO in 124 children with CKD not on dialysis found no significant difference in maintaining haemoglobin (Warady 2006).
There were limited data available that compared long‐acting ESA. We identified one study in dialysis patients which compared darbepoetin and CERA and included 313 participants (STRIATA Study 2008). This study identified a small (0.3 g/dL) statistically significant, but not clinically significant, benefit from CERA over darbepoetin with no differences in adverse event profiles. However, further studies comparing different frequencies are required to determine if these drugs provide similar clinical benefits. PATRONUS Study 2009, which did not match our inclusion criteria for this review, found that CERA was superior to darbepoetin when both drugs were given at four weekly intervals.
Patient‐centred outcomes were generally poorly reported. We found that 13/33 studies recorded all‐cause mortality; only four reported cardiovascular mortality data; and none reported on cardiovascular morbidity. AMICUS Study 2007, which compared CERA with rHuEPO, provided limited quality of life data. Six older studies (Lee 2008; Leung 1995; Lui 1991; Lui 1992; Paganini 1991; Weiss 2000) that evaluated different frequencies of rHuEPO were less likely to report adverse effects data, and none reported on all‐cause mortality.
Although hypertension and vascular access complications are known to be associated with ESA administration, these were reported in only 14 and 11 studies respectively. It is widely accepted that ESA therapy reduces transfusion requirements, however, 22 studies failed to report on transfusion events.
ESA doses in the CERA studies could not be meta‐analysed because data were reported as medians and IQR. Furthermore, ESA doses in other studies could not be meta‐analysed because of different reporting methods. Nagaya 2010 and Locatelli 2004 compared different frequencies of darbepoetin administration, but there was marked heterogeneity in the analyses for haemoglobin (Analysis 3.1) and darbepoetin dose (Analysis 3.2). Reasons for heterogeneity were unclear, but may be related to different schedules used to alter darbepoetin dose in these studies.
Quality of the evidence
We included 33 studies that involved 5526 participants in this review. Of these, 13 were available only in abstract form.
We found that nine (27%) studies demonstrated adequate random sequence generation, and 14 (42%) were assessed as showing allocation procedures at low risk of bias. These parameters were found to be less commonly reported in earlier studies that compared erythropoietin administration frequencies. Blinding was reported in only a few studies, but because outcome measures were reported as laboratory results, this was considered to confer a low risk of bias. Attrition bias (incomplete outcome data) was identified in 50% of the included studies, and reporting bias (selective reporting) was identified or unclear in fewer than 15% of studies (Figure 2).
Quality of studies comparing CERA and rHuEPO was considered high for the outcome of final haemoglobin (Table 1), but a small sample size meant that quality was moderate in the study that compared CERA and darbepoetin. Small numbers of events meant that study quality was assessed as moderate for all‐cause mortality. The quality of studies reporting on adverse events was considered to be moderate for all outcomes, except hypertension, where quality was high. These studies were all well‐funded, large multicentre studies, with potentially better study design and reporting.
Overall, quality of studies comparing different durations of CERA administration was considered to be high or moderate for all outcomes (Table 2).
In the studies comparing darbepoetin and rHuEPO for outcomes of final haemoglobin and all‐cause mortality, overall quality was assessed as moderate (Table 3). In relation to adverse effects, overall quality was assessed as moderate or low because of heterogeneity and high risk of bias in individual studies.
In studies that compared different frequencies of rHuEPO administration, quality of evidence was considered to be low for all outcomes (Table 4). Many of these studies were available only in abstract form. Among these earlier studies participant numbers were low, important outcomes such as mortality were not reported, and components of study design were poorly reported or at high risk of bias.
Only 17/33 studies could be included in Summary of findings tables because other comparisons involved single studies only or data could not be included in meta‐analyses.
Potential biases in the review process
A comprehensive search of the Cochrane Renal Group's Specialised Register was undertaken; the last search was completed in March 2013. This decreased the likelihood that published eligible studies were omitted from our review. However, more recently published eligible studies and eligible studies in congress proceedings not searched could have been missed. We found that 40% of included studies were available only in the abstract form that provided limited information on study methods and results; inclusion of these data could be a source of bias. Two potentially eligible studies in progress were identified from study registries.
This was an extensive review completed independently by two authors who both participated in all steps of the review. This limited the risk of errors in determining study eligibility, data extraction, risk of bias assessment and data synthesis.
Many of the earlier rHuEPO studies were small with incomplete information on study methods and results. Further information could not be obtained about these studies from investigators or the literature.
Among the more recent studies of longer‐acting ESA per‐protocol data were meta‐analysed for the primary outcome of haemoglobin level in three (MAXIMA Study 2007; PROTOS Study 2007; RUBRA Study 2008) of the four studies; ITT data were either not provided or were presented in graphical form only. We did not receive responses from authors we contacted with requests for ITT data. Sensitivity analysis that excluded the study that provided ITT data (AMICUS Study 2007) did not alter the MD and 95% CI. This suggested that use of per‐protocol data did not bias the results.
Agreements and disagreements with other studies or reviews
Carrera 2007 published a narrative review that assessed different frequencies of darbepoetin or comparisons of darbepoetin with rHuEPO in dialysis patients and patients not yet on dialysis. Among dialysis patients, the review included 13 studies of which seven were RCTs. We included six of those RCTs in our review; the other study compared different routes of administration, and therefore was not eligible for inclusion in this review.
A narrative review by Foley 2010 included 10 studies that evaluated darbepoetin or CERA. Of those, seven involved dialysis patients and were included in this review.
Our review results concur with these earlier reviews that longer‐acting ESA administered at less frequent intervals are non‐inferior in RCTs to rHuEPO without differences in adverse events.
The Anaemia Working Group of the European Renal Best Practice (Locatelli 2009) recommended that CERA be administered every two weeks for anaemia correction, and then every four weeks. The Group considered that the safety and tolerability of CERA was similar to other ESA.
The KDIGO Working Group identified no evidence that any given type of ESA was superior to another in terms of efficacy and safety (KDIGO 2012). The KDIGO Working Group concluded that differences in efficacy among ESA were unlikely and made no recommendations about specific types of ESA. The Working Group further suggested that ESA choice would depend on patient and country‐specific issues such as availability and cost.
Authors' conclusions
Implications for practice.
Longer‐acting ESA are now standard practice for management of anaemia among people with CKD. This review identified evidence to demonstrate that darbepoetin and CERA administered at one to four weekly intervals are non‐inferior to rHuEPO given one to three times/week in terms of achieving haemoglobin targets without any significant differences in adverse events.
As well as greater convenience offered by extended dosing intervals for both patients and healthcare providers, longer‐acting ESA use may also result in improved cost efficiency. A recent audit of 82 patients (Summers 2005) demonstrated that switching from a more frequent rHuEPO dose regimen to once or every two weeks, darbepoetin significantly decreased ESA acquisition costs. The MERCURIUS project assessed costs in 21 haemodialysis centres in eight European countries. Administration of ESA at two weekly intervals resulted in significant reductions in pharmacy and dialysis unit staff and equipment costs (Burnier 2009). In many countries, costs of newer longer‐acting but more costly ESA would have to be balanced against costs associated with more frequent rHuEPO administration because agents provide similar degrees of efficacy and safety. Less frequent administration could also reduce needle stick injuries potentially further reducing costs. A possible downside of less frequent administration of ESA could be that doses are missed, although there are no data to support or refute this possibility.
Implications for research.
Additional large, well designed, RCTs are required in the following areas:
Comparisons of different longer‐acting ESA (e.g. darbepoetin compared with CERA)
Comparisons of different frequencies of ESA administration among peritoneal dialysis and paediatric dialysis patients.
Studies should include patient‐centred outcomes including all‐cause mortality, cardiovascular mortality and morbidity, and quality of life assessment. They should also include an estimate of patient and carer satisfaction related to different frequencies of administration.
Additional studies of cost‐effectiveness of different frequencies of administration should be undertaken, particularly in the developing world.
What's new
| Date | Event | Description |
|---|---|---|
| 12 November 2014 | Amended | Minor edit to study numbers in "Quality of the evidence" section |
History
Protocol first published: Issue 4, 2002 Review first published: Issue 4, 2002
| Date | Event | Description |
|---|---|---|
| 2 June 2014 | Amended | Minor copy edit made to search strategies and study name |
| 16 May 2014 | New search has been performed | Methods updated, new search performed, title changed to reflect current terminology and the new scope of the review |
| 16 May 2014 | New citation required and conclusions have changed | New studies and interventions included |
| 30 September 2008 | Amended | Converted to new review format. |
| 25 May 2005 | New citation required and conclusions have changed | Substantive amendment |
Acknowledgements
We are grateful to Conal Day, Marion Campbell, Cam Donaldson, Adrian Grant, Luke Vale, Sheila Wallace and Alison MacLeod who were authors on the original and first update of this review (Cody 2002; Cody 2005a), contributing to the design, quality assessment, data collection, entry, analysis and interpretation, and writing.
We wish to thank the referees for their comments and feedback during the preparation of these reviews.
The initial review (Cody 2002) was supported by a grant from the Chief Scientist's Office, Scottish Office Department of Health.
We would like to thank Dr Daniel Coyne, Dr Marian Klinger, Dr Agnieszka Zolkiewicz, and Dr Anatole Besarab who provided additional information on their studies.
Appendices
Appendix 1. Electronic search strategies
| Database | Search terms |
| CENTRAL |
|
| MEDLINE |
|
| EMBASE |
|
Appendix 2. Risk of bias assessment tool
| Potential source of bias | Assessment criteria |
|
Random sequence generation Selection bias (biased allocation to interventions) due to inadequate generation of a randomised sequence |
Low risk of bias: Random number table; computer random number generator; coin tossing; shuffling cards or envelopes; throwing dice; drawing of lots; minimization (minimization may be implemented without a random element, and this is considered to be equivalent to being random). |
| High risk of bias: Sequence generated by odd or even date of birth; date (or day) of admission; sequence generated by hospital or clinic record number; allocation by judgement of the clinician; by preference of the participant; based on the results of a laboratory test or a series of tests; by availability of the intervention. | |
| Unclear: Insufficient information about the sequence generation process to permit judgement. | |
|
Allocation concealment Selection bias (biased allocation to interventions) due to inadequate concealment of allocations prior to assignment |
Low risk of bias: Randomisation method described that would not allow investigator/participant to know or influence intervention group before eligible participant entered in the study (e.g. central allocation, including telephone, web‐based, and pharmacy‐controlled, randomisation; sequentially numbered drug containers of identical appearance; sequentially numbered, opaque, sealed envelopes). |
| High risk of bias: Using an open random allocation schedule (e.g. a list of random numbers); assignment envelopes were used without appropriate safeguards (e.g. if envelopes were unsealed or non‐opaque or not sequentially numbered); alternation or rotation; date of birth; case record number; any other explicitly unconcealed procedure. | |
| Unclear: Randomisation stated but no information on method used is available. | |
|
Blinding of participants and personnel Performance bias due to knowledge of the allocated interventions by participants and personnel during the study |
Low risk of bias: No blinding or incomplete blinding, but the review authors judge that the outcome is not likely to be influenced by lack of blinding; blinding of participants and key study personnel ensured, and unlikely that the blinding could have been broken. |
| High risk of bias: No blinding or incomplete blinding, and the outcome is likely to be influenced by lack of blinding; blinding of key study participants and personnel attempted, but likely that the blinding could have been broken, and the outcome is likely to be influenced by lack of blinding. | |
| Unclear: Insufficient information to permit judgement | |
|
Blinding of outcome assessment Detection bias due to knowledge of the allocated interventions by outcome assessors. |
Low risk of bias: No blinding of outcome assessment, but the review authors judge that the outcome measurement is not likely to be influenced by lack of blinding; blinding of outcome assessment ensured, and unlikely that the blinding could have been broken. |
| High risk of bias: No blinding of outcome assessment, and the outcome measurement is likely to be influenced by lack of blinding; blinding of outcome assessment, but likely that the blinding could have been broken, and the outcome measurement is likely to be influenced by lack of blinding. | |
| Unclear: Insufficient information to permit judgement | |
|
Incomplete outcome data Attrition bias due to amount, nature or handling of incomplete outcome data. |
Low risk of bias: No missing outcome data; reasons for missing outcome data unlikely to be related to true outcome (for survival data, censoring unlikely to be introducing bias); missing outcome data balanced in numbers across intervention groups, with similar reasons for missing data across groups; for dichotomous outcome data, the proportion of missing outcomes compared with observed event risk not enough to have a clinically relevant impact on the intervention effect estimate; for continuous outcome data, plausible effect size (difference in means or standardized difference in means) among missing outcomes not enough to have a clinically relevant impact on observed effect size; missing data have been imputed using appropriate methods. |
| High risk of bias: Reason for missing outcome data likely to be related to true outcome, with either imbalance in numbers or reasons for missing data across intervention groups; for dichotomous outcome data, the proportion of missing outcomes compared with observed event risk enough to induce clinically relevant bias in intervention effect estimate; for continuous outcome data, plausible effect size (difference in means or standardized difference in means) among missing outcomes enough to induce clinically relevant bias in observed effect size; ‘as‐treated’ analysis done with substantial departure of the intervention received from that assigned at randomisation; potentially inappropriate application of simple imputation. | |
| Unclear: Insufficient information to permit judgement | |
|
Selective reporting Reporting bias due to selective outcome reporting |
Low risk of bias: The study protocol is available and all of the study’s pre‐specified (primary and secondary) outcomes that are of interest in the review have been reported in the pre‐specified way; the study protocol is not available but it is clear that the published reports include all expected outcomes, including those that were pre‐specified (convincing text of this nature may be uncommon). |
| High risk of bias: Not all of the study’s pre‐specified primary outcomes have been reported; one or more primary outcomes is reported using measurements, analysis methods or subsets of the data (e.g. subscales) that were not pre‐specified; one or more reported primary outcomes were not pre‐specified (unless clear justification for their reporting is provided, such as an unexpected adverse effect); one or more outcomes of interest in the review are reported incompletely so that they cannot be entered in a meta‐analysis; the study report fails to include results for a key outcome that would be expected to have been reported for such a study. | |
| Unclear: Insufficient information to permit judgement | |
|
Other bias Bias due to problems not covered elsewhere in the table |
Low risk of bias: The study appears to be free of other sources of bias. |
| High risk of bias: Had a potential source of bias related to the specific study design used; stopped early due to some data‐dependent process (including a formal‐stopping rule); had extreme baseline imbalance; has been claimed to have been fraudulent; had some other problem. | |
| Unclear: Insufficient information to assess whether an important risk of bias exists; insufficient rationale or evidence that an identified problem will introduce bias. |
Data and analyses
Comparison 1. CERA versus other ESA.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Final Hb | 5 | Mean Difference (IV, Random, 95% CI) | Subtotals only | |
| 1.1 CERA every 2 weeks versus rHuEPO | 4 | 1126 | Mean Difference (IV, Random, 95% CI) | 0.08 [‐0.04, 0.21] |
| 1.2 CERA every 4 weeks versus rHuEPO | 2 | 672 | Mean Difference (IV, Random, 95% CI) | ‐0.03 [‐0.17, 0.12] |
| 1.3 CERA every 2 weeks versus darbepoetin | 1 | 249 | Mean Difference (IV, Random, 95% CI) | 0.30 [0.05, 0.55] |
| 2 All‐cause mortality | 5 | Risk Ratio (IV, Random, 95% CI) | Subtotals only | |
| 2.1 CERA every 2 weeks versus rHuEPO | 4 | 1341 | Risk Ratio (IV, Random, 95% CI) | 1.03 [0.67, 1.57] |
| 2.2 CERA every 4 weeks versus rHuEPO | 2 | 827 | Risk Ratio (IV, Random, 95% CI) | 1.15 [0.70, 1.89] |
| 2.3 CERA every 2 weeks versus darbepoetin | 1 | 313 | Risk Ratio (IV, Random, 95% CI) | 0.93 [0.44, 1.97] |
| 3 Number of adverse events due to hypertension | 5 | Risk Ratio (IV, Random, 95% CI) | Subtotals only | |
| 3.1 CERA every 2 weeks versus rHuEPO | 4 | 1341 | Risk Ratio (IV, Random, 95% CI) | 0.93 [0.69, 1.26] |
| 3.2 CERA every 4 weeks versus rHuEPO | 2 | 827 | Risk Ratio (IV, Random, 95% CI) | 1.00 [0.71, 1.40] |
| 3.3 CERA every 2 weeks versus darbepoetin | 1 | 309 | Risk Ratio (IV, Random, 95% CI) | 0.91 [0.43, 1.92] |
| 4 Transfusions | 5 | 2481 | Risk Ratio (IV, Random, 95% CI) | 0.93 [0.72, 1.20] |
| 4.1 CERA every 2 weeks versus rHuEPO | 4 | 1341 | Risk Ratio (IV, Random, 95% CI) | 0.92 [0.64, 1.32] |
| 4.2 CERA every 4 weeks versus rHuEPO | 2 | 827 | Risk Ratio (IV, Random, 95% CI) | 1.01 [0.65, 1.57] |
| 4.3 CERA every 2 weeks versus darbepoetin | 1 | 313 | Risk Ratio (IV, Random, 95% CI) | 0.79 [0.42, 1.51] |
| 5 Number of adverse events due to access thrombosis | 5 | 2477 | Risk Ratio (IV, Random, 95% CI) | 0.99 [0.69, 1.42] |
| 5.1 CERA every 2 weeks versus rHuEPO | 4 | 1341 | Risk Ratio (IV, Random, 95% CI) | 0.96 [0.56, 1.65] |
| 5.2 CERA every 4 weeks versus rHuEPO | 2 | 827 | Risk Ratio (IV, Random, 95% CI) | 1.16 [0.53, 2.54] |
| 5.3 CERA every 2 weeks versus darbepoetin | 1 | 309 | Risk Ratio (IV, Random, 95% CI) | 0.51 [0.05, 5.56] |
Comparison 2. CERA frequencies.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Final haemoglobin | 2 | 675 | Mean Difference (IV, Random, 95% CI) | ‐0.11 [‐0.35, 0.14] |
| 2 All‐cause mortality | 2 | 822 | Risk Ratio (M‐H, Random, 95% CI) | 1.03 [0.60, 1.78] |
| 3 Number of adverse effects due to hypertension | 2 | 822 | Risk Ratio (M‐H, Random, 95% CI) | 1.18 [0.83, 1.67] |
| 4 Transfusion | 2 | 822 | Risk Ratio (M‐H, Random, 95% CI) | 1.11 [0.52, 2.37] |
| 5 Number of adverse events due to access thrombosis | 2 | 822 | Risk Ratio (M‐H, Random, 95% CI) | 0.96 [0.64, 1.43] |
Comparison 3. Darbepoetin every 2 weeks versus once/week.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Final/change in Hb | 2 | 252 | Mean Difference (IV, Random, 95% CI) | 0.04 [‐0.45, 0.52] |
| 2 Final ESA/change in dose | 2 | 252 | Mean Difference (IV, Random, 95% CI) | ‐8.03 [‐21.64, 5.59] |
| 3 All‐cause‐mortality | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 4 Adverse events | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 4.1 Total treatment‐related adverse events | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 4.2 Transfusions | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] |
Comparison 4. Darbepoetin every 2 weeks versus once/month.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Final/change in HB | 1 | Mean Difference (IV, Random, 95% CI) | Totals not selected | |
| 2 Adverse events | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 2.1 Transfusions | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] |
Comparison 5. Darbepoetin versus rHuEPO.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Final/change in Hb | 6 | 1245 | Mean Difference (IV, Random, 95% CI) | 0.02 [‐0.09, 0.12] |
| 2 Final/change in ESA dose | 3 | 757 | Mean Difference (IV, Random, 95% CI) | ‐12.27 [‐21.72, ‐2.82] |
| 3 All‐cause mortality | 5 | 1596 | Risk Ratio (M‐H, Random, 95% CI) | 1.29 [0.82, 2.02] |
| 4 Hypertension | 4 | 1475 | Risk Difference (M‐H, Random, 95% CI) | ‐0.01 [‐0.06, 0.05] |
| 5 Transfusion | 3 | 1069 | Risk Difference (M‐H, Random, 95% CI) | ‐0.02 [‐0.05, ‐0.00] |
| 6 Total treatment‐related adverse events | 3 | 570 | Risk Difference (M‐H, Random, 95% CI) | 0.01 [‐0.03, 0.05] |
| 7 Access thrombosis/vascular complication | 4 | 1475 | Risk Difference (M‐H, Random, 95% CI) | ‐0.01 [‐0.03, 0.00] |
Comparison 6. rHuEPO once/week versus 2 to 3 times/week.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Final/change in Hb or HCT | 7 | 363 | Std. Mean Difference (IV, Random, 95% CI) | ‐0.17 [‐0.39, 0.05] |
| 2 Final/change in EPO dose | 5 | 217 | Mean Difference (IV, Random, 95% CI) | 8.47 [‐1.01, 17.95] |
| 3 Adverse effects | 5 | Risk Difference (IV, Random, 95% CI) | Subtotals only | |
| 3.1 Hypertension | 4 | 175 | Risk Difference (IV, Random, 95% CI) | ‐0.00 [‐0.12, 0.11] |
| 3.2 Transfusions | 1 | 173 | Risk Difference (IV, Random, 95% CI) | ‐0.02 [‐0.10, 0.06] |
| 3.3 Access problems | 1 | 173 | Risk Difference (IV, Random, 95% CI) | ‐0.01 [‐0.06, 0.04] |
Comparison 7. rHuEPO once/week versus every 2 weeks.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Final/change in Hb | 1 | Mean Difference (IV, Random, 95% CI) | Totals not selected | |
| 2 Final rHuEPO dose | 1 | Mean Difference (IV, Random, 95% CI) | Totals not selected | |
| 3 Systolic blood pressure | 1 | Mean Difference (IV, Random, 95% CI) | Totals not selected | |
| 4 Diastolic blood pressure | 1 | Mean Difference (IV, Random, 95% CI) | Totals not selected |
Comparison 8. rHuEPO daily versus weekly.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Final Hb | 1 | Mean Difference (IV, Random, 95% CI) | Totals not selected | |
| 2 Final/change in EPO dose | 1 | Mean Difference (IV, Random, 95% CI) | Totals not selected |
Characteristics of studies
Characteristics of included studies [ordered by study ID]
AMICUS Study 2007.
| Methods |
|
|
| Participants |
|
|
| Interventions | Treatment group 1
Treatment group 2
Co‐interventions
|
|
| Outcomes |
|
|
| Notes |
|
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Central randomisation, computer generated |
| Allocation concealment (selection bias) | Low risk | Central allocation with IVRS |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | Open label study but primary outcome was laboratory based and unlikely to be influenced by blinding |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | Open label study but primary outcome was laboratory based and unlikely to be influenced by blinding |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Efficacy data and safety data available for all patients |
| Selective reporting (reporting bias) | Low risk | Study protocol not available but published results include all expected outcomes |
| Other bias | High risk | Roche sponsored |
BA16285 Study 2007.
| Methods |
|
|
| Participants |
|
|
| Interventions | Treatment group A
Treatment group B
Treatment group C
Other information
Co‐interventions
|
|
| Outcomes |
|
|
| Notes |
|
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Central randomisation, computer generated |
| Allocation concealment (selection bias) | Low risk | Central randomisation, computer generated |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | No blinding but outcome of Hb unlikely to be influenced by lack of blinding |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | No blinding but outcome of Hb unlikely to be influenced by lack of blinding |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | 10/91 did not complete core period but all patients included in ITT analysis. Reasons for withdrawal: adverse events, treatment refusal, inadvertent coadministration of EPO, insufficient therapeutic response, transplant, anaemia not related to CKD |
| Selective reporting (reporting bias) | High risk | Published results did not include ESA dosage. Results of ITT analysis only available graphically and cannot be entered in meta‐analysis. |
| Other bias | High risk | Funded F. Hoffman La Roche Ltd |
BA16286 Study 2007.
| Methods |
|
|
| Participants |
|
|
| Interventions | Treatment group A
Treatment group B
Treatment group C
Other information
Co‐interventions
|
|
| Outcomes |
|
|
| Notes |
|
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | No information provided: "phase II, randomised, open label" |
| Allocation concealment (selection bias) | Unclear risk | No information provided: "assignment in sequential fashion" |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | No blinding but outcome of Hb unlikely to be influenced by lack of blinding |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | No blinding but outcome of Hb unlikely to be influenced by lack of blinding |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | 11/137 withdrawn; 2 died, 2 withdrew because of adverse events, 3 insufficient therapeutic response, 1 transplant, 1 failure to return, 1 holiday, 1 hospitalisation; all patients included in ITT analysis |
| Selective reporting (reporting bias) | High risk | No outcome data provided on ESA dosage. Data on ITT analysis only available graphically and could not be included in meta‐analysis |
| Other bias | High risk | Funded by F. Hoffmann La Roche Ltd |
Brahm 1999.
| Methods |
|
|
| Participants |
|
|
| Interventions | EPO (SC)
|
|
| Outcomes |
|
|
| Notes | ||
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Said to be randomised |
| Allocation concealment (selection bias) | Unclear risk | NS |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | Primary outcome was laboratory based and not likely to be influenced by blinding |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | Primary outcome was laboratory based and not likely to be influenced by blinding |
| Incomplete outcome data (attrition bias) All outcomes | High risk | 27% not analysed and of 22, only 13 completed all 4 cross‐over periods; loss to follow‐up: 8/30 (27%) dropped out during correction period 13/22 (59%) completed all four frequency regimens |
| Selective reporting (reporting bias) | High risk | Cross‐over study; data reported for correction periods and not patients so data cannot be included in meta‐analysis |
| Other bias | Unclear risk | Funding: NS |
Canaud 1995.
| Methods |
|
|
| Participants |
|
|
| Interventions | Treatment group 1
Treatment group 2
Treatment group 3
Treatment group 4
Other information
Co‐interventions
|
|
| Outcomes |
|
|
| Notes |
|
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Stated to be randomised but no other information provided: "open, randomised, multicentre study" |
| Allocation concealment (selection bias) | Unclear risk | NS |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | Open‐labelled study. Primary outcome was laboratory based and not likely to be influenced by blinding |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | Open‐labelled study. Primary outcome was laboratory based and not likely to be influenced by blinding |
| Incomplete outcome data (attrition bias) All outcomes | High risk | 40 (34%) of 116 patients withdrawn from final analysis and this loss could influence the results: transplantation (7), death (1), lost to follow‐up (3), unable to self‐inject (1), final Hb at week 16 higher than target Hb > 12 g/dL (11) |
| Selective reporting (reporting bias) | Low risk | Specified essential outcomes included |
| Other bias | High risk | Study overseen by Cilag laboratories |
Carrera 2003.
| Methods |
|
|
| Participants |
|
|
| Interventions | Treatment group 1
Treatment group 2
Co‐interventions
|
|
| Outcomes |
|
|
| Notes | ||
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Study stated to be randomised but no other information provided: "randomised study" |
| Allocation concealment (selection bias) | Unclear risk | Study stated to be randomised but no other information provided |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | Primary outcome was laboratory based and not likely to be influenced by blinding. Study participants and personnel not blinded |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | Primary outcome was laboratory based and not likely to be influenced by blinding. Study participants and personnel not blinded |
| Incomplete outcome data (attrition bias) All outcomes | High risk | Data only reported on patients, who completed study. 13 (29.5%) did not complete the study; DA group: died (2), transplants (2), surgery (2); rHuEPO group: died (3), transplant (1), surgery (3) |
| Selective reporting (reporting bias) | Low risk | Study protocol not available but published results include all expected outcomes |
| Other bias | Unclear risk | Funding: NS |
Coyne 2000.
| Methods |
|
|
| Participants |
|
|
| Interventions | Treatment group 1
Treatment group 2
Other information
Co‐interventions
|
|
| Outcomes |
|
|
| Notes |
|
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | "Randomization was centrally performed using an IVRS system" (information from author) |
| Allocation concealment (selection bias) | Low risk | "Randomization was centrally performed using an IVRS system" (information from author) |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | Primary outcome was laboratory based and not likely to be influenced by blinding |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | Primary outcome was laboratory based and not likely to be influenced by blinding |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk | NR; data provided as % and unclear whether all patients completed study |
| Selective reporting (reporting bias) | Unclear risk | Abstract only, not all results available |
| Other bias | Unclear risk | NS |
Coyne 2006a.
| Methods |
|
|
| Participants |
|
|
| Interventions | Treatment group 1
Treatment group 2
Other information
Co‐interventions
|
|
| Outcomes |
|
|
| Notes |
|
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | "Randomization was centrally performed using an IVRS system" (information from author) |
| Allocation concealment (selection bias) | Low risk | "Randomization was centrally performed using an IVRS system" (information from author) |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | DA group received placebo IV twice weekly |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | DA group received placebo IV twice weekly |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | 406/407 included in safety analysis. 363, who had at least one evaluation Hb, were included in efficacy analysis |
| Selective reporting (reporting bias) | Low risk | Includes expected outcomes |
| Other bias | High risk | Grant/research support: Amgen |
Frifelt 1996.
| Methods |
|
|
| Participants |
|
|
| Interventions | Treatment group 1
Treatment group 2
Other information
Co‐interventions
|
|
| Outcomes |
|
|
| Notes | ||
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | "randomised, prospective study" |
| Allocation concealment (selection bias) | Unclear risk | Not mentioned |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | Primary outcome was laboratory based and not likely to be influenced by blinding |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | Primary outcome was laboratory based and not likely to be influenced by blinding |
| Incomplete outcome data (attrition bias) All outcomes | High risk | 15% (6/39) excluded and missing data could have influenced final result; peritonitis (4); cerebral ischaemia (1); death due to AMI (1) |
| Selective reporting (reporting bias) | High risk | No reports on mortality and incomplete reporting of adverse effects. Results available only as median and IQR and cannot be entered in meta‐analyses |
| Other bias | High risk | Funded study by Ercopharm, Kvistgaard, Denmark |
Hori 2004.
| Methods |
|
|
| Participants |
|
|
| Interventions | Treatment group 1
Treatment group 2
Co‐interventions
|
|
| Outcomes |
|
|
| Notes |
|
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Study stated to be randomised but no further information given |
| Allocation concealment (selection bias) | Unclear risk | Information not provided |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | Primary outcome was laboratory based and unlikely to be influenced by blinding |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | Primary outcome was laboratory based and unlikely to be influenced by blinding |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk | No information on completeness of follow‐up |
| Selective reporting (reporting bias) | Unclear risk | Abstract only |
| Other bias | High risk | Funding Kirin Brewery Company |
Kwan 2005.
| Methods |
|
|
| Participants |
|
|
| Interventions | Treatment group 1
Treatment group 2
Other information
Co‐interventions
|
|
| Outcomes |
|
|
| Notes |
|
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Insufficient information about sequence generation process to permit judgement |
| Allocation concealment (selection bias) | Unclear risk | Randomisation stated but no information on method used is available |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | Not blinded but primary outcome is laboratory based and unlikely to be subject to bias |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | No blinding of outcome assessment, and outcome unlikely to be influenced by lack of blinding |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk | Insufficient information to permit judgement; 9 (14%) excluded from evaluation but group and reasons not stated |
| Selective reporting (reporting bias) | Unclear risk | Insufficient information to permit judgement |
| Other bias | High risk | Grant/research support (Kirin). Drug supplied by Kirin |
Lago 1996.
| Methods |
|
|
| Participants |
|
|
| Interventions | Treatment group 1
Treatment group 2
Other information
Co‐interventions
|
|
| Outcomes |
|
|
| Notes |
|
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Insufficient information about the sequence generation process to permit judgement |
| Allocation concealment (selection bias) | Unclear risk | Study stated to be randomised but no further information given |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | No blinding, but outcome unlikely to be influenced by lack of blinding |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | No blinding of outcome, but unlikely to be influenced by lack of blinding |
| Incomplete outcome data (attrition bias) All outcomes | High risk | 6/21 in experimental group and 2/9 in control group did not complete study |
| Selective reporting (reporting bias) | Unclear risk | Letter only. Only Hb levels reported |
| Other bias | Unclear risk | No mention of funding source |
Lee 2008.
| Methods |
|
|
| Participants |
|
|
| Interventions | Treatment group 1
Treatment group 2
Other information
Co‐interventions
|
|
| Outcomes |
|
|
| Notes | ||
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Insufficient information about sequence generation process to permit judgement |
| Allocation concealment (selection bias) | Low risk | Randomised by central randomisation. Stratified for EPO dose |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | Open‐label study but primary outcome was laboratory measure and unlikely to be subject to bias |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | Open‐label study but primary outcome was laboratory measure and unlikely to be subject to bias |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Reported ITT and PP data. Loss to follow‐up/withdrawal post randomisation: 4 in each group (protocol violation; dose change in study period; Hb ≥ 12 at randomisation). Loss to follow‐up unlikely to influence results |
| Selective reporting (reporting bias) | High risk | No information on mortality and incomplete information on adverse effects |
| Other bias | High risk | Supported/funded by LG Life Sciences Co Ltd |
Leung 1995.
| Methods |
|
|
| Participants |
|
|
| Interventions | Treatment group 1
Treatment group 2
Other information
Co‐interventions
|
|
| Outcomes |
|
|
| Notes |
|
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Insufficient information about sequence generation to permit judgement |
| Allocation concealment (selection bias) | Unclear risk | Randomisation stated but no information on method used is available |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | Primary outcome was laboratory measure and unlikely to be subject to bias |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | Primary outcome was laboratory measure and unlikely to be subject to bias |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Missing data unlikely to influence results; 2/20 in each group, no reasons given. 10% participants excluded from analysis |
| Selective reporting (reporting bias) | Unclear risk | No information provided on mortality and incomplete information on adverse effects. All data provided without standard deviations so cannot be included in meta‐analyses |
| Other bias | Unclear risk | Funding sources not mentioned |
Locatelli 2002.
| Methods |
|
|
| Participants |
|
|
| Interventions | Treatment group 1
Treatment group 2
Other information
Co‐interventions
|
|
| Outcomes |
|
|
| Notes | ||
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Stratified by centre, but no further information provided |
| Allocation concealment (selection bias) | Low risk | Central randomisation |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | Open‐label study but primary outcome was laboratory measure and unlikely to be subject to bias |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | Open‐label study but primary outcome was laboratory measure and unlikely to be subject to bias |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | 16% (27/173) lost to follow‐up or withdrawn but reasons for missing data unlikely to be related to true outcome: death (6), adverse events (5), protocol violations (4), improvement in anaemia (2), refusal of treatment (2), administrative reasons (7), loss to follow‐up (1) |
| Selective reporting (reporting bias) | High risk | Reported outcomes consistent with expected but data not provided in format that can be entered in meta‐analysis |
| Other bias | High risk | Supported in part by Hoffmann‐La Roche Ltd, Switzerland |
Locatelli 2004.
| Methods |
|
|
| Participants |
|
|
| Interventions | Treatment group 1
Treatment group 2
Other information
Co‐interventions
|
|
| Outcomes |
|
|
| Notes |
|
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Sequence generation not stated. Stratified by centre according to EPO dose "randomly assigned" |
| Allocation concealment (selection bias) | Unclear risk | Randomisation stated but no information on method used available |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | Blinding maintained by placebo injections weekly in patients on 2 weekly regimen. Unclear whether both participants and personnel blinded. Primary outcome is laboratory measurement |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | Blinding maintained by placebo injections weekly in patients on 2 weekly regimen |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk | Unclear. Abstracts only; 213/307 (69%) were included in primary analysis; deaths (19), adverse events (5) |
| Selective reporting (reporting bias) | High risk | Important outcomes provided but data for primary outcome only provided for PP population |
| Other bias | High risk | Funding Amgen Inc, Thousand Oaks, CA |
Lui 1991.
| Methods |
|
|
| Participants |
|
|
| Interventions | Treatment group 1
Treatment group 1
Other information
Co‐interventions
|
|
| Outcomes |
|
|
| Notes | ||
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Said to be randomised. No other information provided |
| Allocation concealment (selection bias) | Unclear risk | Said to be randomised. No other information provided |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | Laboratory outcome, blinding unlikely to influence outcome |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | Laboratory outcome, blinding unlikely to influence outcome |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Reasons for missing data unlikely to be related to true outcome; loss to follow‐up/withdrawal: 3/20 (15%) excluded; peritonitis (2), failure to respond to therapy (1) |
| Selective reporting (reporting bias) | High risk | No report of mortality and incomplete reporting of adverse events |
| Other bias | High risk | Epoetin supplied by Jansen Cilag. Supported by Cilag Research |
Lui 1992.
| Methods |
|
|
| Participants |
|
|
| Interventions | Treatment group 1
Treatment group 2
Other information
Co‐interventions
|
|
| Outcomes |
|
|
| Notes | ||
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Patients said to be randomised. No other information provided |
| Allocation concealment (selection bias) | Unclear risk | Patients said to be randomised. No other information provided |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | Laboratory outcome, blinding unlikely to influence outcome |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | Laboratory outcome, blinding unlikely to influence outcome |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Numbers and reasons for withdrawals stated and unlikely to affect results |
| Selective reporting (reporting bias) | High risk | No information on mortality and limited information on adverse effects |
| Other bias | High risk | Supported by Cilag Research |
MAXIMA Study 2007.
| Methods |
|
|
| Participants |
|
|
| Interventions | Treatment group 1
Treatment group 2
Treatment group 3
Other information
Co‐interventions
|
|
| Outcomes |
|
|
| Notes |
|
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Random computer numbers generated by computer at coordinating centre |
| Allocation concealment (selection bias) | Low risk | Randomisation at coordinating centre |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | Laboratory endpoint, therefore unlikely to be influenced by blinding |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | Laboratory endpoint, therefore unlikely to be influenced by blinding |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | All patients accounted; loss to follow‐up/excluded: adverse events (8), deaths (26), insufficient therapy (1), refused treatment (12), other (36), failure to return for follow‐up (2) |
| Selective reporting (reporting bias) | High risk | Expected outcomes reported but ITT data only shown in graph. PP data in text and tables |
| Other bias | High risk | Industry funded, Hoffman La Roche |
Miranda 1990.
| Methods |
|
|
| Participants |
|
|
| Interventions | Treatment group 1
Treatment group 2
Co‐interventions
|
|
| Outcomes |
|
|
| Notes |
|
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | NS |
| Allocation concealment (selection bias) | Unclear risk | Unclear |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | Laboratory outcome, unlikely to be influenced by blinding |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | Laboratory outcome, unlikely to be influenced by blinding |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk | Unclear whether all patients completed study |
| Selective reporting (reporting bias) | High risk | Information provided graphically or for combined groups of patients so could not be included in meta‐analyses |
| Other bias | Unclear risk | Funding sources NS |
Mircescu 2006.
| Methods |
|
|
| Participants |
|
|
| Interventions | Treatment group 1
Treatment group 2
Other information
Co‐interventions
|
|
| Outcomes |
|
|
| Notes | ||
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Renal Registry Romania. Numbered containers |
| Allocation concealment (selection bias) | Low risk | Central randomisation. Renal Registry Romania |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | No blinding but outcome of Hb unlikely to be influenced by lack of blinding |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | No blinding but outcome of Hb unlikely to be influenced by lack of blinding |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | 4/207 excluded from analyses (4 withdrew after randomisation (2 from each group)), unlikely to influence results |
| Selective reporting (reporting bias) | High risk | No information on mortality |
| Other bias | High risk | Funding for laboratory tests from F. Hoffman la Roche Ltd |
Muirhead 1989.
| Methods |
|
|
| Participants |
|
|
| Interventions | Treatment group 1
Treatment group 2
Co‐interventions
|
|
| Outcomes |
|
|
| Notes |
|
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | NS |
| Allocation concealment (selection bias) | Unclear risk | NS |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | Placebo given to weekly rHuEPO group. Patients and staff did not know frequency |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | Unclear if outcome assessors blinded but outcome of Hb unlikely to be influenced by lack of blinding |
| Incomplete outcome data (attrition bias) All outcomes | High risk | 31% did not complete the study |
| Selective reporting (reporting bias) | High risk | Data only available for combined groups |
| Other bias | Unclear risk | NS |
Murtagh 2000.
| Methods |
|
|
| Participants |
|
|
| Interventions | Treatment group 1
Treatment group 2
Co‐interventions
|
|
| Outcomes |
|
|
| Notes |
|
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | NS |
| Allocation concealment (selection bias) | Unclear risk | NS |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | No blinding but outcome of Hb unlikely to be influenced by lack of blinding |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | No blinding but outcome of Hb unlikely to be influenced by lack of blinding |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk | All 10 patients completed the initial 16 weeks |
| Selective reporting (reporting bias) | High risk | Data only provided for combined groups |
| Other bias | Unclear risk | Funding source: NS |
Nagaya 2010.
| Methods |
|
|
| Participants |
|
|
| Interventions | Treatment group 1
Treatment group 2
Co‐interventions
|
|
| Outcomes |
|
|
| Notes | ||
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Insufficient information about sequence generation process to permit judgement "prospective and randomised" |
| Allocation concealment (selection bias) | Unclear risk | Study stated to be randomised but further information not given |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | No blinding but outcome is unlikely to be influenced by lack of blinding |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | No blinding of outcome assessment, but outcome assessment unlikely to be influenced by lack of blinding |
| Incomplete outcome data (attrition bias) All outcomes | High risk | Patients excluded: 48 randomised, PP 39 (loss to follow‐up/withdrawal: 9/48 (18.8%); surgery (4), transfer to another institution (2), death (10), haematologic disease (1), nasal bleeding (1); missing data may have influenced outcome |
| Selective reporting (reporting bias) | High risk | Insufficient information on adverse effects |
| Other bias | Low risk | Grant from Japan Dialysis outcome Research Group |
Nissenson 2002.
| Methods |
|
|
| Participants |
|
|
| Interventions | Treatment group 1
Treatment group 2
Other information
Co‐interventions
|
|
| Outcomes |
|
|
| Notes | ||
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Stratified by centre. No information provided on sequence generation |
| Allocation concealment (selection bias) | Low risk | Central computer allocation |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | DA group received placebo twice per week |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | Placebo‐controlled. Primary outcome was laboratory‐based and unlikely to be influenced by lack of blinding |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | 4/507 excluded from ITT analysis and unlikely to influence results; loss to follow‐up/withdrawal: 81/507. Protocol violation (1), intolerable AE (24), withdrawal requested (11), death (28), kidney transplant (15), administration decision (2), change in dialysis modality (1) |
| Selective reporting (reporting bias) | High risk | Information provided on all expected outcomes but ITT data available from figures and could not be included in meta‐analyses |
| Other bias | High risk | Supported by Amgen |
Paganini 1991.
| Methods |
|
|
| Participants |
|
|
| Interventions | Treatment group 1
Treatment group 2
Co‐interventions
|
|
| Outcomes |
|
|
| Notes |
|
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | NS |
| Allocation concealment (selection bias) | Unclear risk | NS |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | No blinding, unlikely to be influenced by lack of blinding |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | No blinding but outcome of Hb unlikely to be influenced by lack of blinding |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk | Unclear if all patients completed the study |
| Selective reporting (reporting bias) | Unclear risk | No report of mortality or adverse effects |
| Other bias | Unclear risk | No information |
PROTOS Study 2007.
| Methods |
|
|
| Participants |
|
|
| Interventions | Treatment group 1
Treatment group 2
Treatment group 3
Other information
Co‐interventions
|
|
| Outcomes |
|
|
| Notes | ||
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Central randomisation with geographic stratification |
| Allocation concealment (selection bias) | Low risk | Central randomisation |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | Open‐label study but laboratory endpoint unlikely to be influenced by lack of blinding |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | Open‐label study but laboratory endpoint unlikely to be influenced by lack of blinding |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | All participants accounted |
| Selective reporting (reporting bias) | High risk | Data provided for PP not ITT population. ITT data only available graphically so PP data only included in meta‐analyses (98 excluded from ITT population). Withdrawal/loss to follow‐up: 111/572. Death (41), kidney transplantation (39), treatment refusal (13), AEs (2), insufficient therapeutic response (2), other (13) |
| Other bias | High risk | Funding F. Hoffman La Roche Ltd |
RUBRA Study 2008.
| Methods |
|
|
| Participants |
|
|
| Interventions | Treatment group 1
Treatment group 2
Other information
Co‐interventions
|
|
| Outcomes |
|
|
| Notes |
|
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | NS |
| Allocation concealment (selection bias) | Unclear risk | Stratified by geographical region and route of administration |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | No blinding but outcome of Hb unlikely to be influenced by lack of blinding |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | No blinding but outcome of Hb unlikely to be influenced by lack of blinding |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | All patients accounted; 54/282 (19%) withdrew due to AEs (4), death (16), transplantation (16), insufficient therapeutic response (1), refusal of treatment (1), failure to return (1), other (12) |
| Selective reporting (reporting bias) | High risk | All important outcomes provided. ITT data only available graphically and PP data entered in meta‐analyses |
| Other bias | High risk | Funded by Roche |
STRIATA Study 2008.
| Methods |
|
|
| Participants |
|
|
| Interventions | Treatment group 1
Treatment group 2
Other information
Co‐interventions
|
|
| Outcomes |
|
|
| Notes |
|
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Randomization numbers "allocated sequentially for each study centre" |
| Allocation concealment (selection bias) | Low risk | "Central randomization centre" |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | No blinding but outcome of Hb unlikely to be influenced by lack of blinding |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | No blinding but outcome of Hb unlikely to be influenced by lack of blinding |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | All patients accounted for. Loss to follow‐up/withdrawals: death (22), transplantation (20), refusal of treatment (7), adverse events (2), failure to return (2), patient vacation, patient decision, patient instability, protocol violation, discontinuation of dialysis (11) |
| Selective reporting (reporting bias) | High risk | ITT data only provided graphically and PP data entered in meta‐analyses |
| Other bias | High risk | Funded by F.Hoffman‐ La Roche Ltd. Basel Switzerland; data analyses conducted by sponsor |
Tessitore 2008.
| Methods |
|
|
| Participants |
|
|
| Interventions | Treatment group 1
Treatment group 2
Other information
Co‐interventions
|
|
| Outcomes |
|
|
| Notes |
|
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | NS |
| Allocation concealment (selection bias) | Low risk | Coin toss |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | No blinding but outcome of Hb unlikely to be influenced by lack of blinding |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | No blinding but outcome of Hb unlikely to be influenced by lack of blinding |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk | Unclear if all patients included in analysis |
| Selective reporting (reporting bias) | Unclear risk | Insufficient information to permit judgement |
| Other bias | Unclear risk | Information not provided |
Vanrenterghem 2002.
| Methods |
|
|
| Participants |
|
|
| Interventions | Treatment group 1
Treatment group 2
Co‐interventions
|
|
| Outcomes |
|
|
| Notes | ||
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | NS |
| Allocation concealment (selection bias) | Low risk | Central randomisation. Stratified by centre and EPO dose at study entry |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | No blinding but outcome of Hb unlikely to be influenced by lack of blinding |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | No blinding but outcome of Hb unlikely to be influenced by lack of blinding |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | All patients accounted; loss to follow‐up/withdrawals: 133 did not complete 52 weeks of study. Main reason was death (52), transplant and withdrawal requested; rates were similar between groups during evaluation period |
| Selective reporting (reporting bias) | High risk | Results provided for per protocol population with all results only provided graphically |
| Other bias | High risk | Amgen Inc funded study |
Weiss 2000.
| Methods |
|
|
| Participants |
|
|
| Interventions | Treatment group 1
Treatment group 2
Co‐interventions
|
|
| Outcomes |
|
|
| Notes | ||
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | NS |
| Allocation concealment (selection bias) | Low risk | Central randomisation 3:1 ratio. Stratified by sex and age |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | No blinding but outcome of Hb unlikely to be influenced by lack of blinding |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | No blinding but outcome of Hb unlikely to be influenced by lack of blinding |
| Incomplete outcome data (attrition bias) All outcomes | High risk | 25% withdrew from each group before evaluation; withdrawals 30 and 10 patients respectively in once weekly and control groups withdrew prior to week 16 |
| Selective reporting (reporting bias) | Low risk | Expected outcomes reported |
| Other bias | High risk | Funded by Amgen |
Yoon 2004.
| Methods |
|
|
| Participants |
|
|
| Interventions | Treatment group 1
Treatment group 2
Other information
Co‐interventions
|
|
| Outcomes |
|
|
| Notes |
|
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | NS |
| Allocation concealment (selection bias) | Unclear risk | NS |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | No blinding but outcome of Hb unlikely to be influenced by lack of blinding |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | No blinding but outcome of Hb unlikely to be influenced by lack of blinding |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk | Insufficient data to enable assessment |
| Selective reporting (reporting bias) | Unclear risk | Insufficient data to enable assessment |
| Other bias | Unclear risk | Funding: NS |
AE, adverse event; AMI ‐ acute myocardial infarction; AUC ‐ area under the curve; AV ‐ arteriovenous; BP ‐ blood pressure; CAD ‐ coronary artery disease; CAPD ‐ continuous ambulatory peritoneal dialysis; CCF ‐ chronic coronary failure; CKD ‐ chronic kidney disease; CHF ‐ coronary heart failure; CRP ‐ C‐reactive protein; DA ‐ darbepoetin; DBP ‐ diastolic blood pressure; ESA ‐ erythropoietin‐stimulating agent; ESKD ‐ end‐stage kidney disease; GI ‐ gastrointestinal; Hb ‐ haemoglobin; HCT ‐ haematocrit; HD ‐ haemodialysis; ITT ‐ intention‐to‐treat analysis; IV ‐ intravenous; IVI ‐ intravenous injection; IVRS ‐ interactive voice response system; MI ‐ myocardial infarction; NS ‐ not stated; PD ‐ peritoneal dialysis; PP ‐ per protocol analysis; PRCA ‐ pure red cell aplasia; RA ‐ rheumatoid arthritis; RBC ‐ red blood cell; SAE ‐ serious adverse event; SC ‐ subcutaneous; SLE ‐ systemic lupus erythematosus; TSAT ‐ transferrin saturation; URR ‐urea reduction ratio
Characteristics of excluded studies [ordered by study ID]
| Study | Reason for exclusion |
|---|---|
| Allon 2002 | Pharmacokinetic study |
| BA16260 Study 2006 | Dose finding study |
| Bennett 1991 | Participants not on dialysis |
| Besarab 1998 | Haematocrit target study |
| Bhuiyan 2004 | Dose varied, not frequency |
| Brandt 1999 | Dose varied, not frequency |
| Brown 1988 | Haematocrit target study |
| Canadian EPO Study 1990 | Injection frequency was not randomised; participants were randomised to placebo (Group 1) and two groups maintaining Hb from 95 to 110 g/L (Group 2) or 115 to 130 g/L (Group 3) |
| Castro 1994 | Unclear how patients were allocated to groups; no reply from study author |
| Chazot 2009 | Dose study |
| Dougherty 2004 | Dose dependent study not on dialysis |
| Fan 1992 | Unclear how patients were allocated to groups, wrote to authors, no response |
| Hirakata 2010 | Participants not on dialysis |
| Icardi 1990 | Different routes of administration (not frequency) |
| Ifudu 1998 | No control group, no change in frequency |
| Iwasaki 2008 | Examined conversion ratios for EPO to darbepoetin |
| Kawanishi 2005 | Dose escalation study |
| Kim 2009a | Compared different routes of administration |
| Knebel 2008 | Pharmacokinetic study |
| Locatelli 2008 | Control group received either weekly darbepoetin or rHuEPO 2 to 3 times weekly; numbers receiving each control intervention could not be separated. |
| Macdougall 2003 | Dose escalation study |
| Macdougall 2006 | Participants not on dialysis |
| Macdougall 2007a | Study terminated by sponsor for commercial reasons. No results available |
| Martin 2007 | Dose only varied, not frequency |
| Moiz 2000 | Only varied dosage, not frequency |
| Muirhead 1992 | Injection frequency was not randomised; participants randomised to placebo (Group 1) and two groups maintaining Hb from 95 to 110 g/L (Group 2) or 115 to 130 g/L (Group 3) |
| Nissenson 1995 | Safety and efficacy study of EPO vs. placebo |
| Parfrey 2005 | Hb target study |
| PATRONUS Study 2009 | Compared CERA and darbepoetin at same dose intervals |
| Pawlak 2007 | Study of metalloproteinases with ESA |
| Provenzano 2006 | Described two phase‐2 studies. Dialysis and non‐dialysis patients not separated |
| Raftery 2000 | Not RCT |
| Schmitt 2006 | Not relevant. Focus on pain |
| Smith 2007 | Pharmacokinetic study |
| Smyth 2006 | Not a frequency study |
| Spaia 1995 | No mention of randomisation. Changed both dose and frequency |
| Spinowitz 2006 | Dosage, no change in frequency |
| Stockenhuber 1990 | Study of EPO effect on stem cells |
| Tan 1990a | Not a frequency study |
| Tolman 2005 | Computer‐assisted anaemia management |
| Wang 2000 | Not clear how patients were allocated to treatment. Cross‐over study, wrote to authors to establish if patients were randomised ‐ no reply |
| Yalcinkaya 1997 | Primary randomisation was to two different doses of rHuEPO |
Characteristics of studies awaiting assessment [ordered by study ID]
EMERALD 1 Study 2013.
| Methods |
|
| Participants | Inclusion criteria
Exclusion criteria
|
| Interventions | Peginesatide
Epoetin alfa
|
| Outcomes |
|
| Notes |
EMERALD 2 Study 2013.
| Methods |
|
| Participants | Inclusion criteria
Exclusion criteria
|
| Interventions | Peginesatide
Epoetin alfa or beta
|
| Outcomes |
|
| Notes |
CKD ‐ chronic kidney disease; ESA ‐ erythropoiesis stimulating agent; Hb ‐ haemoglobin; HD ‐ haemodialysis; IV ‐ intravenous; RBC ‐ red blood cell; RCT ‐ randomised controlled trial; SC ‐ subcutaneous
Characteristics of ongoing studies [ordered by study ID]
NCT00436748.
| Trial name or title | A multi‐center, double‐blind, randomized study evaluating de novo weekly and once every two week darbepoetin alfa dosing for the correction of anemia in pediatric subjects with chronic kidney disease receiving and not receiving dialysis |
| Methods |
|
| Participants | Countries: USA, Belgium, Latvia, Lithuania, Mexico, Poland, Puerto Rico, Russian Federation, Slovakia, UK Inclusion criteria
Exclusion criteria
|
| Interventions | Treatment group 1
Treatment group 2
|
| Outcomes | Primary outcome measures
Secondary outcome measures
|
| Starting date | August 2008 |
| Contact information | Amgen Call Centre |
| Notes |
NCT00717821.
| Trial name or title | A randomized, controlled, open label, french multicenter parallel group study to compare the hemoglobin maintenance with once monthly administration of mircera versus epoetin beta or darbepoetin alfa in patients with chronic kidney disease on hemodialysis |
| Methods |
|
| Participants | Inclusion criteria
Exclusion criteria
|
| Interventions | Treatment group 1
Treatment group 2
|
| Outcomes | Primary outcome measures
Secondary outcome measures
|
| Starting date | October 2008 |
| Contact information | Hoffmann‐La Roche |
| Notes |
CKD ‐ chronic kidney disease; ESA ‐ erythropoiesis stimulating agent; GI ‐ gastrointestinal; IV ‐ intravenous; RCT ‐ randomised controlled trial; SC ‐ subcutaneous; TSAT ‐ transferrin saturation
Differences between protocol and review
Newer agents have been added to the 2014 review update; methods updated; title changed to reflect new focus of the review.
Contributions of authors
Two reviewers (JC, CD) independently undertook study quality assessment For the initial review and first update. Data extraction was performed by the lead author (JC) and a sample was double‐checked by another author (CD). Sheila Wallace performed extensive literature searched. The lead author entered data and wrote the text of the review. Marion Campbell and Adrian Grant provided statistical and methodological advice. Alison MacLeod, Conal Daly and Izhar Khan provided clinical perspectives. Cam Donaldson and Luke Vale provided health economics perspectives. All authors commented on the review.
For this 2013 update two authors (DH, EH) undertook all stages of the review and wrote the text of the review.
Declarations of interest
This systematic review was initially one of six funded (in 1996) by Janssen Cilag who manufacture of Eprex (erythropoietin‐α) a recombinant human erythropoietin. Our contract stated that the University of Aberdeen owned the intellectual property rights and we had the right to publish the results without restriction.
No industry funding was sought for the updates of this review.
Edited (no change to conclusions)
References
References to studies included in this review
AMICUS Study 2007 {published data only}
- Chanu P, Gieschke R, Reigner B, Dougherty FC. Pharmacokinetic parameters of C.E.R.A. and are not affected by age in patients with chronic kidney disease on dialysis [abstract no: SaP351]. Nephrology Dialysis Transplantation 2007;22(Suppl 6):vi351. [CENTRAL: CN‐00757500] [Google Scholar]
- Chanu P, Gieschke R, Reigner B, Dougherty FC. Pharmacokinetics of C.E.R.A. and stable maintenance of haemoglobin (Hb) levels with once‐monthly dosing in patients with chronic kidney disease (CKD) [abstract no: SaP325]. Nephrology Dialysis Transplantation 2007;22(Suppl 6):vi342. [CENTRAL: CN‐00757502] [Google Scholar]
- Fishbane S, Dalton C, Beswick R, Dutka P, Schmidt R. Efficacy of C.E.R.A., a continuous erythropoietin receptor activator, in the treatment of renal anemia: overview of 6 global phase 3 trials [abstract no: 59]. American Journal of Kidney Diseases 2007;49(4):A39. [CENTRAL: CN‐00756571] [Google Scholar]
- Klinger M, Arias M, Vargemezis V, Besarab A, Sulowicz W, Gerntholtz T, et al. C.E.R.A. (Continuous Erythropoietin Receptor Activator) administered at extended intervals corrects Hb levels in patients with CKD on dialysis [abstract no: SA‐PO212]. Journal of the American Society of Nephrology 2006;17(Abstracts):620A. [CENTRAL: CN‐00644218] [Google Scholar]
- Klinger M, Arias M, Vargemezis V, Besarab A, Sulowicz W, Gerntholtz T, et al. Efficacy of intravenous methoxy polyethylene glycol‐epoetin beta administered every 2 weeks compared with epoetin administered 3 times weekly in patients treated by hemodialysis or peritoneal dialysis: a randomized trial. American Journal of Kidney Diseases 2007;50(6):989‐1000. [MEDLINE: ] [DOI] [PubMed] [Google Scholar]
- Provenzano R, Macdougall IC, Law A, Ouyang Y, Bexon M. Anemia correction with C.E.R.A. in patients (pts) with chronic kidney disease (CKD) is unaffected by baseline hemoglobin (Hb) level [abstract no: SU‐PO796]. Journal of the American Society of Nephrology 2007;18(Abstracts Issue):760A. [CENTRAL: CN‐00747311] [Google Scholar]
BA16285 Study 2007 {published data only}
- Besarab A, Beyer U, Dougherty FC. Long‐term intravenous CERA (Continuous Erythropoietin Receptor Activator) maintains hemoglobin concentrations in hemodialysis patients [abstract no: W‐PO40127]. Nephrology 2005;10(Suppl 1):A312‐3. [CENTRAL: CN‐00747326] [Google Scholar]
- Besarab A, Salifu MO, Lunde NM, Bansal V, Fishbane S, Dougherty FC, et al. Efficacy and tolerability of intravenous continuous erythropoietin receptor activator: a 19‐week, phase II, multicenter, randomized, open‐label, dose‐finding study with a 12‐month extension phase in patients with chronic renal disease. Clinical Therapeutics 2007;29(4):626‐39. [MEDLINE: ] [DOI] [PubMed] [Google Scholar]
- Dougherty FC, Beyer U. No changes in blood pressure in dialysis patients after 12 months of treatment with IV/SC CERA (Continuous Erythropoietin Receptor Activator) [abstract no: W‐PO40131]. Nephrology 2005;10(Suppl 1):A313‐4. [CENTRAL: CN‐00602138] [Google Scholar]
- Dougherty FC, Beyer U. Safety and tolerability profile of continuous erythropoietin receptor activator (CERA) with extended dosing intervals in patients with chronic kidney disease on dialysis [abstract no: W‐PO40130]. Nephrology 2005;10(Suppl 1):A313. [CENTRAL: CN‐00602137] [Google Scholar]
- Dougherty FC, Loghman‐Adham M, Schultze N, Beyer U. Adequate hemoglobin levels are maintained with continuous erythropoietin receptor activator (CERA) in dialysis patients regardless of gender, age, race and diabetic status [abstract no: MP206]. Nephrology Dialysis Transplantation 2005;20(Suppl 5):v269. [CENTRAL: CN‐00602143] [Google Scholar]
- Dutka P, Tilocca P, BA16285 and BA16286 Study Groups. CERA (Continuous Erythropoietin Receptor Activator) maintains stable hemoglobin concentrations in dialysis patients irrespective of gender, age, race or diabetic status [abstract]. Nephrology Nursing Journal 2006;33(2):138. [CENTRAL: CN‐00602144] [Google Scholar]
- Salifu M, Villa G, Dougherty FC. Adequate hemoglobin levels are maintained with continuous erythropoietin receptor activator (CERA) in dialysis patients with different ranges of iron status and pre‐existing conditions [abstract no: 138]. American Journal of Kidney Diseases 2006;47(4):A53. [CENTRAL: CN‐00602141] [Google Scholar]
BA16286 Study 2007 {published data only}
- Dougherty FC, Beyer U. No changes in blood pressure in dialysis patients after 12 months of treatment with IV/SC CERA (Continuous Erythropoietin Receptor Activator) [abstract no: W‐PO40131]. Nephrology 2005;10(Suppl 1):A313‐4. [CENTRAL: CN‐00602138] [Google Scholar]
- Dougherty FC, Beyer U. Safety and tolerability profile of continuous erythropoietin receptor activator (CERA) with extended dosing intervals in patients with chronic kidney disease on dialysis [abstract no: W‐PO40130]. Nephrology 2005;10(Suppl 1):A313. [CENTRAL: CN‐00602137] [Google Scholar]
- Dougherty FC, Loghman‐Adham M, Schultze N, Beyer U. Adequate hemoglobin levels are maintained with continuous erythropoietin receptor activator (CERA) in dialysis patients regardless of gender, age, race and diabetic status [abstract no: MP206]. Nephrology Dialysis Transplantation 2005;20(Suppl 5):v269. [CENTRAL: CN‐00602143] [Google Scholar]
- Dutka P, Tilocca P, BA16285 and BA16286 Study Groups. CERA (Continuous Erythropoietin Receptor Activator) maintains stable hemoglobin concentrations in dialysis patients irrespective of gender, age, race or diabetic status [abstract]. Nephrology Nursing Journal 2006;33(2):138. [CENTRAL: CN‐00602144] [Google Scholar]
- Locatelli F, Villa G, Arias M, Marchesi D, Dougherty FC, Beyer U, et al. CERA (Continuous Erythropoietin receptor activator) maintains hemoglobin levels in dialysis patients when administered subcutaneously up to once every 4 weeks. [abstract no: SU‐PO051]. Journal of the American Society of Nephrology 2004;15(Oct):543A. [CENTRAL: CN‐00626008] [Google Scholar]
- Locatelli F, Villa G, Beyer U, Dougherty FC. Subcutaneous CERA (Continuous Erythropoietin Receptor Activator) maintains hemoglobin concentrations with dosing intervals up to 4 weeks in dialysis patients [abstract no: MP183]. Nephrology Dialysis Transplantation 2005;20(Suppl 5):v261. [CENTRAL: CN‐00602142] [Google Scholar]
- Locatelli F, Villa G, Francisco AL, Albertazzi A, Adrogue HJ, Dougherty FC, et al. Effect of a continuous erythropoietin receptor activator (C.E.R.A.) on stable haemoglobin in patients with CKD on dialysis: once monthly administration. Current Medical Research & Opinion 2007;23(5):969‐79. [MEDLINE: ] [DOI] [PubMed] [Google Scholar]
- Salifu M, Villa G, Dougherty FC. Adequate hemoglobin levels are maintained with continuous erythropoietin receptor activator (CERA) in dialysis patients with different ranges of iron status and pre‐existing conditions. [abstract no: 138]. American Journal of Kidney Diseases 2006;47(4):A53. [CENTRAL: CN‐00602141] [Google Scholar]
Brahm 1999 {published data only}
- Brahm M. Subcutaneous treatment with recombinant human erythropoietin‐‐the influence of injection frequency and skin‐fold thickness. Scandinavian Journal of Urology & Nephrology 1999;33(3):192‐6. [MEDLINE: ] [DOI] [PubMed] [Google Scholar]
Canaud 1995 {published data only}
- Canaud B, Bennhold I, Delons S, Donnadieu P, Foret M, Franz H, et al. What is the optimum frequency of administration of r‐HuEPO for correcting anemia in hemodialysis patients?. Dialysis & Transplantation 1995;24(6):306‐29. [EMBASE: 1995169525] [Google Scholar]
Carrera 2003 {published data only}
- Carrera F, Anunciada AI, Nogueira C, Silva JG. Comparison of HB levels in dialysis patients receiving three‐times weekly rHuepo switched to once‐weekly darbepoetin alfa: results of a randomized study [abstract]. Nephrology Dialysis Transplantation 2003;18(Suppl 4):164. [CENTRAL: CN‐00444694] [Google Scholar]
Coyne 2000 {published data only}
- Coyne DW, Ling BN, Toto R, McDermott‐Vitak AD, Trotman ML, Jackson L. Novel erythropoiesis stimulating protein (NESP) corrects anemia in dialysis patients when administered at reduced dose frequency compared with recombinant‐human erythropoietin (r‐HuEPO) [abstract no:1380]. Journal of the American Society of Nephrology 2000;11(Sept):263A. [CENTRAL: CN‐00583382] [Google Scholar]
Coyne 2006a {published data only}
- Coyne D, Zeig R, Benz R, Berns J, Varma N, Nakanishi A, et al. A randomized, double‐blind study comparing darbepoetin alfa and recombinant human erythropoietin (rHuEPO) in the treatment of anemia in African‐American (AA) subjects with chronic kidney disease (CKD) receiving hemodialysis (HD) [abstract no: TH‐PO365]. Journal of the American Society of Nephrology 2006;17(Abstracts):184A. [CENTRAL: CN‐00740574] [Google Scholar]
Frifelt 1996 {published data only}
- Frifelt JJ, Tvedegaard E, Bruun K, Steffensen G, Cintin C, Breddam M, et al. Efficacy of recombinant human erythropoietin administered subcutaneously to CAPD patients once weekly. Peritoneal Dialysis International 1996;16(6):594‐8. [MEDLINE: ] [PubMed] [Google Scholar]
Hori 2004 {published data only}
- Hori K, Tsujimoto Y, Ohmori H, Nakamura H, Suga A, Iwasaki M, et al. Randomized, double‐blind, comparative study of intravenous KRN321 (darbepoetin alfa) compared to intravenous recombinant human erythropoietin (rHuEPO) for treatment of anemia in subjects with chronic renal failure (CRF) receiving hemodialysis in Japan [abstract no: F‐PO502]. Journal of the American Society of Nephrology 2004;15(Oct):177A. [CENTRAL: CN‐00740529] [Google Scholar]
Kwan 2005 {published data only}
- Kwan BC, Leung C, Szeto C, Li PK. Efficacy of bi‐weekly versus monthly subcutaneous darbepoetin‐alpha for the maintenance treatment of anemia in peritoneal dialysis patients ‐ an open‐label randomized study [abstract no: SA‐PO805]. Journal of the American Society of Nephrology 2005;16:732A. [CN‐00740570] [Google Scholar]
Lago 1996 {published data only}
- Lago M, Perez‐Garcia R, Garcia De Vinuesa MS, Anaya F, Valderrabano F. Efficiency of once‐weekly subcutaneous administration of recombinant human erythropoietin versus three times a week administration in hemodialysis patients. Nephron 1996;72(4):723‐4. [MEDLINE: ] [DOI] [PubMed] [Google Scholar]
Lee 2008 {published data only}
- Lee YK, Kim SG, Seo JW, Oh JE, Yoon JW, Koo JR, et al. A comparison between once‐weekly and twice‐ or thrice‐weekly subcutaneous injection of epoetin alfa: results from a randomized controlled multicentre study. Nephrology Dialysis Transplantation 2008;23(10):3240‐6. [MEDLINE: ] [DOI] [PubMed] [Google Scholar]
Leung 1995 {published data only}
- Leung CB, Lam TY, Chan CM, Wang A, Li P, Lai KN, et al. Pharmacodynamics of twice weekly versus thrice weekly subcutaneous administration of recombinant human erythropoietin (rHuEPO) for patients on CAPD [abstract]. 6th Asian Pacific Congress of Nephrology; 1995 Dec 5‐9; Hong Kong. 1995:179. [CENTRAL: CN‐00461161]
Locatelli 2002 {published data only}
- Locatelli F, Baldamus C, Villa G, Ganea A, Francisco AM. A rationale for an individualized administration frequency of epoetin beta: a pharmacological perspective. Nephrology Dialysis Transplantation 2002;17 Suppl 6:13‐6. [MEDLINE: ] [DOI] [PubMed] [Google Scholar]
- Locatelli F, Baldamus CA, Villa G, Ganea A, Martin de Francisco AL. Once‐weekly compared with three‐times‐weekly subcutaneous epoetin beta: results from a randomized, multicenter, therapeutic‐equivalence study. American Journal of Kidney Diseases 2002;40(1):119‐25. [MEDLINE: ] [DOI] [PubMed] [Google Scholar]
- Locatelli F, Baldamus CA, Villa G, Martinez F, Francisco ALM, on behalf of the collaborative group. Therapeutic equivalence of once versus thrice weekly subcutaneous injection of epoetin beta in patients with chronic renal failure [abstract]. Nephrology Dialysis Transplantation 2001;16(6):A136. [CENTRAL: CN‐00446451] [Google Scholar]
Locatelli 2004 {published data only}
- Locatelli F, Backs W, Pino MD, Martin A, Guillaume B, Vikydal R, et al. Control of anaemia with Aranesp®: haemoglobin stability achieved with fewer patients requiring dose adjustments after switching to Aranesp® every 2‐weeks [abstract no: SP411]. Nephrology Dialysis Transplantation 2006;21(Suppl 4):iv152. [Google Scholar]
- Locatelli F, Villa G, Backs W, Pino MD. A phase 3, multicentre, randomised, double‐blind, non‐inferiority trial to evaluate the efficacy of Aranesp® (darbepoetin alfa) once every 2 weeks (Q2W) vs Aranesp® once weekly (QW) in patients on haemodialysis (HD) [abstract no: MP181]. Nephrology Dialysis Transplantation 2005;20(Suppl 5):v260. [Google Scholar]
- Locatelli F, Villa G, Backs W, Pino MD. Aranesp® (darbepoetin alfa) maintains hemoglobin (Hb) levels in hemodialysis (HD) patients at extended dosing intervals regardless of previous recombinant human erythropoietin (rHuEPO) dosing frequency. [abstract no: PUB254]. Journal of the American Society of Nephrology 2004;15(Oct):816A. [Google Scholar]
Lui 1991 {published data only}
- Lui SF, Law CB, Ting SM, Li P, Lai KN. Once weekly versus twice weekly subcutaneous administration of recombinant human erythropoietin in patients on continuous ambulatory peritoneal dialysis. Clinical Nephrology 1991;36(5):246‐51. [MEDLINE: ] [PubMed] [Google Scholar]
Lui 1992 {published data only}
- Lui SF, Wong KC, Li PK, Lai KN. Once weekly versus twice weekly subcutaneous administration of recombinant human erythropoietin in haemodialysis patients. American Journal of Nephrology 1992;12(1‐2):55‐60. [MEDLINE: ] [DOI] [PubMed] [Google Scholar]
MAXIMA Study 2007 {published data only}
- Barany P, Besarab A, Macdougall IC, Law A, Ouyang Y, Heifets M. Median hemoglobin (Hb) decline following C.E.R.A. dose interruption is similar to that with other erythropoiesis stimulating agents (ESAs) [abstract no: SU‐PO795]. Journal of the American Society of Nephrology 2007;18(Abstracts Issue):760A. [CENTRAL: CN‐00794522] [Google Scholar]
- Chanu P, Gieschke R, Reigner B, Dougherty FC. Pharmacokinetic parameters of C.E.R.A. and are not affected by age in patients with chronic kidney disease on dialysis [abstract no: SaP351]. Nephrology Dialysis Transplantation 2007;22(Suppl 6):vi351. [CENTRAL: CN‐00757500] [Google Scholar]
- Chanu P, Gieschke R, Reigner B, Dougherty FC. Pharmacokinetics of C.E.R.A. and stable maintenance of haemoglobin (Hb) levels with once‐monthly dosing in patients with chronic kidney disease (CKD) [abstract no: SaP325]. Nephrology Dialysis Transplantation 2007;22(Suppl 6):vi342. [CENTRAL: CN‐00757502] [Google Scholar]
- Fishbane S, Bernardo M, Locatelli F, delAguila M, Edwardes M, Bexon M. Once‐monthly intravenous (IV) C.E.R.A. maintains stable hemoglobin (Hb) in dialysis patients (pts), irrespective of age or gender [abstract no: SU‐PO779]. Journal of the American Society of Nephrology 2007;18(Abstracts Issue):756A. [CENTRAL: CN‐00757405] [Google Scholar]
- Fishbane S, Dalton C, Beswick R, Dutka P, Schmidt R. Efficacy of C.E.R.A., a continuous erythropoietin receptor activator, in the treatment of renal anemia: overview of 6 global phase 3 trials [abstract no: 59]. American Journal of Kidney Diseases 2007;49(4):A39. [CENTRAL: CN‐00756571] [Google Scholar]
- Fishbane S, Levin NW, Mann JF, Lewis JL, Bernardo M, Lunde NM, et al. IV C.E.R.A. (Continuous Erythropoietin Receptor Activator) once every 2 weeks or once monthly maintains stable Hb levels after converting directly from IV epoetin 1‐3 times per week in patients with CKD on dialysis [abstract no: SA‐PO205]. Journal of the American Society of Nephrology 2006;17(Abstracts):618A. [CENTRAL: CN‐00757097] [Google Scholar]
- Imbasciati E, Bernardo M, Caraman P, David‐Neto E, Harris K, Law A, et al. Stable haemoglobin (Hb) levels are maintained with once‐monthly C.E.R.A. in dialysis patients with varying C‐reactive protein (CRP), albumin or dialysis adequacy [abstract no: SuO005]. Nephrology Dialysis Transplantation 2007;22(Suppl 6):vi412. [CENTRAL: CN‐00758076] [Google Scholar]
- Levin NW, Fishbane S, Canedo FV, Zeig S, Nassar GM, Moran JE, et al. Intravenous methoxy polyethylene glycol‐epoetin beta for haemoglobin control in patients with chronic kidney disease who are on dialysis: a randomised non‐inferiority trial (MAXIMA). Lancet 2007;370(9596):1415‐21. [MEDLINE: ] [DOI] [PubMed] [Google Scholar]
- Levin NW, Imbasciati E, Combe C, Rocco MV, Lok CE, Donnelly SM, et al. Adequate Hb levels are maintained with IV C.E.R.A. (Continuous Erythropoietin Receptor Activator) administered up to once monthly in dialysis patients irrespective of age, gender or diabetic status [abstract no: SA‐PO206]. Journal of the American Society of Nephrology 2006;17(Abstracts):619A. [CENTRAL: CN‐00755210] [Google Scholar]
- Mann J, Locatelli F, Sulowicz W, Nissenson A, Portoles J, Levin N, et al. C.E.R.A. provides stable haemoglobin (Hb) levels in CKD patients on dialysis with and without coronary artery disease (CAD) or diabetes mellitus (DM) when administered once monthly [abstract no: SuO002]. Nephrology Dialysis Transplantation 2007;22(Suppl 6):vi411. [CENTRAL: CN‐00791257] [Google Scholar]
- Ryckelynck JP, Valdes Canedo F, Riella M, Lempert K, Donnelly S, Adrogue H, et al. Once‐monthly C.E.R.A. maintains stable haemoglobin concentrations in dialysis patients regardless of gender or age [abstract no: SUO001]. Nephrology Dialysis Transplantation 2007;22(Suppl 6):vi411. [CENTRAL: CN‐00644372] [Google Scholar]
Miranda 1990 {published data only}
- Miranda B, Selgas R, Rinon C, Fernandez‐Zamorano A, Borrego F, Ortuno F, et al. Treatment of the anemia with human recombinant erythropoietin in CAPD patients. Advances in Peritoneal Dialysis 1990;6:296‐301. [MEDLINE: ] [PubMed] [Google Scholar]
Mircescu 2006 {published data only}
- Covic A, Garneata L, Ciocalteu A, Golea O, Gherman‐Capriora M, Capsa D, et al. Are once‐fortnightly and once‐weekly epoetin beta regimens equivalent in haemodialyzed patients? [abstract no: SP407]. Nephrology Dialysis Transplantation 2006;21(Suppl 4):iv150. [CENTRAL: CN‐00583595] [Google Scholar]
- Mircescu G, Garneata L, Ciocalteu A, Golea O, Gherman M, Capsa D, et al. Haemoglobin variability in the maintenance phase of anaemia treatment in haemodialyzed patients ‐ does the epoetin dosing frequency matter? [abstract no: SP409]. Nephrology Dialysis Transplantation 2006;21(Suppl 4):iv151. [CENTRAL: CN‐00583597] [Google Scholar]
- Mircescu G, Garneata L, Ciocalteu A, Golea O, Gherman‐Caprioara M, Capsa D, et al. Once‐every‐2‐weeks and once‐weekly epoetin beta regimens: equivalency in hemodialyzed patients. American Journal of Kidney Diseases 2006;48(3):445‐55. [MEDLINE: ] [DOI] [PubMed] [Google Scholar]
Muirhead 1989 {published data only}
- Muirhead N, Keown PA, Slaughter D, Hodsman AB, Cordy PE, Clark WF, et al. A double‐blind, randomised dose‐finding study of recombinant human erythropoietin in the anaemia of chronic renal failure [abstract]. Nephrology Dialysis Transplantation 1989;4(5):477. [CENTRAL: CN‐00260451] [Google Scholar]
Murtagh 2000 {published data only}
- Murtagh H, Brown JH, Maxwell AP. Experience with novel erythropoiesis stimulating protein (NESP) in the treatment of anaemia in haemodialysis patients [abstract]. Irish Journal of Medical Science 2000;169(4):321. [CENTRAL: CN‐00740599] [Google Scholar]
Nagaya 2010 {published data only}
- Inaguma D, Nagaya H, Hamaguchi K, Tatematsu M, Suzuki S, Kurata K. Intravenous darbepoetin alfa once a week could maintain hemoglobin level more efficiently than once every 2 weeks in Japanese patients on hemodialysis [abstract no: TH‐PO741]. Journal of the American Society of Nephrology 2008;19(Abstracts Issue):275A. [CENTRAL: CN‐00740572] [DOI] [PubMed] [Google Scholar]
- Nagaya H, Inaguma D, Kitagawa A, Murata M, Kamimura Y, Hamaguchi K, et al. Intravenously administered darbepoetin alfa once a week could maintain hemoglobin level more efficiently than once every 2 weeks in patients on hemodialysis. Clinical & Experimental Nephrology 2010;14(2):158‐63. [MEDLINE: ] [DOI] [PubMed] [Google Scholar]
Nissenson 2002 {published data only}
- Nissenson A, Krishnan M, Liu W, McCary L, Stehman‐Breen C, Mix C. Hemoglobin (Hb) variability does not differ between hemodialysis (HD) patients treated with epoetin alfa and darbepoetin alfa [abstract no: TH‐PO360]. Journal of the American Society of Nephrology 2006;17(Abstracts):183A. [CENTRAL: CN‐00740539] [Google Scholar]
- Nissenson AR. Dosing darbepoetin alfa. American Journal of Kidney Diseases 2002 Oct;40(4):872. [MEDLINE: ] [DOI] [PubMed] [Google Scholar]
- Nissenson AR, Swan SK, Lindberg JS, Soroka SD, Beatey R, Wang C, et al. Randomized, controlled trial of darbepoetin alfa for the treatment of anemia in hemodialysis patients. American Journal of Kidney Diseases 2002;40(1):110‐8. [MEDLINE: ] [DOI] [PubMed] [Google Scholar]
- Nissenson AR, Swan SK, Lindberg JS, Soroka SD, McDermott‐Vitak AD, Wang C, et al. Novel Erythropoiesis stimulating protein (NESP) safely maintains hemoglobin concentration levels in hemodialysis patients as effectively as r‐HuEPO when administered once weekly [abstract no: A1326]. Journal of the American Society of Nephrology 2000;11(Sept):252A. [CENTRAL: CN‐00794722] [Google Scholar]
- Nissenson AR, US/Canadian NESP 980117 Study Group. Once weekly IV novel erythoropoiesis stimulating protein (NESP) effectively maintains Hb in hemodialysis patients [abstract]. Nephrology Dialysis Transplantation 2001;16(6):A92. [CENTRAL: CN‐00446969] [Google Scholar]
Paganini 1991 {published data only}
- Paganini EP, Miller T, Eschbach J, Gimenez LF, Graber S, Lazarus M, et al. A prospective randomized multi‐center study of subcutaneous erythropoietin administration [abstract]. Journal of the American Society of Nephrology 1991;2(3):54P. [Google Scholar]
PROTOS Study 2007 {published data only}
- Barany P, Besarab A, Macdougall IC, Law A, Ouyang Y, Heifets M. Median hemoglobin (Hb) decline following C.E.R.A. dose interruption is similar to that with other erythropoiesis stimulating agents (ESAs) [abstract no: SU‐PO795]. Journal of the American Society of Nephrology 2007;18(Abstracts Issue):760A. [CENTRAL: CN‐00794522] [Google Scholar]
- Chanu P, Gieschke R, Reigner B, Dougherty FC. Pharmacokinetic parameters of C.E.R.A. and are not affected by age in patients with chronic kidney disease on dialysis [abstract no: SaP351]. Nephrology Dialysis Transplantation 2007;22(Suppl 6):vi351. [CENTRAL: CN‐00757500] [Google Scholar]
- Chanu P, Gieschke R, Reigner B, Dougherty FC. Pharmacokinetics of C.E.R.A. and stable maintenance of haemoglobin (Hb) levels with once‐monthly dosing in patients with chronic kidney disease (CKD) [abstract no: SaP325]. Nephrology Dialysis Transplantation 2007;22(Suppl 6):vi342. [CENTRAL: CN‐00757502] [Google Scholar]
- Fishbane S, Dalton C, Beswick R, Dutka P, Schmidt R. Efficacy of C.E.R.A., a continuous erythropoietin receptor activator, in the treatment of renal anemia: overview of 6 global phase 3 trials [abstract no: 59]. American Journal of Kidney Diseases 2007;49(4):A39. [CENTRAL: CN‐00756571] [Google Scholar]
- Imbasciati E, Bernardo M, Caraman P, David‐Neto E, Harris K, Law A, et al. Stable haemoglobin (Hb) levels are maintained with once‐monthly C.E.R.A. in dialysis patients with varying C‐reactive protein (CRP), albumin or dialysis adequacy [abstract no: SuO005]. Nephrology Dialysis Transplantation 2007;22(Suppl 6):vi412. [CENTRAL: CN‐00758076] [Google Scholar]
- Locatelli F, Sulowicz W, Harris K, Selgas R, Kaufman J, Klinger M, et al. SC C.E.R.A. (Continuous Erythropoietin Receptor Activator) once every 2 weeks or once monthly maintains stable Hb levels after converting directly from SC epoetin 1‐3 times per week in patients with CKD on dialysis [abstract no: SA‐PO207]. Journal of the American Society of Nephrology 2006;17:619A. [CENTRAL: CN‐00626009] [Google Scholar]
- Mann J, Locatelli F, Sulowicz W, Nissenson A, Portoles J, Levin N, et al. C.E.R.A. provides stable haemoglobin (Hb) levels in CKD patients on dialysis with and without coronary artery disease (CAD) or diabetes mellitus (DM) when administered once monthly [abstract no: SuO002]. Nephrology Dialysis Transplantation 2007;22(Suppl 6):vi411. [CENTRAL: CN‐00791257] [Google Scholar]
- Ryckelynck JP, Harris K, Selgas R, Stompor T, Ladanyi E, Opatrna S, et al. SC C.E.R.A. (Continuous Erythropoietin Receptor Activator) administered up to once monthly in patients with CKD on dialysis maintains adequate Hb levels regardless of age, gender or diabetic status [abstract no: SA‐PO210]. Journal of the American Society of Nephrology 2006;17(Abstracts):620A. [CENTRAL: CN‐00644371] [Google Scholar]
- Ryckelynck JP, Valdes Canedo F, Riella M, Lempert K, Donnelly S, Adrogue H, et al. Once‐monthly C.E.R.A. maintains stable haemoglobin concentrations in dialysis patients regardless of gender or age [abstract no: SUO001]. Nephrology Dialysis Transplantation 2007;22(Suppl 6):vi411. [CENTRAL: CN‐00644372] [Google Scholar]
- Sulowicz W, Locatelli F, Balla J, Csiky B, Rikker C, Aldigier J, et al. Subcutaneous (SC) C.E.R.A. (Continuous Erythropoietin Receptor Activator) administered once every 2 weeks or once monthly maintains haemoglobin (Hb) levels in patients with chronic kidney disease (CKD) on dialysis [abstract no: SP424]. Nephrology Dialysis Transplantation 2006;21(Suppl 4):iv156‐7. [CENTRAL: CN‐00601914] [Google Scholar]
- Sulowicz W, Locatelli F, Ryckelynck JP, Balla J, Csiky B, Harris K, et al. Once‐monthly subcutaneous C.E.R.A. maintains stable hemoglobin control in patients with chronic kidney disease on dialysis and converted directly from epoetin one to three times weekly. Clinical Journal of the American Society of Nephrology: CJASN 2007;2(4):637‐46. [MEDLINE: ] [DOI] [PubMed] [Google Scholar]
RUBRA Study 2008 {published data only}
- Barany P, Besarab A, Macdougall IC, Law A, Ouyang Y, Heifets M. Median hemoglobin (Hb) decline following C.E.R.A. dose interruption is similar to that with other erythropoiesis stimulating agents (ESAs) [abstract no: SU‐PO795]. Journal of the American Society of Nephrology 2007;18(Abstracts Issue):760A. [CENTRAL: CN‐00794522] [Google Scholar]
- Fishbane S, Dalton C, Beswick R, Dutka P, Schmidt R. Efficacy of C.E.R.A., a continuous erythropoietin receptor activator, in the treatment of renal anemia: overview of 6 global phase 3 trials [abstract no: 59]. American Journal of Kidney Diseases 2007;49(4):A39. [CENTRAL: CN‐00756571] [Google Scholar]
- Spinowitz B, Coyne DW, Fraticelli M, Azer M, Dalal S, Villa G, et al. CERA (continuous erythropoietin receptor activator) administered once every 2 weeks via pre‐filled syringe (PFS) maintains stable Hb levels in patients with CKD on dialysis [abstract no: PUB376]. Journal of the American Society of Nephrology 2006;17(Abstracts):895A. [CENTRAL: CN‐00653773] [Google Scholar]
- Spinowitz B, Coyne DW, Lok CE, Fraticelli M, Azer M, Dalal S, et al. CERA maintains stable control of hemoglobin in patients with chronic kidney disease on dialysis when administered once every two weeks. American Journal of Nephrology 2008;28(2):280‐9. [MEDLINE: ] [DOI] [PubMed] [Google Scholar]
STRIATA Study 2008 {published data only}
- Barany P, Besarab A, Macdougall IC, Law A, Ouyang Y, Heifets M. Median hemoglobin (Hb) decline following C.E.R.A. dose interruption is similar to that with other erythropoiesis stimulating agents (ESAs) [abstract no: SU‐PO795]. Journal of the American Society of Nephrology 2007;18(Abstracts Issue):760A. [CENTRAL: CN‐00794522] [Google Scholar]
- Canaud B, Braun J, Locatelli F, Villa G, Vlem B, Sanz Guajardo D, et al. Intravenous (IV) C.E.R.A. (continuous erythropoietin receptor activator) administered once every 2 weeks maintains stable haemoglobin (Hb) levels in patients with chronic kidney disease (CKD) on dialysis [abstract no: SP425]. Nephrology Dialysis Transplantation 2006;21(Suppl 4):iv157. [CENTRAL: CN‐00690645] [Google Scholar]
- Canaud B, Mingardi G, Braun J, Aljama P, Kerr PG, Locatelli F, et al. Intravenous C.E.R.A. maintains stable haemoglobin levels in patients on dialysis previously treated with darbepoetin alfa: results from STRIATA, a randomized phase III study. Nephrology Dialysis Transplantation 2008;23(11):3654‐61. [MEDLINE: ] [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fishbane S, Dalton C, Beswick R, Dutka P, Schmidt R. Efficacy of C.E.R.A., a continuous erythropoietin receptor activator, in the treatment of renal anemia: overview of 6 global phase 3 trials [abstract no: 59]. American Journal of Kidney Diseases 2007;49(4):A39. [CENTRAL: CN‐00756571] [Google Scholar]
Tessitore 2008 {published data only}
- Tessitore N, Mantovani W, Bedogna V, Loss R, Melilli E, Poli A, et al. Cost analysis of switching from epoetin alfa to darbepoetin alfa in chronic hemodialysis patients (HD Pts): a long‐term, randomized, open‐label, cross‐over, pilot study [abstract no: SA‐PO2645]. Journal of the American Society of Nephrology 2008;19(Abstracts Issue):706A. [CENTRAL: SA‐PO2645] [Google Scholar]
Vanrenterghem 2002 {published data only}
- Canaud B. Darbepoetin alfa dose requirements for IV and SC administration are equivalent in anaemic dialysis patients [abstract no: M319]. Nephrology Dialysis Transplantation 2002;17(Suppl 1):137. [CENTRAL: CN‐00550478] [Google Scholar]
- Kerr PG, Harris D, Hawley C, Walker R, European/Australian NESP 970290 Study Group. Novel erythropoiesis stimulating protein (NESP) maintains haemoglobin in ESRD patients with administered once weekly or once every other week [abstract no: 181]. Nephrology 2000;5(3):A112. [CENTRAL: CN‐00509271] [Google Scholar]
- Vanrenterghem Y, Barany P, Mann J, European/Australian NESP 970290 Study Group. Novel erythropoiesis stimulating protein (NESP) maintains hemoglobin (hgb) in ESRD patients when administered once weekly or once every other week [abstract]. Journal of the American Society of Nephrology 1999;10(Program & Abstracts):270A. [CENTRAL: CN‐00583820] [Google Scholar]
- Vanrenterghem Y, Barany P, Mann JF, Kerr PG, Wilson J, Baker NF, et al. Randomized trial of darbepoetin alfa for treatment of renal anemia at a reduced dose frequency compared with rHuEPO in dialysis patients. Kidney International 2002;62(6):2167‐75. [MEDLINE: ] [DOI] [PubMed] [Google Scholar]
Weiss 2000 {published data only}
- Weiss L, Svensson B, Clyne N, Devino J, Frisenette‐Fich C, Kurkus J. Is subcutaneous (s.c.) erythropoietin (EPO) administered once a week for stable hemodialysis (HD) patients sufficient? Results from a Swedish multicenter trial. [abstract no: A1099]. Journal of the American Society of Nephrology 1996;7(9):1468‐9. [CENTRAL: CN‐00583180] [Google Scholar]
- Weiss LG, Clyne N, Divino Fihlho J, Frisenette‐Fich C, Kurkus J, Svensson B. The efficacy of once weekly compared with two or three times weekly subcutaneous epoetin beta: results from a randomized controlled multicentre trial. Swedish Study Group. Nephrology Dialysis Transplantation 2000;15(12):2014‐9. [MEDLINE: ] [DOI] [PubMed] [Google Scholar]
Yoon 2004 {published data only}
- Yoon SY, Bang BK, Kim SG, Han DC, Hwang SD, Park JS, et al. Randomized, controlled trial of darbepoetin alfa for the treatment of renal anemia in hemodialysis patients [abstract no: MP271]. 41st Congress. European Renal Association. European Dialysis and Transplantation Association; 2004 May 15‐18; Lisbon, Portugal. 2004:322. [CENTRAL: CN‐00509573]
References to studies excluded from this review
Allon 2002 {published data only}
- Allon M, Kleinman AK, Walczyk M, Kaupke C, Maroni BJ, Heatherington A, et al. The pharmacokinetics of novel erythropoiesis stimulating protein (NESP) following chronic intravenous administration is time‐and dose‐linear [abstract no: A1308]. Journal of the American Society of Nephrology 2000;11(Sept):248A. [CENTRAL: CN‐00626053] [Google Scholar]
- Allon M, Kleinman K, Walczyk M, Kaupke C, Messer‐Mann L, Olson K, et al. Pharmacokinetics and pharmacodynamics of darbepoetin alfa and epoetin in patients undergoing dialysis. Clinical Pharmacology & Therapeutics 2002;72(5):546‐55. [MEDLINE: ] [DOI] [PubMed] [Google Scholar]
BA16260 Study 2006 {published data only}
- Francisco AL, Sulowicz W, Dougherty FC. Subcutaneous CERA (continuous erythropoiesis receptor activator) has potent erythropoietic activity in dialysis patients with chronic renal anemia: an exploratory multiple‐dose study [abstract]. Journal of the American Society of Nephrology 2003;14(Nov):27A‐8A. [CENTRAL: CN‐00550366] [Google Scholar]
- Francisco AL, Sulowicz W, Klinger M, Niemczyk S, Vargemezis V, Metivier F, et al. Continuous Erythropoietin Receptor Activator (C.E.R.A.) administered at extended administration intervals corrects anaemia in patients with chronic kidney disease on dialysis: a randomised, multicentre, multiple‐dose, phase II study. International Journal of Clinical Practice 2006;60(12):1687‐96. [MEDLINE: ] [DOI] [PubMed] [Google Scholar]
Bennett 1991 {published data only}
- Bennett WM. A multicenter clinical trial of epoetin beta for anemia of end‐stage renal disease. Journal of the American Society of Nephrology 1991;1(7):990‐8. [MEDLINE: ] [DOI] [PubMed] [Google Scholar]
Besarab 1998 {published data only}
- Besarab A, Bolton WK, Browne JK, Egrie JC, Nissenson AR, Okamoto DM, et al. The effects of normal as compared with low hematocrit values in patients with cardiac disease who are receiving hemodialysis and epoetin. New England Journal of Medicine 1998;339(9):584‐90. [MEDLINE: ] [DOI] [PubMed] [Google Scholar]
- Besarab A, Goodkin DA, Nissenson AR, Normal Hematocrit Cardiac Trial Authors. The normal hematocrit study‐‐follow‐up. New England Journal of Medicine 2008;358(4):433‐4. [MEDLINE: ] [DOI] [PubMed] [Google Scholar]
- Conlon P, Kovalik E, Minda SN, Schumm D, Gutman R, Schwab SJ. Normalizing hematocrit in hemodialysis patients does not increase blood pressure [abstract]. Journal of the American Society of Nephrology 1995;6(3):526. [Google Scholar]
- Conlon PJ, Kovalik E, Schumm D, Minda S, Schwab SJ. Normalization of hematocrit in hemodialysis patients does not affect silent ischemia. Renal Failure 2000;22(2):205‐11. [MEDLINE: ] [DOI] [PubMed] [Google Scholar]
- Coyne DW. The health‐related quality of life was not improved by targeting higher hemoglobin in the Normal Hematocrit Trial. Kidney International 2012;82(2):235‐41. [MEDLINE: ] [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goodkin DA. The Normal Hematocrit Cardiac Trial revisited. Seminars in Dialysis 2009;22(5):495‐502. [MEDLINE: ] [DOI] [PubMed] [Google Scholar]
- Kilpatrick R, Critchlow C, Besarab A, Fishbane S, Stehman‐Breen C, Krishnan M, et al. Epoetin alfa (EPO) responsiveness predicts survival in the normal hematocrit study (NHS) [abstract no: TH‐PO382]. Journal of the American Society of Nephrology 2006;17(Abstracts):188A. [CENTRAL: CN‐00615892] [Google Scholar]
- Kilpatrick RD, Critchlow CW, Fishbane S, Besarab A, Stehman‐Breen C, Krishnan M, et al. Greater epoetin alfa responsiveness is associated with improved survival in hemodialysis patients. Clinical Journal of the American Society of Nephrology: CJASN 2008;3(4):1077‐83. [MEDLINE: ] [DOI] [PMC free article] [PubMed] [Google Scholar]
Bhuiyan 2004 {published data only}
- Bhuiyan FK, Rashid HU, Ahmed S, Alam MR. Comparative study of two different dosages of erythropoietin in respect of response to anemia in patients with ESRD on MHD. Bangladesh Renal Journal 2004;23(2):42‐51. [EMBASE: 2006528465] [Google Scholar]
Brandt 1999 {published data only}
- Brandt JR, Avner ED, Hickman RO, Watkins SL. Safety and efficacy of erythropoietin in children with chronic renal failure. Pediatric Nephrology 1999;13(2):143‐7. [MEDLINE: ] [DOI] [PubMed] [Google Scholar]
Brown 1988 {published data only}
- Brown CD, Kieran M, Dosunmu BV, Zhao Z, Larson RH, Friedman EA. Raised hematocrit (HCT) persists six‐weeks after stopping treatment with human recombinant erythropoietin (r‐HuEPO) in azotemic anemic patients [abstract]. Kidney International 1988;33(1):184. [CENTRAL: CN‐00602117] [Google Scholar]
Canadian EPO Study 1990 {published data only}
- Anonymous. Effect of recombinant human erythropoietin therapy on blood pressure in hemodialysis patients. Canadian Erythropoietin Study Group. American Journal of Nephrology 1991;11(1):23‐6. [MEDLINE: ] [DOI] [PubMed] [Google Scholar]
- Canadian ESG. Association between recombinant human erythropoietin and quality of life and exercise capacity of patients receiving haemodialysis. Canadian Erythropoietin Study Group. BMJ 1990;300(6724):573‐8. [MEDLINE: ] [DOI] [PMC free article] [PubMed] [Google Scholar]
- Canadian ESG. The effect of recombinant human erythropoietin (EPO) upon quality of life and exercise capacity of anemic patients on chronic hemodialysis. [abstract]. Kidney International 1990;37(1):278. [Google Scholar]
- Canadian Erythropoietin Study Group. The clinical effects and side‐effects of recombinant human erythropoietin (EPO) in anaemic patients on chronic hemodialysis [abstract]. Kidney International 1990;37(1):278. [CENTRAL: CN‐00583134] [Google Scholar]
- Keown PA. Quality of life in end‐stage renal disease patients during recombinant human erythropoietin therapy. The Canadian Erythropoietin Study Group. Contributions to Nephrology 1991;88:81‐9. [MEDLINE: ] [PubMed] [Google Scholar]
- Keown PA, Canadian ESG. The effect of recombinant human erythropoietin (EPO) upon quality of life (QL) and functional capacity (FC) of anemic patients on chronic hemodialysis [abstract]. Kidney International 1989;35(1):195. [Google Scholar]
- Keown PA, Churchill DN, Poulin‐Costello M, Lei L, Gantotti S, Agodoa I, et al. Dialysis patients treated with Epoetin alfa show improved anemia symptoms: A new analysis of the Canadian Erythropoietin Study Group trial. Hemodialysis International 2010;14(2):168‐73. [MEDLINE: ] [DOI] [PubMed] [Google Scholar]
- Laupacis A. A randomized double‐blind study of recombinant human erythropoietin in anaemic hemodialysis patients. Canadian Erythropoietin Study Group. Transplantation Proceedings 1991;23(2):1825‐6. [MEDLINE: ] [PubMed] [Google Scholar]
- Laupacis A. Changes in quality of life and functional capacity in hemodialysis patients treated with recombinant human erythropoietin. The Canadian Erythropoietin Study Group. Seminars in Nephrology 1990;10(2 Suppl 1):11‐9. [MEDLINE: ] [PubMed] [Google Scholar]
- Laupacis A, Wong C, Churchill D. The use of generic and specific quality‐of‐life measures in hemodialysis patients treated with erythropoietin. The Canadian Erythropoietin Study Group. Controlled Clinical Trials 1991;12(4 Suppl):168S‐79S. [MEDLINE: ] [DOI] [PubMed] [Google Scholar]
- Muirhead N, Keown P, Churchill DN, Lei L, Gitlin M, Mayne TJ. An intent‐to‐treat (ITT) analysis of anemia symptoms in the Canadian erythropoietin study group (CESG) [abstract no: PUB537]. Journal of the American Society of Nephrology 2008;19(Abstracts Issue):932A. [CENTRAL: CN‐00790629] [Google Scholar]
- Muirhead N, Keown P, Gitlin M, Mayne TJ, Churchill DN. A reanalysis of the Canadian erythropoietin study group (CESG) patient‐reported outcomes (PRO) trial [abstract no: 177]. American Journal of Kidney Diseases 2008;51(4):A72. [CENTRAL: CN‐00790972] [Google Scholar]
- Muirhead N, Keown P, Lei L, Gitlin M, Mayne TJ, Churchill D. The relationship between achieved hemoglobin (HB) & exercise tolerance [abstract no: 161]. American Journal of Kidney Diseases 2008;51(4):A68. [CENTRAL: CN‐00796608] [Google Scholar]
- Muirhead N, Keown PA, Churchill DN, Poulin‐Costello M, Gantotti S, Lei L, et al. Dialysis patients treated with Epoetin alpha show improved exercise tolerance and physical function: a new analysis of the Canadian Erythropoietin Study Group trial. Hemodialysis International 2011;15(1):87‐94. [EMBASE: 2011063916] [DOI] [PubMed] [Google Scholar]
- Muirhead N, Laupacis A, Wong C. Erythropoietin for anaemia in haemodialysis patients: results of a maintenance study (the Canadian Erythropoietin Study Group). Nephrology Dialysis Transplantation 1992;7(8):811‐6. [MEDLINE: ] [PubMed] [Google Scholar]
Castro 1994 {published data only}
- Castro MC, Abensur H, Centeno JR, Romao Jr JE, Marcondes M, Sabbaga E. Comparison between thrice‐weekly intravenous and once‐weekly subcutaneous administration of rHuEPO in hemodialysis patients. Dialysis & Transplantation 1994;23(3):132‐43. [EMBASE: 1994307692] [Google Scholar]
Chazot 2009 {published data only}
- Chazot C, Dumoulin A, Ang KS, Paul GJ, Bernard C, Conference Medicale Group. Haemodialysis patients under subcutaneous recombinant human erythropoietin can be directly switched to intravenous Aranesp® [abstract no: F‐PO682]. Journal of the American Society of Nephrology 2005;16:483A. [CENTRAL: CN‐00740583] [Google Scholar]
- Chazot C, Terrat JC, Ang K, Gassia JP, Chedid K, Maurice F, et al. Intravenous darbepoetin A maintains hemoglobin level in hemodialysis patients converting from subcutaneous recombinant human erythropoietin [abstract no: F‐PO501]. Journal of the American Society of Nephrology 2004;15(Oct):177A. [CENTRAL: CN‐00583168] [Google Scholar]
- Chazot C, Terrat JC, Dumoulin A, Ang KS, Gassia JP, Chedid K, et al. Randomized equivalence study evaluating the possibility of switching hemodialysis patients receiving subcutaneous human erythropoietin directly to intravenous darbepoetin alfa. Annals of Pharmacotherapy 2009;43(2):228‐34. [MEDLINE: ] [DOI] [PubMed] [Google Scholar]
Dougherty 2004 {published data only}
- Dougherty FC, Reigner B, Beyer U. Dose‐dependent erythropoietic responses to subcutaneous CERA (continuous erythropoiesis receptor activator) in multiple‐dose study of dialysis patients with chronic renal anaemia [abstract]. 41st Congress. European Renal Association. European Dialysis and Transplantation Association; 2004 May 15‐18; Lisbon, Portugal. 2004:325. [CENTRAL: CN‐00509162]
Fan 1992 {published data only}
- Fan CD, Tsang KK, Wong CM, Chang HC. Therapeutic evaluation of once versus thrice weekly subcutaneous (sc) application of low dose recombinant human erythropoietin (rHuEPO) in hemodialysis patients [abstract]. 9th Asian Colloquium in Nephrology; 1992 May 17‐21; Seoul, Korea. 1992:184. [CENTRAL: CN‐00460720]
Hirakata 2010 {published data only}
- Hirakata H, Gejyo F, Suzuki M, Saito A, Lino Y, Watanabe Y, et al. Effect of darbepoetin alfa (KRN321) subcutaneous treatment on hemoglobin levels, health‐related QOL (HRQOL) and left ventricular mass index (LVMI) in patients with chronic kidney disease (CKD) not on dialysis [abstract no: SA‐PO204]. Journal of the American Society of Nephrology 2006;17(Abstracts):618A. [CENTRAL: CN‐00740524] [Google Scholar]
- Hirakata H, Tsubakihara Y, Gejyo F, Nishi S, Iino Y, Watanabe Y, et al. Maintaining high hemoglobin levels improved the left ventricular mass index and quality of life scores in pre‐dialysis Japanese chronic kidney disease patients. Clinical & Experimental Nephrology 2010;14(1):28‐35. [MEDLINE: ] [DOI] [PubMed] [Google Scholar]
- Inaguma D, Tsubakihara Y, Hirakata H, Hiroe M, Hada Y, Akizawa T, et al. Monthly subcutaneous treatment of darbepoetin alfa (KRN321) could maintain higher Hb safely and have beneficial effects on cardiac function of Japanese CKD patients not on dialysis [abstract no: SaP353]. Nephrology Dialysis Transplantation 2007;22(Suppl 6):vi352. [CENTRAL: CN‐00740573] [Google Scholar]
- Suzuki M, Hada Y, Akaishi M, Hiroe M, Aonuma K, Tsubakihara Y, et al. Effects of anemia correction by erythropoiesis‐stimulating agents on cardiovascular function in non‐dialysis patients with chronic kidney disease. International Heart Journal 2012;53(4):238‐43. [MEDLINE: ] [DOI] [PubMed] [Google Scholar]
Icardi 1990 {published data only}
- Icardi A, Paoletti E, Molinelli G. Efficacy of recombinant erythropoietin after subcutaneous or intraperitoneal administration to patients on CAPD. Advances in Peritoneal Dialysis 1990;6:292‐5. [MEDLINE: ] [PubMed] [Google Scholar]
Ifudu 1998 {published data only}
- Ifudu O, Dawood M, Homel P. Erythropoietin‐induced elevation in blood pressure is immediate and dose dependent. Nephron 1998;79(4):486‐7. [MEDLINE: ] [DOI] [PubMed] [Google Scholar]
Iwasaki 2008 {published data only}
- Iwasaki T, Kimata N, Sugi O, Sawara Y, Jinnai H, Kikuchi K, et al. Difference in reactivity became more evident by changing of the erythropoiesis‐stimulating agent (ESA) [abstract no: PUB462]. Journal of the American Society of Nephrology 2008;19(Abstracts Issue):914A. [CENTRAL: CN‐00792600] [Google Scholar]
Kawanishi 2005 {published data only}
- Kawanishi H, Iwasaki M, Akizawa T, Koshikawa S, KRN321 SG. Dose‐finding and long‐term studies of intravenous KRN321 (darbepoetin alfa) in chronic renal failure patients (CRF) on hemodialysis (HD) in Japan [abstract no: SA‐PO943]. Journal of the American Society of Nephrology 2005;16:763A. [CENTRAL: CN‐00740571] [Google Scholar]
Kim 2009a {published data only}
- Kim CD, Park SH, Kim DJ, Do JY, Shin SK, Kim BS, et al. Randomized trial to compare the dosage of darbepoetin alfa by administration route in hemodialysis patient [abstract no: TH‐PO370]. Journal of the American Society of Nephrology 2006;17(Abstracts):185A. [CENTRAL: CN‐00740537] [DOI] [PubMed] [Google Scholar]
- Kim CD, Park SH, Kim DJ, Park JW, Do JY, Shin SK, et al. Randomized trial to compare the dosage of darbepoetin alfa by administration route in haemodialysis patients. Nephrology 2009;14(5):482‐7. [MEDLINE: ] [DOI] [PubMed] [Google Scholar]
Knebel 2008 {published data only}
- Knebel W, Palmen M, Dowell JA, Gastonguay M. Population pharmacokinetic modeling of epoetin delta in pediatric patients with chronic kidney disease. Journal of Clinical Pharmacology 2008 Jul;48(7):837‐48. [MEDLINE: ] [DOI] [PubMed] [Google Scholar]
Locatelli 2008 {published data only}
- Locatelli F, Villa G, Messa P, Filippini A, Cannella G, Ferrari G, et al. Efficacy and safety of once‐weekly intravenous epoetin alfa in maintaining hemoglobin levels in hemodialysis patients. Journal of Nephrology 2008;21(3):412‐20. [MEDLINE: ] [PubMed] [Google Scholar]
Macdougall 2003 {published data only}
- Macdougall IC. Novel erythropoiesis stimulating protein (NESP) for the treatment of anaemia in ESRD patients [abstract]. 37th Congress. European Renal Association. European Dialysis and Transplantation Association. European Kidney Research Organisation; 2000 Sept 17‐20; Nice, France. 2000:237. [CENTRAL: CN‐00461229]
- Macdougall IC. Novel erythropoiesis stimulating protein (NESP) for the treatment of anaemia in ESRD patients [abstract]. Nephrology Dialysis Transplantation 2000;15(9):A160. [Google Scholar]
- Macdougall IC, Matcham J, Gray SJ, NESP Study Group. Correction of anaemia with darbepoetin alfa in patients with chronic kidney disease receiving dialysis. Nephrology Dialysis Transplantation 2003 Mar;18(3):576‐81. [MEDLINE: ] [DOI] [PubMed] [Google Scholar]
- Macdougall IC, UK NESP Study Group. Novel erythropoiesis stimulating protein (NESP) for the treatment of renal anaemia [abstract]. Journal of the American Society of Nephrology 1998;9(Program & Abstracts):258A‐9A. [CENTRAL: CN‐00446518] [Google Scholar]
Macdougall 2006 {published data only}
- Macdougall IC, Reigner B, Dougherty FC. Consistent pharmacokinetic properties of CERA (Continuous Erythropoietin Receptor Activator) in healthy volunteers and in patients with CKD [abstract no: 89]. American Journal of Kidney Diseases 2006;47(4):A41. [CENTRAL: CN‐00583919] [Google Scholar]
- Macdougall IC, Robson R, Opatrna S, Liogier X, Pannier A, Jordan P, et al. Pharmacokinetics and Pharmacodynamics of Intravenous and Subcutaneous Continuous Erythropoietin Receptor Activator (C.E.R.A.) in Patients with Chronic Kidney Disease. Clinical Journal of The American Society of Nephrology: CJASN 2006;1(6):1211‐5. [MEDLINE: ] [DOI] [PubMed] [Google Scholar]
- Macdougall IC, Robson R, Opatrna S, Liogier X, Pannier A, Reigner B, et al. Pharmacologic profile of CERA (continuous erythropoietin receptor activator) in chronic kidney disease (CKD) patients (pts) following intravenous (IV) and subcutaneous (SC) administration [abstract no: SA‐PO926]. Journal of the American Society of Nephrology 2005;16:759A. [CENTRAL: CN‐00795029] [DOI] [PubMed] [Google Scholar]
Macdougall 2007a {published data only}
- Macdougall IC, Ruiz EF. Extending dosing regimens with epoetin delta: a randomized controlled trial [abstract no: SU‐PO785]. Journal of the American Society of Nephrology 2007;18(Abstracts Issue):758A. [CENTRAL: CN‐00765131] [Google Scholar]
Martin 2007 {published data only}
- Martin KJ. Epoetin delta for the management of renal anemia: a one year study [abstract no: TH‐PO374]. Journal of the American Society of Nephrology 2006;17(Abstracts):186A. [CENTRAL: CN‐00765049] [Google Scholar]
- Martin KJ. The first human cell line‐derived erythropoietin, epoetin‐delta (Dynepo), in the management of anemia in patients with chronic kidney disease. Clinical Nephrology 2007;68(1):26‐31. [MEDLINE: ] [DOI] [PubMed] [Google Scholar]
- Martin KJ, ED3001 Study Group. Epoetin delta in the management of renal anaemia: results of a 6‐month study. Nephrology Dialysis Transplantation 2007;22(10):3052‐4. [MEDLINE: ] [DOI] [PubMed] [Google Scholar]
Moiz 2000 {published data only}
- Moiz B, Hashmi KZ, Yazdani I. Erythropoietin induced hypertension. Journal of the College of Physicians & Surgeons Pakistan 2000;10(9):317‐20. [EMBASE: 2000359629] [Google Scholar]
Muirhead 1992 {published data only}
- Muirhead N. Changes in quality of life in chronic renal failure patients treated with recombinant human erythropoietin [abstract]. 9th Asian Colloquium in Nephrology; 1992 May 17‐21; Seoul, Korea. 1992:45‐6. [CENTRAL: CN‐00461371]
Nissenson 1995 {published data only}
- Nissenson AR, Korbet S, Faber M, Burkart J, Gentile D, Hamburger R, et al. Multicenter trial of erythropoietin in patients on peritoneal dialysis. Journal of the American Society of Nephrology 1995;5(7):1517‐29. [MEDLINE: ] [DOI] [PubMed] [Google Scholar]
Parfrey 2005 {published data only}
- Foley RN, Curtis BM, Parfrey PS. Erythropoietin therapy, hemoglobin targets, and quality of life in healthy hemodialysis patients: a randomized trial. Clinical Journal of the American Society of Nephrology: CJASN 2009;4(4):726‐33. [MEDLINE: ] [DOI] [PMC free article] [PubMed] [Google Scholar]
- Foley RN, Curtis BM, Parfrey PS. Hemoglobin targets and blood transfusions in hemodialysis patients without symptomatic cardiac disease receiving erythropoietin therapy. Clinical Journal of the American Society of Nephrology: CJASN 2008;3(6):1669‐75. [MEDLINE: ] [DOI] [PMC free article] [PubMed] [Google Scholar]
- Foley RN, Curtis BM, Parfrey PS. Hemoglobin targets, blood transfusions and quality of life in hemodialysis patients without symptomatic cardiac disease [abstract no: SA‐PO2745]. Journal of the American Society of Nephrology 2008;19(Abstracts Issue):730A. [CENTRAL: CN‐00756852] [DOI] [PMC free article] [PubMed] [Google Scholar]
- Foley RN, Curtis BM, Randell EW, Parfrey PS. Left ventricular hypertrophy in new hemodialysis patients without symptomatic cardiac disease. Clinical Journal of the American Society of Nephrology: CJASN 2010;5(5):805‐13. [MEDLINE: ] [DOI] [PMC free article] [PubMed] [Google Scholar]
- Foley RN, Parfrey PS, Wittreich BH, Sullivan DJ, Zagari MJ, Frei D, et al. The effect of higher haemoglobin levels on left ventricular vacity volume in patients starting haemodialysis: a blinded, randomised, controlled trial in 596 patients without symptomatic cardiac disease [abstract]. 41st Congress. European Renal Association. European Dialysis and Transplantation Association; 2004 May 15‐18; Lisbon, Portugal. 2004:217. [CENTRAL: CN‐00509197]
- Parfrey PS, Foley RN, Wittreich BH, Sullivan DJ, Zagari MJ, Frei D. Double‐blind comparison of full and partial anemia correction in incident hemodialysis patients without symptomatic heart disease. Journal of the American Society of Nephrology 2005;16(7):2180‐9. [MEDLINE: ] [DOI] [PubMed] [Google Scholar]
- Parfrey PS, Foley RN, Wittreich BH, Sullivan DJ, Zagari MJ, Frei D, et al. Double‐blind comparison of full and partial anemia correction with erythropoietin in incident hemodialysis patients without symptomatic heart disease [abstract no: PUB002]. Journal of the American Society of Nephrology 2004;15(Oct):762A. [CENTRAL: CN‐00583759] [DOI] [PubMed] [Google Scholar]
- Parfrey PS, Foley RN, Wittreich BH, Sullivan DJ, Zagari MJ, Frei D, et al. The effect of higher haemoglobin levels on quality of life in patients starting haemodialysis: a blinded, randomised, controlled trial in 596 patients without symptomatic cardiac disease [abstract]. 41st Congress. European Renal Association. European Dialysis and Transplantation Association; 2004 May 15‐18; Lisbon, Portugal. 2004:229. [CENTRAL: CN‐00509402]
PATRONUS Study 2009 {published data only}
- Besarab A, Canaud B, Francisco ALM, Kerr P, Locatelli F, Lok CE, et al. Randomized comparison of IV C.E.R.A. (Continuous Erythropoietin Receptor Activator) and Darbepoetin Alfa (DA) at extended administration intervals for the maintenance of Hb levels in patients with CKD on dialysis: rationale and design [abstract no: PUB377]. Journal of the American Society of Nephrology 2006;17(Abstracts):896A. [CENTRAL: CN‐00740564] [Google Scholar]
- Carrera F. CERA vs darbepoetin alfa as maintenance therapy for anaemia in patients with chronic kidney disease (CKD): The PATRONUS Study [abstract no: M558]. World Congress of Nephrology; 2009 May 22‐26; Milan, Italy. 2009.
- Carrera F, Lok CE, Francisco A, Locatelli F, Mann JF, Canaud B, et al. Maintenance treatment of renal anaemia in haemodialysis patients with methoxy polyethylene glycol‐epoetin beta versus darbepoetin alfa administered monthly: a randomized comparative trial. Nephrology Dialysis Transplantation 2010;25(12):4009‐17. [MEDLINE: ] [DOI] [PMC free article] [PubMed] [Google Scholar]
- Locatelli F, PATRONUS Study Group. Once‐monthly C.E.R.A. is superior to darbepoetin alfa for maintaining hemoglobin levels in hemodialysis patients regardless of age, gender, diabetic status or presence of hyperlipidemia [abstract no: SA‐PO2412]. Journal of the American Society of Nephrology 2009;20:663A. [CENTRAL: CN‐00747328] [Google Scholar]
- Mann JF, PATRONUS Study Group. Risk factors for vascular events in hemodialysis patients receiving once‐monthly C.E.R.A. or once‐monthly darbepoetin alfa: post hoc analysis of the PATRONUS Study [abstract no: PUB414]. Journal of the American Society of Nephrology 2009;20:921A. [CENTRAL: CN‐00747329] [Google Scholar]
- Francisco AL, PATRONUS Study Group. Significantly higher hemoglobin response rates are achieved in hemodialysis patients with once‐monthly C.E.R.A. compared with darbepoetin alfa in hemodialysis, regardless of etiology of chronic kidney disease [abstract no: SA‐PO2411]. Journal of the American Society of Nephrology 2009;20:663A. [CENTRAL: CN‐00747327] [Google Scholar]
Pawlak 2007 {published data only}
- Pawlak K, Pawlak D, Mysliwiec M. Long‐term erythropoietin therapy does not affect metalloproteinases and their inhibitor levels, oxidative stress and inflammation in hemodialyzed patients. American Journal of Nephrology 2007;27(3):221‐5. [MEDLINE: ] [DOI] [PubMed] [Google Scholar]
Provenzano 2006 {published data only}
- Provenzano R, Dougherty FC. Subcutaneous (SC) CERA (Continuous Erythropoietin Receptor Activator) administered once every 2 weeks effectively corrects anemia in patients with chronic kidney disease (CKD) on dialysis and not on dialysis [abstract no: 126]. American Journal of Kidney Diseases 2006;47(4):A50. [CENTRAL: CN‐00747315] [Google Scholar]
Raftery 2000 {published data only}
- Raftery MJ, Auinger M, Hertlova M. Safety and tolerability of a multidose formulation of epoetin beta in dialysis patients. Collaborative Study Group. Clinical Nephrology 2000;54(3):240‐5. [MEDLINE: ] [PubMed] [Google Scholar]
Schmitt 2006 {published data only}
- Schaefer F, Brummer C, Rosenkranz J, Schmitt CP. Increased subcutaneous injection pain with darbepoetin‐A compared with epoetin‐b in children: results of a randomized, double‐blind study [abstract no: P399]. Pediatric Nephrology 2004;19(9):C191. [CENTRAL: CN‐00740567] [Google Scholar]
- Schmitt CP, Brummer C, Rosenkranz J, Schaefer F. Increased injection pain with darbepoetin‐a compared to epoetin‐b: a double‐blind study in pediatric patients [abstract no: SU‐PO068]. Journal of the American Society of Nephrology 2004;15(Oct):547A. [CENTRAL: CN‐00740568] [Google Scholar]
- Schmitt CP, Brummer C, Rosenkranz J, Schaefer F. Injection pain is increased with darbepoetin‐a compared to epoetin‐b [abstract no: W‐PO40120]. Nephrology 2005;10(Suppl):A311. [CENTRAL: CN‐00740569] [Google Scholar]
- Schmitt CP, Nau B, Brummer C, Rosenkranz J, Schaefer F. Increased injection pain with darbepoetin‐alpha compared to epoetin‐beta in paediatric dialysis patients. Nephrology Dialysis Transplantation 2006;21(12):3520‐4. [MEDLINE: ] [DOI] [PubMed] [Google Scholar]
Smith 2007 {published data only}
- Pratt R. Pharmacokinetics of erythropoietin by a human cell line (Epoetin I): subcutaneous vs intravenous dosing in patients with chronic kidney disease [abstract no: 1137]. Haematologica 2006;91(Suppl 1):414. [CENTRAL: CN‐00716122] [Google Scholar]
- Pratt RD, Dowell J. Pharmacokinetics of epoetin delta: a new erythropoietin produced by gene‐activation in a human cell line [abstract no: TH‐PO378]. Journal of the American Society of Nephrology 2006;17(Abstracts):187A. [CENTRAL: CN‐00644158] [Google Scholar]
- Smith WB, Dowell JA, Pratt RD. Pharmacokinetics and pharmacodynamics of epoetin delta in two studies in healthy volunteers and two studies in patients with chronic kidney disease. Clinical Therapeutics 2007;29(7):1368‐80. [MEDLINE: ] [DOI] [PubMed] [Google Scholar]
Smyth 2006 {published data only}
- Smyth M, Pratt RD. Epoetin delta, erythropoietin produced in a human cell line, is as effective as epoetin alfa in the treatment of anemia [abstract no: 1296]. Blood 2006;108(11):380a. [CENTRAL: CN‐00740519] [Google Scholar]
Spaia 1995 {published data only}
- Spaia S, Gouva H, Petridou P, Kokolou E, Mavropoulou E, Pazarloglu M, et al. Once versus thrice weekly administration of maintenance dose of erythropoietin in haemodialysis patients [abstract]. ISN XIII International Congress of Nephrology; 1995 Jul 2‐6; Madrid, Spain. 1995:508. [CENTRAL: CN‐00509487]
Spinowitz 2006 {published data only}
- Pratt R. Epoetin delta, erythropoietin produced by a human cell line, is effective in the treatment of renal anemia [abstract no: 0576]. Haematologica 2006;91(Suppl 1):213. [CENTRAL: CN‐00625355] [Google Scholar]
- Spinowitz BS, Pratt RD. Epoetin delta is effective for the management of anaemia associated with chronic kidney disease. Current Medical Research & Opinion 2006;22(12):2507‐13. [MEDLINE: ] [DOI] [PubMed] [Google Scholar]
Stockenhuber 1990 {published data only}
- Stockenhuber F, Geissler K, Sunder‐Plassmann G, Kurz RW, Jahn C, Hinterberger W, et al. Recombinant human erythropoietin (r‐HuEPO) activates a broad spectrum of hematopoietic stem cells [abstract]. Kidney International 1989;35(1):321. [CENTRAL: CN‐00766226] [DOI] [PubMed] [Google Scholar]
- Stockenhuber F, Kurz RW, Geissler K, Jahn C, Hinterberger W, Balcke P, et al. Recombinant human erythropoietin activates a broad spectrum of progenitor cells. Kidney International 1990;37(1):150‐6. [MEDLINE: ] [DOI] [PubMed] [Google Scholar]
Tan 1990a {published data only}
- Tan A. Recombinant human erythropoietin (r‐HuEPO): quality of life and other considerations. CANNT Journal 1990:13‐4. [MEDLINE: ] [PubMed] [Google Scholar]
Tolman 2005 {published data only}
- Tolman C, Richardson D, Bartlett C, Will E. A randomised study of weekly subcutaneous aranesp and neorecormon in a large unselected haemodialysis cohort, managed with computer assisted anaemia alogrithms [abstract]. 41st Congress. European Renal Association. European Dialysis and Transplantation Association; 2004 May 15‐18; Lisbon, Portugal. 2004:229‐30. [CENTRAL: CN‐00509514]
- Tolman C, Richardson D, Bartlett C, Will E. Application of computer assisted anaemia management algorithms in haemodialysis patients produces predictable haemoglobin outcomes regardless of the erythropoietic agent or frequency of administration: results of a randomised study [abstract]. 41st Congress. European Renal Association. European Dialysis and Transplantation Association; 2004 May 15‐18; Lisbon, Portugal. 2004:110. [CENTRAL: CN‐00509513]
- Tolman C, Richardson D, Bartlett C, Will E. Structured conversion from thrice weekly to weekly erythropoietic regimens using a computerized decision‐support system: a randomized clinical study. Journal of the American Society of Nephrology 2005;16(5):1463‐70. [MEDLINE: ] [DOI] [PubMed] [Google Scholar]
- Tolman C, Richardson D, Bartlett C, Will EJ. Dose conversion ratio (DCR) from subcutaneous epoetin‐b (EPO) to weekly darbepoetin‐a (DA) is dependent on baseline sensitivity: results from a randomised study [abstract no: SA‐PO461]. Journal of the American Society of Nephrology 2004;15(Oct):404A. [CENTRAL: CN‐00583282] [Google Scholar]
- West RM, Harris K, Gilthorpe MS, Tolman C, Will EJ. Functional data analysis applied to a randomized controlled clinical trial in hemodialysis patients describes the variability of patient responses in the control of renal anemia. Journal of the American Society of Nephrology 2007;18(8):2371‐6. [MEDLINE: ] [DOI] [PubMed] [Google Scholar]
Wang 2000 {published data only}
- Wang AYM, Yu AWY, Lai KN, Lui SF. Dosage requirement of erythropoietin can be reduced with more frequent administration. Nephrology 2000;5(1‐2):33‐8. [EMBASE: 2000165219] [Google Scholar]
Yalcinkaya 1997 {published data only}
- Yalcinkaya F, Tumer N, Cakar N, Ozkaya N. Low‐dose erythropoietin is effective and safe in children on continuous ambulatory peritoneal dialysis. Pediatric Nephrology 1997;11(3):350‐2. [MEDLINE: ] [DOI] [PubMed] [Google Scholar]
References to studies awaiting assessment
EMERALD 1 Study 2013 {published data only}
- Besarab A, Schiller B, Macdougall IC, Locatelli F, Wiecek A, Covic A, et al. Comparison of IV and SC peginesatide and epoetin doses in hemodialysis (HD) patients [abstract]. Hemodialysis International 2012;16(1):139‐40. [EMBASE: 70725373] [Google Scholar]
- Fishbane S, Schiller B, Locatelli F, Covic AC, Provenzano R, Wiecek A, et al. Peginesatide in patients with anemia undergoing hemodialysis. New England Journal of Medicine 2013;368(4):307‐19. [MEDLINE: ] [DOI] [PubMed] [Google Scholar]
- Levin N, Fishbane S, Provenzano R, Kaplan M, Francisco C, Gao H, et al. Dose relationship between peginesatide and epoetin alfa/beta in chronic kidney disease patients on hemodialysis [abstract]. Blood Purification 2012;33(1‐3):213. [EMBASE: 70723737] [Google Scholar]
- Schiller B, Locatelli F, Fishbane S, Macdougall IC, Wiecek A, Covic A, et al. Peginesatide safety results from two studies of hemodialysis (HD) patients [abstract]. Hemodialysis International 2012;16(1):137. [EMBASE: 70725367] [Google Scholar]
- Sharma A, Fishbane S, Provenzano R, Kaplan M, Schiller B, Besarab A, et al. Peginesatide and epoetin‐A/B dose relationship in two studies of chronic kidney disease (CKD) patients on hemodialysis (HD) [abstract]. Hemodialysis International 2012;16(1):138. [EMBASE: 70725370] [Google Scholar]
EMERALD 2 Study 2013 {published data only}
- Besarab A, Schiller B, Macdougall IC, Locatelli F, Wiecek A, Covic A, et al. Comparison of IV and SC peginesatide and epoetin doses in hemodialysis (HD) patients [abstract]. Hemodialysis International 2012;16(1):139‐40. [EMBASE: 70725373] [Google Scholar]
- Fishbane S, Schiller B, Locatelli F, Covic AC, Provenzano R, Wiecek A, et al. Peginesatide in patients with anemia undergoing hemodialysis. New England Journal of Medicine 2013;368(4):307‐19. [MEDLINE: ] [DOI] [PubMed] [Google Scholar]
- Levin N, Fishbane S, Provenzano R, Kaplan M, Francisco C, Gao H, et al. Dose relationship between peginesatide and epoetin alfa/beta in chronic kidney disease patients on hemodialysis [abstract]. Blood Purification 2012;33(1‐3):213. [EMBASE: 70723737] [Google Scholar]
- Schiller B, Locatelli F, Fishbane S, Macdougall IC, Wiecek A, Covic A, et al. Peginesatide safety results from two studies of hemodialysis (HD) patients [abstract]. Hemodialysis International 2012;16(1):137. [EMBASE: 70725367] [Google Scholar]
- Sharma A, Fishbane S, Provenzano R, Kaplan M, Schiller B, Besarab A, et al. Peginesatide and epoetin‐A/B dose relationship in two studies of chronic kidney disease (CKD) patients on hemodialysis (HD) [abstract]. Hemodialysis International 2012;16(1):138. [EMBASE: 70725370] [Google Scholar]
References to ongoing studies
NCT00436748 {published data only}
- NCT00436748. A multi‐center, double‐blind, randomized study evaluating de novo weekly and once every two week darbepoetin alfa dosing for the correction of anemia in pediatric subjects with chronic kidney disease receiving and not receiving dialysis.. clinicaltrials.gov/ct2/show/NCT00436748 (accessed 18 May 2014).
NCT00717821 {published data only}
- NCT00717821. A randomized, controlled, open label, French multicenter parallel group study to compare the hemoglobin maintenance with once monthly administration of mircera versus epoetin beta or darbepoetin alfa in patients with chronic kidney disease on hemodialysis. clinicaltrials.gov/ct2/show/NCT00717821 (accessed 18 May 2014).
Additional references
Burnier 2009
- Burnier M, Douchamps JA, Tanghe A, Demey D, Perrault L, Foster CF, et al. Less frequent dosing of erythropoiesis stimulating agents in patients undergoing dialysis: a European multicentre cost study. Journal of Medical Economics 2009;12(2):77‐86. [MEDLINE: ] [DOI] [PubMed] [Google Scholar]
Canadian EPO 1990a
- Canadian Erythropoietin Study Group. Association between recombinant erythropoietin and quality of life and exercise capacity of patients receiving haemodialysis. BMJ 1990;300(6724):573‐8. [MEDLINE: ] [DOI] [PMC free article] [PubMed] [Google Scholar]
Cannella 1990
- Cannella G, Canna G, Sandrini M, Gaggiotti M, Nordio G, Movilli E, et al. Renormalization of high cardiac output and of left ventricular size following long‐term recombinant erythropoietin treatment of anemic dialyzed uremic patients. Clinical Nephrology 1990;34(6):272‐8. [PubMed] [Google Scholar]
CARI 2011
- McMahon L, MacGinley R. Biochemical and haematological targets. http://www.cari.org.au/Dialysis/dialysis%20biochemical%20hematological/KHA_CARI_Anaemia_guidelines_November_%202011.pdf (accessed 18 May 2014).
Carrera 2007
- Carrera F, Disney A, Molina M. Extended dosing intervals with erythropoiesis‐stimulating agents in chronic kidney disease: a review of clinical data. Nephrology Dialysis Transplantation 2007;22 Suppl 4:iv19‐iv30. [MEDLINE: ] [DOI] [PubMed] [Google Scholar]
Cody 2005b
- Cody JD, Daly C, Campbell MK, Khan I, Rabindranath KS, Vale L, et al. Recombinant human erythropoietin for chronic renal failure anaemia in pre‐dialysis patients. Cochrane Database of Systematic Reviews 2005, Issue 3. [DOI: 10.1002/14651858.CD003266.pub2] [DOI] [PubMed] [Google Scholar]
Eschbach 1987
- Eschbach JW, Egrie JC, Downing MR, Browne JK, Adamson JW. Correction of the anemia of end‐stage renal disease with recombinant human erythropoietin. Results of a combined phase I and II clinical trial. New England Journal of Medicine 1987;316(2):73‐8. [MEDLINE: ] [DOI] [PubMed] [Google Scholar]
Foley 2010
- Foley RN. Emerging erythropoiesis‐stimulating agents. Nature Reviews Nephrology 2010;6(4):218‐23. [MEDLINE: ] [DOI] [PubMed] [Google Scholar]
Higgins 2003
- Higgins JP, Thompson SG, Deeks JJ, Altman DG. Measuring inconsistency in meta‐analyses. BMJ 2003;327(7414):557‐60. [MEDLINE: ] [DOI] [PMC free article] [PubMed] [Google Scholar]
Higgins 2011
- Higgins JP, Green S (editors). Cochrane Handbook for Systematic Reviews of Interventions Version 5.1.0 [updated March 2011]. The Cochrane Collaboration, 2011. Available from www.cochrane‐handbook.org.
Jensen 1994
- Jensen JD, Madsen JK, Jensen LW, Pedersen EB. Reduced production, absorption, and elimination of erythropoietin in uremia compared with healthy volunteers. Journal of the American Society of Nephrology 1994;5(2):177‐85. [MEDLINE: ] [DOI] [PubMed] [Google Scholar]
KDIGO 2012
- KDIGO Workgroup. KDIGO clinical practice guideline for anemia in chronic kidney disease. Kidney International ‐ Supplement 2012;2(4):279‐335. [Google Scholar]
Koch 1991
- Koch KM, Frei U. Treatment of renal anemia, 1960‐1990. Advances in Nephrology From the Necker Hospital 1991;20:19‐30. [MEDLINE: ] [PubMed] [Google Scholar]
Lin 1985
- Lin FK, Suggs S, Lin CH, Browne JK, Smalling R, Egrie JC, et al. Cloning and expression of the human erythropoietin gene. Proceedings of the National Academy of Sciences of the United States of America 1985;82(22):7580‐4. [MEDLINE: ] [DOI] [PMC free article] [PubMed] [Google Scholar]
Locatelli 2009
- Locatelli F, Covic A, Eckhardt KU, Holder R, ERA‐EDTA ERBP Advisory Board. Anaemia management in patients with chronic kidney disease: a position statement by the Anaemia Working Group of the European Renal Best Practice (ERBP). Nephrology Dialysis Transplantation 2009;24(2):348‐54. [MEDLINE: ] [DOI] [PubMed] [Google Scholar]
Locatelli 2011
- Locatelli F, Vecchio L. Erythropoiesis‐stimulating agents in renal medicine. Oncologist 2011;16(Suppl 3):19‐24. [MEDLINE: ] [DOI] [PubMed] [Google Scholar]
Lundin 1989
- Lundin AP. Quality of life: subjective and objective improvements with recombinant human erythropoietin therapy. Seminars in Nephrology 1989;9 Suppl(1):22‐9. [MEDLINE: ] [PubMed] [Google Scholar]
Macdougall 1999
- MacdougallI IC, Gray SJ, Elston O, Breen C, Jenkins B, Browne J, et al. Pharmacokinetics of novel erythropoiesis stimulating protein compared with epoetin alfa in dialysis patients. Journal of the American Society of Nephrology 1999;10(11):2392‐5. [MEDLINE: ] [DOI] [PubMed] [Google Scholar]
Macdougall 2005
- Macdougall IC. CERA (continuous erythropoietin receptor activator): a new erythropoiesis‐stimulating agent for the treatment of anemia. Current Hematology Reports 2005;4(6):436‐40. [MEDLINE: ] [PubMed] [Google Scholar]
Ortho Biotech Jansen Cilag 2001
- Raritan NJ. Division of Jansen Cilag. Important safety message. Eprex. Epoetin alfa: reports of pure red cell aplasia (PRCA) [memorandum]. Package insert warning 2001.
Palmer 2010
- Palmer SC, Navaneethan SD, Craig JC, Johnson DW, Tonelli M, Garg AX, et al. Meta analysis: erythropoiesis stimulating agents in patients with chronic kidney disease. Annals of Internal Medicine 2010;153(1):23‐33. [MEDLINE: ] [DOI] [PubMed] [Google Scholar]
Pappas 2008
- Pappas KD, Gouva CD, Katopodis KP, Nikolopoulos PM, Korantzopoulos PG, Michalis LK, et al. Correction of anemia with erythropoietin in chronic kidney disease (stage 3 or 4): effects on cardiac performance. Cardiovascular Drugs & Therapy 2008;22(1):37‐44. [MEDLINE: ] [DOI] [PubMed] [Google Scholar]
Summers 2005
- Summers S, Winnet G, Matijevic A, Carmichael D, Almond M. An audit comparing subcutaneous epoetin alfa to intravenous darbepoetin alfa in chronic hemodialysis patients. Dialysis & Transplantation 2005;34(6):358‐62. [EMBASE: 2005248464] [Google Scholar]
Warady 2006
- Warady BA, Arar MY, Lerner G, Nakanishi AM, Stehman‐Breen C. Darbepoetin alfa for the treatment of anemia in pediatric patients with chronic kidney disease. Pediatric Nephrology 2006;21(8):1144‐52. [MEDLINE: ] [DOI] [PubMed] [Google Scholar]
Ward 1990
- Ward HJ. Implications of recombinant erythropoietin therapy for renal transplantation. American Journal of Nephrology 1990;10 Suppl(2):44‐52. [MEDLINE: ] [DOI] [PubMed] [Google Scholar]
Winearls 1986
- Winearls CG, Oliver DO, Pippard MJ, Reid C, Downing MR, Cotes PM. Effects of human erythropoietin derived from recombinant DNA on the anaemia of patients maintained by chronic haemodialysis. Lancet 1986;2(8157):1175‐8. [MEDLINE: ] [DOI] [PubMed] [Google Scholar]
Wolcott 1989
- Wolcott DL, Marsh JT, Rue A, Carr C, Nissenson AR. Recombinant human erythropoietin treatment may improve quality of life and cognitive function in chronic hemodialysis patients. American Journal of Kidney Diseases 1989;14(6):478‐85. [MEDLINE: ] [DOI] [PubMed] [Google Scholar]
References to other published versions of this review
Cody 2002
- Cody J, Daly C, Campbell M, Donaldson C, Grant A, Khan I, et al. Frequency of administration of recombinant human erythropoietin for anaemia of end‐stage renal disease in dialysis patients. Cochrane Database of Systematic Reviews 2002, Issue 2. [DOI: 10.1002/14651858.CD003895] [DOI] [PubMed] [Google Scholar]
Cody 2005a
- Cody JD, Daly C, Campbell MK, Khan I, Rabindranath KS, Vale I, et al. Recombinant human erythropoietin for chronic renal failure anaemia in pre‐dialysis patients. Cochrane Database of Systematic Reviews 2005, Issue 3. [DOI: 10.1002/14651858.CD003266.pub2] [DOI] [PubMed] [Google Scholar]