

Abstract

An efficient approach for the synthesis of 1,2-diaryl diketones was developed from readily available α-methylene ketones by catalysis of I2. In the same oxidation system, a novel one-pot procedure was established for the construction of antiviral and anticancer quinoxalines. The reactions proceeded well with a wide variety of substrates and good functional group tolerance, affording desired compounds in moderate to excellent yields. Quinoxalines 4ca and 4ad inhibited viral entry of SARS-CoV-2 spike pseudoviruses into HEK-293T-ACE2h host cells as dual blockers of both human ACE2 receptor and viral spike RBD with IC50 values of 19.70 and 21.28 μM, respectively. In addition, cytotoxic evaluation revealed that 4aa, 4ba, 4ia, and 4ab suppressed four cancer cells with IC50 values ranging from 6.25 to 28.55 μM.
1. Introduction
1,2-Diaryl diketones are a vital class of organic compounds frequently found in structural units of natural products and bioactive molecules.1−9 They are also versatile intermediates used in the synthesis of biologically active heterocyclics, such as imidazoles,10−13 quinoxalines,14−16 indoles,17 pyrazines,18 and triazines.19−21 Furthermore, 1,2-diaryl diketones could be transformed into greatly important functional materials with electronic and photochemical properties.22−27 Therefore, various traditional and modern methods for the formation of 1,2-diaryl diketones have been reported, involving the oxidation of α-halo and α-hydroxyl ketones,28−32 alkene and alkyne oxidation,33−43 oxidative cleavage of 1,3-diketones,44−48 benzyl phenyl ketone oxidation,49−54 oxidative decarboxylative coupling of arylpropiolic/cinnamic acids with arylboronic acids or aryl halides,55−57 and oxidative rearrangement of α,β-unsaturated diaryl ketones.58 Among the above methods, the direct oxidation of benzyl phenyl ketone has emerged as a powerfully straightforward strategy to synthesize 1,2-diaryl diketones. In 2012, Alimohammadi et al.59 reported a chromium reagent 2,6-dicarboxypyridinium fluorochromate-promoted (2,6-DCPFC) method for the oxidation of benzylic C–H bonds into the corresponding carbonyl compounds, but the 2,6-DCPFC was prepared by treatment of aqueous solution of CrO3 and HF with pyridine 2,6-dicarboxylic acid. Then, Goggiamani et al.60 disclosed a novel copper-catalyzed oxidation of deoxybenzoins to benzils using PPh3 as the ligand. Subsequently, a similar copper-catalyzed direct oxidation of C–H bond to synthesize benzils was developed by Wang et al.,61 in which K2CO3 was required as the additive to accelerate the reaction. Additionally, Ravikumar et al.62 utilized base/DMSO catalytic system for the oxidation of methylene compounds to afford 1,2-diaryl diketones, releasing the environmentally unfriendly and foul-smelling dimethyl sulfide.
Although these methods have generated smoothly the desired compounds, there are still several drawbacks, including the use of noble transition-metal catalysts, complex ligands, high catalyst loading (≥15 mol %), lengthy reaction times, and the tedious preparation of the starting materials. Therefore, from the view of atomic economy and green chemistry, it is still extremely desirable to develop new synthetic approaches for the preparation of 1,2-diaryl diketones under catalytic and metal-free conditions.
To our delight, we developed an efficient and practical method for the oxidative carbonylation of α-methylene ketones into the desired 1,2-diaryl diketones using friendly catalytic system I2-TBHP (tert-butyl hydroperoxide) in DMF (N,N-dimethylformamide). Gratifyingly, this catalytic transformation was further applied to construct antiviral and anticancer quinoxalines from α-methylene ketones and o-phenylenediamines in one pot without any modified reaction conditions, showing good to excellent yields, wide substrate scope, and high functional group tolerance.
2. Results and Discussion
2.1. Chemistry
Initially, benzyl phenyl ketone 1a was chosen as the model substrate to explore the oxidation reaction, and the results are shown in Table 1. When the reaction of 1a was carried out in the presence of 10 mol % TBAI using 3.0 equiv of TBHP as the oxidant in DMF at 100 °C for 5 h, the intended benzil 2a was obtained in only 29% yield (Table 1, entry 1). Subsequently, both NIS and I2 were examined as catalysts, and it was discovered that utilizing 10 mol % I2 as a catalyst produced the desired product 2a in outstanding yield (87%) (Table 1, entries 2–3). The increase of the loading of I2 afforded a slightly higher yield (88%); however, the decrease of the loading of I2 gave the product 2a in a lower yield (Table 1, entries 4–5). From the point of view of economy, 10 mol % I2 was selected as the catalyst to further optimize the reaction conditions. When the amount of TBHP was 2.0 or 4.0 equiv, the yields of 2a were decreased to 70 and 77%, respectively (Table 1, entries 6–7). Then, other solvents were also investigated, including DMAc (N,N-dimethylacetamide), NMP (N-methyl-2-pyrrolidone), DMSO (dimethyl sulfoxide), toluene, 1,4-dioxane, and H2O. It was found that DMF was the most effective one among all tested solvents for this reaction (Table 1, entry 3 vs entries 8–13). Moreover, the yield of 2a dramatically decreased to 61% at a relatively low temperature of 80 °C (Table 1, entry 14). Notably, no desired product 2a was obtained in the absence of either TBHP or I2 (Table 1, entries 15 and 16). Thus, the reaction conditions were optimized as 10 mol % I2 and 3.0 equiv TBHP in DMF at 100 °C for 5 h (Table 1, entry 3). In comparison to the previously reported literatures,53,54 our current work had two improvements. One difference is that the amount of catalyst I2 was reduced from 50 mol % to 10 mol % in the preceding literature. The other advantage is that using DMF as the solvent instead of DMSO avoids the release of the environmentally unfriendly and foul-smelling dimethyl sulfide.
Table 1. Optimization of the Conditions for the Oxidative Carbonylation of 1aa.

| entry | catalyst (mol %) | [O] (equiv) | solvent | T (°C) | yieldb (%) |
|---|---|---|---|---|---|
| 1 | TBAI (10) | TBHP (3) | DMF | 100 | 29 |
| 2 | NIS (10) | TBHP (3) | DMF | 100 | 63 |
| 3 | I2(10) | TBHP (3) | DMF | 100 | 87 |
| 4 | I2(50) | TBHP (3) | DMF | 100 | 88 |
| 5 | I2(5) | TBHP (3) | DMF | 100 | 82 |
| 6 | I2(10) | TBHP (2) | DMF | 100 | 70 |
| 7 | I2(10) | TBHP (4) | DMF | 100 | 77 |
| 8 | I2(10) | TBHP (3) | DMAc | 100 | trace |
| 9 | I2(10) | TBHP (4) | NMP | 100 | trace |
| 10 | I2(10) | TBHP (3) | DMSO | 100 | 51 |
| 11 | I2(10) | TBHP (3) | toluene | 100 | NR |
| 12 | I2(10) | TBHP (3) | 1,4-dioxane | 100 | NR |
| 13 | I2(10) | TBHP (3) | H2O | 100 | trace |
| 14 | I2(10) | TBHP (3) | DMF | 80 | 61 |
| 15 | I2(10) | DMF | 100 | trace | |
| 16 | TBHP (3) | DMF | 100 | NR |
Unless otherwise specified, reactions were carried out using 1a (0.3 mmol), catalyst (10 mol %), and oxidant (0.9 mmol) in solvent (2.0 mL) at 100 °C.
Isolated yields.
With the optimized reaction conditions in hand (Table 1, entry 3), the scope of the α-methylene ketones was explored (Scheme 1). As a result, various substrates with R1 bearing electron-donating groups or electron-withdrawing groups were found to generate smoothly the expected products. When the substrates substituted with electron-donating groups R1 (such as -CH3, -OCH3, -tBu, -3,5-diCH3) were initially carried out under the standard conditions, the desired products 2b–2g were obtained in 71%–92% yields. It was worth noting that the substrates containing −CH3 group (R1) at the meta- and ortho-positions afforded rather good yields of the corresponding products 2e (86%) and 2f (81%). Furthermore, the substrate 1h with phenyl group could be transformed into product 2h in 80% yield. When the electron-withdrawing groups (R1 = -Cl, -Br, -F, -COCH3) at the para-positions of aryl rings (Ar1) were examined, the desired products 2i–2l were generated in moderate to good yields (48–75%). 2k (R1 = F, R2 = H) and 2l were, respectively, obtained in only 54 and 48% yields, probably due to the strong electron-withdrawing effect of the -F and -COCH3. Additionally, the effects of -Cl group at meta and ortho substitutions of aryl rings (Ar1) revealed that the yields gradually fell with the trend of meta and ortho substitutions (2m, 2n vs 2i). Delightfully, substrates containing naphthalene (1o, 1p), furan (1q), and thiophene (1r) were also oxidized to the corresponding products 2o, 2p, 2q, and 2r in 61, 71, 49, and 90% yields, respectively.
Scheme 1. Substrate Scope of the α-Methylene Ketones.
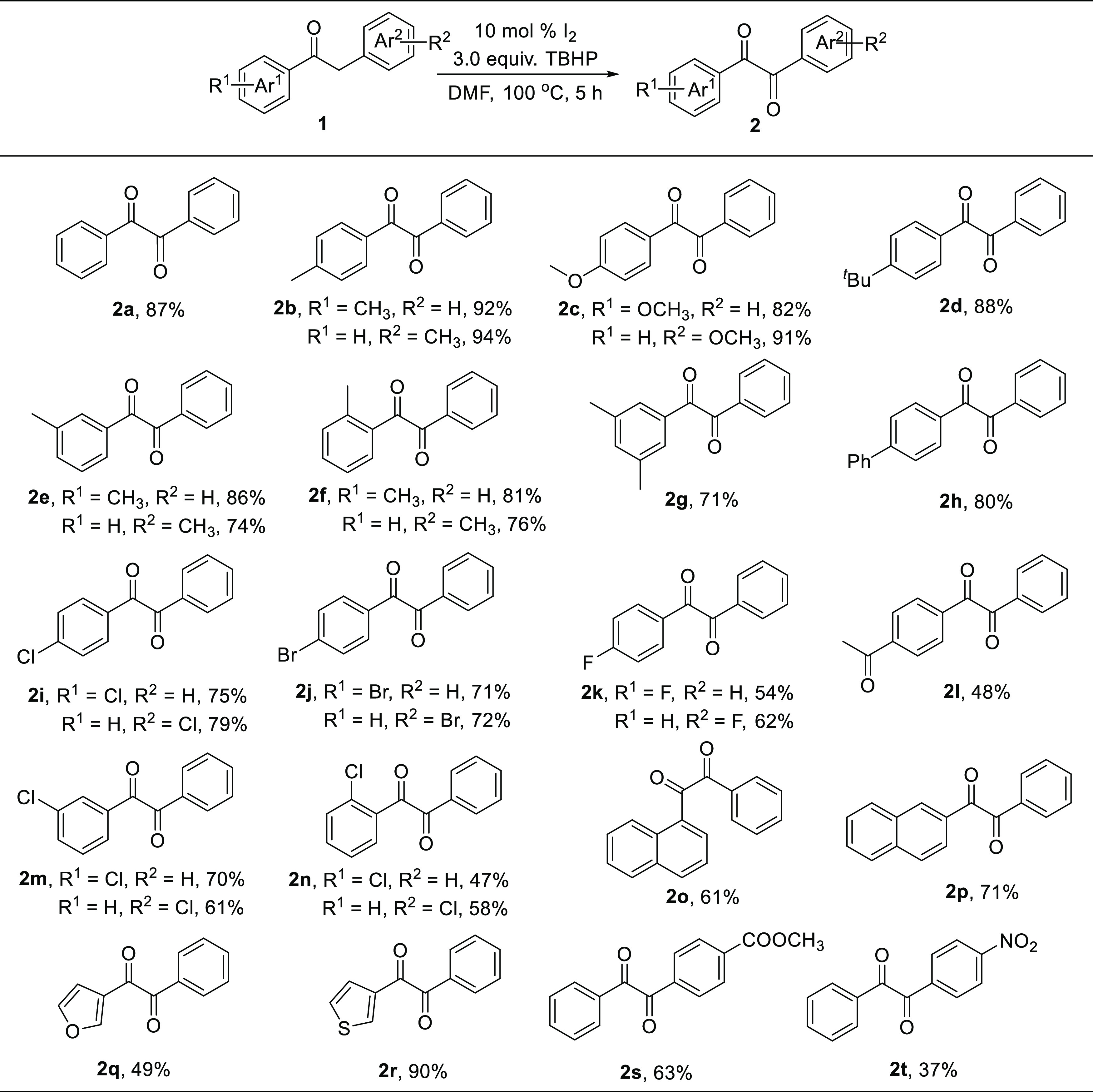
Subsequently, we also examined substrates with various functional groups R2 (-CH3, -OCH3, -Cl, -Br, -F, -COOCH3, -NO2) in this reaction. The results showed that electron-donating groups were more reactive than electron-withdrawing groups substituted on the aryl rings Ar2 (2b–2c, 2e–2f, 2i–2k, 2m–2n, 2s–2t). When the substrates with electron-donating groups -CH3 and -OCH3 (R2) were subjected to the reaction conditions, the intended products 2b and 2c were acquired in outstanding yields of 94 and 91%, respectively. Further substrate investigations demonstrated that the -CH3 group at the m- and o-substituents of the Ar2 produced the respective products 2e and 2f in slightly lower yields of 74 and 76%, probably due to the high steric hindrance. While electron-withdrawing functional groups (-Cl, -Br, -F, -COOCH3) on the aryl rings Ar2 were studied under the standard conditions, the corresponding products (2i–2k, 2s) were prepared in good yields (62–79%). It should be noted that nitro (-NO2) was well tolerated, affording the expected product 2t in only 37% yield due to the strong electron-withdrawing effect of the -NO2. In addition, substrates with -Cl group (R2) at the m- and o-positions of aryl rings Ar2 were also smoothly converted into the desired products 2m and 2n in 61 and 58% yields, respectively. Unfortunately, no desired products were observed when the substrates of acetophenone, acetone, and 2-butanone were used to carry out the reactions under the optimized conditions.
To expand the synthetic potential of our present catalytic protocol, a gram-scale reaction of 1a was carried out under the optimized conditions, delivering the desired product 2a in 62% yield (Scheme 2).
Scheme 2. Gram-Scale Synthesis of Benzil 2a.

Moreover, several control experiments were conducted to explore the mechanism of the reaction (Scheme 3). When the substrates 5 and 6 were employed under the standard reaction conditions, no desired products were observed (Scheme 3a,b), indicating that the presence of the aryl rings in 1 is crucial for the success of this reaction. Under optimized conditions, the reaction was additionally performed in the presence of 1.5 equiv 2,2,6,6-tetramethylpiperidine-1-oxyl (TEMPO) (Scheme 3c) as a radical scavenger without producing any other side products, indicating that no reaction occurred. This result demonstrated that the reaction might include a radical process. The involvement of a radical process was different from that reported in the literature.37
Scheme 3. Control Experiments.

Based on the above control experiments and previous literature reports, a possible mechanism was proposed in Scheme 4. Initially, the tert-butoxyl and tert-butylperoxy radicals (tBuO• and tBuOO•) were formed via the redox cycle reaction of I2 and TBHP.70−72 Subsequently, the generated tert-butoxyl radical (tBuO•) abstracted hydrogen atom from benzyl phenyl ketone 1a to afford the benzylic radical I,73,74 and it was followed by the reaction of tert-butylperoxy radical (tBuOO•) with the intermediate I to generate the peroxide intermediate II.75,76 Finally, the intermediate II underwent rapid oxidation or a Kornblum–DeLaMare rearrangement to give the desired product 2a with the elimination of tert-butyl alcohol.70,77−79
Scheme 4. Proposed Mechanism.

Recently, the synthesis of quinoxalines has also received great attention due to their biological activities, including anti-infectious,80,81 antidepressant,82 antitumor,83−86 anti-inflammatory,87,88 antibacterial,89,90 and antimalarial effects.91 Inspired by the success of the oxidation of α-methylene ketones to benzils 2, we further tested its applications to construct quinoxaline compounds. Fortunately, quinoxalines92 were synthesized from α-methylene ketones and o-phenylenediamines in one pot under the above standard conditions. To obtain quinoxalines with potential antiviral entry activities against SARS-CoV-2, we initially utilized the molecular docking study to predict the binding interaction of 20 quinoxalines (4aa–4ta in Scheme S1) with both human ACE2 and SARS-CoV-2 Spike RBD proteins. As a result of the calculated binding energy (Table S1), 4ca has the lowest binding energy with both ACE2 and Spike RBD proteins while 4ra possesses the highest energy with both proteins among all 20 quinoxalines. A lower binding energy suggested a more stable interaction between the molecule and the protein. Based on the above molecular docking results, nine representative quinoxalines including 4ca and 4ra were selected to be synthesized not only for examining the application of benzils but also for the acquirement of quinoxalines with antiviral entry activities against SARS-CoV-2.
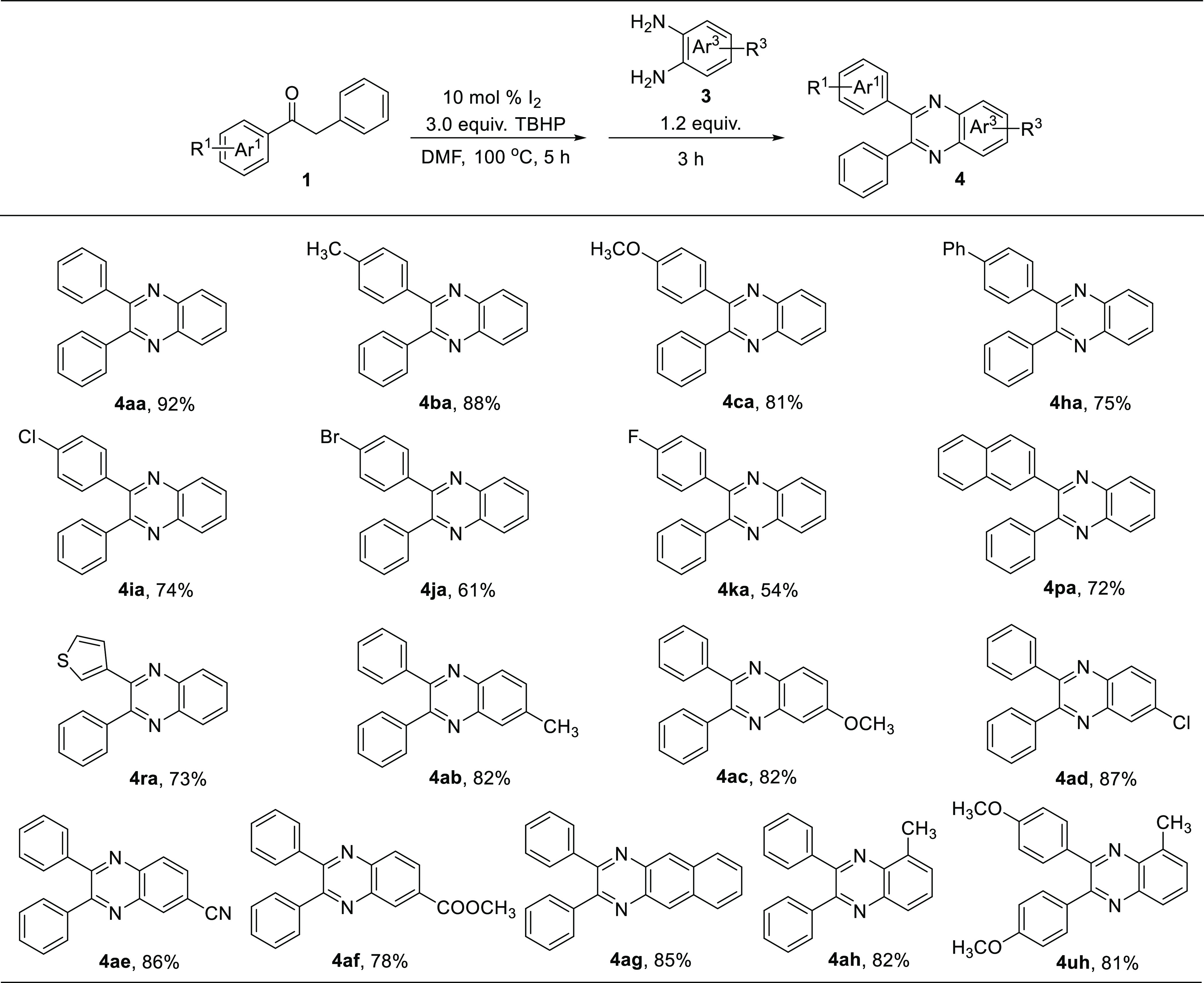
Then, we investigated the scope of the α-methylene ketones and o-phenylenediamines, and the results are shown in Scheme 5. Both electron-donating (-CH3, -OCH3, -Ph) and electron-withdrawing groups (-Cl, -Br, -F) at the p-positions of aryl ring Ar1 underwent smoothly the oxidation and cyclization reactions in one pot, producing the desired products (4ba, 4ca, 4ha, 4ia, 4ja, 4ka) in moderate to excellent yields (54–88%). To our delight, the substrates bearing naphthalene and thiophene rings proceeded well to generate the expected products (4pa, 4ra) in good yields (72%–73%). Furthermore, the compatibility and scope of o-phenylenediamines 3 were also explored. The reactions of the substrates 3 with electron-donating groups (-CH3, -OCH3) and electron-withdrawing groups (-Cl, -CN, -COOCH3) produced the corresponding products 4ab–4af in 78–87% yields. Gratifyingly, the benzyl phenyl ketone 1a reacted smoothly with 2,3-diaminonapthalene 3g, providing 4ag in 85% yield. Finally, the reactions of benzyl phenyl ketone 1a and 1,2-bis(4-methoxyphenyl)ethenone 1u with 3-methylbenzene-1,2-diamine 3h were explored under the standard conditions, smoothly giving the desired products 4ah and 4uh in 82 and 81% yields, respectively.
Scheme 5. One-Pot Synthesis of Quinoxalines from α-Methylene Ketones and o-Phenylenediamines.
2.2. Biological Evaluation
All synthesized quinoxalines 4aa–4ca, 4ha, 4ia–4ka, 4pa, 4ra, and 4ab–4ag were evaluated for their potential to inhibit viral entry of a SARS-CoV-2 spike pseudotyped virus bearing luciferase reporter into HEK-293T-ACE2h host cell that highly expresses human ACE2 (angiotensin-converting enzyme 2) receptor (Table 2).63 Initially, the maximum nontoxic concentration (CC0) values of all quinoxalines were examined on the host cells of HEK-293T-ACE2h by MTT,93,94 and their antiviral entry activities were subsequently evaluated under the compounds’ concentrations no more than the CC0 values. SARS-CoV-2 antibody was used as a positive inhibitor. 5′,7-Diallyl-3,3′-bis((5-(4-methoxyphenyl)-1,3,4-oxadiazol-2-yl)methyl)-[5,7′-bibenzo[d]oxazole]-2,2′(3H,3′H)-dione (6p), a recently reported ACE2 blocker,63 was employed as a positive antiviral entrance reference. As a result (Table 2 and Figure 1), 4ca, 4ha, and 4ad exhibited antiviral entry effect on SARS-CoV-2 spike pseudoviruses with IC50 values of 19.70, 142.50, and 21.28 μM, respectively. Quinoxaline 4ca exhibited a selectivity index (SI) of 14.05, whereas the SI of 4ad was higher than 23.49, indicating that 4ad had a larger safety window between cytotoxicity and antiviral activity than 4ca. The binding behaviors of 4ca, 4ha, and 4ad with both SARS-CoV-2 spike RBD and human ACE2 protein were also examined by biolayer interferometry (BLI) binding assay (Table 3 and Figure 2). It was found that 4ca, 4ha, and 4ad bind to human ACE2 protein with KD values of 3.67, 237.90, and 4.02 μM, respectively. With SARS-CoV-2 spike RBD, quinoxalines 4ca, 4ha, and 4ad had KD values of 15.48, 135.25, and 8.16 μM, respectively, indicating that both 4ca and 4ad had stronger binding affinity with human ACE2 protein than that with spike RBD. Molecular docking study (Table 3 and Figure 3) revealed that 4ca interacts with the amino acid residues of ASN 394 and TRP 349 in human ACE2 receptor with a calculated CDOCKER interaction energy of −38.4377 kcal/mol, and 4ad interacts with human ACE2 receptor via the sites of TRP 349, TYR 510, and HIS 345 with a CDOCKER interaction energy of −28.8546 kcal/mol. The computed CDOCKER interaction energies of 4ca and 4ad with SARS-CoV-2 spike RBD protein were −26.2545 and −22.9192 kcal/mol, respectively. Taken together, quinoxalines 4ca and 4ad could prevent SARS-CoV-2 from entering the host cell by dual blocking of both human ACE2 receptor and viral spike RBD.
Table 2. Antiviral Entry Activities of Quinoxalines for SARS-CoV-2 Spike Pseudovirus into HEK-293T-ACE2h Host Cells.
| compounds | CC0a (μM) | TC50b (μM) | IC50c (μM) | SId |
|---|---|---|---|---|
| 4aa | >250 | >25 | NDe | NDe |
| 4ba | 125 | 271.67 ± 13.00 | ND | ND |
| 4ca | 125 | 276.90 ± 13.45 | 19.70 ± 1.23 | 14.05 |
| 4ha | >250 | >500 | 142.50 ± 12.45 | >3.50 |
| 4ia | 25 | 115.20 ± 9.72 | ND | ND |
| 4ja | 25 | >100 | ND | ND |
| 4ka | 25 | >100 | ND | ND |
| 4pa | >25 | >250 | ND | ND |
| 4ra | 62.5 | 337.06 ± 15.49 | ND | ND |
| 4ab | 62.5 | 288.40 ± 17.65 | ND | ND |
| 4ac | 50 | 116.03 ± 4.28 | ND | ND |
| 4ad | >25 | >500 | 21.28 ± 0.44 | >23.49 |
| 4ae | 125 | 303.27 ± 9.51 | ND | ND |
| 4af | 100 | >100 | ND | ND |
| 4ag | 50 | 107.20 ± 1.64 | ND | ND |
| 6pf | >100 | >100 | 11.90 ± 0.59 | >8.40 |
| antibodyg | 0.10 | ND | 0.032 ± 0.001 | ND |
CC0 represents the maximum nontoxic concentration of compounds.
TC50 means the concentration that caused 50% cells’ death.
IC50 stands for the compounds’ concentration that suppressed 50% viral entry into HEK-293T-ACE2h host cells.
SI represents selectivity index that indicates compounds’ window between cytotoxicity and antiviral activity by the ratio of TC50/IC50.
ND means not determined.
honokiol derivative 6p (a reported ACE2 blocker).
SARS-CoV-2 antibody was utilized as a positive control compound for antiviral entry evaluation.
Figure 1.

Inhibitory effect of 4ca and 4ad on the entry of SARS-CoV-2 pseudovirus to host cells. The luciferase luminescence value was defined as 100% for the pseudovirus control. The values of luminescence of compound-treated groups were normalized accordingly. Data were expressed as mean ± standard error (n = 3). *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001 compared with pseudovirus control group.
Table 3. Binding Parameters of Selected Quinoxalines with Human ACE2 and SARS-CoV-2 Spike RBD Proteins.
| binding
with ACE2 |
binding
with spike RBD |
|||
|---|---|---|---|---|
| compounds | KD valuea (μM) | energyb (kcal/mol) | KD valuea(μM) | energyb (kcal/mol) |
| 4aa | 5891.00 ± 1674.28 | NCc | 1000.45 ± 760.90 | NCc |
| 4ca | 3.67 ± 0.66 | –38.4377 | 15.48 ± 1.14 | –26.2545 |
| 4ha | 237.90 ± 57.28 | NC | 135.25 ± 17.75 | NC |
| 4ad | 4.02 ± 2.03 | –28.8546 | 8.16 ± 1.34 | –22.9192 |
KD means equilibrium dissociation constant of compounds with protein by BLI binding assay.
Energy is the calculated CDOCKER interaction energy.
NC means not computed.
Figure 2.

Binding curves of 4ca and 4ad with human ACE2 and SARS-CoV-2 spike RBD proteins by BLI binding assay. (A) 4ca with ACE2. (B) 4ca with SARS-CoV-2 spike RBD. (C) 4ad with ACE2. (D) 4ad with SARS-CoV-2 spike RBD.
Figure 3.

Docking simulation of quinoxalines 4ad (A, C) and 4ca (B) in the active sites of human ACE2 (PDB code: 1R4L) and SARS-CoV-2 spike RBD (PDB code: 6M0J). (A) 3D and 2D interaction model of 4ad (in gray) with human ACE2 receptor. (B) 2D interaction model of 4ca with human ACE2 receptor. (C) 3D and 2D interaction model of 4ad (in gray) with SARS-CoV-2 spike RBD protein.
In addition, all 13 synthesized quinoxalines were further evaluated for their cytotoxicity against four cancer cell lines (MDA-MB-231, MCF-7, T47D, and A549) by the MTT method. As presented in Table 4, it was found that compounds 4aa, 4ba, 4ia and 4ab exhibited cytotoxic effects on four cancer cell lines of MCF-7, T47D, MDA-MB-231, and A549 with IC50 values ranging from 6.25 to 28.55 μM after 72 h treatment. Compound 4ag displayed IC50 values of 20.52, 45.01, and 44.45 μM against T47D, MDA-MB-231, and A549 cancer cells, respectively. Of note, the cytotoxic effect of 4ia on MCF-7 and T47D cells was 1.96- and 4.27-fold more potent than that of the positive control of berberine,93 respectively. Quinoxaline 4ab also displayed 3.94 times stronger cytotoxicity than berberine against T47D cancer cells.
Table 4. Cytotoxic Activities of 13 Quinoxalines against Four Human Cancer Cell Lines for 72 h.
| IC50 (μM) |
||||
|---|---|---|---|---|
| compounds | MCF-7 | T47D | MDA-MB-231 | A549 |
| 4aa | 16.68 ± 1.34 | 11.36±1.17 | 28.55 ± 1.85 | 26.31 ± 1.96 |
| 4ba | 15.65 ± 1.18 | 9.76 ± 1.30 | 21.45 ± 1.19 | 24.06 ±1.11 |
| 4ca | >50 | >50 | 48.84 ± 1.01 | >50 |
| 4ha | >50 | >50 | >50 | >50 |
| 4ia | 7.30 ± 1.10 | 6.25 ± 0.73 | 17.75 ±0.69 | 18.55 ±0.25 |
| 4pa | >50 | >50 | >50 | >50 |
| 4ra | >50 | >50 | >50 | >50 |
| 4ab | 10.01 ± 0.55 | 6.77 ± 0.57 | 18.21 ± 1.34 | 19.72 ± 1.28 |
| 4ac | >50 | >50 | >50 | >50 |
| 4ad | >50 | >50 | >50 | >50 |
| 4ae | >50 | >50 | >50 | >50 |
| 4af | >50 | >50 | >50 | >50 |
| 4ag | >50 | 20.52 ± 1.64 | 45.01 ± 1.57 | 44.45 ± 3.55 |
| berberinea | 14.32 ± 1.44 | 26.69 ± 2.45 | 17.96 ± 1.91 | 5.22 ± 0.44 |
Berberine was used as the positive control.
3. Conclusions
In summary, we have developed a novel I2-catalyzed oxidative carbonylation of the benzylic Csp3-H bonds using TBHP as the green oxidant. This method provided rapid and efficient access to 1,2-diaryl diketones from the α-methylene ketones, exhibiting several advantages including easily accessible starting materials, broad substrate scope, and environmentally friendly catalytic system. Moreover, we also established an efficient one-pot process for the construction of biologically active quinoxalines in the same oxidation system. A series of desired compounds were synthesized in good to excellent yields with good functional group tolerance. Quinoxalines 4ca and 4ad exhibited potential in suppressing viral entry for SARS-CoV-2 as dual blockers of both human ACE2 receptor and SARS-CoV-2 spike RBD. In addition, quinoxalines 4ia and 4ab exhibited cytotoxic effects that were comparable to or better than berberine on three human breast cancer cell lines of MDA-MB-231, MCF-7, and T47D. Further studies are in progress to elucidate the reaction mechanism and applications of the approaches. The inhibitory effect of 4ca and 4ad on SARS-CoV-2 viral strains needs further study in a biosafety level 3 facility.
4. Experimental Section
4.1. General Details
All reactions were performed in flame-dried glassware in an air condition, unless otherwise noted. Column chromatographic purification of products was carried out using silica gel (200–300 mesh). The commercial reagents were used without further purification. 1H NMR spectra were recorded at 600 MHz, and 13C NMR spectra were measured at 150 MHz in CDCl3 (containing 0.03% TMS) solutions. Tetramethylsilane (δ = 0.00 ppm) and CDCl3 (δ = 77.00 ppm) were used as internal references for 1H NMR and 13C NMR spectra, respectively. High-resolution mass spectra (HRMS) were performed on either an electrospray ionization (ESI) mass spectrometer with a time-of-flight (TOF) analyzer (Agilent 6545 QTOF-MS) or an electrospray ionization (ESI) Fourier transform mass spectrometer (FTMS, Thermo Q-Exactive Focus).
4.2. General Procedure for the Synthesis of Products 2 and 4
Benzyl phenyl ketone 1a (58.9 mg, 0.3 mmol), I2 (7.6 mg, 0.03 mmol), and DMF (2 mL) were placed in a Schlenk tube; then, TBHP (70% in water, 124 μL, 0.9 mmol) was added into the above mixture under air. After the completion of the addition, the reaction mixture was allowed to react at 100 °C (oil bath) for 5 h. Subsequently, the reaction mixture was cooled to room temperature, treated with H2O, saturated sodium thiosulfate solution, and then extracted with ethyl acetate. The combined organic layers were washed with brine and dried over anhydrous Na2SO4. After removal of the ethyl acetate under reduced pressure, the residue was purified by column chromatography on silica gel (petroleum ether/ethyl acetate = 20:1) to afford benzil 2a.
Benzyl phenyl ketone 1a (58.9 mg, 0.3 mmol), I2 (7.6 mg, 0.03 mmol), and DMF (2 mL) were added in a Schlenk tube; then, TBHP (70% in water, 124 μL, 0.9 mmol) was supplemented under air. After the completion of the addition, the reaction mixture was allowed to react at 100 °C (oil bath) for 5 h. Subsequently, o-phenylenediamine 3a (38.9 mg, 0.36 mmol) was added at room temperature. Then, the reaction mixture was stirred at 100 °C (oil bath) for 3 h. After the reaction was completed, the reaction mixture was quenched with H2O and saturated sodium thiosulfate solution and then extracted with ethyl acetate. The combined organic layers were washed with brine and dried over anhydrous Na2SO4. After removal of the ethyl acetate under reduced pressure, the residue was purified by column chromatography on silica gel (petroleum ether/ethyl acetate = 50:1–20:1) to afford 2,3-diphenylquinoxaline 4aa.
4.2.1. Benzil (2a)
Yellow solid (54.8 mg, 87% yield), purified by column chromatography on silica gel (petroleum ether/ethyl acetate = 20:1). 1H NMR (600 MHz, CDCl3) δ 7.49–7.52 (m, 4H), 7.63–7.67 (m, 2H), 7.97 (dd, J1 = 8.4 Hz, J2 = 1.2 Hz, 4H); 13C NMR (150 MHz, CDCl3) δ 128.99, 129.84, 132.94, 134.86, 194.55. HRMS (ESI) m/z [M + H]+ calcd for C14H11O2 211.0754, found 211.0761.
4.2.2. 1-Phenyl-2-(p-tolyl)ethane-1,2-dione (2b)
Yellow oil (61.9 mg, 92% yield, from 1, R1 = CH3, R2 = H), (63.5 mg, 94% yield, from 1, R1 = H, R2 = CH3), purified by column chromatography on silica gel (petroleum ether/ethyl acetate = 50:1-20:1). 1H NMR (600 MHz, CDCl3) δ 2.42 (s, 3H), 7.30 (d, J = 8.4 Hz, 2H), 7.48–7.51 (m, 2H), 7.62–7.65 (m, 1H), 7.86 (d, J = 8.4 Hz, 2H), 7.96 (dd, J1 = 8.1 Hz, J2 = 1.2 Hz, 2H); 13C NMR (150 MHz, CDCl3) δ 21.85, 128.92, 129.70, 129.81, 129.95, 130.52, 133.03, 134.74, 146.18, 194.28, 194.74. HRMS (ESI) m/z [M + H]+ calcd for C15H13O2 225.0910, found 225.0918.
4.2.3. 1-(4-Methoxyphenyl)-2-phenylethane-1,2-dione (2c)
Yellow oil (58.9 mg, 82% yield, from 1, R1 = OCH3, R2 = H), (65.9 mg, 91% yield, from 1, R1 = H, R2 = OCH3), purified by column chromatography on silica gel (petroleum ether/ethyl acetate = 50:1–20:1). 1H NMR (600 MHz, CDCl3) δ 3.87 (s, 3H), 6.97 (d, J = 8.4 Hz, 2H), 7.47–7.51 (m, 2H), 7.61–7.65 (m, 1H), 7.93–7.98 (m, 4H); 13C NMR (150 MHz, CDCl3) δ 55.56, 114.30, 125.97, 128.88, 129.78, 132.28, 133.10, 134.66, 164.94, 193.12, 194.82. HRMS (ESI) m/z [M + H]+ calcd for C15H13O3 241.0859, found 241.0866.
4.2.4. 1-(4-(tert-Butyl)phenyl)-2-phenylethane-1,2-dione (2d)
Yellow oil (70.4 mg, 88% yield), purified by column chromatography on silica gel (petroleum ether/ethyl acetate = 50:1–20:1). 1H NMR (600 MHz, CDCl3) δ 1.34 (s, 9H), 7.48–7.51 (m, 2H), 7.53 (d, J = 8.4 Hz, 2H), 7.62–7.65 (m, 1H), 7.91 (d, J = 8.4 Hz, 2H), 7.96–7.98 (m, 2H); 13C NMR (150 MHz, CDCl3) δ 30.90, 35.32, 126.00, 128.92, 129.83, 129.84, 130.42, 133.05, 134.72, 158.97, 194.26, 194.75. HRMS (ESI) m/z [M + H]+ calcd for C18H19O2 267.1380, found 267.1390.
4.2.5. 1-Phenyl-2-(m-tolyl)ethane-1,2-dione (2e)
Yellow solid (58.1 mg, 86% yield, from 1, R1 = CH3, R2 = H), (49.8 mg, 74% yield, from 1, R1 = H, R2 = CH3), purified by column chromatography on silica gel (petroleum ether/ethyl acetate = 50:1–20:1). 1H NMR (600 MHz, CDCl3) δ 2.40 (s, 3H), 7.37–7.40 (m, 1H), 7.46 (d, J = 7.8 Hz, 1H), 7.48–7.52 (m, 2H), 7.63–7.66 (m, 1H), 7.76 (d, J = 7.8 Hz, 1H), 7.78 (s, 1H), 7.95–7.98 (m, 2H); 13C NMR (150 MHz, CDCl3) δ 21.18, 127.15, 128.87, 128.95, 129.83, 130.13, 132.94, 132.97, 134.80, 135.71, 138.95, 194.67, 194.81. HRMS (ESI) m/z [M + H]+ calcd for C15H13O2 225.0910, found 225.0916.
4.2.6. 1-Phenyl-2-(o-tolyl)ethane-1,2-dione (2f)
Yellow soild (54.7 mg, 81% yield, from 1, R1 = CH3, R2 = H), (51.2 mg, 76% yield, from 1, R1 = H, R2 = CH3), purified by column chromatography on silica gel (petroleum ether/ethyl acetate = 50:1–20:1). 1H NMR (600 MHz, CDCl3) δ 2.70 (s, 3H), 7.24–7.27 (m, 1H), 7.34 (d, J = 7.2 Hz, 1H), 7.46–7.52 (m, 3H), 7.63–7.66 (m, 2H), 7.96–7.98 (m, 2H); 13C NMR (150 MHz, CDCl3) δ 21.85, 125.98, 128.97, 129.87, 131.72, 132.53, 133.00, 133.06, 133.75, 134.66, 141.31, 194.81, 196.76. HRMS (ESI) m/z [M + H]+ calcd for C15H13O2 225.0910, found 225.0917.
4.2.7. 1-(3,5-Dimethylphenyl)-2-phenylethane-1,2-dione (2g)
Yellow solid (50.9 mg, 71% yield), purified by column chromatography on silica gel (petroleum ether/ethyl acetate = 50:1–20:1). 1H NMR (600 MHz, CDCl3) δ 2.36 (s, 6H), 7.29 (s, 1H), 7.49–7.53 (m, 2H), 7.57 (s, 2H), 7.64–7.67 (m, 1H), 7.95–7.98 (m, 2H); 13C NMR (150 MHz, CDCl3) δ 21.11, 127.58, 128.97, 129.90, 133.02, 133.07, 134.78, 136.72, 138.85, 194.83, 195.11. HRMS (ESI) m/z [M + H]+ calcd for C16H15O2 239.1067, found 239.1073.
4.2.8. 1-([1,1′-Biphenyl]-4-yl)-2-phenylethane-1,2-dione (2h)
Yellowish solid (68.8 mg, 80% yield), purified by column chromatography on silica gel (petroleum ether/ethyl acetate = 50:1–20:1). 1H NMR (600 MHz, CDCl3) δ 7.38–7.41 (m, 1H), 7.44–7.47 (m, 2H), 7.49–7.52 (m, 2H), 7.59-7.62 (m, 2H), 7.63–7.66 (m, 1H), 7.70-7.72 (m, 2H), 7.99–8.01 (m, 2H), 8.03–8.05 (m, 2H); 13C NMR (150 MHz, CDCl3) δ 127.28, 127.58, 128.59, 128.98, 129.88, 130.42, 131.62, 132.97, 134.83, 139.38, 147.52, 194.09, 194.51. HRMS (ESI) m/z [M + H]+ calcd for C20H15O2 287.1067, found 287.1069.
4.2.9. 1-(4-Chlorophenyl)-2-phenylethane-1,2-dione (2i)
Yellow solid (55.4 mg, 75% yield, from 1, R1 = Cl, R2 = H), (57.7 mg, 79% yield, from 1, R1 = H, R2 = Cl), purified by column chromatography on silica gel (petroleum ether/ethyl acetate = 50:1–20:1). 1H NMR (600 MHz, CDCl3) δ 7.47–7.53 (m, 4H), 7.65–7.68 (m, 1H), 7.91–7.94 (m, 2H), 7.96 (dd, J1 = 8.4 Hz, J2 = 1.2 Hz, 2H); 13C NMR (150 MHz, CDCl3) δ 129.04, 129.41, 129.90, 131.18, 131.30, 132.72, 135.03, 141.55, 193.04, 193.85. HRMS (ESI) m/z [M + H]+ calcd for C14H10ClO2 245.0364, found 245.0366.
4.2.10. 1-(4-Bromophenyl)-2-phenylethane-1,2-dione (2j)
Yellow solid (61.7 mg, 71% yield, from 1, R1 = Br, R2 = H), (62.5 mg, 72% yield, from 1, R1 = H, R2 = Br), purified by column chromatography on silica gel (petroleum ether/ethyl acetate = 50:1-20:1). 1H NMR (600 MHz, CDCl3) δ 7.50–7.53 (m, 2H), 7.64–7.68 (m, 3H), 7.82–7.86 (m, 2H), 7.96 (dd, J1 = 8.4 Hz, J2 = 1.2 Hz, 2H); 13C NMR (150 MHz, CDCl3) δ 129.04, 129.90, 130.45, 131.19, 131.68, 132.38, 132.71, 135.03, 193.23, 193.80. HRMS (ESI) m/z [M + H]+ calcd for C14H10BrO2 288.9859, found 288.9862.
4.2.11. 1-(4-Fluorophenyl)-2-phenylethane-1,2-dione (2k)95
Yellow solid (36.7 mg, 54% yield, from 1, R1 = F, R2 = H), (42.5 mg, 62% yield, from 1, R1 = H, R2 = F), purified by column chromatography on silica gel (petroleum ether/ethyl acetate = 50:1-20:1). 1H NMR (600 MHz, CDCl3) δ 7.17–7.21 (m, 2H), 7.50–7.54 (m, 2H), 7.65–7.69 (m, 1H), 7.96–7.98 (m, 2H), 8.00–8.04 (m, 2H); 13C NMR (150 MHz, CDCl3) δ 116.37 (JC-F = 21.6 Hz), 129.04, 129.47 (JC-F = 2.4 Hz), 129.91, 132.71 (JC-F = 9.9 Hz), 132.80, 134.99, 166.76 (JC-F = 256.8 Hz), 192.72, 194.06. HRMS (ESI) m/z [M + H]+ calcd for C14H10FO2 229.0659, found 229.0661.
4.2.12. 1-(4-Acetylphenyl)-2-phenylethane-1,2-dione (2l)
Yellow solid (36.1 mg, 48% yield), purified by column chromatography on silica gel (petroleum ether/ethyl acetate = 20:1–10:1). 1H NMR (600 MHz, CDCl3) δ 2.66 (s, 3H), 7.52–7.55 (m, 2H), 7.67–7.70 (m, 1H), 7.97–7.99 (m, 2H), 8.07 (brs, 4H); 13C NMR (150 MHz, CDCl3) δ 26.91, 128.68, 129.10, 129.94, 130.07, 132.65, 135.15, 135.94, 141.29, 193.59, 193.74, 197,21. HRMS (ESI) m/z [M + H]+ calcd for C16H13O3 253.0859, found 253.0863.
4.2.13. 1-(3-Chlorophenyl)-2-phenylethane-1,2-dione (2m)
Yellow solid (51.4 mg, 70% yield, from 1, R1 = Cl, R2 = H), (44.9 mg, 61% yield, from 1, R1 = H, R2 = Cl), purified by column chromatography on silica gel (petroleum ether/ethyl acetate = 50:1-20:1). 1H NMR (600 MHz, CDCl3) δ 7.44–7.47 (m, 1H), 7.51–7.54 (m, 2H), 7.61–7.64 (m, 1H), 7.66–7.69 (m, 1H), 7.83–7.85 (m, 1H), 7.96–7.98 (m, 3H); 13C NMR (150 MHz, CDCl3) δ 128.09, 129.08, 129.53, 129.94, 130.34, 132.63, 134.46, 134.76, 135.12, 135.39, 192.94, 193.59. HRMS (ESI) m/z [M + H]+ calcd for C14H10ClO2 245.0364, found 245.0368.
4.2.14. 1-(2-Chlorophenyl)-2-phenylethane-1,2-dione (2n)
Yellow oil (34.2 mg, 47% yield, from 1, R1 = Cl, R2 = H), (42.3 mg, 58% yield, from 1, R1 = H, R2 = Cl), purified by column chromatography on silica gel (petroleum ether/ethyl acetate = 50:1-20:1). 1H NMR (600 MHz, CDCl3) δ 7.42–7.45 (m, 2H), 7.52–7.56 (m, 3H), 7.64–7.68 (m, 1H), 7.89–7.92 (m, 1H), 8.02–8.05 (m, 2H); 13C NMR (150 MHz, CDCl3) δ 127.35, 128.87, 130.19, 130.49, 132.09, 132.41, 133.83, 133.97, 134.51, 134.57, 192.05, 193.67. HRMS (ESI) m/z [M + H]+ calcd for C14H10ClO2 245.0364, found 245.0367.
4.2.15. 1-(Naphthalen-1-yl)-2-phenylethane-1,2-dione (2o)
Yellow solid (47.5 mg, 61% yield), purified by column chromatography on silica gel (petroleum ether/ethyl acetate = 50:1–20:1). 1H NMR (600 MHz, CDCl3) δ 7.47–7.53 (m, 3H), 7.61–7.67 (m, 2H), 7.73–7.76 (m, 1H), 7.91 (dd, J1 = 7.2 Hz, J2 = 1.2 Hz, 1H), 7.94 (d, J = 7.8 Hz, 1H), 8.02-8.04 (m, 2H), 8.12 (d, J = 7.8 Hz, 1H), 9.31 (d, J = 9.0 Hz, 1H); 13C NMR (150 MHz, CDCl3) δ 124.41, 125.94, 127.11, 128.62, 128.78, 129.03, 129.45, 130.02, 130.94, 133.36, 134.08, 134.73, 135.06, 135.96, 194.58, 197.16. HRMS (ESI) m/z [M + H]+ calcd for C18H13O2 261.0910, found 261.0916.
4.2.16. 1-(Naphthalen-2-yl)-2-phenylethane-1,2-dione (2p)
Yellow solid (55.8 mg, 71% yield), purified by column chromatography on silica gel (petroleum ether/ethyl acetate = 50:1–20:1). 1H NMR (600 MHz, CDCl3) δ 7.51–7.57 (m, 3H), 7.62–7.68 (m, 2H), 7.88–7.92 (m, 2H), 7.96 (d, J = 8.4 Hz, 1H), 8.02–8.04 (m, 2H), 8.10 (dd, J1 = 9.0 Hz, J2 = 1.8 Hz, 1H), 8.41 (s, 1H); 13C NMR (150 MHz, CDCl3) δ 123.64, 127.16, 127.94, 129.03, 129.16, 129.53, 129.92, 129.98, 130.31, 132.33, 133.12, 133.53, 134.89, 136.38, 194.64. HRMS (ESI) m/z [M + H]+ calcd for C18H13O2 261.0910, found 261.0918.
4.2.17. 1-(Furan-3-yl)-2-phenylethane-1,2-dione (2q)
Yellow oil (29.4 mg, 49% yield), purified by column chromatography on silica gel (petroleum ether/ethyl acetate = 50:1–20:1). 1H NMR (600 MHz, CDCl3) δ 6.94 (dd, J1 = 1.8 Hz, J2 = 0.6 Hz, 1H), 7.50–7.54 (m, 3H), 7.64–7.68 (m, 1H), 8.03–8.06 (m, 2H), 8.19–8.20 (m, 1H); 13C NMR (150 MHz, CDCl3) δ 108.59, 123.98, 128.87, 130.33, 132.49, 134.76, 144.60, 151.33, 187.01, 191.84. HRMS (ESI) m/z [M + H]+ calcd for C12H9O3 201.0546, found 201.0552.
4.2.18. 1-Phenyl-2-(thiophen-3-yl)ethane-1,2-dione (2r)
Yellow oil (58.4 mg, 90% yield), purified by column chromatography on silica gel (petroleum ether/ethyl acetate = 50:1–20:1). 1H NMR (600 MHz, CDCl3) δ 7.39–7.41 (m, 1H), 7.49–7.52 (m, 2H), 7.63–7.67 (m, 2H), 7.99–8.02 (m, 2H), 8.21–8.22 (m, 1H); 13C NMR (150 MHz, CDCl3) δ 127.04, 127.18, 128.88, 130.05, 132.62, 134.76, 136.98, 137.97, 187.24, 193.21. HRMS (ESI) m/z [M + H]+ calcd for C12H9O2S 217.0318, found 217.0324.
4.2.19. Methyl 4-(2-oxo-2-phenylacetyl)benzoate (2s)
Yellow solid (50.4 mg, 63% yield), purified by column chromatography on silica gel (petroleum ether/ethyl acetate = 50:1–20:1). 1H NMR (600 MHz, CDCl3) δ 3.96 (s, 3H), 7.51–7.55 (m, 2H), 7.66–7.70 (m, 1H), 7.97–7.99 (m, 2H), 8.04 (d, J = 8.4 Hz, 2H), 8.15–8.18 (m, 2H); 13C NMR (150 MHz, CDCl3) δ 52.57, 129.08, 129.73, 129.92, 130.05, 132.64, 135.11, 135.27, 136.00, 165.83, 193.63, 193.75. HRMS (ESI) m/z [M + H]+ calcd for C16H13O4 269.0808, found 269.0809.
4.2.20. 1-(4-Nitrophenyl)-2-phenylethane-1,2-dione (2t)
Yellow solid (28.4 mg, 37% yield), purified by column chromatography on silica gel (petroleum ether/ethyl acetate = 50:1–20:1). 1H NMR (600 MHz, CDCl3) δ 7.54–7.57 (m, 2H), 7.70–7.73 (m, 1H), 7.98–8.00 (m, 2H), 8.18 (d, J = 9.0 Hz, 2H), 8.36 (d, J = 8.4 Hz, 2H); 13C NMR (150 MHz, CDCl3) δ 124.13, 129.24, 130.06, 130.97, 132.38, 135.47, 137.31, 151.16, 192.08, 192.86. HRMS (ESI) m/z [M + Na]+ calcd for C14H9NO4Na 278.0424, found 278.0428.
4.2.21. 2,3-Diphenylquinoxaline (4aa)
White solid (77.6 mg, 92% yield), purified by column chromatography on silica gel (petroleum ether/ethyl acetate = 50:1–20:1). 1H NMR (600 MHz, CDCl3) δ 7.29–7.36 (m, 6H), 7.50–7.53 (m, 4H), 7.71–7.75 (m, 2H), 8.15–8.19 (m, 2H); 13C NMR (150 MHz, CDCl3) δ 128.17, 128.70, 129.10, 129.75, 129.85, 138.98, 141.13, 153.35. HRMS (ESI) m/z [M + H]+ calcd for C20H15N2 283.1230, found 283.1229.
4.2.22. 2-Phenyl-3-(p-tolyl)quinoxaline (4ba)
White solid (78.1 mg, 88% yield), purified by column chromatography on silica gel (petroleum ether/ethyl acetate = 50:1–20:1). 1H NMR (600 MHz, CDCl3) δ 2.34 (s, 3H), 7.12 (d, J = 7.8 Hz, 2H), 7.31–7.37 (m, 3H), 7.41 (d, J = 7.8 Hz, 2H), 7.52–7.55 (m, 2H), 7.71–7.75 (m, 2H), 8.14–8.17 (m, 2H); 13C NMR (150 MHz, CDCl3) δ 21.25, 128.19, 128.66, 128.91, 129.08, 129.67, 129.73, 129.78, 136.11, 138.76, 139.23, 141.02, 141.21, 153.40. HRMS (ESI) m/z [M + H]+ calcd for C21H17N2 297.1386, found 297.1387.
4.2.23. 2-(4-Methoxyphenyl)-3-phenylquinoxaline (4ca)
Yellowish solid (76.2 mg, 81% yield), purified by column chromatography on silica gel (petroleum ether/ethyl acetate = 50:1–20:1). 1H NMR (600 MHz, CDCl3) δ 3.77 (s, 3H), 6.82–6.85 (m, 2H), 7.31–7.37 (m, 3H), 7.45–7.49 (m, 2H), 7.52–7.55 (m, 2H), 7.68–7.73 (m, 2H), 8.12-8.15 (m, 2H); 13C NMR (150 MHz, CDCl3) δ 55.14, 113.60, 128.19, 128.62, 128.92, 129.01, 129.48, 129.63, 129.73, 131.26, 139.30, 140.86, 141.17, 152.89, 153.27, 160.10. HRMS (ESI) m/z [M + H]+ calcd for C21H17N2O 313.1335, found 313.1334.
4.2.24. 2-([1,1′-Biphenyl]-4-yl)-3-phenylquinoxaline (4ha)
White solid (80.6 mg, 75% yield), purified by column chromatography on silica gel (petroleum ether/ethyl acetate = 50:1–20:1). 1H NMR (600 MHz, CDCl3) δ 7.32–7.39 (m, 4H), 7.40–7.44 (m, 2H), 7.55–7.62 (m, 8H), 7.73–7.77 (m, 2H), 8.16–8.20 (m, 2H); 13C NMR (150 MHz, CDCl3) δ 126.88, 127.02, 127.56, 128.31, 128.76, 128.81, 129.14, 129.81, 129.90, 129.92, 130.28, 137.90, 139.08, 140.29, 141.15, 141.24, 141.43, 152.99, 153.39. HRMS (ESI) m/z [M + H]+ calcd for C26H19N2 359.1543, found 359.1544.
4.2.25. 2-(4-Chlorophenyl)-3-phenylquinoxaline (4ia)
White solid (70.3 mg, 74% yield), purified by column chromatography on silica gel (petroleum ether/ethyl acetate = 50:1–20:1). 1H NMR (600 MHz, CDCl3) δ 7.28–7.31 (m, 2H), 7.33–7.39 (m, 3H), 7.45–7.48 (m, 2H), 7.49–7.52 (m, 2H), 7.74–7.78 (m, 2H), 8.13–8.17 (m, 2H); 13C NMR (150 MHz, CDCl3) δ 128.40, 128.46, 128.94, 129.09, 129.16, 129.73, 130.07, 130.14, 131.19, 135.05, 137.43, 138.76, 141.11, 141.21, 152.07, 153.17. HRMS (ESI) m/z [M + H]+ calcd for C20H14ClN2 317.0840, found 317.0843.
4.2.26. 2-(4-Bromophenyl)-3-phenylquinoxaline (4ja)
White solid (65.7 mg, 61% yield), purified by column chromatography on silica gel (petroleum ether/ethyl acetate = 50:1–20:1). 1H NMR (600 MHz, CDCl3) δ 7.35–7.42 (m, 5H), 7.46–7.48 (m, 2H), 7.50–7.52 (m, 2H), 7.76–7.79 (m, 2H), 8.15–8.19 (m, 2H); 13C NMR (150 MHz, CDCl3) δ 123.45, 128.46, 128.99, 129.15, 129.22, 129.76, 130.12, 130.20, 131.45, 131.47, 137.95, 138.78, 141.18, 141.26, 152.15, 153.19. HRMS (ESI) m/z [M + H]+ calcd for C20H1479BrN2 361.0335, found 361.0321; C20H1481BrN2 363.0314, found 363.0302.
4.2.27. 2-(4-Fluorophenyl)-3-phenylquinoxaline (4ka)95
White solid (49.1 mg, 54% yield), purified by column chromatography on silica gel (petroleum ether/ethyl acetate = 50:1–20:1). 1H NMR (600 MHz, CDCl3) δ 7.00–7.04 (m, 2H), 7.33–7.38 (m, 3H), 7.49–7.53 (m, 4H), 7.75–7.79 (m, 2H), 8.14-8.18 (m, 2H); 13C NMR (150 MHz, CDCl3) δ 115.33 (JC-F = 21.5Hz), 128.38, 128.90, 129.09, 129.19, 129.73, 130.04, 130.05, 131.79 (JC-F = 8.3Hz), 135.07 (JC-F = 3.2Hz), 138.92, 141.15, 141.20, 152.29, 153.28, 163.14 (JC-F = 248.1 Hz). HRMS (ESI) m/z [M + H]+ calcd for C20H14FN2 301.1136, found 301.1125.
4.2.28. 2-(Naphthalen-2-yl)-3-phenylquinoxaline (4pa)
White solid (72.1 mg, 72% yield), purified by column chromatography on silica gel (petroleum ether/ethyl acetate = 50:1–20:1). 1H NMR (600 MHz, CDCl3) δ 7.27–7.30 (m, 2H), 7.31–7.35 (m, 1H), 7.43–7.50 (m, 2H), 7.51 (dd, J1 = 8.4 Hz, J2 = 1.8 Hz, 1H), 7.53–7.56 (m, 2H), 7.73 (d, J = 8.4 Hz, 1H), 7.74–7.78 (m, 2H), 7.79 (d, J = 7.8 Hz, 2H), 8.14 (s, 1H), 8.18–8.22 (m, 2H); 13C NMR (150 MHz, CDCl3) δ 126.22, 126.77, 127.01, 127.59, 127.62, 128.31, 128.58, 128.81, 129.16, 129.18, 129.77, 129.80, 129.95, 133.10, 133.18, 136.48, 139.04, 141.19, 141.29, 153.22, 153.53. HRMS (ESI) m/z [M + H]+ calcd for C24H17N2 333.1386, found 333.1387.
4.2.29. 2-Phenyl-3-(thiophen-3-yl)quinoxaline (4ra)
White solid (63.4 mg, 73% yield), purified by column chromatography on silica gel (petroleum ether/ethyl acetate = 50:1–20:1). 1H NMR (600 MHz, CDCl3) δ 7.21–7.26 (m, 2H), 7.41–7.44 (m, 4H), 7.55–7.59 (m, 2H), 7.71–7.76 (m, 2H), 8.11–8.16 (m, 2H); 13C NMR (150 MHz, CDCl3) δ 125.18, 127.46, 128.38, 128.65, 128.94, 128.95, 129.04, 129.37, 129.68, 129.89, 139.23, 140.05, 140.77, 141.17, 148.43, 153.31. HRMS (ESI) m/z [M + H]+ calcd for C18H13N2S 289.0794, found 289.0793.
4.2.30. 6-Methyl-2,3-diphenylquinoxaline (4ab)
White solid (72.5 mg, 82% yield), purified by column chromatography on silica gel (petroleum ether/ethyl acetate = 50:1–20:1). 1H NMR (600 MHz, CDCl3) δ 2.58 (s, 3H), 7.28–7.35 (m, 6H), 7.48–7.51 (m, 4H), 7.56 (d, J = 8.4 Hz, 1H), 7.94 (s, 1H), 8.05 (d, J = 8.4 Hz, 1H); 13C NMR (150 MHz, CDCl3) δ 21.79, 127.91, 128.10, 128.51, 128.57, 128.58, 129.73, 129.74, 132.18, 139.11, 139.59, 140.35, 141.17, 152.44, 153.19. HRMS (ESI) m/z [M + H]+ calcd for C21H17N2 297.1386, found 297.1387.
4.2.31. 6-Methoxy-2,3-diphenylquinoxaline (4ac)
White solid (76.7 mg, 82% yield), purified by column chromatography on silica gel (petroleum ether/ethyl acetate = 50:1–20:1). 1H NMR (600 MHz, CDCl3) δ 3.95 (s, 3H), 7.28–7.35 (m, 6H), 7.40 (dd, J1 = 9.0 Hz, J2 = 3.0 Hz, 1H), 7.45 (d, J = 2.4 Hz, 1H), 7.48–7.52 (m, 4H), 8.04 (d, J = 8.4 Hz, 1H); 13C NMR (150 MHz, CDCl3) δ 55.70, 106.35, 123.23, 128.10, 128.13, 128.34, 128.55, 129.71, 130.04, 137.28, 139.11, 139.17, 142.65, 150.80, 153.22, 160.77. HRMS (ESI) m/z [M + H]+ calcd for C21H17N2O 313.1335, found 313.1335.
4.2.32. 6-Chloro-2,3-diphenylquinoxaline (4ad)
Yellowish solid (82.6 mg, 87% yield), purified by column chromatography on silica gel (petroleum ether/ethyl acetate = 50:1–20:1). 1H NMR (600 MHz, CDCl3) δ 7.29–7.37 (m, 6H), 7.48–7.51 (m, 4H), 7.65–7.68 (m, 1H), 8.07 (d, J = 9.0 Hz, 1H), 8.15 (d, J = 3.0 Hz, 1H); 13C NMR (150 MHz, CDCl3) δ 127.97, 128.21, 128.22, 128.92, 129.01, 129.72, 129.76, 130.33, 130.83, 135.54, 138.55, 138.62, 139.60, 141.37, 153.49, 154.16. HRMS (ESI) m/z [M + H]+ calcd for C20H14ClN2 317.0840, found 317.0842.
4.2.33. 2,3-Diphenylquinoxaline-6-carbonitrile (4ae)
White solid (79.6 mg, 86% yield), purified by column chromatography on silica gel (petroleum ether/ethyl acetate = 50:1–20:1). 1H NMR (600 MHz, CDCl3) δ 7.33–7.36 (m, 4H), 7.38–7.42 (m, 2H), 7.51–7.55 (m, 4H), 7.85–7.88 (m, 1H), 8.23 (dd, J1 = 9.0 Hz, J2 = 0.6 Hz, 1H), 8.51 (s, 1H); 13C NMR (150 MHz, CDCl3) δ 113.06, 118.04, 128.30, 129.42, 129.52, 129.70, 129.76, 130.49, 130.62, 134.97, 137.99, 138.04, 140.08, 142.42, 155.20, 155.78. HRMS (ESI) m/z [M + H]+ calcd for C21H14N3 308.1182, found 308.1183.
4.2.34. Methyl 2,3-diphenylquinoxaline-6-carboxylate (4af)
White solid (79.7 mg, 78% yield), purified by column chromatography on silica gel (petroleum ether/ethyl acetate = 50:1–20:1). 1H NMR (600 MHz, CDCl3) δ 4.00 (s, 3H), 7.31–7.35 (m, 4H), 7.35–7.39 (m, 2H), 7.52–7.55 (m, 4H), 8.18 (d, J = 9.0 Hz, 1H), 8.33 (dd, J1 = 8.7 Hz, J2 = 2.4 Hz, 1H), 8.88 (d, J = 2.4 Hz, 1H); 13C NMR (150 MHz, CDCl3) δ 52.48, 128.22, 129.03, 129.16, 129.30, 129.34, 129.74, 129.78, 131.08, 131.80, 138.51, 140.28, 143.04, 154.29, 155.00, 166.20. HRMS (ESI) m/z [M + H]+ calcd for C22H17N2O2 341.1285, found 341.1286.
4.2.35. 2,3-Diphenylbenzo[g]quinoxaline (4ag)
Yellow solid (85.2 mg, 85% yield), purified by column chromatography on silica gel (petroleum ether/ethyl acetate = 50:1–20:1). 1H NMR (600 MHz, CDCl3) δ 7.30–7.37 (m, 6H), 7.48–7.51 (m, 2H), 7.54–7.56 (m, 4H), 8.03–8.06 (m, 2H), 8.70 (s, 2H); 13C NMR (150 MHz, CDCl3) δ 126.58, 127.42, 128.14, 128.40, 128.89, 129.75, 133.92, 137.82, 139.04, 154.01. HRMS (ESI) m/z [M + H]+ calcd for C24H17N2 333.1386, found 333.1388.
4.2.36. 5-Methyl-2,3-diphenylquinoxaline (4ah)
Yellow solid (73.3 mg, 82% yield), purified by column chromatography on silica gel (petroleum ether/ethyl acetate = 50:1–20:1). 1H NMR (600 MHz, CDCl3) δ 2.84 (s, 3H), 7.28–7.36 (m, 6H), 7.51–7.54 (m, 2H), 7.55–7.58 (m, 3H), 7.60–7.63 (m, 1H), 7.99 (dd, J1 = 8.4 Hz, J2 = 0.6 Hz, 1H); 13C NMR (150 MHz, CDCl3) δ 17.07, 126.89, 128.03, 128.22, 128.61, 129.61, 129.72, 129.75, 130.08, 137.54, 139.30, 139.35, 140.32, 141.09, 151.69, 152.74. HRMS (ESI) m/z [M + H]+ calcd for C21H17N2 297.1386, found 297.1386.
4.2.37. 2,3-Bis(4-methoxyphenyl)-5-methylquinoxaline (4uh)
Yellowish solid (86.9 mg, 81% yield), purified by column chromatography on silica gel (petroleum ether/ethyl acetate = 50:1–20:1). 1H NMR (600 MHz, CDCl3) δ 2.82 (s, 3H), 3.79 (s, 3H), 3.80 (s, 3H), 6.83–6.88 (m, 4H), 7.48–7.52 (m, 3H), 7.52–7.58 (m, 3H), 7.94 (dd, J1 = 8.1 Hz, J2 = 1.2 Hz, 1H); 13C NMR (150 MHz, CDCl3) δ 17.01, 55.16, 55.19, 113.50, 113.69, 126.65, 129.12, 129.32, 131.09, 131.39, 131.93, 137.21, 140.07, 140.85, 151.18, 152.20, 159.99, 160.02. HRMS (ESI) m/z [M + H]+ calcd for C23H21N2O2 357.1598, found 357.1594.
4.3. Cell Viability Assay
The MTT assay was utilized to examine the cytotoxic effects of quinoxalines on both HEK-293T-ACE2h cells and four human cancer cell lines mentioned below. HEK-293T-ACE2h cells were constructed by Sino Biological (Beijing, China) and cultured in Dulbecco’s modified Eagle’s medium (DMEM) supplemented with 10% fetal bovine serum and the antibiotics of penicillin (50 U/mL), streptomycin (50 μg/mL, Invitrogen, Paisley, Scotland, U.K.), and hygromycin (100 μg/mL) at 37 °C in a humidified atmosphere of 5% CO2. HEK-293T-ACE2h cells were seeded into a 96-well plate at a density of 1 × 104 cells per well and then treated with different concentrations of compounds for 4 h to measure the maximum nontoxic concentration (CC0) of quinoxalines. The CC0 values of quinoxalines for 4 h were determined as the highest tested concentration at which cell viability was 100%. The TC50 value was determined as the concentration that caused 50% cells’ death for 4 h quinoxalines’ treatment. The human breast cancer cell lines (MCF-7 and T47D) and human lung carcinoma cell line (A549) were purchased from the American Type Culture Collection (ATCC, Rockville, MD), and cultivated in DMEM containing 10% fetal bovine serum (FBS) and 1% penicillin–streptomycin. The triple-negative breast cancer cell line (MDA-MB-231) was provided by Cell Bank of Chinese Academy of Sciences (Shanghai, China) and cultured in RPMI 1640 medium supplemented with 10% FBS and 1% penicillin–streptomycin. First, cancer cells were seeded in 96-well plates at a density of 2 × 103–4 × 103 cells per well and then incubated for 24 h at 37 °C with 5% CO2. Subsequently, various concentrations (0–40 μM) of quinoxalines and berberine (as the positive control alkaloid) were added to each well. After further incubation for 72 h, 10 μL of MTT (5 mg/mL) was administered to each well and incubated for another 4 h. DMSO (100 μL) was then added to each well, and the plate was agitated for 10 min to dissolve the formazan completely. Finally, the absorbance value of each well was determined by a microplate reader at 570 nm. The IC50 values were calculated with the SPSS 20.0 software. All of the data were obtained in triplicate and presented as mean ± SD.
4.4. Detection of SARS-CoV-2 Spike Pseudovirus Entry into HEK-293T-ACE2h Cells
In HEK-293T cells, pNL4-3.Luc.R-E- plasmid (from the NIH AIDS repository) and pcDNA3.1-SARS-CoV-2-Spike-Myc plasmid (from Beyotime, Shanghai, China) were utilized to produce SARS-CoV-2 spike pseudotyped viruses according to the published protocol.63 The infectious competency of 30 μL generated pseudovirus was quantified in hexaplicate to be equivalent to that of 8.05 ± 0.39 × 107 copies of SARS-CoV-2 pseudovirus (1010 copies/mL, PSV001) from Sino Biological by a viral infection equation: Y = 197.06X – 87.679, R2 = 0.9997. HEK-293T-ACE2h cells (1 × 104 in 100 μL DMEM) were seeded in 96-well plates and cultured for 24 h. The culture medium was then replaced with 100 μL of compound-containing DMEM for a 2 h incubation at 37 °C. Following that, 30 μL of produced pseudovirus (MOI = 100 virus particles/cell), 20 μL of media, and 50 μL of twofold concentration of compounds were simultaneously supplemented to each well for another 2 h incubation at 37 °C. After removing the inoculum, the cells were overlaid with 100 μL of fresh DMEM and continuously incubated at 37 °C for 48 h. The culture media were discarded, and the cells were washed with PBS once. The plate was then agitated for 10 min after 100 μL of cell lysate solution was supplemented to each well. The cell lysate was then transferred to an opaque 96-well white solid plate, and 100 μL of luminescence solution was filled into each well according to instructions of the Firefly Luciferase Reporter Gene Assay Kit (Beyotime, China). Immediately, the luciferase luminescence in each well was determined at 578 nm by a microplate reader (SpectraMax iD5 Multi-Mode Microplate Reader, Molecular Devices). The control group was HEK-293T-ACE2h cells infected only with SARS-CoV-2 spike pseudovirus, and the luciferase luminescence value was set as 100% for the control group. The luminescence values of compound-treated groups were normalized according to the control group.64 The inhibitory activity of the compounds tested against pseudovirus entry was presented as an IC50 value, which is defined as the concentration that prevents 50% of the pseudovirus from entering the target cell. GraphPad Prism 8.0 software (La Jolla, CA) was used to compute the IC50 values. All of the data came from three separate experiments.
4.5. Biolayer Interferometry (BLI) Binding Assay
Biolayer interferometry was used to examine the binding kinetics of compounds with the proteins of recombinant histidine-tagged SARS-CoV-2 spike RBD and recombinant histidine-tagged human ACE2 (Sino Biological, Beijing, China) by an Octet system (Octet RED96, ForteBio).65,66 A protein-loading program of the instrument immobilized either histidine-tagged SARS-CoV-2 spike RBD protein (25 g/mL aqueous solution) or histidine-tagged human ACE2 protein (25 g/mL aqueous solution) on nickel charged nitrilotriacetic acid (Ni-NTA) biosensors (Fortebi). Each compound’s stock solution (10 mM in DMSO) was serially diluted with PBS to a final DMSO concentration of 1.0%. Before data acquisition, both protein-immobilized and empty biosensors were equilibrated in PBS for 10 min at room temperature and all experiments were carried out at 30 °C. The protein-coated and uncoated biosensors were both dipped in wells containing serially diluted samples. By dipping a protein-immobilized biosensor in the blank buffer, the background signal was subtracted from all samples. Empty biosensors were also dipped in serially diluted samples for reference subtraction under the same conditions. The subtracted sensorgrams were then fitted to a 1:1 binding model with Octet Data Analysis Software v11.1 (ForteBio), and the resulting equilibrium dissociation constant (KD) values were calculated.
4.6. Molecular Docking Study
CDOCKER of Discovery Studio software was utilized to conduct molecular docking analysis on the structure of proteins of spike RBD from SARS-CoV-2 (PDB ID: 6M0J) and human ACE2 bound with its inhibitor (PDB ID: 1R4L), respectively. The active contact sites for compounds were determined to be Tyr449, Gln493, Gln498, Thr500, Asn501, Gly502, and Tyr505 of the spike RBD according to the reported literature.67,68 A docking pocket was produced based on the position of the ACE2 inhibitor.69 Discovery Studio software was utilized to analyze the binding modes.
4.7. Statistical analysis
Statistical comparisons were calculated by Student’s t-test analysis, and p < 0.05 was regarded as statistically significant.
Acknowledgments
This research was funded by The Science and Technology Development Fund, Macau SAR (File no. 0065/2020/A2, 0074/2019/AMJ, 0023/2019/AKP, and 0002/2019/APD) and the Natural Science Foundation of Shandong Province (ZR2020QB013). This work was also supported by the Emergency Key Program of Guangzhou Laboratory (grant no. EKPG21-06). The authors also thank the Department of Science and Technology of Guangdong Province for the support of Guangdong-Hong Kong-Macao Joint Laboratory of Respiratory Infectious Disease.
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acsomega.1c06017.
Quinoxalines 4aa–4ta with calculated binding energy with both human ACE2 and SARS-CoV-2 spike RBD proteins by molecular docking study, 1H-NMR and 13C-NMR spectra of products 2 and 4, and HRMS spectra of products 2 and 4 (PDF)
The authors declare no competing financial interest.
Supplementary Material
References
- Maurya R.; Singh R.; Deepak M.; Handa S. S.; Yadav P. P.; Mishra P. K. Constituents of Pterocarpus marsupium: an ayurvedic crude drug. Phytochemistry 2004, 65, 915–920. 10.1016/j.phytochem.2004.01.021. [DOI] [PubMed] [Google Scholar]
- Mahabusarakam W.; Deachathai S.; Phongpaichit S.; Jansakul C.; Taylor W. C. A benzil and isoflavone derivatives from Derris scandens Benth. Phytochemistry 2004, 65, 1185–1191. 10.1016/j.phytochem.2004.03.006. [DOI] [PubMed] [Google Scholar]
- Wadkins R. M.; Hyatt J. L.; Wei X.; Yoon K. J. P.; Wierdl M.; Edwards C. C.; Morton C. L.; Obenauer J. C.; Damodaran K.; Beroza P. J. J.; et al. Identification and characterization of novel benzil (diphenylethane-1, 2-dione) analogues as inhibitors of mammalian carboxylesterases. J. Med. Chem. 2005, 48, 2906–2915. 10.1021/jm049011j. [DOI] [PubMed] [Google Scholar]
- Hyatt J. L.; Stacy V.; Wadkins R. M.; Yoon K. J. P.; Wierdl M.; Edwards C. C.; Zeller M.; Hunter A. D.; Danks M. K.; Crundwell G. J. J.; et al. Inhibition of carboxylesterases by benzil (diphenylethane-1, 2-dione) and heterocyclic analogues is dependent upon the aromaticity of the ring and the flexibility of the dione moiety. J. Med. Chem. 2005, 48, 5543–5550. 10.1021/jm0504196. [DOI] [PubMed] [Google Scholar]
- Mousset C.; Giraud A.; Provot O.; Hamze A.; Bignon J.; Liu J. M.; Thoret S.; Dubois J.; Brion J. D.; Alami M. Synthesis and antitumor activity of benzils related to combretastatin A-4. Bioorg. Med. Chem. Lett. 2008, 18, 3266–3271. 10.1016/j.bmcl.2008.04.053. [DOI] [PubMed] [Google Scholar]
- Ganapaty S.; Srilakshmi G. V.; Pannakal S. T.; Rahman H.; Laatsch H.; Brun R. Cytotoxic benzil and coumestan derivatives from Tephrosia calophylla. Phytochemistry 2009, 70, 95–99. 10.1016/j.phytochem.2008.10.009. [DOI] [PubMed] [Google Scholar]
- Harada T.; Nakagawa Y.; Wadkins R. M.; Potter P. M.; Wheelock C. E. Comparison of benzil and trifluoromethyl ketone (TFK)-mediated carboxylesterase inhibition using classical and 3D-quantitative structure-activity relationship analysis. Bioorg. Med. Chem. 2009, 17, 149–164. 10.1016/j.bmc.2008.11.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Young B. M.; Hyatt J. L.; Bouck D. C.; Chen T.; Hanumesh P.; Price J.; Boyd V. A.; Potter P. M.; Webb T. R. Structure-activity relationships of substituted 1-pyridyl-2-phenyl-1,2-ethanediones: potent, selective carboxylesterase inhibitors. J. Med. Chem. 2010, 53, 8709–8715. 10.1021/jm101101q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen Y.; Feng Z. M.; Jiang J. S.; Yang Y. N.; Zhang P. C. Dibenzoyl and isoflavonoid glycosides from Sophora flavescens: inhibition of the cytotoxic effect of D-galactosamine on human hepatocyte HL-7702. J. Nat. Prod. 2013, 76, 2337–2345. 10.1021/np400784v. [DOI] [PubMed] [Google Scholar]
- Wolkenberg S. E.; Wisnoski D. D.; Leister W. H.; Wang Y.; Zhao Z.; Lindsley C. W. Efficient synthesis of imidazoles from aldehydes and 1,2-diketones using microwave irradiation. Org. Lett. 2004, 6, 1453–1456. 10.1021/ol049682b. [DOI] [PubMed] [Google Scholar]
- Deng X.; Mani N. S. An efficient route to 4-aryl-5-pyrimidinylimidazoles via sequential functionalization of 2, 4-dichloropyrimidine. Org. Lett. 2006, 8, 269–272. 10.1021/ol052663x. [DOI] [PubMed] [Google Scholar]
- Yu X.; Guttenberger N.; Fuchs E.; Peters M.; Weber H.; Breinbauer R. Diversity-Oriented Synthesis of a Library of Star-Shaped 2H-Imidazolines. ACS Comb. Sci. 2015, 17, 682–690. 10.1021/acscombsci.5b00107. [DOI] [PubMed] [Google Scholar]
- Pradhan K.; Tiwary B. K.; Hossain M.; Chakraborty R.; Nanda A. K. A mechanistic study of carbonyl activation under solvent-free conditions: evidence drawn from the synthesis of imidazoles. RSC Adv. 2016, 6, 10743–10749. 10.1039/C5RA16386B. [DOI] [Google Scholar]
- Zhao Z.; Wisnoski D. D.; Wolkenberg S. E.; Leister W. H.; Wang Y.; Lindsley C. W. General microwave-assisted protocols for the expedient synthesis of quinoxalines and heterocyclic pyrazines. Tetrahedron Lett. 2004, 45, 4873–4876. 10.1016/j.tetlet.2004.04.144. [DOI] [Google Scholar]
- Bhosale R. S.; Sarda S. R.; Ardhapure S. S.; Jadhav W. N.; Bhusare S. R.; Pawar R. P. An efficient protocol for the synthesis of quinoxaline derivatives at room temperature using molecular iodine as the catalyst. Tetrahedron Lett. 2005, 46, 7183–7186. 10.1016/j.tetlet.2005.08.080. [DOI] [Google Scholar]
- Hui X.; Desrivot J.; Bories C.; Loiseau P. M.; Franck X.; Hocquemiller R.; Figadere B. Synthesis and antiprotozoal activity of some new synthetic substituted quinoxalines. Bioorg. Med. Chem. Lett. 2006, 16, 815–820. 10.1016/j.bmcl.2005.11.025. [DOI] [PubMed] [Google Scholar]
- Cabrera A.; Sharma P.; Ayala M.; Rubio-Perez L.; Amézquita-Valencia M. [(S)-BINAP]PdBr2-catalyzed direct synthesis of 2,3-disubstituted indoles via a tandem reaction between arylamines and α-diketones. Tetrahedron Lett. 2011, 52, 6758–6762. 10.1016/j.tetlet.2011.10.025. [DOI] [Google Scholar]
- Boström J.; Berggren K.; Elebring T.; Greasley P. J.; Wilstermann M. Scaffold hopping, synthesis and structure-activity relationships of 5,6-diaryl-pyrazine-2-amide derivatives: a novel series of CB1 receptor antagonists. Bioorg. Med. Chem. 2007, 15, 4077–4084. 10.1016/j.bmc.2007.03.075. [DOI] [PubMed] [Google Scholar]
- Zhao Z.; Leister W. H.; Strauss K. A.; Wisnoski D. D.; Lindsley C. W. Broadening the scope of 1, 2, 4-triazine synthesis by the application of microwave technology. Tetrahedron Lett. 2003, 44, 1123–1127. 10.1016/S0040-4039(02)02845-9. [DOI] [Google Scholar]
- Wang G.; Wang J.; He D.; Li X.; Li J.; Peng Z. Synthesis and biological evaluation of novel 1,2,4-triazine derivatives bearing carbazole moiety as potent α-glucosidase inhibitors. Bioorg. Med. Chem. Lett. 2016, 26, 2806–2809. 10.1016/j.bmcl.2016.04.071. [DOI] [PubMed] [Google Scholar]
- Kucukkilinc T. T.; Yanghagh K. S.; Ayazgok B.; Roknipour M. A.; Moghadam F. H.; Moradi A.; Emami S.; Amini M.; Irannejad H. Synthesis and neuroprotective activity of novel 1,2,4-triazine derivatives with ethyl acetate moiety against H2O2 and Aβ-induced neurotoxicity. Med. Chem. Res. 2017, 26, 3057–3071. 10.1007/s00044-017-2003-x. [DOI] [Google Scholar]
- Ren H.; Li J.; Zhang T.; Wang R.; Gao Z.; Liu D. Synthesis and properties of novel perylenebisimide-cored dendrimers. Dyes Pigm. 2011, 91, 298–303. 10.1016/j.dyepig.2011.04.008. [DOI] [Google Scholar]
- Li H.; Tam T. L.; Lam Y. M.; Mhaisalkar S. G.; Grimsdale A. C. Synthesis of low band gap [1,2,5]-thiadiazolo [3,4-g] quinoxaline and pyrazino [2,3-g] quinoxaline derivatives by selective reduction of benzo [1,2-c;4,5-c′] bis [1,2,5] thiadiazole. Org. Lett. 2011, 13, 46–49. 10.1021/ol102465a. [DOI] [PubMed] [Google Scholar]
- Yamada H.; Ohashi C.; Aotake T.; Katsuta S.; Honsho Y.; Kawano H.; Okujima T.; Uno H.; Ono N.; Seki S.; Nakayama K. FET performance and substitution effect on 2,6-dithienylanthracene devices prepared by photoirradiation of their diketone precursors. Chem. Commun. 2012, 48, 11136–11138. 10.1039/c2cc35439j. [DOI] [PubMed] [Google Scholar]
- Suzuki M.; Aotake T.; Yamaguchi Y.; Noguchi N.; Nakano H.; Nakayama K.-i.; Yamada H. Synthesis and photoreactivity of α-diketone-type precursors of acenes and their use in organic-device fabrication. J. Photochem. Photobiol., C 2014, 18, 50–70. 10.1016/j.jphotochemrev.2013.10.003. [DOI] [Google Scholar]
- Marin L.; Kudrjasova J.; Verstappen P.; Penxten H.; Robeyns K.; Lutsen L.; Vanderzande D. J.; Maes W. Quinoxaline-based cyclo(oligophenylenes). J. Org. Chem. 2015, 80, 2425–2430. 10.1021/jo502771a. [DOI] [PubMed] [Google Scholar]
- Huang S.; Gao T.; Bi A.; Cao X.; Feng B.; Liu M.; Du T.; Feng X.; Zeng W. Revealing aggregation-induced emission effect of imidazolium derivatives and application for detection of Hg2+. Dyes Pigm. 2020, 172, 107830 10.1016/j.dyepig.2019.107830. [DOI] [Google Scholar]
- Su Y.; Zhang L.; Jiao N. Utilization of natural sunlight and air in the aerobic oxidation of benzyl halides. Org. Lett. 2011, 13, 2168–2171. 10.1021/ol2002013. [DOI] [PubMed] [Google Scholar]
- Chen C.-T.; Kao J.-Q.; Salunke S. B.; Lin Y.-H. Enantioselective aerobic oxidation of α-hydroxy-ketones catalyzed by oxidovanadium (V) methoxides bearing chiral, N-salicylidene-tert-butylglycinates. Org. Lett. 2011, 13, 26–29. 10.1021/ol1024053. [DOI] [PubMed] [Google Scholar]
- Muthupandi P.; Sekar G. Zinc-catalyzed aerobic oxidation of benzoins and its extension to enantioselective oxidation. Tetrahedron Lett. 2011, 52, 692–695. 10.1016/j.tetlet.2010.12.004. [DOI] [Google Scholar]
- Urgoitia G.; Martin R. S.; Herrero M. T.; Domínguez E. Palladium NCN and CNC pincer complexes as exceptionally active catalysts for aerobic oxidation in sustainable media. Green Chem. 2011, 13, 2161–2166. 10.1039/c1gc15390k. [DOI] [Google Scholar]
- Urgoitia G.; Maiztegi A.; Martin R. S.; Herrero M. T.; Domínguez E. Aerobic oxidation at benzylic positions catalyzed by a simple Pd(OAc)2/bis-triazole system. RSC Adv. 2015, 5, 103210–103217. 10.1039/C5RA22251F. [DOI] [Google Scholar]
- Chen S.; Liu Z.; Shi E.; Chen L.; Wei W.; Li H.; Cheng Y.; Wan X. Ruthenium-catalyzed oxidation of alkenes at room temperature: a practical and concise approach to α-diketones. Org. Lett. 2011, 13, 2274–2277. 10.1021/ol200716d. [DOI] [PubMed] [Google Scholar]
- Schmidt B.; Krehl S.; Hauke S. Assisted tandem catalytic cross metathesis-oxidation: in one flask from styrenes to 1,2-diketones and further to quinoxalines. J. Org. Chem. 2013, 78, 5427–5435. 10.1021/jo4005684. [DOI] [PubMed] [Google Scholar]
- Su Y.; Sun X.; Wu G.; Jiao N. Catalyst-controlled highly selective coupling and oxygenation of olefins: a direct approach to alcohols, ketones, and diketones. Angew. Chem. Int. Ed. 2013, 52, 9808–9812. 10.1002/anie.201303917. [DOI] [PubMed] [Google Scholar]
- Song T.; Ma Z.; Ren P.; Yuan Y.; Xiao J.; Yang Y. A Bifunctional iron nanocomposite catalyst for efficient oxidation of alkenes to ketones and 1,2-diketones. ACS Catal. 2020, 10, 4617–4629. 10.1021/acscatal.9b05197. [DOI] [Google Scholar]
- Saberi D.; Hashemi H.; Niknam K. One-pot solvent-free synthesis of diaryl 1,2-diketones by the sequential heck oxidation reaction of aryl halides with styrenes. Asian J. Org. Chem. 2017, 6, 169–173. 10.1002/ajoc.201600529. [DOI] [Google Scholar]
- Jung M. E.; Deng G. Synthesis of α-diketones from alkylaryl- and diarylalkynes using mercuric salts. Org. Lett. 2014, 16, 2142–2145. 10.1021/ol500592m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu X.; Li P.; Shi Q.; Wang L. Thiyl radical catalyzed oxidation of diarylalkynes to α-diketones by molecular oxygen under visible-light irradiation. Green Chem. 2016, 18, 6373–6379. 10.1039/C6GC01487A. [DOI] [Google Scholar]
- Zhou J.; Tao X. Z.; Dai J. J.; Li C. G.; Xu J.; Xu H. M.; Xu H. J. Electrochemical synthesis of 1,2-diketones from alkynes under transition-metal-catalyst-free conditions. Chem. Commun. 2019, 55, 9208–9211. 10.1039/C9CC03996A. [DOI] [PubMed] [Google Scholar]
- Charpe V. P.; Sagadevan A.; Hwang K. C. Visible light-induced aerobic oxidation of diarylalkynes to α-diketones catalyzed by copper-superoxo at room temperature. Green Chem. 2020, 22, 4426–4432. 10.1039/D0GC00975J. [DOI] [Google Scholar]
- Dubovtsev A. Y.; Shcherbakov N. V.; Dar’in D. V.; Kukushkin V. Y. Nature of the nucleophilic oxygenation reagent is key to acid-free gold-catalyzed conversion of terminal and internal alkynes to 1,2-dicarbonyls. J. Org. Chem. 2020, 85, 745–757. 10.1021/acs.joc.9b02785. [DOI] [PubMed] [Google Scholar]
- Shen D.; Wang H.; Zheng Y.; Zhu X.; Gong P.; Wang B.; You J.; Zhao Y.; Chao M. Catalyst-free and transition-metal-free approach to 1,2-diketones via aerobic alkyne oxidation. J. Org. Chem. 2021, 86, 5354–5361. 10.1021/acs.joc.0c03010. [DOI] [PubMed] [Google Scholar]
- Tada N.; Shomura M.; Nakayama H.; Miura T.; Itoh A. Direct synthesis of 1,2-diketones by catalytic aerobic oxidative decarboxylation of 1,3-diketones with iodine and base under irradiation of fluorescent light. Synlett 2010, 2010, 1979–1983. 10.1055/s-0030-1258134. [DOI] [Google Scholar]
- Huang L.; Cheng K.; Yao B.; Xie Y.; Zhang Y. Iron-promoted C-C bond cleavage of 1,3-diketones: a route to 1,2-diketones under mild reaction conditions. J. Org. Chem. 2011, 76, 5732–5737. 10.1021/jo200840y. [DOI] [PubMed] [Google Scholar]
- Yuan Y.; Zhu H. Iodine-catalyzed synthesis of 1,2-diaryldiketones by oxidative cleavage of 1,3-diaryldiketones with DMSO. Eur. J. Org. Chem. 2012, 2012, 329–333. 10.1002/ejoc.201101028. [DOI] [Google Scholar]
- Zhou P. J.; Li C. K.; Zhou S. F.; Shoberu A.; Zou J. P. Copper-catalyzed TEMPO oxidative cleavage of 1,3-diketones and β-keto esters for the synthesis of 1,2-diketones and α-keto esters. Org. Biomol. Chem. 2017, 15, 2629–2637. 10.1039/C7OB00241F. [DOI] [PubMed] [Google Scholar]
- Chen L.-S.; Zhang L.-B.; Tian Y.; Li J.-H.; Liu Y.-Q. Copper/Iodine-cocatalyzed C-C cleavage of 1,3-dicarbonyl compounds toward 1,2-dicarbonyl compounds. Eur. J. Org. Chem. 2020, 2020, 5523–5526. 10.1002/ejoc.202000795. [DOI] [Google Scholar]
- Lee J. C.; Park H.-J.; Park J. Y. Rapid microwave-promoted solvent-free oxidation of α-methylene ketones to α-diketones. Tetrahedron Lett. 2002, 43, 5661–5663. 10.1016/S0040-4039(02)01130-9. [DOI] [Google Scholar]
- Ghazanfari D.; Najafizadeh F.; Khosravi F. Selective oxidation of aryl ketones to α-diketones with 4-aminoperoxybenzoic acid supported on silica gel in presence of air. Monatsh. Chem. 2004, 135, 1409–1413. 10.1007/s00706-004-0199-1. [DOI] [Google Scholar]
- Park B.-S.; Lee H.-M.; Cho S.-S. Facile α-ketonization of carbonyl compounds utilizing CuBr2 on alumina. Bull. Korean Chem. Soc. 2007, 28, 871–872. 10.5012/bkcs.2007.28.5.871. [DOI] [Google Scholar]
- Qi C.; Jiang H.; Huang L.; Chen Z.; Chen H. DABCO-catalyzed oxidation of deoxybenzoins to benzils with air and one-pot synthesis of quinoxalines. Synthesis 2010, 2011, 387–396. 10.1055/s-0030-1258375. [DOI] [Google Scholar]
- Jayram J.; Jeena V. An iodine/DMSO-catalyzed sequential one-pot approach to 2,4,5-trisubstituted-1H-imidazoles from α-methylene ketones. RSC Adv. 2018, 8, 37557–37563. 10.1039/C8RA07238H. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jayram J.; Xulu B. A.; Jeena V. Iodine/DMSO promoted oxidation of benzylic Csp3–H bonds to diketones-A mechanistic investigation. Tetrahedron 2019, 75, 130617 10.1016/j.tet.2019.130617. [DOI] [Google Scholar]
- Min H.; Palani T.; Park K.; Hwang J.; Lee S. Copper-catalyzed direct synthesis of diaryl 1,2-diketones from aryl iodides and propiolic acids. J. Org. Chem. 2014, 79, 6279–6285. 10.1021/jo501089k. [DOI] [PubMed] [Google Scholar]
- Lv W.-X.; Zeng Y.-F.; Zhang S.-S.; Li Q.; Wang H. Mild Mn(OAc)3-mediated aerobic oxidative decarboxylative coupling of arylboronic acids and arylpropiolic acids: direct access to diaryl 1,2-diketones. Org. Lett. 2015, 17, 2972–2975. 10.1021/acs.orglett.5b01265. [DOI] [PubMed] [Google Scholar]
- Chand S.; Pandey A. K.; Singh R.; Singh K. N. Visible-light-induced photocatalytic oxidative decarboxylation of cinnamic acids to 1,2-diketones. J. Org. Chem. 2021, 86, 6486–6493. 10.1021/acs.joc.1c00322. [DOI] [PubMed] [Google Scholar]
- Bansode A. H.; Suryavanshi G. Iodine-mediated oxidative rearrangement of α,β-unsaturated diaryl ketones: a facile access to 1,2-diaryl diketones. ACS Omega 2019, 4, 9636–9644. 10.1021/acsomega.9b00833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sarrafi Y.; Tajbakhsh M.; Hosseinzadeh R.; Sadatshahabi M.; Alimohammadi K. 2,6-Dicarboxypyridinium fluorochromate–promoted oxidation of alkyl-arenes into carbonyl compounds under nonaqueous and aprotic conditions. Synth. Commun. 2012, 42, 678–685. 10.1080/00397911.2010.529228. [DOI] [Google Scholar]
- Cacchi S.; Fabrizi G.; Goggiamani A.; Iazzetti A.; Verdiglione R. Copper-catalyzed oxidation of deoxybenzoins to benzils under aerobic conditions. Synthesis 2013, 45, 1701–1707. 10.1055/s-0033-1338451. [DOI] [Google Scholar]
- Yu J.-W.; Mao S.; Wang Y.-Q. Copper-catalyzed base-accelerated direct oxidation of C–H bond to synthesize benzils, isatins, and quinoxalines with molecular oxygen as terminal oxidant. Tetrahedron Lett. 2015, 56, 1575–1580. 10.1016/j.tetlet.2015.02.019. [DOI] [Google Scholar]
- Chebolu R.; Bahuguna A.; Sharma R.; Mishra V. K.; Ravikumar P. C. An unusual chemoselective oxidation strategy by an unprecedented exploration of an electrophilic center of DMSO: a new facet to classical DMSO oxidation. Chem. Commun. 2015, 51, 15438–15441. 10.1039/C5CC05713B. [DOI] [PubMed] [Google Scholar]
- Guo Y.; Meng J.-R.; Liu J.-Z.; Xu T.; Zheng Z.-Y.; Jiang Z.-H.; Bai L.-P. Synthesis and biological evaluation of honokiol derivatives bearing 3-((5-phenyl-1,3,4-oxadiazol-2-yl)methyl)oxazol-2(3H)-ones as potential viral entry inhibitors against SARS-CoV-2. Pharmaceuticals 2021, 14, 885 10.3390/ph14090885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang N.; Han S.; Liu R.; Meng L.; He H.; Zhang Y.; Wang C.; Lv Y.; Wang J.; Li X.; Ding Y.; Fu J.; Hou Y.; Lu W.; Ma W.; Zhan Y.; Dai B.; Zhang J.; Pan X.; Hu S.; Gao J.; Jia Q.; Zhang L.; Ge S.; Wang S.; Liang P.; Hu T.; Lu J.; Wang X.; Zhou H.; Ta W.; Wang Y.; Lu S.; He L. Chloroquine and hydroxychloroquine as ACE2 blockers to inhibit viropexis of 2019-nCoV Spike pseudotyped virus. Phytomedicine 2020, 79, 153333 10.1016/j.phymed.2020.153333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen R. H.; Yang L. J.; Hamdoun S.; Chung S. K.; Lam C. W.; Zhang K. X.; Guo X.; Xia C.; Law B. Y. K.; Wong V. K. W. 1,2,3,4,6-Pentagalloyl glucose, a RBD-ACE2 binding inhibitor to prevent SARS-CoV-2 infection. Front. Pharmacol. 2021, 12, 634176 10.3389/fphar.2021.634176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang G.; Pomplun S.; Loftis A. R.; Tan X.; Loas A.; Pentelute B. L., Investigation of ACE2 N-terminal fragments binding to SARS-CoV-2 Spike RBD. BioRxiv 2020. [Google Scholar]
- Lan J.; Ge J.; Yu J.; Shan S.; Zhou H.; Fan S.; Zhang Q.; Shi X.; Wang Q.; Zhang L.; Wang X. Structure of the SARS-CoV-2 spike receptor-binding domain bound to the ACE2 receptor. Nature 2020, 581, 215–220. 10.1038/s41586-020-2180-5. [DOI] [PubMed] [Google Scholar]
- Ding X.; Wu Y.; Wang Y.; Vilseck J. Z.; Brooks C. L. III, Accelerated CDOCKER with GPUs, parallel simulated annealing, and fast fourier transforms. J. Chem. Theory Comput. 2020, 16, 3910–3919. 10.1021/acs.jctc.0c00145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Towler P.; Staker B.; Prasad S. G.; Menon S.; Tang J.; Parsons T.; Ryan D.; Fisher M.; Williams D.; Dales N. A.; Patane M. A.; Pantoliano M. W. ACE2 X-ray structures reveal a large hinge-bending motion important for inhibitor binding and catalysis. J. Biol. Chem. 2004, 279, 17996–18007. 10.1074/jbc.M311191200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rao S. N.; Reddy N. N. K.; Samanta S.; Adimurthy S. I2-Catalyzed oxidative amidation of benzylamines and benzyl cyanides under mild conditions. J. Org. Chem. 2017, 82, 13632–13642. 10.1021/acs.joc.7b02211. [DOI] [PubMed] [Google Scholar]
- Aruri H.; Singh U.; Kumar S.; Kushwaha M.; Gupta A. P.; Vishwakarma R. A.; Singh P. P. I2/Aqueous TBHP-catalyzed coupling of amides with methylarenes/aldehydes/alcohols: metal-free synthesis of imides. Org. Lett. 2016, 18, 3638–3641. 10.1021/acs.orglett.6b01684. [DOI] [PubMed] [Google Scholar]
- Meesin J.; Pohmakotr M.; Reutrakul V.; Soorukram D.; Leowanawat P.; Saithong S.; Kuhakarn C. TBAI/TBHP-mediated cascade cyclization toward sulfonylated indeno[1,2-c]quinolines. Org. Lett. 2017, 19, 6546–6549. 10.1021/acs.orglett.7b03246. [DOI] [PubMed] [Google Scholar]
- Yan Y.; Zhang Y.; Zha Z.; Wang Z. Mild metal-free sequential dual oxidative amination of C (sp3)-H bonds: efficient synthesis of imidazo [1,5-a] pyridines. Org. Lett. 2013, 15, 2274–2277. 10.1021/ol4008487. [DOI] [PubMed] [Google Scholar]
- Lai J.; Chang L.; Yuan G. I2/TBHP mediated C-N and C-H bond cleavage of tertiary amines toward selective synthesis of sulfonamides and β-arylsulfonyl enamines: the solvent effect on reaction. Org. Lett. 2016, 18, 3194–3197. 10.1021/acs.orglett.6b01412. [DOI] [PubMed] [Google Scholar]
- Wei W.-T.; Zhu W.-M.; Bao W.-H.; Chen W.-T.; Huang Y.-L.; Gao L.-H.; Xu X.-D.; Wang Y.-N.; Chen G.-P. Metal-free C(sp3)-H amination of 2-oxindoles in water: facile synthesis of 3-substituted 3-aminooxindoles. ACS Sustainable Chem. Eng. 2018, 6, 5615–5619. 10.1021/acssuschemeng.8b00641. [DOI] [Google Scholar]
- He J.; Dong J.; Su L.; Wu S.; Liu L.; Yin S. F.; Zhou Y. Selective oxidative cleavage of 3-methylindoles with primary amines affording quinazolinones. Org. Lett. 2020, 22, 2522–2526. 10.1021/acs.orglett.0c00271. [DOI] [PubMed] [Google Scholar]
- Luo W.-K.; Shi X.; Zhou W.; Yang L. Iodine-catalyzed oxidative functionalization of azaarenes with benzylic C(sp(3))-H bonds via N-alkylation/amidation cascade: two-step synthesis of isoindolo[2,1-b]isoquinolin-7(5H)-one. Org. Lett. 2016, 18, 2036–2039. 10.1021/acs.orglett.6b00646. [DOI] [PubMed] [Google Scholar]
- Dhineshkumar J.; Gadde K.; Prabhu K. R. Substituent-directed regioselective azidation: copper-catalyzed C-H azidation and iodine-catalyzed dearomatizative azidation of indole. J. Org. Chem. 2018, 83, 228–235. 10.1021/acs.joc.7b02591. [DOI] [PubMed] [Google Scholar]
- Zhang J.; Xiao D.; Tan H.; Liu W. Highly selective synthesis of 2-tert-butoxy-1-arylethanones via copper(I)-catalyzed oxidation/tert-butoxylation of aryl olefins with TBHP. J. Org. Chem. 2020, 85, 3929–3935. 10.1021/acs.joc.9b03156. [DOI] [PubMed] [Google Scholar]
- Montana M.; Montero V.; Khoumeri O.; Vanelle P. Quinoxaline derivatives as antiviral agents: a systematic review. Molecules 2020, 25, 2784 10.3390/molecules25122784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khatoon H.; Abdulmalek E. Novel synthetic routes to prepare biologically active quinoxalines and their derivatives: a synthetic review for the last two decades. Molecules 2021, 26, 1055 10.3390/molecules26041055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sarges R.; Howard H. R.; Browne R. G.; Lebel L. A.; Seymour P. A.; Koe B. K. 4-Amino[1,2,4]triazolo[4,3-a]quinoxalines. A novel class of potent adenosine receptor antagonists and potential rapid-onset antidepressants. J. Med. Chem. 1990, 33, 2240–2254. 10.1021/jm00170a031. [DOI] [PubMed] [Google Scholar]
- Hazeldine S. T.; Polin L.; Kushner J.; White K.; Bouregeois N. M.; Crantz B.; Palomino E.; Corbett T. H.; Horwitz J. P. II. Synthesis and biological evaluation of some bioisosteres and congeners of the antitumor agent, 2-{4-[(7-chloro-2-quinoxalinyl) oxy] phenoxy} propionic acid (XK469). J. Med. Chem. 2002, 45, 3130–3137. 10.1021/jm0200097. [DOI] [PubMed] [Google Scholar]
- Kim Y. B.; Kim Y. H.; Park J. Y.; Kim S. K. Synthesis and biological activity of new quinoxaline antibiotics of echinomycin analogues. Bioorg. Med. Chem. Lett. 2004, 14, 541–544. 10.1016/j.bmcl.2003.09.086. [DOI] [PubMed] [Google Scholar]
- Undevia S. D.; Innocenti F.; Ramirez J.; House L.; Desai A. A.; Skoog L. A.; Singh D. A.; Karrison T.; Kindler H. L.; Ratain M. J. A phase I and pharmacokinetic study of the quinoxaline antitumour Agent R(+)XK469 in patients with advanced solid tumours. Eur. J. Cancer. 2008, 44, 1684–1692. 10.1016/j.ejca.2008.05.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Unzue A.; Dong J.; Lafleur K.; Zhao H.; Frugier E.; Caflisch A.; Nevado C. Pyrrolo[3,2-b]quinoxaline derivatives as types I1/2 and II Eph tyrosine kinase inhibitors: structure-based design, synthesis, and in vivo validation. J. Med. Chem. 2014, 57, 6834–6844. 10.1021/jm5009242. [DOI] [PubMed] [Google Scholar]
- Smits R. A.; Lim H. D.; Hanzer A.; Zuiderveld O. P.; Guaita E.; Adami M.; Coruzzi G.; Leurs R.; de Esch I. J. Fragment based design of new H4 receptor-ligands with anti-inflammatory properties in vivo. J. Med. Chem. 2008, 51, 2457–2467. 10.1021/jm7014217. [DOI] [PubMed] [Google Scholar]
- Abu-Hashem A. A.; Gouda M. A.; Badria F. A. Synthesis of some new pyrimido[2′,1′:2,3]thiazolo[4,5-b]quinoxaline derivatives as anti-inflammatory and analgesic agents. Eur. J. Med. Chem. 2010, 45, 1976–1981. 10.1016/j.ejmech.2010.01.042. [DOI] [PubMed] [Google Scholar]
- Moreno E.; Ancizu S.; Pérez-Silanes S.; Torres E.; Aldana I.; Monge A. Synthesis and antimycobacterial activity of new quinoxaline-2-carboxamide 1,4-di-N-oxide derivatives. Eur. J. Med. Chem. 2010, 45, 4418–4426. 10.1016/j.ejmech.2010.06.036. [DOI] [PubMed] [Google Scholar]
- Hu Y.; Wang K.; MacMillan J. B. Hunanamycin A, an antibiotic from a marine-derived Bacillus hunanensis. Org. Lett. 2013, 15, 390–393. 10.1021/ol303376c. [DOI] [PubMed] [Google Scholar]
- Guillon J.; Mouray E.; Moreau S.; Mullié C.; Forfar I.; Desplat V.; Belisle-Fabre S.; Pinaud N.; Ravanello F.; Le-Naour A.; Leger J. M.; Gosmann G.; Jarry C.; Deleris G.; Sonnet P.; Grellier P. New ferrocenic pyrrolo[1,2-a]quinoxaline derivatives: synthesis, and in vitro antimalarial activity-Part II. Eur. J. Med. Chem. 2011, 46, 2310–2326. 10.1016/j.ejmech.2011.03.014. [DOI] [PubMed] [Google Scholar]
- Niknam K.; Saberi D.; Mohagheghnejad M. Silica bonded S-sulfonic acid: a recyclable catalyst for the synthesis of quinoxalines at room temperature. Molecules 2009, 14, 1915–1926. 10.3390/molecules14051915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu T.; Zheng Z.; Guo Y.; Bai L. P. Semisynthesis of novel magnolol-based Mannich base derivatives that suppress cancer cells via inducing autophagy. Eur. J. Med. Chem. 2020, 205, 112663 10.1016/j.ejmech.2020.112663. [DOI] [PubMed] [Google Scholar]
- Zhou X.; Chen M.; Zheng Z.; Zhu G.-Y.; Jiang Z.-H.; Bai L.-P. Synthesis and evaluation of novel 12-aryl berberine analogues with hypoxia-inducible factor-1 inhibitory activity. RSC Adv. 2017, 7, 26921–26929. 10.1039/C7RA02238G. [DOI] [Google Scholar]
- Dubovtsev A. Y.; Dar’in D. V.; Krasavin M.; Kukushkin V. Y. Gold-catalyzed oxidation of internal alkynes into benzils and its application for one-pot synthesis of five-, six-, and seven-membered azaheterocycles. Eur. J. Org. Chem. 2019, 2019, 1856–1864. 10.1002/ejoc.201900108. [DOI] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.