

Abstract
Poor delivery efficiency continues to hamper the effectiveness of cancer therapeutics engineered to destroy solid tumors using different strategies such as nanocarriers, targeting agents, and matching treatments to specific genetic mutations. All contemporary systemic anti-cancer agents are dependent upon passive transvascular mechanisms for their delivery into solid tumors. The therapeutic efficacies of our current drug arsenal could be significantly improved with an active delivery strategy. Here, we discuss how drug delivery and therapeutic efficacy are greatly hindered by barriers presented by the vascular endothelial cell layer and by the aberrant nature of tumor blood vessels in general. We describe mechanisms by which molecules cross endothelial cell (EC) barriers in normal tissues and in solid tumors, including paracellular and transcellular pathways that enable passive or active transport. We also discuss specific obstacles to drug delivery that make solid tumors difficult to treat, as well strategies to overcome them and enhance drug penetration. Finally, we describe the caveolae pumping system, a promising active transport alternative to passive drug delivery across the endothelial cell barrier. Each strategy requires further testing to define its therapeutic applicability and clinical utilities.
Keywords: Caveolae, Endothelial cells, Drug delivery, Solid Tumors, Tumor targeting, Tumor vasculature
1. Introduction
Major breakthroughs in systemic therapies for the treatment of solid tumors have long been anticipated to come from innovative approaches such as those utilizing a new generation of nanodrugs that optimize drug stability and retention, the large scale genomic profiling of cancers, and high throughput screening of libraries to identify very specific therapeutic agents. Despite preclinical successes, the vast majority of experimental therapeutics based on these powerful approaches, however, have failed to offer much, if any, significant benefit for the treatment of solid tumors in clinical trials [1–5]. The few exceptions exhibiting anti-tumor efficacy tend to offer modest, incremental improvements at best over standard treatment regimens. None approach the groundbreaking successes of treatments for hematological “liquid” tumors such as imatinib in the first line treatment of chronic myeloid leukemia which resulted in a 10 year overall survival rate of ~84% [6].
Regardless of whether they are clinically treated with antibodies, small molecule drugs, nanomedicines or even antibody drug conjugates, solid tumors consistently seem to show poorer therapeutic responses to approved drug regimens than liquid tumors. The question of why this may be the case seems underappreciated and underexplored. For example, the nanoparticle Doxil, a pegylated liposomal doxorubicin, offers an overall median survival benefit of just over 15 months for recurrent ovarian cancer as a monotherapy [7]. Abraxane, a nanoparticle albumin–bound paclitaxel, created total sales measured in billions but only offers overall median survival benefits of just 11–12 months for NSCLC or pancreatic cancer patients treated not with the single agent but in combination with chemotherapies [8,9]. Though Doxil and Abraxane only marginally improve the therapeutic potencies of the chemotherapeutic agents they carry, they do, however, reduce their relative toxicities. Recent critiques that question the utility of nanomedicines may be somewhat harsh as their rather modest clinical impact is not unique to this class of agents [2,10].
Successful “personalized” trials matching treatments to specific cancer mutations such as BRAFV600E-mutant melanoma [11,12] and epidermal growth factor receptor (EGFR)-mutant (NSCLC) [13] show short lived or partial responses. The impact of even new modern targeted therapies on solid tumors seems rather modest and well below theoretical expectations for both potency and safety. For example, the antibody drug conjugate ado-trastuzumab-emtansine (Kadcyla), a targeted therapy carrying a highly potent microtubule toxin, offers an overall survival benefit of 30.9 months as a monotherapy in the treatment of HER2-positive advanced breast cancer patients after prior treatment with trastuzumab and a taxane [14]. Erbitux, a monoclonal antibody directed against the epidermal growth factor receptor, offers only an overall median survival benefit of 9.5 months for patients with wild-type K-Ras colorectal cancers [15]. Although these drugs are among the few cancer therapies that ultimately achieve FDA approval (5%), they exemplify different classes of therapeutic agents applied to the treatment of solid tumors that do not approach the level of success observed in the treatment of hematological malignancies. Even immune checkpoint inhibitors, deemed by some to show curative potential in the treatment of metastatic solid cancers, unlike any regimen currently available [16], offer limited overall survival benefits in addition to inducing very serious side effects. Ipilimumab, a fully human monoclonal antibody inhibitor of cytotoxic T-lymphocyte-associated antigen 4 (CTLA-4), offers a median OS of 11.4 months in the treatment of unresectable or metastatic melanoma [17]. In patients with advanced, previously treated NSCLC, overall survival was 12.6 months in a clinical trial testing atezolizumab, a humanized monoclonal antibody against programmed death ligand 1 (PD-L1) [18].
Despite revolutionary technologies enabling the discovery of novel targets and targeted therapeutic agents, why have the new age of targeted-by-design drugs not met theoretical expectations? Why have they increased patient survival benefit by mere months—not years and not cures? In addition to suboptimal preclinical tumor models [19,20], innate and acquired drug resistance [21], and poor therapeutic target identification, insufficient specific penetrance of anticancer drugs into tumors remains a key obstacle preventing the full clinical realization of many innovative therapies [22,23]. Modern drugs are clearly well-designed to recognize a single target molecule; they can do so with great specificity and thus are clearly targeted by intent. However, when administered intravenously, they frequently lack sufficient access to solid tumors where the intended target can be bound to elicit maximum pharmacological effect. Unlike liquid tumors, access to the actual malignant cells inside solid tumors is impeded by a formidable vascular wall formed by a monolayer of endothelial cells (ECs) that is further fortified and surrounded by perivascular cells and extracellular matrix. Each of these vascular components—the endothelial cells, the perivascular cells, and the extracellular matrix— contributes directly and/or indirectly in varying degrees to restricting solid tumor penetration by systemic drugs. They ultimately impact drug access to the inside of the tumor and the actual intended target(s) of the drug. Modest or absent antitumor activity exhibited by the most promising therapies in clinical trials could be improved markedly with better delivery systems that overcome vascular barriers to deliver more drug inside solid tumors. Drugs cannot be fully effective unless they can engage their pharmaco-targets and accumulate at local concentrations high enough to have the greatest therapeutic impact possible.
All current systemically delivered anti-cancer molecular therapeutics rely on passive transvascular mechanisms to penetrate malignant primary and metastatic solid tumors. Passive transendothelial delivery, however, tends to be slow and inefficient and requires a high drug concentration gradient across the tumor EC barrier to drive any drug into solid tumors. Consequently, the unfortunate reality for patients is that only a very small amount of a given drug (usually given at fairly high doses) actually reaches tumors (< 0.1–1%) [2,24–33]. Hence, to achieve therapeutically effective drug concentrations in neoplastic sites, ever-increasing drug doses (approaching the maximum tolerated dose) are necessary to create sufficient blood-to-tumor concentration gradients across the EC barrier to drive uptake and see some level of efficacy. As such, poor delivery efficiency and drug toxicity continue to hamper the effectiveness of multiple classes of therapeutics [2,24–31,34–42].
Passive transvascular delivery also thwarts the ability of even the best antibody-based medicines engineered to selectively seek and destroy cancer cells to achieve their full therapeutic potentials. This is most clearly illustrated by nuclear imaging studies using antibodies to target radionuclides into tumor sites. Most radiolabeled antibodies injected intravenously not only enter and accumulate in tumors quite slowly but clear from the blood slowly as well. Therefore, they require several days to weeks before a clear tumor signal can be properly imaged. Prolonged radioimmunoconjugate circulation in the blood renders radiosensitive tissues such as the bone marrow and liver vulnerable to toxic side effects and may be the principle reason why intravenously injected radioimmunotherapy has not been clinically effective for the treatment of solid tumors [5,43–46]. Even the newest immunotherapies such as immune checkpoint inhibitors could benefit greatly from specific and efficient tumor delivery that maximizes the immune response inside tumors while reducing or avoiding inflammatory and other toxic autoimmune processes in other organs [47].
As poor drug delivery can limit and even prevent therapeutic efficacy in the treatment of solid tumors, new, radically different perspectives are urgently needed to first understand and then hopefully overcome these significant biological barriers. In this review, we begin to describe major barriers obstructing therapeutic drug access to solid tumors and key pathways mediating passive transvascular delivery as well as examine a few different strategies that are being explored currently to improve tumor drug delivery and therapeutic efficacy. We also discuss how to get beyond passive transvascular delivery by harnessing a recently discovered active transport mechanism to achieve rapid tumor penetration that may someday help solid tumor treatments.
2. Vascular barriers
2.1. Normal blood capillary vessels
Systemic drugs administered via intravenous injection must overcome significant vascular barriers upon entering circulation to reach their intended tumor targets. Blood vessel permeability is regulated by a layer of endothelial cells that line the walls of capillaries, the primary site of exchange of material between blood and underlying tissues. In normal tissues, vascular barrier function is dependent upon the stability and integrity of these endothelial cells, their intercellular junctions, their attachments to surrounding basement membranes. Barrier function is also shaped by the back and forth communication between endothelial cells, their surrounding vascular cells such as pericytes as well as the parenchymal cells of the tissue itself. Variations in the presence or absence of these elements give rise to 3 broad types of capillary endothelium that are highly specific to certain types of organs: continuous, discontinuous and fenestrated [48] (See Fig. 1).
Fig. 1.
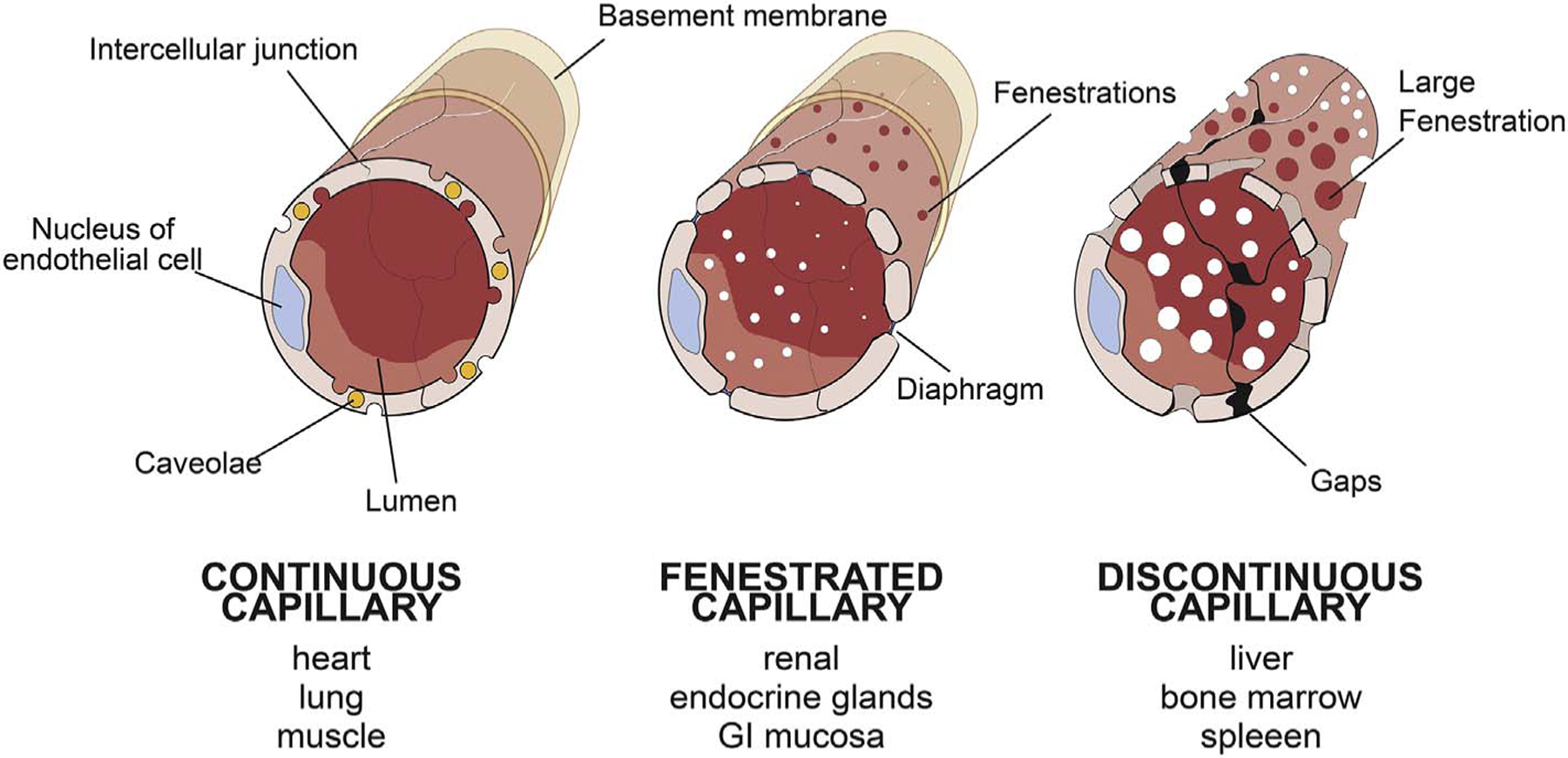
Major types of endothelial cells.
(A) Continuous endothelium.
(B) Fenestrated endothelium.
(C) Discontinuous endothelium.
2.1.1. Continuous endothelium
Continuous endothelium is the most restrictive of the endothelial types and is most prevalent among organs such as heart, lung, and muscle. It has an abundant population of specialized plasmalemmal invaginations called caveolae and intercellular junctions tight enough to restrict the exchange of macromolecules, usually over 2 nm. In a smaller subset of organs requiring special protection or a controlled internal environment such as the blood-brain barrier, testis, and retina, the continuous endothelia are even more restrictive and on par with the tightest epithelial cell barriers. In such endothelia, caveolae are absent and intercellular junctions are especially tight, only permitting the passage of water and very small solutes. They are so restrictive that they must express specific cell surface proteins to transport even molecules as small as glucose.
2.1.2. Fenestrated endothelium
The fenestrated endothelium is moderately restrictive to molecules and are designed for the more rapid, convective exchange of molecules required for proper organ function. They have abundant ~70 nm circular windows spanning the attenuated endothelial cells themselves which are usually sealed by a thin diaphragm. These fenestrae enable high water and small solute fluxes with a minimal permeability change to macromolecules and are found in the renal glomerulus and endocrine tissues.
2.1.3. Discontinuous endothelium
The discontinuous endothelium is characterized by large 100 to 200 nm wide gaps in between endothelial cells and minimal basement membranes. It is highly permeable to essentially all molecules but not cells. This endothelial type supplies organs with extreme filtering and defense functions such as the liver, spleen, and bone marrow.
It is quite apparent that the passage of molecules across blood vessel walls into normal organs and tissues is fine tuned, highly regulated and dependent upon morphological and molecular features of different classes of endothelial cells. Each endothelium is optimally designed to meet organ function needs and can actually be induced to switch from one subtype to another when vascularizing grafted tissues originating from different organs [49]. Specific factors have been identified that can modify endothelial permeability or even induce specific structures like fenestrations in continuous endothelia, as in the case of VEGF [50].
The vascular endothelium can thus be viewed as a master gatekeeper controlling passage of blood molecules and cells into the inside of the tissue (interstitium and parenchyma). As such, the many different types of endothelia in the body performs their regulatory gatekeeping duties in response to meeting the vastly different metabolic and homeostatic needs of resident host organs or tissues.
2.2. Tumor vasculature and vascular determinants of drug penetration and efficacy
Several structural and functional abnormalities in the tumor vasculature impede the systemic delivery of therapeutic agents into solid tumors and/or affect their overall efficacy [22,51,52]. At a gross level, tumor blood vessels lack the hierarchical and evenly spaced organization of well differentiated arteries, arterioles, capillaries, venules and veins of their normal counterparts. Chaotic, disorganized and morphologically torturous, tumor vessels also tend to be enlarged and dilated. The increased vascular surface area of tumor vessels contributes to increased vascular leakiness by creating more access points by which molecules can pass into the tumor interstitium. They also exhibit irregular branching patterns that include excessive loops and arteriovenous shunts. Consequently, blood flow patterns in tumor vessels tend to be erratic, sluggish, and lack a consistent unidirectional path which impacts the efficiency and uniformity of drug delivery.
2.2.1. Tumor endothelia and sprouting angiogenesis
Tumor endothelia may exhibit varying degrees of permeability to drugs that reflect different aspects of the 3 major endothelial types in a heterogeneous and limited manner. Many preclinical tumor models, especially subcutaneous ones, predominantly feature blood vessels derived from the proliferation and migration of endothelial cells from preexisting vessels in the surrounding microenvironment via a process called sprouting angiogenesis [53]. Angiogenic blood vessels in experimental tumor models may exhibit widened interendothelial cell junctions, lack organized basement membranes, feature ample fenestrations, and exhibit tenuous pericyte contact, all leading to increased permeability. Described by some to be a hallmark of cancer [54,55], sprouting angiogenic growth is induced by factors in the tumor microenvironment, the most important of which is vascular endothelial growth factor (VEGF). Through its actions on VEGF receptors, VEGF is critical for both inducing vascular sprouting to create new blood vessels to support tumor growth [56,57]. Importantly for drug access, VEGF also increases tumor vascular permeability by inducing fenestrations in endothelial cells and increasing transcellular pores through vesiculo-vacuolar organelles (VVOs, see section 4.1) which enables increased extravasation of fluids and nutrients as well as therapeutic agents across the vascular wall [50,58].
Sprouting angiogenesis, however, does not reflect the full range of vasculatures possible in human solid tumors [59–63] as the genetic and epigenetic profiles of tumor endothelial cells are influenced by extracellular signals originating from the tumor cells and the local stroma. Heterogeneity in tumor vasculature may be a major factor contributing to the disappointing clinical performances of contemporary anti-angiogenic drugs that are typically designed to block tumor angiogenesis by inhibiting VEGF, specific VEGF receptors or key downstream targets in VEGF receptor signaling pathways [56,57].
2.2.2. Vessel co-option
In addition to sprouting new blood vessels via angiogenesis, tumors can hijack and take over existing vasculature by migrating along the blood vessels of host organs in a VEGF-independent process called vascular or vessel co-option [61,64]. Vessel co-option may underlie some forms of intrinsic and acquired anti-angiogenic drug resistance in human cancer patients [59,60,65,66]. Mature vessels co-opted by tumors may lack the excessive leakiness of sprouting angiogenic vessels and thus reflect more the basal permeabilities of the host organ vasculature (our unpublished observations). Identifying specific functional or molecular biomarkers that define clinical tumor vessel phenotypes such as co-opted vessels will be critical in order to model them preclinically, to understand their underlying mechanisms of development, and to learn how they impact extravasation of drugs and therapeutic efficacy. As significant pools of human cancer patients with tumors using co-opted vessels may not be represented in many current tumor models that rely on angiogenic sprouting processes to develop blood supply, the inclusion of tumor models that utilize co-opted vessels will be very important in preclinical drug testing.
2.2.3. Heterogeneity in VEGF dependence and permeability among sprouting angiogenic vessels
Sprouting angiogenic vessels themselves are dynamic structures and can eventually lose their hyperpermeability and become VEGF-independent [52]. Early angiogenic vessels, referred to as mother vessels, tend to be lined with endothelial cells that are structurally abnormal with VEGF induced fenestrations and widened interendothelial gaps. This leads to hyperpermeability which can increase drug extravasation into tumors. Mother vessels, however, are transient structures that can evolve into one of three different daughter vessels—glomeruloid microvascular proliferations (GMPs), vascular malformations, and capillaries [52]. Resembling renal glomeruli, GMPs are VEGF-dependent and hyperpermeable to plasma and plasma proteins like mother vessels. In contrast, vascular malformations form when mother vessels acquire an asymmetrical coat of smooth muscle cells and eventually become impermeable to macromolecules and able to persist in the absence of VEGF. Capillaries evolved from mother vessels are VEGF-independent as well and form by intraluminal bridging from endothelial cell projections. Like normal capillaries, they are not hyperpermeable. Thus, early angiogenic mother vessels can lose responsiveness to anti-angiogenic therapies by evolving into VEGF-independent capillaries or vascular malformations. In addition, these classes of daughter vessels are relatively impermeable to macromolecules and may exhibit reduced drug extravasation.
2.2.4. Vasculogenic mimicry, intussusception, and vasculogenesis
Other forms of tumor vascularization may occur through processes such as vasculogenic mimicry, intussusception, and vasculogenesis. Vessels may be created through vasculogenic mimicry when tumor cells take on the properties of endothelial cells and are resistant to anti-angiogenic therapies.
Intussusception or non-sprouting angiogenesis is a process of vascular remodeling and expansion that subdivides pre-existing vessels to form new vessels [67–69]. The resulting vessels have normal physiological permeabilities and are not hyperpermeable like sprouting angiogenic vessels. Little is known about the molecular basis of intussusception but it has been suggested that it may represent an adaptive response to treatment with various antitumor and anti-angiogenic compounds [70].
Finally, tumor vascularization may also arise from vasculogenesis, a process formerly believed to only occur during embryogenesis where endothelial cells coalesce to form vascular (capillary) tubes that ultimately fuse and grow in a branching pattern to yield an ever-expanding capillary plexus. This plexus expands, connects with patent functional vessels, and matures with the addition of perivascular cells. In tumors, however, vasculogenesis has also been shown to occur through circulating bone marrow derived endothelial progenitor cells in the blood that differentiate into endothelial cells and incorporate into tumor sites [71]. Further study is needed to assess the impact of these forms of tumor vascularization on drug extravasation and/or tumor responsiveness to anti-angiogenic therapies.
2.2.5. Insufficient vascular density
Tumor cells often proliferate faster than the endothelial cells supporting their neovascularization. The resulting reductions in tumor vascular density lead to neoplastic cell populations situated at distances well over 100 μm from the nearest blood vessel. This contrasts sharply with normal cells of the human body which are typically within 50–100 μm of a capillary. Loss of proximity to the vasculature not only contributes to tumor cell hypoxia and metastasis but also to uneven drug penetration and distribution. Thus, insufficient drug penetration may underlie some forms of drug resistance observed in human patients as even hypoxic regions of solid tumors have been shown to house cancer stem cells with the potential to repopulate tumors after their destruction by therapeutics [72].
2.3. High interstitial fluid pressures
Drug penetration can also be impeded by increased interstitial fluid pressures (IFP) in solid tumors created by a combination of vessel compression by proliferating cancer cells, insufficient lymphatic drainage and/or increased extravasation of proteins and fluids from leaky tumor blood vessels. A reported feature of many types of solid human tumors including breast and colorectal carcinomas and metastatic melanoma, interstitial pressures tend to be uniformly elevated throughout the center of the tumor and drop precipitously in the periphery [73,74]. In normal tissues, transcapillary pressure gradients tend to be slightly negative or fairly low and enable outward flow into tissues. The convective force from water flux drags other molecules across the EC barrier. In contrast, high IFPs in solid tumors reduce hydrodynamic driving forces on drugs moving passively across the EC barrier and thus can impede drug penetration and reduce therapeutic efficiency. Higher IFPs tend to correlate with poorer overall prognosis for human patients [75], however, this is not always the case [76,77]. IFP can vary considerably and may be abnormally high in many current tumor models in part because fast tumor growth may impair the proper and synchronized development of vascular and lymphatic vessels. Whether desynchronized vessel development is widely prevalent in the more slowly growing tumors of many human patients remains to be determined.
3. Paracellular transport across the endothelium
Transport across the EC barrier is possible via routes between the cells (paracellular) or through the cell (transcellular). In normal tissues, the paracellular route mediates most of the diffusive and convective transport of fluids, solutes, and in some cases small macromolecules through interendothelial cell junctions. In normal endothelium under basal conditions, the intercellular junctions restrict macromolecular exchange in a size-dependent manner that can be modelled as cylindrical pores. In solid tumors, most therapeutic agents (including targeted antibodies, radioimmunotherapies, nanoparticles, and small molecule drugs) are dependent upon passive diffusive delivery in part through paracellular transport (Fig. 2).
Fig. 2.
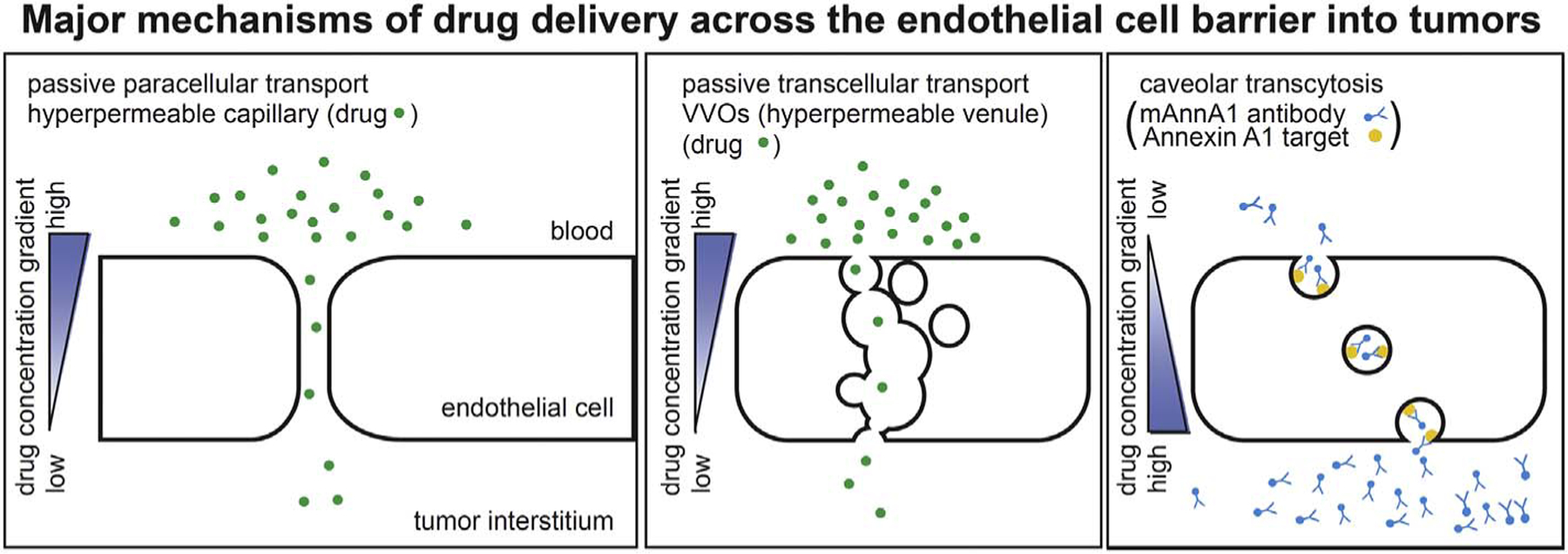
Major mechanisms of drug delivery across the endothelial cell barrier into tumors.
Schematic representations of passive paracellular drug transport via interendothelial junctions, passive transcellular drug transport via vesiculo-vacuolar organelles (VVOs), and active drug transport via caveolar transcytosis. Note these schematic diagrams depict tumor continuous endothelial cells lacking fenestrations.
At least 2 types of junctional structures—adherens and tight junctions— form between endothelial cells. Endothelial cell-cell junctions are composed of different adhesive transmembrane proteins that are expressed in adjacent endothelial cells and are anchored to the actin cytoskeleton via cytoplasmic molecules. Tight junctions tend to be located more apically than adherens junctions and are composed of transmembrane adhesive proteins such as the occludins, claudins, and junctional adhesion molecule-1 (JAM-1) (see review [78]). Adherens junctions are comprised of cadherin family members such as vascular endothelial cadherin (VE-cadherin) and anchor to the actin cytoskeleton via members of the catenin family [78]. These adhesive molecules create junctions when they link with each other across the intercellular space between adjacent endothelial cells. They create a molecular sieve that regulates the paracellular passage of molecules and are subject to upstream regulatory control by molecules such as VEGF [79].
3.1. Paracellular transport into tumors
The tightness of endothelial cell intercellular junctions varies considerably among normal tissues, from extremely restrictive in brain, testis and retina to wide open and very leaky in liver and bone marrow. This wide variability is reflected in the vasculatures of solid tumors. Though tumor blood vessels tend to be leakier than their normal tissue counterparts, they do not usually reach the levels of the leakiest vessels supplying normal tissues (i.e. liver and bone marrow). The tight and adherens junctions in tumor vessels of some animal models are aberrant, and even wide-gapped, and thereby exhibit increased endothelial permeability permitting passage of endogenous macromolecules. This increased permeability is the primary mechanism by which most anti-cancer agents enter the tumor interstitium. This passive transvascular drug delivery requires a concentration gradient between the blood and the tumor interstitium to passively drive drugs across the endothelial barrier to be eventually taken up into the tumor parenchyma—the tumor tissue itself. In normal tissues, low interstitial pressures enable convective movement of fluids and solutes into tissues. In tumors, however, high interstitial fluid pressures probably prevent convection, rendering delivery primarily, if not solely, by diffusion. Poor blood flow can also slow down passive drug extravasation from blood vessels. The need to create a concentration gradient large enough to move drugs by diffusion across interendothelial junctions is the reason why increasingly high dosages are required and why high potency anti-cancer drugs are sought to effectively treat human tumors. Even antibody-drug conjugates intending to target tumor cell surface antigens require some of the most potent toxins known. However, as discussed above, not all tumors have a preponderance of sprouting angiogenic blood vessels that exhibit increased vascular permeability. In such cases, a dependence on passive mechanisms to deliver drugs would lead to poor and insufficient drug accumulation in tumors.
A sub-mechanism of passive transvascular delivery that has gained popularity is the so-called enhanced permeability and retention (EPR) effect. First described by Maeda, the EPR effect posits that the high vascular permeability of solid tumors enables the selective entry and accumulation of compounds of a certain size range preferentially in tumors over normal organs. Furthermore, the tumor retention of these compounds is facilitated by poor to absent lymphatic drainage that limits egress [80]. The EPR effect is a major consideration in the contemporary design of nanoparticles carrying anti-cancer agents and is proposed as a mechanism that promotes selective drug delivery and targeting into tumors over other organs. It has also been commonly invoked to justify passive delivery strategies for a wide spectrum of therapeutics including antibodies, antibody-drug conjugates, and even smaller compounds.
According to this model, if properly sized, compounds will selectively enter solid tumors due to high vascular permeabilities. They will then be retained inside the tumor because of poor lymphatic drainage. The classic EPR perspective predicts tumor uptake and accumulation will not benefit from binding to specific targets inside tumors; “targeted” and “untargeted” probes of the same size will thus be retained similarly, regardless. Theoretically, the optimal size of a candidate nano-drug needs to be large enough to avoid renal excretion but small enough to avoid liver uptake and sequestration. EPR may be a simple and unrealistic model of drug delivery, however, as tumors vessels are not as leaky as those in the liver or reticuloendothelial system (RES). In addition, tumor vessels tend to be far more leaky in most standard tumor models than in many solid tumors in humans. Thus, a truly perfect window for nanoparticle size is unlikely ever to be defined for actual human tumors. Indeed, the high failure rate of experimental nano-cancer therapeutics in clinical trials [2] and the high variability in permeabilities across different types of tumor vasculatures together suggest that the EPR effect may be insufficient to achieve the levels of selective tumor targeting and penetration necessary to generate therapeutically optimal drug concentrations in tumors without comparable non-toxic concentrations in other organs or even in blood. In a recent analysis of delivery efficiency from over 200 preclinical data sets, a median of 0.7% of the injected nanoparticle dose actually ever reaches a solid tumor [2]. Thus, while targeting drugs to tumors may reduce some toxic side effects, current targeted anticancer therapies rely upon a high concentration gradient to enter tumor sites and are thus subject to the same delivery constraints as their untargeted counterparts.
4. Transcellular transport across the endothelium
Transcellular routes mediate the passage of molecules through the endothelial cell itself and are an alternative strategy for cancer drug delivery that needs broader consideration among current therapeutics. Transcellular transport occurs through five different mechanisms:
Transmembrane movement of water and small molecules either passively across the lipid membrane or via specific membrane proteins that facilitate or even actively transport (e.g. ion pumps, glucose transporters).
Fenestrations in endothelial cells, as described in the previous section 2.12, Passage across fenestrations is a passive process.
Transendothelial channels which form transiently and infrequently from the union of several caveolae in a string extending across the endothelium’s luminal to abluminal surface. These transendothelial channels essentially mimic a cylindrical pore from the luminal surface to the abluminal surface of the cell and exhibit selective permeability based on the size of the pore itself. They constitute a passive transvascular delivery route.
Vesiculo-vacuolar organelles (VVOs) are grape-like clusters of interconnected vesicles and vacuoles that span the cytosolic spaces of individual endothelial cells lining capillary venules [58]. They create a convoluted pathway across the cell which resemble bloated transendothelial channels but are not necessarily composed of caveolae. Requiring a specific signal to open, VVOs constitute a passive transvascular delivery route by which even nano-sized cargo can easily enter and move passively through the endothelial cell to reach the tissue interstitium. This passive form of transport is driven by a gradient across the EC barrier created by higher concentrations of molecules in the circulating blood relative to concentrations found inside the tissue.
Caveolae are flask-shaped plasmalemmal invaginations which mediate vesicular membrane trafficking or transport of molecules across the cell through invagination, budding, docking, and fusion [91,92,101,136]. For decades, caveolae were initially associated with classic pinocytosis (fluid phase endocytosis) in many cell types. As transcytotic vesicles in endothelium, caveolae can mediate fluid phase transcytosis and even contribute to the passive transvascular transport of large macromolecules. These macromolecules enter via caveolae ostia (openings at the membrane) and travel through a 20 nm neck region to the bulb of the caveolae. In addition, a more selective receptor-mediated transcytosis can occur when ligands bind “receptors” in caveolae. Recently, antibodies targeting proteins highly enriched in caveolae revealed the additional role of caveolae as a pumping system that can traffic caveolae targeting antibodies and their cargo actively across the EC barrier, even against a concentration gradient [37–39,81].
These modes of transcellular transport can all mediate passive transendothelial delivery and are all regulated to some degree, however, only caveolae and certain transmembrane pumps actively transport molecules as well. In terms of tumor drug delivery, routes via caveolae or VVOs are highly relevant and applicable but are not currently utilized as major drug delivery strategies in the clinic (Fig. 2).
4.1. Vesiculo-vacuolar organelles (VVOs)
Passive transcellular passage of anti-cancer drugs into tumors is theoretically possible through structures discovered by Drs. Harold and Ann Dvorak and their colleagues termed vesiculo-vacuolar organelles (VVOs). Found in both human and animal tumors as well as normal tissues, VVOs have been described as largely “sessile structures” that are unlikely to shuttle back and forth across the endothelial cell cytoplasm like caveolae [58]. Electron micrograph (EM) serial sections show that most VVO clusters extend from the luminal to the abluminal plasma membrane of venular endothelial cells and thus, in principle could serve as a pore-like conduit through which molecules can diffuse or hydrodynamically flow across the EC to access the tumor interstitium [82]. Electron microscopy experiments using horseradish peroxidase and ferritin tracers show clear transvascular movement of large macromolecules down a concentration gradient where concentrations are high in the vessel lumen, lower inside the VVO progressively and even lower exiting the VVO and in the interstitium. These studies also show the thin stomatal diaphragms separating each vesicle-vacuole can be in either an open or closed state and thus affect the passage of molecules through the VVO network [82–84]. VVOs of normal tissues frequently exhibit closed diaphragms and permit minimal passage of molecular tracers whereas those from angiogenic tumor models or ad-VEGF-induced tissue models are hyperpermeable and have been observed to rapidly release tracers into the tumor interstitium [82–84].
Interestingly, Dvorak and colleagues posit that, in tumors, VVO stomatal diaphragms may be opened by VEGF secretion and may be a significant contributing factor to tumor vessel leakiness in some tumor models. This hypothesis is supported by the observations that 1) VVOs from normal tissues can be induced to become hyperpermeable to tracers with the application of VEGF (also histamine and serotonin) in animal models [82,85], and 2) VEGFR2 localizes to tumor VVOs [86]. In addition, the large amount of membrane stored in VVOs appears to contribute to the formation of mother vessels, early angiogenic structures, as venules enlarge significantly [85,87,88].
Beyond a general morphological characterization, little is known about the molecular composition of VVOs and whether they are related to caveolae. Although subsets of VVOs have been detected to colocalize with caveolin-1, caveolin-1 knockout mice have no caveolae but normal numbers of VVOs, suggesting that VVOs are not structurally similar to caveolae. However, caveolin-1 knockout mice also appear to exhibit reductions in permeability in certain tissues and reduced numbers of mother vessels, suggesting that VVOs may be functionally dependent on caveolin-1. In terms of transport mechanisms, it is not entirely clear how the entry of molecules into VVOs and across the endothelial cell monolayer is controlled. Given their predominance in tumor venules, and robust ability to extravasate macromolecules across endothelial cell barriers, further molecular and functional characterization of VVOs is warranted and would potentially enable a way to harness this important transcellular route to improve drug delivery.
4.2. Transcytosis via caveolae, an active transport mechanism
Active transvascular delivery mechanisms require energetic processes to move molecules from the vascular side of the endothelial cell barrier into the tissue interstitium and can do so against a concentration gradient. This is an important consideration, because efficient active transport does not require high drug concentrations to drive transvascular delivery into tumors via diffusion or hydrodynamic drag. Active transport also does not require intravascular fluid pressures exceeding interstitial fluid pressures to push delivery. In fact, the elevated interstitial pressures often seen in solid tumors need not deter active transport mechanisms such as those utilizing caveolae. Caveolar trafficking into and across the endothelial cell can occur uncoupled and independently from hydrodynamic and convective transvascular forces.
Over the course of 30 years, our group has been studying the caveolar transport pathway, a major transcellular route in endothelium. First discovered more than a half century ago by George Palade [89,90], noncoated plasmalemmal vesicles now termed caveolae are ~70 nm flask-shaped membrane invaginations that are distinct from clathrin-coated vesicles and act as dynamic transport vesicles [91–95]. They mediate endocytosis in many cell types and transcytosis, particularly in endothelium [34,93–96]. Numerous in quantity, caveolae can constitute up to 50–70% of the endothelial cell surface membrane and 15% of the total endothelial cell volume in vivo [97]. A distinctive feature of most continuous endothelia such as those of the lungs and heart, caveolae move across the endothelial cell, from the luminal (blood) side to the abluminal (tissue) side, to transport cargo from the blood to reach the inside of the interstitium and parenchyma of target organs and tissues. Initially predicted by Palade to be able to engulf and carry a “quanta” of plasma cargo [98,99], caveolae can carry proteinsized molecules across the EC into tissues by budding away from the luminal EC surface to reattach with the plasma membrane on the other side of the EC (abluminal surface) and release their contents.
Early debates over whether caveolae were static or dynamic structures extended over many decades until experiments showed that caveolae could be induced to bud from the plasma membrane via specific molecules mediating vesicle budding, docking and fusion [92,100]. We have shown that: i) caveolae are released from the plasma membrane to form free caveolae through a membrane fission process that utilizes energy from GTP hydrolysis [92,101]; this process is mediated by dynamin, a GTPase which forms an oligomeric spring-like coil at the neck of caveolae ii) both nonhydrolyzable GTP (GTPɣs) or a mutant dynamin that is unable to hydrolyze GTP prevent caveolae budding and trafficking, and (iii) caveolae trafficking requires specific SNARE/SNAP proteins that mediate docking and fusion of the budded caveolae with their target membranes [91]. Thus, caveolae harbor specific molecular machinery for their own trafficking and utilize cellular energy to move molecules into and across the endothelial cell. Like caveolae, clathrin-coated vesicles can also mediate endocytosis; they are well known to transport maternal IgG in the blood across the placental endothelium and tissue to reach the fetal blood. However, many endothelial cells, especially those of continuous endothelium, harbor few clathrin-coated vesicles but have an abundance of caveolae. Caveolae-mediated transcytosis bypasses lysosomes and their degradative enzymes, an important consideration for therapeutic drug delivery. Thus, given these properties, caveolar transport could have significant potential as a transendothelial drug delivery mechanism for therapeutic agents.
4.3. Kinetics of caveolar transport into normal tissues
Extensive studies of caveolar transport in normal lung tissues provided the first demonstrations that caveolae targeting is a viable strategy to actively transport cargo across the endothelial cell barrier [37,38,81]. In uncovering the caveolae pumping system, these studies also showed caveolae carry molecular cargos that are highly specific for different tissues. Active transcytosis cannot be properly studied in vitro, because cultured endothelial cells phenotypically drift and develop fewer caveolae. They also lose tissue-specific protein expression due to the absence of the tissue microenvironment. Therefore, we realized after a decade of EC culture studies that caveolae expression and transport studies must be done in vivo, requiring the development of new technology. Our subsequent transport studies into lung tissues relied upon a sophisticated in vivo imaging system and a monoclonal antibody we developed that specifically recognizes aminopeptidase P2 (mAPP2). Using mass spectrometry and western blotting techniques, in vivo proteomic analysis of caveolae and the luminal plasma membranes of vascular endothelium—each isolated directly from rat lungs—discovered that aminopeptidase 2 (APP2) is expressed on EC surfaces abundantly in lung and not other organs; APP2 is also highly concentrated in lung EC caveolae [38]. EM analysis confirmed the caveolar localization of APP2 and showed mAPP2 specifically targets caveolae in lung endothelium (Fig. 3) [37].
Fig. 3.
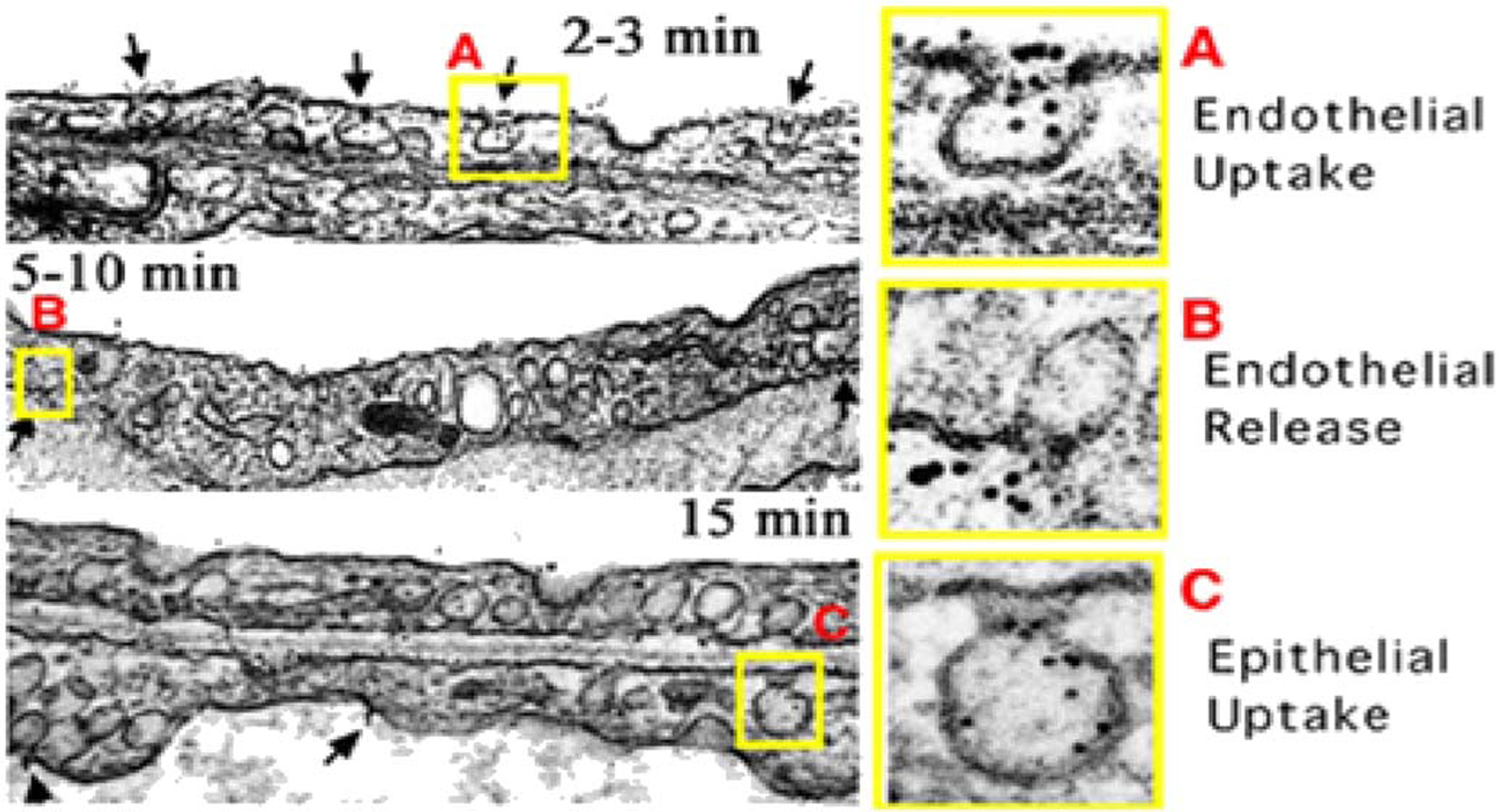
Sequential transcytosis of caveolae-specific antibodies across the vascular endothelium in vivo.
(A) Electron micrographs showing the gold particle labeled mAPP2 antibody entering and targeting luminal caveolae within 2–3 min after perfusion through isolated lungs.
(B) Transcytosis with clear endothelial release of gold particles from abluminal caveolae into the underlying perivascular space and lung interstitium.
(C) Gold particles taken up by epithelial caveolae, transcytosed across the epithelium, and released into airways. (Reprinted with permission from Ref. [36]. Copyright 2002 National Academy of Sciences [Proceedings of the National Academy of Sciences of the USA]).
To move beyond static EM images of caveolae targeting at fixed time points, we used high resolution intravital microscopy (IVM) to visualize in real-time, EC binding and processing of fluorophore-labeled mAPP2 in lung tissue after intravenous injection of live mice (Fig. 4a). These dynamic imaging studies revealed rapid delivery of mAPP2 into lung tissue. First, EC surface binding of the mAPP2 probe was readily observed within 10 s after intravenous injection. Next, transport of the probe from the bloodstream and across the EC barrier occurred within the first minute. This event was followed by the mAPP2 probe flooding the lung tissue thoroughly in < 10 min [37]. In line with the nano-molar affinity of these antibodies, low injection doses, measured in μg/kg (< < 1 nmol/kg) and yielding low nM blood concentrations, showed ample EC binding and pumping. This antibody cleared from the blood and concentrated inside the lung tissue within minutes of injection. In contrast, control IgG antibodies remained in the blood circulation; and antibodies to the EC surface marker CD34 bound and concentrated at the EC surface without tissue entry. At 1 h post-injection, neither antibody extravasated into lung tissue, even at 10 times the mAPP2 dose.
Fig. 4.
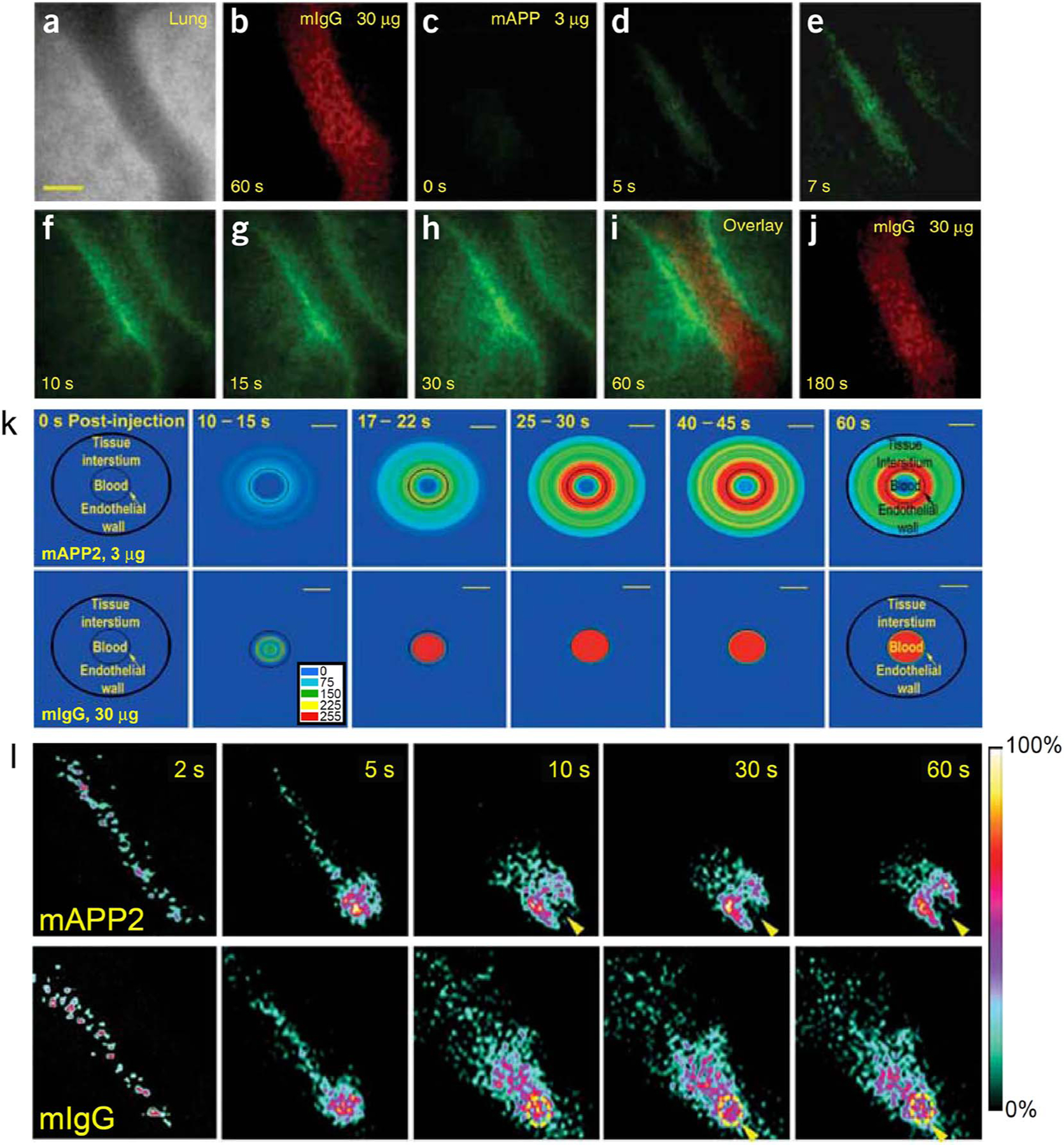
Rapid and specific mAPP2 delivery into in vivo lung tissues via caveolae targeting.
(A–J) High-magnification intravital fluorescence microscopy of solitary lung microvessels. Athymic nude mice with implanted lung tissue were injected through the tail vein with indicated fluorophore-labeled antibodies (30 μg of control antibody, mIgG-A568, followed 1 min later with 3 μg mAPP-A488). (a) Phase-contrast image of microvessel. (b) Fluorescence imaging of control mIgG-A568 60 s after injection showing ample intravessel signal but no detectable extravasation. (c–h) Fluorescence imaging at the indicated times after mAPP-A488 injection. Despite a minimal intravessel signal, the low injected dose produces a progressively concentrated signal first at the endothelial cell surface and then within the perivascular space inside the tissue. (i) Coregistered dual fluorescence image taken 125 s after mIgG-A568 injection and 60 s after mAPP-A488 injection. (j) Imaging of control mIgG-A568 at 180 s after injection shows signal in the microvessel lumen but not in the tissue interstitium.
(K) Polar plots of processed fluorescence images in tissue cross-section. As measured from IVM images of solitary lung microvessels collected after intravenous injection of fluorophore-labeled mAPP2 or IgG antibodies, fluorescence intensities were converted to polar coordinates using the central axis of the blood microvessel as the zero-reference point. The average intensity for each antibody is represented in pseudo-color (see embedded colorimetric scale) at the post-injection times indicated. The outer border of the tissue space examined is represented by a black circle. The blood-endothelial cell interface is represented by the thin lined, smaller circle. Scale bar, 20 μm.
(L) Dynamic and planar ɣ-scintigraphic live imaging of rats injected with 125I–labeled mAPP2 or mIgG antibodies, as indicated. Representative static frames captured at the indicated times show rapid and specific accumulation of mAPP2 but not mIgG within the rat lung. For mAPP2, arrowhead denotes heart shaped cavity rendered by apparent lack of signal in blood-engorged heart and strong signal in lung. For mIgG, dotted yellow circle and arrowhead indicates heart with significant circulating signal from control IgG. (Reprinted with permission from Ref. [37]. Copyright 2007 Nature Publishing Group [Nature Biotechnology]).
Even when blood levels of mAPP2 were lower than its tissue concentration, the antibody still continued to move across the vascular endothelial cell barrier and accrue inside lung tissue despite a considerable transvascular concentration gradient. Unlike standard or facilitated diffusion, active transport is the movement of a substance against its concentration gradient (from low to high concentration). Thus, by definition we have observed active transport or pumping of the caveolae targeting antibody probe across the vascular wall of the lung. This process of caveolar trafficking requires energy. Our basic studies on molecular mechanisms of caveolae trafficking in EC show the energy source, guanosine-triphosphate, must be hydrolyzed by a specific GTPase dynamin to induce caveolar budding and delivery [92,101]. This transvascular pumping also requires caveolae and is absent in the vascular endothelium of lung tissue from caveolin-1 knock-out mice [37]. Without caveolae, mAPP2 only bound the EC surface and went no futher into or across the cell to penetrate the lung tissue.
Noninvasive dynamic ɣ-scintigraphy and SPECT-CT imaging studies of the whole rat verified the rapid kinetics and speed of mAPP2 targeting observed by IVM and established mAPP2 tissue uptake is very specific for lung (Fig. 4b). Rapid and specific lung targeting was readily observed with radiolabeled mAPP2 injected IV [37,81]. A lung silhouette was first discernible 10 s post-injection, and the heart could be seen as a dark, signal-free shadow. By 1 min, the lung signal was already clear and specific. In contrast, control mIgG was detected throughout the entire animal and exhibited little organ targeting with > 100-fold less lung uptake. A comprehensive biodistribution analysis of a radiolabeled APP2 antibody showed that > 70% of the injected antibody dose (%ID) accumulates in the lungs as early as 5 min after IV injection. Blood levels dropped concomitantly and very rapidly. Uptake in other organs was minimal [81]. Targeting indices at 1 h were unprecedented and included a mean tissue targeting index of 556 (TTI = antibody in tissue per g of tissue per antibody in blood per g of blood), a tissue selectivity index of 39,180 (TSI = TTI for targeting IgG per TTI for control IgG) as well as 446-fold more mAPP2 antibody in the lung than control mIgG; these indices indicated specific, rapid and significant lung delivery [37,81]. High levels of signal in lung tissue were observed over a prolonged period with significant levels present even 30 days after injection [81].
Further analysis showed that cumulative residence of mAPP2 was substantially higher in the lungs compared to all other organs and tissues [81]. Within minutes of IV injection, the mAPP2 lung concentration greatly exceeds its levels in other organs, even the blood. Consistent with an active transport mechanism, peak mAPP2 levels in the lungs even exceeded its peak blood concentration at the time of injection. Passive transvascular delivery mechanisms cannot achieve these levels of antibody accumulation on these rapid timescales. Other targeted antibodies typically take days for tissue accumulation to exceed concomitant blood levels and certainly never approach peak blood levels. In contrast, mAPP2 can concentrate specifically in the lung 10 or more times greater than the maximum blood concentration and > 500 times more than the concomitant blood level; this extraordinary feat has yet to be accomplished with any other probe.
These studies illustrate how the caveolae pumping system fundamentally differs from passive transvascular delivery in its robust ability to move cargo against a concentration gradient even with low doses of a caveolae targeting agent (e.g. APP2 antibody). Active delivery via caveolae exceeds passive transvascular delivery into lung by > 100-fold. mAPP2 is the first probe to be pumped across vascular endothelium and actively penetrate a single tissue of the body very specifically. Approaching the theoretical ideal of targeting, mAPP2 concentrates very rapidly and robustly in the lungs and is cleared by pumping from the blood within minutes of IV injection.
5. Strategies to improve passive drug delivery into tumors
There are multiple pathways mediating passive transvascular transport across structures such as EC junctions and VVOs that can be regulated by specific factors and signaling pathways. This has led to several interesting new strategies to enhance drug delivery and therapeutic efficacy in solid tumors.
5.1. Vessel normalization
The vessel normalization hypothesis defines a strategy to increase passive transvascular drug delivery into tumors through the use of anti-angiogenic therapies that target VEGF and its downstream signaling partners [102]. VEGF was initially discovered as a vascular permeability factor by Harold Dvorak [103]. It was later cloned by Napoleon Ferrara [104], who led the development of the first anti-angiogenic directed at VEGF, bevacizumab. A humanized monoclonal VEGF antibody that blocked the growth factor’s ability to draw and maintain tumor blood vessels, bevacizumab dramatically decreased vessel density and inhibited tumor growth in preclinical tests [105]. As a first line monotherapy in clinical trials, however, the VEGF antibody offered no additional survival benefit to human patients. Instead, a modest improvement in the treatment of different cancers was observed when it was co-administered with standard chemotherapy agents [106]. The underlying mechanism for this clinical synergistic combination, was not immediately obvious—how was it possible to increase the efficacy of therapeutic drugs if the leaky blood vessels required to deliver them were being destroyed?
In studies of first generation experimental anti-angiogenic drugs such as TNP-470 and minocycline, Beverly Teicher presaged these clinical effects in experimental tumor models and proposed that anti-angiogenics might potentiate chemotherapies by affecting blood vessels and the surrounding microenvironment and thereby improving drug delivery [107–109]. To explain the clinical findings with bevacizumab, Jain further refined this idea and championed the concept of vessel normalization, a still controversial and counterintuitive working model that specifically posits anti-angiogenics increase drug delivery into tumors by making vessels appear and function more normally [102]. In this process, loss of VEGF signaling enhances tumor blood flow and drug delivery by pruning abnormal, poorly functioning vessels, normalizing endothelial cell function, and ultimately reducing high tumor interstitial fluid pressures. Thus, anti-angiogenics such as bevacizumab may exert a spectrum of effects on tumor vessels. At high doses or with long term use, they may rapidly prune away tumor blood vessels and effectively shut down tumor growth, but as a result, also reduce drug access to tumors in the process. At lower doses or when used on shorter timescales, anti-angiogenic drugs may create a brief window of opportunity that may improve delivery by normalizing the tumor vasculature, alleviating interstitial fluid pressures, and thus enhancing blood flow and drug delivery into tumors.
Vessel normalization is a counterintuitive idea, because VEGF signaling increases vessel permeability through multiple pathways (e.g. VVOs, fenestrations, and intercellular junctions) and thus promotes passive paracellular delivery of drugs into tumors. Blocking VEGF signaling in the tumor microenvironment would therefore reduce drug accumulation in tumors. However, vascular hyperpermeability can also lead to excessive tumor interstitial hypertension and indirectly stall drug passage by eliminating gradients between vascular and interstitial pressures. In this scenario, loss of VEGF signaling through anti-angiogenic treatment would then improve drug passage into tumors. The conditions under which anti-angiogenics might improve drug delivery, therefore, are highly specific and require high tumor interstitial pressures and poor lymphatic drainage– conditions that do not necessarily apply to all tumors. Indeed, based on early preclinical investigations, Teicher and colleagues predicted heterogeneity in the responsiveness of tumors to anti-angiogenics [109].
This prediction appears to have been borne out in in clinical trials. Application of vessel normalization strategies by pretreating tumors with anti-angiogenics prior to delivery of chemotherapeutic agents has yielded mixed results both in terms of improving treatment outcome and/or blood perfusion of tumors [110,111]. In one clinical study, Van der Veldt and colleagues directly observed reductions in drug delivery into non-small cell lung cancer (NSCLC) tumors several days after administration of an induction dose of bevacizumab [111]. This finding was supported in a separate imaging study by Jain and colleagues where overall reduced blood flow, volume, and permeability were also observed in a group of NSCLC patients after anti-angiogenic induction [110]. This latter study extended its observations by also showing that patients could be stratified based on changes in blood flow patterns after treatment. The best responders to the bevacizumab and chemotherapy combination were those few patients who showed improved blood perfusion of tumors after anti-angiogenic induction.
Concurrent delivery of anti-angiogenics with other therapeutic agents also has led to mixed clinical results (see review [106]). Many of these combination therapies have not improved patient survival outcomes in clinical trials. For some observed failures, it has been suggested that optimal scheduling and dosing for anti-angiogenic and chemotherapy delivery needs to be further tested [112,113]. The wide variability in the clinical literature, however, highlight the subtleties and difficulties inherent in the application of vessel normalization strategies. The optimum anti-angiogenic dose and drug regimen for combination therapies incorporating anti-angiogenics are likely to differ among different cancer types and even within individual patient groups. In addition, unlike many current tumor models, a significant percentage of human solid tumors may obtain blood supply through non-angiogenic, VEGF-independent processes such as vessel co-option and thus are inherently resistant to anti-angiogenic therapies [59, 61, 65, 66]. Identifying biomarkers that will help predict which patients will likely benefit most from this approach is a critical step towards more consistent outcomes.
5.2. Vascular promotion
Vascular promotion is another strategy designed to facilitate passive transvascular drug delivery into solid tumors by increasing vessel density, vascular permeability, as well as enhancing tumor blood flow. It combines the pro-angiogenic properties of low dose cilengitide, an integrin inhibitor, with the vasodilating properties of verapamil, a calcium channel antagonist. Verapamil, a drug approved for treating high blood pressure and angina pectoris, increases the supply of blood and oxygen to tissues by relaxing smooth muscle lining blood vessels and has previously been shown to increase tumor blood flow in vivo in a preclinical model for mammary adenocarcinoma [114]. Cilengitide is an RGD-based cyclic peptide that was designed to function as an anti-angiogenic agent through the inhibition of αvβ3 integrins which are highly upregulated in tumor endothelial cells [115,116]. It also has affinities for the related integrins αvβ5 and α5β1 [115]. The RGD motif (Arg-Gly-Asp) is a minimal binding site that enables endogenous extracellular matrix proteins such as fibronectin to bind to integrins [117]. Despite relatively low toxicities in early clinical testing and strong preclinical efficacy, cilengitide (at high doses) failed to demonstrate therapeutic benefit in the treatment of glioblastoma in a large, randomised Phase 3 clinical trial [118]. Though its further clinical development as an anticancer drug was subsequently terminated, cilengitide may still be repurposed as part of a vascular promotion therapy.
The Hodivala-Dilke research group has observed that, at low doses, RGD-based integrin inhibitors like cilengitide become paradoxically pro-angiogenic and increase tumor vascular density and growth in preclinical tumor models reportedly by attenuating VEGFR2 degradation and promoting VEGR2 recycling [119]. Using this property of low dose cilengitide, they demonstrated a triple cilengitide/verapamil/gemcitabine combination therapy outperforms gemcitabine alone in orthotopic models for pancreatic cancer and human lung cancer as well as a genetically engineered mouse model for pancreatic cancer [120]. Improvements in gemcitabine’s therapeutic efficacy were also accompanied by significant increases in tumor blood vessel densities and reduced levels of tumor hypoxia [120].
Testing the ability of vascular promotion therapy to improve blood perfusion in human tumors and enhance the effects of other classes of anticancer agents in preclinical models will be a rational step. Though therapeutic doses of verapamil or cilengitide are both well tolerated by human patients, their combined efforts could still potentially impact patients presenting with other health conditions like hypertension or cardiac disease. Therefore, determining the tumor specificity of cilengitide through complete imaging analyses as well as the toxicity of its combination with verapamil will also be important. Like vessel normalization, however, this approach is likely only applicable to VEGF-dependent vessels and may benefit from efforts to identify imaging and molecular biomarkers to identify cancers that are most likely to respond to anti-angiogenics. In addition, it opens the question of whether other anti-angiogenic drugs elicit similar dosage dependent effects on tumor vasculature.
5.3. iRGDs
iRGDs are unique peptides that are designed to penetrate tumors by incorporating two separate peptides sequences that were both identified through phage library screens–the integrin binding Arg-Gly-Asp (RGD) motif and a cell penetrating CendR element [117,121–123]. Integrins such as αvβ3 and αvβ5 are expressed at low levels in various cell types but highly upregulated on tumor endothelium during angiogenesis. CendR peptides contain the consensus sequence R/KXXR/K and are reported to mainly target neuropilin-1 [122]. A co-receptor for VEGF and semaphorin, neuropilin-1 plays important roles in angiogenesis, regulating vascular permeability, and axon guidance and is expressed broadly in many tissues.
In iRGDs, tumor specificity is designed to be conferred to the CendR element by incorporating RGD as a tumor-targeting peptide [121]. Direct conjugation of doxorubicin and other payloads up to nanoparticle size to the N terminus of the iRGD peptide can be accomplished with all functionalities preserved. These drug conjugates are proposed to be delivered by iRGDs into tumor tissue through a multistep mechanism. First, the iRGD peptide binds αvβ3 and αvβ5 integrins expressed on tumor endothelial cells. Next, a so far undefined cell surface associated protease cleaves the iRGD peptide to expose the CendR element at the C terminus. Finally, upon binding neuropilin-1, the CendR element triggers entry of the peptide and its cargo into tumors. Conjugation of therapeutic agents to iRGD peptides has been shown to improve efficacy in a number of preclinical models. In addition, combinations of the CendR peptide with other tumor targeting peptides such as the lymphatic targeting sequence LyP-1 [124] and the tumor vasculature targeting peptides F3 [125] and NGR [123] have also been shown to improve therapeutic efficacy in preclinical tests. Activation of neuropilin-1 by CendR peptides is reported to increase vascular permeability through an intracellular domain [126]. Consistent with this increased passive transvascular leakiness from neuropilin engagement, iRGD peptides do not have to be conjugated to the drug to elicit the benefit [127]. Uncoupled iRGD peptides appear to enhance the therapeutic efficacies of multiple drug classes simply by systemic co-injection [127]. These results have been questioned in a cancer biology reproducibility project which failed to observe key iRGD effects [128]. Some labs, however, have detected enhanced drug delivery and therapeutic efficacy with iRGDs but at levels ranging from 40% to 5-fold over background delivery [129,130]; these levels are much lower than the 7–40-fold originally reported [127]. Discrepancies in iRGD therapeutic efficacy in the published literature may stem from differences in the quality of the peptides used in experiments [131]. Clearer protocols on iRGD synthesis and in vitro verification processes with standardized activity assays may help to clear up questions about their functionality.
The transport mechanism by which the CendR motif in iRGDs induces the increased passage of attached and unattached cargo across the endothelial cell barrier remains unclear and has been speculated to occur through clathrin-mediated endocytosis or a process similar to macropinocytosis largely based on in vitro studies of tumor cell cultures; these findings have not been verified in vivo [132,133]. Another logical mechanism that, thus far, has not been ruled out is neuropilin-1 mediated increases in passive delivery through paracellular routes. To avoid many of the artifacts or caveats associated with cell culture techniques, future efforts to characterize iRGD transport mechanisms must be performed in vivo, most easily using intravital microscopy to follow transport under in vivo conditions. Immunohistochemical and EM techniques are also needed to visualize the delivery of fluorophore-labeled, biotinylated or gold particle labeled iRGDs into in vivo tumor endothelia.
A full evaluation of iRGD tumor targeting properties is also warranted preclinically as a prelude to clinical testing which is a logical step after several reported preclinical successes with a wide array of therapeutic peptides. Many normal endothelial cells express both neuropilin-1 and many integrins. Therefore, well-performed targeting studies including both classic quantitative biodistribution analysis and whole-body imaging are needed, and so far, lacking. The only SPECT/CT imaging study to assess iRGD specificity provided a single over-saturated image post-injection that showed a clear tumor signal. However, by far, most of the detected dose still went to the liver and spleen in a manner that did not differ substantially from the unconjugated nanoparticle imaging agent alone [134]. We still do not know key absolute (not relative) tissue uptakes and what percentage of an intravenously injected iRGD drug dose accumulates in tumors versus other organs, regardless of whether the drug is iRGD-coupled or coad-ministered with the peptide. Hence, the actual tumor delivery, efficiency, and specificity of the iRGD-mediated delivery system is unknown and not properly quantified.
6. Active transvascular delivery using caveolae pumping
6.1. Subtractive proteomic mapping and imaging to identify targets in tumor and organ-specific endothelial membranes and caveolae
In an effort to overcome the vascular barrier and the problems associated with passive transvascular drug delivery, work from our group in recent years has focused on ways to harness the caveolar transport pathway. Vascular endothelia are fine-tuned and adapted to support the functions of their host organ by factors in the surrounding tissue microenvironment. This led us to hypothesize that each organ’s microenvironment modulates protein expression in vascular endothelial cells and in EC caveolae, specifically [135]. In our early investigations into endothelial cell surface molecular heterogeneity, we developed a silica nanoparticle coating technique to physically isolate the luminal, blood accessible plasma membranes of vascular endothelium and its caveolae directly from multiple organs and tumors. These EC surface proteins were analyzed by large scale mass spectrometry and cataloged in a database that provides comprehensive, in vivo vascular proteomic maps that can be mined for molecularly distinct information. We discovered that EC plasma membranes and even caveolae of different organs and tissues have distinct proteomic signatures.
Our subtractive mapping identified tumor- or organ-specific proteins concentrated in EC caveolae that could provide a novel way to overcome the restrictive EC barrier, ultimately for the delivery and concentration of both large and small molecules within a specific tissue or tumor [34,36–39,94,136–140]. Applying subcellular fractionation and mass spectrometry techniques, we characterized the protein constituents of caveolae isolated from rat lungs carrying metastatic mammary tumors as well as caveolae from healthy lungs [38]. By subtracting the EC caveolar proteome in diseased tissues from healthy ones, we discovered tumor caveolae were unique in that they carry a truncated form of Annexin A1 (AnnA1). By screening hybridomas, we developed a series of monoclonal antibodies for this target including one, mAnnA1, that specifically recognizes AnnA1 across multiple species (human, rat, mouse).
6.2. Annexin A1 a tumor-induced endothelial cell surface protein concentrated in caveolae
Annexin A1 is a member of the annexin family of proteins which bind to phospholipids in a calcium-dependent manner [141–143]. Initially studied for its role in inflammation, Annexin A1 has since been shown to be important for tumor growth, angiogenesis and metastasis [144] and is believed to block apoptosis by binding to p65 in the NF-kappaβ signal transduction pathway (for a review see [143]). The modified 34 kDa form of Annexin found through our subtractive proteomic analysis is truncated at Lys26 in the N-terminus, a modification that has been shown to be promoted by EGFR kinase phosphorylation of the protein [145,146]. We observed expression of AnnA1 in caveolae of multiple human tumors including breast, lung, liver, kidney, brain and prostate tumors, but not healthy tissues [38,39]. Its absence from the vascular endothelium of multiple normal organs concurs with previously published expression analyses, including tissue immunostaining, by other labs [147–149].
6.3. Dynamic imaging of caveolar transport into normal and tumor tissues
6.3.1. Kinetics of caveolar transport into tumor tissue
To define in greater detail how mAnnA1 is processed by normal and tumor EC in vivo, we developed and applied a new IVM tumor model imaging system to observe directly and continuously the binding and delivery of caveolae targeting antibodies carrying fluorescent reporters across the EC barrier [39]. This system includes computational algorithms we developed to assess and quantify EC surface binding and transvascular fluxes from digital movies [37,39]. IVM changes the status of tumors from a “black box” with little knowledge of their internal workings to a microscopically accessible biological system in which drug processing, targeting, kinetics and cellular events can all be observed and recorded in real time in live animals. It offers an unparalleled view into tumor development with dynamic, high resolution and continuous in vivo imaging of molecular and cellular events. It changes the status of tumors from a “black box” with little knowledge of their internal workings to a microscopically accessible biological system in which drug processing, targeting, kinetics and cellular events can all be observed and recorded in real time in live animals. By applying IVM to the dorsal skinfold window chamber, we were able to perform live dynamic imaging of implanted tumors for several weeks [39, 150]. We also developed novel tumor models that have orthotopic tissue microenvironments and are amenable to IVM to allow us to open the “black box” of the solid tumor and see inside.
Because subcutaneously implanted tumors, the most popular and current IVM standard, lack proper orthotopic stroma and lack fidelity with human tumors, we expanded the preclinical utility of the IVM tumor imaging system by creating an orthotopic tissue environment for tumors in the ectopic dorsal skin. We co-implanted different types of tumor cell spheroids with their corresponding minced, orthotopic tissues into dorsal skin window chambers to create an ectopic-orthotopic (EO) model [39,150]. Using various GFP expressing transgenic mice, we have shown that the implanted orthotopic tissue becomes the stroma of the new tumor and provides the co-opted blood vessels supplying it. Vascular tubes in the minced tissue fuse to form patent tumor blood vessels that anastomose with vessels of the mouse host to supply blood to the growing tumors [150].
Following IV injection of fluorophore-labeled mAnnA1 into mice, dynamic and continuous in vivo transport studies using IVM of EO tumors show rapid EC binding and robust movement of the fluorophore cargo across the endothelium with release into the tumor interstitium; this caveolae targeting agent rapidly penetrates and achieves signal saturation in solid tumors within 1–2 h [39]. Despite the very low IV dose (1–3 μg fluoro-conjugate mAnnA1) and the resultant low to nil signal detected inside the blood vessels (observed throughout the duration of the experiment), the signal inside the tumor tissue continued to increase within minutes of injection until approaching image saturation at 60 min (Fig. 5A). In addition, by incorporating a red fluorescent reporter into tumor cells and labeling mAnnA1 with a green one, we observed the two distinct signals colocalize. The antibody flooded the tumor throughout, but not the surrounding tissue.
Fig. 5.
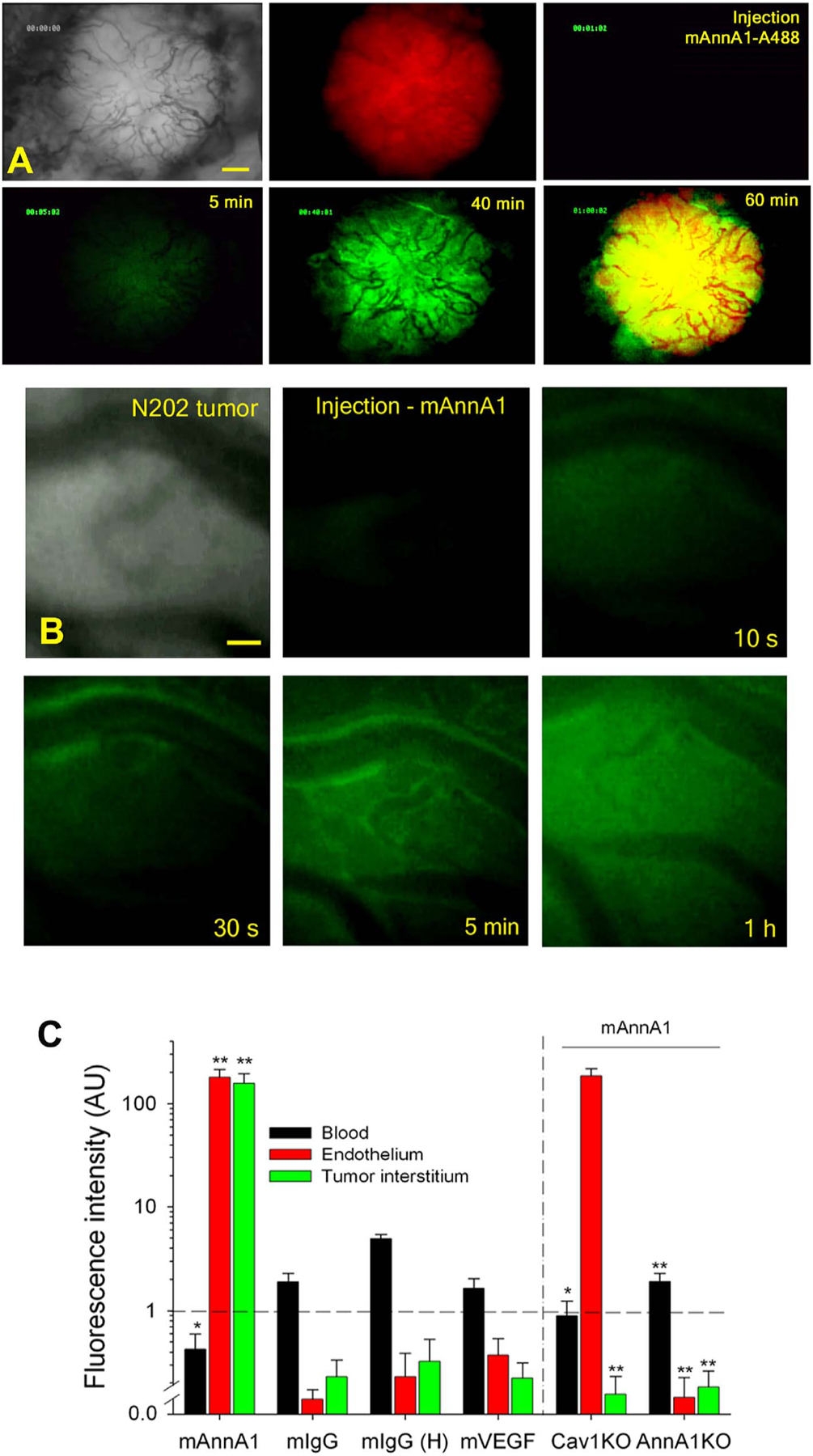
Rapid and specific mAnnA1 delivery into in vivo IVM tumors via caveolae targeting.
(A) Fluorescence IVM imaging of mammary tumor after intravenous injection of mAnnA1 shows rapid and pervasive tumor penetration. Low-magnification images captured from light and fluorescence video microscopy through window chambers of H2B-mCherry–expressing N202 mammary tumors (red) in mice after tail vein injection of 4 μg of mAnnA1 conjugated to Alexa Fluor 488 fluorophore. Light phase image shows the tumor and its vasculature. The static images were captured in the red or green fluorescence channel at the indicated times. The overlap of red- and green-channel images (tumor and mAnnA1 probe, respectively) is shown at the 60 min timepoint (last panel). Scale bar, 300 μm.
(B) Static images captured through window chambers of mammary tumors in mice injected intravenously with fluorophore conjugated antibodies (mAnnA1, IgG, or mVEGF) and imaged continuously with fluorescence video microscopy at high magnification. The first frame is a light image of the tumor and vasculature. Subsequent fluorescence image frames were captured at the indicated times after injection of 2 μg of the Alexa 488 fluorophore-conjugated mAnnA1 (green). Scale bar, 20 μm.
(C) Quantification of fluorescence uptake in three compartments of tumors. Region-of-interest analysis of blood, endothelium and tumor interstitium was performed to measure the fluorescence intensity detected at 60 min post-injection of the indicated antibodies (mAnnA1, mIgG, mIgG (H) and mVEGF), all at 3 μg except for an additional dose at 10 μg for mIgG –designated mIgG (H)–in WT mice and in Cav1KO and AnnA1KO mice. Data are presented as the mean ± s.d. for n = 3. *P < 0.05, **P < 0.01, each compartment signal for mAnnA1 was compared to all other antibodies. The mAnnA1 compartment signal for Cav1 KO or AnnA1 KO was compared to WT mice. Ranked ANOVA with Tukey’s post-hoc test. (Reprinted with permission from Ref. [39]. Copyright 2014 Nature Publishing Group [Nature Medicine]).
An analysis of fluorescence signal from 3 major compartments (blood, endothelium, and tumor interstitium) was used to quantify mAnnA1 processing [39]. The first event, observed within minutes of fluoro-mAnnA1 IV injection, was rapid endothelial cell surface binding that peaked between 10 and 20 min. It was followed by a rising tumor interstitial signal that grew most rapidly after 30 min and matched the signal in the vascular wall by 60 min. At 1 h, > 98% of total fluorescence signal in the tumor was in the tumor interstitium (Fig. 5B). This high magnification imaging revealed rapid mAnnA1 binding to the luminal EC surface with subsequent transendothelial pumping concentrating the fluorescent probe inside the tumor within 20–60 min of IV injection. Blood clearance of mAnnA1 was equally rapid.
This rapid tumor uptake of mAnnA1 exceeded uptake of control IgG or VEGF antibody probes by > 100-fold in three tumor types (prostate, lung and mammary). Although mAnnA1 concentrations in the tumor interstitium rapidly exceeded its levels in the blood, the antibody’s signal continued to rise. This is, by definition, active transport or in other words, pumping. Caveolae pumping transported the antibody across the EC barrier against a concentration gradient and a sizeable opposing diffusional force, opposite to the observed flux [38,39]. The antibody signal inside the tumor at 1 h post-injection is not only 50–100 times greater than the declining signal in the blood but also exceeds the maximum blood signal that occurs at the time of injection by 5–10-fold, again consistent with active transport into tumors (Fig. 5B). Targeting tumors via the caveolae pumping system thus enables unprecedentedly rapid and active transcytosis into tumors, yielding extraordinarily high intratumoral concentrations despite the very low antibody dose given.
If caveolae pumping truly delivers fluorophore-labeled mAnnA1 into tumors, then we reasoned knocking out caveolin-1 should block transport. This was indeed the case. Genetically knocking out caveolin-1 stalled mAnnA1 transport at the endothelial surface. While its binding to the endothelium proceeded in the absence of caveolin-1, mAnnA1’s entry into the tumor interstitium and parenchyma was blocked (Fig. 5B) [39]. In contrast, knocking out the annexin A1 target prevented mAnnA1’s binding to the endothelium and also prevented its tumor uptake (Fig. 5B) [39]. This genetic evidence supports the transport data in confirming the caveolar basis of mAnnA1 cargo delivery into tumors and demonstrates the ability of this antibody to actively penetrate solid tumors requires caveolae and the target protein, AnnA1.
6.3.2. Differences between caveolar transport into normal and tumor tissues
Our studies of transvascular transport into the normal lung and tumor endothelia have enabled the observation that caveolar pumping is more rapid and robust in normal tissues than in tumors. The lung caveolae targeting antibody mAPP2 crosses the normal lung endothelium and approaches peak levels in the lungs within a few minutes. In contrast, mAnnA1 takes significantly longer to flood tumors (1 h). The slower kinetics of caveolar transport into tumors compared to normal tissues may be due to a number of factors including poor tumor blood flow and heterogeneity in tumor ECs leading to fewer caveolae or increased distances for transcytosis. In addition, tumors receive less of the cardiac output than the lungs and therefore less of the circulating antibody. Despite these differences, IVM imaging reveals caveolae can traffic rapidly and act as an efficient pumping system across endothelium in both normal and diseased tissues. mAPP2 was the first caveolae targeting antibody and probe to be 1) pumped across the EC barrier, 2) concentrate to unprecedented levels in a single tissue, and 3) target and actively penetrate lung tissue specifically. Likewise, mAnnA1 is the first agent to actively penetrate solid tumors and concentrate to levels exceeding peak blood levels. We have demonstrated the caveolae pumping system can successfully drive mAnnA1 conjugated probes directly into tumors.
Together, these results indicate that mAnnA1 recognizes a highly viable delivery target in blood vessels of rodent and human tumors, one that is inherently accessible and potentially useful for the imaging and treatment of a diverse array of tumors. By attaching therapeutic cargo to the mAnnA1 antibody, the caveolae pumping system could be utilized to concentrate toxic agents specifically in solid tumors while sparing healthy tissues. EM shows that caveolae targeting agents can enter and bind caveolae in tumor or vascular endothelium, respectively, and can carry cargo, even 10–15 nm gold particles, across the EC barrier [36,37]. Rather than relying on a passive concentration gradient to drive transendothelial transport into tumors, mAnnA1, with its attached cargo, piggybacks onto caveolae and uses the caveolae pumping system to access the tumor interstitium.
Thus, this technology embodies elements of an ideal targeted delivery system in its ability to pump cargo against a concentration gradient into the tumor interstitium. This caveolae targeting strategy must undergo extensive preclinical and clinical testing to determine whether caveolae pumping can enhance therapeutic agent delivery and anti-cancer efficacy. Detailed preclinical biodistribution and whole-body imaging studies, like the ones conducted for the lung caveolae targeting antibody mAPP2 [37,81], are absolutely necessary to evaluate the specificity of mAnnA1 for tumors in vivo. It will also be critical to evaluate the toxicity of the AnnA1 antibody and learn whether its ability to actively transport cargo into human solid tumors is as effective as it is in experimental tumor models. Although its potential for clinical and diagnostic applications appear tremendous, only future clinical imaging and therapy trials will define its ultimate benefits and challenges.
7. Concluding remarks
The last half century of research has slowly revealed that endothelial cells do not simply form a static, passive, cellophane-type permeability barrier that is uniform in all tissues. Rather, they constitute a dynamic range of morphologically and molecularly distinct barriers in normal organs and in solid tumors. As such, we are only beginning to unmask the many secrets underlying how the endothelium functions to restrict and regulate the passage of molecules. Several vascular obstacles impede drug efficacy and delivery into solid tumors including poor blood flow, poor vascularization leading to hypoxic areas, high interstitial fluid pressures, and heterogeneity in the permeability and VEGF dependence of tumor vasculature.
In this review, we discuss the current state of a few vascular strategies designed to improve passive transvascular drug delivery into solid tumors and the challenges that remain to translate them into clinical benefit. Vascular promotion therapy is a fairly new drug delivery strategy that has yet to undergo clinical evaluation for both toxicity as well as efficacy. For vessel normalization as well as vascular promotion strategies, the identification of imaging or molecular biomarkers that predict which patients’ tumors will exhibit anti-angiogenic sensitivity would be a critical step towards achieving more consistent clinical outcomes. For iRGD based drug delivery strategies, full characterizations of iRGD tumor targeting that include measurements of absolute tissue uptake and tumor specificity are warranted in addition to clinical testing.
To maximize the potential of precision oncology, nanodrugs, and new generations of experimental therapeutics, as well as bolster the effectiveness of the current cancer drug arsenal, we may need to move beyond a reliance on passive delivery strategies and develop other strategies that allow for the rapid, specific, and active delivery of systemically administered agents directly into solid tumors. Akin to ion gradients across the plasma membrane driving passive transmembrane transport, passive transvascular transport relies on the potential energy stored in large concentration gradients between the blood and tumor interstitium to push drugs across the endothelial cell barrier and into the tumor. Molecules diffuse from high to low concentration in passive transvascular transport. In contrast, a truly active transport mechanism can utilize stored cellular energy in the form of ATP or GTP to force select molecules to be transported across a barrier in a direction opposite to the concentration gradient, namely low to high. The caveolae pumping system requires ATP and GTP driven membrane fission and fusion to traffic the release of specific bound cargo across the endothelial cell and thereby concentrate cargo inside the tissue. It can do so in vivo against a large concentration gradient in a very tissue specific manner. Our work with the AnnA1 antibody demonstrate the feasibility of harnessing the caveolae pumping system as an active delivery strategy to move cargo into tumor sites. Other imaging or therapeutic agents using this pathway, even at low intravenous doses, could also target tumor caveolae, be pumped across the endothelium, and concentrate inside solid tumors. Going beyond the current passive delivery paradigm may revolutionize how solid tumors and other diseases are imaged and treated in the future. More detailed targeting, imaging and therapeutic studies are needed to define the benefits and limitations of the caveolae pumping system.
In our work, we have demonstrated how proteomic imaging approaches to identify targets in transcytotic pathways such as AnnA1 in tumor caveolae and APP2 in lungs may be important ways to explore alternatives to passive transvascular targeting and bypass the need to create large concentration gradients between blood and tumor interstitium to deliver drugs. Given the immense plasticity of vascular endothelia and their sensitivity to their microenvironment, there are likely many more caveolar targets waiting to be identified that may be specific to certain types of tumors, other diseases, or organs. We believe new active delivery strategies for therapeutic agents would open a new chapter in the long-running war against cancer and expand the current therapeutic repertoire used for the treatment of solid tumors. Much work is needed to convert promise into clinical reality for the many patients suffering from cancer. Towards this end, we strongly advocate for more research into ways to exploit both passive and active delivery routes to maximize therapeutic delivery and potency.
Acknowledgments
This work was supported by the National Institutes of Health grants U01CA193787 and R01CA169644
References
- [1].Le Tourneau C, Delord JP, Goncalves A, Gavoille C, Dubot C, Isambert N, Campone M, Tredan O, Massiani MA, Mauborgne C, Armanet S, Servant N, Bieche I, Bernard V, Gentien D, Jezequel P, Attignon V, Boyault S, Vincent-Salomon A, Servois V, Sablin MP, Kamal M, Paoletti X, Molecularly targeted therapy based on tumour molecular profiling versus conventional therapy for advanced cancer (SHIVA): a multicentre, open-label, proof-of-concept, randomised, controlled phase 2 trial, Lancet Oncol. 16 (2015) 1324–1334. [DOI] [PubMed] [Google Scholar]
- [2].Wilhelm S, Tavares AJ, Dai Q, Ohta S, Audet J, Dvorak HF, Chan WCW, Analysis of nanoparticle delivery to tumours, Nat. Rev. Mater 1 (2016) 16014. [Google Scholar]
- [3].Friedman AA, Letai A, Fisher DE, Flaherty KT, Precision medicine for cancer with next-generation functional diagnostics, Nat. Rev. Cancer 15 (2015) 747–756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Arnedos M, Vicier C, Loi S, Lefebvre C, Michiels S, Bonnefoi H, Andre F, Precision medicine for metastatic breast cancer[mdash]limitations and solutions, Nat. Rev. Clin. Oncol 12 (2015) 693–704. [DOI] [PubMed] [Google Scholar]
- [5].Larson SM, Carrasquillo JA, Cheung NK, Press OW, Radioimmunotherapy of human tumours, Nat. Rev. Cancer 15 (2015) 347–360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Hochhaus A, Larson RA, Guilhot F, Radich JP, Branford S, Hughes TP, Baccarani M, Deininger MW, Cervantes F, Fujihara S, Ortmann C-E, Menssen HD, Kantarjian H, O’Brien SG, Druker BJ, Long-term outcomes of Imatinib treatment for chronic myeloid leukemia, N. Engl. J. Med 376 (2017) 917–927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Gordon AN, Tonda M, Sun S, Rackoff W, Long-term survival advantage for women treated with pegylated liposomal doxorubicin compared with topotecan in a phase 3 randomized study of recurrent and refractory epithelial ovarian cancer, Gynecol. Oncol 95 (2004) 1–8. [DOI] [PubMed] [Google Scholar]
- [8].Von Hoff DD, Ervin T, Arena FP, Chiorean EG, Infante J, Moore M, Seay T, Tjulandin SA, Ma WW, Saleh MN, Harris M, Reni M, Dowden S, Laheru D, Bahary N, Ramanathan RK, Tabernero J, Hidalgo M, Goldstein D, Van Cutsem E, Wei X, Iglesias J, Renschler MF, Increased survival in pancreatic cancer with nab-paclitaxel plus gemcitabine, N. Engl. J. Med 369 (2013) 1691–1703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Socinski MA, Bondarenko I, Karaseva NA, Makhson AM, Vynnychenko I, Okamoto I, Hon JK, Hirsh V, Bhar P, Zhang H, Iglesias JL, Renschler MF, Weekly nab-paclitaxel in combination with carboplatin versus solvent-based paclitaxel plus carboplatin as first-line therapy in patients with advanced non-small-cell lung cancer: final results of a phase III trial, J. Clin. Oncol. Off. J. Am. Soc. Clin. Oncol 30 (2012) 2055–2062. [DOI] [PubMed] [Google Scholar]
- [10].Shi J, Kantoff PW, Wooster R, Farokhzad OC, Cancer nanomedicine: progress, challenges and opportunities, Nat. Rev. Cancer 17 (2017) 20–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Chapman PB, Hauschild A, Robert C, Haanen JB, Ascierto P, Larkin J, Dummer R, Garbe C, Testori A, Maio M, Hogg D, Lorigan P, Lebbe C, Jouary T, Schadendorf D, Ribas A, O’Day SJ, Sosman JA, Kirkwood JM, Eggermont AMM, Dreno B, Nolop K, Li J, Nelson B, Hou J, Lee RJ, Flaherty KT, McArthur GA, Improved survival with vemurafenib in melanoma with BRAF V600E mutation, N. Engl. J. Med 364 (2011) 2507–2516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Flaherty KT, Puzanov I, Kim KB, Ribas A, McArthur GA, Sosman JA, O’Dwyer PJ, Lee RJ, Grippo JF, Nolop K, Chapman PB, Inhibition of mutated, activated BRAF in metastatic melanoma, N. Engl. J. Med 363 (2010) 809–819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Mok TS, Wu Y-L, Thongprasert S, Yang C-H, Chu D-T, Saijo N, Sunpaweravong P, Han B, Margono B, Ichinose Y, Nishiwaki Y, Ohe Y, Yang J-J, Chewaskulyong B, Jiang H, Duffield EL, Watkins CL, Armour AA, Fukuoka M, Gefitinib or carboplatin–paclitaxel in pulmonary adenocarcinoma, N. Engl. J. Med 361 (2009) 947–957. [DOI] [PubMed] [Google Scholar]
- [14].Verma S, Miles D, Gianni L, Krop IE, Welslau M, Baselga J, Pegram M, Oh D-Y, Diéras V, Guardino E, Fang L, Lu MW, Olsen S, Blackwell K, Trastuzumab emtansine for HER2-positive advanced breast cancer, N. Engl. J. Med 367 (2012) 1783–1791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Karapetis CS, Khambata-Ford S, Jonker DJ, O’Callaghan CJ, Tu D, Tebbutt NC, Simes RJ, Chalchal H, Shapiro JD, Robitaille S, Price TJ, Shepherd L, Au HJ, Langer C, Moore MJ, Zalcberg JR, K-ras mutations and benefit from cetuximab in advanced colorectal cancer, N. Engl. J. Med 359 (2008) 1757–1765. [DOI] [PubMed] [Google Scholar]
- [16].Rosenberg SA, Raising the bar: the curative potential of human cancer immunotherapy, Sci. Transl. Med 4 (2012) 127ps128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Schadendorf D, Hodi FS, Robert C, Weber JS, Margolin K, Hamid O, Patt D, Chen TT, Berman DM, Wolchok JD, Pooled analysis of long-term survival data from phase II and phase III trials of Ipilimumab in unresectable or metastatic melanoma, J. Clin. Oncol. Off. J. Am. Soc. Clin. Oncol 33 (2015) 1889–1894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Fehrenbacher L, Spira A, Ballinger M, Kowanetz M, Vansteenkiste J, Mazieres J, Park K, Smith D, Artal-Cortes A, Lewanski C, Braiteh F, Waterkamp D, He P, Zou W, Chen DS, Yi J, Sandler A, Rittmeyer A, Atezolizumab versus docetaxel for patients with previously treated non-small-cell lung cancer (POPLAR): a multicentre, open-label, phase 2 randomised controlled trial, Lancet (London, England) 387 (2016) 1837–1846. [DOI] [PubMed] [Google Scholar]
- [19].Day CP, Merlino G, Van Dyke T, Preclinical mouse cancer models: a maze of opportunities and challenges, Cell 163 (2015) 39–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Francia G, Cruz-Munoz W, Man S, Xu P, Kerbel RS, Mouse models of advanced spontaneous metastasis for experimental therapeutics, Nat. Rev. Cancer 11 (2011) 135–141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Holohan C, Van Schaeybroeck S, Longley DB, Johnston PG, Cancer drug resistance: an evolving paradigm, Nat. Rev. Cancer 13 (2013) 714–726. [DOI] [PubMed] [Google Scholar]
- [22].Jain RK, Stylianopoulos T, Delivering nanomedicine to solid tumors, Nat. Rev. Clin. Oncol 7 (2010) 653–664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Minchinton AI, Tannock IF, Drug penetration in solid tumours, Nat. Rev. Cancer 6 (2006) 583–592. [DOI] [PubMed] [Google Scholar]
- [24].Dvorak HF, Nagy JA, Dvorak AM, Structure of solid tumors and their vasculature: implications for therapy with monoclonal antibodies, Cancer Cells 3 (1991) 77–85. [PubMed] [Google Scholar]
- [25].Huang X, Molema G, King S, Watkins L, Edgington TS, Thorpe PE, Tumor infarction in mice by antibody-directed targeting of tissue factor to tumor vasculature, Science 275 (1997) 547–550. [DOI] [PubMed] [Google Scholar]
- [26].Bae YH, Park K, Targeted drug delivery to tumors: myths, reality and possibility, J. Control. Release 153 (2011) 198–205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Wang AZ, Langer R, Farokhzad OC, Nanoparticle delivery of cancer drugs, Annu. Rev. Med 63 (2012) 185–198. [DOI] [PubMed] [Google Scholar]
- [28].Park K, Facing the truth about nanotechnology in drug delivery, ACS Nano 7 (2013) 7442–7447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Xu J, Zhou X, Li Y, Tian Y, Cancer nanotechnology: recent trends and developments in strategies for targeting cancer cells to improve cancer imaging and treatment, Curr. Drug Metab (2017). [DOI] [PubMed] [Google Scholar]
- [30].Sousa F, Castro P, Fonte P, Kennedy PJ, Neves-Petersen MT, Sarmento B, Nanoparticles for the delivery of therapeutic antibodies: dogma or promising strategy? Expert Opin. Drug Deliv (2016) 1–14. [DOI] [PubMed] [Google Scholar]
- [31].Weathers SS, Gilbert MR, Toward personalized targeted therapeutics: an overview, Neurotherapeutics (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Sykes EA, Chen J, Zheng G, Chan WC, Investigating the impact of nanoparticle size on active and passive tumor targeting efficiency, ACS Nano 8 (2014) 5696–5706. [DOI] [PubMed] [Google Scholar]
- [33].Ruoslahti E, Bhatia SN, Sailor MJ, Targeting of drugs and nanoparticles to tumors, J. Cell Biol 188 (2010) 759–768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Schnitzer JE, Vascular targeting as a strategy for cancer therapy, N. Engl. J. Med 339 (1998) 472–474. [DOI] [PubMed] [Google Scholar]
- [35].Chrastina A, Massey KA, Schnitzer JE, Overcoming in vivo barriers to targeted nanodelivery, Wiley Interdiscip. Rev. Nanomed. Nanobiotechnol 3 (2011) 421–437. [DOI] [PubMed] [Google Scholar]
- [36].McIntosh DP, Tan X-Y, Oh P, Schnitzer JE, Targeting endothelium and its dynamic caveolae for tissue-specific transcytosis in vivo: a pathway to overcome cell barriers to drug and gene delivery, Proc. Natl. Acad. Sci. U. S. A 99 (2002) 1996–2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Oh P, Borgstrom P, Witkiewicz H, Li Y, Borgstrom BJ, Chrastina A, Iwata K, Zinn KR, Baldwin R, Testa JE, Schnitzer JE, Live dynamic imaging of caveolae pumping targeted antibody rapidly and specifically across endothelium in the lung, Nat. Biotechnol 25 (2007) 327–337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Oh P, Li Y, Yu JY, Durr E, Krasinska KM, Carver LA, Testa JE, Schnitzer JE, Subtractive proteomic mapping of the endothelial surface in lung and solid tumours for tissue-specific therapy, Nature 429 (2004) 629–635. [DOI] [PubMed] [Google Scholar]
- [39].Oh P, Testa JE, Borgstrom P, Witkiewicz H, Li Y, Schnitzer JE, In vivo proteomic imaging analysis of caveolae reveals pumping system to penetrate solid tumors, Nat. Med 20 (2014) 1062–1068. [DOI] [PubMed] [Google Scholar]
- [40].Bommareddy PK, Silk AW, Kaufman HL, Intratumoral approaches for the treatment of melanoma, Cancer J. 23 (2017) 40–47. [DOI] [PubMed] [Google Scholar]
- [41].Kota J, Hancock J, Kwon J, Korc M, Pancreatic cancer: stroma and its current and emerging targeted therapies, Cancer Lett. 391 (2017) 38–49. [DOI] [PubMed] [Google Scholar]
- [42].Zhu L, Staley C, Kooby D, El-Rays B, Mao H, Yang L, Current status of biomarker and targeted nanoparticle development: the precision oncology approach for pancreatic cancer therapy, Cancer Lett. 388 (2017) 139–148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Sultana A, Shore S, Raraty MG, Vinjamuri S, Evans JE, Smith CT, Lane S, Chauhan S, Bosonnet L, Garvey C, Sutton R, Neoptolemos JP, Ghaneh P, Randomised Phase I/II trial assessing the safety and efficacy of radiolabelled anti-carcinoembryonic antigen I(131) KAb201 antibodies given intra-arterially or intravenously in patients with unresectable pancreatic adenocarcinoma, BMC Cancer 9 (2009) 66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Divgi CR, Bander NH, Scott AM, O’Donoghue JA, Sgouros G, Welt S, Finn RD, Morrissey F, Capitelli P, Williams JM, Deland D, Nakhre A, Oosterwijk E, Gulec S, Graham MC, Larson SM, Old LJ, Phase I/II radioimmunotherapy trial with iodine-131-labeled monoclonal antibody G250 in metastatic renal cell carcinoma, Clin. Cancer Res 4 (1998) 2729–2739. [PubMed] [Google Scholar]
- [45].Mahe MA, Fumoleau P, Fabbro M, Guastalla JP, Faurous P, Chauvot P, Chetanoud L, Classe JM, Rouanet P, Chatal JF, A phase II study of intraperitoneal radioimmunotherapy with iodine-131-labeled monoclonal antibody OC-125 in patients with residual ovarian carcinoma, Clin. Cancer Res 5 (1999) 3249s–3253s. [PubMed] [Google Scholar]
- [46].Mulligan T, Carrasquillo JA, Chung Y, Milenic DE, Schlom J, Feuerstein I, Paik C, Perentesis P, Reynolds J, Curt G, et al. , Phase I study of intravenous Lu-labeled CC49 murine monoclonal antibody in patients with advanced adenocarcinoma, Clin. Cancer Res 1 (1995) 1447–1454. [PubMed] [Google Scholar]
- [47].Postow MA, Callahan MK, Wolchok JD, Immune checkpoint blockade in cancer therapy, J. Clin. Oncol. Off. J. Am. Soc. Clin. Oncol 33 (2015) 1974–1982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Bennett HS, Luft JH, Hampton JC, Morphological classifications of vertebrate blood capillaries, Am. J. Phys 196 (1959) 381–390. [DOI] [PubMed] [Google Scholar]
- [49].Stewart PA, Wiley MJ, Developing nervous tissue induces formation of blood-brain barrier characteristics in invading endothelial cells: a study using quail—-chick transplantation chimeras, Dev. Biol 84 (1981) 183–192. [DOI] [PubMed] [Google Scholar]
- [50].Roberts WG, Palade GE, Neovasculature induced by vascular endothelial growth factor is fenestrated, Cancer Res. 57 (1997) 765–772. [PubMed] [Google Scholar]
- [51].Aird WC, Endothelial cell heterogeneity, Cold Spring Harbor Perspect. Med 2 (2012) a006429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [52].Nagy JA, Chang SH, Shih SC, Dvorak AM, Dvorak HF, Heterogeneity of the tumor vasculature, Semin. Thromb. Hemost 36 (2010) 321–331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [53].Folkman J, What is the evidence that tumors are angiogenesis dependent? JNCI 82 (1990) 4–7. [DOI] [PubMed] [Google Scholar]
- [54].Hanahan D, Weinberg RA, Hallmarks of cancer: the next generation, Cell 144 (2011) 646–674. [DOI] [PubMed] [Google Scholar]
- [55].Carmeliet P, Jain RK, Angiogenesis in cancer and other diseases, Nature 407 (2000) 249–257. [DOI] [PubMed] [Google Scholar]
- [56].Ferrara N, Gerber HP, LeCouter J, The biology of VEGF and its receptors, Nat. Med 9 (2003) 669–676. [DOI] [PubMed] [Google Scholar]
- [57].Nagy JA, Dvorak AM, Dvorak HF, VEGF-A and the induction of pathological angiogenesis, Annu. Rev. Pathol 2 (2007) 251–275. [DOI] [PubMed] [Google Scholar]
- [58].Dvorak AM, Feng D, The vesiculo-vacuolar organelle (VVO). A new endothelial cell permeability organelle, J. Histochem. Cytochem 49 (2001) 419–432. [DOI] [PubMed] [Google Scholar]
- [59].Frentzas S, Simoneau E, Bridgeman VL, Vermeulen PB, Foo S, Kostaras E, Nathan MR, Wotherspoon A, Gao ZH, Shi Y, Van den Eynden G, Daley F, Peckitt C, Tan X, Salman A, Lazaris A, Gazinska P, Berg TJ, Eltahir Z, Ritsma L, van Rheenen J, Khashper A, Brown G, Nystrom H, Sund M, Van Laere S, Loyer E, Dirix L, Cunningham D, Metrakos P, Reynolds AR, Vessel co-option mediates resistance to anti-angiogenic therapy in liver metastases, Nat. Med 22 (2016) 1294–1302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [60].Stessels F, Van den Eynden G, Van der Auwera I, Salgado R, Van den Heuvel E, Harris AL, Jackson DG, Colpaert CG, Van Marck EA, Dirix LY, Vermeulen PB, Breast adenocarcinoma liver metastases, in contrast to colorectal cancer liver metastases, display a non-angiogenic growth pattern that preserves the stroma and lacks hypoxia, Br. J. Cancer 90 (2004) 1429–1436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [61].Donnem T, Hu J, Ferguson M, Adighibe O, Snell C, Harris AL, Gatter KC, Pezzella F, Vessel co-option in primary human tumors and metastases: an obstacle to effective anti-angiogenic treatment? Cancer Med. 2 (2013) 427–436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [62].Pezzella F, Pastorino U, Tagliabue E, Andreola S, Sozzi G, Gasparini G, Menard S, Gatter KC, Harris AL, Fox S, Buyse M, Pilotti S, Pierotti M, Rilke F, Non-small-cell lung carcinoma tumor growth without morphological evidence of neo-angiogenesis, Am. J. Pathol 151 (1997) 1417–1423. [PMC free article] [PubMed] [Google Scholar]
- [63].Eberhard A, Kahlert S, Goede V, Hemmerlein B, Plate KH, Augustin HG, Heterogeneity of angiogenesis and blood vessel maturation in human tumors: implications for antiangiogenic tumor therapies, Cancer Res. 60 (2000) 1388–1393. [PubMed] [Google Scholar]
- [64].Holash J, Maisonpierre PC, Compton D, Boland P, Alexander CR, Zagzag D, Yancopoulos GD, Wiegand SJ, Vessel cooption, regression, and growth in tumors mediated by angiopoietins and VEGF, Science 284 (1999) 1994–1998. [DOI] [PubMed] [Google Scholar]
- [65].Kuczynski EA, Yin M, Bar-Zion A, Lee CR, Butz H, Man S, Daley F, Vermeulen PB, Yousef GM, Foster FS, Reynolds AR, Kerbel RS, Co-option of liver vessels and not sprouting angiogenesis drives acquired sorafenib resistance in hepatocellular carcinoma, J. Natl. Cancer Inst 108 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [66].Bridgeman VL, Vermeulen PB, Foo S, Bilecz A, Daley F, Kostaras E, Nathan MR, Wan E, Frentzas S, Schweiger T, Hegedus B, Hoetzenecker K, Renyi-Vamos F, Kuczynski EA, Vasudev NS, Larkin J, Gore M, Dvorak HF, Paku S, Kerbel RS, Dome B, Reynolds AR, Vessel co-option is common in human lung metastases and mediates resistance to anti-angiogenic therapy in preclinical lung metastasis models, J. Pathol 241 (2017) 362–374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [67].Short RHD, Alveolar epithelium in relation to growth of the lung, Philos. Trans. R. Soc. Lond. Ser. B Biol. Sci 235 (1950) 35–86. [DOI] [PubMed] [Google Scholar]
- [68].Patan S, Haenni B, Burri PH, Implementation of intussusceptive microvascular growth in the chicken chorioallantoic membrane (CAM): 1. Pillar formation by folding of the capillary wall, Microvasc. Res 51 (1996) 80–98. [DOI] [PubMed] [Google Scholar]
- [69].Patan S, Munn LL, Jain RK, Intussusceptive microvascular growth in a human colon adenocarcinoma xenograft: a novel mechanism of tumor angiogenesis, Microvasc. Res 51 (1996) 260–272. [DOI] [PubMed] [Google Scholar]
- [70].Ribatti D, Djonov V, Intussusceptive microvascular growth in tumors, Cancer Lett. 316 (2012) 126–131. [DOI] [PubMed] [Google Scholar]
- [71].Asahara T, Masuda H, Takahashi T, Kalka C, Pastore C, Silver M, Kearne M, Magner M, Isner JM, Bone marrow origin of endothelial progenitor cells responsible for postnatal vasculogenesis in physiological and pathological neovascularization, Circ.Res 85 (1999) 221–228. [DOI] [PubMed] [Google Scholar]
- [72].Dean M, Fojo T, Bates S, Tumour stem cells and drug resistance, Nat. Rev. Cancer 5 (2005) 275–284. [DOI] [PubMed] [Google Scholar]
- [73].Heldin CH, Rubin K, Pietras K, Ostman A, High interstitial fluid pressure - an obstacle in cancer therapy, Nat. Rev. Cancer 4 (2004) 806–813. [DOI] [PubMed] [Google Scholar]
- [74].Boucher Y, Baxter LT, Jain RK, Interstitial pressure gradients in tissue-isolated and subcutaneous tumors: implications for therapy, Cancer Res. 50 (1990) 4478–4484. [PubMed] [Google Scholar]
- [75].Milosevic M, Fyles A, Hedley D, Pintilie M, Levin W, Manchul L, Hill R, Interstitial fluid pressure predicts survival in patients with cervix cancer independent of clinical prognostic factors and tumor oxygen measurements, Cancer Res. 61 (2001) 6400–6405. [PubMed] [Google Scholar]
- [76].Curti BD, Urba WJ, Alvord WG, Janik JE, Smith II JW, Madara K, Longo DL, Interstitial pressure of subcutaneous nodules in melanoma and lymphoma patients: changes during treatment, Cancer Res. 53 (1993) 2204–2207. [PubMed] [Google Scholar]
- [77].Herrera FG, Chan P, Doll C, Milosevic M, Oza A, Syed A, Pintilie M, Levin W, Manchul L, Fyles A, A prospective phase I-II trial of the cyclooxygenase-2 inhibitor celecoxib in patients with carcinoma of the cervix with biomarker assessment of the tumor microenvironment, Int. J. Radiat. Oncol. Biol. Phys 67 (2007) 97–103. [DOI] [PubMed] [Google Scholar]
- [78].Dejana E, Spagnuolo R, Bazzoni G, Interendothelial junctions and their role in the control of angiogenesis, vascular permeability and leukocyte transmigration, Thromb. Haemost 86 (2001) 308–315. [PubMed] [Google Scholar]
- [79].Kevil CG, Payne DK, Mire E, Alexander JS, Vascular permeability factor/vascular endothelial cell growth factor-mediated permeability occurs through disorganization of endothelial junctional proteins, J. Biol. Chem 273 (1998) 15099–15103. [DOI] [PubMed] [Google Scholar]
- [80].Matsumura Y, Maeda H, A new concept for macromolecular therapeutics in cancer chemotherapy: mechanism of tumoritropic accumulation of proteins and the antitumor agent smancs, Cancer Res. 46 (1986) 6387–6392. [PubMed] [Google Scholar]
- [81].Chrastina A, Valadon P, Massey KA, Schnitzer JE, Lung vascular targeting using antibody to aminopeptidase P: CT-SPECT imaging, biodistribution and pharmacokinetic analysis, J. Vasc. Res 47 (2010) 531–543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [82].Feng D, Nagy JA, Hipp J, Dvorak HF, Dvorak AM, Vesiculo-vacuolar organelles and the regulation of venule permeability to macromolecules by vascular permeability factor, histamine, and serotonin, J. Exp. Med 183 (1996) 1981–1986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [83].Kohn S, Nagy JA, Dvorak HF, Dvorak AM, Pathways of macromolecular tracer transport across venules and small veins. Structural basis for the hyperpermeability of tumor blood vessels, Lab. Investi 67 (1992) 596–607. [PubMed] [Google Scholar]
- [84].Dvorak AM, Kohn S, Morgan ES, Fox P, Nagy JA, Dvorak HF, The vesiculo-vacuolar organelle (VVO): a distinct endothelial cell structure that provides a transcellular pathway for macromolecular extravasation, J. Leukoc. Biol 59 (1996) 100–115. [PubMed] [Google Scholar]
- [85].Feng D, Nagy JA, Dvorak AM, Dvorak HF, Different pathways of macromolecule extravasation from hyperpermeable tumor vessels, Microvasc. Res 59 (2000) 24–37. [DOI] [PubMed] [Google Scholar]
- [86].Feng D, Nagy JA, Brekken RA, Pettersson A, Manseau EJ, Pyne K, Mulligan R, Thorpe PE, Dvorak HF, Dvorak AM, Ultrastructural localization of the vascular permeability factor/vascular endothelial growth factor (VPF/VEGF) receptor-2 (FLK-1, KDR) in normal mouse kidney and in the hyperpermeable vessels induced by VPF/VEGF-expressing tumors and adenoviral vectors, J. Histochem. Cytochem 48 (2000) 545–556. [DOI] [PubMed] [Google Scholar]
- [87].Pettersson A, Nagy JA, Brown LF, Sundberg C, Morgan E, Jungles S, Carter R, Krieger JE, Manseau EJ, Harvey VS, Eckelhoefer IA, Feng D, Dvorak AM, Mulligan RC, Dvorak HF, Heterogeneity of the angiogenic response induced in different normal adult tissues by vascular permeability factor/vascular endothelial growth factor, Laboratory investigation; a journal of technical methods and pathology 80 (2000) 99–115. [DOI] [PubMed] [Google Scholar]
- [88].Sundberg C, Nagy JA, Brown LF, Feng D, Eckelhoefer IA, Manseau EJ, Dvorak AM, Dvorak HF, Glomeruloid microvascular proliferation follows adenoviral vascular permeability factor/vascular endothelial growth factor-164 gene delivery, Am. J. Pathol 158 (2001) 1145–1160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [89].Bruns RR, Palade GE, Studies on blood capillaries: I. general organization of blood capillaries in muscle, J. Cell Biol 37 (1968) 244–276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [90].Palade GE, Fine structure of blood capillaries, J. Appl. Phys 24 (1953) 1424. [Google Scholar]
- [91].Schnitzer JE, Oh P, Caveolae are dynamic carriers that bud from plasma membranes in a GTP-dependent manner, Mol. Biol. Cell 6 (1995) 402a. [Google Scholar]
- [92].Schnitzer JE, Oh P, McIntosh DP, Role of GTP hydrolysis in fission of caveolae directly from plasma membranes [publisher’s erratum appears in Science 1996 Nov 15; 274(5290):1069], Science 274 (1996) 239–242. [DOI] [PubMed] [Google Scholar]
- [93].Schnitzer JE, Oh P, Jacobson BS, Dvorak AM, Caveolin-enriched caveolae purified from endothelium in situ are transport vesicles for albondin-mediateed transcytosis of albumin, Mol. Biol. Cell 5 (1994) A75. [Google Scholar]
- [94].Schnitzer JE, Caveolae: from basic trafficking mechanisms to targeting transcytosis for tissue-specific drug and gene delivery in vivo, Adv. Drug Deliv. Rev 49 (2001) 265–280. [DOI] [PubMed] [Google Scholar]
- [95].Schnitzer JE, The endothelial cell surface and caveolae in health and disease, in: Born GVR, Schwartz CJ (Eds.), Vascular Endothelium: Physiology, Pathology and Therapeutic Opportunities, Schattauer, Stuttgart, 1997, pp. 77–95. [Google Scholar]
- [96].Carver LA, Schnitzer JE, Tissue-specific pharmacodelivery and overcoming key cell barriers in vivo: Vascular targeting of caveolae, in: Muzykantov V, Torchilin B (Eds.), Biomedical Aspects of Drug Targeting, Kluwer Academic Publishers, Boston, 2002, pp. 107–128. [Google Scholar]
- [97].Bundgaard M, Frøkjaer-Jensen J, Crone C, Endothelial plasmalemmal vesicles as elements in a system of branching invaginations from the cell surface, Proc. Natl. Acad. Sci 76 (1979) 6439–6442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [98].Palade GE, Transport in quanta across the endothelium of blood capillaries, Anat. Rec 136 (1960) 254. [Google Scholar]
- [99].Palade GE, Blood capillaries of the heart and other organs, Circulation 24 (1961) 368–388. [DOI] [PubMed] [Google Scholar]
- [100].Schnitzer JE, Liu J, Oh P, Endothelial caveolae have the molecular transport machinery for vesicle budding, docking, and fusion including VAMP, NSF, SNAP, annexins, and GTPases, J. Biol. Chem 270 (1995) 14399–14404. [DOI] [PubMed] [Google Scholar]
- [101].Oh P, McIntosh DP, Schnitzer JE, Dynamin at the neck of caveolae mediates their budding to form transport vesicles by GTP-driven fission from the plasma membrane of endothelium, J. Cell Biol 141 (1998) 101–114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [102].Jain RK, Normalizing tumor vasculature with anti-angiogenic therapy: a new paradigm for combination therapy, Nat. Med 7 (2001) 987–989. [DOI] [PubMed] [Google Scholar]
- [103].Senger DR, Galli SJ, Dvorak AM, Perruzzi CA, Harvey VS, Dvorak HF, Tumor cells secrete a vascular permeability factor that promotes accumulation of ascites fluid, Science 219 (1983) 983–985. [DOI] [PubMed] [Google Scholar]
- [104].Leung DW, Cachianes G, Kuang WJ, Goeddel DV, Ferrara N, Vascular endothelial growth factor is a secreted angiogenic mitogen, Science 246 (1989) 1306–1309. [DOI] [PubMed] [Google Scholar]
- [105].Kim KJ, Li B, Winer J, Armanini M, Gillett N, Phillips HS, Ferrara N, Inhibition of vascular endothelial growth factor-induced angiogenesis suppresses tumour growth in vivo, Nature 362 (1993) 841–844. [DOI] [PubMed] [Google Scholar]
- [106].Rapisarda A, Melillo G, Overcoming disappointing results with antiangiogenic therapy by targeting hypoxia, Nat. Rev. Clin. Oncol 9 (2012) 378–390. [DOI] [PubMed] [Google Scholar]
- [107].Teicher BA, Holden SA, Ara G, Dupuis NP, Liu F, Yuan J, Ikebe M, Kakeji Y, Influence of an anti-angiogenic treatment on 9L gliosarcoma: oxygenation and response to cytotoxic therapy, Int. J. Cancer 61 (1995) 732–737. [DOI] [PubMed] [Google Scholar]
- [108].Teicher BA, Holden SA, Ara G, Sotomayor EA, Huang ZD, Chen YN, Brem H, Potentiation of cytotoxic cancer therapies by TNP-470 alone and with other anti-angiogenic agents, Int. J. Cancer 57 (1994) 920–925. [DOI] [PubMed] [Google Scholar]
- [109].Teicher BA, Emi Y, Kakeji Y, Northey D, TNP-470/minocycline/cytotoxic therapy: a systems approach to cancer therapy, Eur. J. Cancer 32a (1996) 2461–2466. [DOI] [PubMed] [Google Scholar]
- [110].Heist RS, Duda DG, Sahani DV, Ancukiewicz M, Fidias P, Sequist LV, Temel JS, Shaw AT, Pennell NA, Neal JW, Gandhi L, Lynch TJ, Engelman JA, Jain RK, Improved tumor vascularization after anti-VEGF therapy with carboplatin and nab-paclitaxel associates with survival in lung cancer, Proc. Natl. Acad. Sci. U. S. A 112 (2015) 1547–1552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [111].Van der Veldt AA, Lubberink M, Bahce I, Walraven M, de Boer MP, Greuter HN, Hendrikse NH, Eriksson J, Windhorst AD, Postmus PE, Verheul HM, Serne EH, Lammertsma AA, Smit EF, Rapid decrease in delivery of chemotherapy to tumors after anti-VEGF therapy: implications for scheduling of anti-angiogenic drugs, Cancer Cell 21 (2012) 82–91. [DOI] [PubMed] [Google Scholar]
- [112].Avallone A, Pecori B, Bianco F, Aloj L, Tatangelo F, Romano C, Granata V, Marone P, Leone A, Botti G, Petrillo A, Caraco C, Iaffaioli VR, Muto P, Romano G, Comella P, Budillon A, Delrio P, Critical role of bevacizumab scheduling in combination with pre-surgical chemo-radiotherapy in MRI-defined high-risk locally advanced rectal cancer: results of the BRANCH trial, Oncotarget 6 (2015) 30394–30407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [113].Jain RK, Antiangiogenesis strategies revisited: from starving tumors to alleviating hypoxia, Cancer Cell 26 (2014) 605–622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [114].Kaelin WG Jr., S. Shrivastav, D.G. Shand, R.L. Jirtle, Effect of verapamil on malignant tissue blood flow in SMT-2A tumor-bearing rats, Cancer Res. 42 (1982) 3944–3949. [PubMed] [Google Scholar]
- [115].Mas-Moruno C, Rechenmacher F, Kessler H, Cilengitide: the first anti-angiogenic small molecule drug candidate design, synthesis and clinical evaluation, Anti Cancer Agents Med. Chem 10 (2010) 753–768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [116].Weis SM, Cheresh DA, Tumor angiogenesis: molecular pathways and therapeutic targets, Nat. Med 17 (2011) 1359–1370. [DOI] [PubMed] [Google Scholar]
- [117].Pierschbacher MD, Ruoslahti E, Cell attachment activity of fibronectin can be duplicated by small synthetic fragments of the molecule, Nature 309 (1984) 30–33. [DOI] [PubMed] [Google Scholar]
- [118].Stupp R, Hegi ME, Gorlia T, Erridge SC, Perry J, Hong YK, Aldape KD, Lhermitte B, Pietsch T, Grujicic D, Steinbach JP, Wick W, Tarnawski R, Nam DH, Hau P, Weyerbrock A, Taphoorn MJ, Shen CC, Rao N, Thurzo L, Herrlinger U, Gupta T, Kortmann RD, Adamska K, McBain C, Brandes AA, Tonn JC, Schnell O, Wiegel T, Kim CY, Nabors LB, Reardon DA, van den Bent MJ, Hicking C, Markivskyy A, Picard M, Weller M, European Organisation for R, Treatment of C, Canadian Brain Tumor C, C.s. team, Cilengitide combined with standard treatment for patients with newly diagnosed glioblastoma with methylated MGMT promoter (CENTRIC EORTC 26071–22072 study): a multicentre, randomised, open-label, phase 3 trial, Lancet Oncol. 15 (2014) 1100–1108. [DOI] [PubMed] [Google Scholar]
- [119].Reynolds AR, Hart IR, Watson AR, Welti JC, Silva RG, Robinson SD, Da Violante G, Gourlaouen M, Salih M, Jones MC, Jones DT, Saunders G, Kostourou V, Perron-Sierra F, Norman JC, Tucker GC, Hodivala-Dilke KM, Stimulation of tumor growth and angiogenesis by low concentrations of RGD-mimetic integrin inhibitors, Nat. Med 15 (2009) 392–400. [DOI] [PubMed] [Google Scholar]
- [120].Wong PP, Demircioglu F, Ghazaly E, Alrawashdeh W, Stratford MR, Scudamore CL, Cereser B, Crnogorac-Jurcevic T, McDonald S, Elia G, Hagemann T, Kocher HM, Hodivala-Dilke KM, Dual-action combination therapy enhances angiogenesis while reducing tumor growth and spread, Cancer Cell 27 (2015) 123–137. [DOI] [PubMed] [Google Scholar]
- [121].Sugahara KN, Teesalu T, Karmali PP, Kotamraju VR, Agemy L, Girard OM, Hanahan D, Mattrey RF, Ruoslahti E, Tissue-penetrating delivery of compounds and nanoparticles into tumors, Cancer Cell 16 (2009) 510–520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [122].Teesalu T, Sugahara KN, Kotamraju VR, Ruoslahti E, C-end rule peptides mediate neuropilin-1-dependent cell, vascular, and tissue penetration, Proc. Natl. Acad. Sci. U. S. A 106 (2009) 16157–16162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [123].Arap W, Pasqualini R, Ruoslahti E, Cancer treatment by targeted drug delivery to tumor vasculature in a mouse model, Science 279 (1998) 377–380. [DOI] [PubMed] [Google Scholar]
- [124].Laakkonen P, Porkka K, Hoffman JA, Ruoslahti E, A tumor-homing peptide with a targeting specificity related to lymphatic vessels, Nat. Med 8 (2002) 751–755. [DOI] [PubMed] [Google Scholar]
- [125].Porkka K, Laakkonen P, Hoffman JA, Bernasconi M, Ruoslahti E, A fragment of the HMGN2 protein homes to the nuclei of tumor cells and tumor endothelial cells in vivo, Proc. Natl. Acad. Sci. U. S. A 99 (2002) 7444–7449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [126].Roth L, Prahst C, Ruckdeschel T, Savant S, Westrom S, Fantin A, Riedel M, Heroult M, Ruhrberg C, Augustin HG, Neuropilin-1 mediates vascular permeability independently of vascular endothelial growth factor receptor-2 activation, Sci. Signal 9 (2016) ra42. [DOI] [PubMed] [Google Scholar]
- [127].Sugahara KN, Teesalu T, Karmali PP, Kotamraju VR, Agemy L, Greenwald DR, Ruoslahti E, Coadministration of a tumor-penetrating peptide enhances the efficacy of cancer drugs, Science 328 (2010) 1031–1035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [128].Mantis C, Kandela I, Aird F, Replication study: coadministration of a tumor-penetrating peptide enhances the efficacy of cancer drugs, elife 6 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [129].Zhang Q, Zhang Y, Li K, Wang H, Li H, Zheng J, A novel strategy to improve the therapeutic efficacy of gemcitabine for non-small cell lung cancer by the tumor-penetrating peptide iRGD, PLoS One 10 (2015) e0129865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [130].Schmithals C, Koberle V, Korkusuz H, Pleli T, Kakoschky B, Augusto EA, Ibrahim AA, Arencibia JM, Vafaizadeh V, Groner B, Korf HW, Kronenberger B, Zeuzem S, Vogl TJ, Waidmann O, Piiper A, Improving drug penetrability with iRGD leverages the therapeutic response to Sorafenib and doxorubicin in hepatocellular carcinoma, Cancer Res. 75 (2015) 3147–3154. [DOI] [PubMed] [Google Scholar]
- [131].Kaiser J, Mixed results from cancer replications unsettle field, Science 355 (2017) 234–235. [DOI] [PubMed] [Google Scholar]
- [132].Liu Y, Ji M, Wong MK, Joo KI, Wang P, Enhanced therapeutic efficacy of iRGD-conjugated crosslinked multilayer liposomes for drug delivery, Biomed. Res. Int 2013 (2013) 378380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [133].Pang HB, Braun GB, Friman T, Aza-Blanc P, Ruidiaz ME, Sugahara KN, Teesalu T, Ruoslahti E, An endocytosis pathway initiated through neuropilin-1 and regulated by nutrient availability, Nat. Commun 5 (2014) 4904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [134].Wang CF, Sarparanta MP, Makila EM, Hyvonen ML, Laakkonen PM, Salonen JJ, Hirvonen JT, Airaksinen AJ, Santos HA, Multifunctional porous silicon nanoparticles for cancer theranostics, Biomaterials 48 (2015) 108–118. [DOI] [PubMed] [Google Scholar]
- [135].Schnitzer JE, Update on the cellular and molecular basis of capillary permeability, Trends in Cardiovas. Med 3 (1993) 124–130. [DOI] [PubMed] [Google Scholar]
- [136].Carver LA, Schnitzer JE, Caveolae: mining little caves for new cancer targets, Nat. Rev. Cancer 3 (2003) 571–581. [DOI] [PubMed] [Google Scholar]
- [137].Durr E, Yu J, Krasinska KM, Carver LA, Yates JR, Testa JE, Oh P, Schnitzer JE, Direct proteomic mapping of the lung microvascular endothelial cell surface in vivo and in cell culture, Nat. Biotechnol 22 (2004) 985–992. [DOI] [PubMed] [Google Scholar]
- [138].Valadon P, Garnett JD, Testa JE, Bauerle M, Oh P, Schnitzer JE, Screening phage display libraries for organ-specific vascular immunotargeting in vivo, Proc. Natl. Acad. Sci. U. S. A 103 (2006) 407–412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [139].Chrastina A, Schnitzer JE, 125-Iodine radiolabeling of silver nanoparticles for in vivo SPECT imaging, Int. J. Nanomedicine 5 (2010) 653–659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [140].Carver LA, Schnitzer JE, Proteomic Mapping of Endothelium and Vascular Targeting In Vivo, in: Aird WC (Ed.), Endothelial Biomedicine, 2007, pp. 881–898. [Google Scholar]
- [141].Gerke V, Creutz CE, Moss SE, Annexins: linking Ca2+ signalling to membrane dynamics, Nat. Rev. Mol. Cell Biol 6 (2005) 449–461. [DOI] [PubMed] [Google Scholar]
- [142].Rescher U, Gerke V, Annexins–unique membrane binding proteins with diverse functions, J. Cell Sci 117 (2004) 2631–2639. [DOI] [PubMed] [Google Scholar]
- [143].Boudhraa Z, Bouchon B, Viallard C, D’Incan M, Degoul F, Annexin A1 localization and its relevance to cancer, Clin. Sci. (Lond.) 130 (2016) 205–220. [DOI] [PubMed] [Google Scholar]
- [144].Yi M, Schnitzer JE, Impaired tumor growth, metastasis, angiogenesis and wound healing in annexin A1-null mice, Proc. Natl. Acad. Sci. U. S. A 106 (2009) 17886–17891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [145].Wang W, Creutz CE, Role of the amino-terminal domain in regulating interactions of annexin I with membranes: effects of amino-terminal truncation and mutagenesis of the phosphorylation sites, Biochemistry 33 (1994) 275–282. [DOI] [PubMed] [Google Scholar]
- [146].Haigler HT, Schlaepfer DD, Burgess WH, Characterization of lipocortin I and an immunologically unrelated 33-kDa protein as epidermal growth factor receptor/kinase substrates and phospholipase A2 inhibitors, J. Biol. Chem 262 (1987) 6921–6930. [PubMed] [Google Scholar]
- [147].Dreier R, Schmid KW, Gerke V, Riehemann K, Differential expression of annexins I, II and IV in human tissues: an immunohistochemical study, Histochem. Cell Biol 110 (1998) 137–148. [DOI] [PubMed] [Google Scholar]
- [148].Ahn SH, Sawada H, Ro JY, Nicolson GL, Differential expression of annexin I in human mammary ductal epithelial cells in normal and benign and malignant breast tissues, Clin. Exp. Metastasis 15 (1997) 151–156. [DOI] [PubMed] [Google Scholar]
- [149].Eberhard DA, Brown MD, VandenBerg SR, Alterations of annexin expression in pathological neuronal and glial reactions. Immunohistochemical localization of annexins I, II (p36 and p11 subunits), IV, and VI in the human hippocampus, Am. J. Pathol 145 (1994) 640–649. [PMC free article] [PubMed] [Google Scholar]
- [150].Borgstrom P, Oh P, Czarny M, Racine B, Schnitzer JE, Co-implanting orthotopic tissue creates stroma microenvironment enhancing growth and angiogenesis of multiple tumors, F1000Research v2, (2013) 2–129. [DOI] [PMC free article] [PubMed] [Google Scholar]