

SUMMARY:
The genetic interferonopathies are a heterogeneous group of disorders thought to be caused by the dysregulated expression of interferons and are now commonly considered in the differential diagnosis of children presenting with recurrent or persistent inflammatory phenotypes. With emerging therapeutic options, recognition of these disorders is increasingly important, and neuroimaging plays a vital role. In this article, we discuss the wide spectrum of neuroradiologic features associated with monogenic interferonopathies by reviewing the literature and illustrate these with cases from our institutions. These cases include intracerebral calcifications, white matter T2 hyperintensities, deep WM cysts, cerebral atrophy, large cerebral artery disease, bilateral striatal necrosis, and masslike lesions. A better understanding of the breadth of the neuroimaging phenotypes in conjunction with clinical and laboratory findings will enable earlier diagnosis and direct therapeutic strategies.
Interferons (IFNs) are a family of cytokines that are induced through the stimulation of pattern-recognition receptors that sense pathogen-derived nucleic acids.1-3 They exert their activity through a complex network of regulatory pathways (Fig 1) for modulating innate/acquired immunity and resistance to viral infections.2-4 These tightly regulated mechanisms protect against inappropriate immune activation triggered by endogenous nucleic acids while maintaining a rapid and effective response to exogenous nucleic acids derived from pathogens.3,4 The interferonopathies are an expanding group of disorders that are thought to disrupt this delicate equipoise by overactivation of the IFN response, resulting in pathology from chronic inflammation.2-5 In their most distinctive form, these diseases are monogenic neuroinflammatory disorders of infancy.3-5
FIG 1.

A highly simplified diagram highlighting key pathways affected in type 1 interferonopathies, in which neuroimaging findings have been described. The black bars indicate inhibition, and the arrows, activation. ? indicates that the mechanism of IFN induction is unclear. CGAS indicates cyclic GMP-AMP synthase; TBK1, TANK-binding kinase 1; IRF3, IFN regulatory factor 3; MAVS, mitochondrial antiviral-signaling protein; IFNAR, IFN α/β receptor; JAK1, Janus kinase 1; TYK2, tyrosine kinase 2; PSMB8, proteasome subunit β type-8; TREX1, three prime repair exonuclease 1; RNASE H, ribonuclease H; SAMHD1, SAM And HD domain-containing deoxynucleoside triphosphate triphosphohydrolase 1; IFIH1, interferon induced with helicase C domain 1; PNPT1, polyribonucleotide nucleotidyltransferase 1; DNASE2, deoxyribonuclease 2; ACP5, acid phosphatase 5; ISG15, interferon-stimulated gene 15; RNU7-1, RNA, U7 small nuclear 1; USP18, ubiquitin-specific peptidase 18; STAT2, signal transducer and activator of transcription 2; ER, endoplasmic reticulum; mtDNA, mitochondrial DNA.
In 1984, Aicardi and Goutières6 described a disorder with progressive encephalopathy, CSF lymphocytosis, and neuroradiologic findings that mimic transplacental infection but test serologically negative. We now know that this disorder, which we refer to as Aicardi-Goutières syndrome (AGS), is characterized by genetic mutations that disrupt mechanisms that protect against inappropriate immune activation triggered by endogenous nucleic acids.1,3,4 The subsequent dysregulated expression of IFNs is thought to promote unwanted inflammatory responses through their pleiotropic effects.2-4 This interferonopathy hypothesis is supported by the fact that elevated IFN levels in the CSF and blood have long been described in patients with AGS.1,7 More recently, upregulation of interferon-stimulated genes, an “interferon signature,” which can be detected in peripheral blood, has been reported to be present in most patients with AGS and other interferonopathies.1,3,4 As more mutations in these genes are described, a broad spectrum of phenotypes with considerable overlap has been revealed, many of which have prominent systemic manifestations, including inflammatory vasculopathy and systemic autoimmune disease.3,4,8-10 Recognition of these diseases has become increasingly important not only for more reliable genetic counseling but because of the emergence of promising therapeutic strategies.3,11,12 Neuroimaging often provides a valuable clue to the diagnosis of this expanding group of disorders3,5,11,13-15 and will be the focus of this article.
Search Strategy
MEDLINE and EMBASE were searched from inception to week 1 of 2021 using the Ovid online portal. We used the following keywords: 1) “interferonopathy” (n = 245), 2) “Aicardi-Goutières syndrome” (n = 501), 3) 1 and “intracranial calcification” (n = 10), 4) 1 and “brain” (n = 34), 5) 1 and “neuroimaging”, 5) 2 and “neuroimaging” (n = 5), and 6) 1 and 2 (n = 79). The abstracts were reviewed to determine whether the article should be included. The search was supplemented by searching the bibliographies of relevant articles.
Monogenic Interferonopathies and Neuroimaging
The Online Supplemental Data provide a list of interferonopathies in which neuroimaging findings have been described; Fig 1 illustrates the key pathways involved. Common neuroimaging findings across the interferonopathies include intracerebral calcifications and T2 white matter hyperintensities (WMH) (Online Supplemental Data).
AGS, the best-described group of interferonopathies, is characterized by mutations in any of several genes (including TREX1, RNASEH2A/B/C, SAMHD1, ADAR1, IFIH1),3 which are involved in the metabolism of nucleic acids or their recognition.1 Recent additions to this are LSM11 and RNU7-1, which encode small nuclear ribonucleoproteins, which are key components of the U7 replication-dependent histone pre-messenger RNA–processing complex, which is thought to help maintain the structure of nucleosomes, preventing activation of cyclic GMP-AMP synthase by nuclear DNA.16
TREX1 mutations are associated with a true neonatal presentation (Figs 2 and 3) and, therefore, mimic transplacental infection.17 Most patients, however, present a little later, with the most frequent mutations occurring in RNASEH2B (Fig 4).7,18 The neuroimaging phenotypes continue to expand as these mutations are better described and more are discovered. For example, intracranial vasculopathy has been described with SAMHD1 mutations,19 and bilateral striatal necrosis, with ADAR1 mutations (Online Supplemental Data).20
FIG 2.

T2, gradient recalled-echo (GRE) and T1-weighted images of a 9-day-old boy with a TREX1 mutation demonstrating basal ganglia and periventricular calcifications.
FIG 3.

CT, SWI, and T2-weighted images of a boy with a TREX1 mutation. The upper row shows imaging at 2 months of age, and the lower row, at 2 years of age. There are periventricular, basal ganglia, brainstem, and cerebellar WM calcifications, which progress with time. Note the interval volume loss with WM rarefaction, most marked in the anterior temporal lobes.
FIG 4.

A 1-year-old girl with a RNASEH2B mutation. There are patchy WMH throughout the brain. Symmetric T2-signal abnormality is noted in the globus pallidus, thalamus, and dentate nucleus. There is global cerebral volume loss. CT confirms basal ganglia calcifications.
Similar destructive neuroinflammatory conditions have been documented in genetic disorders in which other components of the IFN signaling pathway are affected (Online Supplemental Data).1,3,13 For example, loss-of-function mutations in ubiquitin-specific peptidase 18 (USP18), which is a negative regulator of type I IFN signaling, is also associated with intracerebral calcification and WMH (Online Supplemental Data).13,21
In some interferonopathies, systemic findings are a more prominent feature, such as interstitial lung disease in STING-associated vasculopathy of infancy (SAVI) or skin lesions in proteasome-associated autoinflammatory syndromes (Online Supplemental Data). Intracranial calcifications have also been described in a few of these cases and can, on occasion, provide an important clue to the diagnosis.
Due to the considerable overlap in the neuroimaging phenotypes, it is seldom possible to make a diagnosis on the basis of neuroradiologic findings alone, and these should always be considered in conjunction with clinical and laboratory findings (Fig 5). Indeed, neuroimaging can sometimes have normal findings, and the lack of neuroimaging findings, like intracranial calcifications, does not exclude an interferonopathy, certainly early on in the disease. In the rest of this article, we describe both characteristic and more unusual neuroradiologic features associated with interferonopathies, including findings that could lead one to suspect certain mutations. These include intracerebral calcifications, WMH, deep WM cysts, cerebral atrophy, large cerebral artery disease, bilateral striatal necrosis, and masslike lesions.
FIG 5.

A potential route to the diagnosis of monogenic type 1 interferonopathies. The red boxes contain clinical, laboratory, and imaging findings that lead one to suspect an interferonopathy. The green boxes contain diagnostic tests performed when a monogenic type 1 interferonopathy is strongly suspected. ESR indicates erythrocyte sedimentation rate; WBC, white blood cells; ISG, interferon-stimulated genes; TORCH, (T)oxoplasmosis, (O)ther Agents, (R)ubella (also known as German Measles), (C)ytomegalovirus, and (H)erpes Simplex; mRNA, messenger RNA.
Intracerebral Calcification
Intracerebral calcifications (ICC) have been reported to be present in >90% of subjects with AGS.18 ICC are also described in most other interferonopathies, even when systemic features predominate such as proteasome-associated autoinflammatory syndromes and SAVI (Online Supplemental Data).3 Calcifications tend to be punctate or linear, reflecting their relationship to microvascular structures. Often sagittal or coronal reconstructions make it easier to appreciate the relationship with the deep, perforating vessels or deep medullary veins/arteries (Fig 6). The calcifications (Figs 2 and 3) are most commonly symmetric and seen in the basal ganglia and deep WM and dentate nuclei of the cerebellum18 but can be seen throughout the brain. The morphology of the calcifications varies from fine/punctate to larger conglomerates. TREX1 mutations are usually associated with more severe calcifications.18 Neuropathologic studies show calcifications in the walls of medium/small vessels and perivascular spaces.22-24 One study has suggested that IFNs act directly on vascular smooth-muscle cells, inducing a calcifying microangiopathy.24 The increasing use of MR imaging as the first imaging technique in clinical practice may reduce the identification or recognition of intracranial calcifications, but the sensitivity can be improved by using gradient-echo or susceptibility-weighted sequences. CT is recommended in patients in whom interferonopathies are suspected and no calcifications are seen on MR imaging.18
FIG 6.
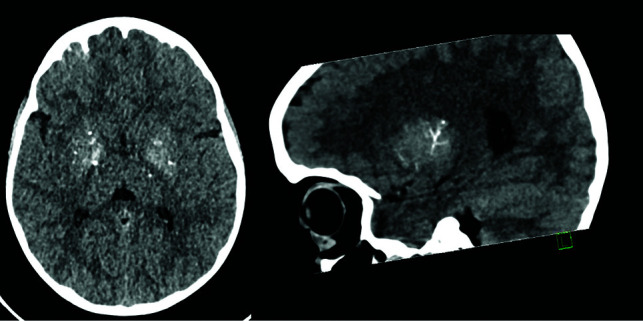
A CT study in axial and sagittal reconstructions shows punctate and linear branching calcifications along the deep perforators in a 7-year-old boy with a SAMHD1 mutation.
WM Abnormalities
WMH have been described in most interferonopathies (Online Supple-mental Data) and appear to be the most common finding in those with a later disease onset when ICC are less common.25 Neuropathologic studies in AGS show inhomogeneous demyelination and astrogliosis, which is, again, supportive of a microangiopathy.22,23 WM involvement is most commonly diffuse or with a frontotemporal predominance, but involvement can also be periventricular or patchy.18 TREX1 mutations are associated with more severe involvement, causing WM rarefaction in the anterior frontal and temporal lobes and deep WM cysts.15,18,26 Delayed myelination can be associated with RNASEH2B mutations (Fig 7) but progresses with time because it is not a primary disorder of myelination.18 Interferonopathies should also be considered in patients with MR imaging findings interpreted as periventricular leukomalacia.18,23
FIG 7.

A 24-week-old boy with a RNASEH2C mutation with WM abnormalities in the frontal and temporal lobes and generalized volume loss.
Brain Volume Loss
The degree of brain volume loss (Figs 3 and 7) depends on when the patient presents during the course of the disease, and it can be progressive. It is likely to be due to a combination of gray and WM volume loss. Although not always obvious on imaging, the neocortex and cerebellar cortex can be involved with numerous microinfarcts seen on some neuropathologic studies, which are likely to contribute to brain volume loss.23,24 Pontocerebellar hypoplasia has also been reported, but this is relatively infrequent and is likely subsequent to the supratentorial brain volume loss.15,18,26,27
Large Cerebral Artery Disease
Intracerebral large-artery involvement, including stroke, Moyamoya-type vasculopathy, aneurysms, and stenosis of single vessels, has most often been reported with SAMHD1 mutations (Fig 8)18,19 and less frequently with TREX1 mutations.28 Neuropathology in a patient with a SAMHD1 mutation also showed calcifying microangiopathy,24 which suggests a disease process similar to the that in other interferonopathies (Fig 6). The reason for the increased frequency of large-vessel disease in these mutations, however, is uncertain.
FIG 8.
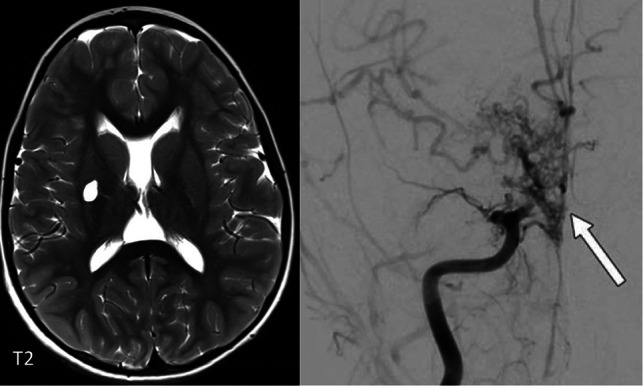
Lacunar infarct in the right putamen in a 7-year-old boy with a SAMDH1 mutation. The angiogram on the right demonstrates a Moyamoya-type vasculopathy with occlusion of the right M1 and A1 (arrow).
Bilateral Striatal Necrosis
Bilateral striatal necrosis has been reported in the context of ADAR1 (Fig 9) and PNPT1 (Fig 10) mutations in addition to WMH and ICC.20,29,30 Imaging can show bilateral striatal swelling and signal abnormality with cavitation/atrophy on follow-up imaging. Globus pallidus swelling and signal abnormality can also be seen (Fig 11). The mechanism remains unclear, and metabolic disorders should be ruled out. These findings can sometimes be seen in a para-/postinfectious setting, and it has been hypothesized that local neuroinflammation may contribute to synaptic dysfunction and excitotoxicity.31 Freckle-like skin lesions are characteristic of ADAR1 mutations.29
FIG 9.

A, Bilateral putaminal signal abnormality in a 4-year-old boy with a confirmed ADAR1 mutation. There was no diffusion restriction. B, A different patient also with an ADAR1 mutation showing cavitation in both putamina.
FIG 10.

An 18-month-old girl with a PNTP1 mutation with signal abnormality and decreased volume of both the putamina and globi pallidi. Diffusion restriction is seen in the globi pallidi.
FIG 11.
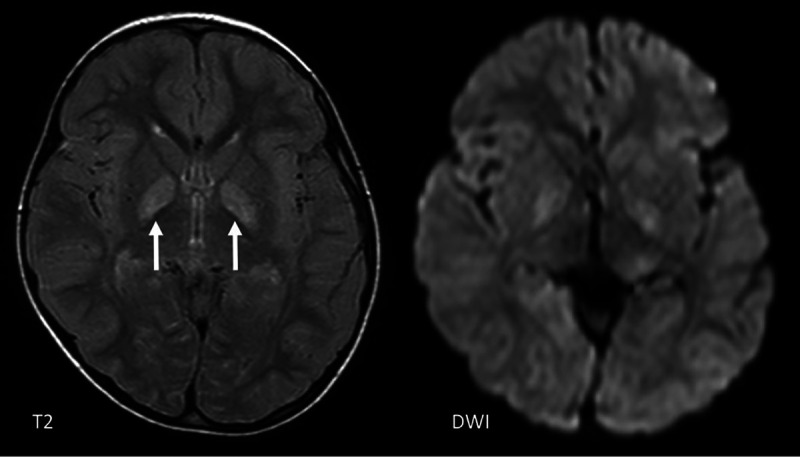
Atypical imaging features in a 4-year-old girl with an ADAR1 mutation showing a T2 signal abnormality and mild diffusion restriction in the globi pallidi bilaterally (arrows), but not in the putamina.
Masslike Lesions
Masslike or ring-enhancing lesions have been described in heterozygous C-terminal frameshift mutations in TREX1 (causing a disorder called retinal vasculopathy with cerebral leukodystrophy), which occur in conjunction with ICC and scattered WMH, though this disorder tends to present later in life.32 These lesions are often misdiagnosed as tumors or as other inflammatory diseases such as sarcoidosis. Neuropathology can show necrotizing granulomatous inflammation.11 The presence of coexistent neuroimaging features such as ICC should alert the clinician to the potential diagnosis of interferonopathy.
Hemorrhage
Deep GM, cerebellar, and intraventricular hemorrhage has been described in patients with USP18 deficiency11,33 and TREX1 mutations.18 Although these patients had coexistent coagulopathies, the location of the hemorrhage is reminiscent of that seen with COL4A1 mutations and could conceivably be due to an underlying vasculopathy. Subdural and subarachnoid hemorrhage has also been reported in patients with a STAT2 mutation, which disrupts STAT2 interaction with USP18, a key negative regulator of IFN signaling.34
Some Differential Diagnoses to Consider
Due to the nonspecific nature of the neuroradiologic findings, the differential diagnosis is wide, including other inflammatory and neurogenetic disorders. In cases of neonatal presentation, congenital infections should first be ruled out. It has been suggested that “true periventricular” calcification in the ependymal/subependymal regions is more suggestive of infection.35 Other features that make infection more likely include malformations of cortical development and anterior temporal cysts as seen in congenital cytomegalovirus (Fig 12).35
FIG 12.

T2-weighted MR imaging of a 9-day-old girl with congenital cytomegalovirus infection showing polymicrogyria (white arrow), periventricular calcifications (gray arrow), and anterior temporal lobe cysts (black arrow).
Other conditions known to be associated with an increased expression of IFN-regulated genes include systemic lupus erythematosus (SLE)36 and juvenile dermatomyositis.37 These, however, tend to present later in life, and systemic features usually predominate. The most common neuroimaging findings described with SLE are also of a microangiopathy, including nonspecific focal WMH,38 brain atrophy, intracranial calcifications, and hemorrhage,39 though other inflammatory findings such as leptomeningeal enhancement, large-vessel vasculitis, and masslike lesions have also been described.40
Hyperinflammatory clinical syndromes like hemophagocytic lymphohistiocytosis should also be considered. Hemophagocytic lymphohistiocytosis can be primary, resulting from genetic defects or secondary to infection, malignancy, immunosuppression, or autoimmune diseases such as SLE. CNS involvement is common, and the neuroimaging findings are variable, including focal/diffuse WMH, perivascular enhancement (particularly in the posterior fossa), meningeal enhancement, masslike lesions, and hemorrhage (Fig 13).41
FIG 13.

A 6 month-old girl with perforin-deficient hemophagocytic lymphohistiocytosis. There is diffuse WM signal abnormality seen on the T2-weighted image and multiple enhancing pseudotumoural lesions seen on the T1 postcontrast image (T1 + C) in the left thalamus and posterior temporal and occipital lobes bilaterally.
Some leukodystrophies, like Cockayne syndrome, can have a very similar appearance with diffuse WMH and ICC. Unlike AGS, there is a severe delay or an arrest in myelination, and the calcifications can be cortical/leptomeningeal (Fig 14).42
FIG 14.

A 6-year-old boy with Cockayne syndrome showing subcortical and basal ganglia calcifications on SWI and diffuse WM signal abnormality seen on the T2-weighted image. There is marked WM volume loss.
Genetic microangiopathies such as collagen IV (COL4A1/A2) related disease can also show similar imaging findings.35 They are characteristically associated with porencephalic cysts or periventricular leukomalacia.43 It has been suggested that the calcification is more subtle, involving subependymal/periventricular regions (Fig 15).43
FIG 15.

Hemosiderin-lined cavities in the basal ganglia and thalamus and subtle periventricular calcification on CT (white arrows) in a 3-year-old boy with a COL4A1 mutation.
Calcifications of the cerebral microvessels are also seen in primary familial brain calcification, a group of neurogenetic disorders associated with diverse motor, cognitive, and psychiatric symptoms.44 Calcifications are typically symmetric, commonly involving the deep GM structures. The autosomal dominant form can be caused by mutations in genes encoding phosphate transporters (SLC20A2, XPR1) or platelet-derived growth factor B and its receptor (PDGFB, PDGFRB). An autosomal recessive form has been described with mutations in encoding junctional adhesional molecules (JAM2, JAM3), myogenesis-regulating glycosidase (MYORG), and occludin (OCLN).45 Calcification is more severe in the autosomal recessive form involving the subcortical WM. Brainstem calcification may be more common with MYORG mutations.44
Some conditions with clinical and imaging overlap to transplacental infection may ultimately turn out to be interferonopathies. One such example is RNaseT2 mutations, which result in loss of ribonuclease T2 protein function, causing accumulation of ribosomal RNA.46 RNaseT2 mutation causes cystic leukoencephalopathy without megalencephaly. The imaging findings are very similar to those of congenital cytomegalovirus infection and are characterized by anterior temporal lobe cysts and extensive WM hyperintensities in a normal-sized brain.
CONCLUSIONS
Neuroimaging is essential in interferonopathies, in which neuroinflammation is a predominant feature, as in AGS. Even when systemic features predominate, neuroimaging can often provide an important clue to the diagnosis. However, the spectrum of findings is wide and should always be considered in conjunction with clinical and laboratory findings. A common feature on neuroimaging seems to be evidence of calcifying microangiopathy characterized by WMH and ICC, which is best picked up on susceptibility-weighted MR imaging sequences or CT, though large-vessel disease, bilateral striatal necrosis, hemorrhage, and masslike lesions are also seen with some mutations. A CT scan is recommended when type 1 interferonopathies are suspected and no calcifications are seen on MR imaging. The list of interferonopathies and associated neuroimaging findings will, no doubt, continue to grow as more genotype-phenotype correlations are identified.
As a common feature, all interferons activate the Janus kinase signal transducers of activation and the transcription signaling pathway (Fig 1).47 There are now several encouraging reports, with Janus kinase inhibitor therapy resulting in a prompt and sustained recovery, sometimes with an improvement or stabilization in imaging appearance.11,33,34 Reverse-transcriptase inhibitors have also been shown to reduce interferon signaling in patients with AGS.48 Clinical trials are currently underway. These promising new therapeutic strategies make recognition of these disorders all the more important. As this field continues to grow, neuroimaging is likely to play an increasing role in diagnosis, monitoring, and prognostication.
ABBREVIATIONS:
- AGS
Aicardi-Goutières syndrome
- ICC
intracranial calcifications
- IFN
interferon
- SLE
systemic lupus erythematosus
- SAVI
STING-associated vasculopathy of infancy
- WMH
white matter hyperintensities
Footnotes
Disclosures: Felice D’Arco—UNRELATED: Expert Testimony: expert witness in UK and Ireland; Payment for Lectures Including Service on Speakers Bureaus: European Society of Neuroradiology, Comments: honoraria for lectures in the European courses of neuroradiology. Cheryl Hemingway—UNRELATED: Consultancy: pharmaceutical companies, Comments: I have provided expert advice on steering groups for the following companies: Roche, Biogen, UCB, Viela Bio, Novartis; Grants/Grants Pending: Medical Research Council, Comments: I have a research grant for pediatric MS research for some of my salary; Payment for Lectures Including Service on Speakers Bureaus: pharmaceutical, Comments: I have received money from Novartis for delivering a lecture on Pediatric MS. Kshitij Mankad—UNRELATED: Employment: Great Ormond Street Hospital, Comments: regular post; Expert Testimony: medicolegal reporting in the UK; Payment for Lectures Including Service on Speakers Bureaus: speaker honoraria, Comments: Novartis, Siemens.
References
- 1.Crow YJ, Manel N. Aicardi–Goutières syndrome and the type I interferonopathies. Nat Rev Immunol 2015;15:429–40 10.1038/nri3850 [DOI] [PubMed] [Google Scholar]
- 2.Lee-Kirsch MA. The type I interferonopathies. Annu Rev Med 2017;68:297–315 10.1146/annurev-med-050715-104506 [DOI] [PubMed] [Google Scholar]
- 3.Eleftheriou D, Brogan PA. Genetic interferonopathies: an overview. Best Pract Res Clin Rheumatol 2017;31:441–59 10.1016/j.berh.2017.12.002 [DOI] [PubMed] [Google Scholar]
- 4.Volpi S, Picco P, Caorsi R, et al. Type I interferonopathies in pediatric rheumatology. Pediatr Rheumatol Online 2016;14:35 10.1186/s12969-016-0094-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.McGlasson S, Jury A, Jackson A, et al. Type I interferon dysregulation and neurological disease. Nat Rev Neurol 2015;11:515–23 10.1038/nrneurol.2015.143 [DOI] [PubMed] [Google Scholar]
- 6.Aicardi J, Goutières F. A progressive familial encephalopathy in infancy with calcifications of the basal ganglia and chronic cerebrospinal fluid lymphocytosis. Ann Neurol 1984;15:49–54 10.1002/ana.410150109 [DOI] [PubMed] [Google Scholar]
- 7.Crow YJ, Chase DS, Schmidt JL, et al. Characterization of human disease phenotypes associated with mutations in TREX1, RNASEH2A, RNASEH2B, RNASEH2C, SAMHD1, ADAR, and IFIH1. Am J Med Genet 2015;167:296–312 10.1002/ajmg.a.36887 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Clarke SL, Robertson L, Rice GI, et al. Type 1 interferonopathy presenting as juvenile idiopathic arthritis with interstitial lung disease: report of a new phenotype. Pediatr Rheumatol Online 2020;18:37 10.1186/s12969-020-00425-w [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Clarke SL, Pellowe EJ, de Jesus AA, et al. Interstitial lung disease caused by STING-associated vasculopathy with onset in infancy. Am J Respir Crit Care Med 2016;194:639–42 10.1164/rccm.201510-2102LE [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Li J, An S, Du Z. Familial interstitial lung disease caused by mutation of the STING1 gene. Front Pediatr 2020;8:543 10.3389/fped.2020.00543 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Mulhern CM, Hong Y, Omoyinmi E, et al. Janus kinase 1/2 inhibition for the treatment of autoinflammation associated with heterozygous TNFAIP3 mutation. J Allergy Clin Immunol 2019;144:863–66.e5 10.1016/j.jaci.2019.05.026 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Schwartz DM, Blackstone SA, Sampaio-Moura N, et al. Type I interferon signature predicts response to Janus kinase inhibition in haploinsufficiency of A20. Ann Rheum Dis 2020;79:429–31 10.1136/annrheumdis-2019-215918 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Meuwissen ME, Schot R, Buta S, et al. Human USP18 deficiency underlies type 1 interferonopathy leading to severe pseudo-TORCH syndrome. J Exp Med 2016;213:1163–74 10.1084/jem.20151529 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Uzgil B, Scherr EH. Neuroimaging in Aicardi-Goutieres syndrome. Neurology 2016;86:15–16 10.1212/WNL.0000000000002227 [DOI] [PubMed] [Google Scholar]
- 15.Uggetti C, Piana RL, Orcesi S, et al. Aicardi-Goutières syndrome: neuroradiologic findings and follow-up. AJNR Am J Neuroradiol 2009;30:1971–76 10.3174/ajnr.A1694 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Uggenti C, Lepelley A, Depp M, et al. cGAS-mediated induction of type I interferon due to inborn errors of histone pre-mRNA processing. Nat Genet 2020;52:1364–72 10.1038/s41588-020-00737-3 [DOI] [PubMed] [Google Scholar]
- 17.Livingston J, Crow Y. Neurologic phenotypes associated with mutations in TREX1, RNASEH2A, RNASEH2B, RNASEH2C, SAMHD1, ADAR1, and IFIH1: Aicard-Goutières syndrome and beyond. Neuropediatrics 2016;47:355–60 10.1055/s-0036-1592307 [DOI] [PubMed] [Google Scholar]
- 18.La Piana R, Uggetti C, Roncarolo F, et al. Neuroradiologic patterns and novel imaging findings in Aicardi-Goutières syndrome. Neurology 2016;86:28–35 10.1212/WNL.0000000000002228 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ramesh V, Bernardi B, Stafa A, et al. Intracerebral large artery disease in Aicardi-Goutières syndrome implicates SAMHD1 in vascular homeostasis: SAMHD1 and intracerebral large artery disease. Dev Med Child Neurol 2010;52:725–32 10.1111/j.1469-8749.2010.03727.x [DOI] [PubMed] [Google Scholar]
- 20.Livingston JH, Lin JP, Dale RC, et al. A type I interferon signature identifies bilateral striatal necrosis due to mutations in ADAR1. J Med Genet 2014;51:76–82 10.1136/jmedgenet-2013-102038 [DOI] [PubMed] [Google Scholar]
- 21.Ma L, Zhou Y, Zhang Y, et al. Role of A20 in interferon-α-mediated functional restoration of myeloid dendritic cells in patients with chronic hepatitis C. Immunology 2014;143:670–78 10.1111/imm.12350 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Gilani A, Adang LA, Vanderver A, et al. Neuropathological findings in a case of IFIH1-related Aicardi-Goutières syndrome. Pediatr Dev Pathol 2019;22:566–70 10.1177/1093526619837797 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Barth PG. The neuropathology of Aicardi-Goutières syndrome. Eur J Paediatr Neurol 2002;6(Suppl A):A27–31; discussion A37–39, A77–86 10.1053/ejpn.2002.0570 [DOI] [PubMed] [Google Scholar]
- 24.Klok MD, Bakels HS, Postma NL, et al. Interferon-α and the calcifying microangiopathy in Aicardi-Goutières syndrome. Ann Clin Transl Neurol 2015;2:774–79 10.1002/acn3.213 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Piccoli C, Bronner N, Gavazzi F, et al. Late-onset Aicardi Goutières syndrome: a characterization of presenting clinical features. Pediatr Neurol 2021;115:1–6 10.1016/j.pediatrneurol.2020.10.012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Vanderver A, Prust M, Kadom N, et al. Early-onset Aicardi-Goutières Syndrome: magnetic resonance imaging (MRI) pattern recognition. J Child Neurol 2015;30:1343–48 10.1177/0883073814562252 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Abdel-Salam GM, Abdel-Hamid MS, Mohammad SA, et al. Aicardi-Goutières syndrome: unusual neuro-radiological manifestations. Metab Brain Dis 2017;32:679–83 10.1007/s11011-017-9993-4 [DOI] [PubMed] [Google Scholar]
- 28.Wu CC, Peng SS, Lee WT. Intracerebral large artery disease in Aicardi-Goutières syndrome with TREX1 mutation: a case report. Neurol Sci 2020;41:3353–56 10.1007/s10072-020-04516-0 [DOI] [PubMed] [Google Scholar]
- 29.La Piana R, Uggetti C, Olivieri I, et al. Bilateral striatal necrosis in two subjects with Aicardi-Goutières syndrome due to mutations in ADAR1 (AGS6). Am J Med Genet A 2014;164A:815–19 10.1002/ajmg.a.36360 [DOI] [PubMed] [Google Scholar]
- 30.Vedrenne V, Gowher A, De Lonlay P, et al. Mutation in PNPT1, which encodes a polyribonucleotide nucleotidyltransferase, impairs RNA import into mitochondria and causes respiratory-chain deficiency. Am J Hum Genet 2012;91:912–18 10.1016/j.ajhg.2012.09.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Cirillo G, Cirillo M, Panetsos F, et al. Selective vulnerability of basal ganglia: insights into the mechanisms of bilateral striatal necrosis. J Neuropathol Exp Neurol 2019;78:123–19 10.1093/jnen/nly123 [DOI] [PubMed] [Google Scholar]
- 32.Weerasekare J, Kassa R, Keegan BM. Retinal vasculopathy with cerebral leukoencephalopathy due to TREX-1 mutation: an important mimicker of CNS inflammatory disease. Mult Scler Relat Disord 2021;47:102639 10.1016/j.msard.2020.102639 [DOI] [PubMed] [Google Scholar]
- 33.Alsohime F, Martin-Fernandez M, Temsah MH, et al. Janus kinase inhibitor therapy in a child with inherited USP18 deficiency. N Engl J Med 2020;382:256–65 10.1056/NEJMoa1905633 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Duncan CJ, Thompson B, Chen R, et al. Severe type I interferonopathy and unrestrained interferon signaling due to a homozygous germline mutation in STAT2. Sci Immunol 2019;4:eaav7501 10.1126/sciimmunol.aav7501 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Livingston JH, Stivaros S, Warren D, et al. Intracranial calcification in childhood: a review of aetiologies and recognizable phenotypes. Dev Med Child Neurol 2014;56:612–26 10.1111/dmcn.12359 [DOI] [PubMed] [Google Scholar]
- 36.Rönnblom L, Leonard D. Interferon pathway in SLE: one key to unlocking the mystery of the disease. Lupus Sci Med 2019;6:e000270 10.1136/lupus-2018-000270 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Kim H, Gunter-Rahman F, McGrath JA, et al. Expression of interferon-regulated genes in juvenile dermatomyositis versus Mendelian autoinflammatory interferonopathies. Arthritis Res Ther 2020;22:69 10.1186/s13075-020-02160-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Al-Obaidi M, Saunders D, Brown S, et al. Evaluation of magnetic resonance imaging abnormalities in juvenile onset neuropsychiatric systemic lupus erythematosus. Clin Rheumatol 2016;35:2449–56 10.1007/s10067-016-3376-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Lalani TA, Kanne JP, Hatfield GA, et al. Imaging findings in systemic lupus erythematosus. Radiographics 2004;24:1069–86 10.1148/rg.244985082 [DOI] [PubMed] [Google Scholar]
- 40.Panagopoulos D, Themistocleous M. Central nervous system manifestation of lupus erythematosus resembling brain abscess. Int J Pediatr Adolesc Med 2019;6:29–37 10.1016/j.ijpam.2018.11.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Klein C, Kleinschmidt-DeMasters BK, Liang X, et al. A review of neuropathological features of familial and adult hemophagocytic lymphohistiocytosis. J Neuropathol Exp Neurol 2019;78:197–208 10.1093/jnen/nlz001 [DOI] [PubMed] [Google Scholar]
- 42.Koob M, Laugel V, Durand M, et al. Neuroimaging in Cockayne syndrome. AJNR Am J Neuroradiol 2010;31:1623–30 10.3174/ajnr.A2135 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Livingston J, Doherty D, Orcesi S, et al. COL4A1 mutations associated with a characteristic pattern of intracranial calcification. Neuropediatrics 2011;42:227–33 10.1055/s-0031-1295493 [DOI] [PubMed] [Google Scholar]
- 44.Grangeon L, Wallon D, Charbonnier C, et al. ; French PFBC study group. Biallelic MYORG mutation carriers exhibit primary brain calcification with a distinct phenotype. Brain 2019;142:1573–86 10.1093/brain/awz095 [DOI] [PubMed] [Google Scholar]
- 45.LeBlanc MA, Penney LS, Gaston D, et al. A novel rearrangement of occludin causes brain calcification and renal dysfunction. Hum Genet 2013;132:1223–34 10.1007/s00439-013-1327-y [DOI] [PubMed] [Google Scholar]
- 46.Tonduti D, Orcesi S, Jenkinson EM, et al. Clinical, radiological and possible pathological overlap of cystic leukoencephalopathy without megalencephaly and Aicardi-Goutières syndrome. Eur J Paediatr Neurol 2016;20:604–10 10.1016/j.ejpn.2016.03.009 [DOI] [PubMed] [Google Scholar]
- 47.Prinz M, Knobeloch KP. Type I interferons as ambiguous modulators of chronic inflammation in the central nervous system. Front Immun 2012;3:67 10.3389/fimmu.2012.00067 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Rice GI, Meyzer C, Bouazza N, et al. Reverse-transcriptase inhibitors in the Aicardi–Goutières syndrome. N Engl J Med 2018;379:2275–77 10.1056/NEJMc1810983 [DOI] [PubMed] [Google Scholar]