

ABSTRACT
The majority of neoantigens arise from unique mutations that are not shared between individual patients, making neoantigen-directed immunotherapy a fully personalized treatment approach. Novel technical advances in next-generation sequencing of tumor samples and artificial intelligence (AI) allow fast and systematic prediction of tumor neoantigens. This study investigates feasibility, safety, immunity, and anti-tumor potential of the personalized peptide-based neoantigen vaccine, EVX-01, including the novel CD8+ T-cell inducing adjuvant, CAF®09b, in patients with metastatic melanoma (NTC03715985). The AI platform PIONEERTM was used for identification of tumor-derived neoantigens to be included in a peptide-based personalized therapeutic cancer vaccine. EVX-01 immunotherapy consisted of 6 administrations with 5–10 PIONEERTM-predicted neoantigens as synthetic peptides combined with the novel liposome-based Cationic Adjuvant Formulation 09b (CAF®09b) to strengthen T-cell responses. EVX-01 was combined with immune checkpoint inhibitors to augment the activity of EVX-01-induced immune responses. The primary endpoint was safety, exploratory endpoints included feasibility, immunologic and objective responses. This interim analysis reports the results from the first dose-level cohort of five patients. We documented a short vaccine manufacturing time of 48–55 days which enabled the initiation of EVX-01 treatment within 60 days from baseline biopsy. No severe adverse events were observed. EVX-01 elicited long-lasting EVX-01-specific T-cell responses in all patients. Competitive manufacturing time was demonstrated. EVX-01 was shown to be safe and able to elicit immune responses targeting tumor neoantigens with encouraging early indications of a clinical and meaningful antitumor efficacy, warranting further study.
KEYWORDS: Personalized therapy, neoantigen, immune response, cancer vaccine, immunotherapy
Introduction
In the past decade, immunotherapy has revolutionized cancer treatment. Especially immune checkpoint inhibitors (CPI) targeting the programmed cell death protein 1 (PD-1) have shown remarkable clinical results in many cancers, including advanced melanoma.1
More recently, it was shown that durable tumor regression after CPI was mediated primarily by T-cells that recognized antigens derived from somatic mutations (neoantigens) expressed by tumors and that CPI non-responders did not mediate a tumor-specific T-cell response.2–6 A tumor contains up to thousands of somatic genomic alterations that are genuinely unique to the individual patient.7 Some of these genes are translated into mutant proteins, which the immune system may recognize as “foreign” neoantigens and act as a potential target for immune-mediated tumor killing.8
There have been numerous efforts to develop therapeutic cancer vaccines, but their translation into efficacious clinical therapies has been challenging9 potentially due to many of these vaccines targeting tumor-associated antigens (TAA) not specific to the tumor cells nor the patients. Hence, to exploit the full potential of cancer vaccination, personalized neoantigen vaccines have been introduced.10 Potentially that would improve the tumor-specificity and lower the levels of normal-cell toxicity. Furthermore, high-avidity T-cells targeting neoantigens are more likely to exist because this T-cell repertoire has not been deleted in the thymus.11 Therefore, neoantigen vaccines represent an attractive approach for therapeutic cancer vaccines.
Early clinical trials in melanoma, non-small lung cancer, bladder cancer, and glioblastoma patients have demonstrated that neoantigen T-cell responses can be induced by vaccination, highlighting their potential as cancer immunotherapy.12–17 The neoantigen-based vaccines were found to be safe and capable of eliciting a T-cell response, and a few clinical studies have also demonstrated anti-tumor potential.17–19
Although initial clinical test of neoantigen vaccines has shown promising results, neoantigen targeting treatments have some inherent complications that needs to be addressed to secure future success: i) Manufacturing timelines. As cancer mutations are rarely shared between any two tumors and patients’ immune system are unique; neoantigen vaccines has to be patient specific and manufactured specifically for each patients. In general, the neoantigen vaccine studies published so far have been using vaccine protocols and technologies with a production timeline between 12–24 weeks.16,19,20 This only enables addition of vaccination long after initiation of CPI treatment, whereas earlier onset of simultaneous treatment with CPI and the vaccine most likely would lead to higher likelihood of treatment synergy. Hence, production timeline must be shortened and cost per production batch reduced compared to traditional production of bulk product, ii) Identification of neoepitopes. Depending on the specific tumor and its inherent mutation rate, the number of potential neoepitopes vary from a few hundred to thousands. These numbers of identified neo-peptides are not feasible for injection into patients, why identification of the most efficacies neoepitopes are crucial for clinical affect. Multiple different methods exists for detecting neoepitopes including immuno-peptidomics, T-cell assays and bioinformatics methods based on sequencing data (DNA and mRNA).21 As both immuno-peptidomics and T-cell assays needs considerable patient material and are time and labor intensive most clinical trials use a bioinformatic pipeline to identify neoepitopes.16,19,20 Immunogenicity readouts from treated patients have shown that immunogenic neoepitopes can be identified with success by bioinformatics methods, however improvement is needed to increase precision and the number of effective neoepitopes in each treatment, iii) Induction of neoepitope specific CD8+ and CD4 + T-cells in patients. A strong vaccine system is needed to ensure potent responses to identified neoepitopes. To date neoantigen vaccines have been generated using different technologies including dendritic cell-based vaccines,22 mRNA-based vaccines,23 viral vector-based vaccines24 and peptide-based vaccines.14–16 All methods show induction of neoepitope T-cells however with varying success.
To address these challenges in neoepitope vaccine treatment, we designed a neoantigen vaccine production, distribution and administration protocol (EVX-01) using the PIONEERTM neoepitope identification platform, fast solid-state peptide synthesis, and mixing of the neopeptides with vaccine adjuvant CAF®09b. Peptide synthesis was selected to ensure fast production timelines and low risk of manufacturing failure, the PIONEERTM system as it rapidly (within 24 hours) identifies neoepitopes from DNA and mRNA sequencing data, and CAF®09b as it has been developed to induce strong CD4+ and CD8+ T-cells against peptides and proteins when admixed before injection. PIONEERTM is a fully automated GxP immuno-oncology platform developed by Evaxion Biotech that allows rank potential neoepitopes based on their likely to generate a profound anti-tumor immune response. The ranking ensures that selected neo-peptides have the following characteristics: 1) Epitopes residing in the neo-peptide are only present in tumor cells, 2) Consist of at least one CD4+ or CD8+ epitope – preferably both, 3) Neopeptides originate from proteins expressed in the tumor cell.
CAF®09b is a novel liposome-based vaccine adjuvant developed by Statens Serum Institut (SSI), Denmark, based on cationic surfactant dimethyl-dioctadecyl ammonium (DDA) combined with C-type lectin agonist monomycoloyl glycerol (MMG) and TLR3 agonist poly I:C. CAF®09b has in preclinical models shown superior ability to induce CD8+ T-cell responses when administrated intraperitoneal (i.p.) compared to traditional administration routes.25–27,28 Ideally, the CAF®09b adjuvant aims to increase the uptake of peptides by antigen-presenting cells (APCs) and promotes their activation status to induce cross-presentation and proinflammatory signaling. Thereby enhancing peptide-presentation on MHC class I and II by APCs, activating both vaccine-specific CD4+ and CD8+ T-cells, and optimizing an anti-tumor effect.
We developed a fully integrated Good Manufacturing Practice (GMP) manufacturing pipeline based on an optimized predefined workflow for each activity in the manufacturing process from tumor biopsy to fill/finish of final product. Activities was performed in parallel when possible and feasible and clear communication and oversight was imposed track progress and ensure each activity was initiated at the right time with the right input. This ensured as fast turnaround time as currently possible.
To evaluate the efficacy of co-administration of the EVX-01 vaccine with CPI treatment, we conducted a phase I study in patients with advanced melanoma, investigating the feasibility of manufacturing the neopeptide-vaccine, along with safety, immunogenicity, and clinical responses.
Here we report the data from a planned interim analysis on five patients treated with EVX-01 at the first safety dose level.
Methods and material
Trial design
This is a first-in-man phase I clinical trial conducted at the National Center for Cancer Immune Therapy (CCIT-DK) and the Department of Oncology, Copenhagen University Hospital, Herlev, Denmark (Figure 1a). The study protocol was approved by the Danish Medicines Agency and The Ethics Committee and conducted following the Helsinki agreement29 and guidelines for Good Clinical Practice (GCP).30 Legal approvals from the Ethical Committee and Medicines Agency were obtained. The trial is registered at clinicaltrial.gov (NCT03715985) and clinicaltrialsregister.eu (EudraCT No. 2018–002892-16). All patients had signed informed consent before inclusion.
Figure 1.
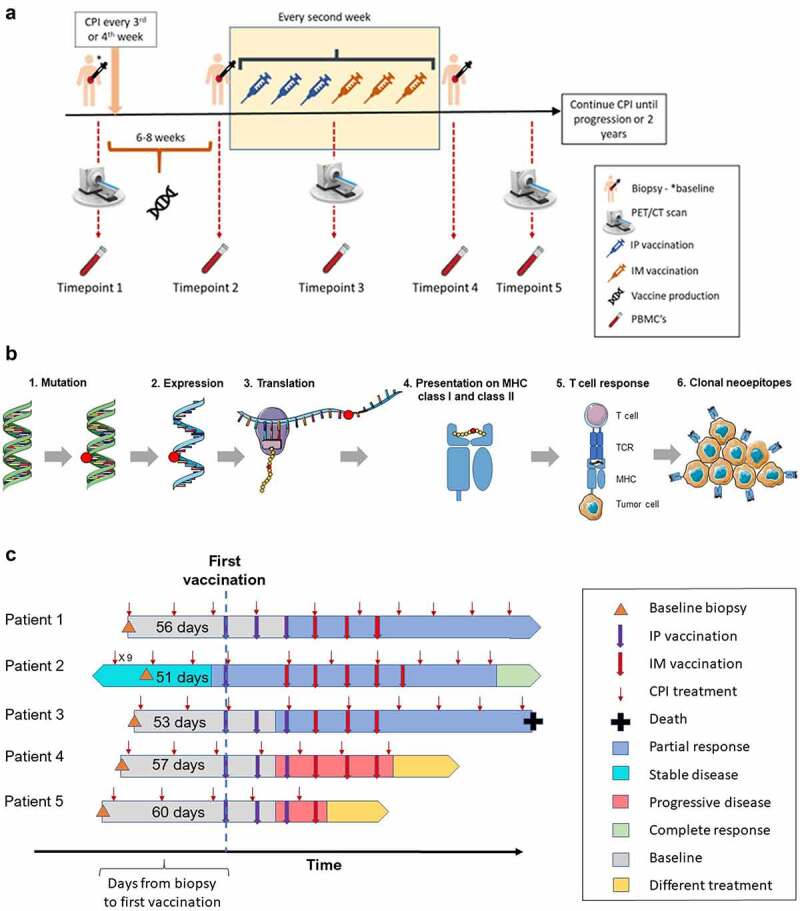
Study timelines. (A) Biopsy, PET/CT scan, and blood samples was collected at baseline. Treatment with CPI was either initiated shortly after biopsy or had already been initiated for at least 4 months before biopsy. EVX-01 vaccination was administered around week 6–8 (as quickly as possible) and every second week for a total of 6 vaccinations. Tumor biopsies were performed again if possible, at TP2 and TP4. Radiographic imaging was performed every 12 weeks, and blood samples were collected TP 1 to TP4 and every time a scan was performed. (B) An overview of the mechanisms in tumor cells and surrounding immune cells that are believed to be required/desirable for a neoepitope to have a clinical effect. 1) Tumor-specific mutations are detected using WES sequencing data from the tumor sample and normal sample. 2) The expression levels of each mutation are determined by analyzing tumor RNA sequencing data. 3) The tumor-specific mutations are translated into protein space, generating neopeptide sequences. 4–5) Neopeptide sequences predicted to be presented by the patient’s specific HLA class I and class II molecules are identified. Neoepitopes must be presented by MHC class I and class II in order to be recognized by T-cells. 6) The subset of neoepitopes that are clonal, meaning present in all tumor cells, are prioritized as this allows the elicited immune response to eradicate the whole tumor, as well as potential metastases in the patient. (Arts in 1B obtained from https://smart.servier.com/). (C) Overview of patient inclusion, CPI initiation, baseline biopsy, time before vaccine treatment and follow-up information of the first five patients. The blue and red arrows indicate time points for either i.p vaccinations or i.m vaccinations, respectively. The depiction of disease condition and patient status are indicated with various colors.
For the second and third patient included in this study, a minimum of two vaccines had to be administered to the previous patient before treatment could commence. The primary endpoints of the study were safety and tolerability of the treatment based on the occurrence of adverse events (AE) according to the NCI Common Terminology Criteria for Adverse Events (CTCAE version 4.0). The secondary endpoints were the feasibility to manufacture a personalized neoantigen vaccine within 6–8 weeks of enrollment with the PIONEERTM based production pipeline and to evaluate the immune response before, during, and after treatment with the personalized neoantigen vaccine. The manufacturing feasibility was assessed by whether neoantigen could be identified and the vaccines could be synthesized and formulated for clinical use. The tertiary endpoints were efficacy based on best overall response (BOR), progression-free survival (PFS), and overall survival (OS).
Assessments in the study included physical examination, ECOG performance, vital signs (before and after treatment), and blood samples to ensure the safety of the participants. Imaging (PET-CT or CT scan) was performed at baseline, after three, and after six vaccinations followed by imaging every 12 weeks to assess clinical efficacy of the treatment. Tumors were assessed by the investigators according to the RECIST version 1.1 criteria. Tumor biopsies were obtained at baseline (obligatory), and right before the first vaccination and after the last vaccination (voluntary).
We complied with the laboratory health and safety procedures during the course of conducting the experimental work.
Patients
Eligible patients were at least 18 years old with advanced unresectable melanoma confirmed cytologically or histologically. The patients should be candidates for treatment with an anti-PD-1 CPI and not previously treated with CPI in the metastatic/unresectable setting. Patients on therapy with an anti-PD-1 agent in the metastatic/unresectable setting for at least 4 months without unequivocal objective response or disease progression and who qualified for continued treatment with the same agent were also eligible. Additional inclusion criteria included; at least one measurable lesion as per investigator-assessed RECIST version 1.1; an Eastern Cooperative Oncology Group (ECOG) performance status of 0 or 1; adequate organ function; and ability to provide sufficient tumor tissue for whole-exome sequencing (WES).
Key exclusion criteria included: Active, known, or suspected autoimmune disease; a history of life-threatening or severe immune-related adverse events on treatment with another immunotherapy; or severe allergy or anaphylactic reactions earlier in life.
Design and manufacturing of personalized neoantigen vaccines
All personalized neoantigen vaccines were designed using the PIONEER™ platform developed by Evaxion Biotech based on whole-exome sequencing (WES) data from tumor and healthy tissue, mRNA sequencing data from tumor tissue and HLA typing data from healthy tissue. Approximately 1,2 mm (needle biopsy) tissue was needed to give 250 ng purified DNA to perform WES. NGS data were preprocessed to remove adapter sequences and low-quality bases. The WES reads were mapped to the GRCh38 human reference genome and used to detect tumor-specific somatic variants and their clonality. Variants resulting in altered amino acid sequences were introduced into the corresponding reference protein sequences and translated into “neopeptides” by extracting 13 amino acids (AA)(when possible) up and downstream of the variant resulting in neopeptides with lengths of 15–27 AAs. The neopeptide sequences were subjected to prediction for both MHC class I and -class II ligands presented by the patient’s HLAs. The mRNA sequencing reads were mapped to GRCh38 and used to calculate the expression levels of transcript isoforms hosting a somatic variant. Neopeptides were ranked based on the likelihood of containing an MHC ligand, host transcript expression levels and the clonality of the identified somatic variants. The top 20–25 ranked neopeptides were selected for manufacturing.
Design of personalized neoantigen vaccines: PIONEERTM
All personalized neoantigen vaccines were designed using the PIONEER™ platform developed by Evaxion Biotech (Figure 1b). PIONEER™ runs an automated bioinformatics pipeline starting from raw NGS reads in FASTQ format and ending in a list of neoepitope sequences sent for manufacturing. The ranking ensures that selected neo-peptides have the following characteristics: 1) Consist of at least one CD4+ or CD8+ epitope – preferably both, 2) Epitopes residing in the neo-peptide are only present in tumor cells 3) Neopeptides originate from proteins expressed in the tumor cell and not normal cells. The latter criteria reduce the risk of autoimmune reactions in patients and hence is an important safety measure. All computational analyses were run on the Danish National Supercomputer for Life Sciences (Computerome) hosted at the Technical University of Denmark.
Manufacturing of personalized neoantigen vaccine
Five of the PIONEERTM predicted neopeptides where deselected due to the low likelihood of successful synthesis before manufacturing, based on their sequence-derived biophysical compositions. All peptides (intermediates) were manufactured by Pepscan using an endotoxin-free process, according to cGMP where applicable, relying on Fmoc-based solid state synthesis on a SYMPHONY® X (Gyros Protein Technologies, US). Purification was performed using a linear gradient with a C18 reverse phase preparatory HPLC followed by lyophilization to remove trace volatiles resulting in TFA-salt peptides. Quality control was performed on all manufactured single peptides evaluating residual ACN, TFA content, identity (Mw, Retention), purity (>95%) and solubility at 0.1 and 1.0 mg/mL in aqueous buffer using a turbidimetry assay (Ph. Eur. 2.2.1). The drug substance (NPV_DS001) was prepared from successfully manufactured peptides that passed all QC requirements, by dissolving individual peptides at 20 mg/mL in 100% DSMO, with subsequent equivolumetric pooling to a final concentration of 2 mg/mL pr. peptide. The resulting NPV_DS001 was controlled for appearance, identity, purity, concentration, and solubility of the single peptides followed by QC/QA release. The final investigational drug product (NPV_DP001) for vaccine formulation was prepared by manual 0.2 filtration and filling of bulk NPV_DS001 with following endotoxin and sterility testing before final release for human dosing. Stability studies where performed “live” for all manufacturing runs with monthly pull-points until T = 6 months at −20°C and 5–8°C respectively.
To support the rapid vaccine manufacturing, a Formulation Standard Operating Procedure (SOP) was developed by SSI, Evaxion and CCIT to ensure a replicable vaccine formulation method suitable for use in the clinical setting. The Formulation SOP was tested in feasibility studies covering the varying physicochemical properties e.g. solubility and charge of the predicted peptides in the formulation matrix. Different vaccine formulation matrices and peptide:adjuvant ratios were tested, and a vaccine formulation was identified that ensured a physically stable vaccine independent of specific peptide properties.
CAF®09b supply and final vaccine formulation
Each EVX-01 vaccine comprised 5–10 synthetically manufactured peptides formulated with the CAF®09b adjuvant. Reconstitution of each EVX-01 vaccine with CAF®09b was performed at CCIT immediately before administration to the patients as follows; a total of 1.08 mL sterile filtered Tris reconstitution buffer was added to an ampule with 0.12 mL sterile filtered NPV-dp001 and mixed thoroughly. Subsequently, 1 mL of this peptide solution was added to a 2 R vial containing 1.0 mL CAF®09b 2500/500/125 (batch 728301). After thoroughly mixing, the final vaccine could be drawn into a syringe.
Initial individual peptide dose was chosen based on experience from a similar peptide-based therapeutic prostate cancer vaccine, Bcl-XL_42-CAF®09b (EudraCT No.: 2015–003719-39) (manuscript in preparation) and supportive tox with this vaccine (manuscript in preparation) Both CAF®09b alone as well as the Bcl-Xl_42 CAF®09b vaccine was investigated in a repeated dose toxicology study in rabbits, and the vaccine was evaluated to be safe for clinical trial. The immunogenicity profile has been described in different publications since 2014.27,31,32
Treatment
Included patients were treated with an anti-PD-1 agent according to local guidelines consisting of i.v. infusions of the approved dose of anti-PD-1 q3w or q4w. When ready, the personalized EVX-01 vaccine was added to the treatment schedule. The patients received EVX-01 vaccination every two weeks for a total of 6 treatments. The first three vaccines were administered by i.p injection, and the last three vaccines were administered by i.m. injections (Figure 1a). At the lowest dose level, from which this interim analysis reports, the vaccine consisted of 5–10 peptides (in total 500 μg/dose) derived from personalized neoantigens mixed with the adjuvant, CAF®09b (625 μg DDA/dose, 125 μg MMG/dose, and 31 μg poly I:C/dose).
Peripheral blood mononuclear cells
Peripheral blood samples were collected at different time points: Prior to anti-PD-1 therapy (T1), prior to first vaccination (T2), after three (T3) and six vaccinations (T4) and during follow-up (T5). After a maximum of 3 hours PBMCs were separated with gradient-centrifugation using Lymphoprep (Takeda) density gradient and cryopreserved in 90% human AB serum (Sigma.Aldrich, Ref. No H4522-100 ml) and 10% DMSO using controlled-rate freezing (Cool-Cell, Biocision) in −80°C. The following day they were moved to −140°C freezer until used for analysis.
Skin-test infiltrating lymphocytes
A voluntary delayed-type hypersensitivity (DTH) skin test was performed after six vaccinations with two intradermal injections of the EVX-01 peptides and one injection with saline solution as a negative control. After 48 hours, the maximum diameter of induration was measured, and 5mm punch biopsies were taken from each injection site. After collection, the fresh tissue was directly transported to the laboratory for skin test-infiltrating lymphocytes (SKILs) culture. In short, minimally expanded SKILs were expanded from tissue fragments for 3 to 6 weeks. Tissue biopsies were chopped into 1–3 mm3 fragments and placed in separate wells of a 24 well-culture plate (Thermo Fisher Scientific) containing 2 ml of medium consisting of 90% RPMI-1640 plus GlutaMAX and 25 mM HEPES (Thermo Fisher), 10% heat inactivated AB Human serum (HS; Sigma-Aldrich), 100 U/ml penicillin and 100 µg/ml streptomycin (Pen Strep, Thermo Fisher Scientific), 1.25 μg/ml Amphotericin B (Fungizone, Bristol-Myers Squibb) and 100 IU/ml rhIL-2 (Proleukin, Novartis). The plates were incubated at 37°C with 5% CO2, and half of the medium was replaced at day 5 and subsequently 3 times weekly. After 3 to 6 weeks pooled SKILs were cryopreserved.
PBMCs prestimulation
PBMCs were prestimulated with the EVX-01 peptides in addition to IL-2 (120 U/ml), IL-15 (10 ng/ml), and IL-21 (50 ng/ml) (Preprotech) for ten to 13 days before screening for peptide recognition of T-cells using IFNγ ELISPOT assay or intracellular cytokine staining. The CEFT pool (Pepscan) was used as a positive control and consists of peptides from Cytomegalovirus, Epstein-Barr virus, Influenza virus and Clostridium tetani. After pre-stimulation, cells were rested overnight in X–Vivo 15 medium (Lonza) without peptides or interleukins.
T-cell activation assay by IFNγ ELISPOT
Screening for peptide recognition of T-cells was carried out with IFNγ ELISPOT assays following BD manufacture protocol. Per well, 300,000 EVX-01 prestimulated PBMCs were added; or 100,000 SKILs with 10,000 autologous monocytes. Monocytes were isolated from PBMCs with positive enrichment using CD14 microbeads (Miltenyi Biotec) according to manufacturer’s instruction. The EVX-01 single peptides and/or EVX-01 peptide pool were added with a final concentration of 0.5 µg/mL alongside a positive control (PHA; 1.5–3 µg/mL) and an irrelevant peptide comprising multiple HLA epitopes (EV09; GenScript; 0.5–1.5 µg/mL) as negative control) with sequence: GDVKIHAHKVVLANISPYFKAMFTGNL. The experiments were run in duplicates (PBMCs) or triplicates (SKILs) and analyzed after overnight incubation. The spots were counted using the AID multiSpot Reader (Autoimmun Diagnostika GmbH; PBMCs) or the ImmunoSpot Series 2.0 Analyzer (CTL Analyzer; Bonn, Germany; SKILs). A positive ELISPOT response was defined when the number of spots for tested peptides exceeded the background spot number plus 3 times the standard deviation of the background (irrelevant peptide) and at least 10 spots over the background. This definition was set based on previous recommendations33 and procedures used.17 Due to limitations of available PBMCs and a technical failure during initial analyses, no valid ELISPOT data is available for patient 4.
T-cell activation assays by flow cytometry
PBMCs
After prestimulation, PBMCs were rested only in X–Vivo 15 medium without peptides or interleukins overnight. Either the vaccine pool peptides or CEFT pool peptides (dependent on pre-stimulation) or irrelevant peptide were added to the cells. Cells were then incubated in a 37°C incubator for 2 hours. Anti-human CD107a antibody was added into the medium together with the peptides. After 2 hours, Brefeldin A and Monensin (GolgiPlug™ and GolgiStop™ respectively; dilution of 1:1000, BD Biosciences) were added to the cells and they were incubated for another 6 hours. PBMCs from a healthy donor were incubated with Leukocyte activation cocktail (BD Bioscience) for 8 hours as the assay positive control. After 8 hours incubation time in total, cells were washed and stained for Live/Dead Fixable Dead Cell Stain Near-IR (Thermo Fisher) and antibodies for surface markers (Supplementary Table S1). Cells were then fixed and permeabilized using Foxp3/Transcription Factor Staining Buffer Set (Invitrogen). Then they were intracellularly stained for TNF and IFNγ. Cells were analyzed using LSRFortessa (BD) or a NovoCyte Quanteon™ Flow Cytometer (Agilent). PBMCs from patients 1, 2, 3 and 4 were stained with Panel 1 while PBMCs from patient 5 were stained with Panel 2 (Supplementary Table S1). Flow cytometry data were analyzed with FlowJo version 10 (Becton Dickinson). T-cell reactivity was defined as the percentage of live CD8+ or CD4+ T-cells staining positive for at least two of three markers (TNF, IFNγ, CD107a). Effector cells with irrelevant peptide served as an unstimulated control. A specific response was defined as the detection of a response greater than twice the unstimulated control and a minimum of 50 positive flow cytometry events after subtraction of the background.
SKILs
SKILs were thawed and rested overnight prior to the initiation of the assays. EVX-01-specific T-cell activation was assessed with 8-hour co-culture at 37°C of SKILs and peptides in the presence of autologous monocytes. Monocytes were isolated from PBMCs with positive enrichment using CD14 microbeads according to manufacturer’s instruction. The SKIL to monocyte ratio was 10:1. The single peptides and peptide pool were added with a final concentration of 0.5 µg/mL alongside a positive control (PMA; 25 ng/mL + Ionomycin; 0.5 µM) and a negative control (irrelevant peptide 1.5 µg/mL). Anti-human CD107a antibody, Brefeldin A (dilution of 1:1000, GolgiPlug™) and Monensin (dilution of 1:1000, GolgiStop™) were added after 2 hours of co-culture. After a total of 8 hours of incubation, the cells were washed twice with DPBS (Sigma-Aldrich/Merck KGaA) and stained with live/dead reagent and antibodies for surface markers (Panel 2, Supplementary Table S1). The cells were then washed, fixed, and permeabilized overnight at 4°C using the FoxP3/Transcription Factor Staining Buffer Set (eBiosciences, Thermo Fisher Scientific). The cells were subsequently stained with antibodies binding intracellular targets (Panel 2, Supplementary Table S1). After staining and washing, cells were analyzed on a NovoCyte Quanteon™ Flow Cytometer. Flow cytometry data were analyzed with FlowJo version 10 (Becton Dickinson). T-cell reactivity was defined as the percentage of live CD8+ or CD4+ T-cells staining positive for at least two of four markers (TNF, IFNγ, CD107a, CD137) minus the background (unstimulated control). SKILs with monocytes and irrelevant peptide served as an unstimulated control while SKILs in the presence of PMA/Ionomycin were used as a positive control. A specific response was defined as the detection of a response greater than twice the background and a minimum of 50 positive flow cytometry events after subtraction of the background.
Details on the antibodies for flow cytometry used in the study can be found in the Supplementary Table S1.
Results
Patients and demographics
Eight patients with advanced stage of melanoma were enrolled between January and September 2019. Of these, five patients were treated at the first dose level and deemed evaluable.
Two patients were excluded from the trial before receiving EVX-01-treatment: One patient had rapid progressive disease before the vaccine production was finished and switched to second-line treatment. Another patient did not meet inclusion criteria for measurable lesions at reevaluation of the baseline scan. A third patient was excluded retrospectively due to a technical issue during DNA sequencing. No vaccine-related adverse events were observed in this patient.
Of the evaluable patients, four patients were candidates to begin standard treatment with CPI as monotherapy and one patient (patient 2) had stable disease during standard treatment with CPI (9 cycles) for at least 4 months (Figure 1c). The patient demographics and baseline disease characteristics are listed in Table 1.
Table 1.
Patient overview
| Patient | Age | Sex | AJCCM-stage at entry | Site of metastatic disease | LDH at baseline | Biomarkers | CPI(dosage/number of treatments before vaccination) | Vaccinenumber of IP | Vaccinenumberof IM | BOR(PR, CR, PD)/days after first vaccination |
|---|---|---|---|---|---|---|---|---|---|---|
| 1 | 81 | M | M1b | IntramuscularPleuraLungLymp nodes | 259 | PD-L1>1%<50% BRAF mut |
Pembrolizumab(175 mg/3) | 3 | 3 | PR35 |
| 2 | 49 | F | M1c | SubcutaneousUterus | 147 | PD-L1 > 1% BRAF mut |
Pembrolizumab(125 mg/11) | 1 | 5 | CR78 |
| 3 | 61 | M | M1c | Lymph nodesGall bladder | 118 | PD-L1 < 1% BRAF WT |
Nivolumab(480 mg/2) | 3 | 3 | PR24 |
| 4 | 84 | F | M1a | Lymph nodesSubcutaneous | 184 | PD-L1 5% BRAF mut |
Pembrolizumab(100 mg/3) | 3 | 3 | PD24 |
| 5 | 79 | F | M1b | LungLymph nodes | 835 | PD-L1 < 1% BRAF WT |
Pembrolizumab(175 mg/3) | 3 | 1 | PD24 |
Feasibility of EVX-01 manufacturing and vaccination
All five patient-specific vaccine productions were completed within a 48–55-day time frame. Details for each manufacturing run are compiled in Table 2. The vaccines were composed of 5 to 10 patient-specific neopeptides at a dose level of 50–100 ug pr. peptide.
Table 2.
Peptide overview
| Trial IDPatient | Tumor CDS Coverage | Normal CDS Coverage | mRNA reads | Somatic mutations (no) | Manufactured peptides (no) | Included peptides (no) | Peptide dose (ug) | Turn-around (days) |
|---|---|---|---|---|---|---|---|---|
| 1 | 308 X | 119 X | 79.3 M | 12179 | 5 | 5 | 50 ug for 1 peptides (CA063)100 ug for 4 peptides (CA064,70–73) | 50 |
| 2 | 111 X | 44 X | 94.1 M | 1360 | 12 | 10 | 50 ug for 10 peptides | 48 |
| 3 | 217 X | 84 X | 129.4 M | 5689 | 10 | 8 | 50 ug for 7 peptides100 ug for 1 peptides (CA462) | 48 |
| 4 | 256 X | 105 X | 102.3 M | 933 | 14 | 10 | 50 ug for 10 peptides | 55 |
| 5 | 262 X | 124 X | 149.3 M | 339 | 8 | 8 | 50 ug for 7 peptides100 ug for 1 peptides (CA581) | 55 |
The duration from baseline biopsy to the first i.p. vaccination was from 51 to 60 days; administration of the vaccine was performed approximately 3–5 days after completed production due to combined administration with the anti-PD-1 agent. Four patients received all six vaccines. Patient 2 received only one vaccine i.p. and the remaining five vaccinations were given i.m. due to anxiety toward receiving i.p. injections causing a delay of 2.5 weeks between the first and second vaccination. Patient 5 was discontinued from the trial after receiving four vaccinations due to disease progression (Figure 1c).
Safety, tolerability, and clinical monitoring
All patients began CPI at least 6 weeks before the first vaccination. All treatment-related AEs are presented in Table 3. All AEs were grade 1 except for one patient experiencing grade 2 fatigue. The most frequent events were fatigue, pain at the injection site, and stomachache. In summary, the treatment was very well tolerated. No treatment-related serious AE (SAE) was observed.
Table 3.
Toxicity
| Trial IDPatient | Vaccine 1 | Vaccine 2 | Vaccine 3 | Vaccine 4 | Vaccine 5 | Vaccine 6 | follow-up |
|---|---|---|---|---|---|---|---|
| 1 | - Pain injection site G1 | - Rash G1 | - Pain injection site G1-Pain at jaw G1 | - Oral mucositis G1- Dry mouth G1- Productive cough G1 | - Pain injection site G1- Stomach pain G1- Groin pain G1 | - Pain injection site G1 | -None |
| 2 | - Stomach pain G1-Fatigue G2 | - Pain injection site G1- Stomach pain G1 | - Fatigue G1 | None | - Fatigue G1- Pain injection site G1- Dry skin G1- Mucitis G1 | - Fatigue G1- Mucositis G1- Dry skin G1 | - Fatigue G1 |
| 3 | - Pain injection site G1- Fever G1 | - Pain injection site G1- Fatigue G1- Cough G1- Fever G1 | - Pain injection side G1 | - Pain injection site G1 | -None | - Papular eczema G1 | - |
| 4 | - Hematoma G1- Edema under eye G1 | - Stomach pain G1 | - None | - None | - None | - None | - None |
| 5 | -None | - None | - None | - None | - | - | - |
The best overall tumor response (BOR) comprised one patient (patient 2) with complete response (CR) and two patients, patient 1 and 3, achieving a partial response (PR) with 54% and 77% tumor regression, respectively. Furthermore, the only sign of disease in patient 3 was a 11 mm lymph node lesion. At the time of inclusion, patient 2 had stable disease after nine pembrolizumab administrations. However, the scan just before the first treatment with EVX-01 showed PR with 44% size decrease in target lesions and the following scan after three vaccinations showed CR.
Patients 4 and 5 had progressive disease (PD) after two vaccinations. To rule out pseudo-progression, treatment was continued but PD was confirmed at the next scan (after four and two additional vaccines, respectively).
EVX-01-specific T-cells responses in PBMCs after vaccination
The ability of EVX-01 immunotherapy to induce EVX-01-specific T-cell responses was evaluated by both IFNγ ELISpot and T-cell activation assays by flow-cytometry ICS in PBMCs after prestimulation with the EVX-01 peptides.
The five evaluated patients were vaccinated with 5–10 neopeptides, hence a total of 31 neopeptides were included in EVX-01 across this patient group. T-cell reactivity to the vaccine was evaluated at baseline (T1), after CPI initiation (T2), after administration of vaccine EVX-01 (T3 = 3 vaccine doses), (T4 = 6 vaccine doses) and at follow-up (TP5). Before administration of EVX-01 (timepoint 1 and 2), minor T-cell responses were observed toward the EVX-01 included peptides (peptide 4 in patient 1 and peptide 7 in patient 3). Post vaccination (timepoint 3 and 4), T-cell responses were observed in all patients, ranging from 3 to 7 peptide-specific T-cell responses induced in each patient (Figure 2). In total, T-cell response was detected toward 23 of the 31 (74%) neopeptides included. The majority of the T-cell responses were de novo T-cell responses, not observed prior to EVX-01 administration. De novo responses were detected in all patients. The level of these T-cells responses ranged in intensity, with patient 5 showing a strong T-cell induction, while T-cell responses in patient 3 were substantially lower. The EVX-01 induced T-cell responses, were for most of the patients detectable throughout T3 and T4. Long-term follow-up samples (4 to 16 months post EVX-01 treatment initiation) displayed persistence in T-cell responses to vaccine-peptide in the 3 patients evaluated. For patient 1 we detected a specific T-cell response in 4 out of 5 peptides (80%), which was detectable at follow-up at 14 months. Peptide-specific T-cell responses in patient 2 were seen for 3 peptides (peptide 2, 5 and 6) at TP3 (30%) and 7 peptides (peptide 2, 3, 4, 5, 6, 8, and 10) at TP4 (70%). At follow-up 10 months after the last vaccination 7 peptides (peptide 1, 2, 4, 6, 7, 8, 10) still showed positive responses (70%).
Figure 2.
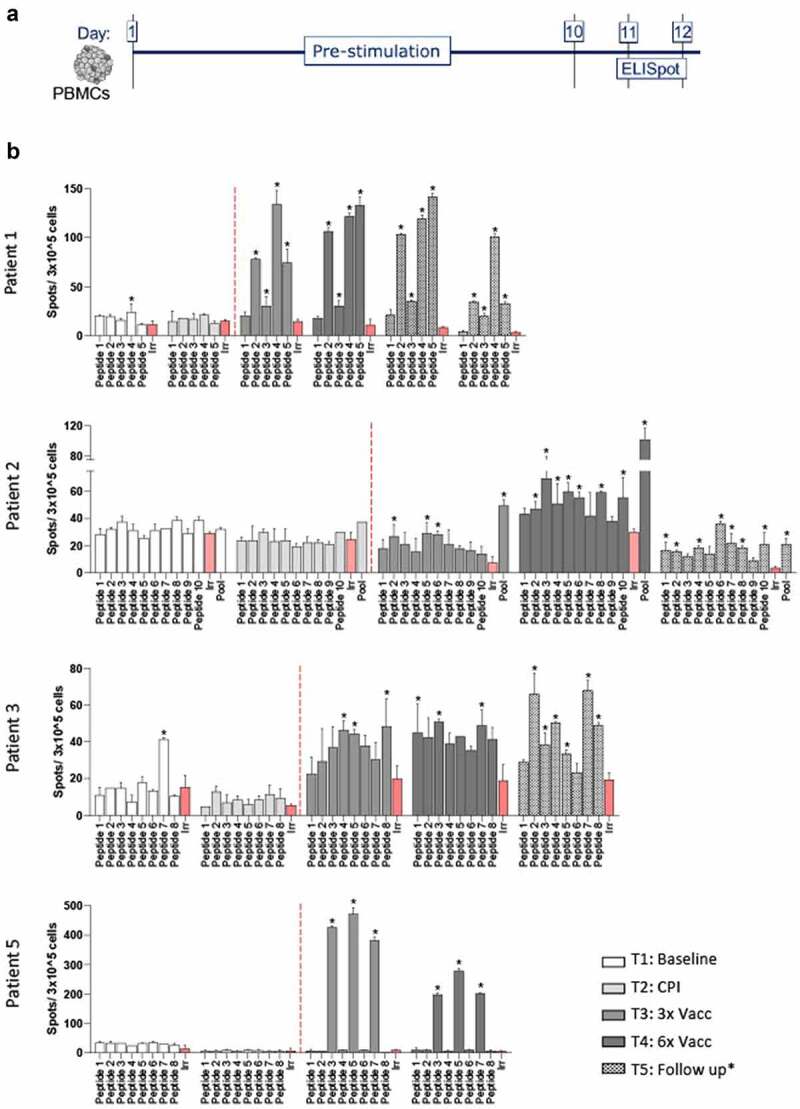
EVX-01-specific T-cell responses shown by Elispot on PBMCs.(A) PBMCs were prestimulated with the EVX-01 peptides in addition to IL-2 for ten to thirteen days before screening for peptide recognition of T-cells using IFNγ ELISPOT assay. After 10 days EVX-01 single peptides and/or EVX-01 peptide pool were added to prestimulated PBMCs. A positive ELISPOT response was defined when the number of spots for tested peptides exceeded the background spot number plus 3 times the standard deviation of the background (irrelevant peptide) and at least 10 spots over background (*).(B) Peptide specific T-cell response was determined for patient 1, 2, 3 and 5. Patient 4 was left out due to technical consideration and insufficient PBMC availability. The red columns represent irrelevant peptides. Highlighted columns represent positive ELISPOT response. For each patient we observed specific vaccine induced T-cell response after both 3 and 6 vaccinations. Patient 1, 2 and 3 continued to show T-cell activation at follow-up (14, 10 and 1,5 months after the last vaccination respectively).
One peptide response in patient 3 was seen at baseline but not at TP2 (CPI alone). After the first 3 vaccinations 3 peptides (peptide 4, 5 and 8) out of 8 (37,5%) showed a positive response and after 6 vaccinations 3 different peptides (peptide 1, 3 and 7) showed a positive response. At follow-up (1,5 months after last vaccination) 6 peptides (peptide 2, 3, 4, 5, 7 and 8) still showed a response (75%).
In patient 5 we found response against 3 peptides (peptide 3, 5 and 7) with a positive response at TP3 and TP4. Importantly, we further evaluated whether EVX-01-specific T-cell responses were dominated by CD4+ or CD8+ T-cell reactivity. Here, prestimulated PBMCs from all five patients were analyzed for reactivity against the EVX-01 peptide pool in a T-cell activation assay by flow cytometry. Neoantigen-specific CD4+ T-cells were detected in all patients throughout T3 and T4. In patient 5, both CD4+ and CD8+ neoantigen-specific T-cells were detected after co-culture with EVX-01 full length 27 mer peptides, although the CD4+ T-cell response was more pronounced (Figure 3, Supplementary Figure S2 and S3). Of relevance, EVX-01 seemed to induce stable CD4+ responses over time, as shown for the 3 evaluated patients (1, 2 and 3) at multiple follow-up timepoints (1,5 to 14 months from the last vaccination) after completion of the EVX-01 treatment (Supplementary Figure S2).
Figure 3.
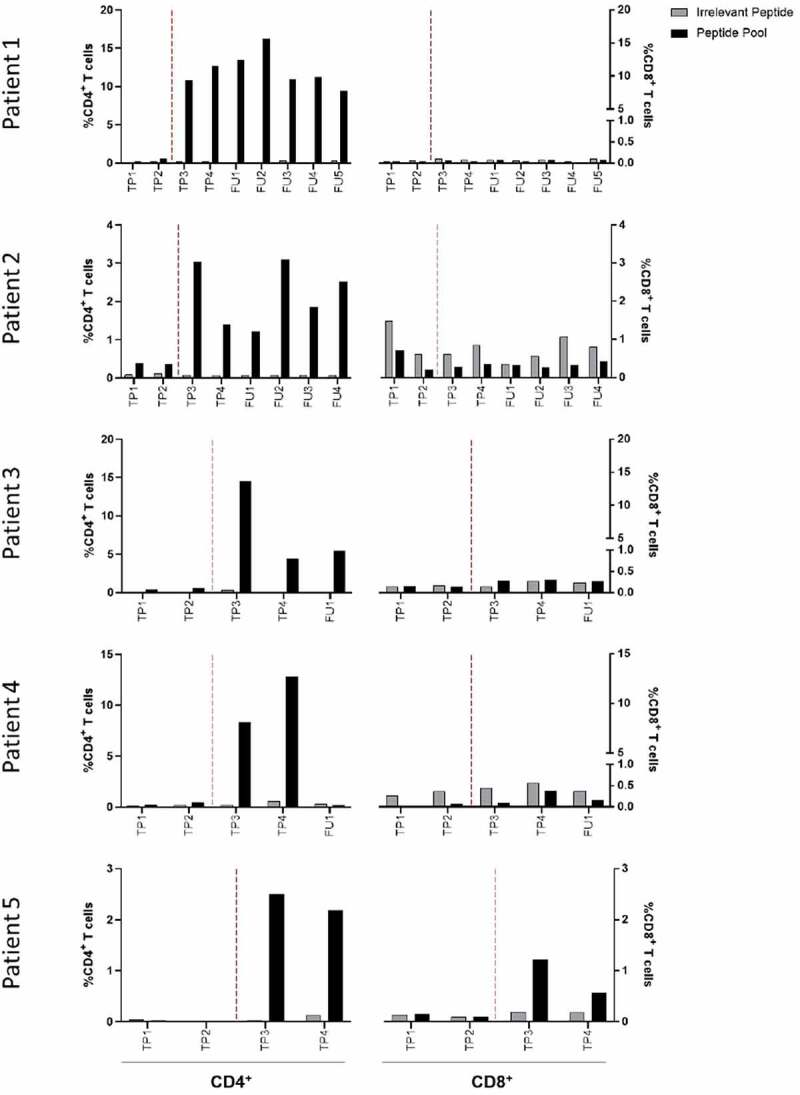
EVX-01 induced mainly CD4+ T-cell responses in PBMCs after vaccination. EVX-01-specific CD4+ T-cells were identified in all five patients at multiple timepoints after vaccination. EVX-01-specific CD8+ T-cells were identified in patient 5 at time point 3 (TP3). Prestimulated PBMCs were restimulated with EVX-01 peptide pool or irrelevant peptide (negative control) for 8 hours and subsequently analyzed by flow cytometry. T-cell reactivity was defined as the percentage of live CD8+ or CD4+ T-cells staining positive for at least two of three markers (TNF, IFNγ, CD107a). TP1 = Baseline; TP2 = CPI; TP3 = 3x vaccination; TP4 = 6x vaccination; FU = follow-up. Vertical hatched line separates timepoints before and after vaccinations.
EVX-01-specific T-cells home into solid tissue
To test whether the EVX-01-specific T-cells as detected in the peripheral blood could migrate toward the neoantigens, a DTH skin tests was performed in patient 2. The SKILs grown from the skin biopsies showed a robust immune response against the EVX-01 peptide pool without prior prestimulation (Figure 4a and 4b). A clear immune response against peptide 3, 5, 6, and 10 could be detected in the SKILs. The T-cell activation assay showed that the response observed in the SKILs was dominated by CD4+ T-cell reactivity; no EVX-01-specific CD8+ T-cell response could be detected in the SKILs (Figure 4c).
Figure 4.
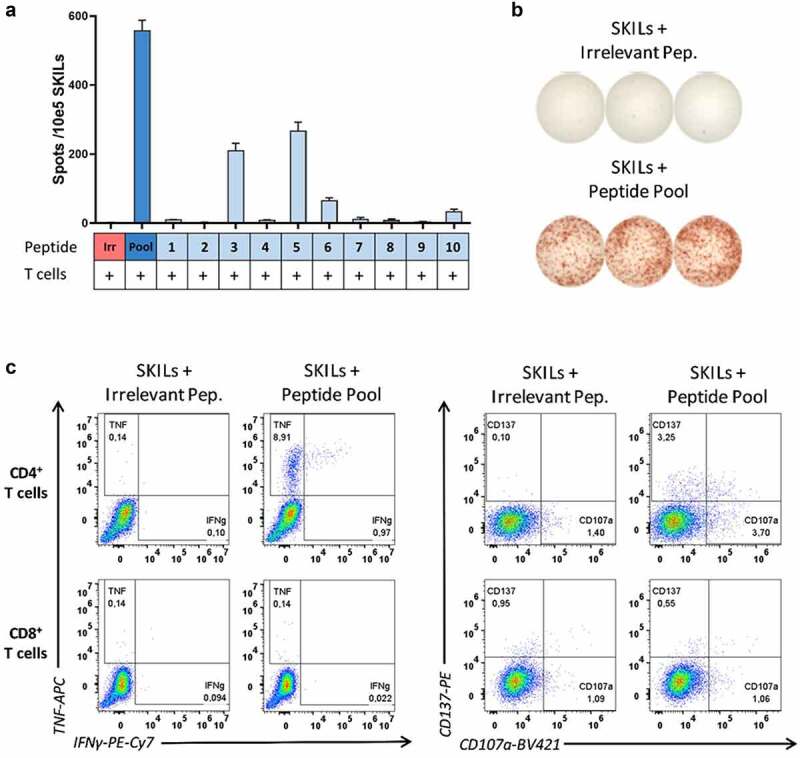
EVX-01-specific T-cell responses were detected in SKILs (skin-test infiltrating lymphocytes) after vaccination of patient 2. (A) IFNγ-ELISPOT responses were detected in SKILs isolated from patient 2 (TP4, after 6 vaccinations) after overnight co-culture with EVX-01 peptide pool and individual peptides. 100,000 SKILs with 10,000 autologous monocytes were seeded per well. (B) Representative example of ELISPOT-wells. (C) EVX-01-specific CD4+ T-cells were identified in the SKILs isolated from patient 2 (TP4, after 6 vaccinations). SKILs were restimulated with EVX-01 peptide pool or irrelevant peptide (negative control) for 8 hours. T-cell reactivity was defined as the percentage of live CD8+ or CD4+ T-cells staining positive for at least two of four markers (CD137, TNF, IFNγ, CD107a).
Discussion
Five patients with metastatic melanoma were treated with CPI combined with a personalized neopeptide vaccine, EVX-01, and the novel adjuvant CAF®09b. The EVX-01 vaccine was developed based on the identification of neoantigens by PIONEERTM. We found that the vaccine-production was feasible and fast, and the treatment was safe, with only few non-severe side effects observed. Personalized EVX-01 treatment induced de novo EVX-01-specific T-cell responses, and durable clinical responses were observed at the lowest dose level reported here. Higher dose levels are currently being investigated and will be reported later.
In this preplanned interim analysis, we showed that it was possible to design and prepare a personal vaccine in less than 8 weeks from biopsy to final product (48–55 days). Manufacturing speed is particularly relevant in the metastatic setting to avoid unnecessary treatment delay. Compared to other similar neopeptide vaccines tested in humans, our production time is currently one of the fastest reported.14,16,17,19,34 To minimize vaccine production time a tight working chain was established covering the whole production course from tumor biopsy to vaccine administration. All potential points of delay were identified and addressed in advance. As it was not feasible to setup a dedicated manufacturing site covering everything from DNA/mRNA sequencing to fill/finish several partners was needed for manufacturing of each individual patient-specific batch. Hence, material and information had to be transferred from one party to the other causing natural delays and added risk of mistakes. In the future, having one single site capable of performing both sequencing, peptide synthesis, purification and fill/finish will significantly reduce manufacturing timelines as well as reduce risk of delays. In the presented study the patients received standard treatment with CPI during the manufacturing time as not to “wait” for treatment. Still, the practical aspects of personalized vaccine manufacturing require ongoing optimization to shorten the time from biopsy to treatment to be beneficial to more patients in the future.
These early data indicate that EVX-01+ CAF®09b combined with CPI in patients with disseminated melanoma is safe, as no SAEs were reported for any of the included patients. However, given the small sample size safety remains to be further investigated in a larger cohort and at higher dose levels. If the combination indeed proves to add only mild side effects compared to anti-PD-(L)1 treatment alone, it would be an important advantage over the known high risk of severe toxicity (>50% grade 3–4 AEs) for the ipilimumab and nivolumab combination.35
Despite strong de novo EVX-01-specific T-cell responses patients, patient 2 showed weak reactivity (Figure 2), with an increase in response at timepoint 3 and 4. Furthermore, in this patient we were able to evaluate T-cell reactivity in SKILs which showed a clear response toward peptide 3, 5, 6, and 10.
We aimed for an optimal strategy to boost neoantigen-specific CD4+ and CD8+ T-cell responses by use of the novel CAF®09b adjuvant with initial i.p. administration followed by i.m. to induce a balanced CD4+ and CD8+ T-cell responses.26,27,28 Indeed, at the lowest dose level reported here, we observed EVX-01-specific CD4+ T-cell responses in all five patients, and a CD8+ T-cell response in one of the patients after three i.p. administrations. However, the CD4+ and CD8+ T-cell immune responses observed do not yet allow a firm conclusion whether the i.p. route boosts vaccine immunity more effectively than the i.m. vaccinations. CCIT-DK is currently investigating the different administrations route and T-cell responses to CAF®09b-peptide-vaccine in patient with prostate cancer (NCT03412786).
The dominant observation of CD4+ T-cell responses is aligned with observations from other neoantigen vaccine studies.14,17,19,23 However, a number of factors could influence the observed CD4+ dominance; i) an assay bias toward CD4+ stimulation related to the use of full-length peptides for prestimulation, ii) the dominating CD4+ T-cell response may be a true reflection of the immune responses induced by EVX-01 at the lowest dose level reported here – higher dose levels might change the CD4+/CD8 + T-cell balance toward CD8 + T-cells as observed in preclinical models36 or iii) prediction models could be optimized for better CD8+ antigen prediction. For EVX-01 long peptides of 15–27 amino acids were selected as they might be better at overcoming immune tolerance than short peptides,37 and it has been suggested that long peptides may induce both CD4+ and CD8+ T-cell responses,37 but shorter peptides might be better at inducing CD8+ T-cell responses.10 Long peptides also have the advantages that they are processed in APCs and therefore less likely to be degraded before reaching the APC and probably more immunogenic compared to short peptides.38,39 Furthermore, it is well known that CD8+ T-cell responses are best obtained by restimulation with minimal epitopes, and that full-length peptides and proteins better detect CD4+ T-cell responses. Work is therefore in progress to optimize the pre- and re-stimulation procedures to better accommodate CD4+ as well as CD8+ T-cell analysis. follow-up studies will investigate this subject in more detail.
Out of the five patients, three patients showed an objective response (2 PR, 1 CR). As all the patients received both EVX-01 immunotherapy and anti-PD-1 treatment we are unable to tell if the added EVX-01 vaccination was responsible for the responses, especially since patient 2 already showed regression before start of EVX-01. Similarly, Ott et al.17 observed radiographic responses in melanoma patients treated with both anti-PD-1 and their neoantigen vaccine (NEO-PV-01), but they also conclude that the same responses could possibly be seen with anti-PD-1 as monotherapy. Comparative studies in mice have shown that combined treatment with a tumor vaccine and CPI is more effective than monotherapy.40,41 However, these vaccines did not consist of neopeptides. The inclusion of more patients with stable disease before EVX-01 treatment would probably help us appraise the effectiveness of the neopeptide vaccinations. After vaccine optimization, a larger trial is needed to investigate superiority over CPI alone.
Some challenging aspects of neoantigen-targeting vaccines comprise the optimal identification of immunogenic neoepitopes and the intrinsic personalized nature of the vaccine. Thus, development of accurate epitope predicting algorithms and efficient validation tools are important for personalized neoantigen-based cancer immunotherapy. To this end, it still remains a challenge to effectively target multiple clonal neoantigens to create a broad and potent T-cell response toward the tumor.
In conclusion, personalized immunotherapy with neoantigens is a promising approach in cancer treatment, and precise identification of immunogenic neoantigens are required for effective neoantigen-based cancer immunotherapy.
Here, we demonstrate that EVX-01, a personal neoantigen vaccine, is safe, feasible, and capable of eliciting T-cell responses in a clinical setting where the patients received concurrent standard immune therapy. Our pipeline with improved technologies allows for fast identification of immunogenic neoantigens and rapid manufacturing of the peptide vaccine, which is required to make neoantigen-based cancer immunotherapy applicable in larger cohorts. Additional vaccine dose levels have been added to the trial and patient enrollment is presently ongoing. If a favorable safety profile is confirmed at the higher dose levels, the objective responses warrants further investigation of the efficacy of EVX-01.
Supplementary Material
Acknowledgments
We thank all the patients and their families who participated in the study. We are grateful for the staff at Kennedy center (Genomic Center) for support and assistance. We thank Torben Lorentzen (MD) and the staff at Gastroenheden Herlev for performing needle biopsies; Nis Nørgaard (MD) and the staff at the Department of Urology at Herlev Hospital for performing ultrasound-guided intraperitoneal vaccinations; Christian Skjødt Steenmans for his contributions to developing and GAMP5 validating the PIONEER system; the PIONEER operations team for designing the personalized neoantigen vaccines; the staff at Evaxion for assay development support and data interpretation; the laboratory technicians for invaluable help with the vaccine preparation; the staff at SSI for data analysis, data interpretation and writing of the report; the Department of Oncology, Herlev Hospital, physicians and nurses for their support in taking care of the patients.
Funding Statement
These works were supported by research grants by Innovation Fond Denmark, and the Danish Cancer Society (Knæk Cancer).
Disclosure statement
Marco Donia has received honoraria for lectures from Roche and Novartis (past 2 years). Inge Marie Svane has received honoraria for consultancies and lectures from Novartis, Roche, MSD, Pierre Fabre, and BMS. CCIT-DK has received economic support for trial personal wages from Evaxion Biotech A/S, Denmark. SRH is the cofounder of Tetramer-shop and PokeACell and is the co-inventor of a number of licensed patents. ABS, TT, CG, JFN and JVK are employees of Evaxion Biotech A/S and have financial interest in the company. All other authors declare that they have no conflict of interest.
Translational Relevance
Several studies have demonstrated the potentials of neoantigen-based personalized vaccines in different types of cancers. To further investigate the predictive quality of the AI platform PIONEERTM for the identification of immune stimulatory neoantigens, we conducted a clinical study evaluating a PIONEER-guided neoantigen vaccine (EVX-01) administered concurrently with standard checkpoint inhibitors to patients with advanced melanoma. Overall, EVX-01 was safe and well tolerated. EVX-01 elicited T-cell mediated immune responses targeting tumor neoantigens with encouraging indications of immunogenicity and clinical relevance.
Supplementary material
Supplemental data for this article can be accessed on the publisher’s website
References
- 1.Robert C A decade of immune-checkpoint inhibitors in cancer therapy. Nature Communications [Internet]. Springer US; 2020;11:10–15 10.1038/s41467-020-17670-y. Available from: 10.1038/s41467-020-17670-y 1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Snyder A, Makarov V, Merghoub T, Yuan J, Zaretsky JM, Desrichard A, Walsh LA, Postow MA, Wong P, Ho TS, et al. Genetic Basis for Clinical Response to CTLA-4 Blockade in Melanoma. N Engl J Med. 2014;371(23):2189–2199. doi: 10.1056/NEJMoa1406498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Park YJ, Kuen DS, Chung Y. Future prospects of immune checkpoint blockade in cancer: from response prediction to overcoming resistance. Exp Mol Med [Internet]. Springer US; 2018;50:1–13. Available from: 10.1038/s12276-018-0130-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Schumacher TN, Hacohen N.. Neoantigens encoded in the cancer genome. Curr Opin Immunol. 2016;41:98–103. [Internet]. Elsevier Ltd; 2016;:. Available from. doi: 10.1016/j.coi.2016.07.005. [DOI] [PubMed] [Google Scholar]
- 5.Luksza M, Riaz N, Makarov V, Balachandran VP, Hellmann MD, Solovyov A, Rizvi NA, Merghoub T, Levine AJ, Chan TA, et al. A neoantigen fitness model predicts tumor response to checkpoint blockade immunotherapy. Nature. 2017;551(7738):517–520. doi: 10.1038/nature24473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Mc Granahan N, Furness AJS, Rosenthal R, Lyngaa R, Saini SK, Jamal-Hanjani M, Jamal-Hanjani M, Wilson GA, Birkbak NJ, Hiley CT, et al. Clonal neoantigens elict T cell immunoreactivity and sensitivity to immune checkpoint blockade. Science. 2016;351(6280):1463–1469. doi: 10.1126/science.aaf1490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Alexandrov LB, Nik-Zainal S, Wedge DC, Aparicio SAJR, Behjati S, V. BA, Bignell GR, Bolli N, Borg A, Børresen-Dale A-L, et al. Signatures of mutational processes in human cancer. Nature. 2013;500(7463):415–421. doi: 10.1038/nature12477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Wells DK, van Buuren MM, Dang KK, Hubbard-Lucey VM, Sheehan KCF, Campbell KM, Lamb A, Ward JP, Sidney J, Blazquez AB, et al. Key Parameters of Tumor Epitope Immunogenicity Revealed Through a Consortium Approach Improve Neoantigen Prediction. Cell. 2020;183(3):818–834.e13. doi: 10.1016/j.cell.2020.09.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Jou J, Harrington KJ, Zocca MB, Ehrnrooth E, Cohen EEW. The changing landscape of therapeutic cancer vaccines-novel platforms and neoantigen identification. Clin Cancer Res. 2021;27(3):689–703. doi: 10.1158/1078-0432.CCR-20-0245. [DOI] [PubMed] [Google Scholar]
- 10.Aldous AR, Dong JZ. Personalized neoantigen vaccines: a new approach to cancer immunotherapy. Bioorganic Med Chem [Internet]. Elsevier Ltd; 2018;26:2842–2849. Available from: 10.1016/j.bmc.2017.10.021 10 [DOI] [PubMed] [Google Scholar]
- 11.Hurwitz AA, Cuss SM, Stagliano KE, Zhu Z. T cell avidity and tumor immunity: problems and solutions. Cancer Microenviron. 2014;7(1–2):1–9. doi: 10.1007/s12307-013-0143-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Carreno BM, Magrini V, Becker-Hapak M, Kaabinejadian S, Hundal J, Petti AA, Magrini V, Becker-Hapak M, Kaabinejadian S, Hundal J, et al. A dendritic cell vaccine increases the breadth and diversity of melanoma neoantigen-specific T cells. Science. 2015;348(6230):80. doi: 10.1126/science.aaa4972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Sahin U, Oehm P, Derhovanessian E, Jabulowsky RA, Vormehr M, Gold M, Maurus D, Schwarck-Kokarakis D, Kuhn AN, Omokoko T, et al. An RNA vaccine drives immunity in checkpoint-inhibitor-treated melanoma. Nature [Internet]. Springer US; 2020;585:107–112. Available from: 10.1038/s41586-020-2537-9 7823 [DOI] [PubMed] [Google Scholar]
- 14.Ott PA, Hu Z, Keskin DB, Shukla SA, Sun J, Bozym DJ, Zhang W, Luoma A, Giobbie-Hurder A, Peter L, et al. An immunogenic personal neoantigen vaccine for patients with melanoma. Nature [Internet]. Macmillan Publishers Limited, part of Springer Nature. All rights reserved.; 2017;547:217. Available from: 10.1038/nature22991 7662 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hilf N, Kuttruff-Coqui S, Frenzel K, Bukur V, Stevanović S, Gouttefangeas C, Plattern, M, Tabatabai, G, Dutoit, V, Van der Burg, SH et al Actively personalized vaccination trial for newly diagnosed glioblastoma. Nature. 2019;565(7738): 240–245. doi: 10.1038/s41586-018-0810-y. [DOI] [PubMed] [Google Scholar]
- 16.Keskin DB, Anandappa AJ, Sun J, Tirosh I, Mathewson ND, Li S, Oliveira G, Giobbie-Hurder A, Felt K, Gjini E, et al. Neoantigen vaccine generates intratumoral T cell responses in phase Ib glioblastoma trial. Nature [Internet]. Springer US; 2019;565:234–239. Available from: 10.1038/s41586-018-0792-9 7738 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ott PA, Hu-Lieskovan S, Chmielowski B, Govindan R, Naing A, Bhardwaj N, Margolin K, Awad MM, Hellmann MD, Lin JJ, et al. A Phase Ib Trial of Personalized Neoantigen Therapy Plus Anti-PD-1 in Patients with Advanced Melanoma, Non-small Cell Lung Cancer, or Bladder Cancer. Cell. 2020;183(2):347–362.e24. doi: 10.1016/j.cell.2020.08.053. [DOI] [PubMed] [Google Scholar]
- 18.Vermaelen K. Vaccine strategies to improve anticancer cellular immune responses. Front Immunol. 2019;10:1–17. doi: 10.3389/fimmu.2019.00008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Fang Y, Mo F, Shou J, Wang H, Luo K, Zhang S, Han N, Li H, Ye S, Zhou Z, et al. A pan-cancer clinical study of personalized neoantigen vaccine monotherapy in treating patients with various types of advanced solid tumors. Clin Cancer Res. clincanres.2881.2019, 2020;clincanres.2881.2019. doi: 10.1158/1078-0432.CCR-19-2881. [DOI] [PubMed] [Google Scholar]
- 20.Ott PA, Hu-Lieskovan S, Chmielowski B, Govindan R, Naing A, Bhardwaj N, Margolin K, Awad MM, Hellmann MD, and Lin JJet al. A Phase Ib Trial of Personalized Neoantigen Therapy Plus Anti-PD-1 in Patients with Advanced Melanoma, Non-small Cell Lung Cancer, or Bladder Cancer. Cell. 2020;183(2):347–362.e24. doi: 10.1016/j.cell.2020.08.053 [DOI] [PubMed] [Google Scholar]
- 21.Garcia-Garijo A, Fajardo CA, Gros A. Determinants for neoantigen identification. Front Immunol. 2019;10:1–19. doi: 10.3389/fimmu.2019.01392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Palucka K, Banchereau J.. Dendritic-Cell-Based Therapeutic Cancer Vaccines. CellPress Immunity. 2013;39(1):38–48. doi: 10.1016/j.immuni.2013.07.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Sahin U, Derhovanessian E, Miller M, Kloke BP, Simon P, Löwer M, Bukur, V, Tadmor, A, Luxemburger, U, Schrörs, B, et al. Personalized RNA mutanome vaccines mobilize poly-specific therapeutic immunity against cancer. Nature. 2017;547(7662):222–226. doi: 10.1038/nature23003. [DOI] [PubMed] [Google Scholar]
- 24.D’Alise AM, Leoni G, Cotugno G, Troise F, Langone F, Fichera I, De Lucia M, Avalle L, Vitale R, Leuzzi A, et al. Adenoviral vaccine targeting multiple neoantigens as strategy to eradicate large tumors combined with checkpoint blockade. Nature Communications [Internet]. Springer US; 2019;10:1–12. Available from: 10.1038/s41467-019-10594-2 1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Filskov J, Andersen P, Agger EM, Bukh J. HCV p7 as a novel vaccine-target inducing multifunctional CD4+ and CD8+ T-cells targeting liver cells expressing the viral antigen. Sci Rep. 2019;9(1):1–14. doi: 10.1038/s41598-019-50365-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Overgaard NH, Frøsig TM, Jakobsen JT, Buus S, Andersen MH, Jungersen G. Low antigen dose formulated in CAF09 adjuvant Favours a cytotoxic T-cell response following intraperitoneal immunization in Göttingen minipigs. Vaccine [Internet]. The Author(s); 2017;35:5629–5636. Available from: 10.1016/j.vaccine.2017.08.057 42 [DOI] [PubMed] [Google Scholar]
- 27.Schmidt ST, Khadke S, Korsholm KS, Perrie Y, Rades T, Andersen P, Foged C, Christensen D. The administration route is decisive for the ability of the vaccine adjuvant CAF09 to induce antigen-specific CD8+ T-cell responses: the immunological consequences of the biodistribution profile. J Control Release [Internet]. The Authors; 2016;239:107–117. Available from: 10.1016/j.jconrel.2016.08.034 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Korsholm KS, Hansen J, Karlsen K, Filskov J, Mikkelsen M, Lindenstrøm T, Schmidt ST, Andersen P, and Christensen D.. Induction of CD8+ T-cell responses against subunit antigens by the novel cationic liposomal CAF09 adjuvant. Vaccine. 2014;32(31):3927–3935. doi: 10.1016/j.vaccine.2014.05.050 [DOI] [PubMed] [Google Scholar]
- 29.WMA. WMA Helsinki Agreement [Internet]. Available from: https://www.wma.net/policies-post/wma-declaration-of-helsinki-ethical-principles-for-medical-research-involving-human-subjects/ Accessed 19 October 2021
- 30.GCP. GCP guidelines [Internet]. Available from: https://gcp-enhed.dk/. Accessed 20 October 2021
- 31.Schmidt ST, Christensen D, Perrie Y. Applying microfluidics for the production of the cationic liposome-based vaccine adjuvant caf09b. Pharmaceutics. 2020;12(12):1–17. doi: 10.3390/pharmaceutics12121237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Korsholm KS, Hansen J, Karlsen K, Filskov J, Mikkelsen M, Lindenstrøm T, Schmidt ST, Andersen P, Christensen D. Induction of CD8+ T-cell responses against subunit antigens by the novel cationic liposomal CAF09 adjuvant. Vaccine [Internet]. Elsevier Ltd; 2014;32:3927–3935. Available from: 10.1016/j.vaccine.2014.05.050 31 [DOI] [PubMed] [Google Scholar]
- 33.Moodie Z, Price L, Gouttefangeas C, Mander A, Janetzki S, Löwer M, Welters MJP, Ottensmeier C, van der Burg SH, Britten CM, et al. Response definition criteria for ELISPOT assays revisited. Cancer Immunol Immunother. 2010;59(10):1489–1501. doi: 10.1007/s00262-010-0875-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Hellmann MD, and Snyder A.. Making It Personal: neoantigen Vaccines in Metastatic Melanoma. CellPress. 2017;47:221–223. doi: 10.1016/j.immuni.2017.08.001. [DOI] [PubMed] [Google Scholar]
- 35.Wolchok JD, Kluger H, Callahan MK, Postow MA, Rizvi NA, Lesokhin AM, Segal NH, Ariyan CE, Gordon R-A, Reed K, et al. Safety and clinical activity of combined PD-1 (nivolumab) and CTLA-4 (ipilimumab) blockade in advanced melanoma patients. N Engl J Med. 2013;369(2):122–133. doi: 10.1056/NEJMoa1302369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Billeskov R, Wang Y, Solaymani-Mohammadi S, Frey B, Kulkarni S, Andersen P, Agger EM, Sui Y, Berzofsky JA. Low Antigen Dose in Adjuvant-Based Vaccination Selectively Induces CD4 T Cells with Enhanced Functional Avidity and Protective Efficacy. J Immunol. 2017;198(9):3494–3506. doi: 10.4049/jimmunol.1600965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Chen X, Yang J, Wang L, Liu B. Personalized neoantigen vaccination with synthetic long peptides: recent advances and future perspectives. Theranostics. 2020;10(13):6011–6023. doi: 10.7150/thno.38742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Bijker MS, van Den Eeden Sjf, Franken KL, Melief CJM, Offringa R, van der Burg SH, van Den Eeden SJF. CD8 + CTL Priming by Exact Peptide Epitopes in Incomplete Freund’s Adjuvant Induces a Vanishing CTL Response, whereas Long Peptides Induce Sustained CTL Reactivity. J Immunol. 2007;179(8):5033–5040. doi: 10.4049/jimmunol.179.8.5033. [DOI] [PubMed] [Google Scholar]
- 39.Slingluff CL Jr. The Present and Future of Peptide Vaccines for Cancer: single or Multiple, Long or Short, Alone or in Combination? The Cancer Journal. 2011;17(5):343–350. doi: 10.1097/PPO.0b013e318233e5b2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Ali OA, Lewin SA, Dranoff G, Mooney DJ. Vaccines combined with immune checkpoint antibodies promote cytotoxic T-cell activity and tumor eradication. Cancer Immunol Res. 2016;4(2):95–100. doi: 10.1158/2326-6066.CIR-14-0126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Karyampudi L, Lamichhane P, Scheid AD, Kalli KR, Shreeder B, Krempski JW, Behrens, MD, Knutson, KL, et al. Accumulation of memory precursor cd8 t cells in regressing tumors following combination therapy with vaccine and anti-pd-1 antibody. Cancer Research. 2014;74(11):2974–2985. doi: 10.1158/0008-5472.CAN-13-2564. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.