

Abstract
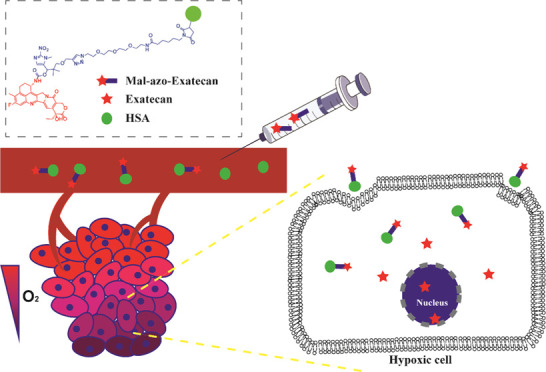
As an effective drug delivery strategy for traditional antitumor drugs, the stimulus-responsive albumin-based prodrugs are getting more and more attention. These prodrugs only release drugs in specific tumor microenvironments, which can prevent premature release of the drug in the circulation. Tumor hypoxia is a fundamental feature of the solid tumor microenvironment. As a hypoxia-activated linker, the 5-position branched linker of 1-methyl-2-nitro-5-hydroxymethylimidazole can be a trigger for albumin-based prodrugs. In this study, we report the synthesis and biological evaluation of the hypoxia-activated albumin-binding prodrug Mal-azo-Exatecan. After intravenous administration, the maleimide on the side chain can rapidly bind to endogenous albumin, enabling the prodrugs to accumulate in tumors, where tumor-associated hypoxia microenvironments trigger the selective release of Exatecan. The 5-position branched linker of 1-methyl-2-nitro-5-hydroxymethylimidazole as a cleavable linker has high plasma stability and does not cause Exatecan release from HSA-azo-Exatecan during circulation in vivo, avoiding systemic side effects caused by Exatecan.
Introduction
Human serum albumin (HSA) is one of the most important proteins in plasma with multiple functions.1 It is also an ideal candidate for drug delivery due to its lack of toxicity and immunogenicity. As a carrier, albumin can provide tumor specificity, reduce drug-related toxicity by altering drug distribution in vivo and enhancing cellular uptake, and maintain therapeutic concentrations of therapeutic agents over time.2 It also has the potential to extend the half-life of the drug.3 Nowadays, drug delivery systems that use albumin as a drug carrier include drug conjugates, drug adducts, albumin-binding derivatives, and nanoparticles.4−6 Among them, albumin-binding derivatives especially the albumin-binding prodrugs receive much attention.7,8 Albumin-binding prodrugs are designed with the concept of rapid and selective covalent binding to the cysteine 34 position (Cys-34) of serum albumin after intravenous administration to form a macromolecular drug delivery system.9 Since covalent binding of a prodrug to albumin greatly reduces the antitumor activity, most of the current research is focused on stimulus-responsive albumin-binding prodrugs.10,11 A stimulus-responsive albumin-binding delivery system can effectively accumulate at the tumor site, respond to internal or external stimuli, and release drugs. An advantage of the stimulus-responsive albumin-binding delivery system is that it prevents premature release of the drug in the circulation and avoids systemic toxic effects.12
By now, stimulus-responsive albumin-binding prodrugs include bioresponsive, pH-activated, and enzyme-activated albumin-bound prodrugs. For bioresponsive albumin-bound prodrugs, they can be triggered to release the parent drug in a strongly reductive environment in tumor cells.13 Xu et al. described bioresponsive albumin-conjugated paclitaxel prodrugs that improved biodistribution and tumor accumulation of paclitaxel and the tumor inhibition rate of this prodrug was approximately 3 times higher than that of the parent drug paclitaxel in vivo.14 For pH-activated albumin-bound prodrugs, rapid proliferation of cancer cells triggers glycolysis and lowers the pH in the tumor microenvironment, which would facilitate the controlled release of drugs.15 Among them, INNO-206, which is an acid cleaved albumin-bound adriamycin prodrug, is currently being assessed in phase III studies for use against sarcoma and gastric cancer.16 Enzyme-activated albumin-bound prodrugs rely on the high expression of specific enzymes in tumor tissue, which can be triggered to release the drug in the presence of the corresponding enzymes.17 Kratz et al. synthesized a large number of enzymes corresponding to albumin-binding prodrugs including urokinase,18 cathepsin B,19 and plasmin.20 Papot et al. focused on the β-glucuronidase-reactive albumin-binding prodrugs.21,22 All in all, the release of stimulus-responsive albumin-binding prodrugs depends on the tumor microenvironment.
Tumor hypoxia is a fundamental feature of the solid tumor microenvironment.23,24 The oxygen content in most solid tumors is much lower than that in normal tissues. Treatment of hypoxic tumors becomes one of the most important directions in oncology treatment, including molecular target drugs that act on HIFs and their related signaling pathways and hypoxia-activated prodrugs. At present, the most researched is the hypoxia-activated prodrug, which uses the tumor hypoxic environment to activate the inactive prodrug to release the antitumor drug.25 Expression of reductases such as nitroreductase and quinone reductase was much higher in hypoxic tumor tissue than in normal tissue.26 In our previous work, we designed a series of hypoxia-sensitive linker chains using 2-nitroimidazole as a framework and found that the 5-position branched linker of 1-methyl-2-nitro-5-hydroxymethylimidazole27 was sensitive to the hypoxic environment in solid tumors and had excellent stability in PBS, which has the potential as a trigger for albumin-binding prodrugs.
Exatecan is a class of camptothecin analogue (Figure 1), which shows excellent antitumor activities in many types of tumors. However, phase II studies and phase III studies do not show ideal antitumor effects due to the ineffective delivery of drugs to tumor tissues.28 For the past two years, DS-8201a, an antibody drug conjugate (ADC), using Exatecan derivatives Dxd as effector molecules, has been on the market, resulting in drug delivery systems of Exatecan and its derivatives receiving a lot of attention.29−31
Figure 1.

Exatecan, Dxd, and the hypoxia-activated trigger.
In this study, we reported the synthesis and biological evaluation of a hypoxia-activated albumin-binding prodrug Mal-azo-Exatecan (Figure 2). We used the 5-position branched linker of 1-methyl-2-nitro-5-hydroxymethylimidazole as a trigger for hypoxic cleavage, which is bound to the potent camptothecin analogue Exatecan using a carbamate bond. The maleimide on the side chain, which could rapidly bind to endogenous albumin after intravenous administration,32 formed a large molecule albumin carrier system HSA-azo-Exatecan (Figure 3). HSA-azo-Exatecan accumulated in tumor tissue through the enhanced permeability and retention (EPR) effect33 and the interaction of albumin receptor (gp60),34 which could release Exatecan triggered by nitroreductase in a hypoxic environment. Meanwhile, we introduced PEG chains to increase the water solubility of the prodrug. The 5-position branched linker of 1-methyl-2-nitro-5-hydroxymethylimidazole as a cleavable linker has high plasma stability and does not cause Exatecan release from HSA-azo-Exatecan during circulation in vivo, avoiding systemic side effects caused by Exatecan.
Figure 2.
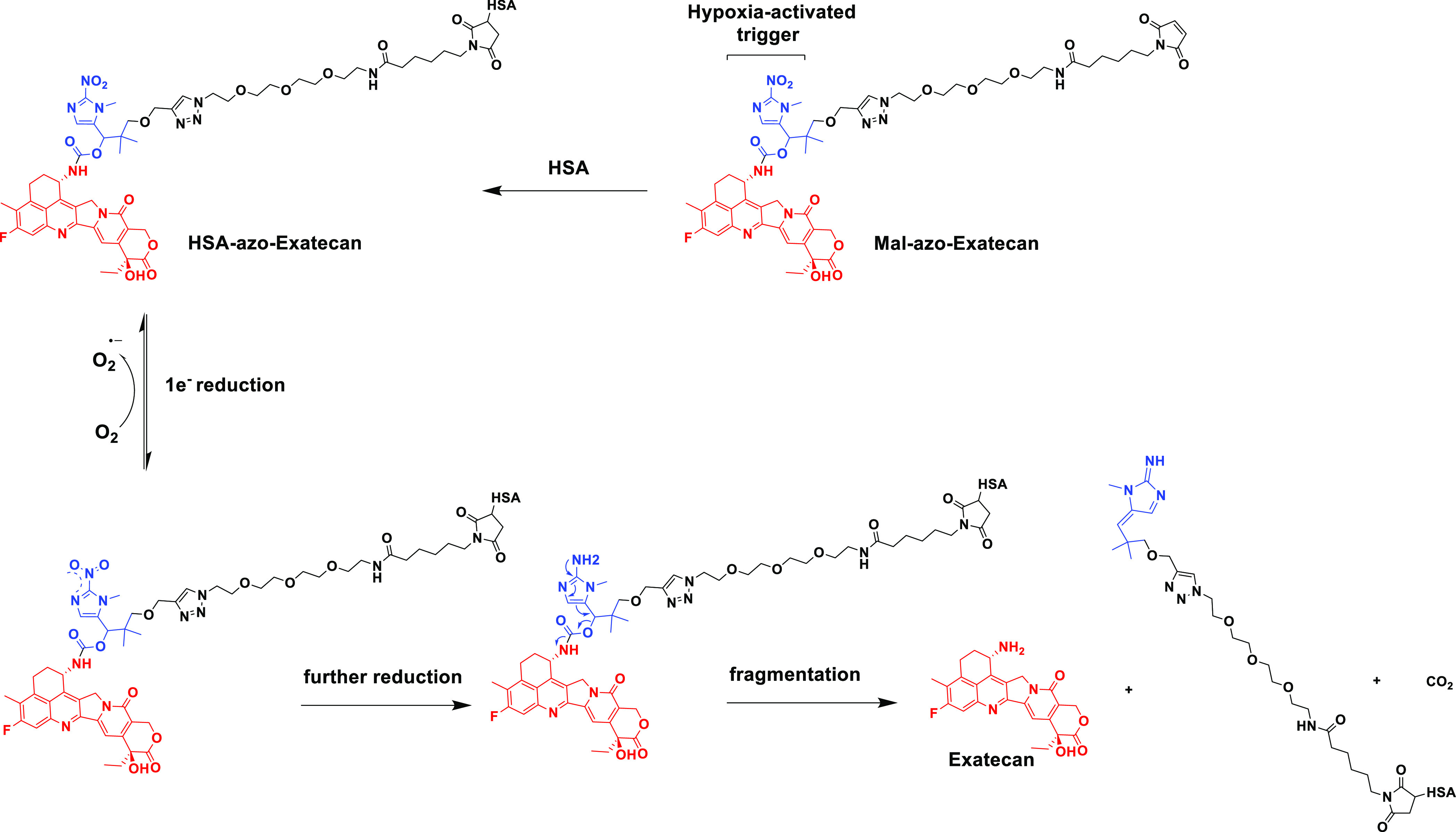
Production of Mal-azo-Exatecan and its drug release mechanism.
Figure 3.

Schematic diagram of the Mal-azo-Exatecan therapeutic strategy.
Results and Discussion
Binding of Mal-azo-Exatecan to Albumin
The rate of binding of Mal-azo-Exatecan to the circulating albumin in mouse and human plasma was evaluated in vitro using HPLC. As shown in Table 1, 100% of Mal-azo-Exatecan was bound to albumin within 2 min postincubation, confirming the fast rate of Michael addition (Table 1 and Figure S1). The binding of Mal-azo-Exatecan to HSA was accomplished within 8 min.
Table 1. Rate of Binding to Albumin of the Prodrug Mal-azo-Exatecan in Murine, Rat, and Human Plasma.
| albumin
binding |
||
|---|---|---|
| time (min) | bound (%) | |
| murine plasma | 2 | 95.81 ± 0.06 |
| 8 | 99.55 ± 0.11 | |
| rat plasma | 2 | 96.77 ± 0.15 |
| 8 | 99.38 ± 0.12 | |
| human plasma | 0.25 | 99.51 ± 0.01 |
| 2 | 100 ± 0.00 | |
| HSA | 2 | 89.41 ± 0.05 |
| 8 | 99.50 ± 0.13 | |
The molecular weights of HSA-azo-Exatecan and HSA were determined by MALDI-TOF MS (Figure S2A). After combining with Mal-azo-Exatecan (molecular weight 1140.1934), the maximum strength mass value of HSA changed from 66699.0153 to 67829.8742, and the mass difference was 1130.8589, indicating that a single molecule of Mal-azo-Exatecan was bound to a molecule of HSA.
The hydrodynamic diameters and size distribution of HSA-azo-Exatecan were measured by dynamic light scattering (DLS). The Z-average size of HSA-azo-Exatecan was 2.79 nm, and the Z-average size of HSA was 2.40 nm (Figure S2B). The ζ-potentials of HSA-azo-Exatecan and HSA were similar, which were −11.9 ± 1.7 mV (n = 3) and −13.6 ± 0.7 mV (n = 3). These data indicate that albumin-bound Mal-azo-Exatecan did not change the nano-sized structure of native serum albumin.
Stability of Albumin Conjugates
The rat plasma albumin conjugate of Mal-azo-Exatecan had extremely high stability in rat plasma, and no drug release was detected within 7 days (Table 2 and Figure S3). The conjugate of Mal-azo-Exatecan and human plasma albumin also had high stability in human plasma, and no drug release was detected within 7 days. The stability of Mal-azo-Exatecan and mouse plasma albumin conjugate was slightly poor in mouse plasma and could release 0.68% Exatecan in 7 days. In short, the albumin conjugates formed by the covalent binding of Mal-azo-Exatecan with various plasmas showed excellent plasma stability and minimized premature release of payloads.
Table 2. Plasma Stability of the Albumin-Drug Conjugatesa.
| % drug
released |
|||
|---|---|---|---|
| 1d | 2d | 7d | |
| murine plasma | 0.04 ± 0.02 | 0.44 ± 0.01 | 0.68 ± 0.02 |
| rat plasma | / | / | / |
| human plasma | / | / | / |
Note: / indicates that no free Exatecan was detected.
In Vitro Drug Release
The in vitro drug release profiles of Mal-azo-Exatecan and HSA-azo-Exatecan were investigated under hypoxic conditions (Figure 4). In the presence of nitroreductase, Mal-azo-Exatecan released 17.5% of Exatecan in 1 h. After 4 h, Mal-azo-Exatecan released only 23.1% of Exatecan. After binding with HSA, HSA-azo-Exatecan released 10.1% of Exatecan in 1 h. After 4 h, HSA-azo-Exatecan released only 19% of Exatecan. In summary, under hypoxic conditions, HSA-azo-Exatecan could release Exatecan mediated by nitroreductase.
Figure 4.

Kinetics of Exatecan release from Mal-azo-Exatecan and HSA-azo-Exatecan in the presence of nitroreductase.
In Vitro Efficacy of Mal-azo-Exatecan
We examined the antiproliferative activity of Mal-azo-Exatecan against human H460, HT29, A549, HepG2, MCF-7, and Mia PaCa-2 tumor cell lines (Table 3 and Figure S4). Under normal conditions, Mal-azo-Exatecan had lower cytotoxic activity than Exatecan. Under hypoxic conditions, Exatecan had the same cytotoxic activity as normal conditions. However, Mal-azo-Exatecan had more potent cytotoxic activity than that under normal conditions, indicating that Mal-azo-Exatecan had some hypoxic selectivity. Among them, Mal-azo-Exatecan had the highest hypoxia selectivity in NCI-H460 and the HCR (hypoxic cytotoxicity ratio) of 14 times.
Table 3. IC50 Values of Exatecan and Mal-azo-Exatecana.
| exatecan |
Mal-azo-Exatecan |
|||||
|---|---|---|---|---|---|---|
| cell lines | IC50(air) (nM) | IC50(N2) (nM) | HCR | IC50(air) (μM) | IC50(N2) (μM) | HCR |
| H460 | 1.47 ± 0.40 | 1.18 ± 0.10 | 1.25 | 49.86 ± 12.91 | 3.45 ± 0.32 | 14.43 |
| HT29 | 13.24 ± 2.00 | 18.31 ± 6.32 | 0.72 | 95.42 ± 4.87 | 51.33 ± 6.65 | 1.86 |
| A549 | 10.82 ± 0.90 | 11.93 ± 0.77 | 0.91 | >100 | 17.22 ± 1.59 | >5.88 |
| HepG2 | 96.48 ± 7.02 | 154.86 ± 25.21 | 0.62 | >100 | 32.52 ± 7.84 | >3.08 |
| MCF-7 | 38.34 ± 5.88 | 87.04 ± 4.83 | 0.44 | >100 | 71.97 ± 9.33 | >1.39 |
| Mla PaCa-2 | 0.72 ± 0.23 | 0.25 ± 0.05 | 2.88 | 13.38 ± 2.10 | 4.99 ± 1.14 | 2.68 |
Note: HCR, hypoxic cytotoxicity ratio; HCR= IC50 (Air)/IC50 (N2).
In Vivo Drug Accumulation and Biodistribution Study
The in vivo distribution properties and tumor selectivity of Exatecan and Mal-azo-Exatecan were evaluated in a mouse model bearing H460-transplanted tumors (Figure S5). Free Exatecan was mainly in the livers and spleens, and the content of Exatecan in tumors was low. In contrast, Mal-azo-Exatecan showed a higher tumor accumulation than free Exatecan in tumors. At 2, 6, 24, and 48 h, the cumulative amount of Exatecan in the tumor tissue of the Mal-azo-Exatecan group was 6-, 21-, 46-, and 72-fold higher than that of the Exatecan group, respectively. These results suggested that this prodrug enhances the selectivity in vivo and improves the preferential accumulation of the prodrug in tumors.
Meanwhile, the cumulative amount of Mal-azo-Exatecan increased over time. At 6, 24, and 48 h, the cumulative amount of Mal-azo-Exatecan in the tumor tissue of the Mal-azo-Exatecan group was 1.5-, 2.8-, 3.4-fold higher than that of the cumulative amount of Mal-azo-Exatecan at 2 h (Figure 5). Unfortunately, the release of the prodrug in the tumor is not ideal. At 2 and 6 h, the release of Exatecan in the tumor tissue of the Mal-azo-Exatecan group was lower than the cumulative amount of the Exatecan group. As time went on, the content of Exatecan of the Exatecan group gradually reduced. However, at 24 and 48 h, the release of Exatecan in the tumor tissue of the Mal-azo-Exatecan group increased. These results suggested that Mal-azo-Exatecan could improve the preferential accumulation of the prodrug in tumors, but the rate of drug release was low.
Figure 5.
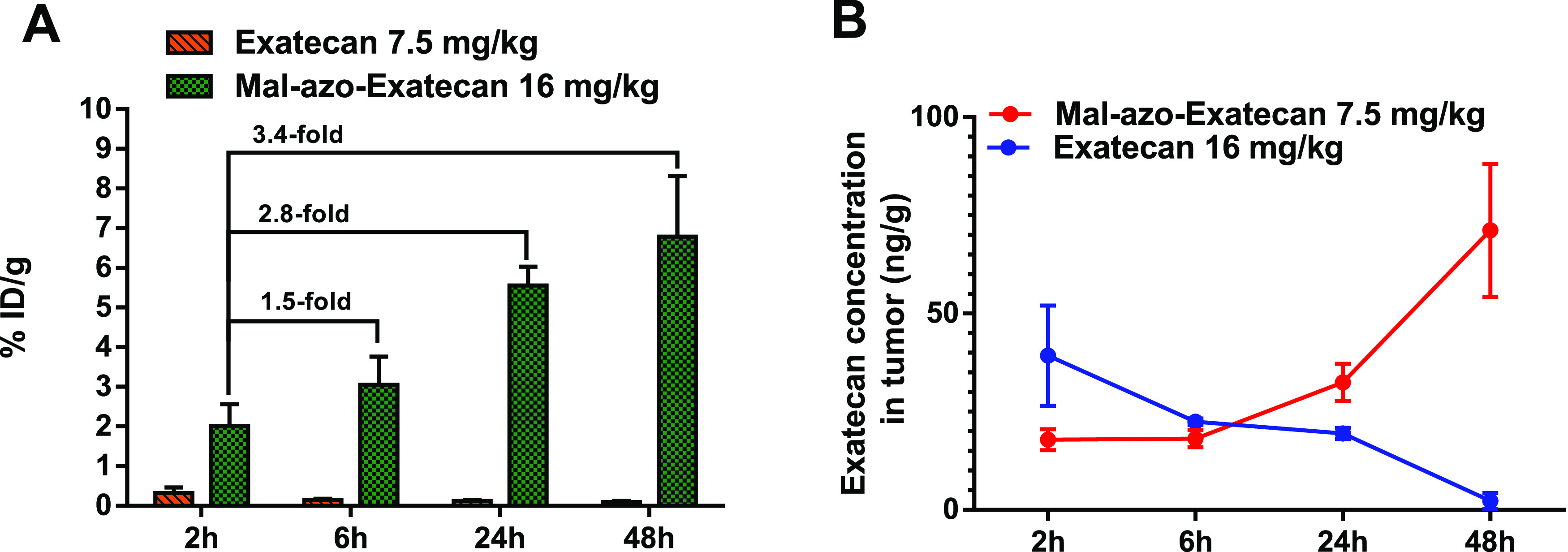
(A) Tumour accumulation by Exatecan (7.5 mg/kg) and Mal-azo-Exatecan (16 mg/kg) in the H460 transplant tumor model. (B) Release of Exatecan in tumors.
Inhibition effect of Mal-azo-Exatecan
Due to the highest hypoxic selectivity of Mal-azo-Exatecan in the H460 cell line, the in vivo antitumor activity of Mal-azo-Exatecan was assessed in H460-transplanted tumor-bearing BALB/C nude mice. Mice in the saline, Exatecan, and Mal-azo-Exatecan groups were given tail vein injections every three days. Mice in the saline and Mal-azo-Exatecan groups were given five consecutive doses and then stopped for a fortnight. Mice in the Exatecan group lost more than 20% of their body weight before the third dose and were stopped once. Mice in the Exatecan group were stopped for a fortnight after a total of four doses. The tumor volume of the saline group grew rapidly over time (Figure 6A). However, the antitumor effects of the Mal-azo-Exatecan group were similar to those of the Exatecan group. On day 30, the tumor volume of the Mal-azo-Exatecan group was 961 ± 297 mm3 and the tumor volume of the Exatecan group was 1281 ± 666 mm3. The weight of the mice in the Exatecan group decreased significantly and gradually recovered after stopping the drug once. Meanwhile, the body weight of the mice in the Mal-azo-Exatecan group increased relatively steadily (Figure 6B), indicating that Mal-azo-Exatecan was less toxic than Exatecan.
Figure 6.

(A) Tumor volume of H460-transplanted tumor-bearing BALB/C nude mice of the three groups over 18 days. (B) Body weight of the H460-transplanted tumor-bearing BALB/C nude mice of the three groups over 18 days.
Histological Analysis
Potential toxicities were evaluated by histological evaluation of hematoxylin and eosin (H&E)-stained tissues (Figure 7). The H&E staining images of major organs indicated that the Exatecan group exhibited massive hepatocyte necrosis with nuclei fragmentation and lysis and no clear pathologic changes were detected in the Mal-azo-Exatecan group. These results suggest that Mal-azo-Exatecan has low pathological toxicity.
Figure 7.

H&E staining of the main organs of H460-transplanted tumor-bearing BALB/C nude mice from each group after treatment.
Conclusions
In this study, we developed a new hypoxia-activated albumin-binding prodrug Mal-azo-Exatecan leading to the in vivo formation of the corresponding albumin conjugate and the release of the camptothecin derivative Exatecan in the tumor tissue. In hypoxic conditions, Mal-azo-Exatecan showed more potent antitumor activities than the normal conditions, which indicated that the prodrug had some hypoxic selectivity. In the antitumor effect evaluation in H460-transplanted tumor-bearing BALB/C nude mice, Mal-azo-Exatecan had lower toxicity and better antitumor effect than Exatecan, but it did not successfully inhibit tumor growth. The in vivo distribution experiment showed that the prodrug did not release Exatecan well in the tumor, leading to its poor antitumor effect in vivo. This phenomenon might be due to the fact that the level of hypoxia in this tumor model was not very high, resulting in a slow release of the prodrug after accumulation, which was not sufficient to inhibit tumor growth. Therefore, more consideration needs to be given to whether the tumour hypoxia microenvironment can release the drug rapidly in subsequent design.
Experimental Section
Materials
Methanol (MeOH), ethanol (EtOH), ethyl acetate (EA), anhydrous sodium sulfate (Na2SO4), dichloromethane (DCM), hydrochloric acid (HCl), tetrakis (acetonitrile) copper(I) hexafluoroho phosphate (Cu(MeCN)4PF6), petroleum ether (PE, 60–90), sodium bicarbonate (NaHCO3), and N,N-diisopropylethylamine (DIPEA) were obtained from Sinopharm Chemical Reagent Co., Ltd. (Shanghai, China). 4-Dimethylaminopyridine (DMAP) and N-succinimidyl 6-maleimidohexanoate (EMCS) were obtained from Suzhou Highfine Biological Co., Ltd. (Suzhou, China). N,N-Dimethylacetamide (DMF), deuterated chloroform (CDCl3, 99.8%), deuterium dimethyl sulfoxide (DMSO-d6, 99.8%), β-glucuronidase from Escherichia coli (5000 units), 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT), and albumin from human serum were purchased from Sigma-Aldrich (St. Louis, MO). Acetonitrile (ACN, HPLC grade) and trifluoroacetic acid (TFA, HPLC grade) were purchased from J&K Scientific Ltd. (Beijing, China). Culture media, penicillin–streptomycin, and 4% paraformaldehyde solution were obtained from HyClone (Logan, Utah). Foetal bovine serum (FBS) and trypsin were obtained from Gibco (BRL, MD). An Amicon Ultra-4 centrifugal unit (MWCO 10 kDa) was obtained from Merck Chemicals Co., Ltd. (Shanghai, China).
Synthesis of Mal-azo-Exatecan
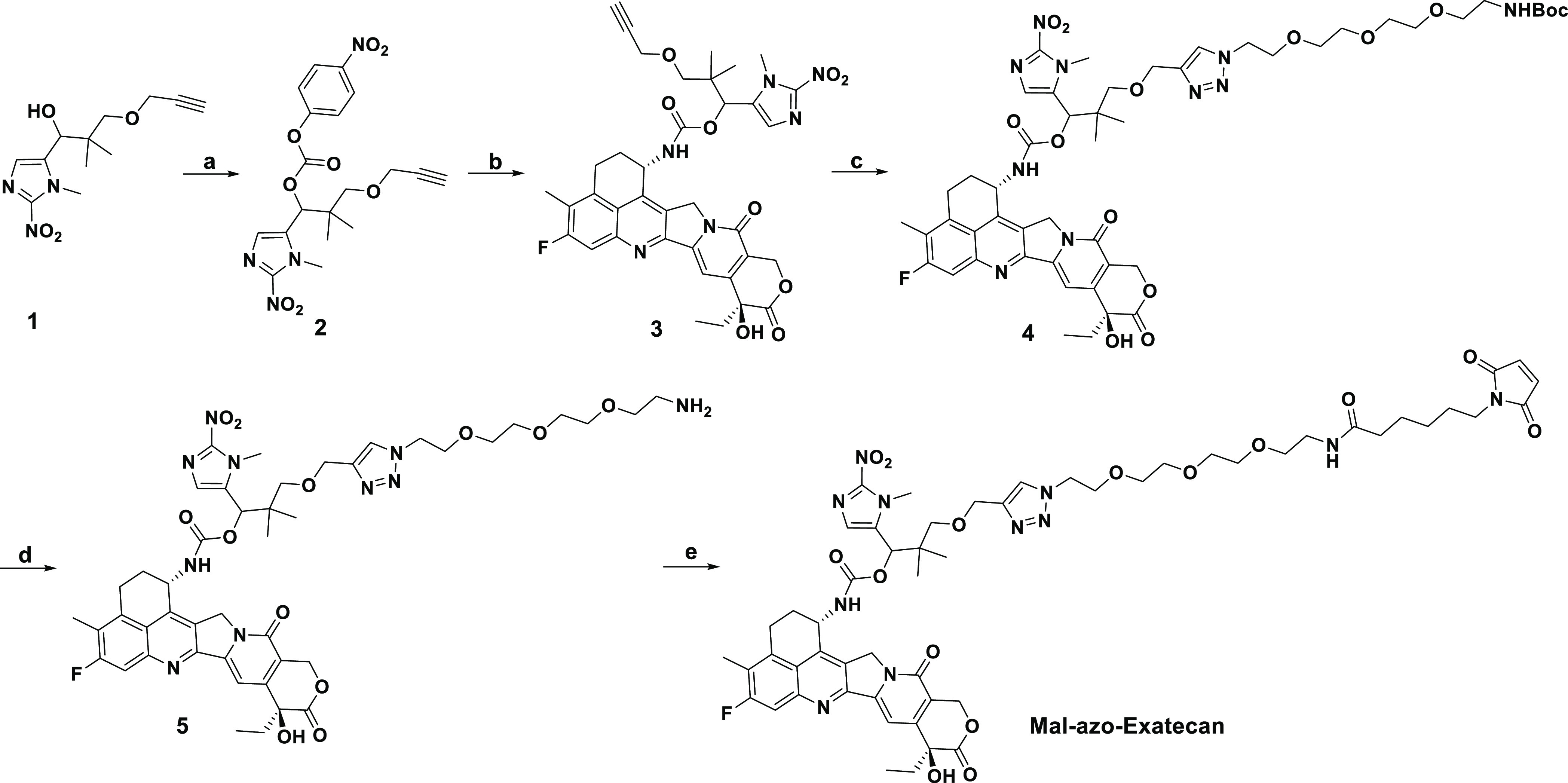
The detailed synthesis of the Mal-azo-Exatecan is illustrated in Scheme 1. The starting material compound 1 prepared according to previous literature was reacted with 4-nitrophenyl chloroformate in the presence of pyridine to yield compound 2. Compound 2 was reacted with Exatecan in the presence of DMAP in DMF to yield compound 3. Compound 3 was reacted with N3-PEG4-NH-BOC to form compound 4 by a click reaction. After complete deprotection using TFA and reaction with EMCS, Mal-azo-Exatecan was obtained. More details can be found in the Supporting Information.
Scheme 1. Reagents and Conditions: (a) 4-Nitrophenyl Chloroformate, Pyridine, DCM, rt; (b) Exatecan, DMAP, DMF, rt; (c) N3PEG4NHBoc, Cu(MeCN)4PF6, DCM, rt; (d) TFA, DCM, rt; and (e) EMCS, DIPEA, DCM, rt.
Cell Lines and Culture
A549 (human lung cancer cell line) and H460 (human large cell lung cancer cell line) cells were cultured in RPMI-1640 with 10% FBS and 1% Pen-Strep. The human colon cancer cell line HT29 was cultured in McCoy’s 5A with 10% FBS and 1% Pen-Strep. The human breast cancer cell line MCF-7 was cultured in MEM with 10% FBS and 1% Pen-Strep. The human hepatocarcinoma cell line HepG2 and human pancreatic cancer cell line were cultured in DMEM with 10% FBS and 1% Pen-Strep. All of the cell lines were obtained from the Chinese Academy of Science Cell Bank for Type Culture Collection (Shanghai, China) and maintained in a humidified atmosphere at 37 °C and 5% CO2.
HPLC Determination of Nitroreductase-Dependent Drug Release
Nitroreductase (50 U, purchased from Sigma-Aldrich) and NADPH (1 mM, purchased from Sigma-Aldrich) were added to a solution of Mal-azo-Exatecan or HSA-azo-Exatecan (100 μM in PBS, pH 7.4) and incubated in the three-gas incubator (1% O2) at 37 °C. The samples (100 μL) were taken at a specific point, immediately added to 100 μL of cold methanol, vortexed for 1 min, and then centrifuged for 30 min at 4 °C. The release of Exatecan over time was detected by HPLC (Method 1).
In Vitro MTT Cytotoxicity Assay
HT460, HT29, A549, MCF-7, Mia PaCa-2 (4 × 103 cells/well), and HepG2 (7 × 103 cells/well) cells were seeded in a 96-well plate. Twenty-four hours later, cells were exposed to Exatecan or Mal-azo-Exatecan and incubated in a normal and anaerobic incubator, which was further incubated for 72 h. Cell viability and proliferation behavior were assessed by the MTT assay. The absorbance was measured at 570 nm using an automated microplate reader (Spectra Max M5, Molecular Devices). The cell viability was calculated using eq 1 as follows
| 1 |
The ODsample is the optical density (OD) value of cells treated with various formulations, the ODcontrol is the OD value of cells incubated with culture media, and the ODblank is the OD value of the culture media alone.
In Vivo Antitumor Efficacy
When the tumor sizes reached ∼100 mm3, the H460 tumor-bearing mice were randomly assigned into control and treatment groups, with six mice per group. Control groups were given vehicle alone, and treatment groups received the indicated compounds (iv). Three groups were designed and received intravenous injections of 0.9% physiological saline solution (control group), 7.5 mg/kg Exatecan hydrochloride, or 16 mg/kg Mal-azo-Exatecan twice per week for 2 weeks (five doses in total). The length and width of the tumor volume were determined using Verniers calipers twice a week. The tumor volume (V) was calculated using eq 2 as follows
| 2 |
The body weight was recorded twice a week. At the experimental endpoint, all of the mice were sacrificed and tumors and major tissues were excised for weight measurement and further examination. The tumor growth inhibition rate (IR) was calculated based on the weight of the tumor on the last day.
Histological Analysis
The main organs (heart, liver, spleen, lung, and kidney) and tumors were excised, fixed in 4% formaldehyde, embedded in paraffin, and sectioned into slices at a thickness of 3 μm for further hematoxylin and eosin (H&E) staining. The H&E-stained samples were observed under an optical microscope.
Acknowledgments
This work was supported by the National Natural Science Foundation of China (No. 22077034 and No. 82104000). Xiaofan Gu is thanked for help in antitumor effect experiments in vivo.
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acsomega.1c05671.
Methods: general methods for synthesis and HPLC methods; In vitro data: binding kinetics of Mal-azo-Exatecan to albumin in murine, rat, and human plasma, stability of albumin conjugates in murine, rat, and human plasma, characterization of HSA-azo-Exatecan, albumin binding study using HPLC analysis, stability of albumin conjugates in murine, rat, and human plasma, IC50 data; In vivo data: xenografts model, in vivo biodistribution study, in vivo distribution properties and tumor selectivity, and statistical analysis; and experimental: synthesis of Mal-azo-Exatecan, NMR raw spectra, and high-resolution mass spectra of Mal-azo-Exatecan (PDF)
Author Contributions
† Z.C. and Y.H. contributed equally to this work.
The authors declare no competing financial interest.
Supplementary Material
References
- Kudarha R. R.; Sawant K. K. Albumin Based Versatile Multifunctional Nanocarriers for Cancer Therapy: Fabrication, Surface Modification, Multimodal Therapeutics and Imaging Approaches. Mater. Sci. Eng., C 2017, 81, 607–626. 10.1016/j.msec.2017.08.004. [DOI] [PubMed] [Google Scholar]
- Roy A. S.; Tripathy D. R.; Chatterjee A.; Dasgupta S. A Spectroscopic Study of the Interaction of the Antioxidant Naringin with Bovine Serum Albumin. Biophys. Chem. 2010, 01, 141–152. 10.4236/jbpc.2010.13017. [DOI] [Google Scholar]
- Carvalho J. R.; Machado M. V. New Insights about Albumin and Liver Disease. Ann. Hepatol. 2018, 17, 547–560. 10.5604/01.3001.0012.0916. [DOI] [PubMed] [Google Scholar]
- Kratz F. Albumin as a Drug Carrier: Design of Prodrugs, Drug Conjugates and Nanoparticles. J. Controlled Release 2008, 132, 171–183. 10.1016/j.jconrel.2008.05.010. [DOI] [PubMed] [Google Scholar]
- Teran-Saavedra N. G.; Sarabia-Sainz J. A.; Velázquez-Contreras E. F.; Ramos-Clamont Montfort G.; Pedroza-Montero M.; Vazquez-Moreno L. Albumin-Albumin/Lactosylated Core-Shell Nanoparticles: Therapy to Treat Hepatocellular Carcinoma for Controlled Delivery of Doxorubicin. Molecules 2020, 25, 5432 10.3390/molecules25225432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cai G.; Wang S.; Zhao L.; Sun Y.; Yang D.; Lee R. J.; Zhao M.; Zhang H.; Zhou Y. Thiophene Derivatives as Anticancer Agents and Their Delivery to Tumor Cells Using Albumin Nanoparticles. Molecules 2019, 24, 192 10.3390/molecules24010192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chubarov A.; Spitsyna A.; Krumkacheva O.; Mitin D.; Suvorov D.; Tormyshev V.; Fedin M.; Bowman M. K.; Bagryanskaya E. Reversible Dimerization of Human Serum Albumin. Molecules 2021, 26, 108 10.3390/molecules26010108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mehtala J. G.; Kulczar C.; Lavan M.; Knipp G.; Wei A. Cys34-PEGylated Human Serum Albumin for Drug Binding and Delivery. Bioconjugate Chem. 2015, 26, 941–949. 10.1021/acs.bioconjchem.5b00143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mayr J.; Heffeter P.; Groza D.; Galvez L.; Koellensperger G.; Roller A.; Alte B.; Haider M.; Berger W.; Kowol C. R.; Keppler B. K. An Albumin-Based Tumor-Targeted Oxaliplatin Prodrug with Distinctly Improved Anticancer Activity in Vivo. Chem. Sci. 2017, 8, 2241–2250. 10.1039/c6sc03862j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gu M.; Wang X.; Toh T. B.; Chow E. K. H. Applications of Stimuli-Responsive Nanoscale Drug Delivery Systems in Translational Research. Drug Discovery Today 2018, 23, 1043–1052. 10.1016/j.drudis.2017.11.009. [DOI] [PubMed] [Google Scholar]
- Taghizadeh B.; Taranejoo S.; Monemian S. A.; Moghaddam Z. S.; Daliri K.; Derakhshankhah H.; Derakhshani Z. Classification of Stimuli-Responsive Polymers as Anticancer Drug Delivery Systems. Drug Delivery 2015, 22, 145–155. 10.3109/10717544.2014.887157. [DOI] [PubMed] [Google Scholar]
- Kim Y. R.; Hwang J.; Koh H. J.; Jang K.; Lee J. D.; Choi J.; Yang C. S. The Targeted Delivery of the C-Src Peptide Complexed with Schizophyllan to Macrophages Inhibits Polymicrobial Sepsis and Ulcerative Colitis in Mice. Biomaterials 2016, 89, 1–13. 10.1016/j.biomaterials.2016.02.035. [DOI] [PubMed] [Google Scholar]
- Saito G.; Swanson J. A.; Lee K. D. Drug Delivery Strategy Utilizing Conjugation via Reversible Disulfide Linkages: Role and Site of Cellular Reducing Activities. Adv. Drug Delivery Rev. 2003, 55, 199–215. 10.1016/S0169-409X(02)00179-5. [DOI] [PubMed] [Google Scholar]
- Yang J.; Lv Q.; Wei W.; Yang Z.; Dong J.; Zhang R.; Kan Q.; He Z.; Xu Y. Bioresponsive Albumin-Conjugated Paclitaxel Prodrugs for Cancer Therapy. Drug Delivery 2018, 25, 807–814. 10.1080/10717544.2018.1451935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liberti Mv.; Locasale J. W. The Warburg Effect: How Does It Benefit Cancer Cells?. Trends Biochem. Sci. 2016, 41, 211–218. 10.1016/j.tibs.2015.12.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cranmer L. D. Spotlight on Aldoxorubicin (INNO-206) and Its Potential in the Treatment of Soft Tissue Sarcomas: Evidence to Date. OncoTargets Ther. 2019, 12, 2047–2062. 10.2147/OTT.S145539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmid B.; Chung D. E.; Warnecke A.; Fichtner I.; Kratz F. Albumin-Binding Prodrugs of Camptothecin and Doxorubicin with an Ala-Leu-Ala-Leu-Linker That Are Cleaved by Cathepsin B: Synthesis and Antitumor Efficacy. Bioconjugate Chem. 2007, 18, 702–716. 10.1021/bc0602735. [DOI] [PubMed] [Google Scholar]
- Chung D. E.; Kratz F. Development of a Novel Albumin-Binding Prodrug That Is Cleaved by Urokinase-Type-Plasminogen Activator (UPA). Bioorg. Med. Chem. Lett. 2006, 16, 5157–5163. 10.1016/j.bmcl.2006.07.023. [DOI] [PubMed] [Google Scholar]
- Schmid B.; Chung D. E.; Warnecke A.; Fichtner I.; Kratz F. Albumin-Binding Prodrugs of Camptothecin and Doxorubicin with an Ala-Leu-Ala-Leu-Linker That Are Cleaved by Cathepsin B: Synthesis and Antitumor Efficacy. Bioconjugate Chem. 2007, 18, 702–716. 10.1021/bc0602735. [DOI] [PubMed] [Google Scholar]
- Warnecke A.; Fichtner I.; Saß G.; Kratz F. Synthesis, Cleavage Profile, and Antitumor Efficacy of an Albumin-Binding Prodrug of Methotrexate That Is Cleaved by Plasmin and Cathepsin B. Arch. Pharm. Chem. Life Sci. 2007, 340, 389–395. 10.1002/ardp.200700025. [DOI] [PubMed] [Google Scholar]
- Legigan T.; Clarhaut J.; Renoux B.; Tranoy-opalinski I.; Monvoisin A.; Poitiers D.; Poitiers de U.; Brunet R. M. Synthesis and Antitumor Efficacy of a β-Glucuronidase-Responsive albumin-binding prodrug of doxorubicin. J. Med. Chem. 2012, 55, 4516–4520. 10.1021/jm300348r. [DOI] [PubMed] [Google Scholar]
- Renoux B.; Raes F.; Legigan T.; Péraudeau E.; Eddhif B.; Poinot P.; Tranoy-Opalinski I.; Alsarraf J.; Koniev O.; Kolodych S.; Lerondel S.; le Pape A.; Clarhaut J.; Papot S. Targeting the Tumour Microenvironment with an Enzyme-Responsive Drug Delivery System for the Efficient Therapy of Breast and Pancreatic Cancers. Chem. Sci. 2017, 8, 3427–3433. 10.1039/c7sc00472a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harris A. L. Hypoxia - A Key Regulatory Factor in Tumour Growth. Nat. Rev. Cancer 2002, 2, 38–47. 10.1038/nrc704. [DOI] [PubMed] [Google Scholar]
- Brown J. M.; Wilson W. R. Exploiting Tumour Hypoxia in Cancer Treatment. Nat. Rev. Cancer 2004, 4, 437–447. 10.1038/nrc1367. [DOI] [PubMed] [Google Scholar]
- Baran N.; Konopleva M. Molecular Pathways: Hypoxia-Activated Prodrugs in Cancer Therapy. Clin. Cancer Res. 2017, 23, 2382–2390. 10.1158/1078-0432.CCR-16-0895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fitzsimmons S. A.; Workman P.; Grever M.; Paull K.; Camalier R.; Lewis A. D.. Reductase enzyme expression across the national cancer institute tumor cell line panel: correlation with sensitivity to mitomycin C and EO9. J. Natl. Cancer Inst. 1996, 88, 259−269. [DOI] [PubMed]
- Huang Y.; Jin C.; Yu J.; Wang L.; Lu W. A Novel Multifunctional 2-Nitroimidazole-Based Bioreductive Linker and Its Application in Hypoxia-Activated Prodrugs. Bioorg. Chem. 2020, 101, 103975 10.1016/j.bioorg.2020.103975. [DOI] [PubMed] [Google Scholar]
- Verschraegen C. F.; Kudelka A. P.; Hu W.; Vincent M.; Kavanagh J. J.; Loyer E.; Bastien L.; Duggal A.; de Jager R. A Phase II Study of Intravenous Exatecan Mesylate (DX-8951f) Administered Daily for 5 Days Every 3 Weeks to Patients with Advanced Ovarian, Tubal or Peritoneal Cancer Resistant to Platinum, Taxane and Topotecan. Cancer Chemother. Pharmacol. 2004, 53, 1–7. 10.1007/s00280-003-0696-7. [DOI] [PubMed] [Google Scholar]
- Nakada T.; Sugihara K.; Jikoh T.; Abe Y.; Agatsuma T. The Latest Research and Development into the Antibody–Drug Conjugate, [Fam-] Trastuzumab Deruxtecan (DS-8201a), for HER2 Cancer Therapy. Chem. Pharm. Bull. 2019, 67, 173–185. 10.1248/cpb.c18-00744. [DOI] [PubMed] [Google Scholar]
- Li W.; Veale K. H.; Qiu Q.; Sinkevicius K. W.; Maloney E. K.; Costoplus J. A.; Lau J.; Evans H. L.; Setiady Y.; Ab O.; Abbott S. M.; Lee J.; Wisitpitthaya S.; Skaletskaya A.; Wang L.; Keating T. A.; Chari R. V. J.; Widdison W. C. Synthesis and Evaluation of Camptothecin Antibody-Drug Conjugates. ACS Med. Chem. Lett. 2019, 10, 1386–1392. 10.1021/acsmedchemlett.9b00301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wente M. N.; Kleeff J.; Büchler M. W.; Wanders J.; Cheverton P.; Langman S.; Friess H. DE-310, a Macromolecular Prodrug of the Topoisomerase-I-Inhibitor Exatecan (DX-8951), in Patients with Operable Solid Tumors. Investig. New Drugs 2005, 23, 339–347. 10.1007/s10637-005-1442-2. [DOI] [PubMed] [Google Scholar]
- Nicolaou K. C.; Rigol S. The Role of Organic Synthesis in the Emergence and Development of Antibody–Drug Conjugates as Targeted Cancer Therapies. Angew. Chem., Int. Ed. 2019, 58, 11206–11241. 10.1002/anie.201903498. [DOI] [PubMed] [Google Scholar]
- Maeda H.; Sawa T.; Konno T. Mechanism of Tumor-Targeted Delivery of Macromolecular Drugs, Including the EPR Effect in Solid Tumor and Clinical Overview of the Prototype Polymeric Drug SMANCS. J. Controlled Release 2001, 74, 47–61. 10.1016/S0168-3659(01)00309-1. [DOI] [PubMed] [Google Scholar]
- Schnitzer J. E.Gp60 Is an Albumin-Binding Glycoprotein Expressed By Continuous Endothelium Involved in Albumin Transcytosis By Continuous Endothelium Glycoprotein Expressed Involved in Albumin Transcytosis. Am. J. Physiol. 1992, 262, H246−H254. [DOI] [PubMed]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.