

Abstract
Untargeted metabolomics of disease-associated intestinal microbiota can detect quantitative changes in metabolite profiles and complement other methodologies to reveal the full effect of intestinal dysbiosis. Here, we used the T cell transfer mouse model of colitis to identify small-molecule metabolites with altered abundance due to intestinal inflammation. We applied untargeted metabolomics to detect metabolite signatures in cecal, colonic, and fecal samples from healthy and colitic mice and to uncover differences that would aid in the identification of colitis-associated metabolic processes. We provided an unbiased spatial survey of the GI tract for small molecules, and we identified the likely source of metabolites and biotransformations. Several prioritized metabolites that we detected as being altered in colitis were evaluated for their ability to induce inflammatory signaling in cultured macrophages, such as NF-κB signaling and the expression of cytokines and chemokines upon LPS stimulation. Multiple previously uncharacterized anti-inflammatory and inflammation-augmenting metabolites were thus identified, with phytosphingosine showing the most effective anti-inflammatory activity in vitro. We further demonstrated that oral administration of phytosphingosine decreased inflammation in a mouse model of colitis induced by the compound TNBS. The collection of distinct metabolites we identified and characterized, many of which have not been previously associated with colitis, may offer new biological insight into IBD-associated inflammation and disease pathogenesis.
Crohn’s disease (CD) and ulcerative colitis (UC) are two primary forms of inflammatory bowel disease (IBD) that have steadily risen in the Western population over the last several decades (1, 2). Both of these forms of IBD are of considerable biomedical importance because they can result in malnutrition due to inefficient absorption of nutrients (3, 4), increased susceptibility to colorectal cancer (CRC) (5), higher rates of morbidity (6), and substantial decreases in quality of life (7). Many factors govern susceptibility to IBD, and a complex interplay between environmental factors (such as diet and antibiotics), host genetics and immunity, and the commensal gut microbiota is required to maintain proper intestinal homeostasis (3, 8). Changes in these interacting factors can lead to chronic epithelial and mucosal inflammation characteristic of IBD. IBD is typically treated with drugs developed for other diseases, notably antibiotics, anti-inflammatory agents, corticosteroids, and immunosuppressants (9–11). However, the systemic side effects of these agents highlight the need to develop IBD-specific treatments, a task that requires the identification of new therapeutic strategies to target IBD.
Introduction of elemental diets can also ameliorate features of IBD-associated inflammation. In this context, suppression of IBD symptoms is linked to a general reduction in bacterial metabolite production and a surface coating of bacteria with immunoglobulin observable within two weeks of diet initiation (12). These findings suggest that small-molecule food components, combined with the metabolic machinery of the host and the intestinal microbiome, can actively modulate IBD phenotypes. The specific constituents or nutrients in food that are directly or indirectly responsible for modulating intestinal inflammation are largely unknown. It also remains to be elucidated if these diet-derived small molecules are active in their native state or require metabolic modification by the host and/or microbiome, akin to prodrugs, to exert immunoregulatory functions.
To identify IBD-associated dysregulated metabolites and understand their functional relevance for inflammation-related dysbiosis, we performed mass spectrometry-based metabolomic analysis on microbiome samples collected from a murine T cell transfer model of colitis. Using our untargeted metabolomics platform, we quantified and compared metabolite levels extracted from cecal, colonic, and fecal samples collected from wild-type (WT) and isogenic control mice fed identical diets. Selected metabolites whose levels differed substantially between control and IBD-associated samples were further characterized in cell-based functional assays and in an in vivo model to assess their ability to modulate inflammation. Our findings offer both spatial resolution and functional information at a molecular level across the gastrointestinal (GI) tract during colitis. The diversity in the list of metabolites we identified as being altered in IBD demonstrates the range of chemical reactions and biological processes along the GI tract in health and colitis-associated inflammation.
RESULTS
Individual Regions of the GI Tract Have Distinct Compositions of Metabolites
Our metabolomic analyses were focused on the identification of differences in molecular signatures between two control groups, C57BL/6J WT and B6.129S7-Rag1tm1mom/J (Rag1−/−) mice (Rag), and a mouse model of colitis in which naïve T cells introduced retro-orbitally into Rag1−/− mice (RagT) drive intestinal inflammation over the course of 8 weeks. Because chemical transformations catalyzed by the host and/or microbiome are heavily influenced by the environment (such as by stomach pH, anaerobic atmosphere, and others), and given the variability in microbiome composition along the GI tract (13, 14), we analyzed individual samples isolated from the cecum, colon, and feces on day 56 post-T cell transfer, when inflammation is most severe (Fig. 1 and fig. S1). We also profiled the chow provided ad libitum to all mouse cohorts to establish baseline small-molecule levels, and to enable source tracing of potentially important unique and/or shared active metabolites to the cecal, colonic, or fecal samples.
Fig. 1. Experimental design.
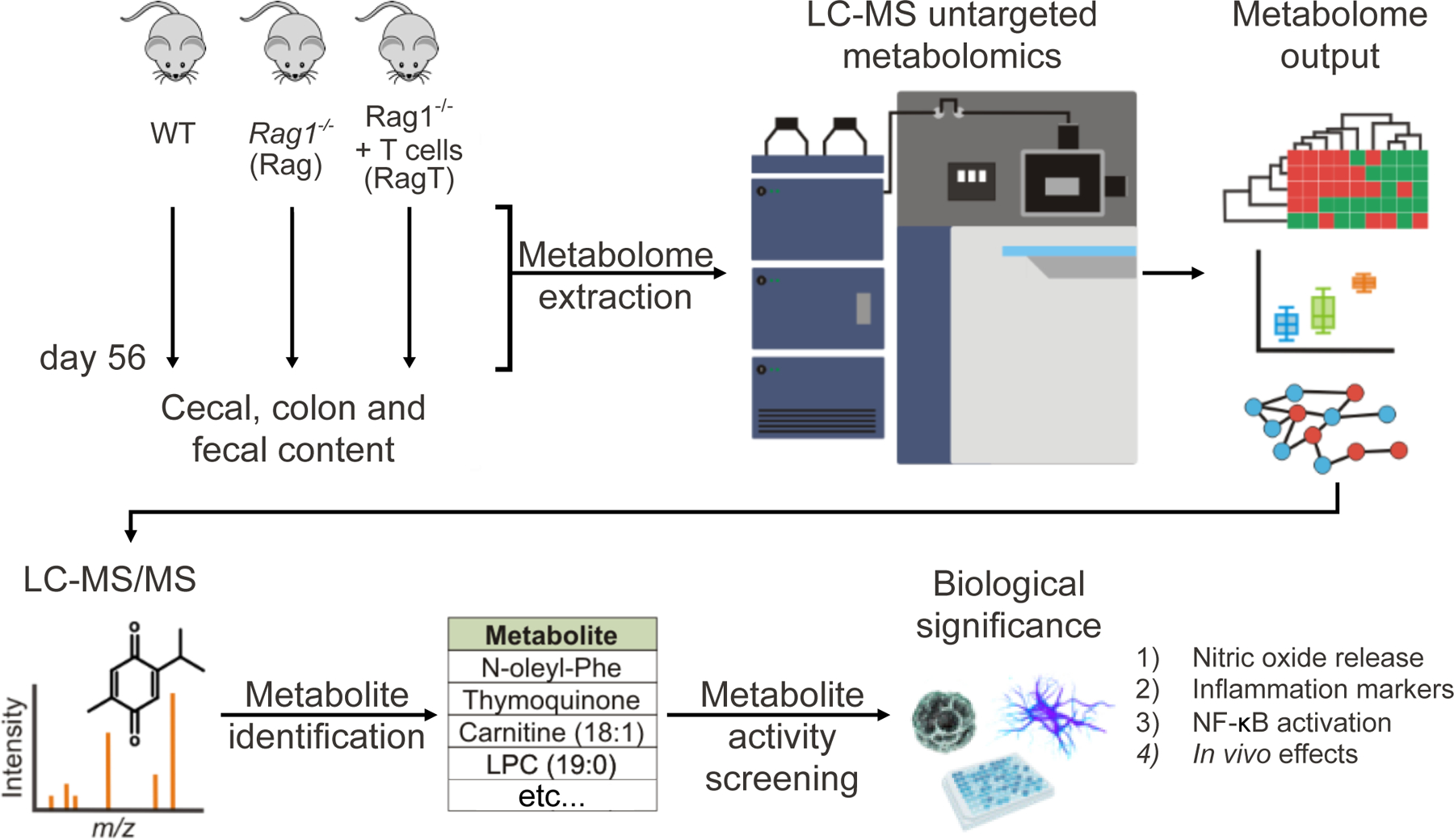
Cecal, colonic, and fecal samples from isogenic controls WT and Rag mice, as well as the colitic RagT group (6 mice per group, 18 total) were collected on day 56 for metabolome extraction and liquid chromatography-mass spectrometry untargeted metabolomics analysis. Following data analysis, features were prioritized for MS/MS experiments, metabolite identification, and differential analysis to find altered levels of molecules in RagT colitis samples. A panel of metabolites with significantly altered levels in IBD, and no prior known role in this disease, were profiled in cell-based assays that can reveal pro-inflammatory and anti-inflammatory properties.
We detected a total of 51,886 distinct metabolic features in samples of chow, control cohorts, and the colitic group. A general trend observed in all three murine cohorts was a substantial increase in metabolic composition along the GI tract, as evidenced by the number of metabolic features detected (specific m/z value with a specific retention time) in chow (~22,000 features), cecum (~25,000 features), and colon (~29,000 features) (Fig. 2A). The originating source (which refers to the first instance of detection) for more than 40% of the total metabolic features was the chow (21,730 features) (Fig. 2B). Furthermore, approximately 28% of all measured metabolic features were only detected in the chow, suggesting that these molecules were likely metabolized or absorbed prior to reaching the cecum (Fig. 2C). Approximately 39% of the total metabolic features were first detected in the cecum, supporting the observations of extensive metabolic transformations in the upper GI tract. On the other hand, in the lower GI tract samples, ~50% of the metabolic features were shared with chow, cecum, colon, and/or feces samples (meaning that metabolic features were detected in 2 or more of the 4 sample types), suggesting that half of the detected small molecules remained largely unmodified throughout passage in the GI tract. Approximately 9,498 metabolic features (18.3% of all metabolic features) were unique to the cecum and colon and undetectable in the chow or fecal samples. These findings highlight the potential limitations of relying on feces-only metabolomic analyses to gain biological insights, for we observed a substantial reduction in the diversity and number of small molecules in the fecal content relative to all other biological samples (Figs. 2A, B, C).
Fig. 2. Distribution of metabolic features across the GI tract.
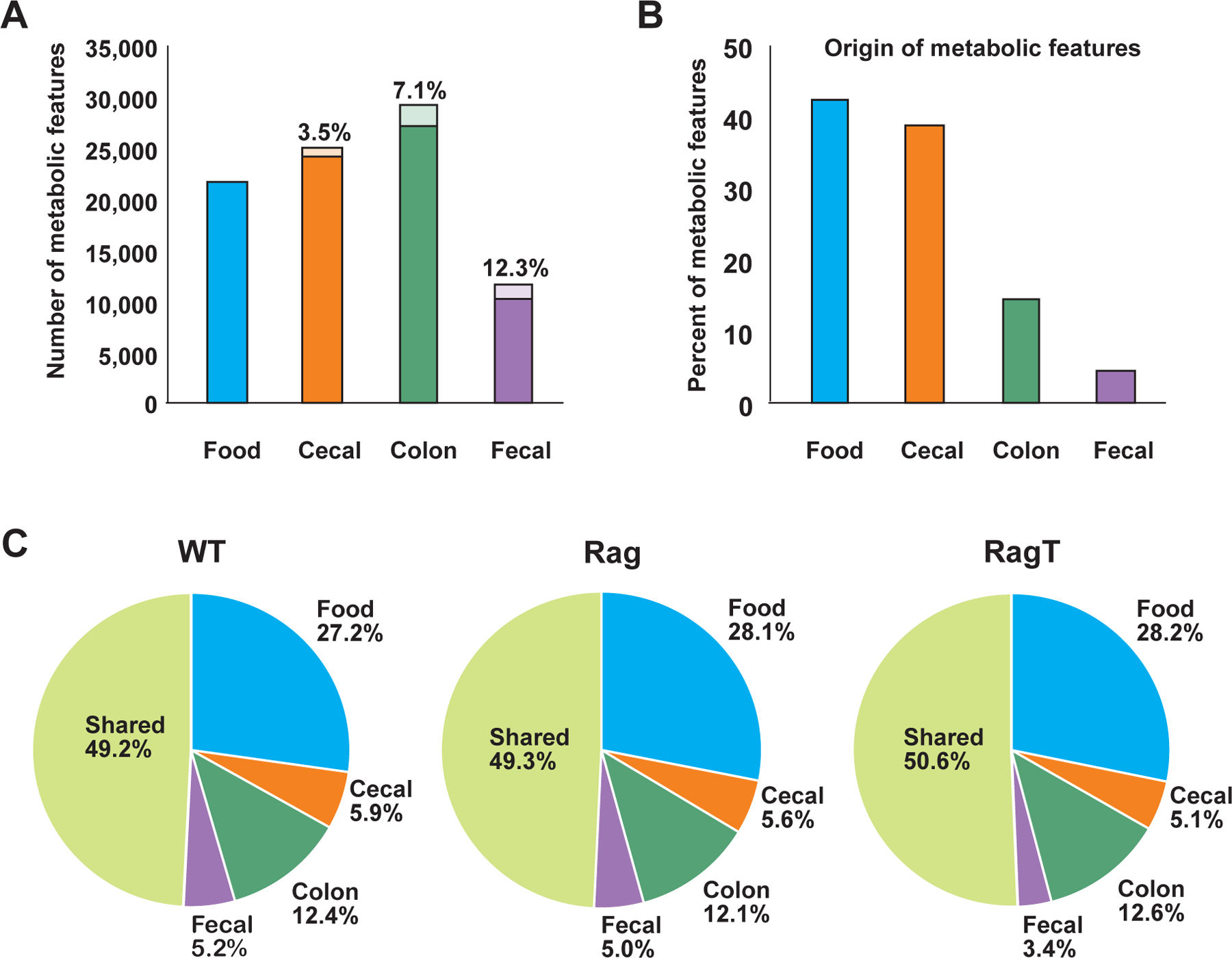
(A) Total number of metabolic features detected in chow, cecal, colonic, and fecal samples. The percentage of dysregulated features (p-value <0.05) in each sample type between the collective controls (WT and Rag) and RagT mice are represented by the lightly colored portion of the bar graph. (B) Source of metabolic features (sample type in which the metabolic features are first detected) across the GI tract is represented as a percentage of the total number of metabolic features (51,886). (C) Overlap of shared and unique metabolic features across the GI tract.
The Polarity of Metabolites Decreases Along the GI Tract
In addition to the distinct distribution of small molecules, the polarity of metabolites detected decreased in the lower GI samples, with the highest number of polar features measured in chow itself. The majority of metabolites identified in chow were detected and quantitated using HILIC (68.3%, chromatography for polar metabolites), and the remaining features (31.7%), consisting primarily of non-polar and hydrophobic metabolites, were detected using reverse-phase (RP) chromatography. Analyses of the cecal and colonic metabolomes revealed an overall increase in non-polar small molecules relative to chow, as demonstrated by an equal ratio (~1:1) of the metabolic features. In feces, the majority of the metabolic features (71.8%) were identified using RP for non-polar metabolites and consisted primarily of lipids, with only 28.2% of metabolites detected using HILIC (Data File S1).
IBD-dysregulated Metabolites Increase Along the GI Tract
Of the 51,886 total measurable metabolic features, more than 4,000 (approximately 7%) differed in abundance between the WT and Rag control groups and the RagT colitic cohort. The number of differentially detected features between the collective controls (WT and Rag) and colitic mice was inversely correlated with metabolic composition: 3.5%, 7.1%, and 12.3% of the IBD-modulated features were dysregulated in the cecum, colon, and feces, respectively (Fig. 2A).
Quantitation and Validation of Previously Reported IBD-associated Metabolites
Several metabolites we identified as being dysregulated in our colitic group have previously been described in other IBD studies, offering confidence of the robustness of our untargeted metabolomics data collection and analyses. For instance, we found a 4.5-fold change (FC) increase in taurocholic acid in the colitic RagT mice, a finding consistent with prior reports of bile acids being dysregulated in IBD (Fig. 3 and Data File S2) (15–17). Linolenic acid has been reported to be an anti-inflammatory agent in the context of IBD (18), and we observed a 1.8 FC increase in linolenic acid in the feces of the colitis cohort relative to both control groups. Similarly, large differences in oleoylethanolamide (OEA) were detected, with a significant 2.4 FC increase in this lipid in RagT fecal samples (Fig. 3 and Data File S2), a finding that agrees with a study that detected increased OEA levels in IBD patient plasma (19). Studies have shown that administration of OEA has beneficial effects on intestinal microbiota composition and anti-inflammatory effects in DSS-induced colitis (20–22).
Fig. 3. IBD-altered metabolites in cecal, colonic, and fecal samples.
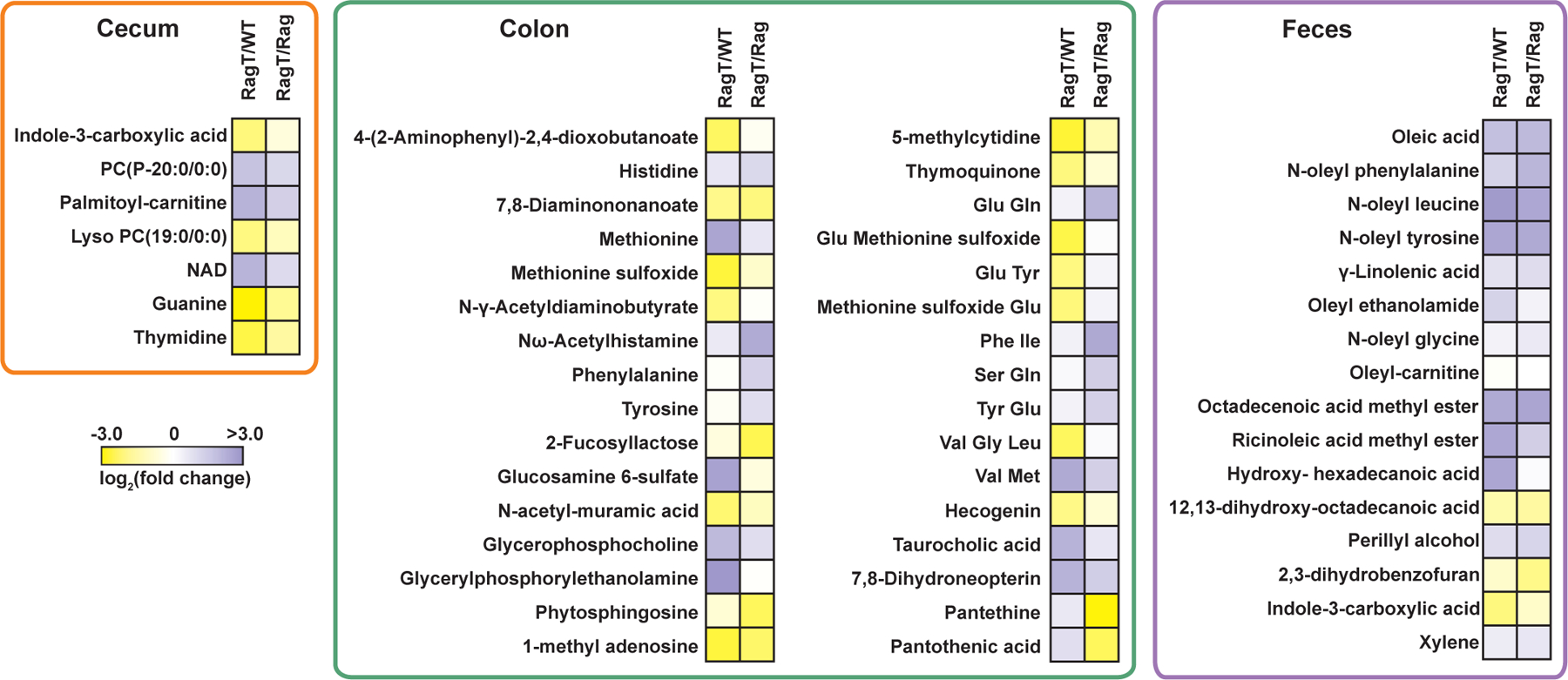
Heatmap of statistically significant (q-value <0.05, one-way ANOVA) metabolites represented as fold-change of RagT relative to WT and Rag using a logarithmic scale (log2). Yellow and purple shading indicate down- and up-regulation in the RagT cohort, respectively.
Several small-molecule metabolites whose levels are decreased or absent in IBD were also diminished in our colitic cohort. For example, methionine sulfoxide levels were decreased by a 4.8 FC in RagT mice relative to WT, consistent with prior studies that correlated aberrant methionine metabolism with IBD (Fig. 3 and Data File S2) (23–25). Conversely, levels of the reduced form (methionine) were significantly increased in colitic RagT mice by a 5.9 FC relative to WT. We also observed a smaller but statistically significant increase in reduced methionine in Rag colon samples relative to those of WT mice by a 1.6 FC, hinting that Rag mice may intrinsically have elevated levels of methionine (Fig. 3 and Data File S2). Our quantitative analyses also revealed a significant upregulation of the methylated nucleosides 1-methyladenosine and 5-methylcytidine by 2.8 and 5.2 FC, respectively, in WT colonic samples over the RagT group (Fig. 3 and Data File S2). Methylated nucleosides likely protect the mucosal epithelia, because inhibition of cellular methylation in a murine model of colitis exacerbates disease (26). Finally, amino acids (including histidine, tyrosine, and phenylalanine) were dysregulated in RagT mice and several of these metabolites are associated with IBD (27).
Functional Characterization of New IBD-associated Metabolites
We set out to evaluate the immunomodulatory properties of differentially regulated metabolites to identify compounds that could potentially play a role in modifying IBD progression. We first profiled thirty small molecules (Fig. 3) in a cell viability screen to exclude toxic metabolites prior to analysis in functional assays in primary macrophages to uncover metabolite-dependent innate inflammatory responses. These thirty significantly altered molecules and four “control” metabolites (which were molecules of similar classes found in comparable levels across all cohorts) were selected based on availability, ease of treatment, and relative abundance. Treatment-challenging metabolites (dipeptides) and highly abundant metabolites in both control and colitis cohorts (amino acids) were excluded (Data File S2). To evaluate cytotoxicity and define working concentrations for subsequent assays, primary mouse peritoneal macrophages were treated with metabolites and cell viability determined (fig. S2). Next, we tested the ability of metabolites to modulate macrophage nitric oxide (NO) production, measured as the breakdown product nitrite, upon stimulation with E. coli lipopolysaccharide (LPS) (28). Primary macrophages were preincubated with the maximum tolerated concentration (that did not alter cell viability) of individual metabolites prior to stimulation with 20 ng/ml LPS. Nitrite levels in media were measured 36 h later to identify inflammation-augmenting or anti-inflammatory agents (compared to LPS-induced inflammation). The synthetic glucocorticoid dexamethasone and 100 ng/ml LPS were used as controls for anti-inflammatory and increased inflammatory activity, respectively. We identified 14 metabolites and 4 of the non-differentiated “control” metabolites that significantly increased or decreased NO levels relative to 20 ng/ml LPS (fig. S3). Due to their ability to modulate inflammation, we focused on these 18 compounds in subsequent inflammation regulation assays.
To examine the potential immunomodulatory properties of IBD-regulated metabolites, we tested their ability to alter expression of pro-inflammatory markers in primary mouse macrophages treated with LPS. Cells were preincubated with metabolites active in the NO release assay prior to stimulation with 20 ng/ml LPS and mRNA levels of canonical inflammatory markers were measured 3 or 36 h or both after addition of LPS. Genes examined included those encoding tumor necrosis factor alpha (TNFα), interleukin (IL)-1β, IL-6, inducible nitric oxide synthase (NOS2), and the monocyte chemoattractant protein-1 (MCP-1; also known as CCL2). Gene expression was calculated relative to that measured in cells treated only with 20 ng/ml LPS. Integration of gene expression profiles and levels of NO released allowed us to bin the metabolites into three separate classes: anti-inflammatory, inflammation-augmenting, and mixed response (Fig. 4A).
Fig. 4. Functional characterization of IBD-altered metabolites.
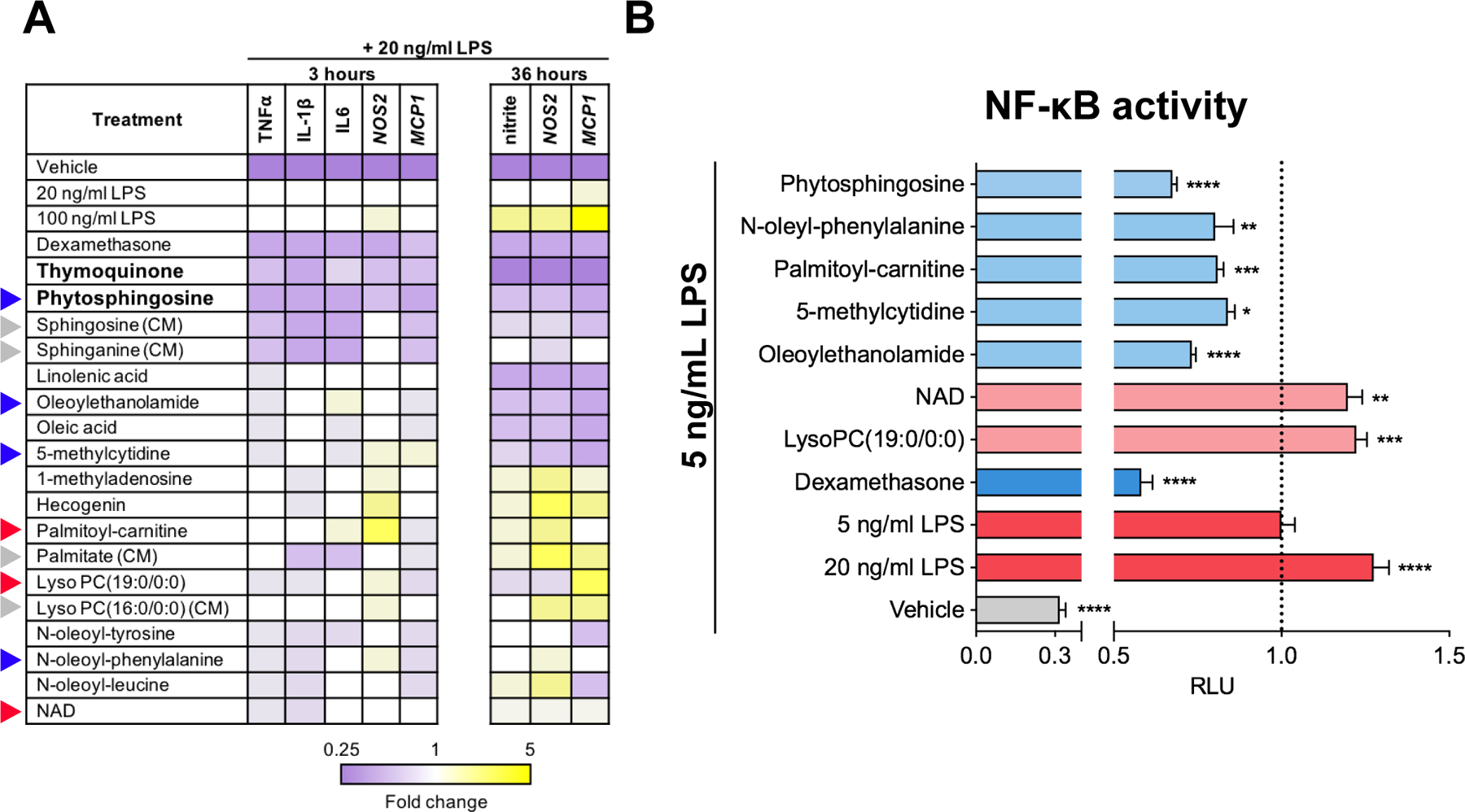
(A) Primary mouse macrophages were preincubated with DMSO control or metabolites prior to exposure to LPS or vehicle control (PBS), and gene expression quantitated by RT-qPCR at 3 and 36 hr after LPS stimulation. Relative RNA levels are shown. NO release was measured at 36 hr as the aqueous breakdown product nitrite. Blue, red, and grey triangles denote anti-inflammatory, pro-inflammatory, and control metabolites, respectively, that were profiled in (B). (B) THP-1 cells stably expressing an NF-κB luciferase reporter were pre-incubated with metabolites or DMSO control prior to stimulation with 5 ng/mL LPS or vehicle control (PBS). Effect on luciferase activity of anti-inflammatory (light blue) and pro-inflammatory (light red) metabolites expressed relative to that elicited by 5 ng/mL LPS. (CM) denotes control compound. *, **, *** and **** indicate p-value <0.05, 0.01. 0.001 and 0.0001, respectively.
The majority of metabolites showed mixed-responses, with variable effects on the selected inflammatory markers. For instance, the anti-inflammatory agent OEA and the pro-inflammatory agent LPC(19:0) showed mixed responses, suggesting that these molecules have a complex, perhaps multi-faceted mechanism of action. Pre-treatment of macrophages with OEA prior to LPS stimulation reduced MCP1 mRNA levels, as would be expected, but also increased expression of the mRNA encoding the pro-inflammatory cytokine IL-6. Similarly, the pro-inflammatory metabolite LPC(19:0) (29) generated a time-dependent shift in mRNA levels of MCP1, reducing its levels at 3 h, but increasing them at 36 h of treatment relative to the corresponding control. Nonetheless, two metabolites – thymoquinone and phytosphingosine – showed consistent, robust suppression of all inflammation markers tested (Fig. 4A).
We next tested the ability of four metabolites that showed broad suppression of pro-inflammatory gene expression (phytosphingosine, oleoylethanolamide, 5-methylcytidine, and N-oleyl-phenylalanine), and three others that increased pro-inflammatory gene expression (palmitoyl-carnitine, lyso-PC, and NAD), to modulate NF-κB signaling, a critical regulator of inflammation. THP-1 macrophages stably transfected with an NF-κB-driven luciferase reporter (30) were pre-incubated with individual metabolites at various concentrations (fig. S2) prior to LPS addition. Phytosphingosine, oleoylethanolamide, N-oleyl-phenylalanine, 5-methylcytidine, and palmitoyl-carnitine all significantly reduced LPS-induced NF-κB signaling, with phytosphingosine showing the greatest anti-inflammatory potential at the concentrations tested (Fig. 4B). Palmitoyl-carnitine was tested based on its ability to increase LPS-induced pro-inflammatory gene expression in mouse primary macrophages (Fig. 4A), but in human THP-1 cells, it dampened LPS-stimulated NF-κB signaling (Fig. 4B). Collectively, these functional assays not only confirmed the previously reported immunomodulatory properties of several metabolites we identified as being altered in IBD (thymoquinone, oleoylethanolamide, and linolenic acid), but they also revealed the anti-inflammatory potential of metabolites such as phytosphingosine, sphingosine, sphinganine, and 5-methylcytidine that have not been previously associated with inflammation.
Phytosphingosine Mitigates Inflammation in TNBS-induced Colitis
To evaluate the extent to which the anti-inflammatory activity of phytosphingosine, the IBD-modulated metabolite with the greatest efficacy in NF-κB reporter assays, might attenuate inflammation in vivo, we tested its effects in an acute TNBS-induced colitis mouse model (15, 31). Phytosphingosine was significantly downregulated in colitis-associated colon content samples, as evidenced by a 3.8 FC and 1.4 FC reduction in levels relative to Rag and WT, respectively. In contrast, phytosphingosine levels were not statistically altered in cecum samples among the treatment groups, nor was the metabolite detected in fecal samples from any mice. WT mice were dosed by oral gavage with vehicle or phytosphingosine 1 h prior to, and 3 days after, rectal administration of TNBS (2,4,6-trinitrobenzenesulfonic acid), as previously described (15). Mice treated only with TNBS showed a trend towards reduced body weight and colon length that did not reach statistical significance (Fig. 5A, B). However, TNBS treatment elicited significant increases in expression of inflammation markers and decreases in intestinal endothelial permeability markers (Fig. 5C). Co-treatment with phytosphingosine suppressed the increase in the expression of the inflammatory marker MCP-1 in colonic tissues relative to treatment with only TNBS. The inhibition of TNFα expression did not reach statistical significance with the group size used. Co-treatment with phytosphingosine also partially restored the expression of genes encoding intestinal epithelial barrier markers suppressed by TNBS treatment, such as tight junction protein ZO-1 (TJP1) and occludin (OCLN) that are responsible for linking tight junction transmembrane proteins (such as claudins, junctional adhesion molecules, occludin) to the actin cytoskeleton and the formation and regulation of the tight junction paracellular barrier. Phytosphingosine co-treatment increased the mRNA levels of TJP1 and OCLN relative to mice treated only with TNBS and resulted in a 30% recovery in TJP1 and 20% recovery in OCLN expression (Fig. 5C). Phytosphingosine could originate from a combination of host and/or microbial sources. In the host, the phytosphingosine biosynthetic pathway includes a penultimate reaction catalyzed by sphingolipid Δ(4)-desaturase (DEGS2) that acts as a sphingolipid C4-monooxygenase to generate phytoceramide, the substrate that is ultimately converted to phytosphingosine by ceramidase (CerS) (Fig. 5D). DEGS2 is mostly highly expressed in the intestinal epithelium relative to other tissues (32). Consistent with the lower levels of phytosphingosine we detected in IBD samples, we found that TNBS treatment decreased expression of DEGS2 in the intestine (Fig. 5D).
Fig. 5. Phytosphingosine ameliorates TNBS-induced colitis.
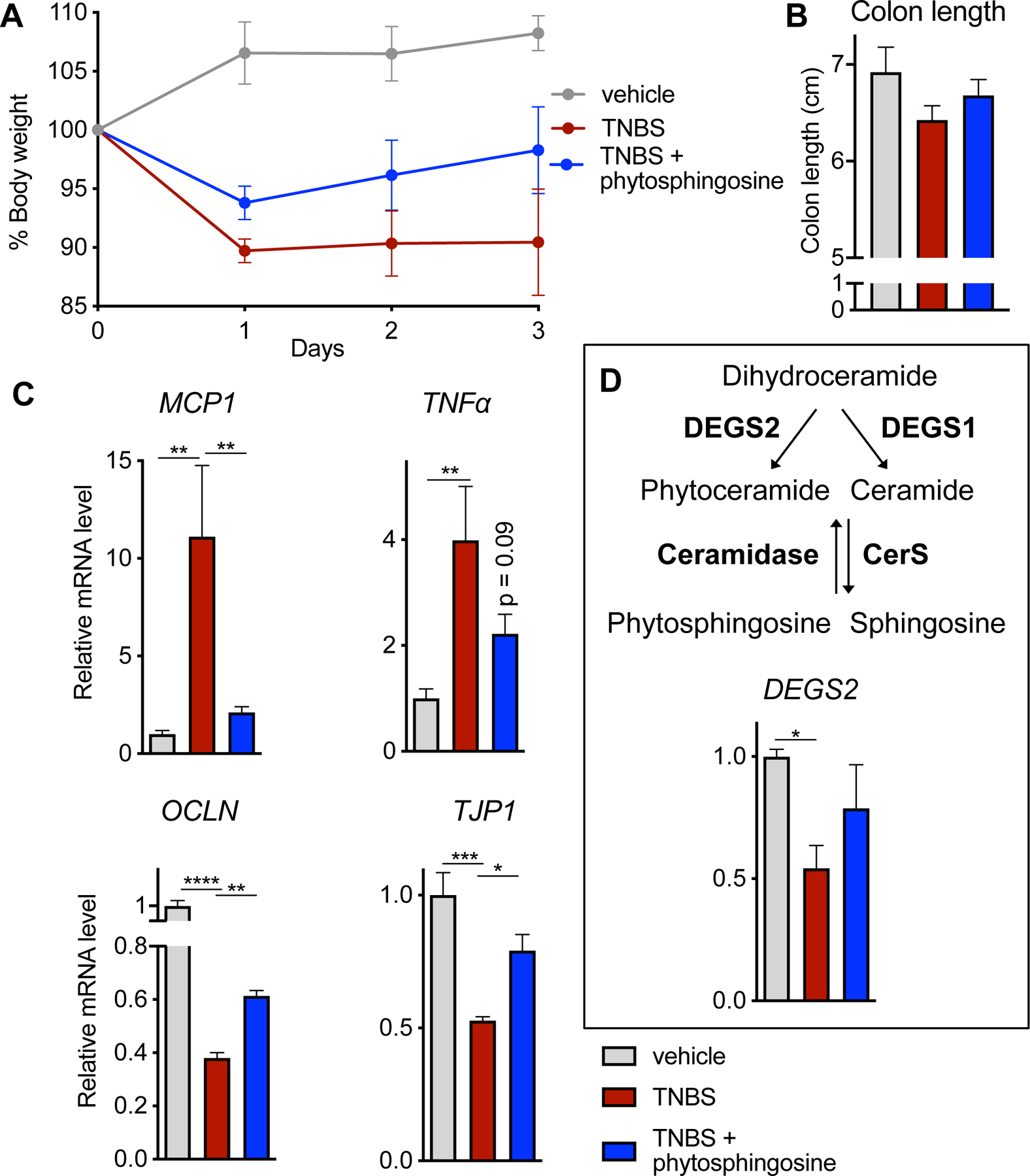
WT mice were dosed orally with vehicle or 25 mg/kg phytosphingosine prior to treatment with TNBS. Phytosphingosine treatment was continued daily. (A) Body weight evolution, (B) colon length, and (C) expression of inflammation and (MCP1 and TNFα) and barrier permeability markers (OCLN and TJP1) in treated mice. (D) Levels of mRNA encoding DEGS2, the upstream enzyme required for phytosphingosine production in the host. p-value <0.05 denoted by * and <0.01 denoted by **. Veh n=5 mice, TNBS n=4 mice, and TNBS + phytosphingosine n=5 mice.
We also tested in this model the effects of thymoquinone, a metabolite with decreased levels in the IBD-associated colon samples relative to controls, which has been previously shown to reduce gut inflammation (33, 34). Thymoquinone also acted as a robust anti-inflammatory agent in LPS-treated primary macrophages (Fig. 4A). Pre-exposure of mice to 25 mg/kg of thymoquinone prior to TNBS addition and daily oral dosing for three days post-TNBS treatment protected mice from TNBS-induced pro-inflammatory gene expression (fig. S4, A,B). Co- treatment with thymoquinone reduced mRNA levels for MCP-1 by 3.8 FC and TNFα by 2.3 FC relative to the TNBS-alone cohort (fig. S4B). However, thymoquinone did not rescue decreased expression of the membrane permeability markers TJP1 and OCLN at the mRNA level (fig. S4, C), suggesting that phytosphingosine and thymoquinone modulate disease progression through distinct pathways.
DISCUSSION
In this work, we performed untargeted metabolomic analysis to identify metabolites with altered levels in a T-cell transfer mouse model of colitis relative to two control groups (WT and Rag mice) and the standard chow provided ad libitum. The relative abundance of IBD-dysregulated metabolic features for each different GI tract region comprised a broad and chemically diverse metabolome, including (but not limited to) amino acids, carbohydrates, peptides, nucleotides, lipids, vitamins, and sterols. Dysbiosis of the metabolome was rather limited relative to the well-documented large alterations in taxonomic composition found in IBD-associated distal gut microbiomes (35–39). These observations offer some support for the neutral theory of composition, which posits that the fitness of neutral species that share the same general niche changes identically (or similarly) along an environmental axis.
In contrast to the decrease in composition of the metabolome we noted along the GI tract, the percentage of small-molecule features dysregulated between the healthy and colitic groups increased along the GI tract. In the cecum, colon, and feces, 3.5%, 7.1%, and 12.3% of the total detectable metabolites for each sample type were dysregulated, respectively. The cecum samples had the lowest percentage of colitis-altered metabolites; this may be attributable to the large number of unidentifiable metabolic features produced by plants and animals (diet), the intestinal microbiome, and the host that are yet to be described (40). Although we observed differences in metabolite levels between control and colitic mice, the number of metabolic features detected and their relative distribution along the GI tract remained similar in all three different cohorts.
Despite having the lowest diversity in metabolite features, the largest differences between control and IBD groups, as assessed by the percentage of dysregulated metabolic features, were found in the colonic and fecal samples and consisted of a mix of polar/non-polar metabolites in the colon and primarily non-polar compounds in the feces. It is possible that the accumulation of dysregulated metabolites along the GI tract simply reflected a reduced number of metabolic processes contributed by an altered microbiome and/or colitic host. It is also likely that some fraction of metabolites may originate from lysis-susceptible bacteria that have an increased taxonomic footprint in the context of intestinal inflammation (35, 41–43). Many of the lipid-like metabolites upregulated in the colitic fecal samples have been reported in other IBD-associated studies (44). The overrepresentation of hydrophobic lipids in fecal samples relative to cecal and colonic samples likely derives from bacterial cell walls and other intracellular bacterial components, because anaerobic bacteria are susceptible to lysis upon exposure to oxygen (45). The diversity in metabolome physicochemical composition along the GI tract highlights the complexity of chemical reactions and biological processes localized to different regions, as well as the possibility that certain metabolites may have distinct functions and/or biological roles for both the host and the microbiome along the GI tract.
A broad range of metabolite classes, including amino acids, carbohydrates, lipids, dipeptides, sterols, and vitamins were dysregulated in the colon (meaning that they showed increased or decreased abundance in the colitis samples relative to controls) (Fig. 3 and Data File S2) and many have been linked previously with intestinal inflammation. For instance, methionine and methionine sulfoxide (as well as 3 dipeptides) resulting from aberrant methionine metabolism have been associated with IBD and other gut inflammatory phenotypes (23–25). The disruption in methionine metabolism is exemplified by the reduced and oxidized species being inversely dysregulated in colitis. We found an increase of methionine sulfoxide levels in WT mice, whereas those of the reduced form (methionine) were significantly upregulated in colitic RagT mice. Our findings suggest that methionine may act as an inflammatory agent, a notion that is supported by the observation that methionine-reduced diets are protective in IBD, and by the roles of methionine in regulation of reactive oxygen species (ROS) production and NF-κB signaling (46, 47). Similarly, we detected altered levels of taurocholic acid, an observation in line with previous reports of alterations of bile acids levels in IBD. Taurocholic acid levels are substantially upregulated in colitic mice and elevated in human feces from IBD patients, and this bile acid promotes pathobiont expansion and colitis in IL-10−/− mice (48–50). These and other (Data File S2) representative examples of dysregulated molecules previously associated with intestinal inflammation conditions provided known benchmarks to assess the robustness of our untargeted metabolomics methodology. Our ability to detect alterations in these metabolites in our IBD samples provides confidence that the newly identified dysregulated metabolites we report in this study are linked to naïve T cell-induced IBD.
Studying these new IBD-linked metabolites in cellular assays uncovered a set of highly abundant molecules in our microbiome samples that showed a mixed response. We anticipate that these molecules may have important roles in host physiology and microbiome ecology, as may be revealed in future studies. Our mixed response results with the established anti-inflammatory agent OEA and the pro-inflammatory molecule LPC(19:0) demonstrate a complex mechanism of action with potential spatiotemporal regulation of IBD that requires further investigation.
Several metabolites were also identified that showed robust pro-inflammatory effects not previously reported, including hecogenin and 1-methyladenosine. Further study of these molecules may yield new biological insights into innate immune responses. Finally, we also identified metabolites such as phytosphingosine that displayed anti-inflammatory activity in cell-based assays and modest efficacy in an acute mouse model of colitis. This result shows that cell-based functional assays only demonstrate the narrow impact of each metabolite on the phenotype tested, and further analyses in other models, and specifically in vivo assessment, are required to uncover additional processes affected by physiologically active metabolites. In sum, our findings establish the feasibility of our metabolomics activity screening strategy to identify and prioritize for further analysis physiologically relevant metabolites with in vivo activity. Such studies will provide greater understanding of their biological relevance and role in disease.
MATERIALS AND METHODS
Murine Adoptive T Cell Transfer Chronic IBD Model and Cecal, Colon and Fecal Sample Collection
Animal protocols were approved by The Institutional Animal Care and Use Committee (IACUC) at The Scripps Research Institute (TSRI). All mice were purchased from Jackson Laboratories and maintained in a specific pathogen-free barrier facility at TSRI for the duration of the study. We used the T cell transfer model of colitis to induce intestinal inflammation, as previously shown (39, 51). Briefly, 12 6-week-old B6.129S7-Rag1tm1mom/J(Rag1−/−) mice (Rag) and 6 C57BL/6J (WT) mice were cohoused for two weeks to normalize the microbiota. CD3+CD4+CD8–CD25–Foxp3– naïve T cells collected via fluorescence-activated cell sorting from the spleens of donor Foxp3-EGFP reporter mice were transferred retro-orbitally to 6 Rag mice (approximately 5 × 105 T cells per mouse) (referred to as “RagT”). The control Rag and WT mice were injected retro-orbitally with a similar volume of sterile PBS and subsequently separated for the remainder of the time course from the colitic cohort (n = 6 for all three cohorts). The mice were fed standard chow (Harlan Teklad) throughout the 8-week time course and all mice were separated by treatment group for the remainder of the experiment. Mice were euthanized on day 56 post-T cell transfer and all microbiome samples (cecal, colonic, and fecal) were collected and prepared for untargeted metabolomics data collection and histology.
TNBS IBD Model
Colitis was induced in male C57BL/6 mice by a single intracolonic injection of 150 μL 50% ethanol containing 2.5% w/v TNBS, as previously described (52). Briefly, mice were anesthetized with isoflurane and the TNBS solution was administered with a catheter. Mice were held upside down for 30 s to ensure proper TNBS distribution. Mice were dosed with vehicle (ethanol:DMSO:Cremophor EL:saline 7:10:13:70) or compounds prior to TNBS induction and daily thereafter. Mice were euthanized 3 days post-treatment for tissue collection and analysis.
Primary Macrophage Culture Assays
Thioglycollate-elicited peritoneal macrophages (PM) were obtained 4 days after intraperitoneal injection of 3 mL sterile 4% thioglycollate medium in male C57BL/6 mice. The PM were obtained by peritoneal lavage with 50 mL ice-cold PBS, treated with ACK lysis buffer for 3 min on ice and washed twice with PBS prior to resuspending in complete medium (RPMI containing 10% fetal bovine serum (FBS), 2 mM L-glutamine and antibiotic/antimycotic). Cells were plated in 96-well tissue culture plates at 8 × 104 cells/well (53). Cells were treated with DMSO control or metabolites (at the concentrations outlined in fig. S2) for 3 hr in 100 μL RPMI1640 containing 2% charcoal-dextran treated FBS prior to addition of LPS (Sigma) at a final concentration of 20 ng/mL or vehicle control (PBS). Cell viability was determined after a 36-h incubation period using CellTiter-Glo. For transcript levels, cells were either collected at 3 or 36 hr post-LPS treatment for qPCR analysis. Nitrite levels (used as a proxy measurement of NO release) in media were measured 36 hr post-LPS treatment by the Griess test (Sigma). Absorption was measured at 520 nm according to manufacturer’s protocol (28).
Metabolite Extraction
For metabolite extraction, 500 µL of cold acetonitrile/methanol/water (2:2:1, v:v:v) was added to lyophilized fecal, cecal, and colonic content samples per 1.5 mg (solvent volume was adjusted accordingly to sample weight). Protein precipitation was performed by 3 cycles of freeze/thawing and sonication. Each cycle included a flash freezing step in liquid nitrogen for 1 min, vortex for 30 sec and sonication in an ice-cold bath for 15 min. This was followed by incubation at −20 °C for 1 hr and centrifugation at 16000 x g for 15 min at 4 °C. The supernatants were then transferred to another tube and dried down in a vacuum concentrator. The dry metabolite extracts were stored at −80 °C prior to their analysis by liquid chromatography/mass spectrometry (LC-MS). Metabolite extracts were reconstituted with acetonitrile/water (1:1, v:v) (based on the initial weight of the sample used for extraction), vortexed for 1 min, sonicated for 15 min, and centrifuged at 16,000 x g and 4 °C for 15 min to remove non-soluble debris.
Metabolite Measurement by LC-MS
The metabolites extracts were analyzed by both reverse-phase (RP) and hydrophilic interaction liquid chromatography (HILIC) (54). Plasma samples analyses were carried out in a Synapt G2-Si q-ToF mass spectrometer equipped with an I-class UHPLC (Waters Corporation, Milford, MA). Data was acquired over a m/z range 50–1200 Da. The electrospray source conditions were as follows: capillary, +3.00 kV; sampling cone, 40 V; source temperature, 100 °C; desolvation temperature, 250 °C; desolvation gas flow, 600 L/hr; and cone gas flow, 50 L/hr, respectively. Leucine–enkephalin (m/z 556.2771) was used for lock mass correction.
The same mobile phases were used for both RP and HILIC chromatography, consisting of 0.1% formic acid in water (v/v) as phase A and 0.1% formic acid in acetonitrile (v/v) as phase B. The flow rate consisted on 400 µL/min and the columns used were: ACQUITY BEH C18 column (2.1 × 100 mm, 1.7 µm, Water Corporation, Milford, MA) for the RP analysis and an ACQUITY BEH Amide (2.1 × 100 mm, 1.7 µm, Water Corporation, Milford, MA) for the HILIC analysis. Data was acquired in positive ion mode. The injection volume was 2 μL. For identification purposes, putative molecules of interest were fragmented at 3 different collision energies (10, 20 and 40 eV).
Raw LC-MS data were converted to mzXML format using ProteoWizard MS Converter version 3.0.7529 (55). The mzXML files were uploaded to XCMS Online for data processing (56). Peaks were first detected, aligned across samples and integrated. Features were then deisotoped and annotated for adducts formation and common losses (57, 58). Data were processed using the following parameter settings: centWave for feature detection (Δm/z = 15 ppm, minimum peak width = 10 s, and maximum peak width = 60 s); obiwarp settings for retention time correction (profStep = 0.5); and parameters for chromatogram alignment, including mzwid = 0.015, minfrac = 0.5, and bw = 5. Multigroup statistical processing was carried out automatically by XCMS Online using a one-way ANOVA followed by a Kruskal-Wallis non-parametric test. After statistical analysis, only features with q-value <0.05 were selected for identification by MS/MS experiments (56, 59). To identify these metabolites, the resulting MS/MS spectra were matched against the METLIN database and analyzed under the exact same conditions using standard reference material to match both retention time and fragmentation pattern according to MSI guidelines (60, 61).
Histology
Segments of small intestinal and colonic tissues were fixed in 10% formalin for histology and demonstration of the established gross morphological changes associated with naïve T cell-induced colitis (fig. S1).
qPCR of Inflammatory Markers
Total RNA was isolated from cells and tissues using the Direct-zol RNA MiniPrep Plus kit (Zymo Research). Taqman-based quantitative real-time PCR was performed using the SuperScript III Platinum One-Step qRT–PCR reagent (Thermo Fisher Scientific). Samples were run as multiplexed reactions normalized to an internal control (36B4; acidic ribosomal phosphoprotein P0 mRNA). Sequences of primers and probes used are in Data File S3. Relative mRNA expression was determined by the ΔΔ-Ct method.
NF-κB-based Cellular Assays
THP-1 macrophages stably expressing NF-κB-driven luciferase were plated in 96-well white-walled clear-bottomed tissue culture plates at 8 × 104 cells per well in the presence of 5 ng/mL PMA in RPMI1640 containing 10% FBS for 48 hr. Cells were washed and rested for 24 hr in RPMI1640 containing 2% charcoal-dextran treated FBS. The cells were then supplemented with the panel of metabolites for 3 hr as described prior to the addition of 5 ng/mL LPS for an additional 3 hr. Bright-Glo™ Luciferase Assay System (Promega) was used to measure luciferase activity according to manufacturer’s protocol. Dexamethasone (2 μM) and 20 ng/mL LPS were used as positive controls for suppression and induction of NF-κB signaling, respectively.
Supplementary Material
Acknowledgments:
We thank John Teijaro and Nhan Halley Nguyen for assistance with T cell mouse generation and Magan Hall for assistance with histology.
Funding:
This research was partially funded by:
National Institutes of Health grant P30 DK120515 (LE)
National Institutes of Health grant R21 CA181027 (DWW)
National Institutes of Health grant R21 AI139744 (DWW)
National Institutes of Health grant RC2 DK114785 (ES)
National Institutes of Health grant R35 GM130385 (GS)
National Institutes of Health grant R01 GM114368 (GS)
National Institutes of Health grant P30 MH062261 (GS)
National Institutes of Health grant U01 CA235493 (GS)
NIH Cloud Credits Model Pilot, a component of the NIH Big Data to Knowledge (BD2K) program (GS)
Footnotes
Competing interests: The authors declare that they have no competing interests.
Data and materials availability:
Metabolomic data were deposited onto figshare (https://doi.org/10.6084/m9.figshare.16434825.v1.). All other data needed to evaluate the conclusions in the paper are present in the paper or the Supplementary Materials.
References and Notes
- 1.Carter MJ, Lobo AJ, Travis SP, Guidelines for the management of inflammatory bowel disease in adults. Gut 53 Suppl 5, V1–16 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Wong SH, Ng SC, What can we learn from inflammatory bowel disease in developing countries? Curr. Gastroenterol. Rep 15, 313 (2013). [DOI] [PubMed] [Google Scholar]
- 3.Maloy KJ, Powrie F, Intestinal homeostasis and its breakdown in inflammatory bowel disease. Nature 474, 298–306 (2011). [DOI] [PubMed] [Google Scholar]
- 4.Loftus EV Jr., Clinical epidemiology of inflammatory bowel disease: incidence, prevalence, and environmental influences. Gastroenterology 126, 1504–1517 (2004). [DOI] [PubMed] [Google Scholar]
- 5.Ahmadi A, Polyak S, Draganov PV, Colorectal cancer surveillance in inflammatory bowel disease: the search continues. World J. Gastroenterol 15, 61–66 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Loftus EV Jr., The burden of inflammatory bowel disease in the United States: a moving target? Clin. Gastroenterol. Hepatol 5, 1383–1384 (2007). [DOI] [PubMed] [Google Scholar]
- 7.Cohen RD, The quality of life in patients with Crohn’s disease. Aliment. Pharmacol. Ther 16, 1603–1609 (2002). [DOI] [PubMed] [Google Scholar]
- 8.Xavier RJ, Podolsky DK, Unravelling the pathogenesis of inflammatory bowel disease. Nature 448, 427–434 (2007). [DOI] [PubMed] [Google Scholar]
- 9.Kozuch PL, Hanauer SB, Treatment of inflammatory bowel disease: a review of medical therapy. World J. Gastroenterol 14, 354–377 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Greenbloom SL, Steinhart AH, Combination ciprofloxacin and metronidazole for active Crohn’s disease. Canadian J. Gastroenterol 12, 53–56 (1997). [DOI] [PubMed] [Google Scholar]
- 11.Sartor RB, Therapeutic manipulation of the enteric microflora in inflammatory bowel diseases: antibiotics, probiotics, and prebiotics. Gastroenterol 126, 1620–1633 (2004). [DOI] [PubMed] [Google Scholar]
- 12.Hunter J, Elemental diet and the nutritional treatment of Crohn’s disease. Gastroenterol. Hepatol. Bed Bench 8, 4–5 (2015). [PMC free article] [PubMed] [Google Scholar]
- 13.Han X, Shao H, Wang Y, Hu A, Chen R, Chen Q, Composition of the bacterial community in the gastrointestinal tract of Kunming mice. Electronic J. Biotech 43, 16–22 (2020). [Google Scholar]
- 14.Suzuki TA, Nachman MW, Spatial heterogeneity of gut microbial composition along the gastrointestinal tract in natural populations of house mice. PLoS One 11, e0163720 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Sinha SR, Haileselassie Y, Nguyen LP, Tropini C, Wang M, Becker LS, Sim D, Jarr K, Spear ET, Singh G, Namkoong H, Bittinger K, Fischbach MA, Sonnenburg JL, Habtezion A, Dysbiosis-induced secondary bile acid deficiency promotes intestinal inflammation. Cell Host Microbe 27, 659–670 e655 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Duboc H, Rainteau D, Rajca S, Humbert L, Farabos D, Maubert M, Grondin V, Jouet P, Bouhassira D, Seksik P, Sokol H, Coffin B, Sabate JM, Increase in fecal primary bile acids and dysbiosis in patients with diarrhea-predominant irritable bowel syndrome. Neurogastroenterol. Motil 24, 513–520, e246–517 (2012). [DOI] [PubMed] [Google Scholar]
- 17.Tiratterra E, Franco P, Porru E, Katsanos KH, Christodoulou DK, Roda G, Role of bile acids in inflammatory bowel disease. Ann. Gastroenterol 31, 266–272 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Reifen R, Karlinsky A, Stark AH, Berkovich Z, Nyska A, alpha-Linolenic acid (ALA) is an anti-inflammatory agent in inflammatory bowel disease. J. Nutr. Biochem 26, 1632–1640 (2015). [DOI] [PubMed] [Google Scholar]
- 19.Grill M, Hogenauer C, Blesl A, Haybaeck J, Golob-Schwarzl N, Ferreiros N, Thomas D, Gurke R, Trotzmuller M, Kofeler HC, Galle B, Schicho R, Members of the endocannabinoid system are distinctly regulated in inflammatory bowel disease and colorectal cancer. Sci. Rep 9, 2358 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Lama A, Provensi G, Amoriello R, Pirozzi C, Rani B, Mollica MP, Raso GM, Ballerini C, Meli R, Passani MB, The anti-inflammatory and immune-modulatory effects of OEA limit DSS-induced colitis in mice. Biomed. Pharmacother 129, 110368 (2020). [DOI] [PubMed] [Google Scholar]
- 21.Otagiri S, Ohnishi S, Ohara M, Fu Q, Yamamoto K, Yamamoto K, Katsurada T, Sakamoto N, Oleoylethanolamide ameliorates dextran sulfate sodium-induced colitis in rats. Front. Pharmacol 11, 1277 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Di Paola M, Bonechi E, Provensi G, Costa A, Clarke G, Ballerini C, De Filippo C, Passani MB, Oleoylethanolamide treatment affects gut microbiota composition and the expression of intestinal cytokines in Peyer’s patches of mice. Sci. Rep 8, 14881 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Liu Y, Wang X, Hu CA, Therapeutic potential of amino acids in inflammatory bowel disease. Nutrients 9, (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Price ND, Magis AT, Earls JC, Glusman G, Levy R, Lausted C, McDonald DT, Kusebauch U, Moss CL, Zhou Y, Qin S, Moritz RL, Brogaard K, Omenn GS, Lovejoy JC, Hood L, A wellness study of 108 individuals using personal, dense, dynamic data clouds. Nat. Biotechnol 35, 747–756 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Witko-Sarsat V, Descamps-Latscha B, Neutrophil-derived oxidants and proteinases as immunomodulatory mediators in inflammation. Mediators Inflamm 3, 257–273 (1994). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kominsky DJ, Keely S, MacManus CF, Glover LE, Scully M, Collins CB, Bowers BE, Campbell EL, Colgan SP, An endogenously anti-inflammatory role for methylation in mucosal inflammation identified through metabolite profiling. J. Immunol 186, 6505–6514 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Sugihara K, Morhardt TL, Kamada N, The role of dietary nutrients in inflammatory bowel disease. Front. Immunol 9, 3183 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Yang K, Wu Y, Xie H, Li M, Ming S, Li L, Li M, Wu M, Gong S, Huang X, Macrophage-mediated inflammatory response decreases mycobacterial survival in mouse MSCs by augmenting NO production. Sci. Rep 6, 27326 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Dennis EA, Cao J, Hsu YH, Magrioti V, Kokotos G, Phospholipase A2 enzymes: physical structure, biological function, disease implication, chemical inhibition, and therapeutic intervention. Chem. Rev 111, 6130–6185 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Verma S, De Jesus P, Chanda SK, Verma IM, SNW1, a novel transcriptional regulator of the NF-kappaB pathway. Mol. Cell. Biol 39, (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Morris GP, Beck PL, Herridge MS, Depew WT, Szewczuk MR, Wallace JL, Hapten-induced model of chronic inflammation and ulceration in the rat colon. Gastroenterology 96, 795–803 (1989). [PubMed] [Google Scholar]
- 32.Omae F, Miyazaki M, Enomoto A, Suzuki M, Suzuki Y, Suzuki A, DES2 protein is responsible for phytoceramide biosynthesis in the mouse small intestine. Biochem. J 379, 687–695 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Lei X, Liu M, Yang Z, Ji M, Guo X, Dong W, Thymoquinone prevents and ameliorates dextran sulfate sodium-induced colitis in mice. Dig. Dis. Sci 57, 2296–2303 (2012). [DOI] [PubMed] [Google Scholar]
- 34.Subramanya SB, Venkataraman B, Raj V, Al Marzooqi S, Alhassani A, AlHassani A, Ahmed KJ, Attoub S, Thymoquinone, a bioactive phytochemical alleviates colon inflammation through Nrf2/Keap1 system: an experimental study using both in vivo and in vitro model of colon inflammation. FASEB J 33, 764.762–764.762 (2019). [Google Scholar]
- 35.Frank DN, St Amand AL, Feldman RA, Boedeker EC, Harpaz N, Pace NR, Molecular-phylogenetic characterization of microbial community imbalances in human inflammatory bowel diseases. Proc. Natl. Acad. Sci. USA 104, 13780–13785 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Morgan XC, Tickle TL, Sokol H, Gevers D, Devaney KL, Ward DV, Reyes JA, Shah SA, LeLeiko N, Snapper SB, Bousvaros A, Korzenik J, Sands BE, Xavier RJ, Huttenhower C, Dysfunction of the intestinal microbiome in inflammatory bowel disease and treatment. Genome Biol 13, R79 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Alam MT, Amos GCA, Murphy ARJ, Murch S, Wellington EMH, Arasaradnam RP, Microbial imbalance in inflammatory bowel disease patients at different taxonomic levels. Gut Pathog 12, 1 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Mayers MD, Moon C, Stupp GS, Su AI, Wolan DW, Quantitative metaproteomics and activity-based probe enrichment reveals significant alterations in protein expression from a mouse model of inflammatory bowel disease. J. Proteome Res 16, 1014–1026 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Moon C, Stupp GS, Su AI, Wolan DW, Metaproteomics of colonic microbiota unveils discrete protein functions among colitic mice and control groups. Proteomics 18, (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Monge ME, Dodds JN, Baker ES, Edison AS, Fernandez FM, Challenges in identifying the dark molecules of life. Annu. Rev. Anal. Chem 12, 177–199 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Zimmermann J, Hubschmann T, Schattenberg F, Schumann J, Durek P, Riedel R, Friedrich M, Glauben R, Siegmund B, Radbruch A, Muller S, Chang HD, High-resolution microbiota flow cytometry reveals dynamic colitis-associated changes in fecal bacterial composition. Eur. J. Immunol 46, 1300–1303 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Sokol H, Seksik P, Furet JP, Firmesse O, Nion-Larmurier I, Beaugerie L, Cosnes J, Corthier G, Marteau P, Dore J, Low counts of Faecalibacterium prausnitzii in colitis microbiota. Inflamm. Bowel Dis 15, 1183–1189 (2009). [DOI] [PubMed] [Google Scholar]
- 43.Joossens M, Huys G, Cnockaert M, De Preter V, Verbeke K, Rutgeerts P, Vandamme P, Vermeire S, Dysbiosis of the faecal microbiota in patients with Crohn’s disease and their unaffected relatives. Gut 60, 631–637 (2011). [DOI] [PubMed] [Google Scholar]
- 44.Franzosa EA, Sirota-Madi A, Avila-Pacheco J, Fornelos N, Haiser HJ, Reinker S, Vatanen T, Hall AB, Mallick H, McIver LJ, Sauk JS, Wilson RG, Stevens BW, Scott JM, Pierce K, Deik AA, Bullock K, Imhann F, Porter JA, Zhernakova A, Fu J, Weersma RK, Wijmenga C, Clish CB, Vlamakis H, Huttenhower C, Xavier RJ, Gut microbiome structure and metabolic activity in inflammatory bowel disease. Nat. Microbiol 4, 293–305 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Zeng H, Grapov D, Jackson MI, Fahrmann J, Fiehn O, Combs GF, Integrating multiple analytical datasets to compare metabolite profiles of mouse colonic-cecal contents and feces. Metabolites 5, 489–501 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Liu G, Yu L, Fang J, Hu CA, Yin J, Ni H, Ren W, Duraipandiyan V, Chen S, Al-Dhabi NA, Yin Y, Methionine restriction on oxidative stress and immune response in DSS-induced colitis mice. Oncotarget 8, 44511–44520 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Benight N, Burrin D, Stoll B, Puiman P, The role of methionine metabolism in inflammatory bowel disease. Inflamm. Bowel Dis 14, S42–S43 (2008). [Google Scholar]
- 48.Jansson J, Willing B, Lucio M, Fekete A, Dicksved J, Halfvarson J, Tysk C, Schmitt-Kopplin P, Metabolomics reveals metabolic biomarkers of Crohn’s disease. PLoS One 4, e6386 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Duboc H, Rajca S, Rainteau D, Benarous D, Maubert MA, Quervain E, Thomas G, Barbu V, Humbert L, Despras G, Bridonneau C, Dumetz F, Grill JP, Masliah J, Beaugerie L, Cosnes J, Chazouilleres O, Poupon R, Wolf C, Mallet JM, Langella P, Trugnan G, Sokol H, Seksik P, Connecting dysbiosis, bile-acid dysmetabolism and gut inflammation in inflammatory bowel diseases. Gut 62, 531–539 (2013). [DOI] [PubMed] [Google Scholar]
- 50.Devkota S, Wang Y, Musch MW, Leone V, Fehlner-Peach H, Nadimpalli A, Antonopoulos DA, Jabri B, Chang EB, Dietary-fat-induced taurocholic acid promotes pathobiont expansion and colitis in Il10−/− mice. Nature 487, 104–108 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Powrie F, Leach MW, Mauze S, Caddle LB, Coffman RL, Phenotypically distinct subsets of CD4+ T cells induce or protect from chronic intestinal inflammation in C. B-17 scid mice. Int. Immunol 5, 1461–1471 (1993). [DOI] [PubMed] [Google Scholar]
- 52.Singh R, Chandrashekharappa S, Bodduluri SR, Baby BV, Hegde B, Kotla NG, Hiwale AA, Saiyed T, Patel P, Vijay-Kumar M, Langille MGI, Douglas GM, Cheng X, Rouchka EC, Waigel SJ, Dryden GW, Alatassi H, Zhang HG, Haribabu B, Vemula PK, Jala VR, Enhancement of the gut barrier integrity by a microbial metabolite through the Nrf2 pathway. Nat. Commun 10, 89 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Molteni V, Li X, Nabakka J, Liang F, Wityak J, Koder A, Vargas L, Romeo R, Mitro N, Mak PA, Seidel HM, Haslam JA, Chow D, Tuntland T, Spalding TA, Brock A, Bradley M, Castrillo A, Tontonoz P, Saez E, N-Acylthiadiazolines, a new class of liver X receptor agonists with selectivity for LXRβ. J. Med. Chem 50, 4255–4259 (2007). [DOI] [PubMed] [Google Scholar]
- 54.Ivanisevic J, Zhu ZJ, Plate L, Tautenhahn R, Chen S, O’Brien PJ, Johnson CH, Marletta MA, Patti GJ, Siuzdak G, Toward ‘omic scale metabolite profiling: a dual separation-mass spectrometry approach for coverage of lipid and central carbon metabolism. Anal. Chem 85, 6876–6884 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Kessner D, Chambers M, Burke R, Agus D, Mallick P, ProteoWizard: open source software for rapid proteomics tools development. Bioinformatics 24, 2534–2536 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Tautenhahn R, Patti GJ, Rinehart D, Siuzdak G, XCMS Online: a web-based platform to process untargeted metabolomic data. Anal. Chem 84, 5035–5039 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Domingo-Almenara X, Montenegro-Burke JR, Guijas C, Majumder ELW, Benton HP, Siuzdak G, Autonomous METLIN-guided in-source fragment annotation for untargeted metabolomics. Anal. Chem 91, 3246–3253 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Domingo-Almenara X, Montenegro-Burke JR, Benton HP, Siuzdak G, Annotation: a computational solution for streamlining metabolomics analysis. Anal. Chem 90, 480–489 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Story JD, A direct approach to false discovery rates. J. R. Statist. Soc. B 64, 479–498 (2002). [Google Scholar]
- 60.Schymanski EL, Jeon J, Gulde R, Fenner K, Ruff M, Singer HP, Hollender J, Identifying small molecules via high resolution mass spectrometry: communicating confidence. Environ. Sci. Technol 48, 2097–2098 (2014). [DOI] [PubMed] [Google Scholar]
- 61.Montenegro-Burke JR, Guijas C, Siuzdak G, METLIN: a tandem mass spectral library of standards. Methods Mol. Biol 2104, 149–163 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Metabolomic data were deposited onto figshare (https://doi.org/10.6084/m9.figshare.16434825.v1.). All other data needed to evaluate the conclusions in the paper are present in the paper or the Supplementary Materials.