

Summary
Animals traversing different environments encounter both stable background stimuli and novel cues, which are thought to be detected by primary sensory neurons and then distinguished by downstream brain circuits. Here we show that each of the ~1000 olfactory sensory neuron (OSN) subtypes in the mouse harbors a distinct transcriptome whose content is precisely determined by interactions between its odorant receptor and the environment. This transcriptional variation is systematically organized to support sensory adaptation: expression levels of more than 70 genes relevant to transforming odors into spikes continuously vary across OSN subtypes, dynamically adjust to new environments over hours, and accurately predict acute OSN-specific odor responses. The sensory periphery therefore separates salient signals from predictable background via a transcriptional rheostat whose moment-to-moment state reflects the past and constrains the future; these findings suggest a general model in which structured transcriptional variation within a cell type reflects individual experience.
Graphical Abstract
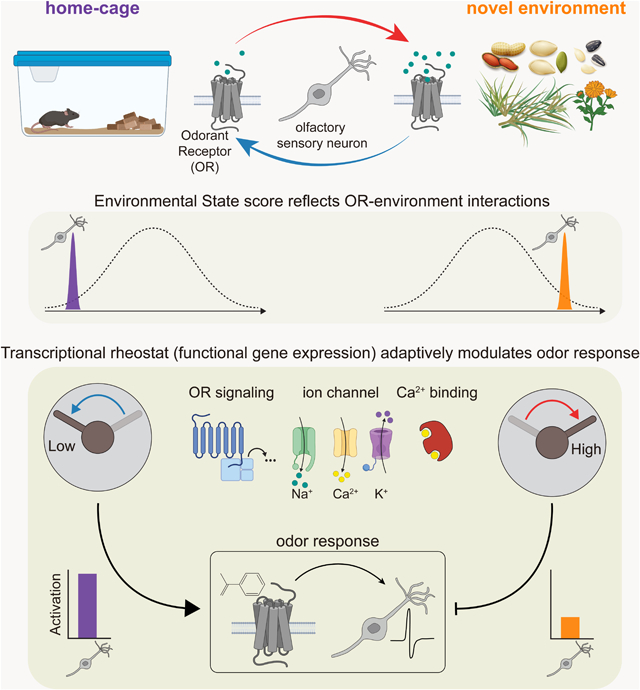
ETOC:
Olfactory sensory neurons in mice adaptively shape odor response via environment-dependent changes in gene expression to separate salient olfactory cues from predictable background.
Olfactory sensory neurons harbor unique, odorant receptor-specific transcriptomes
Receptor engagement by environmental odors drives transcriptional variation
Environment-dependent changes in gene expression adaptively modify odor responses
In vivo imaging reveals that peripheral odor codes are altered across environments
Introduction
Sensory adaptation allows neurons and networks to minimize responses to background stimuli, thereby building more efficient neural codes that emphasize surprising or novel information (Attneave, 1954; Barlow, 1961; Benda, 2021; Weber et al., 2019). Rapid (milliseconds to seconds) sensory adaptation via post-translational mechanisms occurs in most sensory neurons, and plays an important role in shaping peripheral responses to highly dynamic stimuli (Kostal et al., 2008; Martelli and Storace, 2021; Moore, 1994). However, it is not clear how sensory neurons adapt at the longer timescales over which animals traverse different environments — and therefore encounter different background stimuli — as is common during a typical circadian cycle.
Activity-dependent transcription evolves over minutes to hours and therefore in principle could support longer-term adaptation (Yap and Greenberg, 2018). For example, mature brain neurons are thought to use the regulated transcription of synaptic proteins and ion channels to homeostatically maintain their firing rates within a narrow target range (Davis, 2006, 2013; Marder, 2011; Marder and Goaillard, 2006; Turrigiano, 2011; Turrigiano, 1999; Turrigiano, 2017). If similar transcriptional mechanisms underlie adaptation in the periphery, then sensory neurons of a given type would be expected to occupy a range of transcriptional states, with the specific state adopted by any given neuron both reflecting its prior activity history and predicting its future responses to sensory inputs (Tyssowski and Gray, 2019). However, the relationship between activity-dependent gene regulation and functional adaptation remains elusive, in part because it has not yet been shown in either peripheral or central neurons that analog changes in activity yield proportional, bidirectional and systematic changes in the expression of genes that tune neural function (Davis, 2013).
There are ~1000 different olfactory sensory neuron (OSN) subtypes in the mouse, each of which can be identified across individuals based upon its expression of a single odorant receptor (OR) (Monahan and Lomvardas, 2015). ORs are G-protein coupled receptors that transduce odor binding into an influx of calcium, which leads to action potentials. The expressed OR endows each OSN with a specific level of neural activity that depends upon the odors present within a given environment. Although post-translational mechanisms sculpt OSN responses to rapidly fluctuating odor plumes, at longer timescales it is thought that OSNs faithfully report odor-receptor interactions to the brain. For example, OSN responses to odors (as assessed by functional imaging of OSN axons in the olfactory bulb) appear similar in individual mice tested at different times, across different mice, and in mice before and after training to associate a test odorant with a reward (Bozza et al., 2004; Chu et al., 2017; Kato et al., 2012; Rubin and Katz, 1999; Soucy et al., 2009; Spors and Grinvald, 2002; Wachowiak and Cohen, 2001). In contrast, olfactory bulb and cortical neurons adapt to the repeated presentation of an odor on timescales of minutes (Best et al., 2005; Cleland and Linster, 2005; McNamara et al., 2008; Wilson, 1998; Wilson, 2000; Yadon and Wilson, 2005). These findings suggest that ORs confer stable odor response properties upon OSNs, while the brain separates salient odors from background; this idea has pervasively influenced theoretical and computational models of odor coding (Brann and Datta, 2020; Chong et al., 2020; Grabska-Barwińska et al., 2016; Hopfield, 1991; Litwin-Kumar et al., 2017; Schaffer et al., 2018; Teşileanu et al., 2019; Wilson et al., 2017; Zwicker et al., 2016).
Consistent with this view, transcriptional analyses suggest that the genes expressed by mature OSNs are largely similar across subtypes, with the exception of the ORs themselves, a handful of genes related to dorsoventral position in the olfactory epithelium, and ~10 axon guidance genes that enable glomerular targeting (Fletcher et al., 2017; Hanchate et al., 2015; Mori and Sakano, 2011; Nakashima et al., 2013; Scholz et al., 2016; Vihani et al., 2020; Wu et al., 2018). This homogeneity suggests that individual OSN subtypes — which vary widely in their level of activity within a given environment — do not adapt their odor responses by actively modulating gene expression. In contrast, bulk RNA and activity measurements that pool across all OSNs in mice have hinted that OSNs may use transcription to compensate for the chronic absence or presence of activity, although results and interpretations have varied across experiments (Barber and Coppola, 2015; Cadiou et al., 2014; Coppola and Waggener, 2012; Coppola et al., 2006; Fischl et al., 2014; Fitzwater and Coppola, 2020; Hagendorf et al., 2009; Kass et al., 2012; Waggener and Coppola, 2007; Wang et al., 1993; Wang et al., 2017). Resolving these discrepancies — and ultimately testing the hypothesis that dynamic gene expression mediates long-term adaptation in sensory neurons — requires asking how odor-evoked activity influences gene expression within particular OSN subtypes; this level of specificity is essential to clarify whether OSN transcriptomes are changed in an OR- and environment-specific manner; whether these changes are limited to small numbers of genes (as appears to be the case in C. elegans (Cho et al., 2016; Juang et al., 2013; L’Etoile et al., 2002)) or organized into a systematic program of gene expression; and whether environment-dependent transcriptional changes actually adapt OR-specific odor responses.
To address these questions we characterized, via single cell RNA sequencing (scSeq), transcriptional variation across the extraordinarily large array of identifiable OSN subtypes. Our experiments reveal that each OSN subtype expresses a distinguishable set of transcripts that is stereotyped across mice within a given environment. The main axis of transcriptional variability includes more than 70 functionally-relevant genes that adaptively attenuate or amplify the transformation of odors into spikes. Furthermore, the expression of these genes coordinately and continuously varies across OSN subtypes, is modulated by specific interactions between ORs and the environment, and predicts OSN responses to new odors. Transcriptional variation among OSNs is therefore systematically organized through a rheostat-like mechanism (akin to a balance control on a stereo) whose setting in each OSN is defined by OR-environment interactions. Our findings suggest that the olfactory system uses this transcriptional rheostat to proportionally and bidirectionally adapt to persistent background odors — thereby enabling OSNs to engage in a form of sensory predictive coding — before odor information is transmitted to neural circuits responsible for perception. These data reveal that peripheral odor codes are flexible rather than fixed, and support a broad model in which neurons continuously individualize their transcriptomes to facilitate functional adaptation.
Results
To assess relationships between transcription and function in the peripheral olfactory system we performed scSeq on ~770,000 mature mouse OSNs (Table S1). Our initial analysis focused on ~40,000 mature OSNs derived from adult mice housed in a typical home cage environment; collectively these OSNs expressed over 1,000 odorant receptors (ORs), with nearly every mature neuron expressing only one OR (Figures 1A, S1A–D). UMAP embeddings of ~1,350 highly variable genes (HVGs, here and in all analyses excluding the OR genes themselves, see Methods) revealed that OSNs are partitioned into several broad subtypes related to positional identity and the expression of the lipid receptor CD36 (Figure 1B).
Figure 1. Each OR is associated with a distinct OSN transcriptome.
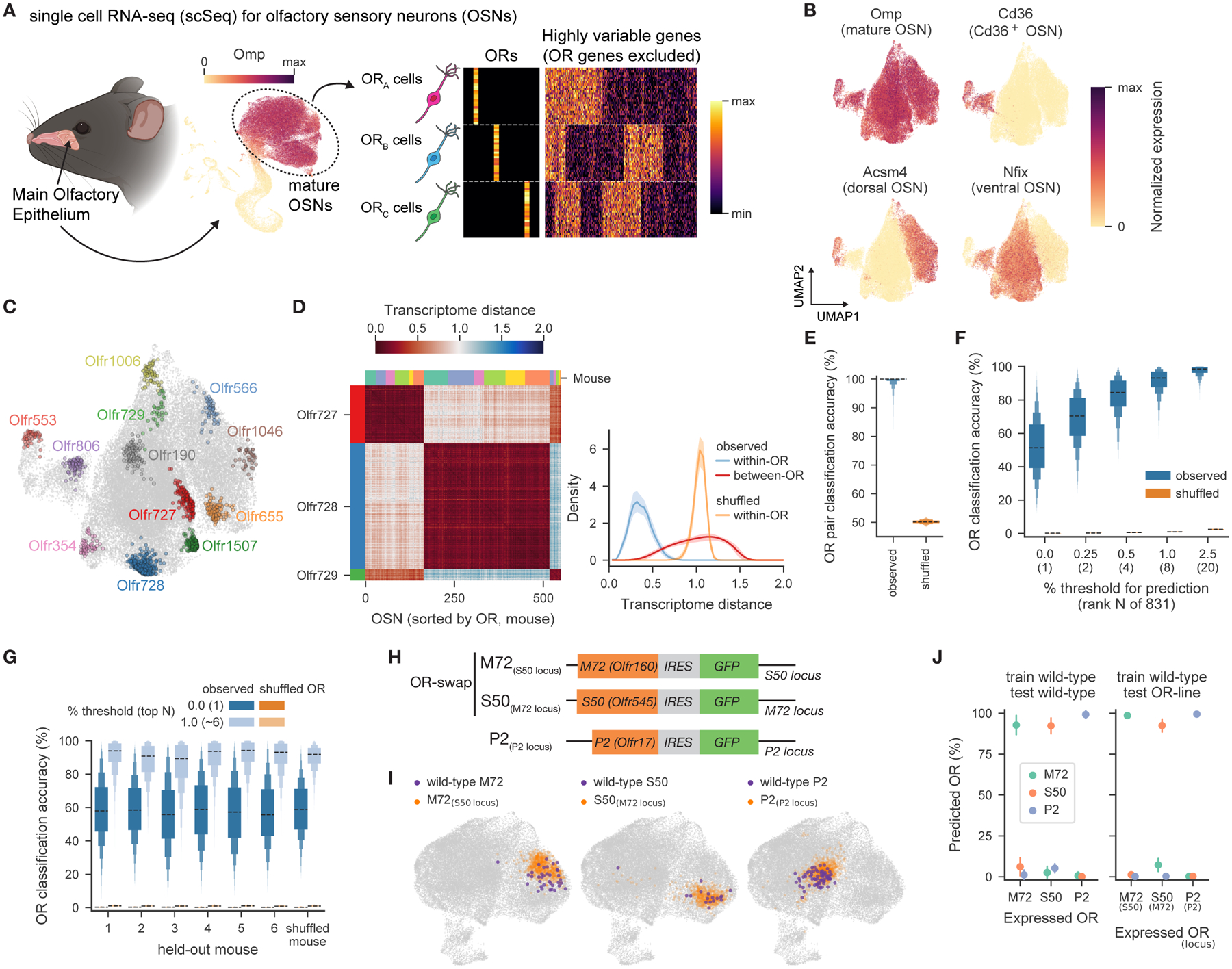
A. Schematic of single cell RNA-seq (scSeq) experiments. Odorant receptor (OR) genes, which identify each OSN subtype, were excluded from the highly variable genes used in downstream analyses of OSN gene expression.
B. UMAP plots visualizing gene expression in mature OSNs, with normalized expression of known identity-related marker genes (38,345 mature OSNs from 6 mice depicted).
C. UMAP plot depicting OSN subtypes expressing the indicated ORs.
D. (left) Transcriptome distances between individual OSNs expressing Olfr727, Olfr728 or Olfr729 (color bars indicate individual mice). (right) Distributions of distances between cells expressing the same OR (within-OR, mean and 2.5–97.5th percentile across 1,000 restarts indicated) or different ORs (between-OR, mean and interquartile range across OSN subtypes of the pairwise distances between a given OSN subtype and all others indicated).
E. Accuracy of pairwise linear classifiers at predicting via OSN transcriptomes which OR is expressed by a given OSN (black dashed line indicates median, boxes represent 25th/75th percentile, 12.5th/87.5th percentile, etc. across 831C2 =344,865 pairs). Classification performance is at chance levels upon shuffling OR labels across OSNs.
F. Accuracy of linear classifiers predicting which OR (out of 831) is expressed, at varying levels of prediction accuracy, from perfect (0.0%) to the rate at which the correct OR was within the top 2.5% of predicted ORs. Distributions depict the mean accuracy across the 831 OR-defined OSN subtypes (each averaged across 1,000 restarts). Performance is at chance levels upon shuffling OR labels across OSNs.
G. Accuracy of linear classifiers predicting which OR is expressed in an OSN from a held-out mouse, with training data provided from 5 separate mice. Both perfect (dark blue) and the top 1% (light blue) of predictions are shown.
H. Schematic of the genomic loci for the OR-swap and P2-IRES-GFP mouse lines.
I. UMAP plots of mature OSNs from wild-type and the mice shown in H. highlighting OSNs expressing M72, S50, or P2 receptors.
J. Accuracy of linear classifiers predicting OR identity for OSNs expressing M72, S50, or P2, trained and tested as indicated.
See also Figure S1.
OSN transcriptomes are stereotyped and uniquely specified by the expressed OR
Given the few known transcriptional differences among mature OSNs, we expected that OR-defined OSN subpopulations would overlap in UMAP space; instead, OSNs expressing each OR were locally clustered and separated from those expressing different ORs, suggesting that each OSN subtype is transcriptionally unique (Figures 1C, S1E). For example, the transcriptomes of OSNs expressing Olfr727 were self-similar, and distinct from OSNs expressing Olfr728 and Olfr729 (Figures 1D). The transcriptomes of all OR-identified OSN subtypes were distinguishable, regardless of how transcriptome distances were computed or the frequency with which each OR was expressed (Figures 1D, S1F–I). Linear classifiers distinguished OSNs expressing different ORs with near perfect accuracy and predicted the specific expressed OR (of the 831 queried) with >50 % accuracy (vs. 0.1 % at chance) (Figures 1E–F, S1J). Approximately 1000 highly variable genes were required for maximum classification accuracy, although individual genes contributed marginally to overall performance (Figures S1K–L). Importantly, classifiers accurately predicted the identity of the expressed OR in OSNs derived from held-out mice, indicating OR-transcriptome relationships generalize across individuals housed in a similar environment (Figure 1G).
The unexpectedly close relationship between OR expression and OSN transcriptomes could result from shared regulatory mechanisms, or from each OR determining its associated transcriptome. To distinguish these possibilities, we performed scSeq in mice with swapped coding regions for the M72 (Olfr160) and S50 (Olfr545) receptor genes (Figure 1H). OSNs expressing M72 receptor from the S50 gene locus (M72(S50 locus)) aligned with wild-type M72 OSNs; likewise, OSNs expressing S50 from the M72 gene locus S50(M72 locus) mice resembled wild-type S50 OSNs (Figures 1I–J). The unique gene expression signature associated with each OSN is therefore largely specified by the expressed OR protein.
OSNs express a limited number of transcription factors and axon guidance genes in an OR-dependent manner during development (Mori and Sakano, 2011; Parrilla et al., 2016). However, genes from these categories were neither required for accurate OR predictions nor were as predictive as either the complete set or the most predictive subset of HVGs (Figure S1M). Each OSN can also be categorized by the expression of CD36, the specific class of its expressed OR (Class I vs Class II), or the dorsal or ventral location of its associated glomeruli in the olfactory bulb; classifiers accurately distinguished ORs from OSNs within or between each of these categories (Figure S1N–O). Furthermore, classifiers distinguished OSNs independently of the genomic or phylogenetic relationships between ORs (Figure S1P–Q). Therefore, known differences relating to OSN identity or genomic/phylogenetic features of ORs do not explain the transcriptional diversity of mature OSNs.
Decomposing OSN transcriptomes into identity and activity gene expression programs
Alternatively, the diversity in OSN transcriptomes might reflect OR-associated differences in neural activity. To test this possibility, we performed consensus non-negative matrix factorization (cNMF), which decomposes gene expression patterns observed across OSNs into sets of co-expressed genes called gene expression programs (GEPs) (Figure 2A, see Methods); importantly, cNMF successfully identified both identity-associated and activity-related GEPs in a prior study (Kotliar et al., 2019). cNMF identified 10 GEPs — each composed of 100s of largely non-overlapping genes — that effectively captured OR-driven transcriptional variability, whose usages varied across OSNs in an OR-specific manner, and which accurately predicted the expressed OR in each OSN (Figures 2B and S2A–D).
Figure 2. OSN transcriptional variation can be decomposed into identity and activity gene expression programs.
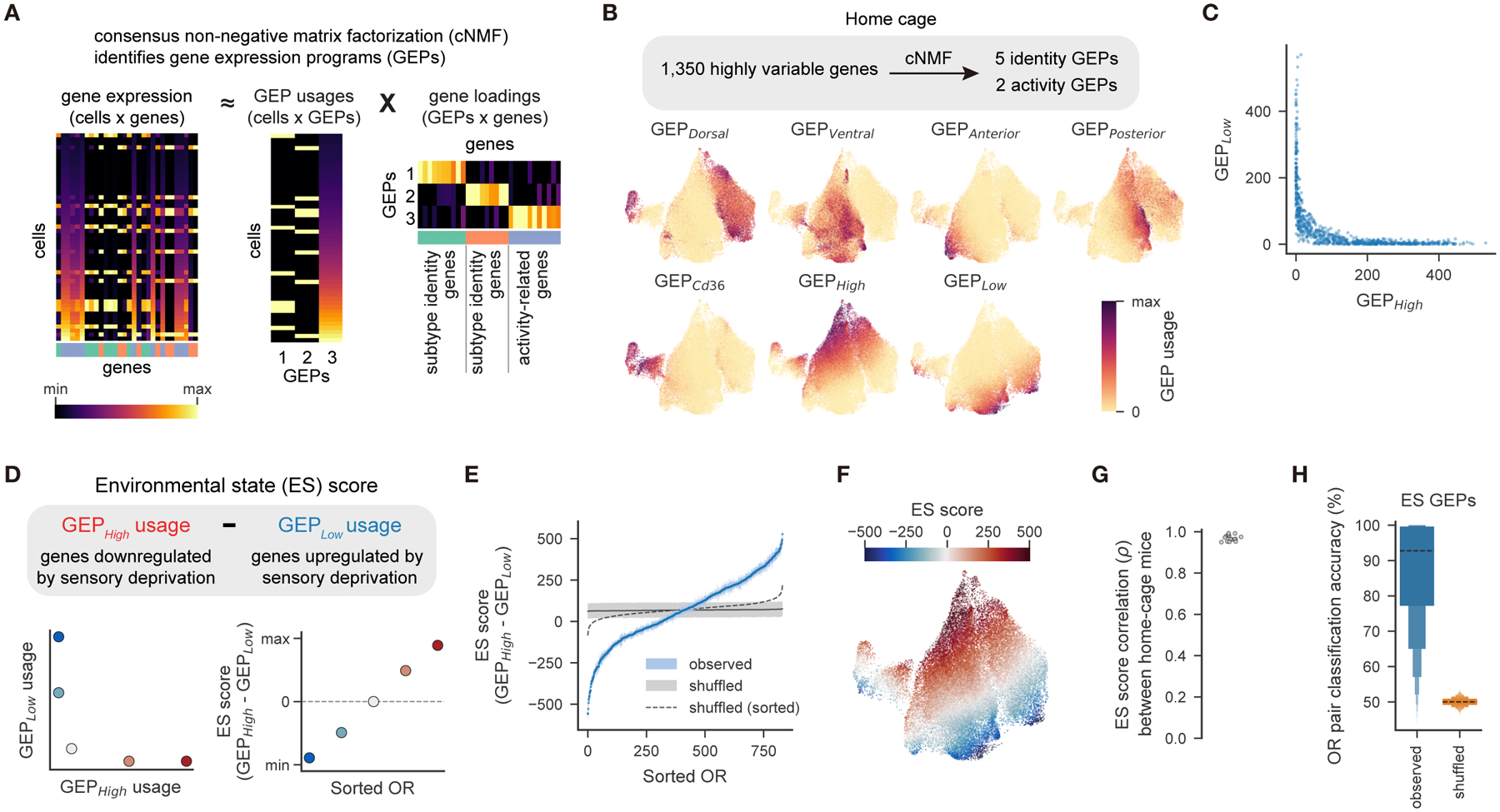
A. Consensus non-negative matrix factorization (cNMF) identifies coherent gene expression programs (GEPs). Gene expression in each cell can be decomposed into a set of usages across a small number of GEPs, each of which are defined by their loadings for each gene.
B. UMAP plots showing the usage of each functionally annotated GEP in mature OSNs from wild-type mice housed in the home cage.
C. GEPHigh and GEPLow usages by the OSNs shown in B. Each point indicates the mean GEP usage for all OSNs expressing the same OR for each of the 831 ORs detected in at least 10 OSNs.
D. Environmental state (ES) scores for each OSN are calculated by taking the difference between GEPHigh and GEPLow.
E. Mean ES score for each OSN subtype. ES scores were shuffled 1,000 times across cells, and the mean interquartile range across shuffles is depicted in gray for each OSN subtype, with the mean across sorted shuffles depicted by the dashed line.
F. As in B. but for ES scores.
G. Between-mouse correlation coefficients of the mean ES score for each OSN subtype.
H. Accuracy of pairwise linear classifiers (as in Fig. 1E) predicting OR identity using only the usages of GEPHigh and GEPLow.
Of the 10 identified GEPs, we focused on the seven whose constituent genes suggested putative functions (Figure S2E). Five GEPs encoded known aspects of OSN identity (dorsal, ventral, anterior, posterior, and CD36-positivity, Figures S2B, S2F–H, Table S2). The other two GEPs, which we refer to as GEPHigh and GEPLow, included genes associated with neural activity in OSNs: GEPHigh included genes downregulated by chronic sensory deprivation (e.g., S100a5, Pcp4l1, and Kirrel2) while GEPLow included genes upregulated after chronic sensory deprivation (e.g., Calb2, Kirrel3, and Ppp3caI) (Fischl et al., 2014; Wang et al., 2017). Each OSN primarily used either GEPLow or GEPHigh, and the usage of each GEP varied continuously across OSN subtypes (Figures 2C, S2B).
We hypothesized that persistent interactions between each OR and the wide variety of odors in a given environment (like the food, bedding and semiochemical odors present in a typical home cage) might explain the continuous variation of GEPLow or GEPHigh across OSN subtypes. To explore this idea, we developed a metric (simply the difference between GEPHigh and GEPLow) that we provisionally refer to as the environmental state (ES) score, (Figure 2D). ES scores for OSNs derived from mice housed in the home cage varied continuously across OSN subtypes; however, OR-specific ES scores were remarkably consistent across mice and enabled accurate predictions of OR identity (Figure 2E–H). Both ES scores and GEPHigh and GEPLow usages aligned with the first principal component of the OSN transcriptomes (Figure S2I–J). GEPHigh and GEPLow genes were not expressed in immature OSNs but their expression increased and diversified after the onset of OR expression (Figure S2K–L).
OSN transcriptomes systematically reflect environment-dependent activity
Consistent with the hypothesis that GEPHigh and GEPLow genes, and thus ES scores, are sensitive to the chronic activity state of each OSN, artificially lowering (via nares occlusion for a month) or raising (via pulsed optogenetic stimulation for 12 hours) OSN activity systematically decreased or increased ES scores, respectively (Figures 3A–H, S3A–E). To address whether OSN ES scores are sensitive to chronic activity levels under more physiological conditions, we compared OSN gene expression between mice housed in either a regular home cage or in two distinct naturalistic odor environments (Figure 3I). ES scores shifted significantly and bidirectionally in ~45 % of OSN subtypes across environments, consistent with specific odor environments differentially ligating or disengaging distinct ORs (Figure 3J–N, S3F–H).
Figure 3. OSN transcriptomes are shaped by the odor environment.
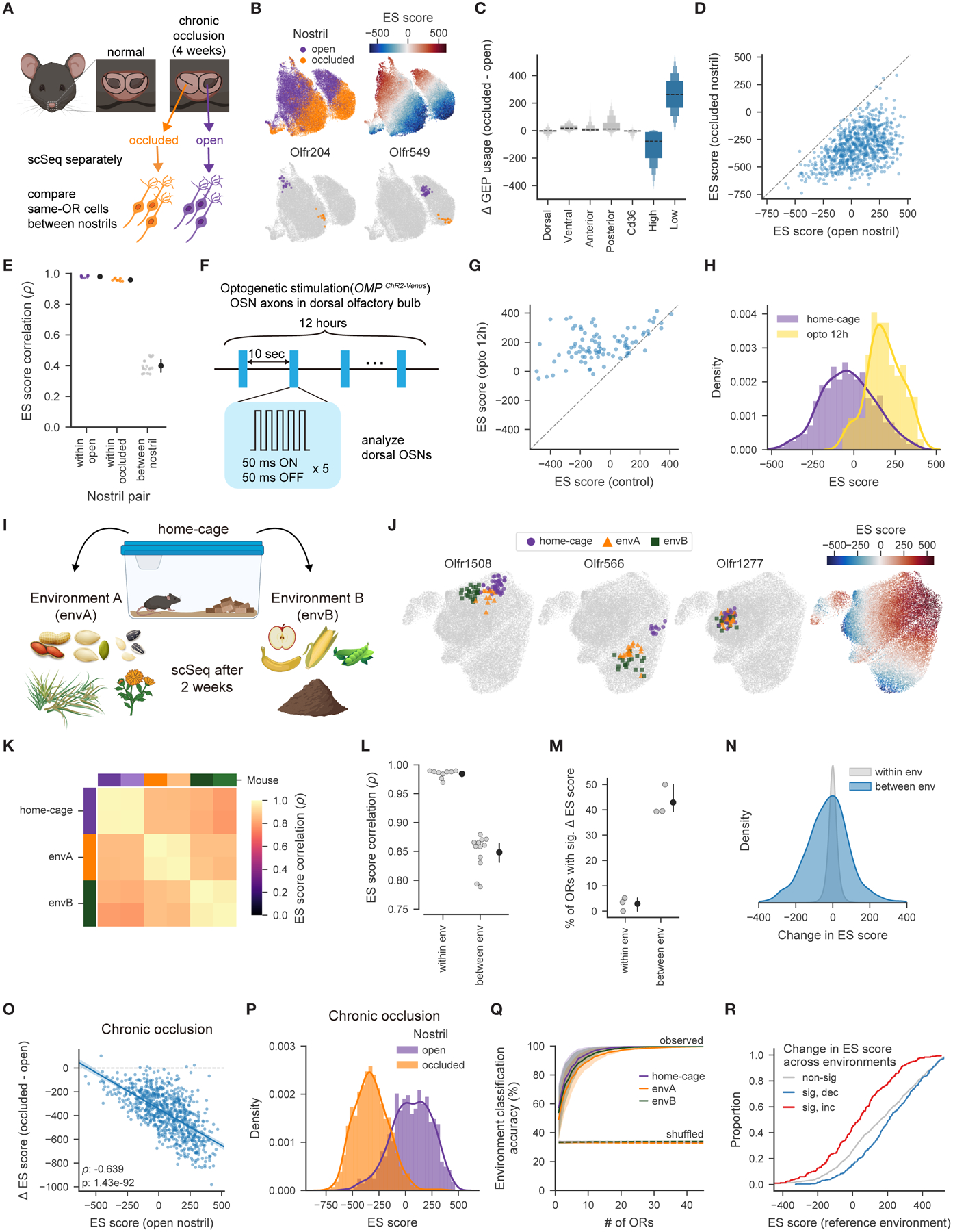
A. Schematic of the chronic naris occlusion experiment.
B. UMAP plots of mature OSNs depicting the nostril of origin, ES scores, and OSNs expressing example ORs.
C. Mean occlusion-dependent changes in GEP usage across OSN subtypes, as defined by OR expression.
D. Mean ES score for each OSN subtype for each nostril. ES scores decreased by occlusion for 770 out of 797 OSN subtypes (FDR ≤ 0.01).
E. Correlation of the mean ES scores for each OSN subtype from the indicated nostril pairs (p=1.68 × 10−5, Mann-Whitney U test, for both within- vs. between-nostril comparisons).
F. Schematic of the chronic optogenetic stimulation experiment.
G. Mean ES score for each dorsal OSN subtype from stimulated and control (no-light) mice. ES scores increased following optogenetic stimulation for 56 out of 90 subtypes (FDR ≤ 0.01).
H. Distribution of mean ES scores across OSN subtypes from each condition. ES scores following optogenetic stimulation are right-shifted (p=1.33 × 10−34, KS-test).
I. Schematic of the environment switch experiment.
J. UMAP plots highlighting (left) OSNs expressing example ORs from mice housed in each of the three environments. (right) OSN ES scores.
K. Pairwise correlations of the mean ES scores for each OSN subtype, across mice from the indicated environments.
L. Pairwise correlations from K. separated by the type of comparison. Correlations are higher for mice housed within the same environment (p=7.17 × 10−7, Mann-Whitney U test).
M. Percent of OSN subtypes whose ES scores change significantly (FDR ≤ 0.01), for animals from the same or different environments.
N. Distribution of ES score changes, for OSN subtypes from the same or different environments. ES score changes are larger between environments (p=5.16 × 10−140, KS-test).
O. The observed occlusion-induced change in ES scores for each OSN subtype, as a function of ES scores from open nostrils.
P. Distribution of the mean ES scores across OSN subtypes from each nostril. ES scores following occlusion are left-shifted (p=3.74 × 10−217, KS-test).
Q. Accuracy of a minimum distance classification procedure (see Methods) to predict in which environment a mouse was housed, based upon ES scores for each OSN subtype (observed or shuffled environment as labeled, curves depict the mean and interquartile range of classification accuracy for each environment across 1000 restarts).
R. Cumulative distribution of ES scores in a given reference environment for OSN subtypes whose ES scores were significantly increased, decreased, or unchanged across environments. Those OSNs whose ES scores decreased tended to have higher ES scores in the reference environment (p=4.03 × 10−8, KS-test); the opposite was true for those OSNs whose scores increased (p=2.32 × 10−14).
See also Figure S3.
Three additional lines of evidence argue that ES scores reflect specific interactions between each OR and each environment. First, occlusion-dependent changes in ES scores for each OR-defined OSN subtype were negatively correlated with the ES score in the open nostril (Figure 3O). ES scores after occlusion were sufficiently diverse to enable accurate predictions of OR identity; this diversity might reflect a variety of OR-specific spontaneous activity patterns (Figures 3P, S3I–K). Second, after switching environments, ES scores for each OSN subtype were consistent across mice and accurately predicted the specific environment in which each mouse was housed (Figures 3Q, S3L-M). Finally, OSN subtypes whose ES scores increased across odor environments tended to have relatively low initial ES scores, and conversely those that fell tended to have relatively high initial ES scores (Figure 3R).
We ruled out several alternative explanations for environment-dependent changes in OSN ES scores. Odor-dependent modulation in OR expression levels or β-Arrestin2-mediated OR endocytosis could, in principle, influence ES scores (Ibarra-Soria et al., 2017; Mashukova et al., 2006; von der Weid et al., 2015). However, across all three of our experimental manipulations, OR gene expression and the usage of identity-related GEPs remained constant (Figures 3C, S3C, S3H, S3N–Q). Furthermore, β-Arrestin2 knockout mice exhibited largely normal ES scores that adapted to a new environment (Figures S3R–T). Taken together, these results reveal that specific interactions between each OR and the environment differentially drive activity in each OSN, which in turn specifies the particular level at which GEPHigh and GEPLow genes are expressed. Our results also demonstrate that, under physiological conditions and in realistic odor environments, odors and air chronically engage the entire OSN array.
Structured correlations and flexibility in genes influencing odor-activity coupling
What functions might environment-dependent gene expression confer upon mature OSNs? Inspection revealed that GEPHigh and GEPLow include 73 genes with known or likely roles in shaping OSN sensory responses (e.g., calcium homeostasis, OR signaling, and intrinsic excitability), and an additional ~40 genes putatively involved in axon guidance or synaptic transmission (Figure 4A, Table S3). The expression of these genes smoothly co-varied across OSN subtypes and exhibited structured correlations and anticorrelations with other GEPHigh and GEPLow genes (Figures 4B, S4A–C). Like the ES score itself, the expression of “functional” genes (here and throughout, defined as the set of 73 GEPHigh and GEPLow genes relevant to sensory responses) both distinguished all OSNs and systematically fell after naris occlusion (Figures S4D–F). The observed pattern of functional gene expression in each OSN suggested a role in compensating for ongoing activity; the expression of genes that likely attenuate odor responses was higher in OSNs with high ES scores than in those with low ES scores, whereas genes that likely amplify responses showed the opposite trend (Figures 4C–D). Furthermore, persistently changing OSN activity by chronic occlusion, optogenetic stimulation, and switching environments caused functional gene expression to change bidirectionally in an OR-dependent manner (Figures 4E–F and S4G–K, Table S4).
Figure 4. Large-scale transcriptional variation is organized into an environment-dependent rheostat.
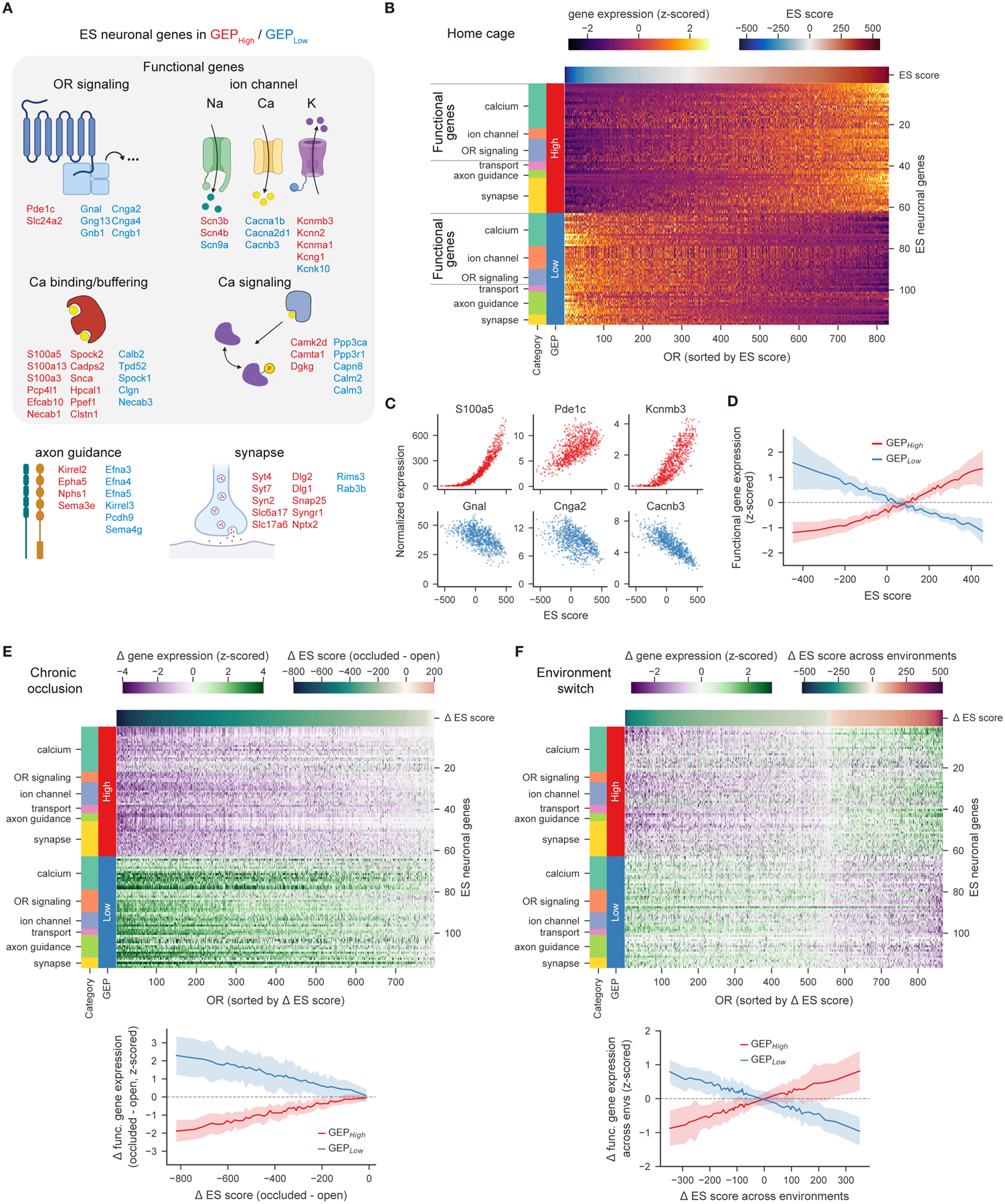
A. Categorization of ES neuronal genes with high loadings in either GEPHigh or GEPLow, including “functional” genes that may regulate sensory responses. A subset of the 117 genes used in B, E, F are shown.
B. Heatmap of the z-scored expression across OSN subtypes of 117 ES neuronal genes (including 73 functional genes).
C. Expression (normalized by the total number of transcripts per cell and averaged across cells for each OSN subtype) of example functional genes associated with either GEPHigh (red) or GEPLow (blue), as a function of ES scores.
D. Expression of 73 functional genes as a function of OSN ES scores (binned into 50 quantiles); plots depict the mean and standard deviation of the z-scored expression of functional genes associated with GEPHigh (red) and GEPLow (blue).
E. (top) Heatmap of the chronic occlusion-dependent change in z-scored expression of ES neuronal genes for each OSN subtype. (bottom) Similar to D., but for changes in both functional gene expression and ES scores.
F. Similar to E. but for environment-dependent changes in functional gene expression.
Act-Seq quantifies odor-evoked OSN activation
To test the possibility that the continuous variation in functional gene expression uniquely adapts the odor responses of each OR-defined OSN subtype to each environment, we subjected the olfactory epithelium to Act-Seq, a scSeq variant in which neural activity is read out as a rapid change in gene expression (Wu et al., 2017). Act-Seq reliably identified a subset of OSNs (and therefore ORs) that acutely responded after a two-hour exposure to the volatile odorants acetophenone and octanal, as assessed by immediate early gene (IEG) expression (with similar results obtained using unsupervised analysis of odor-evoked transcriptional changes; Figures 5A–B and S5A, see Methods). Act-Seq identified a subset of ORs (~12–13 % of total ORs) activated by either acetophenone or octanal; these included most known responsive ORs for each odor, as well as many new acetophenone and octanol receptors (Figures S5B–D) (Bozza et al., 2002; Jiang et al., 2015; Repicky and Luetje, 2009; von der Weid et al., 2015).
Figure 5. Act-Seq identifies odor-responsive receptors and reveals that OSN transcriptomes determine acute in vivo odor responses.
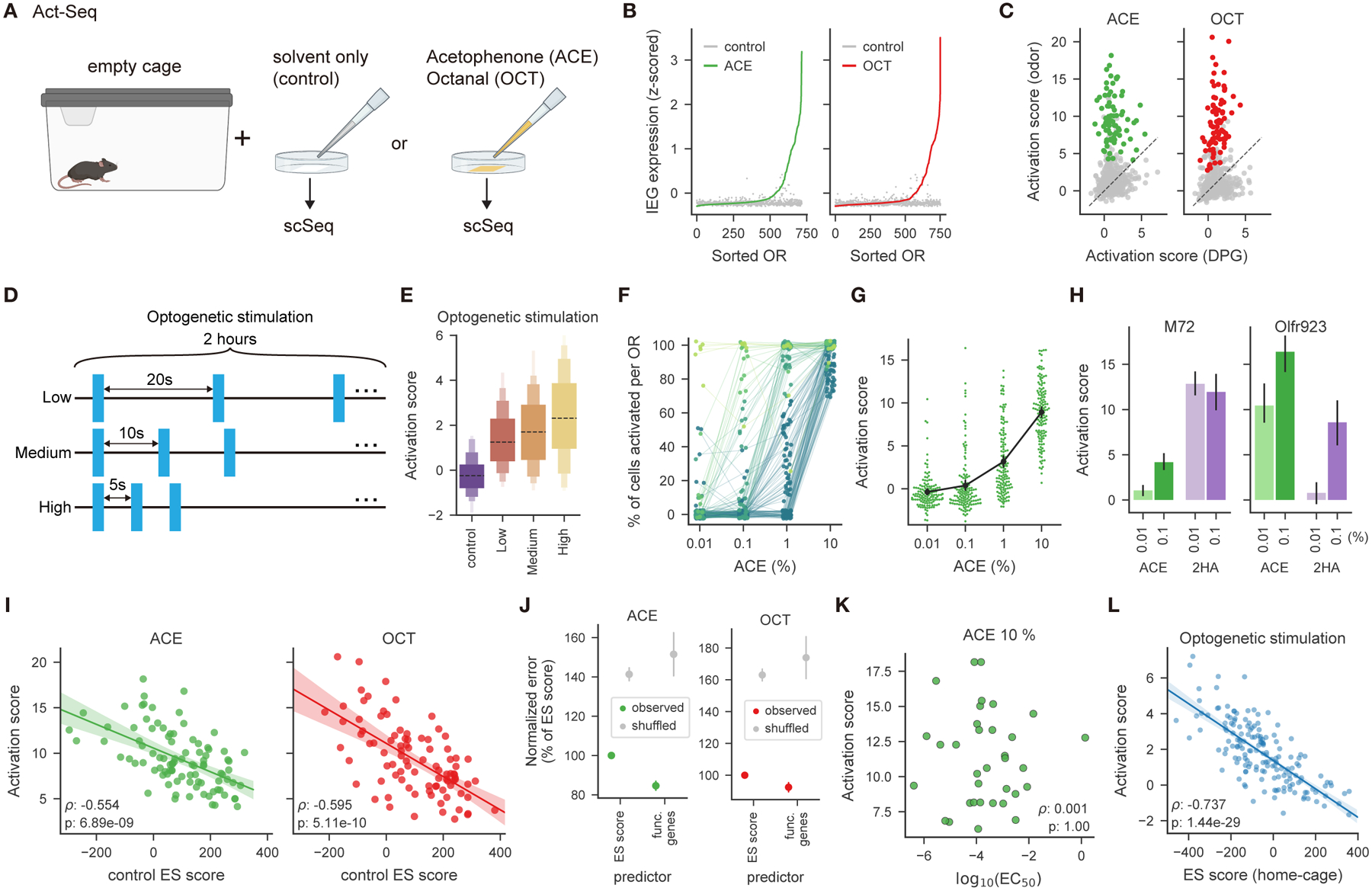
A. Schematic of the Act-Seq experiment.
B. z-scored expression of 10 immediate early genes (IEGs, listed in Methods) for each OSN subtype from control or odor conditions (acetophenone (ACE) or octanal (OCT)), sorted for each odor. IEG expression was higher in the odor conditions (p < 1 × 10−18 for each odor via Wilcoxon signed-rank test).
C. Odor-evoked activation score for each OSN subtype, as a function of that evoked by control solvent (dipropylene glycol, DPG). Odor-evoked activation scores are higher for responsive OSN subtypes (in color, p < 1 × 10−14, one-sided Wilcoxon signed-rank test for each odor).
D. Schematic of the acute (two hour) optogenetic stimulation experiment.
E. Distribution of mean activation scores for OSN subtypes expressing dorsal ORs in optogenetically-stimulated and control conditions. Activation scores increase with stimulation frequency (p < 1 × 10−5, one-sided Jonckheere-Terpstra trend test).
F. Percent of cells activated by acetophenone, for OSN subtypes that were responsive at 10% (and colored dark to light by the number of concentrations that activated each OSN subtype), increase with concentration (p < 1 × 10−5, one-sided Jonckheere-Terpstra trend test).
G. Activation scores, for OSN subtypes that were responsive at 10%, increase with increasing concentrations of acetophenone, (p < 1 × 10−5, one-sided Jonckheere-Terpstra trend test).
H. Activation scores for OSNs expressing either M72 (Olfr160) or Olfr923 to either acetophenone or 2-hydroxy acetophenone (2HA) at the indicated concentrations.
I. Activation scores for each odor as a function of control ES scores, for odor-responsive OSN subtypes.
J. Mean-squared error of predicted activation scores via linear regression models fit on either ES scores or expression of 73 functional genes, for odor-responsive OSN subtypes. Error bars represent mean and SD across 1,000 restarts, normalized to the models fit on ES scores.
K. Activation scores for acetophenone-responsive OSN subtypes whose ORs have known in vitro EC50 values (Jiang et al., 2015; von der Weid et al., 2015).
L. Similar to I. but for optogenetically-activated dorsal OSN subtypes (Medium intensity in D.).
Acetophenone and octanal elicited similar acute transcriptional changes across many genes (Figure S5E). We therefore developed an analog measure of odor response (referred to herein as an activation score) to quantitatively summarize information from the ~500 genes (including IEGs) that were reliably and rapidly changed by odor exposure (and which lacked strong ES score correlations, Figure 5C, and Table S5, see Methods). To test whether activation scores capture meaningful variation in activity we subjected OSNs to optogenetic stimulation, which revealed a tonic relationship between stimulation frequency and activation scores (Figure 5D–E). Similarly, increasing concentrations of acetophenone tonically increased both the number of ORs activated by acetophenone and the activation scores of acetophenone-responsive OSNs (Figures 5F–G, S5F).
To test whether activation scores also capture differences in binding affinities between different ligands for the same receptor, we took advantage of 2-hydroxyacetophenone (2-HA) which, relative to acetophenone, binds more strongly to the M72 receptor (Arneodo et al., 2018; Zhang et al., 2012). Act-Seq revealed that 2-HA indeed elicits higher activation scores than acetophenone in OSNs expressing M72 (Figure 5H). Conversely, activation scores for OSNs expressing Olfr923, a high affinity acetophenone receptor, were higher for acetophenone than 2-HA (Hu et al., 2020; Jiang et al., 2015). Furthermore, 2-HA, acetophenone, and two related odors each elicited a broad range of OR- and odor-specific activation scores that were stereotyped across mice and accurately predicted the specific odor each mouse experienced (Figures S5G–M). Taken together, these results demonstrate that Act-Seq can identify the OSN subtypes (and thus the ORs) that can bind to and respond to any odorant, and that the activation score affords a quantitative transcriptional measure of the degree to which a given OSN has responded to an odor in vivo.
OSN transcriptomes predict acute odor responses
Consistent with the hypothesis that the transcriptome of each OSN shapes its acute odor responses, ES scores were negatively correlated with odor activation: the higher the ES score for a given acetophenone-responsive OSN subtype (identified across experiments via its OR), the lower its acute, acetophenone-evoked response (Figures 5I, S10N–O). This negative correlation was also observed in OSNs responding to octanal and to the three acetophenone-related odors (Figures 5I, S5O–P). The 73 functional genes were as effective as the ES score at predicting acute OSN odor responses, but the in vitro EC50 of each acetophenone OR was unable to support in vivo odor response predictions (Figures 5J–K, S5Q–R) (Jiang et al., 2015; Saito et al., 2009). Interestingly, OSN activation scores following two hours of acute optogenetic stimulation were also negatively correlated with control ES scores, indicating that the ability of light to transcriptionally activate each OSN also depends on the expression levels of functional genes (Figures 5L, S5S).
If functional gene expression indeed plays a causal role in determining odor responses, then experimentally raising or lowering ES scores should predictably change acute odor responses (Figure 6A). We tested this hypothesis by lowering OSN activity via transient unilateral naris occlusion using removable plugs, and then performing Act-Seq after acute odor exposure (Figure 6B). Transient occlusion reduced ES scores in an OR-specific manner but did not change the correlation structure between functional genes (Figures 6C, S6A–B). Similar sets of ORs were activated by acetophenone in control conditions and after occlusion, but acetophenone receptor-expressing OSNs derived from occluded nostrils exhibited higher activation scores (Figures 6D, S6C). Critically, the occlusion-dependent changes in ES scores and functional gene expression predicted the occlusion-dependent changes in odor-evoked activation (Figures 6E, S6D–E).
Figure 6. Changes in OSN transcriptomes predict changes in acute odor responses, and acute odor responses predict long-term transcriptome changes.
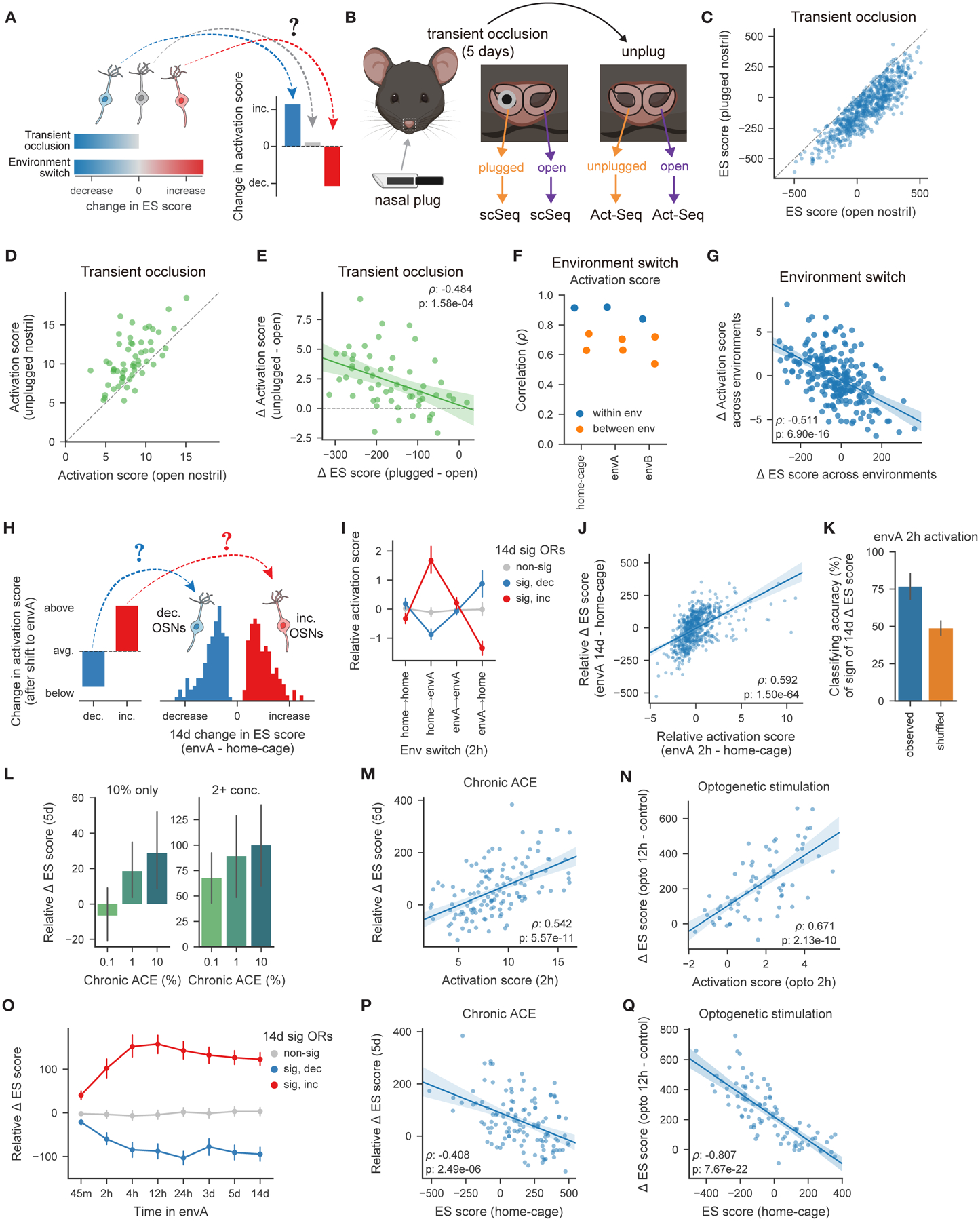
A. Model depicting how occlusion- or environment-dependent changes in ES scores could affect acute odor responses.
B. Schematic of transient naris occlusion followed by Act-Seq.
C. ES score, for the 745 OSN subtypes identified in transiently-occluded (but not unplugged) mice. Occlusion decreased ES scores for 726 OSN subtypes (554 significantly at FDR ≤ 0.01).
D. Activation scores for acetophenone-responsive OSN subtypes are higher in unplugged than open nostrils (p = 2.29 × 10−7, Wilcoxon signed-rank test).
E. Changes in activation scores between acetophenone-responsive OSN subtypes from open or unplugged nostrils, as a function of the occlusion-dependent change for each subtype (as measured in the data in C.).
F. Correlation of the activation scores for acetophenone-responsive OSN subtypes, using data from either the same or different environments. Activation scores are more consistent within a given environment (p = 0.014 Mann-Whitney U test).
G. Changes in activation scores across environments, as a function of changes in ES scores across environments, for acetophenone-responsive OSN subtypes.
H. Model depicting how activation upon a shift to a novel odor environment like environment A (envA) could subsequently change ES scores.
I. Activation scores (relative to the mean across all OSN subtypes) two hours after the indicated environment shift. Compared to shifts between the same environment, home-envA and envA-home shifts induced opposing changes in activation scores, specifically for OSN subtypes with either significant increases or decreases in ES scores after two weeks (p < 1 × 10−5 one-sided Jonckheere-Terpstra trend test for both; p= p = 0.662 for OSN subtypes that remained constant after two weeks).
J. Changes in ES scores as a function of activation scores observed two hours after a shift to envA, for each OSN subtype (relative to their mean across all OSN subtypes).
K. Accuracy of classifiers predicting whether ES scores would rise or fall after two weeks, based upon the activation scores observed two hours after a shift to envA. For 1,000 restarts, classifiers were fit using only OSN subtypes with significant ES scores changes after two weeks in envA, and the mean accuracy was summarized across OSN subtypes (error bars depict the mean and SD across restarts, accuracy is at chance levels after permuting the signs of the ES score changes).
L. Mean change in ES scores after five days of chronic exposure (relative to the mean across all OSN subtypes) to the indicated concentration of acetophenone for OSN subtypes that were acutely responsive to 10% odor (as identified in the experiments depicted in Fig. 5). Subtypes were separated into those responding to either 10% alone or at least two concentrations (significant effect of OR responsivity (10% only vs. at least two concentrations, F(1,270) = 43.04, p = 2.72 × 10−10) and chronic acetophenone concentration (F(2,270) = 4.48, p = 0.012) on ES score changes, but the interaction was not significant (F(2,270) = 0.0085, p = 0.99), via two-way ANOVA).
M. Change in ES scores (relative to the mean across all OSN subtypes) after chronic exposure to acetophenone, as a function of the acute activation score, for all acetophenone-responsive OSN subtypes identified at each concentration.
N. Same as M, but for ES score changes as a function of activation scores after acute optogenetic stimulation (Medium intensity, see Fig. 5D).
O. Change in ES scores (relative to the mean across all OSN subtypes), at various timepoints after a home-envA shift, for OSN subtypes categorized based on the sign and significance of their two-week ES score changes.
P. Similar to M. but for changes in ES scores (relative to the mean across all OSN subtypes) induced by chronic exposure to acetophenone as a function of ES scores from mice housed in the home-cage.
Q. Similar to P. but for optogenetic stimulation and plotting changes in ES scores.
See also Figure S6.
Similarly, switching mice from the home cage into a new odor environment (which bidirectionally modulates ES scores (Figures 3I–N)), altered acetophenone-driven OSN activation scores in an environment-dependent manner (Figure 6F, S6F–I); these scores were sufficiently different across environments and reliable across mice that they could be used to identify the environment in which a given mouse was housed (Figures 6F, S6J–L). Activation scores for acetophenone-responsive OSN subtypes were also negatively correlated with ES scores within a given environment, and the bidirectional changes in ES scores or functional gene expression following environment shifts predicted the corresponding change in the acetophenone response for each OSN subtype (Figures 6G, S6M–N). Together, these results demonstrate that odor-evoked activation is not determined solely by the affinities of odors for their receptors, but rather is flexibly tuned in each OSN by functional genes, which adapt OSNs in an OR-specific manner to the specific contents of each odor environment.
Acute odor responses predict future changes in functional gene expression
We wondered how different environments induce changes in OSN transcriptomes, and hypothesized that acute activity triggered by new odor environments sculpts long term functional gene expression (Figure 6H). For OSN subtypes whose ES scores changed significantly across environments, switching between different environments recruited changes in OSN activation scores, while activation was negligible in mice moved between similar environments, demonstrating that OSN activation captures salient changes in the odor environment (Figure 6I). Furthermore, OSN-specific activation scores measured two hours after an environment shift predicted OSN ES score changes measured after two weeks (Figures 6J–K). Similarly, OSN activation scores observed after acute two-hour exposure to acetophenone or optogenetic stimulation predicted future ES score changes after chronic acetophenone exposure or prolonged optogenetic stimulation, respectively (Figures 6L–N, S6O). These data demonstrate that ES scores adjust proportionally to acute perturbations in activity. Interestingly, much of the long-term change in ES scores and functional genes was apparent four hours after switching environments, suggesting that ES scores integrate activity on timescales of hours (Figures 6O, S6P–Q). In contrast, transient two-hour exposure of mice to acetophenone did not change ES scores after 24 hours (Figure S6R), indicating that the ongoing presence of odors is required for persistent changes in functional gene expression.
We observed that the long-term ES score changes observed after chronic acetophenone exposure could be predicted by the baseline ES score of each acetophenone-responsive OSN subtype in the home cage; a similar relationship was observed following optogenetic stimulation (Figures 6P–Q). Given that baseline ES scores also determine acute responses after odor or light stimulation (Figures 5I, 5L), these results are consistent with odor-evoked activity and ES scores being linked together in a closed loop. We noted that OSNs with high ES scores exhibited some residual IEG expression in the home cage (Figure S6S). This finding suggests that transcription-based adaptation in OSNs compensates for but does not fully eliminate activity driven by background odors, suggesting that under normal circumstances OSNs are continuously re-adapting to their environment.
Odor environments influence odor codes accessed by the brain
To test whether the ambient environment also shapes odor codes in the brain, we performed presynaptic functional imaging of OSN axons in the olfactory bulb (Figure 7A). OSN axons expressing the same OR converge on one or more insular structures known as glomeruli; imaging pre-synaptic OSN neural activity in olfactory bulb glomeruli can therefore characterize odor responses across different OSN subtypes. We imaged responses to a panel of purified odors before, during, and after a two-week period in which mice were housed in one of the alternative odor environments described above (Figures 7B, S7A–B). Response amplitudes to the same odor stimulus changed bidirectionally in ~50 % of glomeruli after mice were switched from the standard home cage into a new environment, consistent with prior work exploring the influence of a single odor on subsequent responses (Kass et al., 2016); these environment-dependent changes were consistent across both mice and odorants, and reverted when mice were later returned to standard home cages (Figures 7C–E, S7C–F).
Figure 7. The ambient odor environment determines functional odor responses.
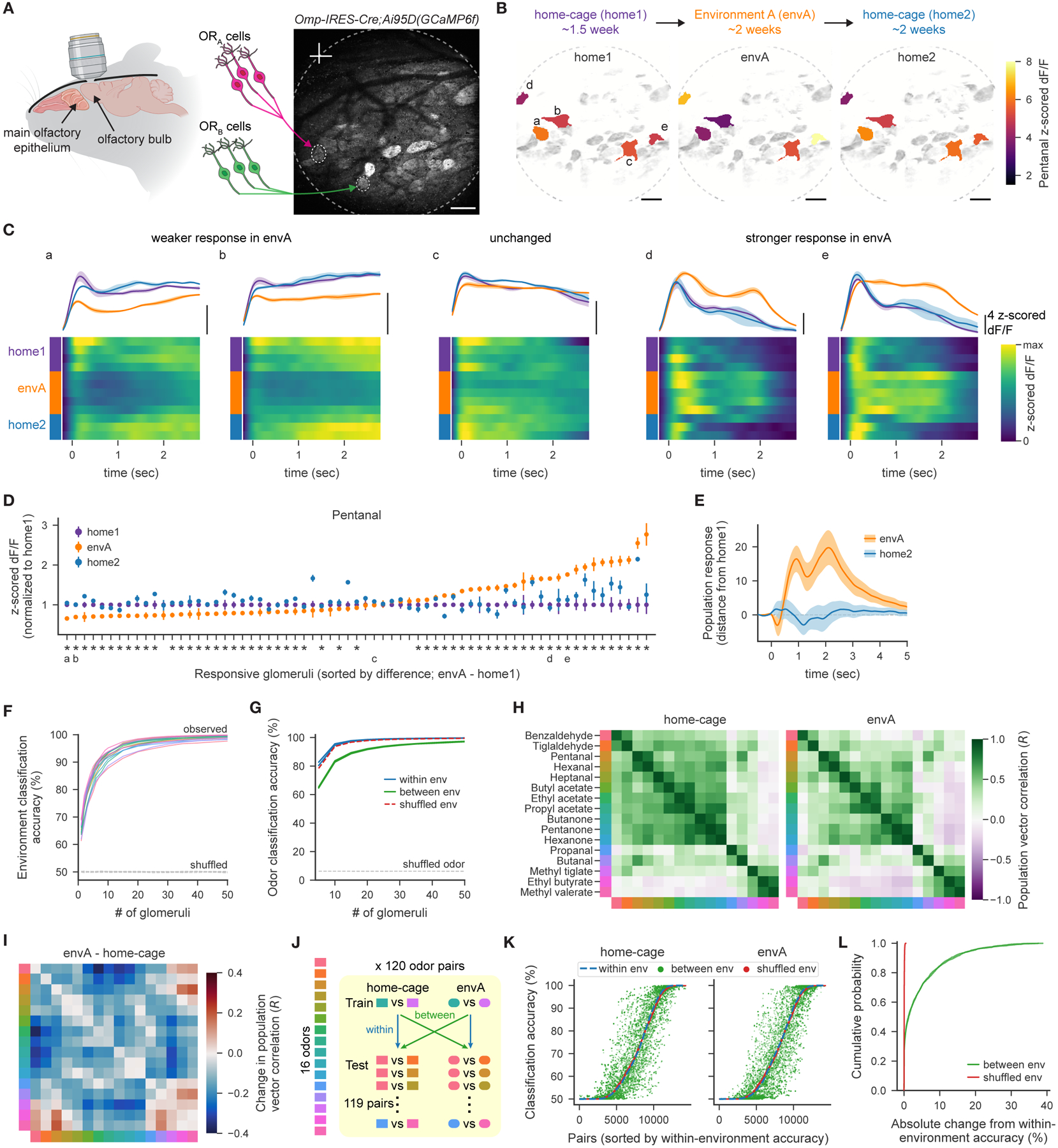
A. Schematic of the olfactory bulb imaging experiment. Scale bar = 200 μm. Data shown in A.-D. are from mouse 1 (see Fig. S7B).
B. Mean z-scored dF/F responses to pentanal for example glomeruli and the mean fluorescence image (in gray) for each environment. Data from glomeruli marked a-e are depicted in C. and D. Scale bars = 200 μm.
C. Z-scored dF/F responses for example glomeruli. Heatmaps depict average across trials within each session, and the mean ± SEM across sessions for each environment are shown above.
D. Glomerular responses to pentanal for each environment (mean ± SEM across sessions), normalized to the home-cage (home1) response, for pentanal-responsive glomeruli. Asterisks indicate glomeruli whose responses in environment A (envA) differ from those of home1 (FDR ≤ 0.01, permutation test).
E. Distance between the population response to pentanal in home1 and the response in either envA or home2, as a function of time (see Methods). Shaded error bars depict the mean and SD across 1000 restarts.
F. Accuracy of pairwise linear classifiers predicting in which environment (home-cage or envA) a mouse was housed from the mean odor responses of increasing populations of glomeruli for each odor (as colored in H.).
G. Accuracy of linear classifiers predicting odor identity from mean glomerular responses, to each of 16 odors, using increasing populations of glomeruli. Classification in which training and test data are from either the same or different environments, and from data with shuffled environment labels, are shown separately.
H. Correlation matrix summarizing pairwise relationships between mean odor responses across the population of glomeruli from mouse 5 (see Fig. S7B).
I. The difference between the correlation matrices shown in H.
J. Procedure for evaluating changes in odor codes across environments, by assessing how well a given pairwise odor classifier generalizes, across all other odor pairs, to test data from the same or different environments.
K. Classification accuracy for each of the test × train pairs (120 × 119 pairs), sorted by the accuracy when training and test data are from the same environment (within-environment). Colors depict whether training and test data are either from same or different environments, or from data with shuffled environment labels.
L. Summary of K. showing the cumulative fraction of the absolute change in generalization accuracy, relative to within-environment accuracy.
See also Figure S7.
To determine how environmental history influences odor coding, we imaged responses to a panel of 16 odors that broadly activated dorsal glomeruli (Figure S7G). A large fraction of glomeruli exhibited environment-specific odor response amplitudes, and linear decoders for each odor accurately distinguished between environments (Figures 7F and S7H). Odor identity could also be accurately predicted based upon glomerular responses within any given environment; however, classifiers trained in one environment performed worse across environments (Figure 7G). Quantification of pairwise correlations in glomerular odor responses revealed that relationships for many odor pairs were modulated across environments, while close odor relationships were often preserved (Figures 7H–I, S7I); similarly, classification analysis designed to quantify the relational structure in odor responses revealed diffuse changes in odor relationships across odor environments (Figures 7J–L, S7J). Taken together, these data demonstrate that changes in OSN transcriptomes evoked by complex naturalistic environments are paralleled by pervasive environment-dependent changes in both pre-synaptic glomerular odor responses and population-level odor codes in the brain.
Discussion
OSNs are thought to primarily communicate information about odor-OR interactions, which is used by the brain to facilitate odor perception (Buck, 1996; Cleland and Linster, 2005; de March et al., 2020; Firestein, 2001; Imai et al., 2010; Sullivan et al., 1995; Touhara, 2002). Here we find that each of the ~1000 OR-defined OSN subtypes in the mouse olfactory system adapt to the environment through a common mechanism: a transcriptional rheostat. This rheostat is composed of more than 70 genes relevant to OSN function, whose expression levels vary continuously across OSNs in an OR-dependent manner and adapt dynamically as mice traverse distinct environments. The specific position adopted by this rheostat (i.e., the OR-specific pattern of functional gene expression) predicts the odor response amplitude of a given OSN; furthermore, acute odor responses triggered by novel odor environments predict future patterns of functional gene expression (i.e., the setting of the rheostat).
These observations suggest a closed regulatory loop in which analog changes in environment-driven activity yield proportional transcriptional changes that predictably influence future neural responses to odors. Thus, rather than faithfully reporting the extent of odor-OR interactions, the peripheral olfactory system uses gene expression to instantiate expectation, thereby building odor codes that are personalized by each animal’s experience. While fast, post-translational mechanisms for sensory adaptation have been observed across modalities — and many circuit-level mechanisms have been characterized that adapt central sensory responses — our data reveal a systematic, large-scale adaptive transcriptional program that operates at the level of sensory neurons themselves (Benda, 2021; Burns and Baylor, 2001; Fettiplace and Ricci, 2003; Kadohisa and Wilson, 2006; Martelli and Storace, 2021; Wark et al., 2007; Zufall and Leinders-Zufall, 2000).
Our data demonstrate that the OR repertoire is densely activated by airflow and odors in our environments, suggesting that every OSN must contend with some degree of chronic activation. Because experience bidirectionally alters ES gene expression on timescales of hours, transcription-mediated adaptation most likely helps to center OSNs in their dynamic range as animals traverse different odor contexts, or as odor environments evolve during circadian cycles. As such, the adaptive mechanism identified here likely serves a distinct purpose from those that rapidly truncate responses to odor filaments (and which support e.g., odor-guided navigation), and from central habituation mechanisms that sparsen odor representations on the minutes-long timescale but cannot restore information that is lost when OSNs operate outside their dynamic range (Kadohisa and Wilson, 2006; Kostal et al., 2008; Lecoq et al., 2009; Martelli and Storace, 2021; Moore, 1994; Nagel and Wilson, 2011; Wilson, 2009).
Transcription factors and axon guidance genes appear insufficient to effectively distinguish OSN transcriptomes or predict OR expression. Although known identity markers have been thought to only assign OSNs to broad dorsal or ventral domains (Bozza et al., 2009; Kobayakawa et al., 2007; Tan and Xie, 2018), the dorsal, ventral, anterior and posterior GEPs are sufficiently diverse and organized to support OR predictions (Figures S2F–H, S3Q). These observations suggest a fine gradation in OSN cell identities organized across the two main anatomical axes (Bozza et al., 2009; Miyamichi et al., 2005; Pacifico et al., 2012). The usages of these identity-related GEPs and OR expression levels remained largely constant across all our experimental manipulations, indicating that functional genes are uniquely sensitive to the environment. Although the mechanisms that render ES genes sensitive to neural activity — and which coordinate expression levels across ES genes — remain to be determined, mature OSNs express several transcription factors (including Ebf1 and Lhx2) that in principle could be sensitive to activity; the prominent role of heterochromatin in regulating OR expression suggests that epigenetic mechanisms involving chromatin or DNA modifications might also coordinate ES gene expression (Monahan and Lomvardas, 2015; Monahan et al., 2017).
Environment-driven variation in functional gene expression both highlights new odor information and dynamically sculpts population-level odor representations. Given that peripheral odors codes are modified by ongoing experience, it is not clear how the brain supports odor identification and generalization across different environments. It is possible that some features of population OSN activity (e.g., timing, dynamics) are relatively insensitive to differences in functional gene expression, and/or that circuit-level attractor dynamics in the olfactory bulb and cortex cause central representations for odors to converge, despite differing inputs. Conversely, the brain may take advantage of environment-specific differences in peripheral odor codes to better understand the context in which a given stimulus was encountered. Transcription-dependent changes in both vomeronasal sensory neurons and fly olfactory receptor neurons renders their odor responses sensitive to sex hormones (Dey et al., 2015; Ng et al., 2019), raising the possibility that internal states may similarly influence conventional mammalian OSNs and thereby further modify odor codes.
There is a close conceptual relationship between the bidirectional sensory adaptation we describe here and firing rate homeostasis (FRH), the process through which neurons in a network maintain stable firing rates by adjusting both synaptic weights and intrinsic neural properties (Davis, 2006, 2013; Marder, 2011; Marder and Goaillard, 2006; Turrigiano, 2011; Turrigiano, 1999; Turrigiano, 2008, 2017). Invertebrate neurons of a given type with identical firing properties can exhibit significant variation in ion channel expression, suggesting that there are many transcriptional means to achieving the same functional end (Goldman et al., 2001; Marder, 2011; O’Leary et al., 2013; Schulz et al., 2007). In contrast, our experiments in OSNs identify a deterministic and proportional interdependence between environment-dependent activity and functional gene expression patterns. We speculate that this tight closed-loop relationship emerges because OSNs (unlike neurons in networks) cannot manipulate synaptic weights to adapt the strength of their inputs, as input strength in OSNs is entirely determined by odor-receptor interactions. It has been proposed that structured correlations in the expression of functional genes can be used to define different cell types (O’Leary et al., 2013; Schulz et al., 2007); OSNs, whose functional genes exhibit near-perfect expression correlations along a continuum of activity levels, clearly meet this operational definition.
scSeq analysis reveals a startling diversity of transcriptomes associated with neurons of a single putative type (Kim et al., 2019; Li et al., 2017; Tasic et al., 2018). It remains unclear whether these transcriptional differences reflect an occult diversity of neural subtypes or if they instead reflect the ongoing activity-dependent modulation of a single underlying cell type (Gouwens et al., 2020; Kim et al., 2020; Scala et al., 2021). Together, our findings — enabled by the ability to query gene expression and activity in ~1000 different OSN subtypes across experiments — are consistent with a general model in which neurons systematically modulate their transcriptomes to continuously adapt to their inputs.
Limitations of the study
Act-Seq can reliably identify analog odor-OR relationships across much of the OR repertoire in vivo, suggesting its general utility for deorphanizing odors and receptors; however, OSNs expressing rare ORs may be missed by scSeq. Our in vivo measures of acute odor-evoked neural activity depend upon transcription, rather than directly querying either calcium or action potentials, and may not fully capture the activity state of each OSN. While our data strongly suggest that functional gene expression alters odor-spike coupling, it may also alter the gain of activity-transcription coupling. Furthermore, the mechanisms through which these functional genes, as well as potentially other genes, adapt OSNs remains to be determined. The significant environment-dependent changes in axonal calcium responses in the olfactory bulb roughly match observed transcriptional changes in OSNs; however, it is possible that some aspect of these changes is caused by pre-synaptic inhibition.
RESOURCE AVAILABILITY
Lead Contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Sandeep R. Datta (srdatta@hms.harvard.edu).
Materials availability
This study did not generate new unique reagents.
Data and code availability
Single-cell RNA-seq data have been deposited at GEO and are publicly available as of the date of publication. The accession number is listed in the key resources table.
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Biological samples | ||
| Mouse olfactory epithelium | This paper | N/A |
| Chemicals, peptides, and recombinant proteins | ||
| Pentanal | Sigma-Aldrich | Cat# 110132 |
| Pentanone | Sigma-Aldrich | Cat# 471194 |
| Ethyl butyrate | Sigma-Aldrich | Cat# W242713 |
| Heptanal | Sigma-Aldrich | Cat# W254002 |
| Hexanal | Sigma-Aldrich | Cat# 115606 |
| Propanal | Sigma-Aldrich | Cat# 538124 |
| Butanal | Sigma-Aldrich | Cat# W217018 |
| Butanone | Sigma-Aldrich | Cat# W217018 |
| Hexanone | Sigma-Aldrich | Cat# 103004 |
| Ethyl acetate | Sigma-Aldrich | Cat# 270989 |
| Propyl acetate | Sigma-Aldrich | Cat# 537438 |
| Butyl acetate | Sigma-Aldrich | Cat# 287725 |
| Benzaldehyde | Sigma-Aldrich | Cat# B1334 |
| Tiglaldehyde | Sigma-Aldrich | Cat# W340707 |
| Methyl tiglate | Thermo Fisher | Cat# T024825ML |
| Methyl valerate | Sigma-Aldrich | Cat# W275204 |
| Acetophonone | Sigma-Aldrich | Cat# A10701 |
| 2-Hydroxy acetophenone | Sigma-Aldrich | Cat# H18607 |
| 4-Methyl acetophenone | Sigma-Aldrich | Cat# W267708 |
| Methyl salicylate | Sigma-Aldrich | Cat# M6752 |
| Octanal | Sigma-Aldrich | Cat# O5608 |
| Mineral oil | Sigma-Aldrich | Cat# M5904 |
| Dipropylene glycol | Sigma-Aldrich | Cat# D215554 |
| Actinomycin D | Sigma-Aldrich | Cat# A9415 |
| Triptolide | Sigma-Aldrich | Cat# T3652 |
| Anisomycin | Sigma-Aldrich | Cat# A9789 |
| Papain | Worthington | Cat# LK003178 |
| DNase I | Worthington | Cat# LK003172 |
| EBSS | Worthington | Cat# LK003188 |
| Hibernate-A | Thermo Fisher | Cat# A1247501 |
| Heat-inactivated Fetal Bovine Serum | Thermo Fisher | Cat# 16140071 |
| Propidium iodide | Thermo Fisher | Cat# P1304MP |
| PBS | Thermo Fisher | Cat# 70011044 |
| Critical commercial assays | ||
| Chromium Next GEM Single Cell 3' Kit v3.1 | 10x genomics | Cat# 1000268 |
| Chromium Next GEM Chip G Single Cell Kit | 10x genomics | Cat# 1000120 |
| Dual Index Kit TT Set A | 10x genomics | Cat# 1000215 |
| Agilent High Sensitivity DNA kit | Agilent | Cat# 5067–4626 |
| KAPA Library Quantification Kit, illumina platform, Complete kit (Universal) | Roche | Cat# KK4824 |
| Deposited data | ||
| Raw and analyzed sequencing data | This paper | GEO: GSE173947 |
| Experimental models: Organisms/strains | ||
| Mouse: C57BL/6J | The Jackson Laboratory | JAX: 000664, RRID:IMSR_JAX:000664 |
| Mouse: OMP-IRES-GFP | The Jackson Laboratory | JAX: 006667, RRID:IMSR_JAX:006667 |
| Mouse: B6J.Cg-Gt(ROSA)26Sortm95.1(CAG-GCaMP6f)Hze/MwarJ | The Jackson Laboratory | JAX:028865, RRID:IMSR_JAX:028865 |
| Mouse: Arrb2tm1Rjl/J | The Jackson Laboratory | JAX:011130, RRID:IMSR_JAX:011130 |
| Mouse: B6;129P2-Olfr160tm11(Olfr545)Mom/MomJ (S50(M72 locus)) | Thomas Bozza | JAX:006714, RRID:IMSR_JAX:006714 |
| Mouse: B6;129P2-Olfr545tm3(Olfr160)Mom/MomJ (M72(S50 locus)) | Thomas Bozza | JAX:006715, RRID:IMSR_JAX:006715 |
| Mouse: Olfr17tm7Mom/MomJ | Stavros Lomvardas | JAX:006669, RRID:IMSR_JAX:006669 |
| Mouse: OMP-IRES-Cre | Richard Axel | MGI:3045687 |
| Mouse: OMP-ChR2(H134R)-Venus | Thomas Bozza and Dmitry Rinberg | Li et al, 2014 |
| Software and algorithms | ||
| Python 3.6–3.8 | Python | https://www.python.org/ |
| MATLAB (2014a, 2015a) | Mathworks | https://www.mathworks.com/ |
| ScanImage 5 | Vidirio Technologies | http://scanimage.vidriotechnologies.com/display/SIH/ScanImage+Home |
| Wavesurfer 0.775 | Howard Hughes Medical Institute Janelia Research Campus | https://wavesurfer.janelia.org/ |
| Suite2p | Howard Hughes Medical Institute Janelia Research Campus | https://github.com/mouseland/suite2p |
| Adobe Illustrator | Adobe | RRID:SCR_010279, https://www.adobe.com/products/illustrator.html |
| Cell Ranger (2.2.0 for v2 and v3 data and 4.0.0 for v3.1 data) | 10x genomics | https://support.10xgenomics.com/single-cell-gene-expression/software/overview/welcome |
| Scanpy 1.6.0 | Wolf et al. 2018 | https://github.com/theislab/scanpy |
| Python analysis scripts | This paper | Zenode: https://doi.org/10.5281/zenodo.5550454 GitHub: https://github.com/dattalab/Tsukahara_Brann_OSN |
| Other | ||
| Teensy 3.2 | Adafruit | Cat# 2756 |
| blue LED | Digikey | Cat# 1416–1052-6-ND |
All original code has been deposited at GitHub and Zenodo, and the links and DOI are listed in the key resources table.
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Mice
C57BL/6J, OMP-IRES-GEP, Ai95D (GCaMP6f) and β-arrestin2 knock out mice were obtained from Jackson Laboratory (stock number 000664, 006667, 028865, 011130). “OR-swap” mice (M72(S50 locus) and S50(M72 locus)) were maintained in the Bozza laboratory (and are available from Jackson Laboratory with stock numbers 006715, 006714) (Bozza et al., 2009). P2-IRES-GFP mice (P2(P2 locus)) were obtained from the Lomvardas laboratory (and are available from Jackson Laboratory with stock number 006669) (Feinstein and Mombaerts, 2004). OMP-IRES-Cre mice were obtained from the Axel laboratory (Eggan et al., 2004). OMP-ChR2(H134R)-Venus mice were maintained in the Bozza laboratory and obtained from the Rinberg laboratory (Li et al., 2014). Mice of either sex between 6–16 weeks-old were used for experiments. Mouse husbandry and experiments were performed following institutional and federal guidelines and were approved by Harvard Medical School’s Institutional Animal Care and Use Committee.
METHOD DETAILS
Chronic naris occlusion
To identify gene expression programs sensitive to ongoing activity from environmental odorants, 7 day old mice were anesthetized on ice, and one of the two nostrils was occluded by cautery, as previously described (Fischl et al., 2014). This age was chosen to minimize deficits in axon targeting that occur from manipulating activity levels at earlier post-natal timepoints (Ma et al., 2014). Unilateral naris occlusion was confirmed using a dissection microscope. After ~1 month, mice were used for scSeq experiments. Cells isolated from occluded and open nostrils were processed and analyzed separately.
Odor exposure (Act-Seq)
Act-Seq (Wu et al., 2017) was performed following odor exposure to quantify odor-evoked responses in each OSN subtype (defined by their expressed OR) across the entire OR repertoire. Mice were habituated to a reversed light cycle for at least for 1 week before odor exposure, transferred to a new disposable cage with regular bedding/food and kept overnight in a satellite animal facility. On the experimental day, each mouse was first transferred to a new empty disposable cage during the dark cycle and habituated for 20 minutes. Empty cages were used to avoid any activation by odorants present within the environments in which the mice were housed. Odors were added to a piece of filter paper (100 μL of odorant) in a 35 mm petri dish and a cotton ball (200 μL of odorant) and placed in each cage. After 2 hours, mice were euthanized, and dissociation was performed as described below. Odors used in this study were as follows. Dipropylene glycol (DPG, control solvent), and 10 % of acetophenone, octanal, 2-hydroxy acetophenone, 4-methyl acetophenone, and methyl salicylate. As a control, mice were exposed to DPG alone for 30 min. This control condition was used to account for any drift in transcriptomes during the overnight housing and habituation periods prior to odor exposure and designed to capture the transcriptome in the state it would have been in the odor-exposed animals prior to odor exposure. The 30 min DPG control was used for all Act-Seq experiments, except following transient naris occlusion (see below); in the Act-Seq experiments our results did not depend upon the specific control condition we used, which likely reflects the relative stability of the transcriptome for each OSN subtype. Act-Seq was also performed using lower concentrations of acetophenone (1.0, 0.1, 0.01%) and 2-hydroxy acetophenone (0.1%, 0.01%). All odor dilutions were made with DPG, and all odors and DPG were obtained from Sigma. To assess the persistence of any transcriptional changes observed as a result of odor exposure, a cohort of mice was exposed to acetophenone or DPG for 2 hours, transferred to new clean regular home cages, and subjected to scSeq after an additional 22 hours.
Transient naris occlusion
Because odor responses in occluded OSNs cannot be examined in the chronic occlusion experiment due to permanent cautery, transient unilateral naris occlusion was performed using removable nasal plugs. Adult mice (6–12 weeks old) were anesthetized by isoflurane and removal nasal plugs were inserted into one of the two nostrils, as previously described (Galliano et al., 2021; Kass et al., 2012). The occlusion was confirmed by measuring airflow from the occluded nostril via thermocouple. After 5 days, a subset of mice was used for scSeq experiment directly (control mice) to assess the effects of naris occlusion on changes in gene expression. Under brief anesthesia by isoflurane, the remaining mice were unplugged and transferred to a new empty disposable cage containing either DPG alone or 10% acetophenone. After 2 hours, mice were subjected to scSeq. Each of the two nostrils was used separately in both control (open and plugged) and odor-exposed mice (open and unplugged). Mice whose nostrils did not experience airflow after unplugged, as assessed transcriptionally, were excluded from any analyses.
Environment switch experiments
To examine environment-dependent changes in gene expression and their effects on odor-evoked responses, Act-Seq was performed 2 weeks after environment switches. Adult mice (6–12 weeks old) were group-housed in the regular home-cage environment for at least one week before the transfer to their novel environments. Individual mice were transferred to a new disposable cage containing materials for a novel environment in the satellite animal facility. Two different novel environments were used, with each containing non-overlapping contents: Environment A (paper bedding, hay, dried flowers (such as marigold), peanuts, seeds (e.g., sunflower seeds, pumpkin seeds), puffed rice), and Environment B (garden soil, aspen shavings, coconut husks, dried fruits (e.g., berries, mango, pineapple), dried vegetables (e.g., green peas, corn, bell peppers), millet, fresh fruits (banana, apple, peach), corn flakes). Mice were singly-housed throughout the entire 2 weeks and were transferred to newly prepared environmental cages every 12 hours to minimize the contributions of any murine odors, which are likely the predominant odorants present within standard home-cage housing environments. After 2 weeks, individual mice were transferred to new empty disposable cages, and Act-Seq using acetophenone was performed as above.
To assess the dynamics of gene expression changes in response to novel environments, mice were housed overnight in a new disposable cage with regular bedding/food in a satellite animal facility. On the experimental day, each mouse was first transferred to Environment A cages and subjected to scSeq after 45 minutes, 2, 4, 12, 24 hours, or 3, 5, 14 days (with cages refreshed every 12 hours for durations longer than 12 hours). To account for any non-specific effects resulting from transferring mice from the animal facility to disposable cages within the satellite facility, data from mice transferred and housed in new disposable cages with regular bedding and food overnight were used as controls for downstream analyses of the effects of Environment A on gene expression.
For β-arrestin2 knock out mice, scSeq was performed on cohorts of mice that were either housed in home cages, housed in new disposable cages with regular bedding and food overnight and then transferred to Environment A cages for 5 days, or housed in new disposable cages with regular bedding and food overnight as controls, as described above. Each of the three knock out datasets was compared to its respective dataset from wild-type mice housed in the same condition.
To determine whether OSN activation reflects salient differences between environments, one set of mice were housed in a new disposable cage with regular bedding/food overnight in a satellite animal facility and then transferred to Environment A for 5 days. On the experimental day, mice were transferred to either a new Environment A cage or a home cage for two hours before being subjected to scSeq. An additional cohort of mice was housed in the home cage. The day before the experiment, they were transferred to a new disposable cage with regular bedding/food overnight in a satellite animal facility. On the experimental day they were transferred to a new home cage for two hours before being subjected to scSeq. These three conditions were compared to mice transferred to Environment A cages for 2 hours as part of the time course described above, thus generating all four combinations of environmental switches between mice housed in Environment A and home cages.
Chronic acetophenone exposure
To compare the effects of chronic odor exposure with the acute activation observed via Act-Seq, scSeq was performed after exposure to DPG alone or 0.1, 1, 10% of acetopheone (diluted by DPG) for 5 days. Adult mice were individually housed in regular home cages in a satellite animal facility overnight, then two cotton balls, each of which was soaked with 300 μL of DPG or diluted acetophenone solution, were added into each cage. The odorized cotton balls were replaced every 12 hours and cages were replaced every 2 days.
Optogenetic stimulation
To characterize quantitative changes in gene expression after activation of OSNs, OMP-ChR2(H134R)-Venus mice were subject to optogenetic activation and analyzed via scSeq. Mice (N=14) were anesthetized with 2% isoflurane and injected with bupivacaine (1.25 mg/kg) under the scalp. An incision was then made to expose the dorsal skull and a scalpel (Aspen Surgical 372615 Bard-Parker) was used to thin the bone overlying the right olfactory bulb, after which cyanoacrylate glue (Loctite Glass Glue) was applied over the thinned bone. A blue LED (470 nm, Lumileds LXZ1-PB01, Digikey) was soldered to a two-pin connector (Millmax, Digikey ED8450-ND) and manually placed over the bulb. A titanium head bar was then placed over the caudal end of the skull, and the whole dorsal surface was covered with dental cement (Metabond, Parkell). After the dental cement dried, a layer of black nail polish was applied to minimize the leak of blue light during stimulation. Postoperative care included a subcutaneous injection of buprenorphine SR (1 mg/kg, given 1 hour prior to surgery start) and carprofen (5 mg/kg) administered through drinking water. Before experiments, mice were tethered to a cable connecting the LED to a microcontroller (Teensy 3.2, Adafruit) that provided a custom stimulation pattern. The LED emitted 100mW at 470nm, measured with a light meter (Thorlabs PM100D).
For chronic activation, OSNs were activated by 5 pulses (50 msec on, 50 msec off cycle) with a 10 second inter-pulse interval for 12 hours before being subjected to scSeq. For acute activation, OSNs were activated by the same cycle as above with a 5, 10, or 20 second inter-pulse interval for 2 hours (denoted as High, Medium, and Low, respectively) and then subjected to scSeq. For both chronic and acute activation, control mice underwent the same surgery and tethering, but were implanted with a dummy connector and did not receive LED stimulation. Home cage control samples for optogenetic experiments were mice that underwent the same surgery but were subjected to scSeq directly from the home cage without any optogenetic stimulation. Because the LED was placed over the right olfactory bulb, only the right nostril was dissociated and used for scSeq for all optogenetic stimulation and control experiments.
Preparation of single cell suspensions for scSeq
The main olfactory epithelium was dissected in Earle’s Balanced Salt Solution (EBSS, pre-treated with carbogen for at least 5 minutes before each use, Worthington), then transferred to a round-bottomed glass dish containing 750 μL of papain solution (one vial of Papain (Worthington) dissolved in 5 mL of EBSS and then equilibrated 10 minutes at 37 °C) and 100 μL of DNase solution (one vial of DNase-I (Worthington) dissolved in 500 μL of EBSS). Bone was removed from the epithelium under a dissecting microscope and the resulting epithelial tissue was placed in a 5 mL tube (Becton Dickinson) with an additional 1.75 mL of Papain solution and 200 μL of DNase-I solution and rocked gently for ~60 minutes at 37 °C. The tissue was then gently triturated with a 5 mL pipette 10–15 times, passed through a 40 μm cell strainer (Becton Dickinson), and washed with 1 mL of Hibernate-A medium containing 10 % Fetal Bovine Serum (FBS, GIBCO). Filtered cells were transferred to a new 5 mL tube and centrifuged 5 minutes at 300 × g. The supernatant was decanted, the cells were washed once with 4 mL Hibernate-A containing 10 % FBS and resuspended in 1 mL Hibernate-A containing 0.2 % FBS. Importantly, each of the solutions above contained transcriptional inhibitors (5 μg/mL of Actinomycin D, 10 μg/mL of Anisomycin and 10 μM of Triptolide, all obtained from Sigma) for all experiments, especially the Act-Seq experiments, except where noted (Table S1).
Fluorescence activated cell sorting
Dissociated cells were stained with propidium iodide (final concentration 1.65 μg/mL) and subjected to FACS to remove dead cells and obvious doublets. For fluorescent reporter-expressing mouse lines (OMP-IRES-GFP, P2(P2 locus), M72(S50 locus), S50(M72 locus), OMP-ChR2(H134R)-Venus), fluorescence positive and negative cells were sorted separately using a FITC filter. Cells were sorted into Hibernate-A containing 5 % FBS (and 0.1x concentration of transcriptional inhibitors), centrifuged for 5 minutes at 300 g, and resuspended with PBS. For samples from OMP-IRES-GFP and OMP-ChR2(H134R)-Venus mice, fluorescence positive cells were used for single cell RNA-seq (scSeq) experiments. For samples from OR lines (P2(P2 locus), M72(S50 locus) and S50(M72 locus)), fluorescence positive and negative cells were combined for scSeq experiments.
Single cell library preparation
Single cell RNA-seq library were prepared from the single cell suspensions via the chromium single cell gene expression system (chromium single cell 3’ reagents and GEM v2, v3 or v3.1 dual index, 10x genomics), using the default protocols provided by 10x genomics. Each replicate, other than GFP-positive ones, was loaded at a concentration predicted to yield 10,000 cells, for which the expected multiplet rate is ~8.0 %.
Sequencing
Sequencing library fragments were examined using the Agilent High Sensitivity DNA kit (Agilent) and quantified via qPCR by the KAPA library quantification kit (Roche). NextSeq and NovaSeq platforms were used for sequencing libraries. 75 cycle High output kit was used for NextSeq (Read1 = 26 cycle, Index (i7) = 8 cycle, Read2 = 58 cycle). Full flowcells of 100 cycle SP/S1/S2 kits, full flowcells or single lanes of 200 cycle S4 kit were used for NovaSeq (minimum read lengths: Read1 = 28 cycle, Index (i7) = 8 cycle, Read2 = 89 cycle for v2 and v3 kits and Read1 = 28 cycle, Index (i7) = 10 cycle, Index (i5) = 10 cycle, Read2 = 77 cycle for v3.1 dual index kits). Demultiplexed fastq files were generated by mkfastq function in Cell Ranger software.
Generating gene expression matrix (raw UMI count) from sequencing data
Demultiplexed fastq files were aligned to the mouse reference genome mm10 (Ensembl 93) and converted into gene expression matrices using the 10x Genomics Cell Ranger software (version 2.2.0 (v2 and v3 samples) or 4.0.0 (v3.1 samples)) with two key modifications. First, despite the fact that the 10x genomic platforms have relatively high rates of intronic priming (La Manno et al., 2018), Cell Ranger by default considers multi-mapped reads that map to a single exonic locus as well as to non-exonic (e.g. intronic) locations as being uniquely mapped to that gene (and modifies the MAPQ scores accordingly), even when the read may have better alignment to the non-exonic loci. These multimapped reads (typically 5–10% of all reads) were filtered out by removing any read in the bam file with a MM:i:1 tag. Second, the output BAM files from Cell Ranger also contained many reads that were uniquely mapped to separate genes despite sharing the same cell barcode and unique molecular identifier (UMI), which is biologically implausible given that the cell barcode and UMI should uniquely identify each individual transcript. This could be either the result of UMI collision or misalignment. In support of the latter possibility, different reads of individual UMIs were often each uniquely aligned to multiple related genes, such as the large family of olfactory receptors (ORs). To avoid double-counting UMIs and inflating the numbers of genes, ORs, or UMIs detected in each cell, all ambiguous UMIs that mapped to multiple genes were removed from the BAM file with custom scripts, using samtools and pysam. The gene expression matrix was then recomputed by counting the number of distinct UMIs for each cell barcode for each gene.
Extracting mature olfactory sensory neurons from all cells
Mature OSNs were identified using an iterative subclustering procedure via the Scanpy python package (Wolf et al., 2018). In each cell, the UMIs were total-count normalized and scaled by 10,000 (TPT normalized). Variable genes that were overdispersed relative to their mean were identified using the SPRING gene filtering function “filter_genes” with parameters (90, 3, 10; see (Weinreb et al., 2018)). Ribosomal, mitrochondrial, and OR genes were excluded from the variable genes. To identify mature OSNs, the gene expression of the set of variable genes was log-normalized, and for each gene, the residuals from linear regression models using the total number of UMIs and percent of UMIs for mitochondrial genes as predictors were then scaled via z-scoring and reduced to a smaller number of dimensions via principal component analysis (PCA). Cells were clustered using the top 35 PCs via the Leiden algorithm (resolution = 1.2). Clusters containing mature OSNs were identified based on their expression of known mature OSN marker genes (e.g. Omp, Stoml3, Cnga2, Adcy3), the presence of cells expressing OR genes, as well as the absence of immature OSN marker genes or genes found in non-neuronal cells (e.g. Sox11, Gap43, Cbr2, C1qb, Aqp3). A small cluster containing the non-canonical Gucy2d (GCD) and Gucy1b2 expressing neurons was routinely identified but not used for any analyses in this paper. Clusters containing dying cells with low numbers of total counts, low numbers of genes, and high percentages of mitochondrial genes were removed. This clustering procedure typically revealed a cluster of OSNs that had higher than average total counts (often close to double the mean of other OSN clusters) and the majority of cells in such clusters often expressed multiple ORs; these cells are likely OSN-OSN doublets and were not considered further. Similarly, clusters containing mixtures of OSN and non-neuronal markers were also likely doublets between OSNs and other cell types and were not considered further. Using only the mature OSN clusters that passed this initial filtering step (~60–65% of cells), the above procedure (starting from variable gene identification but using only 20 PCs) was repeated multiple times to remove any additional immature or unhealthy OSNs (with low counts or >10% of total counts in mitochondrial genes) until stable results were obtained. Importantly, although immature OSNs (e.g. Gap43+ and weakly Omp+) routinely express OR genes and other mature OSN markers, these OSNs were excluded by the conservative subclustering procedure described above so that any comparisons between cells expressing the same OR would not be confounded by any differences in OSN maturity.
OR gene annotation (including functional or pseudogenes, class I or II, dorsal or ventral, and CD36-positive or -negative), OR protein similarity, and OSN positional subtypes
The Ensembl database (Release 93) was used for OR gene annotation. Functional OR genes were identified as genes that are associated with GO term “olfactory receptor activity” (GO:0004984); the TAAR family of receptors was also included (GO:0001594, GO:1990081). To identify the OR class for each mouse OR, phylogenetic analysis was performed using all mouse and zebrafish OR proteins in MEGA7 with default parameters. As previously described, OR proteins cluster into two main phylogenetic clusters (Zhang and Firestein, 2002). The ORs that clustered together with zebrafish ORs are the class I ORs and the remaining ORs are class II ORs. To compare OR protein similarity, the pairwise phylogenetic distance matrix between OR protein sequences was calculated by taking the cophenetic distance of the phylogenetic tree of all ORs. For each pair of ORs, the cophenetic distance is equal to the height of a dendrogram in which the two branches of the phylogenetic tree containing both ORs first merge into a single cluster. Unsupervised graph-based clustering of cells identified a cluster of OSNs expressing Cd36, as previously described (Oberland et al., 2015; Xavier et al., 2016). ORs were identified as Cd36-positive if the majority of OSNs expressing that OR were found within the Cd36-positive clusters. OSNs were categorized as dorsal or ventral based on their expression of known dorsal (Nqo1, Acsm4) and ventral (Ncam2, Nfix) marker genes. ORs expressed in dorsal or ventral OSNs were referred to as dorsal or ventral ORs. With the exception of three known ventral class I ORs, all other class I ORs are dorsal, whereas OSNs expressing individual class II OR expressed either dorsal or ventral marker genes. The OR genes are located in gene clusters across most chromosomes in the genome; OR genes whose transcription start sites (TSS) were within 3 megabases of any other OR gene were considered part of the same gene cluster (Monahan et al., 2017).
Identification of the OR expressed in each OSN
Of the 1172 identified functional ORs, an OR was considered expressed in any given OSN if at least 3 UMIs were detected (except for the experiments using the 10x v2 chemistry, where a threshold of 2 UMIs was used). Mature OSNs expressing single ORs were identified based on the OR that they expressed, and all cells expressing the same OR were considered as the same OSN subtype for downstream analyses. The vast majority of mature OSNs expressed a single OR (Figure S1D). OSNs expressing multiple ORs at high levels typically had higher numbers of total counts and were likely cell doublets that remained even after the previous filtering steps. OSN expressing multiple ORs with higher expression for a single OR gene also sometimes expressed other OR genes at low levels, as seen during development (Hanchate et al., 2015), or due to contamination from ambient RNA or errors in gene alignment. Regardless, any OSN expressing either zero or multiple OR genes was not considered further for any downstream analyses. OR frequency was defined empirically based on the fraction of all mature OSNs expressing a given OR. The expression level for each OR was determined based on the normalized expression (see below) of that OR gene within the OSNs singly-expressing that OR. In the “OR-swap” mice, in which the coding sequence for each OR was knocked into the locus of the other, cells expressing swapped ORs were identified based on the gene counts for the locus rather than the expressed OR (e.g. in M72(S50 locus) mice, OR transcripts that originated from the S50 locus indicate the expression of M72 swap cells while OSNs expressing M72 from the endogenous locus were not considered and vice versa for S50(M72 locus) mice) due to fact that only coding sequences were swapped whereas the 3’ UTR for the original OR remain and are detected given the 3’ bias of the 10x data that arises from the use of oligo(dT) primers. The median number of UMIs/genes detected per mature OSNs and number of mature OSNs used for each replicate are summarized in Table S1.
Accounting for OR frequency
ORs were expressed at a wide range of frequencies, spanning multiple orders of magnitude (Figure S1A–B). For instance, given that the 200 OR genes with the highest frequencies are expressed in ~50% of all OSNs, if standard algorithms like PCA are applied on all OSNs at once, they would weight ORs expressed in 100 cells 100x more than an OR expressed in a single cell, and the resulting top PCs would capture the axes of transcriptional variation that distinguished highly-expressed ORs rather than all OSN subtypes. Similarly, classifiers trained on all cells would more easily distinguish highly-expressed ORs (from their higher influence on the PCs, increased presence in training data, and higher likelihood of accurate predictions by chance), and one could achieve high levels of overall prediction accuracy even with models that were unable to distinguish among the majority of ORs. Therefore, to account for OR frequency, most analyses were performed using equal numbers of OSNs per OR by subsampling from the population of OSNs that expressed the set of ORs each detected in at least a certain number of cells (e.g. at least 4–10, as specified, depending on the analysis). This subsampling procedure was performed 1,000 times and results were summarized for each OSN subtype (as defined by the expressed OR) across the 1,000 restarts. Analyses at the OSN subtype level were performed by first averaging across all OSNs expressing the same OR within a given condition.
Normalization and scaling
The filtered mature OSN datasets containing only cells expressing single ORs were renormalized, starting again from the raw UMI counts for each gene. Normalization was performed by dividing the counts for each gene in each cell by the total counts across all genes (excluding mitochondrial and ribosomal genes, as well as highly-expressed lncRNAs and lincRNAs like Malat1 and Gm42418, and sex-specific transcripts like Xist) in that cell and multiplying by 10,000 to yield tags per ten thousand (TPT) normalized data. Normalized expression in the figures and throughout refers to TPT-normalized data, where a value of 1 indicates on average 1 UMI for that gene per 10,000 UMIs. For analyses that required scaled data (e.g. PCA and classification), the logarithm of the TPT-normalized data was used (log(TPT + 1)). Scaling via z-scoring was performed for each gene, using means and standard deviations identified by taking the mean of the values from 100 restarts containing equal numbers of cells per OR. The home-cage data consisted of six replicates distributed across 2 batches; separate scaling was used for each batch, but similar results were obtained by scaling both batches together. For the “OR-swap” experiments, the means and standard deviations were fit using only cells from wild-type mice collected with the same 10x v2 chemistry platform, and these were applied to the “OR-swap” and P2(P2 locus) OSNs. In the chronic occlusion data, the means and standard deviations were recomputed using data from both nostrils. In the environment switch experiments, the gene scaling was recomputed using data from all environments.
Dataset integration
Importantly, besides scaling and normalizing the gene expression data, no methods for batch correction or dataset integration were performed for any dataset. The mature OSNs were readily identified and separately isolated from each dataset (obviating the need for any additional alignment of cell types across datasets), and batch effects were rarely observed when comparing the normalized expression between datasets (apart from changes in the levels of mitochondrial and ribosomal genes, which were excluded from normalization procedures and downstream analyses). This analytical choice was deliberate, as data integration procedures are affected by changes in OSN composition (due to differences in the set and frequency of ORs detected across datasets and the wide range of OR frequencies), can modify the transcriptomes by averaging and leaking data across cells (which may express different ORs), and could make OSNs transcriptomes more or less similar to each other in a manner that would hinder and bias downstream comparisons between cells expressing the same OR across experimental conditions. For instance, in the Act-Seq experiments, our expectation was that a small fraction of OSNs would exhibit difference in their transcriptomes upon being activated by odorants; however, dataset integration procedures typically attempt to normalize such differences, and thus tend to make control OSNs appear more activated and odor-exposed OSNs appear less activated. Some datasets were collected with different 10x kits (v2, v3, and v3.1; see Table S1); as these kits differ in their sensitivities, each sample was compared with appropriate control samples collected with the same platform.
Gene selection
A consensus set of genes whose expression varied across mature OSNs were identified and used as the basis of most downstream analyses where transcriptomes were compared across OSNs and conditions. After using the OR genes to identify the OR expressed in each cell, the OR genes were not considered further, and were subsequently removed from all downstream analyses. Mitochondrial and ribosomal genes, and the lncRNAs mentioned above were also excluded. Using the home-cage data, the population of OSNs that expressed 831 ORs in at least 10 cells was used. To identify genes whose expression varied across cells expressing different ORs, 10 cells were selected for each OR and the F-score between the scaled gene expression and OR identity was computed for each gene, using the set of ~15,000 genes that were expressed in at least 0.2% of OSNs. This procedure was performed 1000 times and genes that were consistently strongly significant (Benjamini-Hochberg FDR-corrected p-value ≤ 1 × 10−7 for at least 90% of restarts) were considered highly-variable genes (HVGs). This procedure identified a set of 1350 non-OR genes that were used for downstream analyses. Varying thresholds, such as the number of restarts to consider a gene as a HVG, yielded different numbers of HVGs but gave consistent results with respect to other analyses (Figure S1I).
Principal Component Analysis
Principal component analysis (PCA) was performed using equal numbers of cells for each OR to avoid any OR having undue weights on any PC based on its frequency of expression. All versions of PCA shown used the scaled expression of the set of 1350 HVGs, and these HVGs were reduced to the top 20 PCs; however, stable results were obtained with different numbers of HVGs and PCs (Figure S1I). After fitting PCA in this manner using equal numbers of cells for each OR the fitted gene loadings (VT) were applied to the scaled HVG expression for all cells to obtain PCA scores for the remaining cells. In analyses in which this subsampling procedure was repeated across restarts, PCA was fit on the subset of cells sampled for each restart. For UMAP visualization, the results from a single restart are shown (although qualitatively similar restarts were obtained across restarts). In the “OR-swap” experiments, PCA was fit using only OSNs from wild-type mice (with equal numbers of cells per OR) and then applied to the cells from the “OR-swap” and P2(P2 locus) mouse lines. In the chronic occlusion experiments, PCA was refit using equal numbers of cells per each OR for each nostril, and in the environment switch experiments, PCA was fit using equal cells for each OR from each of the DPG-exposed control mice from each of the three environments.
Visualization via UMAP
UMAP was used solely for visualization purposes (McInnes et al., 2018). To generate UMAP visualizations, the gene expression of each cell was further reduced from 20 PCs to 2 dimensions using the Uniform Manifold Approximation and Projection (UMAP) technique with parameters n_neighbors=25, min_dist=0.6, and metric=“euclidean”. As for PCA, UMAP embeddings were fit using balanced numbers of cells for each OR (and only for wild-type OSNs in the “OR-swap” experiments), and these embeddings were then used to transform to the PCA scores for all cells of a given dataset into the same UMAP space. UMAP was fit using 1000 epochs and a learning rate of 0.25 to improve the stability of this transformation step. The UMAP embeddings were refit separately using the PCA scores from each dataset, yielding embeddings that differed qualitatively in appearance yet retained similar features (dorsal, ventral, and Cd36-positive OSNs were separated, the main axis was activity-dependent, cells expressing the same OR were locally clustered). UMAP and PCA were also refit using only cells from the occluded nostrils to determine if OSN subtypes cluster based on intrinsic activity in the absence of odor-evoked activity.
Transcriptome distance
To measure the similarity of cells based on their gene expression, the transcriptome distance was defined as the cosine distance (1 – cosine similarity) between the PC scores of pairs of OSNs. The cosine distance is bounded between 0 (an angle of 0° indicating similar transcriptomes), 1 (an angle of 90° indicating orthogonality) and 2 (an angle of 180° indicating opposing transcriptomes).
Equal numbers of cells (10) were selected for each OR (of the 831 ORs detected in at least 10 cells in the home-cage dataset), PCA was refit based on the scaled HVG expression of these 8310 cells and the pairwise transcriptome distance matrix was then evaluated. Within-OR distances we calculated, for each OR, as the median pairwise distance between all pairs of cells sharing that OR. For between-OR distances, the median pairwise distance between all pairs of ORs was calculated. Then, since each OR is more similar and dissimilar to specific subsets of other ORs, for each OR, the distribution of between-OR distances were summarized across all pairs of ORs sharing that OR. Starting from subsampling OSNs, the within- and between-OR distances were recomputed across 1000 restarts, and the distribution of within-OR distances across ORs was compared to the distribution of pairwise distances between each OR and all other ORs. On each restart, the OR labels were also shuffled across cells, and within-OR distances were recomputed (and were close to 1 indicating that the transcriptomes of random cells were dissimilar). Similar results were also obtained using the Euclidean and Correlation distance metrics. To evaluate whether these distributions of within- and between-OR transcriptome distances remained separable (even though distances increase with increasing dimensionality) across various choices of gene sets and numbers of PCs, the Jensen–Shannon divergence was computed by comparing the discretized versions of these two distributions on each restart (with 111 evenly spaced bins between 0 and 2) and comparing the entropy of the average distribution to that of the average of the entropies of each individual distribution.
k-nearest neighbor analysis
To determine whether cells sharing the same OR are clustered locally within gene expression space, a k-nearest neighbor (KNN) approach was used. For each of 1,000 restarts, using the pairwise distance matrix between cells described above, for each cell for each OR, the fraction of all the other cells that share the same OR within k neighbors was computed empirically for progressively larger groups of k neighbors. The fraction of same-OR cells found within k neighbors was averaged for each OR across restarts and was then summarized across the entire set of 831 ORs. The local clustering of cells sharing the same-OR was not apparent when shuffling the OR labels across the cells from each restart.
Classification of ORs expressed in home cage data
Classification pipeline
In all instances, classification was performed in a cross-validated manner using linear classifiers fit on equal numbers of cells for each OR. Equal numbers of cells were randomly selected for each OR (10 cells for each of the 831 ORs expressed in at least 10 OSNs in the home-cage data). For both pairwise and OR identity classification, the number of cross-validation folds was chosen to be equal to the number of subsampled cells (10) for each OR. For each fold, one cell expressing each OR was held out and the remained cells were used to train linear support vector machine (SVM) models (SVC with kernel = “linear”, decision_function_shape = “one-vs-one”, and regularization parameter C = 0.03). The hyperparameters for the classification pipeline were identified via a random search, but similar results were obtained across a range of hyperparameters. The input to the classifiers was the scaled gene expression (excluding any OR, ribosomal, or mitochondrial genes), using all genes (~13,000) that were expressed in at least 0.5% of all cells. During each cross-validation fold, feature selection was performed by keeping the top 10% of genes with the highest F-score with the OR labels of the training fold cells. Importantly, although this is the same procedure used for HVG selection described above and the 10% threshold was designed to yield a similar number of genes, because feature selection was performed in a cross-validated manner, the resulting set of 1,300 genes identified for each fold may differ slightly from the set of HVGs used for other analyses). Next, the dimensionality of these 1,300 genes was further reduced to 20 dimensions with linear transformations via PCA and Linear Discriminant Analysis (LDA), also using only the cells in the training fold. Importantly, in addition to using equal numbers of cells per OR (to make predictions at similar chance levels for each OR), the feature selection, PCA and LDA transformations, and SVM decision boundaries were all fit using only the data in the training folds and then applied to the held-out cells in the test fold to generate predictions of the expressed OR in each held-out cell. This classification pipeline therefore avoids data leakage (which could aid in the prediction of OR identities and potentially bias results) between training and test folds.
Classification between OR pairs
Pairwise classification was performed using the same classification pipeline. For each training fold, as described above, the gene sets, PCA and LDA transformations were fit using all cells in the training fold, and then applied to transform all held-out cells into the same 20D subspace. Then, using the training data, separate SVM models were fit for each pair of ORs (831C2 = 344,865 pairs) and these decisions boundaries were applied to the held-out cells of that OR pair to predict which OR within that pair each held-out cell expressed. This cross-validation procedure was repeated for each cross-validation fold, and the mean classification accuracy was computed for each OR pair. The entire pairwise classification procedure (starting from subsampling OSNs) was repeated across 1,000 restarts (yielding in total 344,865 × 10 × 1,000 = 3.45 billion models). The mean accuracy for each OR pair was summarized across the 1000 restarts, and the distributions of classifications accuracies were shown for the 344,865 OR pairs. The median prediction accuracy was 100% (compared to a chance accuracy of 50%), indicating that in the majority of OR pairs, the OR identity for every OSN of that OR pair was correctly predicted in each of the 1,000 restarts. The same classification procedure was performed separately for both nostrils for the 436 ORs detected in at least 10 cells in each nostril in the chronic occlusion data.
Classification of OR identity across all ORs
The same cross-validation and classification procedure was used to predict which OR (out of 831) was expressed in each held-out cell. “One-vs-one” SVM models were first fit on the PCA and LDA-transformed gene expression of all OR pairs in the training set and then these transformations and models were applied to each cell in the test fold; this procedure was repeated for each fold. The “decision function” from this procedure thus returned for each held-out cell the predictions from the model fitted on each pair of ORs in the respective training fold, yielding a matrix of 8310 cells × 344,865 models that indicates for each of the 344,865 “one-vs-one” binary pairwise classifiers which OR within that pair was more likely to be expressed. Predictions were generated using the standard voting procedure; for each cell, for each OR, the number of times that the subset of 831 pairwise models containing that OR predicted that specific OR (rather than the other OR in each pair) as more likely to be expressed were summed. The OR with the highest number of “+1” votes (out of a maximum possible 831) was predicted as the OR that was most likely to be expressed in a given cell. Since this procedure calculated the total number of votes for each OR for each cell, it also generated the likelihood (based on the ranks of the number of “+1” votes for each OR) any given OR was expressed in any given cell. Cells whose OR identity was correctly predicted were those in which the actual OR expressed in that cell was the one that received the most votes (or on rare occasions tied for the most votes).
Given that correctly predicting the exact OR (out of 831) of each held-out cell given only 9 training cells expressing the same OR and 7470 (830 × 9) cells expressing other ORs is a difficult task (with chance accuracy 1/831), the frequency in which the OR expressed in each held-out cell was within the top-N% of predicted ORs was also calculated using the ranks of the number of votes for each OR described above. In this case, predictions were considered as accurate at increasing percent thresholds (e.g. 0.25% = the actual OR was within the top 2 ORs with the most votes and 1% = within the top 8 ORs out of 831). For both the exact and top-N% classification procedures, the classification accuracy for each OR was summarized across the 10 cells expressing that OR in each restart and the accuracy for each OR was then averaged across 1000 restarts yielding the average classification accuracy for each of the 831 ORs, which are shown in the figures. Across the 831 ORs, the median classification accuracy was ~50%. Given the balanced number of cells per OR, classification accuracy did not depend on OR frequency (Figure S1J) and was at chance levels for classifiers fit on the same data in each restart but with shuffled OR labels (1/831 = 0.1% for the exact predictions and 1% for the top-1% predictions). This same OR identity classification procedure was performed separately for both nostrils for the 436 ORs detected in at least 10 cells in each nostril in the chronic occlusion data.
Classification with different numbers of genes
OR identity classification was performed as described above, but the % threshold for the F-score feature selection procedure of the classification pipeline was modified to assess the classification performance with varying numbers of genes. First, the high threshold was progressively increased from 0.5 to 12.5% of the ~13,000 expressed genes (compared to the default of 10%). Next, starting from the default threshold of 10%, the most informative genes were progressively excluded by increasing the low threshold in 0.5% intervals such that the top 0.5%, then the top 1%, were excluded until only genes in the top 9.5–10% remained. 10 restarts were performed for each gene threshold, and the mean accuracy across ORs was calculated.
Classification with different gene sets
OR identity classification was performed as above for the different gene sets, using genes from each set that were expressed in at least 0.5% of cells. These gene sets included the set of 543 mouse transcription factors detected in the OSNs (Aibar et al., 2017), and a set of 119 axon guidance genes (defined as known cell adhesion molecules with the following prefixes: Slit, Robo, Epha, Ephb, Efn, Nrp, Sema, Plx, Kirrel, Pcdh, Cdh, Tenm; using these prefixes was important because common GO terms for axon guidance genes also include OSN-specific transcription factors like Lhx2 and Bcl11b and other genes like ion channels that are not directly involved in axon guidance). As a comparison, classification was performed using only the top 0.75% of highly variable genes based on their F-score (rather than the default used above of 10%, thus yielding ~100 rather than 1,300 genes). Optimal hyperparameters for feature selection (based on their F-score in the training folds, as before) for the percent of genes from each gene set to keep, as well as the number of dimensions for PCA and LDA and SVM regularization strength were identified via randomized searches for each gene set. Classification was also performed after removing all transcription factors and axon guidance genes (and increasing the F-score threshold to 10.5% to again use the top ~1,300 genes). Models trained with only axon guidance genes or transcription factors performed substantially worse than those with the top 100 F-score genes, and excluding all axon guidance genes and transcription factors genes had little effect on classification accuracy (Figure S1M).
Classification generalization across mice
Pairwise and OR identity classification was performed with the same pipeline described above. Rather than just generalizing across held-out cells (which may have been from either the same or other mice), here classification was performed by leaving out all of the OSNs from single mouse, while the remaining 5 out of 6 mice were used for training to fit the classification pipeline (gene selection, PCA and LDA transformations, and the SVM decision boundaries). For each mouse, ORs found in at least 10 cells in the 5 training mice and at least 4 cells in the test mouse were used, and classification accuracies for each OR in the test mouse were evaluated across 1,000 restarts. Similar accuracies were observed when shuffling mouse identities, demonstrating that models successful generalize across mice. Generalization performance was at chance levels when trained on models with shuffled OR labels.
Classification of OR identity in “OR-swap” mice.
Classification was performed for the “OR-swap” mice using the PCs fit on the set of 1350 HVGs. The top 10 PCs were used as inputs to “one-vs-one” SVM linear classifiers trained to distinguish between M72, S50, and P2. Using the 42/18/84 cells for M72/S50/P2 in the wild-type data, the models were trained using 15-fold cross validation (subsampling 15 cells for each OR) to evaluate their ability to distinguish between wild-type OSNs expressing the three ORs. Models fit on only the 15 wild-type cells subsampled for each OR also successfully generalized to accurately predict the OR identities in the more than 500 held-out cells for each OR from the “OR-swap” and P2(P2 locus) mouse lines. Classification accuracy was summarized across 1,000 restarts.
Classification of OR classes
Classification of OR class was performed at the OSN subtype level (by taking the means of all the OSNs expressing each OR) between the 504/207/105 ventral class II, dorsal class II, and dorsal class I ORs. For each of 1,000 restarts, 80 OSN subtypes were selected for each OR type and classification was performed using 10-fold cross-validation with a pipeline that used linear SVMs (SVC with kernel = “linear”, decision_function_shape = “one-vs-one”, and regularization parameter C = 0.03) fit on the top 10 PCs in each training fold to predict the OR class of the held-out ORs. Classification accuracies were at chance performance (33.3%) upon shuffling the OR type labels across OSN subtypes.
Consensus Non-negative Matrix Factorization (cNMF)
Non-negative Matrix Factorization (NMF) was used to decompose the transcriptomes of each OSN into a smaller set of interpretable factors. NMF decomposes the gene expression matrix (cells × HVGs) into a combination of two matrices (W * H), one containing gene loadings (H) for each factor and another containing the usages (W) of these factors in each cell (Figure 2A). In contrast to PCA, these NMF factors often represent biologically meaningful and interpretable gene sets, and therefore were referred to as gene expression programs (GEPs). Because NMF is non-deterministic, a recently-described consensus NMF (cNMF) strategy was used, which has successfully found identity- and activity-related GEPs in a range of scSeq datasets (Kotliar et al., 2019). TPT-normalized data rather than raw counts (as used previously) were used as inputs to the cNMF procedure, since biological variation in the number of counts per cell was not expected given that all cells were mature OSNs. Each gene was scaled by its standard deviation using the standard deviations described above.
In brief, as previously described (Kotliar et al., 2019), cNMF applies NMF multiple times and, after excluding outliers (based on their distance to nearest neighbors, density_threshold=0.3), clusters the gene loadings across restarts with k-means clustering and uses the median of these clusters as the new gene loadings. In each of 20 restarts in which 10 cells were resampled for each of the 831 ORs, NMF was refit 150 times across a range of factors. A rank 18 decomposition, which balanced the tradeoff between reconstruction error (as measured by the Frobenius norm) and the consistency of the identified GEPs across restarts (as measured by the silhouette score), was used for the analyses presented here, but similar GEPs were observed with factorizations of other ranks. To match factors across restarts, the 54,000 × 54,000 distance matrix (18 × 150 × 20) was subject to k-means clustering using the value of k (19) that maximized the silhouette score. Clusters for stable factors should have close to 3,000 members (20 × 150). For each of the 19 clusters, the median loading across cluster members for each gene were used as the consensus gene loadings for that GEP. Three of these 19 clusters either appeared in few restarts or had non-zero usages only in small numbers of cells and were not considered further. For the 16 remaining GEPs, the loadings were normalized to sum to 1 for each GEP; these loadings can be found in Table S2. Using this set of 16 GEPs as the new H matrix, the usages of these GEPs for each OSN were calculated by reapplying NMF while keeping the gene loadings constant (with update_H=False).
Identification of OR-specific GEPs, the Environmental State (ES) GEPs, and calculation of the Environmental State (ES) score
Individual GEPs may explain variation across ORs, allowing for accurate classification of OR identities, or they may explain other, independent, aspects of variation. To distinguish between these possibilities, across 10,000 restarts 10 cells were randomly subsampled for each OR, and the correlation between the mean GEP usages for cells expressing each OR (vectors of length 831 for each of the 16 GEPs) were compared between the first and second half of cells for each OR. The 10 GEPs with consistently-high correlations (R > 0.9) were considered “OR-specific”, and each had higher gene loadings in a restricted set of HVGs (Figure S2A–B).
GEPs were identified based on manual inspection of genes with high loadings and high correlations with the usages for that GEP; four GEPs included known dorsal, ventral, anterior or posterior identity marker genes, while one GEP was specific to genes associated with CD36 positivity. One GEP contained genes like Ddit3 (CHOP) that are associated with ER stress and the unfolded protein response, but this GEP was not identified as OR-specific in our mature OSN dataset. GEPs that were not OR-specific (GEPs 11–16) and whose means across cells expressing the same OR are therefore not meaningful, or GEPs whose functions were unable to be identified based on their gene loadings (GEPs 8–10, which also had usages that were weakly correlated with other GEPs) were not considered for downstream analyses.
Two of the OR-specific GEPs encoded many genes associated with neural activity in OSNs (reviewed in (Wang et al., 2017)). One included S100a5, Pcp4l1, and Kirrel2 (which are downregulated by chronic sensory deprivation), while another included Calb2, Kirrel3, Ppp3ca (which are upregulated after chronic sensory deprivation; see Figure 4C). These two GEPs were referred to as GEPHigh and GEPLow. The usages of GEPHigh and GEPLow were anti-correlated and expressed in a mutually-exclusive manner across a continuous range of values (Figures 2 and S2). The difference in their usages was therefore summarized as a single metric to define a single continuous axis of gene expression. This difference (GEPHigh - GEPLow) was referred to as the Environmental State (ES) score, and GEPHigh and GEPLow were collectively referred to as ES GEPs, given the hypothesis that variation in ES GEP usages and thus ES scores across OSNs expressing different ORs is a consequence of the differential contribution of environmental odors on the activity levels and expression of ES-related genes across OSNs.
Among the identity GEPs, GEPDorsal and GEPVentral, as well as GEPAnterior and GEPPosterior also varied smoothly and in a similar mutually-exclusive manner across ORs; their differences in usages were therefor also summarized into single metrics (DV score = GEPDorsal - GEPVentral and AP score = GEPAnterior - GEPPosterior) to capture the continuous variation across these 3 independent axes (DV, AP, and ES; see Figure S2). The DV scores were distributed bimodally, and OSNs whose DV score = GEPDorsal - GEPVentral was at least 40 were considered as dorsal OSNs, yielding consistent results as the manual gene-based identification of dorsal OSNs described above.
Identification of neuronal and functional genes in environmental state (ES) GEPs
Genes associated with each of GEPHigh and GEPLow were identified by two criteria: 1) the gene was within the top 200 genes with the highest loading for that GEP and 2) the expression of that gene was correlated with the GEP usage on OR-by-OR basis in the home cage data (the Spearman’s rank correlation between the vectors of GEP means and gene means across the 831 ORs was 0.25). This approach identified 168 and 177 genes in GEPHigh and GEPLow, respectively. The potential functions/ontologies of these genes were manually annotated, and 63 and 54 “neuronal” genes whose known or proposed function in neurons were identified for the two ES GEPs. As listed in Table S3, these neuronal genes were categorized according to their putative roles in either calcium homeostasis (calcium binding, calcium signaling, inositol phosphatide related) ion transfer (ion channel, ion pump/transporter), axon guidance (axon guidance, cell adhesion), synapse (synapse, secretory peptide), or protein transport (ER/golgi/cilia related). The remaining genes with high loadings and correlations with the ES GEPs had similar patterns of gene expression in OSNs as the above genes, but their functions in OSNs remain unannotated. The “neuronal” genes were further restricted to include only the “functional” gene categories that are predicted to directly impact OSN odor responses, and which included the 73 genes that were part of the calcium homeostasis, ion transfer and OR signaling categories (see Table S3). Of note, while many known components of OR signaling are included in this list, the expression of Ano2 and Adcy3 remained constant across OSN subtypes and did not meet the above correlation thresholds (ρ < 0.2 for GEPHigh, GEPLow, and the ES score for both genes). Furthermore, although Omp was weakly correlated with the ES score (ρ = 0.36), because it was not part of the HVGs it did not contribute to the usages of any GEP.
Applying cNMF from home-cage data to other data sets
Although cNMF gene loadings were identified using the home-cage data, they were subsequently applied to other datasets to thus compare, on an OR-by-OR basis, changes in gene expression and GEP usages across experimental conditions. For all datasets, the TPT-normalized HVG expression from that dataset was scaled by the standard deviations from the home-cage data that were used when initially fitting cNMF. Using the fixed set of gene loadings (H), new usages (W) were solved by refitting NMF (update_H=False). To compare GEP usages across conditions, the mean GEP usage in that condition was calculated for each OSN subtype for all cells expressing that OR, and then differences between conditions were computed for each OSn subtype; distributions depict the change across OSN subtypes.
Comparisons of ES scores across mice and conditions
Changes in ES scores were evaluated by comparing the ES scores for each OSN subtype across experimental conditions. For chronic and transient occlusion ES scores from occluded nostrils were compared to those from open nostrils. For environment switches ES scores were compared for each OSN subtype between pairs of environments. For optogenetic stimulation, the change in ES scores was compared between mice housed in the home cages and those that either were or were not optogenetically-stimulated. In the chronic acetophenone experiments, ES scores were compared between mice that were chronically exposed to various concentrations of acetophenone and those that were exposed to the control solvent DPG. The changes in ES scores and z-scored functional gene expression across conditions for each OSN subtype are described in Table S4. The relative ES score change for each OSN subtype was calculated by subtracting the mean across all OSN subtypes, to normalize for any global changes across conditions that were present across the population of OSNs.
The Spearman’s rank correlation was also used to compare the consistency of ES scores in OSNs expressing the same OR across datasets. ORs identified in each dataset were compared, and the correlation coefficient was calculated on an OR-by-OR basis between the vectors of mean usages for each OSN subtype in each dataset (where each value is the mean ES score across all OSNs that express that OR in each dataset). On-diagonal values for pairwise correlation matrices were calculated via bootstrapping, by resampling with replacement the ES scores for each OSN subtype from the OSNs expressing that OR and taking the mean of the pairwise correlations (of the OR-by-OR ES score means) between all pairs (10,000) from 100 bootstrap restarts.
In all experiments, identity-related GEP usages were stable for each OR across conditions (ρ > 0.95), whereas the ES GEPs and ES scores moved predictably with changes in activity. To assess how the change in ES scores related to the overall change in transcriptomes across conditions, equal numbers of cells were subsampled for each OR, and the difference in the mean ES score for each OR was correlated with the PCs of the Δ HVG vs OR matrix (the difference in mean expression of each HVG between cells expressing each OR in each condition). In the chronic occlusion, optogenetic stimulation, and environment switch experiments, the changes in ES scores were strongly correlated (ρ > 0.8) with the top PC of this matrix, and thus well aligned with the main axis of overall gene expression changes. Given that ES scores are also aligned with the top PC of the transcriptomes in the home cage, these data together suggest that activity-regulated ES genes define a main axis of transcriptional variation, and experimental manipulation causes restricted changes in gene expression along this axis.
Identifying ORs with significant ES score changes across datasets
ORs with significant changes in their associated ES scores across datasets were identified empirically using the d-prime metric, which measured the separability of the ES score distributions for each OSN subtype. Within each OSN subtype, the d-prime statistic was calculated by taking the difference in the mean ES scores for each condition and dividing by the square-root of the average of the variance in the two conditions. This observed d-prime statistic was compared to the distribution of resulting d-prime values from permuting the condition labels across all cells of a given OSN subtype 10,000 times. P-values were calculated empirically, corrections for multiple comparisons were performed via the Benjamini-Hochberg FDR procedure, and FDR-corrected p-values ≤ 0.01 were considered as statistically significant. ORs with significant shifts in the chronic occlusion and environment switch experiments had mean d-prime values of 5.63 and 2.50, respectively, indicating excellent separability. OSN subtypes with significant ES score changes were separated into those whose mean ES scores increased and decreases based on the sign of the difference in mean ES scores across datasets. In the environment switch experiments, each pair of environments was analyzed separately, but results are concatenated across the 3 environment pairs (and therefore in plots depicting OSN subtypes there are 3 datapoints for OSN subtypes identified in all 3 environments).
Classification of OR identity with NMF GEPs
Pairwise OR classification and classification of OR identity was performed using the same cross-validation and balanced subsampling procedures described above. However, rather than performing gene selection and dimensionality reduction during training, the direct input to the linear SVM classifiers was the GEP usages for each OSN for either all 10 OR-specific GEPs or only the two ES GEPs, as specified. Classification was performed using the 831 ORs found in at least 10 cells in the home-cage data, the 436 ORs found in at least 10 cells in both nostrils in the chronic occlusion data, and in the 488/496/522 ORs found in at least 8 cells in home-cage, environment A and environment B datasets from the environment switch experiments. Despite using only 2 or 10 dimensions to distinguish the entire set of ORs, classification accuracies were above chance for all datasets, and at chance levels upon shuffling OR labels for each OSN.
Classification of nostril and environment identities using the NMF GEPs and ES scores
Rather than identifying the OR expressed in each cell, linear SVM classifiers were also used to test whether OSNs expressing the same OR were distinguishable across datasets based on their GEP usages. Separate classifiers were used for each OSN subtype and k-fold cross-validation was used, with k set to be equal to the numbers of cells subsampled per condition. As expected, classification accuracy was significantly above chance for the chronic occlusion and environment switch experiments, especially for the ORs with significant ES shifts (but ORs with smaller but non-significant shifts were still discriminable at above-chance levels). Classification performance was similar using all 10 GEPs or only the 2 ES GEPs, and at chance levels when the dataset label was permuted across OSNs for each of 1,000 restarts, further demonstrating that the main change across conditions is restricted to changes in the ES score axis.
Classification of environment identity was also performed across subpopulations of OSN subtypes, using a minimum distance classification procedure. Across 1,000 restarts, six OSNs were subsampled for each OSN subtype for each environment (from the 477 subtypes present with enough cells in all environments), and five OSNs were used for training. The mean ES score for each subtype for each of the three environments was calculated (yielding three training vectors containing the mean ES scores across the population of 477 subtypes). The ES scores of the held-out cells for each subtype were similarly concatenated into three test vectors. For each of the 1,000 restarts, subpopulations of the OSN subtypes (containing 1–50 subtypes) were randomly sub-selected 1,000 times and the training vector for that subpopulation with the minimum distance to the same subpopulation from each test vector was the predicted environment label; predictions were considered accurate if the training vector with the minimum distance came from the same environment as the test vector. Accuracies were summarized across restarts, and classification performance was at chance levels when the environment labels were shuffled across training cells for each OSN subtype.
Modified ES scores with HVG subsets
Modified versions of the ES GEPs were constructed to assess the contributions of individual genes within the ES GEPs. Although many genes have low loadings for each GEP and a restricted subset have higher loadings, each GEP was defined based on their loadings across the entire set of 1350 HVGs. To test whether the 73 functional genes could substitute for the entire set of HVGs, the loadings for all of the remaining HVGs were set to 0, the total loadings for each GEP were rescaled to sum to 1 across the 73 functional genes, and this new gene loading (H) matrix was used to calculate updated usages (W) by reapplying NMF (with update_H=False).
The “functional” ES score, constructed again as GEPHigh - GEPLow from the new set of usages was highly correlated (ρ > 0.99) with the ES score from all HVGs, decreased as expected upon occlusion, and could distinguish between OSNs expressing different pairs of ORs using the classification procedure described above, thus indicating that the “functional” genes and their expression can substitute for the entire set of HVGs (Figure S4). We also observed that the levels of some of the activation genes varied slightly across environments. To assess the contribution of these activation genes on ES scores and on the ES scores changes measured after environmental shifts, the loadings for the 169 activation genes that were part of the HVGs were set to 0 for all GEPs and the GEP usage and ES score was recalculated as described above. The ES scores and changes in ES scores calculated using all HVGs or excluding the activation genes were highly correlated (ρ > 0.99), suggesting that activation genes (whose expression primarily changes during periods of acute activation) do not account for the significant shifts in ES scores observed across environments.
Identification and pseudotime analysis of immature OSNs
Pseudotime analysis was performed on the immature OSNs identified from mice housed in the home cages. The Leiden clustering procedure that was used to identify mature OSNs also identified clusters of immature OSNs identified, which were used for pseudotime analysis. Diffusion pseudotime (n_dcs=10) was calculated in Scanpy on the nearest neighbor graph (n_pcs=10, n_neighbors=10), and normalized to the lowest pseudotime values of immature OSNs that had undergone OR choice and were expressing ORs. To compare gene expression patterns and GEP usages in immature OSNs expressing given ORs with those of their respective mature OSN subtype, only immature OSNs that had chosen and expressed ORs were used. Gene expression and GEP usage as a function of pseudotime were evaluated using linear generalized additive models (GAMs) in pyGAM. A linear GAM was fit using 15 basis splines (lambda=1) for individual genes and fit using 50 splines (lambda=5) for GEP usages, and the mean and 95% confidence intervals of the mean were evaluated across a grid of 500 evenly-spaced pseudotime values. The mature ES scores across OSN subtypes were discretized into five quantiles, and separate GAMs were fit using the immature OSNs expressing ORs from each mature ES score quantile.
Differential expression testing
Given the wide variation in gene expression across OSNs in the home-cage condition, differential expression testing was performed at the OR-by-OR level to identify genes that consistently changed across multiple OSN subtypes. Differential expression (DE) testing was performed using ORs found in at least 6 cells in each dataset to balance the trade-off between the number of OSN subtypes and within-subtype variance. Testing was performed via the Wilcoxon signed-rank test (comparing, for each gene, the differences between means for each OSN subtype across conditions). P-values were corrected for multiple comparisons across genes via the Benjamini-Hochberg FDR procedure.
For chronic occlusion, the DE genes with the most significant changes were defined as those among the top 500 genes with the lowest p-values that had consistent changes across ORs (same sign of change in at least 50 % of the 673 OSN subtypes detected in at least 6 cells in each nostril and absolute value of the median log2-fold change across OSN subtypes of at least 0.5); however, consistent results were obtained across a wide range of thresholds given that most DE genes changed in the majority of the OSN subtypes. As expected, genes that increased or decreased during occlusion had high loadings for GEPLow and GEPHigh, respectively. DE testing via the Wilcoxon signed-rank test was also performed to identify genes that were acutely responsive to odor exposure.
For each odor, the same DE testing procedure was performed separately for activated and non-activated OSN subtypes (as defined based on the percentage of activated cells expressing that OR; see below). To exclude the small subsets of genes that changed indiscriminately across all OSN subtypes regardless of whether they had been activated by odor, genes whose p-value was higher for non-activated ORs or those whose mean change in expression across OSN subtypes was less than 1.5 times higher in the activated ORs than non-activated ORs were removed and not considered for downstream analyses.
Identification of activated cells and ORs by Act-seq
OSN subtypes that were responsive to odorants were identified via a two-step process. First, individual OSNs (as defined by the expressed OR) were determined as activated based on their mean z-scored immediate early genes (IEG) expression. A set of 10 IEGs were used (Btg2, Egr1, Fos, Fosb, Gm13889, Junb, Nr4a1, Nr4a2, Pcdh10, and Srxn1). These genes were reliably and strongly induced in odor-exposed mice and, unlike many of the activity-dependent genes that are part of the ES GEPs and whose expression varies widely across OSNs expressing different ORs, these IEGs had consistently low expression across OSNs in control mice (average expression ~0.15 in the control conditions and average log2 fold-change of ~5 in the odor conditions). The set of 10 IEGs was used since even though all of the IEGs were specific to acute activation by odors, they were still only detected in a minority of cells due to the sparse nature of scSeq. The means and standard deviations for IEG z-scoring were fit on the TPT-normalized IEG expression (without the log transformation due to the sparsity of expression) across 100 restarts containing equal numbers of subsampled cells for each OR from mice that were exposed to either acetophenone or octanal. OSNs whose mean z-scored expression was above 0.2 (~15% of OSNs in odor-exposed mice but less than 2% of OSNs in control mice exposed to the DPG solvent alone).
Second, as expected, the distribution of the percent of OSNs per OSN subtype was bimodal, with a majority of OSN subtypes showing low percentages of activated OSNs with a smaller subset showing high percentages of activated cells. Using the ORs detected in at least 4 cells in both control and odor-exposed conditions, activated ORs were defined as those in which less than 20% of cells expressing that OR were activated in the control conditions and more than 70% of cells were activated in the odor-exposed conditions, with the additional requirement of non-zero expression of at least 4 IEGs (to avoid false positives from OSNs activated by semiochemicals in the empty cages used for Act-Seq or those that strongly induced a small number of IEGs). Given that the set of ORs and the number of cells expressing each OR differed across datasets, odor-responsive OSN subtypes were re-evaluated for each dataset. Overall, odor-responsive OSN subtypes that were detected in multiple datasets were consistently identified as odor-responsive, though a small fraction of them (and thus their expressed ORs) were identified as activated in some datasets but only partially activated in others. These results are likely due to both the consequences of experimental manipulations, which were designed to affect odor responses, as well as the relatively conservative thresholds used above to identify ORs as activated.
For the environment switch experiments and the Act-Seq experiments using the four acetophenone-related odors, activated ORs were identified as described above, except using an IEG threshold of −0.05 because of the higher levels of IEG expression in the 10x v3.1 data. For the acetophenone concentration series experiment, activated ORs were either identified for each concentration, or the ORs identified as activated at 10% were also examined at lower concentrations, as specified in the text. In the optogenetic stimulation experiments, no IEG thresholds were used to identify activated cells, and instead all dorsal OSN subtypes were used for downstream analyses. In the chronic acetophenone exposure experiments, the set of acetophenone-responsive ORs identified via the acute Act-Seq experiments were used.
Quantifying the magnitude of odor responses via the activation score
Although IEG induction could identify activated OSNs in a binary manner, the sparse nature of the IEGs made them less suitable for measuring differences in the degree of activation. Therefore, a larger set of odor-responsive genes (from the DE genes identified above) were used to construct an analog metric for activation and therefore compare differences in the amount of activation across OSN subtypes (as defined by their expressed ORs). The set of “activation” genes used for this metric satisfied the following criteria 1) significant responses (FDR ≤ 1 × 10−3) to both acetophenone and octanal, and 2) average TPT-expression of at least 0.8 in activated ORs (such that the metric wouldn’t be biased by the presence of zeros). Acetophenone and octanal induced similar changes in gene expression in their respective responsive OSN subtypes. Of the ~500 genes passing these criteria, the small number of genes that had high (ρ > 0.4) correlations with the ES score in the control mice were removed, leaving a set of 472 “activation” genes that were used for all Act-Seq experiments (Table S5).
Consistent with activation score genes generally not being differentially used in the home cage, the majority of these activation genes were not part of the 1350 HVGs in the home-cage data, and thus do not contribute to any GEP usages. The remaining 169 activation genes that were also considered as HVGs often had weak loadings across multiple GEPs. Only 16 of these 169 genes had high loadings for GEPHigh, and removing all activation genes from the HVGs and recomputing GEP usages yielded similar results. Together, these results suggest that activation gene expression is specific to the acute effects of odor exposure and does not contribute significantly to baseline variation in transcriptional identities or ES scores across OSN subtypes. To summarize the effects of odor activation across this set of activation genes, the TPT-expression of these activation genes were z-scored (using means and standard deviations for these genes in the control, acetophenone, and octanal datasets, identified by subsampling equal numbers of cells across all ORs expressed in at least 6 cells across 100 restarts). Next, using the combined set of activated acetophenone- and octanal-responsive OSN subtypes, PCA was performed on the differences in z-scored activation gene expression between control and odor conditions. The top PC of this activation gene delta matrix was kept, and the activation score was defined as the product of the weights for this top PC and the difference in z-scored activation gene expression between odor and control conditions.
Thus, the activation score summarizes for each OSN subtype the overall movement of the activation genes between control and odor conditions along this main activation axis. Although the activation score weights were fit using the combined set of acetophenone- and octanal-responsive OSN subtypes expressed in at least 6 cells for stability purposes, all subsequent Act-Seq results are shown using the larger sets of ORs detected in at least 4 cells in both control and odor conditions. To obtain activation scores for OSN subtypes from other datasets (such as those in the transient occlusion and environment switch experiments), the activation score was evaluated in a similar manner by multiplying the activation PC weights and the delta activation gene expression (odor – control) for each OSN subtype. Only activated OSNs from the odor-exposed mice were used for calculating the activation score. The weights were kept constant across experiments, but three separate versions of the gene scaling was used for the ACE and OCT Act-Seq experiments, the transient occlusion experiments, and all other Act-Seq and environment switch experiments, respectively, to account for changes in the gene means due to differences in 10x kit versions and sequencing depth across these datasets. In all experiments, activation scores were higher in odor-exposed and odor-responsive OSN subtypes. Across replicates from the same condition, OSN subtypes had consistent levels of activation, demonstrating that the activation score successfully captures differences in the amount of activation in each condition across OSNs expressing different ORs.
Unsupervised methods for identifying activated cells and ORs
Two additional complementary methods were used to identify activated cells and measure activation in the ACE and OCT Act-Seq experiments. First unsupervised graph-based Leiden clustering (resolution=0.6) was fit on the top 20 PCs from the scaled HVGs (where PCA was fit using equal numbers of cells from control, acetophenone, and octanal mice and applied to all cells). A single cluster bore the hallmarks of activated cells, and primarily contained cells from the odor condition with increased IEG expression. Additionally, nearly all of the OSNs from odor-exposed mice that expressed activated ORs, as identified via the IEG approach described above, were part of this activated cluster.
Second, activated ORs and weights on genes affected by odor exposure were simultaneously identified via unsupervised tensor component analysis (TCA). Nonnegative tensor decomposition was fit using the hierarchical alternating least-squares algorithm (ncp_hals) in the tensortools python package (Williams et al., 2018), and the tensor of conditions × activation genes × OSN subtypes was reduced with a rank 4 decomposition into 4 rank-1 tensors. One of these tensors represented activation and had 1) high weights in the odor but not control condition, 2) high weights for activation genes affected by acute odor exposure (like Pcdh10 and Fos), as well as 3) high weights for the odor-responsive OSN subtypes identified above based on their IEG expressions; furthermore, the TCA weights for the activation factor each OSN subtype was correlated with the mean activation score for that subtype (ρ > 0.85 for both ACE and OCT).
Comparison of ES scores and activation scores for Act-Seq experiments
ES scores were compared to the activation scores for each OSN subtype to test how OSN transcriptional variation affects odor responses. All correlations were calculated between the ES scores in control mice and the activation scores from separate odor-exposed mice, using the set of odor-responsive OSN subtypes detected in a least 4 cells in both control and odor conditions. Comparisons were made at the OSN subtype level, between the mean ES score from the control condition and that OSN subtype’s activation score. When comparing the change in ES score and change in activation scores across datasets, only OSN subtypes present in at least 4 cells in both control and odor conditions for each dataset being compared were used.
Predictions of activation scores from control ES scores and functional gene expression via linear regression models
Cross-validated linear regression models were constructed using the TPT-normalized expression of the functional genes in the control mice as predictors of the activation score in odor-exposed mice. Regularization was performed using elastic-net regularized linear models, where the optimal hyperparameters (alpha, the l1-ratio, and standardization of regressors) were identified through grid searches for each dataset. 5-fold cross-validation was used, and model performance was evaluated via the mean squared error (MSE) between predicated and observed activation scores from each test fold. This cross-validation procedure was repeated 1,000 times and the distribution of observed MSEs was compared to the predictions obtained when the same models were trained and tested on data in which the activation scores were permuted across OSN subtypes; this shuffling procedure represents performance similar to that of null models that predict the mean activation score for all subtypes. Reported errors were normalized as a percentage of the observed data to facilitate comparisons across datasets. For a subset of datasets, the predictions of the activation score from the functional genes were also compared to predictions from ordinary least squares (OLS) linear regression models that used the control ES score as a single predictor. Predictions of the changes in activation scores were evaluated in a similar way to those of the activation score, except the changes in functional gene expression for each OSN subtype were used as predictors. In addition to shuffling the change in activation score across OSN subtypes, these models were also evaluated on data where the condition label was permuted across OSNs for each OSN subtype before calculating the changes in the functional gene expression for that subtype.
Classification of odors and environments from activation scores
Classification of odor identity for the acetophenone analogs based on activation scores was performed using a minimum distance procedure in a similar manner to classification of environmental identity via ES scores. The input was the set of 106 OSN subtypes whose ORs were activated by at least one of the four acetophenone-related odors and were detected in at least four OSNs for each odor (since few ORs were present and activated in enough cells across all four datasets; but see below). Across 1,000 restarts, three OSNs per odor were used for training and the fourth was used for testing. The mean activation score for each subtype for each odor was calculated. For each of the 1,000 restarts, subpopulations of the OSN subtypes were randomly sub-selected 1,000 times and predictions were considered accurate if, across the subpopulation of activation scores, the training vector with the minimum distance for each test vector shared the same odor label. Predictions were at chance levels upon shuffling odor labels across cells for each OSN subtype. The same classification procedure was also used for each pair of acetophenone odors, but the input was the set of ORs activated by both analogs for each pair to determine if the pattern of activation scores across OSN subtypes can distinguish between odors, even for ORs activated by both odors.
Classification of environments using OSN activation scores after odor exposure as predictors was performed using cross-validated linear SVM classifiers. Activation scores were computed for each OSN (rather than the OSN subtype as described above) by applying the activation gene weights to the difference in expression between the activation gene expression in each cell in the odor condition and that of the mean of its respective OSN subtype in the control condition. Classification was performed as before by subsampling equal numbers of cells per OR on each of 1,000 restarts, and prediction accuracies were at chance levels when the OR labels were shuffled across OR pairs or the environment pair labels were shuffled across the OSNs expression each OR.
in vitro odor-receptor interaction metrics
The set of activated ORs identified via Act-Seq were compared to those identified via the DREAM and phospho-S6 immunoprecipitation methods (Jiang et al., 2015; von der Weid et al., 2015). In vitro EC50 values to acetophenone for its receptors were obtained from (Jiang et al., 2015; Saito et al., 2009). Only ORs from these prior papers that were also identified in the Act-Seq experiments described above were used for comparisons. The 717 ORs detected in four or more OSNs in both DPG and acetophenone-exposed animals included 16/22 DREAM and 25/49 phospho-S6 ACE ORs. ORs that were not identified by Act-Seq were also not identified in more than 4 OSNs in these experiments, suggesting that these receptors might be “missing” because they are expressed in particularly small numbers of OSNs; note that this observation raises the possibility that these receptors are also near the noise floor in the DREAM and pS6 methods, since these approaches rely on bulk sequencing approaches that are also likely affected by OR frequency. In contrast to the relationship observed between the control ES score and activation scores, there was no relationship between in vitro EC50 values and activation scores from any concentration of ACE and OLS linear regression models were unable to predict activation scores from the EC50 values alone.
Comparisons of acute and long-term ES changes upon environment shifts
OSN specific activation scores were measured after mice were shifted from the home-cage to environment A (envA). The activation score at these timepoints was then compared to the two-week changes in the ES score (as measured as the difference in the ES score between home-cage and env-housed mice in the environment switch experiments). The activation score was calculated by applying the activation gene weights to the activation gene deltas between mice from each timepoint and control mice housed overnight. The activation scores were normalized by subtracting the mean activation score across OSN subtypes, to account for any non-specific changes in activation across the entire population (e.g. due to changes in odor sampling that may occur upon the transfer to new environment cages). The resulting “relative activation scores” were compared between OSN subtypes with significant increases and decreases in their ES scores after two weeks in envA, for subtypes present in at least 4 cells in all 4 conditions (home-cage and envA after two-weeks, as well as in the overnight control data and in the envA two hour dataset). As expected, OSN subtypes with significant increases/decreases in their ES scores after two weeks showed increased/decreased activation (relative to the mean) at each timepoint. In contrast, the opposite effects were observed for mice switched from envA back to home cages, and little activation was observed between mice switched to cages of the same environment.
The correlation between the activation scores at two hours and the two-week ES score shifts was evaluated using all OSN subtypes, and the two-week ES score shifts were also compared to the change in the ES score after 24 hours. Predictions of the sign of the two-week ES score based on the activation score at each timepoint was performed using 10-fold cross-validated linear SVMs (fit at the OSN subtype level using a random subset of 80 of the OSN subtypes whose ES scores significantly increased and 80 that decreased after two-weeks for classification on each of 1,000 restarts). Classification accuracy was consistently above chance levels but was at chance levels upon shuffling the sign of the two-week ES score change for each restart.
Craniotomy and cranial window for chronic olfactory bulb imaging
All surgical procedures were performed in accordance with the guidelines provided by IACUC at Harvard Medical School. Prior to surgery, 4–6 weeks old Omp-IRES-Cre;Ai95D (GCaMP6f) mice, were subcutaneously injected with the analgesic Buprenorphine-SR (1mg/kg) and anesthetized 1–2 hours later with isoflurane (3% induction, 1–2% maintenance). The skin overlying the left olfactory bulb was retracted and the underlying periosteum was scraped off with a scalpel. The skull was subsequently cleaned with saline and allowed to dry. A 2.5 mm circular craniotomy was made using a biopsy punch (Integra Miltex) taking care not to damage the underlying dura. Gelfoam saturated with cold saline was placed over the craniotomy to reduce swelling and suppress minor bleeds. A custom cranial window was constructed using two circular coverslips (Warner Instruments, 3mm, #1 thickness). The first coverslip was ground down to 2.5 mm in diameter using a diamond scribe and subsequently bonded to the second 3mm coverslip using Norland 71 optical epoxy. With the smaller coverslip in contact with the dura, the exposed edges of the 3mm coverslip were gently depressed until flush with the surrounding skull and secured in place with glass glue (Loctite, P.N. 233841). This “top hat” design ensured a tight seal around the craniotomy. For head-fixation, a custom titanium head plate was positioned over the skull and, using a manual manipulator, aligned to be coplanar with the cranial window. Following alignment, the head-plate was secured to the skull using dental cement (C&B Metabond, Parkell) and the animal was returned to the home cage for recovery. The supplementary analgesic Carprofen (5mg/kg) was provided in drinking water for 4 days following surgery. Throughout the 1–2 weeks prior to chronic imaging, the craniotomy was regularly inspected for any changes in clarity. Animals with excessive overgrowth of dura and skull tissue were not imaged and were removed from the study. In order to keep the cranial window clear of debris between imaging sessions, the dorsal surface of the skull was covered with silicone sealant (Kwik-Cast, World Precision Instruments).
Odor delivery
A 23-valve olfactometer was used to present odors, as previously described (Island Motion Systems, see (Pashkovski et al., 2020)). In brief, custom Arduino software was used to control valve opening and closing, thereby enabling switching between odor vials and the blank vial. This software also controlled the output of two mass flow controllers (MFC). The first MFC delivered a constant carrier flow at 0.75 L min−1 of purified air into a common channel; the second MFC supplied a constant flow at 0.25 L min−1 of clean air that was injected into an odor vial (see below) and then merged with the carrier flow 1 inch (2.54 cm) in front of the mouse’s nose. A larger exhaust fan drew air from the cage enclosing the imaging rig to prevent any cross-contamination between odors. Monomolecular odors were diluted in Mineral oil (Sigma), and this vapor-phase concentration was further diluted 1:4 by the carrier airflow. Odor presentations lasted for two seconds and were interleaved by 30 seconds of blank (Mineral oil) delivery. The order of presentation of odors was pseudo-randomized for each experiment, such that on any given trial, odors were presented once in no predictable order.
In vivo olfactory bulb imaging
Chronic imaging of the dorsal surface of the olfactory bulb was performed after recovery from the surgery (1–2 weeks) under ketamine (100mg/kg) and xylazine (10 mg/kg) induced anesthesia, to specifically isolate responses in OSN axons with less modulation from local interneurons and long-range cortical feedback. Depth of anesthesia was assessed every 15 minutes via the toe-pinch reflex and intraperitoneal ketamine was administered at 1/3 concentration of the induction dose for anesthesia maintenance. Multiphoton imaging was performed at 60 Hz using a 16 kHz resonant galvo scanner (Cambridge Technologies) with a chameleon laser tuned to 920 nm delivered to 30–50 mW of excited power at the front end of the objective (10X Olympus XLUMPLFL10XW-SP lens, NA 0.6), as previously described (Pashkovski et al. 2020). Emitted fluorescence was detected using Hamamatsu H10770PA-40 PMTs. Scanimage 5 was used for hardware control and image data acquisition; WaveSurfer was used for recording of acquisition, odor and frame triggers, as well as sniffing data in a subset of mice. To obtain consistent field of views across sessions, 0.5 mg of Texas-Red conjugated Dextran (3000 MW, D3328 Thermo Fisher) dye was injected subcutaneously, and the field of view was aligned using both red and green channels based on patterns of vasculature and glomeruli, respectively (using the first session as a template). Animals were monitored throughout each imaging session for any drift in the field of view across trials.
Experimental designs for olfactory bulb imaging across environment shifts
Initial experiments consisted of a cohort of four mice imaged across multiple environment shifts. Each imaging session consisted of at least 8 trials, in which each of the 7 odors were presented. The odors used included 3 odors that activate broad sets of the dorsal glomeruli and which were presented at both a high/low concentration (pentanone (25/2.5 %), ethyl butyrate (2.5/0.25 %), and pentanal (25/2.5 %)) as well as a 7th blank odorant (Mineral oil). When mice were not being imaged, they were housed in either regular home-cage or Environment A (envA) cages, consisting of the same contents as used for the scSeq experiments. Throughout the course of the experiment, mice were sequentially housed in the regular home cages (home1), envA cages (envA), and then regular home cages again (home2). 4–6 sessions were obtained for home1, 4–6 sessions were obtained for envA (across 14 days of housing in envA), and 2–3 sessions were obtained for home2 (across an additional 14 days of housing in the regular home-cage environments).
In an additional cohort of two mice, the responses to a broader panel of 16 odors (to facilitate decoding analyses) that densely activated the dorsal olfactory bulb were imaged. Each session consisted of at least 6 trials, in which a blank odor and each of the 16 odors (2.5% of Pentanal, Pentanone, Ethyl butyrate, Heptanal, Hexanal, Propanal, Butanal, Butanone, Hexanone, Ethyl acetate, Propyl acetate, Butyl acetate, Benzaldehyde, Tiglaldehyde, Methyl tiglate, Methyl valerate, all of which were obtained from Sigma except for Methyl tiglate, which was obtained from Thermo Fisher) were presented pseudo-randomly. Throughout the course of the experiment, mice were sequentially housed in the regular home cages (home-cage) and envA cages (envA).
Registration, ROI detection and signal extraction for olfactory bulb imaging data
For alignment of the FOV across sessions, baseline-subtracted images were downsampled (from 60 Hz to 10 Hz) and the mean image from each session was used to register the field of view across imaging sessions. Registration across sessions was performed using an iterative approach to align each session to a common template session. First, the optimal geometric transformation that maximized the overall cross correlation between mean images was identified using OpenCV (findTransformECC with warp_mode=MOTION_HOMOGRAPHY and gaussFiltSize=1). Next, local deformations were further aligned with Dipy (SymmetricDiffeomorphicRegistration with metric=CCMetric, level_iters = [250, 100, 50, 25], and inv_iter=50). The resulting procedure gave an invertible warping for each session that was subsequently applied to all frames from that session. Throughout a session, the field of view was stable, and no motion correction was applied; therefore, after registration, glomeruli were stably aligned across all trials and sessions. The z-depth of the image plane was aligned for every imaging session based on the specific pattern of vasculature and glomeruli found in each mouse, and this single plane was imaged throughout each session. Figure S7B shows that FOVs are stable across sessions and environments, as assessed by the pairwise enhanced correlation coefficient between mean images of the entire FOVs (computeECC function in OpenCV). Regions of interest (ROIs, glomeruli) were identified via Suite2p (skipping the registration and deconvolution steps) from the entire set of registered baseline-subtracted and down-sampled images from all sessions (Pachitariu et al., 2017). ROIs that covered multiple glomeruli or masks glomeruli split into multiple ROIs were manually corrected. Using the set of identified ROIs, fluorescence traces were manually extracted from the raw movies from each session by applying the session-specific warping described above to each movie and then taking the dot product of the fluorescence in the aligned movies with the pixel weights of each ROI mask. Neuropil subtraction was not performed since annuli from each glomerulus would overlap with other glomeruli and there was little background fluorescence outside of activated glomeruli.
Analysis of GCaMP signals
Normalization of GCaMP signals
For each trial and glomerulus, GCaMP signals were baseline-subtracted and normalized relative to their baseline (dF/F). The 10th percentile of the ten seconds prior to each odor presentation was considered as the baseline. dF/F values were interpolated to 50 Hz and smoothed by convolving with a gaussian filter (length=21, standard deviation = 5). Because of the long duration of the initial cohort, in which mice were imaged across multiple environment shifts over more than a month, dF/F traces were z-scored for each trial; dF/F values were not z-scored for the later cohort, which was imaged for a shorter duration across a single environment shift.
Identifying odor-responsive glomeruli
Responsive glomeruli were identified separately for each environment. Responsive glomeruli were defined as those whose mean response from 0.0 to 3.0 seconds after odor valve onset was significantly higher than that of the prior 10 seconds (p ≤ 1 × 10−3, via the Wilcoxon signed-rank test and adjusted via the Holm–Bonferroni method across odors for each glomerulus) and whose mean z-scored dF/F was greater than 0.5 (or mean dF/F was greater than 0.125 for the second cohort).
Aligning responses to the population mean
Responses were aligned across trials using the population mean to account for the variability in response onset present because odors were given in open loop without controlling for sniffing. The set of glomeruli that responded in at least two environments with a positive mean odor response were used for aligning odor responses across trials. The population mean was computed across these responsive glomeruli for each trial and the population onset was defined as the first timepoint in which the population exceeded a threshold (0.75 for z-scored dF/F data and 0.25 for dF/F); traces were aligned across trials by shifting them based on the identified population onset. This alignment procedure increased the correlation of the odor response across trials for each odor, suggesting that responses are more reliable once aligned to population, rather than odor, onset. Sniffing was also recorded with a nasal pressure sensor (Honeywell AWM3100V) in a subset of mice to further assess the performance of this alignment procedure. Sniffing signals were uncorrelated across trials when using the odor valve times but were correlated across trials when using the shifts identified via the population onset. After alignment, the population response reliably increased following inspiration, further suggesting that this alignment procedure helps to correct to variability in timing of odor responses across trials due to sniffing. After alignment relative to the population mean, responsive glomeruli were re-identified used the same thresholds and procedure described above, except now using the first 3 seconds after population onset, and responsive glomeruli kept for downstream analyses were those that were identified as responsive in at least two environments, and whose response to a given odor was a least twice that of the response to the blank.
Identifying environment-sensitive glomeruli
For the responsive glomeruli for each odor, the mean response amplitudes for each trial (the mean dF/F or z-scored dF/F values in the first 3 seconds following population onset) were averaged across trials within each environment. Glomeruli whose odor response amplitude differed across environment pairs were identified via permutation testing. The observed differences in means across environments was compared to a null distribution calculated by following the same procedure after permuting environment labels across trials 100,000 times. P-values were calculated empirically for each glomerulus-odor pair and were adjusted pairs by the Benjamini-Hochberg FDR procedure. Pairs whose FDR-corrected p-value was ≤ 0.01 were considered as significantly different across environments. Across mice and odors, 20–80% of responsive glomeruli demonstrated significant shifts in response amplitudes across the shift from home-cage to envA. In contrast, fewer glomerular-odor pairs were significantly different between home1 and home2. Glomerular responses were considered as reverted if the response amplitudes in home2 were more similar to home1 than envA. While one would naively expect reversions via regression to the mean if data were independent, no glomeruli were identified as environmentally sensitive when environmental labels were shuffled before calculating the observed difference, indicating that the observed differences in response amplitudes between home1 and envA (as well as those between envA and home2) are not the results of spurious partitioning of responses. Together, these results suggest that a large fraction of glomeruli responsive to a given odor are stable within environments but vary across environments.
Population level analysis
Population odor responses were analyzed using pseudo-populations of glomeruli imaged across mice. Pentanal elicited the broadest responses across the population of glomeruli and was therefore used to assess how the population response differed between environments. To test whether differences at the population level in the pentanal responses in envA compared to home1 were reverted in home2, an “environmental axis” that separated the home1 and envA responses was constructed. For each mouse, two testing sessions were held out for both home1 and envA, and the remaining sessions were used for training, and the response at each timepoint in these training sessions was averaged across all training trials. PCA was fit on the population responses in the training data to reduce the dimensionality of the glomerular population to 10 dimensions, and the mean response was calculated in 30 overlapping windows in time (each 2 seconds long starting from −0.2 s and each subsequent one offset by 0.05 s from odor onset). The differences across the 10 PCs between the response in envA and home1 was calculated for each window, and the weights were averaged across the 30 windows and then normalized by their L-2 norm. This procedure thus returns the projection vector designed to maximally separate the odor responses in the two environments. To test whether this projection generalized to held-out sessions, the PCA transformation was applied to the mean response in the held-out sessions for home1 and envA, as well as to the home2 sessions. The difference between the population responses along this environment axis between the held-out home1 and envA session sessions was compared to the difference between the home2 and held-out home1 sessions. This cross-validation procedure was repeated across 1,000 restarts, and the mean and standard deviations across restarts were evaluated. Across all restarts, the response in envA readily diverged after odor onset, whereas the response in home2 remained similar to that in home1 at all timepoints after odor onset.
Pairwise odor correlations within and across environments
Pairwise correlations for each odor pair were calculated for each environment using the population vectors of mean odor responses (averaged across the first 3 seconds following population onset). For each odor pair, the union of responsive glomeruli was used. The pairwise correlation distance between all trials for each odor was computed and the median across trial pairs was computed for each odor pair. This same procedure also applied to pairs of the same odor, and the on-diagonal of the resulting correlation matrix represents the median across trials pairs and demonstrates the stability of the population response to the same odor across trials. The difference in correlation matrices across environments was computed, and the observed difference in the mean absolute change in pairwise correlations of the lower triangle (excluding the diagonal) was compared to a null distribution of means from shuffling the environment identity of each trial across 10,000 shuffles. For both mice, the observed difference was greater than that observed in any shuffle. Correlation matrices are shown after hierarchical clustering odors using Ward’s method.
Classifying environment and odor identities via glomerular populations
Environment identity classification
Classification of environment identity was performed for each odor separately using the responsive glomeruli for each odor. Classification was performed using pseudo-populations of glomeruli combined across mice, using the mean response for each glomerulus across the first 3 seconds following population onset. For each of 100 restarts, 18 trials were randomly chosen for each mouse for each environment and were split into 12 training and 6 test trials. Glomerular responses were permuted across trials within each mouse and then concatenated across mice to generate pseudo-trials for model training and testing. On each restart, subpopulations of glomeruli were randomly selected 100 times, thus yielding 10,000 models. Standardization across trials for each glomerulus via z-scoring and linear SVM classifiers (regularization parameter C = 0.1) were both fit on the training data and applied to predict the environment label of the held-out test pseudo-trials, and the accuracy was summarized for each subpopulation across restarts. Performance was a chance levels upon shuffling the environment labels of the training and test data.
Odor identity classification and classification generalization
Odor identity was predicted in a similar manner, using the mean responses across the first 3 seconds following population onset in pseudo-populations of glomeruli. For each of 100 restarts, for each environment, 6 test trials (containing the responses to each of the 16 odors) were randomly held out, and the glomeruli were randomly permuted across test trials 10 times to generate pseudo-populations for 60 test pseudo-trials for each of the 16 odors. The remaining training trials were randomly subsampled from each environment to yield 24 training trials for each odor. On each restart, subpopulations of glomeruli were randomly selected 20 times, thus yielding 2,000 (20 × 100) models for each threshold. A classification pipeline was fit on the training data, consisting of standardization across trials for each glomerulus via z-scoring, reducing the dimensionality via PCA (n_components=5 when the number of glomeruli was <10, else n_components=10), and fitting linear SVM classifiers (“one-vs-one” configuration with regularization parameter C = 0.5), and then applied to the test data. The training data from each environment and the test data from held-out trials from each environment were each used for training and testing, thus generating all four combinations (2 × 2) of training and testing data from either the same or different environments. Classification performance was summarized across restarts for each threshold for each of the within-environment and between-environment training/testing combinations and compared to 1) performance when environment labels were shuffled across the training trials and across the test trials from each environment and 2) performance when the odor labels were shuffled. Classification performance quickly saturated with increasing number of glomeruli, was higher for held-out trials from the same environment, and was at chance performance when odor labels were shuffled.
Odor generalization was performed to determine how the changes in pairwise odor correlations observed across environments impacted odor coding. The same pseudo-populations were constructed for training and test trials from each environment as described above for odor decoding. The entire pseudo-population of glomeruli was used without subsampling. The training and test data was first transformed with the z-scoring and PCA transformation (n_components=10) described above and fit on the training data. However, rather than fitting a single classifier to all trials from all odors, a separate classifier was fit on each odor pair in the training data (16C2 = 120 pairs) and applied to the remaining odor pairs (119) in the test data, thus yielding 120 × 119 = 14,280 predictions. This same procedure was repeated for each of the four within- and between-environment training/testing combinations, and the entire procedure was repeated when the environment labels were shuffled across the training trials and across the test trials from each environment. Classification accuracy from classifiers trained and tested on different odor pairs from the same environment (within-environment) was sorted across the 14,280 pairs and the accuracy for each pair in the within-environment condition was compared to that of the between-environment and shuffled-environment conditions. Although the overall distribution of classification accuracies was similar for each of the three conditions, the mean absolute difference between within-environment and between-environment accuracies for each pair was greater for the observed, rather than the shuffled environmental labels, further demonstrating that pairwise relationships between odors are altered across environments.
Software used for data analysis
Single-cell analyses were performed in python (versions 3.6–3.8) using the Scanpy package (Wolf et al., 2018) as well as custom-written scripts using the open-source python scientific stack (pysam, SciPy, NumPy, scikit-learn, umap-learn, pandas, statsmodels, matplotlib, seaborn, numba, dask). cNMF was performed using modified versions of the code described in (Kotliar et al., 2019) and accessed at https://github.com/dylkot/cNMF. Imaging data was analyzed using additional packages, including OpenCV and Suite2p (Pachitariu et al., 2017). Scripts to replicate data analyses are available at https://github.com/dattalab/Tsukahara_Brann_OSN.
QUANTIFICATION AND STATISTICAL ANALYSIS
Hypothesis testing was performed using non-parametric statistical tests, including the Mann-Whitney U Test and the Wilcoxon signed-rank test, except in the cases where p-values were calculated empirically using resampling-based permutation tests. Distributions were compared via the Kolmogorov–Smirnov (KS) test. Experiments with multiple factors were tested with either a two-way ANOVA or with the Jonckheere-Terpstra trend test (calculated empirically via 10,000 permutations), a non-parametric extension of the Kruskal–Wallis test to evaluate trends across groups. Statistical tests that were performed for multiple genes, ORs, GEPs, glomeruli, and odors were corrected for multiple comparisons using the Benjamini-Hochberg FDR Procedure or the Holm–Bonferroni method, as specified in the text. The results of statistical tests are listed in the figure legends. The precision of sample statistics and regression trend lines were evaluated using bootstrapping, and, except where noted, plots and error bars depict the mean and the 95% confidence intervals of the mean across 1,000–10,000 bootstraps. Results from classification, regression, and other analyses were repeated across 1,000 restarts, using balanced subsamples of the data in analyses at the OSN-level to account for differences in OR frequencies. Throughout the paper, a non-parametric version of the box plot (also known as a letter-value plot) was used to represent multiple quantiles and tails of large distributions of data (e.g. to summarize across OSN subtypes or sets of ORs or OR pairs) in an agnostic way that does require setting bandwidth parameters as in violin plots or kernel density estimates. Like a conventional box plot, the largest box represents the interquartile range (25–75 percentile) and the median (dotted-line). Subsequent boxes recursively represent exponentially-smaller quantiles (the 12.5–25 and 75–87.5 percentiles, then 6.25–12.5 and 87.5–93.75 percentiles, then 3.125–6.25 and 93.75–96.875 percentiles, and so forth).
Supplementary Material
Table S1. Summary of single cell RNA-seq datasets for olfactory sensory neurons used in this study, related to Figures 1–6.
Table S2. Loadings of 1,350 highly variable genes for 16 gene expression programs identified by consensus non-negative matrix factorization of the home cage dataset, related to Figure 2.
Table S3. 117 environmental state neuronal genes, including 73 functional genes that may regulate sensory responses, related to Figure 4.
Table S4. Changes in z-scored expression of 117 environmental state neuronal genes across conditions, for each OSN subtype, related to Figure 4.
Table S5. 472 genes used to calculate activation score that were acutely odor-sensitive in responsive OSNs, related to Figure 5. 8 values that characterize 472 genes we used to calculate activation score are shown. Columns are as follows: “activation_score_loading”: gene loading for calculating activation score. “ess_correlation”: Spearman’s rho between normalized expression and environmental state score in the home cage dataset on OR-by-OR basis. “p_adj_ACE2h”: FDR corrected p-values for the differential expression (via Wilcoxon signed-rank test) between control and acetophenone 2h samples for acetophenone responsive ORs on OR-by-OR basis. “ctl_expr_ACE2h”: Normalized mean expression for OSNs expressing-acetophenone responsive ORs in the control data. “expression_ACE2h”: Normalized mean expression for OSNs expressing acetophenone responsive ORs in the acetophenone 2h data. “p_adj_OCT2h”, “ctl_expr_OCT2h”, “expression_OCT2h”: same as the previous 3 columns for acetophenone responsive ORs, but for OSNs expressing octanal-responsive ORs.
Figure S1. OSN transcriptomes are predictable across individuals, and variation is distributed across hundreds of genes, related to Figure 1. A. Histogram of the number of cells expressing each OR, from wild-type mice housed in the home cage.
B. The cumulative percent of ORs observed in OSNs (sorted by how often each OR is observed) as a function of the cumulative fraction of OSNs that express any given OR. The top 200 ORs (of the 1016 detected) account for > 50% of OSNs.
C. The number of OSN subtypes identified, thresholded by the number of OSNs expressing a given OR; colors represent numbers of OSNs used for this analysis; a threshold of 10 OSNs yielded 831 OSN subtypes.
D. Percent of OSNs expressing 0, 1 or >1 ORs thresholded by the number of transcripts (unique molecular identifiers, UMIs) required for an OR gene to be considered “detected”. Many of the rare 2 OR cells were clearly doublets and contained higher than average total numbers of UMIs compared to a typical 1 OR cell. Therefore, downstream analyses used a threshold of 3 UMIs and only considered cells in which only one OR was detected.
E. UMAP plots highlighting OSNs expressing the indicated ORs.
F. The mean fraction of all OSNs expressing a given OR (averaged across 1,000 restarts) as a function of the rank-ordered transcriptome distance from each cell expressing that OR. Shaded error indicates the interquartile range across ORs.
G. Distribution of within-OR transcriptome distances across OSN subtypes using cells from the same or different mice, or after shuffling OR labels.
H. Distribution of within-OR transcriptome distances as a function of the number of OSNs expressing each OR.
I. Separability of the distributions of within-OR and between-OR transcriptome distances, as measured by the Jensen-Shannon Divergence (JSD), for various number of genes and increasing number of principal components. JSD was normalized to the maximum JSD value across all conditions. Highly variable genes (HVGs, used to compute the principal components) were selected based on their variability across OSNs expressing different ORs. Stars represent the number of HVGs used in the analyses shown in the paper. The JSD is low, and separability is at chance levels, when OR labels are shuffled.
J. Distribution of accuracies for linear classifiers predicting which OR is expressed in a given OSN, as a function of the number of OSNs expressing each OR.
K. Mean accuracy across OSN subtypes of linear classifiers predicting which OR is expressed as a function of the number of HVGs included (sorted by their informativeness, see Methods). Colors indicate classification thresholds, and error bars depict the mean and SD across 10 restarts.
L. Similar to K. but as a function of the number of genes removed.
M. (left) Accuracy of linear classifiers predicting which OR (out of 831) is expressed, after excluding all transcription factor (TF) and axon guidance genes. Distributions depict the mean accuracy across the 831 OR-defined OSN subtypes (each averaged across 1,000 restarts). (right) Similar to left, but for various gene sets, including axon guidance genes and TF genes (which both contain HVGs and non-HVGs), and the 100 most predictive or all HVGs (reproduced from Fig. 1F). Classifier hyperparameters were optimized for each gene set.
N. Accuracy of pairwise linear classifiers predicting which OR is expressed, with respect to (left) anatomical identity or (middle) OR class. (right) Accuracy of linear classifiers to predict which of dorsal class I, dorsal class II or ventral class II OR is expressed.
O. Similar to left and middle of N. but with respect to CD36-positivity.
P. Similar to left and middle of N. but with respect to genomic distance (distance between transcriptional start sites (TSS), in base pairs).
Q. Similar to left and middle of N. but with respect to the phylogenetic distance.
Figure S2. GEPHigh and GEPLow are aligned with the main axis of transcriptional variation across OSNs, related to Figure 2. A. Consensus non-negative matrix factorization (cNMF) identifies 10 OR-specific gene expression programs (GEPs) whose usages are correlated among OSNs that express the same OR. OSNs expressing the same OR were randomly split into two populations and the correlation in GEP usage between the two populations was computed for each GEP. Error bars depict the mean correlation with SD across 10,000 restarts. 6 GEPs (including one with many genes related to the unfolded protein response (UPR)) only showed weak correlation, indicating that the usages for such GEPs are not specific to ORs in mature OSNs.
B. UMAP plots showing the usages of identified GEPs, together with the expression of genes with high loadings for each GEP.
C. Gene loadings for the subset of HVGs whose loadings was higher than 2/1350 (total number of HVGs) in any of the 10 OR-specific GEPs; genes were hierarchically clustered by their loadings.
D. Accuracy of linear classifiers predicting OR identity using GEPs across (left) OR pairs or (right) across all ORs. Performance upon shuffling OR labels is at chance = 1/831.
E. Correlation coefficients between mean GEP usages for each OSN subtype. Strong anticorrelation between Dorsal/Ventral (ρ = −0.85), Anterior/Posterior (ρ = −0.70) and High/Low GEPs (ρ = −0.87) is observed.
F. Mean usages of (left) GEPDorsal and GEPVentral or (right) GEPAnterior and GEPPosterior for each OSN subtype, from wild-type mice housed in the home cage.
G. Accuracy of pairwise linear classifiers predicting OR identity using only the usages of either the Dorsal/Ventral (D/V) or the Anterior/Posterior (A/P) GEPs.
H. Correlation coefficients of environmental state (ES), Dorsal/Ventral (DV), and Anterior/Posterior (AP) scores. DV and AP scores are calculated by GEPDorsal - GEPVentral and GEPAnterior - GEPPosterior, respectively. Individual mice are indicated as colored on top.
I. Correlation coefficients, computed across OSN subtypes, between the GEP usages and PC scores (calculated from the top 1,350 HVGs).
J. Absolute values of the correlation coefficient, computed across OSN subtypes, between the ES score and the PC score for each of top 10 PCs. The ES score is highly correlated with the first PC (ρ = 0.89), indicating that the ES score is a major axis of transcriptional variation across OSNs derived from mice housed in the home cage. Error bars depict the mean and SD across 1,000 restarts.
K. Mean z-scored expression of immature (Gng8, Gap43) and mature (Gnal, Omp) OSN marker genes as a function of the developmental pseudotime from the onset of OR expression in late-immature OSNs (see Methods).
L. (left) GEPLow, (middle) GEPHigh, and (right) ES scores as a function of the developmental pseudotime from the onset of OR expression in late-immature OSNs. OSN subtypes are fractionated by their ES scores in mature OSNs.
Figure S3. The dependence of OSN transcriptomes on long-term interactions with odors and air, related to Figure 3. A. Accuracy of linear classifiers predicting OR identity based on HVG expression (see Figs. 1E–F) from open or occluded nostrils across OR pairs (left) or across all ORs (right).
B. Accuracy of linear classifiers predicting whether a given cell for each OSN subtype is from the open or closed nostril, using the usage of either (left) all 10 OR-specific GEPs or (right) only GEPHigh/GEPLow.
C. Gene loadings for each GEP for HVGs that are differentially expressed (DE), or are unchanged (non-DE, in gray) by chronic naris occlusion. HVGs that decrease upon occlusion (in blue) have higher than average loadings compared to non-DE genes for GEPHigh (p < 1 × 10−57, KS-test, adjusted by the Holm-Bonferroni method) and lower than average loadings for GEPLow, and vice versa for genes that increased (in red, p < 1 × 10−48, KS-test, adjusted by the Holm-Bonferroni method).
D. Mean usages of GEPHigh and GEPLow for 797 OSN subtypes for each nostril, using data from all mice.
E. Mean ES score for each OSN subtype for each nostril. Data from each mouse are shown separately. ES scores decreased by occlusion for 314/341, 356/391, and 320/340 of OSN subtypes, respectively (FDR ≤ 0.01).
F. Mean ES scores for each OSN subtype for mouse pairs from the same or different environments.
G. Cumulative percent of OSN subtypes whose ES scores differ significantly (FDR ≤ 0.01) in different numbers of environment pairs.
H. Mean environment-dependent changes in GEP usage across OSN subtypes, categorized based on the sign and significance of their ES score changes across environments.
I. Accuracy of pairwise linear classifiers predicting OR identity using only the usages of the two ES GEPs (GEPHigh and GEPLow) of OSNs from each nostril in the chronic occlusion data.
J. UMAP plot highlighting OSNs expressing the indicated ORs, using only cells from occluded nostrils.
K. UMAP plot depicting the z-scored ES scores, using only cells from occluded nostrils.
L. Accuracy of pairwise linear classifiers predicting OR identity using only the usages of the two ES GEPs of OSNs from each of the three environments.
M. Accuracy of pairwise linear classifiers predicting in which environment a mouse was housed, based upon usages of either all GEPs or ES GEPs. Classification was performed for each OSN subtype, and distributions of the mean accuracies (across 1,000 restarts) across OSN subtypes with or without significant ES score changes across environments are shown (shuffled = environment labels permuted).
N. OR expression levels (as quantified by the number of transcripts of the expressed OR normalized by the total number of transcripts per cell) in each nostril, for each OSN subtype.
O. Similar to N. but for optogenetically-stimulated (12h) and control (no light) mice.
P. Pairwise correlations across all environments of the (left) mean OR expression levels or (right) the number of cells in which each OR was detected.
Q. As in the right panel of F. but for Dorsal/Ventral (DV), and Anterior/Posterior (AP) scores.
R. ES scores for each OSN subtype from wild-type (WT) and β-Arrestin2 mutant mice housed in the home cage.
S. Similar to R. but for OR expression levels.
T. (left) Mean change in ES scores (relative to the mean across all OSN subtypes) 5 days after a shift to environment A (envA), for OSN subtypes from β-Arrestin2 mutant mice. OSN subtypes whose ES score rose or fell after 2 week in envA in WT data show increase or decrease in ES scores in mutants (p < 1 × 10−15 via Wilcoxon signed-rank test). (right) Correlation coefficient of changes in ES score for each OSN subtype between WT and β-Arrestin2 mutant mice (p < 1 × 10−4 via permutation test across 10,000 shuffles).
Figure S4. Functional gene expression exhibits organized correlation structure, related to Figure 4. A. Heatmap showing pairwise correlations between the expression levels, across OSN subtypes, of the 117 ES neuronal genes, in data from mice housed in the home cage.
B. Summary of the correlations between pairs of the 73 functional genes (see Fig. 4A and Table S3), separated by pairs from either GEPHigh alone, GEPLow alone, or both GEPHigh and GEPLow.
C. Correlation of the expression of individual functional genes and the ES score, computed across OSN subtypes, compared to correlations observed upon shuffling ES scores across OSN subtypes.
D. Mean ES score and mean “functional” ES score (using GEPHigh and GEPLow usages calculated from the loadings of the 73 functional genes) for each OSN subtype from mice housed in the home cage.
E. Accuracy of pairwise linear classifiers trained to predict OR identity using the GEPHigh and GEPLow usages calculated from the loadings of the 73 functional genes.
F. Mean occlusion-dependent change in ES score and “functional” ES score for each OSN subtype.
G. Similar to B. but for occlusion-dependent changes in functional gene expression.
H. Similar to C. but for occlusion-dependent changes in ES scores and functional gene expression.
I. Optogenetic stimulation-dependent changes in expression of 73 functional genes as a function of changes in ES scores (binned into 10 quantiles); plots depict the mean and SD of the z-scored expression of functional genes associated with GEPHigh (red) and GEPLow (blue).
J. Similar to B. but for environment-dependent changes in functional gene expression.
K. Similar to C. but for environment-dependent changes in ES scores and functional gene expression.
Figure S5. Act-Seq identified odor-activated OSNs and quantified the degree of transcriptional odor response, related to Figure 5. A. (top left) UMAP plot showing mean z-scored expression of 10 immediate early genes (IEGs, see Methods for list of IEGs). (top right) Fraction of cells per OSN subtype that were activated by acetophenone (ACE) or octacal (OCT) based on IEG expression. OSN subtypes were defined as odor-responsive when at least 70 percent of cells expressing the given OR for that subtype were activated (dashed line), and these ORs were considered as activated. (middle left) UMAP plot highlighting activated cells identified by unsupervised Leiden clustering (see Methods). (middle right) Fraction of cells per OSN subtype that were activated based on Leiden clustering, colored by odor-responsiveness as assessed by IEG expression. (bottom left) UMAP plot depicting odor-driven activation as quantified by unsupervised tensor component analysis (TCA). (bottom right) Distribution of TCA loadings of a factor representing odor activation (with 11-times higher weight on the odor source compared to the control condition) for OSN subtypes for mice exposed to ACE and OCT, colored by odor-responsiveness as assessed by IEG expression.
B. Distribution of the mean expression of 10 IEGs across OSN subtypes, separated by odor-responsiveness.
C. Venn diagram depicting the overlap between ORs identified as acetophenone receptors via Act-seq, the DREAM method, and phospho-S6 immunoprecipitation (from von der Wied et al. 2015, Jiang et al. 2015). For the 8 ORs identified by only the phospho-S6 method, 3 of them were not activated in any cells via Act-Seq and the remaining 5 of them were partially activated (activated in 43.8% of cells per OR on average). This analysis only includes the 717 ORs which were present in four or more OSNs in these odor-activation experiments. Although Act-Seq captured nearly all such receptors that were also captured by DREAM and phospho-S6 (and many missed by both of these techniques), the 717 observed ORs included only 16/22 DREAM and 25/49 phospho-S6 ACE ORs; these “missed” receptors possibly reflect ORs that are expressed in particularly small numbers of OSNs.
D. Venn diagram depicting the overlap between ORs activated by ACE and OCT. The overlap is at chance levels (odds ratio = 1.42, p=0.29 Fisher’s exact test).
E. Comparisons of acute gene expression changes induced by ACE or OCT compared to solvent-only (DPG) condition for 996 genes with significant changes (FDR ≤ 1 × 10−3) in at least one odor.
F. Mean percent of ORs activated by the indicated concentration of acetophenone.
G. Mean percent of ORs activated by 10% acetophenone and its analogs (2-hydroxyacetophenone (2HA), 4-methyl acetophenone (4MA), methyl salicylate (MS)).
H. Heatmap depicting percent overlap of activated ORs among four acetophenone-related odors at 10% concentration. ORs activated by acetophenone analogs are also consistently activated by acetophenone (odds ratio > 22, p < 1 × 10−11 via Fisher’s exact test for each of three analogs).
I. Correlation coefficients across individual mice of activation scores for OSN subtypes responsive to either ACE or OCT (ρ ≥ 0.89 between mice exposed to the same odor).
J. Correlation coefficients comparing activation scores for OSN subtypes activated by acetophenone-related odors, shown for pairs of mice exposed to the same or different odors. The correlation is higher in mice exposed to the same odor (p = 0.002, Mann-Whitney U test).
K. Activation scores for four acetophenone-related odors, for OSN subtypes responsive to each odor.
L. Accuracy of pairwise linear classifiers trained to distinguish pairs of acetophenone-related odors based upon activation scores from odor-responsive OSN subtypes (using only OSN subtypes responsive to both odors in each pair). Observed and shuffled (odor labels are shuffled 1,000 times) classification accuracies are shown.
M. Accuracy of a minimum distance classification procedure to predict odor identity of the four acetophenone-related odors using OSN subtype-specific activation scores (see Methods), as a function of the number of OSN subtypes (from the set of OSN subtypes responsive to any of the four odors). The curves depict the mean and interquartile range of classifier accuracy for each odor across 1,000 restarts for the observed data, and for data in which odor labels were shuffled for each OSN subtype.
N. Correlation coefficient between the activation score and the control ES score for each OSN subtype, evaluated at various thresholds used for identifying activated cells based on IEG expression and for identifying responsive ORs (fraction of cells activated). The threshold used in Fig. 5I (70% of cells per OR and IEG expression ≥ 0.2) is highlighted with stars.
O. Correlation coefficient between the activation score and the control ES score for each OSN subtype, within and between individual mice. For each odor, the observed negative correlation between activation and control ES score is higher (p < 1 × 10−4 via permutation test) than any value in the distribution of correlation coefficients measured after permuting the activation score across OSN subtypes for 10,000 shuffles.
P. Similar to O. but for four acetophenone-related odors (p < 8 × 10−3 via permutation test for each odor).
Q. Mean activation scores at the indicated concentration for each of the acetophenone-responsive OSN subtypes, as a function of the in vitro EC50 for each OR, as reported in Jiang et al. 2015 and Saito et al. 2009 (see also Fig. 5K).
R. Normalized error for predictions (see Methods) of activation scores (both observed and shuffled) for OSN subtypes responsive to ACE at 10%, based on control ES scores, the in vitro EC50 for each OR (from Jiang et al. 2015 and Saito et al. 2009), or both. Error bars represent mean and SD across 1,000 restarts.
S. Similar to O. but for data derived from the acute optogenetic stimulation experiment (p < 1 × 10−4 via permutation test for each stimulation frequency).
Figure S6. Odor responses are determined by OSN Environmental State (ES) scores and changes in ES score are influenced by acute odor responses, related to Figure 6. A. Summary of the correlations between pairs of the 73 functional genes, separated by pairs from either GEPHigh alone, GEPLow alone, or both GEPHigh and GEPLow, using data from transiently-occluded (but not unplugged) nostrils (see Fig. 6B).
B. Mean transient occlusion-dependent changes in GEP usage across OSN subtypes.
C. Fraction of OSN subtypes responsive to acetophenone (ACE) from unplugged nostrils, for OSN subtypes responsive to 10 % ACE (as shown in the top right of Fig. S5A).
D. Mean changes in ES (left) or activation scores (right) between acetophenone-responsive OSN subtypes from open or unplugged nostrils, as a function of the difference in ES scores (binned into quartiles), as measured in control mice that were transiently-occluded (but not unplugged). OSN subtypes whose ES scores fell more showed increased activation (p = 2.63 × 10−4, Mann-Whitney U test).
E. Normalized mean-squared error for predictions of the occlusion-dependent change in activation scores for OSN subtypes responsive to acetophenone, with predictions based upon the occlusion-dependent changes in functional gene expression. Model errors increase if the nostril type (open vs. transiently occluded) of the control mice was shuffled, or if the activation scores are shuffled across OSN subtype. Errors were normalized to the value from the observed data, and the error bars depict the mean and SD across 1,000 restarts.
F. Venn diagram, showing that ORs identified as activated by acetophenone in each environment are largely consistent but not fully overlapped; modulation of odor responses across environments may contribute to the changes in activated ORs identified in each environment via our relatively conservative thresholds (see G.).
G. Activation score distributions for OSN subtypes that were responsive to acetophenone in different numbers of environments (0–3). OSN subtypes that responded to acetophenone in 1–2 environments were still activated more in the other environments in which they were not considered as responsive (shown in gray) than those that were not responsive in any environment, consistent with the possibility that these receptors were activated to some extent in all environments.
H. The overlap in identified OSN subtypes responsive to acetophenone between pairs of mice housed within the same or different olfactory environments. Pairs of mice exposed to acetophenone exhibit activation in overlapping sets of OSN subtypes (positive log-odds), but the overlap is greater between mice that were housed within the same olfactory environment (p=0.026, Mann-Whitney U test). The overlap and log-odds are at chance levels after permuting the identity of ACE ORs across 1,000 restarts for each mouse pair.
I. The cumulative percent of acetophenone-responsive OSN subtypes identified in each environment, colored by the number of environments in which each subtype responded.
J. Mean activation scores for each acetophenone-responsive OSN subtype, sorted by each environment, for OSN subtypes responsive in all three environments.
K. Correlation coefficient between the activation score and the control ES score for each acetophenone-responsive OSN subtypes, using data from mice housed in the indicated environments. The observed value is stronger than any value in the distribution of correlation coefficients measured after permuting the activation score across OSN subtypes for 10,000 shuffles (p < 1 × 10−4 via permutation test for each environment).
L. Classification accuracy for a pairwise linear classifier predicting the environment from which a given set of OSNs derived. Classification was performed for each acetophenone-responsive OSN subtype with significant ES score changes across environments, using the activation scores in each environment for that OSN subtype. Shuffle = permuted environment.
M. Mean environment-dependent changes in activation scores, for acetophenone-responsive OSN subtypes, separated in OSN subtypes whose ES scores rose or fell across environments. OSNs whose ES scores increased in one environment relative to another exhibited lower acetophenone-driven activation scores, while those whose ES scores decreased showed the opposite trend (p = 1.77 × 10−11, Mann-Whitney U test).
N. Similar to E., but for the environment-dependent changes in activation scores.
O. OR expression levels after chronic exposure to the indicated concentration of acetophenone for 5 days, normalized to the control condition (exposure to DPG for 5 days). ORs identified as activated in none of the concentrations, at 10% alone, or at 2 or more concentrations by acute (two-hour) Act-Seq are shown separately.
P. Correlation between environment-dependent changes in ES scores (relative to their mean across all OSN subtypes) after 24 hours and 2 weeks, for individual OSN subtypes.
Q. Mean change in z-scored expression of 73 functional genes (relative to the mean change observed across all OSN subtypes and show separately for genes associated with GEPHigh and GEPLow), at various timepoints after a shift to envA, for OSN subtypes categorized based on the sign and significance of their two-week ES score changes.
R. Mean ES scores for OSN subtypes from mice exposed to acetophenone or DPG for 2h and housed in home cages for 22h. Acetophenone-induced changes in the ES scores were not significantly different for acetophenone-responsive OSN subtypes compared to those that did not respond to acetophenone (Mann-Whitney U test, p = 0.214).
S. Mean expression of Nr4a1 and Fos normalized by the total number of transcripts per cell for OSN subtypes, as a function of the ES scores (binned by steps of 50) for each OSN subtype in mice housed in the home cage, compared to that observed in acetophenone-responsive OSN subtypes following two-hour exposure to acetophenone.
Figure S7. Odor environments determine acute functional odor responses in the olfactory bulb, related to Figure 7. A. Max-normalized mean fluorescence for individual sessions from a single mouse (mouse 1 in B.), imaged for five weeks across environments (rows). Scale bars = 200 μm.
B. Pairwise enhanced correlation coefficients for the entire FOV for all imaging sessions from each mouse (after registering FOVs across sessions, see Methods).
C. Percent of odor-responsive glomeruli (see Methods), for the three indicated odors at two separate concentrations. Each circle indicates a single mouse.
D. Percent of environment-sensitive glomeruli (of the glomeruli responsive to each odor) whose odor responses significantly differed (FDR ≤ 0.01 via permutation test), when mice were housed in home1 compared to environment A (envA). Odors were tested at two concentrations; pentanal-high corresponds to the concentration used in Fig. 7B–E. Each point represents a single mouse.
E. Fold-change in the number of glomeruli that differ significantly when comparing odor responses between home1/envA and home1/home2; each circle indicates a single mouse. A larger number of glomeruli change significantly across environments (see also F.).
F. Percent of glomeruli whose difference in odor response between home1/envA has the opposite sign as that observed between envA/home2, for all glomeruli significantly different between home1/envA (dots refer to individual mice); upon return to the home cage after experiencing envA, the odor responses of nearly all glomeruli are at least partially reverted.
G. Percent of odor-responsive glomeruli for the mice used for decoding analysis (mouse 5 and 6 in B.). Each circle indicates an odor, and colors correspond to those in Fig. 7H.
H. Percent of environment-sensitive glomeruli (of the glomeruli responsive to each odor, colored as in Fig. 7H) whose odor responses significantly differed (FDR ≤ 0.01 via permutation test), when mice were housed in home1 compared to envA.
I. Absolute changes in pairwise correlations for mean odor responses (see Fig. 7H–I). Observed values and distribution of means after permuting environment labels across 10,000 shuffles are shown for each mouse (p < 1 × 10−4 for each mouse).
J. Similar to Fig. 7K. Classification accuracy for each of the test × train pairs (120 × 119 pairs), colored by whether training and test data are from either the same or different environments, or from data with shuffled environment labels (and sorted independently for each of the three conditions).
Acknowledgements
We thank John McGann and Matthew Grubb for technical advice on naris occlusion, Venkatesh Murthy, Dima Rinberg and Catherine Dulac for mice, and Kelsey Tyssowski, Vanessa Ruta, Rachel Wilson, Bernardo Sabatini, David Ginty, Michael Greenberg, Piali Sengupta, and the Datta lab for useful comments on the manuscript. We thank Neha Bhagat for laboratory assistance, and Janet Wallace for administrative assistance. We thank the Bauer Core Facility at Harvard University for sequencing and the Flow Cytometry Core at Beth Israel Deaconess Medical Center. Portions of this research were conducted on the O2 High Performance Compute Cluster at Harvard Medical School. Illustrations were created in part with BioRender.com and Vecteezy.com. SRD is supported by NIH grants R011DC016222 and U19 NS112953 and by grants from the Simons Collaboration on the Global Brain, the Brain Research Foundation, and the Tan Yang Center at Harvard Medical School. TT is supported by postdoctoral fellowships from the Japan Society for the Promotion of Science, the Louis Perry Jones Fund, and the Tan Yang Center. DHB is supported by an NSF Graduate Research Fellowship and by NIH grant F31DC019017.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Declaration of Interests
The authors declare no competing interests.
References
- Aibar S, González-Blas CB, Moerman T, Huynh-Thu VA, Imrichova H, Hulselmans G, Rambow F, Marine J-C, Geurts P, Aerts J, et al. (2017). SCENIC: single-cell regulatory network inference and clustering. Nature Methods 14, 1083–1086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arneodo EM, Penikis KB, Rabinowitz N, Licata A, Cichy A, Zhang J, Bozza T, and Rinberg D (2018). Stimulus dependent diversity and stereotypy in the output of an olfactory functional unit. Nat Commun 9, 1347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Attneave F (1954). Some informational aspects of visual perception. Psychol Rev 61, 183–193. [DOI] [PubMed] [Google Scholar]
- Barber CN, and Coppola DM (2015). Compensatory plasticity in the olfactory epithelium: age, timing, and reversibility. Journal of Neurophysiology 114, 2023–2032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barlow H (1961). Possible principles underlying the transformation of sensory messages. In Sensory Communication (Cambridge, MA: MIT press; ), pp. 217–234. [Google Scholar]
- Benda J (2021). Neural adaptation. Current Biology 31, R110–R116. [DOI] [PubMed] [Google Scholar]
- Best AR, Thompson JV, Fletcher ML, and Wilson DA (2005). Cortical metabotropic glutamate receptors contribute to habituation of a simple odor-evoked behavior. Journal of Neuroscience 25, 2513–2517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bozza T, Feinstein P, Zheng C, and Mombaerts P (2002). Odorant receptor expression defines functional units in the mouse olfactory system. Journal of Neuroscience 22, 3033–3043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bozza T, McGann JP, Mombaerts P, and Wachowiak M (2004). In vivo imaging of neuronal activity by targeted expression of a genetically encoded probe in the mouse. Neuron 42, 9–21. [DOI] [PubMed] [Google Scholar]
- Bozza T, Vassalli A, Fuss S, Zhang J-J, Weiland B, Pacifico R, Feinstein P, and Mombaerts P (2009). Mapping of class I and class II odorant receptors to glomerular domains by two distinct types of olfactory sensory neurons in the mouse. Neuron 61, 220–233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brann DH, and Datta SR (2020). Finding the Brain in the Nose. Annual Review of Neuroscience 43, 277–295. [DOI] [PubMed] [Google Scholar]
- Buck LB (1996). Information coding in the vertebrate olfactory system. Annual Review of Neuroscience 19, 517–544. [DOI] [PubMed] [Google Scholar]
- Burns ME, and Baylor DA (2001). Activation, deactivation, and adaptation in vertebrate photoreceptor cells. Annual Review of Neuroscience 24, 779–805. [DOI] [PubMed] [Google Scholar]
- Cadiou H, Aoude I, Tazir B, Molinas A, Fenech C, Meunier N, and Grosmaitre X (2014). Postnatal Odorant Exposure Induces Peripheral Olfactory Plasticity at the Cellular Level. Journal of Neuroscience 34, 4857–4870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cho CE, Brueggemann C, Noelle DL, and Bargmann CI (2016). Parallel encoding of sensory history and behavioral preference during Caenorhabditis elegans olfactory learning. Elife. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chong E, Moroni M, Wilson C, Shoham S, Panzeri S, and Rinberg D (2020). Manipulating synthetic optogenetic odors reveals the coding logic of olfactory perception. Science 368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chu MW, Li WL, and Komiyama T (2017). Lack of Pattern Separation in Sensory Inputs to the Olfactory Bulb during Perceptual Learning. eNeuro 4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cleland TA, and Linster C (2005). Computation in the olfactory system. Chemical Senses 30, 801–813. [DOI] [PubMed] [Google Scholar]
- Coppola DM, and Waggener CT (2012). The effects of unilateral naris occlusion on gene expression profiles in mouse olfactory mucosa. J Mol Neurosci 47, 604–618. [DOI] [PubMed] [Google Scholar]
- Coppola DM, Waguespack AM, Reems MR, Butman ML, and Cherry JA (2006). Naris occlusion alters transductory protein immunoreactivity in olfactory epithelium. Histol Histopathol 21, 487–501. [DOI] [PubMed] [Google Scholar]
- Davis GW (2006). Homeostatic control of neural activity: from phenomenology to molecular design. Annual Review of Neuroscience 29, 307–323. [DOI] [PubMed] [Google Scholar]
- Davis GW (2013). Homeostatic signaling and the stabilization of neural function. Neuron 80, 718–728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de March CA, Titlow WB, Sengoku T, Breheny P, Matsunami H, and McClintock TS (2020). Modulation of the combinatorial code of odorant receptor response patterns in odorant mixtures. Mol Cell Neurosci 104, 103469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dey S, Chamero P, Pru JK, Chien M-S, Ibarra-Soria X, Spencer KR, Logan DW, Matsunami H, Peluso JJ, and Stowers L (2015). Cyclic Regulation of Sensory Perception by a Female Hormone Alters Behavior. Cell 161, 1334–1344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eggan K, Baldwin K, Tackett M, Osborne J, Gogos J, Chess A, Axel R, and Jaenisch R (2004). Mice cloned from olfactory sensory neurons. Nature 428, 44–49. [DOI] [PubMed] [Google Scholar]
- Feinstein P, and Mombaerts P (2004). A contextual model for axonal sorting into glomeruli in the mouse olfactory system. Cell 117, 817–831. [DOI] [PubMed] [Google Scholar]
- Fettiplace R, and Ricci AJ (2003). Adaptation in auditory hair cells. Curr Opin Neurobiol 13, 446–451. [DOI] [PubMed] [Google Scholar]
- Firestein S (2001). How the olfactory system makes sense of scents. Nature 413, 211–218. [DOI] [PubMed] [Google Scholar]
- Fischl AM, Heron PM, Stromberg AJ, and McClintock TS (2014). Activity-dependent genes in mouse olfactory sensory neurons. Chemical Senses 39, 439–449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fitzwater E, and Coppola DM (2020). Olfactory Deprivation and Enrichment: An Identity of Opposites? Chemical Senses 46. [DOI] [PubMed] [Google Scholar]
- Fletcher RB, Das D, Gadye L, Street KN, Baudhuin A, Wagner A, Cole MB, Flores Q, Choi YG, Yosef N, et al. (2017). Deconstructing Olfactory Stem Cell Trajectories at Single-Cell Resolution. Cell Stem Cell 20, 817–830.e818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Galliano E, Hahn C, Browne LP, R Villamayor P, Tufo C, Crespo A, and Grubb MS (2021). Brief Sensory Deprivation Triggers Cell Type-Specific Structural and Functional Plasticity in Olfactory Bulb Neurons. Journal of Neuroscience 41, 2135–2151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goldman MS, Golowasch J, Marder E, and Abbott LF (2001). Global structure, robustness, and modulation of neuronal models. Journal of Neuroscience 21, 5229–5238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gouwens NW, Sorensen SA, Baftizadeh F, Budzillo A, Lee BR, Jarsky T, Alfiler L, Baker K, Barkan E, Berry K, et al. (2020). Integrated Morphoelectric and Transcriptomic Classification of Cortical GABAergic Cells. Cell 183, 935–953.e919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grabska-Barwińska A, Barthelmé S, Beck J, Mainen ZF, Pouget A, and Latham PE (2016). A probabilistic approach to demixing odors. Nature Neuroscience 20, 98–106. [DOI] [PubMed] [Google Scholar]
- Hagendorf S, Fluegge D, Engelhardt C, and Spehr M (2009). Homeostatic control of sensory output in basal vomeronasal neurons: activity-dependent expression of ether-à-go-go-related gene potassium channels. Journal of Neuroscience 29, 206–221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanchate NK, Kondoh K, Lu Z, Kuang D, Ye X, Qiu X, Pachter L, Trapnell C, and Buck LB (2015). Single-cell transcriptomics reveals receptor transformations during olfactory neurogenesis. Science 350, 1251–1255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hopfield JJ (1991). Olfactory computation and object perception. Proceedings of the National Academy of Sciences 88, 6462–6466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu XS, Ikegami K, Vihani A, Zhu KW, Zapata M, de March CA, Do M, Vaidya N, Kucera G, Bock C, et al. (2020). Concentration-Dependent Recruitment of Mammalian Odorant Receptors. eNeuro 7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ibarra-Soria X, Nakahara TS, Lilue J, Jiang Y, Trimmer C, Souza MA, Netto PH, Ikegami K, Murphy NR, Kusma M, et al. (2017). Variation in olfactory neuron repertoires is genetically controlled and environmentally modulated. Elife 6, 1083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Imai T, Sakano H, and Vosshall LB (2010). Topographic Mapping—The Olfactory System. Cold Spring Harbor Perspectives in Biology 2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang Y, Gong NN, Hu XS, Ni MJ, Pasi R, and Matsunami H (2015). Molecular profiling of activated olfactory neurons identifies odorant receptors for odors in vivo. Nature Neuroscience, 1–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Juang B-T, Gu C, Starnes L, Palladino F, Goga A, Kennedy S, and L’Etoile ND (2013). Endogenous nuclear RNAi mediates behavioral adaptation to odor. Cell 154, 1010–1022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kadohisa M, and Wilson DA (2006). Olfactory cortical adaptation facilitates detection of odors against background. Journal of Neurophysiology 95, 1888–1896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kass MD, Pottackal J, Turkel DJ, and McGann JP (2012). Changes in the Neural Representation of Odorants After Olfactory Deprivation in the Adult Mouse Olfactory Bulb. Chemical Senses 38, 77–89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kato HK, Chu MW, Isaacson JS, and Komiyama T (2012). Dynamic sensory representations in the olfactory bulb: modulation by wakefulness and experience. Neuron 76, 962–975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim D-W, Yao Z, Graybuck LT, Kim T-K, Nguyen TN, Smith KA, Fong O, Yi L, Koulena N, Pierson N, et al. (2019). Multimodal Analysis of Cell Types in a Hypothalamic Node Controlling Social Behavior. Cell 179, 713–728.e717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim EJ, Zhang Z, Huang L, Ito-Cole T, Jacobs MW, Juavinett AL, Senturk G, Hu M, Ku M, Ecker JR, et al. (2020). Extraction of Distinct Neuronal Cell Types from within a Genetically Continuous Population. Neuron 107, 274–282.e276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kostal L, Lansky P, and Rospars J-P (2008). Efficient olfactory coding in the pheromone receptor neuron of a moth. PLOS Computational Biology 4, e1000053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kotliar D, Veres A, Nagy MA, and Tabrizi S (2019). Identifying gene expression programs of cell-type identity and cellular activity with single-cell RNA-Seq. Elife. [DOI] [PMC free article] [PubMed] [Google Scholar]
- L’Etoile ND, Coburn CM, Eastham J, Kistler A, Gallegos G, and Bargmann CI (2002). The cyclic GMP-dependent protein kinase EGL-4 regulates olfactory adaptation in C. elegans. Neuron 36, 1079–1089. [DOI] [PubMed] [Google Scholar]
- La Manno G, Soldatov R, Zeisel A, Braun E, Hochgerner H, Petukhov V, Lidschreiber K, Kastriti ME, Lönnerberg P, Furlan A, et al. (2018). RNA velocity of single cells. Nature 560, 494–498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lecoq J, Tiret P, and Charpak S (2009). Peripheral adaptation codes for high odor concentration in glomeruli. Journal of Neuroscience 29, 3067–3072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li A, Gire DH, Bozza T, and Restrepo D (2014). Precise detection of direct glomerular input duration by the olfactory bulb. Journal of Neuroscience 34, 16058–16064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H, Horns F, Wu B, Xie Q, Li J, Li T, Luginbuhl DJ, Quake SR, and Luo L (2017). Classifying Drosophila Olfactory Projection Neuron Subtypes by Single-Cell RNA Sequencing. Cell 171, 1206–1220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Litwin-Kumar A, Harris KD, Axel R, Sompolinsky H, and Abbott LF (2017). Optimal Degrees of Synaptic Connectivity. Neuron 93, 1153–1164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma L, Wu Y, Qiu Q, Scheerer H, Moran A, and Yu CR (2014). A Developmental Switch of Axon Targeting in the Continuously Regenerating Mouse Olfactory System. Science 344, 194–197. [DOI] [PubMed] [Google Scholar]
- Marder E (2011). Variability, compensation, and modulation in neurons and circuits. Proceedings of the National Academy of Sciences 108 Suppl 3, 15542–15548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marder E, and Goaillard J-M (2006). Variability, compensation and homeostasis in neuron and network function. Nature Reviews Neuroscience 7, 563–574. [DOI] [PubMed] [Google Scholar]
- Martelli C, and Storace DA (2021). Stimulus Driven Functional Transformations in the Early Olfactory System. Frontiers in Cellular Neuroscience 15, 684742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mashukova A, Spehr M, Hatt H, and Neuhaus EM (2006). beta-Arrestin2-Mediated Internalization of Mammalian Odorant Receptors. Journal of Neuroscience 26, 9902 – 9912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McInnes L, Healy J, and Melville J (2018). UMAP: Uniform Manifold Approximation and Projection for Dimension Reduction. arXivorg stat.ML. [Google Scholar]
- McNamara AM, Magidson PD, Linster C, Wilson DA, and Cleland TA (2008). Distinct neural mechanisms mediate olfactory memory formation at different timescales. Learn Mem 15, 117–125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miyamichi K, Serizawa S, Kimura HM, and Sakano H (2005). Continuous and overlapping expression domains of odorant receptor genes in the olfactory epithelium determine the dorsal/ventral positioning of glomeruli in the olfactory bulb. Journal of Neuroscience 25, 3586–3592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Monahan K, and Lomvardas S (2015). Monoallelic expression of olfactory receptors. Annual Review of Cell and Developmental Biology 31, 721–740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Monahan K, Schieren I, Cheung J, Mumbey-Wafula A, Monuki ES, and Lomvardas S (2017). Cooperative interactions enable singular olfactory receptor expression in mouse olfactory neurons. Elife 6, e28620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moore PA (1994). A model of the role of adaptation and disadaptation in olfactory receptor neurons: implications for the coding of temporal and intensity patterns in odor signals. Chemical Senses 19, 71–86. [DOI] [PubMed] [Google Scholar]
- Mori K, and Sakano H (2011). How is the olfactory map formed and interpreted in the mammalian brain? Annual Review of Neuroscience 34, 467–499. [DOI] [PubMed] [Google Scholar]
- Nagel KI, and Wilson RI (2011). Biophysical mechanisms underlying olfactory receptor neuron dynamics. Nature Neuroscience 14, 208–216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakashima A, Takeuchi H, Imai T, Saito H, Kiyonari H, Abe T, Chen M, Weinstein LS, Yu CR, Storm DR, et al. (2013). Agonist-independent GPCR activity regulates anterior-posterior targeting of olfactory sensory neurons. Cell 154, 1314–1325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ng R, Salem SS, Wu S-T, Wu M, Lin H-H, Shepherd AK, Joiner WJ, Wang JW, and Su C-Y (2019). Amplification of Drosophila Olfactory Responses by a DEG/ENaC Channel. Neuron 104, 947–959.e945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Leary T, Williams AH, Caplan JS, and Marder E (2013). Correlations in ion channel expression emerge from homeostatic tuning rules. Proceedings of the National Academy of Sciences 110, E2645–2654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oberland S, Ackels T, Gaab S, Pelz T, Spehr J, Spehr M, and Neuhaus EM (2015). CD36 is involved in oleic acid detection by the murine olfactory system. Frontiers in Cellular Neuroscience 9, 366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pachitariu M, Stringer C, Dipoppa M, Schröder S, Rossi LF, Dalgleish H, Carandini M, and Harris KD (2017). Suite2p: beyond 10,000 neurons with standard two-photon microscopy. bioRxiv. [Google Scholar]
- Pacifico R, Dewan A, Cawley D, Guo C, and Bozza T (2012). An olfactory subsystem that mediates high-sensitivity detection of volatile amines. Cell Reports 2, 76–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parrilla M, Chang I, Degl’Innocenti A, and Omura M (2016). Expression of homeobox genes in the mouse olfactory epithelium. The Journal of Comparative Neurology 524, 2713–2739. [DOI] [PubMed] [Google Scholar]
- Pashkovski SL, Iurilli G, Brann D, Chicharro D, Drummey K, Franks K, Panzeri S, and Datta SR (2020). Structure and flexibility in cortical representations of odour space. Nature 583, 253–258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Repicky SE, and Luetje CW (2009). Molecular receptive range variation among mouse odorant receptors for aliphatic carboxylic acids. Journal of Neurochemistry 109, 193–202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rubin BD, and Katz LC (1999). Optical imaging of odorant representations in the mammalian olfactory bulb. Neuron 23, 499–511. [DOI] [PubMed] [Google Scholar]
- Saito H, Chi Q, Zhuang H, Matsunami H, and Mainland JD (2009). Odor Coding by a Mammalian Receptor Repertoire. Science Signaling 2, ra9–ra9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scala F, Kobak D, Bernabucci M, Bernaerts Y, Cadwell CR, Castro JR, Hartmanis L, Jiang X, Laturnus S, Miranda E, et al. (2021). Phenotypic variation of transcriptomic cell types in mouse motor cortex. Nature 598, 144–150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schaffer ES, Stettler DD, Kato D, Choi GB, Axel R, and Abbott LF (2018). Odor Perception on the Two Sides of the Brain: Consistency Despite Randomness. Neuron 98, 736–742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scholz P, Kalbe B, Jansen F, Altmueller J, Becker C, Mohrhardt J, Schreiner B, Gisselmann G, Hatt H, and Osterloh S (2016). Transcriptome Analysis of Murine Olfactory Sensory Neurons during Development Using Single Cell RNA-Seq. Chemical Senses 41, 313–323. [DOI] [PubMed] [Google Scholar]
- Schulz DJ, Goaillard J-M, and Marder EE (2007). Quantitative expression profiling of identified neurons reveals cell-specific constraints on highly variable levels of gene expression. Proceedings of the National Academy of Sciences 104, 13187–13191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soucy ER, Albeanu DF, Fantana AL, Murthy VN, and Meister M (2009). Precision and diversity in an odor map on the olfactory bulb. Nature Neuroscience 12, 210–220. [DOI] [PubMed] [Google Scholar]
- Spors H, and Grinvald A (2002). Spatio-temporal dynamics of odor representations in the mammalian olfactory bulb. Neuron 34, 301–315. [DOI] [PubMed] [Google Scholar]
- Sullivan SL, Ressler KJ, and Buck LB (1995). Spatial patterning and information coding in the olfactory system. Current Opinion in Genetics & Development 5, 516–523. [DOI] [PubMed] [Google Scholar]
- Tasic B, Yao Z, Graybuck LT, Smith KA, Nguyen TN, Bertagnolli D, Goldy J, Garren E, Economo MN, Viswanathan S, et al. (2018). Shared and distinct transcriptomic cell types across neocortical areas. Nature 563, 72–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Teşileanu T, Cocco S, Monasson R, and Balasubramanian V (2019). Adaptation of olfactory receptor abundances for efficient coding. Elife 8, 308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Touhara K (2002). Odor discrimination by G protein-coupled olfactory receptors. Microsc Res Tech 58, 135–141. [DOI] [PubMed] [Google Scholar]
- Turrigiano G (2011). Too many cooks? Intrinsic and synaptic homeostatic mechanisms in cortical circuit refinement. Annual Review of Neuroscience 34, 89–103. [DOI] [PubMed] [Google Scholar]
- Turrigiano GG (1999). Homeostatic plasticity in neuronal networks: the more things change, the more they stay the same. Trends Neurosci 22, 221–227. [DOI] [PubMed] [Google Scholar]
- Turrigiano GG (2008). The self-tuning neuron: synaptic scaling of excitatory synapses. Cell 135, 422–435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turrigiano GG (2017). The dialectic of Hebb and homeostasis. Philosophical Transactions of the Royal Society B: Biological Sciences 372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tyssowski KM, and Gray JM (2019). The neuronal stimulation-transcription coupling map. Curr Opin Neurobiol 59, 87–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vihani A, Hu XS, Gundala S, Koyama S, Block E, and Matsunami H (2020). Semiochemical responsive olfactory sensory neurons are sexually dimorphic and plastic. Elife 9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- von der Weid B, Rossier D, Lindup M, Tuberosa J, Widmer A, Col JD, Kan C, Carleton A, and Rodriguez I (2015). Large-scale transcriptional profiling of chemosensory neurons identifies receptor-ligand pairs in vivo. Nature Neuroscience, 1–11. [DOI] [PubMed] [Google Scholar]
- Wachowiak M, and Cohen LB (2001). Representation of odorants by receptor neuron input to the mouse olfactory bulb. Neuron 32, 723–735. [DOI] [PubMed] [Google Scholar]
- Waggener CT, and Coppola DM (2007). Naris occlusion alters the electro-olfactogram: evidence for compensatory plasticity in the olfactory system. Neurosci Lett 427, 112–116. [DOI] [PubMed] [Google Scholar]
- Wang HW, Wysocki, and Gold GH (1993). Induction of olfactory receptor sensitivity in mice. Science 260, 998–1000. [DOI] [PubMed] [Google Scholar]
- Wang Q, Titlow WB, McClintock DA, Stromberg AJ, and McClintock TS (2017). Activity-Dependent Gene Expression in the Mammalian Olfactory Epithelium. Chemical Senses 42, 611–624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wark B, Lundstrom BN, and Fairhall A (2007). Sensory adaptation. Curr Opin Neurobiol 17, 423–429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weber AI, Krishnamurthy K, and Fairhall AL (2019). Coding Principles in Adaptation. Annual Review of Vision Science 5, 427–449. [DOI] [PubMed] [Google Scholar]
- Weinreb C, Wolock S, and Klein AM (2018). SPRING: a kinetic interface for visualizing high dimensional single-cell expression data. Bioinformatics 34, 1246–1248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williams AH, Kim TH, Wang F, Vyas S, Ryu SI, Shenoy KV, Schnitzer M, Kolda TG, and Ganguli S (2018). Unsupervised Discovery of Demixed, Low-Dimensional Neural Dynamics across Multiple Timescales through Tensor Component Analysis. Neuron 98, 1099–1115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilson CD, Serrano GO, Koulakov AA, and Rinberg D (2017). A primacy code for odor identity. Nat Commun 8, 1477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilson DA (1998). Habituation of odor responses in the rat anterior piriform cortex. Journal of Neurophysiology 79, 1425–1440. [DOI] [PubMed] [Google Scholar]
- Wilson DA (2000). Comparison of odor receptive field plasticity in the rat olfactory bulb and anterior piriform cortex. Journal of Neurophysiology 84, 3036–3042. [DOI] [PubMed] [Google Scholar]
- Wilson DA (2009). Olfaction as a model system for the neurobiology of mammalian short-term habituation. Neurobiology of Learning and Memory 92, 199–205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wolf FA, Angerer P, and Theis FJ (2018). SCANPY: large-scale single-cell gene expression data analysis. Genome Biology 19, 15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu Y, Ma L, Duyck K, Long CC, Moran A, Scheerer H, Blanck J, Peak A, Box A, Perera A, et al. (2018). A Population of Navigator Neurons Is Essential for Olfactory Map Formation during the Critical Period. Neuron 100, 1066–1082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu YE, Pan L, Zuo Y, Li X, and Hong W (2017). Detecting Activated Cell Populations Using Single-Cell RNA-Seq. Neuron 96, 313–329. [DOI] [PubMed] [Google Scholar]
- Xavier AM, Ludwig RG, Nagai MH, de Almeida TJ, Watanabe HM, Hirata MY, Rosenstock TR, Papes F, Malnic B, and Glezer I (2016). CD36 is expressed in a defined subpopulation of neurons in the olfactory epithelium. Scientific Reports 6, 25507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yadon CA, and Wilson DA (2005). The role of metabotropic glutamate receptors and cortical adaptation in habituation of odor-guided behavior. Learn Mem 12, 601–605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yap E-L, and Greenberg ME (2018). Activity-Regulated Transcription: Bridging the Gap between Neural Activity and Behavior. Neuron 100, 330–348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang J, Huang G, Dewan A, Feinstein P, and Bozza T (2012). Uncoupling stimulus specificity and glomerular position in the mouse olfactory system. Mol Cell Neurosci 51, 79–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang X, and Firestein S (2002). The olfactory receptor gene superfamily of the mouse. Nature Neuroscience 5, 124–133. [DOI] [PubMed] [Google Scholar]
- Zufall F, and Leinders-Zufall T (2000). The cellular and molecular basis of odor adaptation. Chemical Senses 25, 473–481. [DOI] [PubMed] [Google Scholar]
- Zwicker D, Murugan A, and Brenner MP (2016). Receptor arrays optimized for natural odor statistics. Proceedings of the National Academy of Sciences 113, 5560–5575. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1. Summary of single cell RNA-seq datasets for olfactory sensory neurons used in this study, related to Figures 1–6.
Table S2. Loadings of 1,350 highly variable genes for 16 gene expression programs identified by consensus non-negative matrix factorization of the home cage dataset, related to Figure 2.
Table S3. 117 environmental state neuronal genes, including 73 functional genes that may regulate sensory responses, related to Figure 4.
Table S4. Changes in z-scored expression of 117 environmental state neuronal genes across conditions, for each OSN subtype, related to Figure 4.
Table S5. 472 genes used to calculate activation score that were acutely odor-sensitive in responsive OSNs, related to Figure 5. 8 values that characterize 472 genes we used to calculate activation score are shown. Columns are as follows: “activation_score_loading”: gene loading for calculating activation score. “ess_correlation”: Spearman’s rho between normalized expression and environmental state score in the home cage dataset on OR-by-OR basis. “p_adj_ACE2h”: FDR corrected p-values for the differential expression (via Wilcoxon signed-rank test) between control and acetophenone 2h samples for acetophenone responsive ORs on OR-by-OR basis. “ctl_expr_ACE2h”: Normalized mean expression for OSNs expressing-acetophenone responsive ORs in the control data. “expression_ACE2h”: Normalized mean expression for OSNs expressing acetophenone responsive ORs in the acetophenone 2h data. “p_adj_OCT2h”, “ctl_expr_OCT2h”, “expression_OCT2h”: same as the previous 3 columns for acetophenone responsive ORs, but for OSNs expressing octanal-responsive ORs.
Figure S1. OSN transcriptomes are predictable across individuals, and variation is distributed across hundreds of genes, related to Figure 1. A. Histogram of the number of cells expressing each OR, from wild-type mice housed in the home cage.
B. The cumulative percent of ORs observed in OSNs (sorted by how often each OR is observed) as a function of the cumulative fraction of OSNs that express any given OR. The top 200 ORs (of the 1016 detected) account for > 50% of OSNs.
C. The number of OSN subtypes identified, thresholded by the number of OSNs expressing a given OR; colors represent numbers of OSNs used for this analysis; a threshold of 10 OSNs yielded 831 OSN subtypes.
D. Percent of OSNs expressing 0, 1 or >1 ORs thresholded by the number of transcripts (unique molecular identifiers, UMIs) required for an OR gene to be considered “detected”. Many of the rare 2 OR cells were clearly doublets and contained higher than average total numbers of UMIs compared to a typical 1 OR cell. Therefore, downstream analyses used a threshold of 3 UMIs and only considered cells in which only one OR was detected.
E. UMAP plots highlighting OSNs expressing the indicated ORs.
F. The mean fraction of all OSNs expressing a given OR (averaged across 1,000 restarts) as a function of the rank-ordered transcriptome distance from each cell expressing that OR. Shaded error indicates the interquartile range across ORs.
G. Distribution of within-OR transcriptome distances across OSN subtypes using cells from the same or different mice, or after shuffling OR labels.
H. Distribution of within-OR transcriptome distances as a function of the number of OSNs expressing each OR.
I. Separability of the distributions of within-OR and between-OR transcriptome distances, as measured by the Jensen-Shannon Divergence (JSD), for various number of genes and increasing number of principal components. JSD was normalized to the maximum JSD value across all conditions. Highly variable genes (HVGs, used to compute the principal components) were selected based on their variability across OSNs expressing different ORs. Stars represent the number of HVGs used in the analyses shown in the paper. The JSD is low, and separability is at chance levels, when OR labels are shuffled.
J. Distribution of accuracies for linear classifiers predicting which OR is expressed in a given OSN, as a function of the number of OSNs expressing each OR.
K. Mean accuracy across OSN subtypes of linear classifiers predicting which OR is expressed as a function of the number of HVGs included (sorted by their informativeness, see Methods). Colors indicate classification thresholds, and error bars depict the mean and SD across 10 restarts.
L. Similar to K. but as a function of the number of genes removed.
M. (left) Accuracy of linear classifiers predicting which OR (out of 831) is expressed, after excluding all transcription factor (TF) and axon guidance genes. Distributions depict the mean accuracy across the 831 OR-defined OSN subtypes (each averaged across 1,000 restarts). (right) Similar to left, but for various gene sets, including axon guidance genes and TF genes (which both contain HVGs and non-HVGs), and the 100 most predictive or all HVGs (reproduced from Fig. 1F). Classifier hyperparameters were optimized for each gene set.
N. Accuracy of pairwise linear classifiers predicting which OR is expressed, with respect to (left) anatomical identity or (middle) OR class. (right) Accuracy of linear classifiers to predict which of dorsal class I, dorsal class II or ventral class II OR is expressed.
O. Similar to left and middle of N. but with respect to CD36-positivity.
P. Similar to left and middle of N. but with respect to genomic distance (distance between transcriptional start sites (TSS), in base pairs).
Q. Similar to left and middle of N. but with respect to the phylogenetic distance.
Figure S2. GEPHigh and GEPLow are aligned with the main axis of transcriptional variation across OSNs, related to Figure 2. A. Consensus non-negative matrix factorization (cNMF) identifies 10 OR-specific gene expression programs (GEPs) whose usages are correlated among OSNs that express the same OR. OSNs expressing the same OR were randomly split into two populations and the correlation in GEP usage between the two populations was computed for each GEP. Error bars depict the mean correlation with SD across 10,000 restarts. 6 GEPs (including one with many genes related to the unfolded protein response (UPR)) only showed weak correlation, indicating that the usages for such GEPs are not specific to ORs in mature OSNs.
B. UMAP plots showing the usages of identified GEPs, together with the expression of genes with high loadings for each GEP.
C. Gene loadings for the subset of HVGs whose loadings was higher than 2/1350 (total number of HVGs) in any of the 10 OR-specific GEPs; genes were hierarchically clustered by their loadings.
D. Accuracy of linear classifiers predicting OR identity using GEPs across (left) OR pairs or (right) across all ORs. Performance upon shuffling OR labels is at chance = 1/831.
E. Correlation coefficients between mean GEP usages for each OSN subtype. Strong anticorrelation between Dorsal/Ventral (ρ = −0.85), Anterior/Posterior (ρ = −0.70) and High/Low GEPs (ρ = −0.87) is observed.
F. Mean usages of (left) GEPDorsal and GEPVentral or (right) GEPAnterior and GEPPosterior for each OSN subtype, from wild-type mice housed in the home cage.
G. Accuracy of pairwise linear classifiers predicting OR identity using only the usages of either the Dorsal/Ventral (D/V) or the Anterior/Posterior (A/P) GEPs.
H. Correlation coefficients of environmental state (ES), Dorsal/Ventral (DV), and Anterior/Posterior (AP) scores. DV and AP scores are calculated by GEPDorsal - GEPVentral and GEPAnterior - GEPPosterior, respectively. Individual mice are indicated as colored on top.
I. Correlation coefficients, computed across OSN subtypes, between the GEP usages and PC scores (calculated from the top 1,350 HVGs).
J. Absolute values of the correlation coefficient, computed across OSN subtypes, between the ES score and the PC score for each of top 10 PCs. The ES score is highly correlated with the first PC (ρ = 0.89), indicating that the ES score is a major axis of transcriptional variation across OSNs derived from mice housed in the home cage. Error bars depict the mean and SD across 1,000 restarts.
K. Mean z-scored expression of immature (Gng8, Gap43) and mature (Gnal, Omp) OSN marker genes as a function of the developmental pseudotime from the onset of OR expression in late-immature OSNs (see Methods).
L. (left) GEPLow, (middle) GEPHigh, and (right) ES scores as a function of the developmental pseudotime from the onset of OR expression in late-immature OSNs. OSN subtypes are fractionated by their ES scores in mature OSNs.
Figure S3. The dependence of OSN transcriptomes on long-term interactions with odors and air, related to Figure 3. A. Accuracy of linear classifiers predicting OR identity based on HVG expression (see Figs. 1E–F) from open or occluded nostrils across OR pairs (left) or across all ORs (right).
B. Accuracy of linear classifiers predicting whether a given cell for each OSN subtype is from the open or closed nostril, using the usage of either (left) all 10 OR-specific GEPs or (right) only GEPHigh/GEPLow.
C. Gene loadings for each GEP for HVGs that are differentially expressed (DE), or are unchanged (non-DE, in gray) by chronic naris occlusion. HVGs that decrease upon occlusion (in blue) have higher than average loadings compared to non-DE genes for GEPHigh (p < 1 × 10−57, KS-test, adjusted by the Holm-Bonferroni method) and lower than average loadings for GEPLow, and vice versa for genes that increased (in red, p < 1 × 10−48, KS-test, adjusted by the Holm-Bonferroni method).
D. Mean usages of GEPHigh and GEPLow for 797 OSN subtypes for each nostril, using data from all mice.
E. Mean ES score for each OSN subtype for each nostril. Data from each mouse are shown separately. ES scores decreased by occlusion for 314/341, 356/391, and 320/340 of OSN subtypes, respectively (FDR ≤ 0.01).
F. Mean ES scores for each OSN subtype for mouse pairs from the same or different environments.
G. Cumulative percent of OSN subtypes whose ES scores differ significantly (FDR ≤ 0.01) in different numbers of environment pairs.
H. Mean environment-dependent changes in GEP usage across OSN subtypes, categorized based on the sign and significance of their ES score changes across environments.
I. Accuracy of pairwise linear classifiers predicting OR identity using only the usages of the two ES GEPs (GEPHigh and GEPLow) of OSNs from each nostril in the chronic occlusion data.
J. UMAP plot highlighting OSNs expressing the indicated ORs, using only cells from occluded nostrils.
K. UMAP plot depicting the z-scored ES scores, using only cells from occluded nostrils.
L. Accuracy of pairwise linear classifiers predicting OR identity using only the usages of the two ES GEPs of OSNs from each of the three environments.
M. Accuracy of pairwise linear classifiers predicting in which environment a mouse was housed, based upon usages of either all GEPs or ES GEPs. Classification was performed for each OSN subtype, and distributions of the mean accuracies (across 1,000 restarts) across OSN subtypes with or without significant ES score changes across environments are shown (shuffled = environment labels permuted).
N. OR expression levels (as quantified by the number of transcripts of the expressed OR normalized by the total number of transcripts per cell) in each nostril, for each OSN subtype.
O. Similar to N. but for optogenetically-stimulated (12h) and control (no light) mice.
P. Pairwise correlations across all environments of the (left) mean OR expression levels or (right) the number of cells in which each OR was detected.
Q. As in the right panel of F. but for Dorsal/Ventral (DV), and Anterior/Posterior (AP) scores.
R. ES scores for each OSN subtype from wild-type (WT) and β-Arrestin2 mutant mice housed in the home cage.
S. Similar to R. but for OR expression levels.
T. (left) Mean change in ES scores (relative to the mean across all OSN subtypes) 5 days after a shift to environment A (envA), for OSN subtypes from β-Arrestin2 mutant mice. OSN subtypes whose ES score rose or fell after 2 week in envA in WT data show increase or decrease in ES scores in mutants (p < 1 × 10−15 via Wilcoxon signed-rank test). (right) Correlation coefficient of changes in ES score for each OSN subtype between WT and β-Arrestin2 mutant mice (p < 1 × 10−4 via permutation test across 10,000 shuffles).
Figure S4. Functional gene expression exhibits organized correlation structure, related to Figure 4. A. Heatmap showing pairwise correlations between the expression levels, across OSN subtypes, of the 117 ES neuronal genes, in data from mice housed in the home cage.
B. Summary of the correlations between pairs of the 73 functional genes (see Fig. 4A and Table S3), separated by pairs from either GEPHigh alone, GEPLow alone, or both GEPHigh and GEPLow.
C. Correlation of the expression of individual functional genes and the ES score, computed across OSN subtypes, compared to correlations observed upon shuffling ES scores across OSN subtypes.
D. Mean ES score and mean “functional” ES score (using GEPHigh and GEPLow usages calculated from the loadings of the 73 functional genes) for each OSN subtype from mice housed in the home cage.
E. Accuracy of pairwise linear classifiers trained to predict OR identity using the GEPHigh and GEPLow usages calculated from the loadings of the 73 functional genes.
F. Mean occlusion-dependent change in ES score and “functional” ES score for each OSN subtype.
G. Similar to B. but for occlusion-dependent changes in functional gene expression.
H. Similar to C. but for occlusion-dependent changes in ES scores and functional gene expression.
I. Optogenetic stimulation-dependent changes in expression of 73 functional genes as a function of changes in ES scores (binned into 10 quantiles); plots depict the mean and SD of the z-scored expression of functional genes associated with GEPHigh (red) and GEPLow (blue).
J. Similar to B. but for environment-dependent changes in functional gene expression.
K. Similar to C. but for environment-dependent changes in ES scores and functional gene expression.
Figure S5. Act-Seq identified odor-activated OSNs and quantified the degree of transcriptional odor response, related to Figure 5. A. (top left) UMAP plot showing mean z-scored expression of 10 immediate early genes (IEGs, see Methods for list of IEGs). (top right) Fraction of cells per OSN subtype that were activated by acetophenone (ACE) or octacal (OCT) based on IEG expression. OSN subtypes were defined as odor-responsive when at least 70 percent of cells expressing the given OR for that subtype were activated (dashed line), and these ORs were considered as activated. (middle left) UMAP plot highlighting activated cells identified by unsupervised Leiden clustering (see Methods). (middle right) Fraction of cells per OSN subtype that were activated based on Leiden clustering, colored by odor-responsiveness as assessed by IEG expression. (bottom left) UMAP plot depicting odor-driven activation as quantified by unsupervised tensor component analysis (TCA). (bottom right) Distribution of TCA loadings of a factor representing odor activation (with 11-times higher weight on the odor source compared to the control condition) for OSN subtypes for mice exposed to ACE and OCT, colored by odor-responsiveness as assessed by IEG expression.
B. Distribution of the mean expression of 10 IEGs across OSN subtypes, separated by odor-responsiveness.
C. Venn diagram depicting the overlap between ORs identified as acetophenone receptors via Act-seq, the DREAM method, and phospho-S6 immunoprecipitation (from von der Wied et al. 2015, Jiang et al. 2015). For the 8 ORs identified by only the phospho-S6 method, 3 of them were not activated in any cells via Act-Seq and the remaining 5 of them were partially activated (activated in 43.8% of cells per OR on average). This analysis only includes the 717 ORs which were present in four or more OSNs in these odor-activation experiments. Although Act-Seq captured nearly all such receptors that were also captured by DREAM and phospho-S6 (and many missed by both of these techniques), the 717 observed ORs included only 16/22 DREAM and 25/49 phospho-S6 ACE ORs; these “missed” receptors possibly reflect ORs that are expressed in particularly small numbers of OSNs.
D. Venn diagram depicting the overlap between ORs activated by ACE and OCT. The overlap is at chance levels (odds ratio = 1.42, p=0.29 Fisher’s exact test).
E. Comparisons of acute gene expression changes induced by ACE or OCT compared to solvent-only (DPG) condition for 996 genes with significant changes (FDR ≤ 1 × 10−3) in at least one odor.
F. Mean percent of ORs activated by the indicated concentration of acetophenone.
G. Mean percent of ORs activated by 10% acetophenone and its analogs (2-hydroxyacetophenone (2HA), 4-methyl acetophenone (4MA), methyl salicylate (MS)).
H. Heatmap depicting percent overlap of activated ORs among four acetophenone-related odors at 10% concentration. ORs activated by acetophenone analogs are also consistently activated by acetophenone (odds ratio > 22, p < 1 × 10−11 via Fisher’s exact test for each of three analogs).
I. Correlation coefficients across individual mice of activation scores for OSN subtypes responsive to either ACE or OCT (ρ ≥ 0.89 between mice exposed to the same odor).
J. Correlation coefficients comparing activation scores for OSN subtypes activated by acetophenone-related odors, shown for pairs of mice exposed to the same or different odors. The correlation is higher in mice exposed to the same odor (p = 0.002, Mann-Whitney U test).
K. Activation scores for four acetophenone-related odors, for OSN subtypes responsive to each odor.
L. Accuracy of pairwise linear classifiers trained to distinguish pairs of acetophenone-related odors based upon activation scores from odor-responsive OSN subtypes (using only OSN subtypes responsive to both odors in each pair). Observed and shuffled (odor labels are shuffled 1,000 times) classification accuracies are shown.
M. Accuracy of a minimum distance classification procedure to predict odor identity of the four acetophenone-related odors using OSN subtype-specific activation scores (see Methods), as a function of the number of OSN subtypes (from the set of OSN subtypes responsive to any of the four odors). The curves depict the mean and interquartile range of classifier accuracy for each odor across 1,000 restarts for the observed data, and for data in which odor labels were shuffled for each OSN subtype.
N. Correlation coefficient between the activation score and the control ES score for each OSN subtype, evaluated at various thresholds used for identifying activated cells based on IEG expression and for identifying responsive ORs (fraction of cells activated). The threshold used in Fig. 5I (70% of cells per OR and IEG expression ≥ 0.2) is highlighted with stars.
O. Correlation coefficient between the activation score and the control ES score for each OSN subtype, within and between individual mice. For each odor, the observed negative correlation between activation and control ES score is higher (p < 1 × 10−4 via permutation test) than any value in the distribution of correlation coefficients measured after permuting the activation score across OSN subtypes for 10,000 shuffles.
P. Similar to O. but for four acetophenone-related odors (p < 8 × 10−3 via permutation test for each odor).
Q. Mean activation scores at the indicated concentration for each of the acetophenone-responsive OSN subtypes, as a function of the in vitro EC50 for each OR, as reported in Jiang et al. 2015 and Saito et al. 2009 (see also Fig. 5K).
R. Normalized error for predictions (see Methods) of activation scores (both observed and shuffled) for OSN subtypes responsive to ACE at 10%, based on control ES scores, the in vitro EC50 for each OR (from Jiang et al. 2015 and Saito et al. 2009), or both. Error bars represent mean and SD across 1,000 restarts.
S. Similar to O. but for data derived from the acute optogenetic stimulation experiment (p < 1 × 10−4 via permutation test for each stimulation frequency).
Figure S6. Odor responses are determined by OSN Environmental State (ES) scores and changes in ES score are influenced by acute odor responses, related to Figure 6. A. Summary of the correlations between pairs of the 73 functional genes, separated by pairs from either GEPHigh alone, GEPLow alone, or both GEPHigh and GEPLow, using data from transiently-occluded (but not unplugged) nostrils (see Fig. 6B).
B. Mean transient occlusion-dependent changes in GEP usage across OSN subtypes.
C. Fraction of OSN subtypes responsive to acetophenone (ACE) from unplugged nostrils, for OSN subtypes responsive to 10 % ACE (as shown in the top right of Fig. S5A).
D. Mean changes in ES (left) or activation scores (right) between acetophenone-responsive OSN subtypes from open or unplugged nostrils, as a function of the difference in ES scores (binned into quartiles), as measured in control mice that were transiently-occluded (but not unplugged). OSN subtypes whose ES scores fell more showed increased activation (p = 2.63 × 10−4, Mann-Whitney U test).
E. Normalized mean-squared error for predictions of the occlusion-dependent change in activation scores for OSN subtypes responsive to acetophenone, with predictions based upon the occlusion-dependent changes in functional gene expression. Model errors increase if the nostril type (open vs. transiently occluded) of the control mice was shuffled, or if the activation scores are shuffled across OSN subtype. Errors were normalized to the value from the observed data, and the error bars depict the mean and SD across 1,000 restarts.
F. Venn diagram, showing that ORs identified as activated by acetophenone in each environment are largely consistent but not fully overlapped; modulation of odor responses across environments may contribute to the changes in activated ORs identified in each environment via our relatively conservative thresholds (see G.).
G. Activation score distributions for OSN subtypes that were responsive to acetophenone in different numbers of environments (0–3). OSN subtypes that responded to acetophenone in 1–2 environments were still activated more in the other environments in which they were not considered as responsive (shown in gray) than those that were not responsive in any environment, consistent with the possibility that these receptors were activated to some extent in all environments.
H. The overlap in identified OSN subtypes responsive to acetophenone between pairs of mice housed within the same or different olfactory environments. Pairs of mice exposed to acetophenone exhibit activation in overlapping sets of OSN subtypes (positive log-odds), but the overlap is greater between mice that were housed within the same olfactory environment (p=0.026, Mann-Whitney U test). The overlap and log-odds are at chance levels after permuting the identity of ACE ORs across 1,000 restarts for each mouse pair.
I. The cumulative percent of acetophenone-responsive OSN subtypes identified in each environment, colored by the number of environments in which each subtype responded.
J. Mean activation scores for each acetophenone-responsive OSN subtype, sorted by each environment, for OSN subtypes responsive in all three environments.
K. Correlation coefficient between the activation score and the control ES score for each acetophenone-responsive OSN subtypes, using data from mice housed in the indicated environments. The observed value is stronger than any value in the distribution of correlation coefficients measured after permuting the activation score across OSN subtypes for 10,000 shuffles (p < 1 × 10−4 via permutation test for each environment).
L. Classification accuracy for a pairwise linear classifier predicting the environment from which a given set of OSNs derived. Classification was performed for each acetophenone-responsive OSN subtype with significant ES score changes across environments, using the activation scores in each environment for that OSN subtype. Shuffle = permuted environment.
M. Mean environment-dependent changes in activation scores, for acetophenone-responsive OSN subtypes, separated in OSN subtypes whose ES scores rose or fell across environments. OSNs whose ES scores increased in one environment relative to another exhibited lower acetophenone-driven activation scores, while those whose ES scores decreased showed the opposite trend (p = 1.77 × 10−11, Mann-Whitney U test).
N. Similar to E., but for the environment-dependent changes in activation scores.
O. OR expression levels after chronic exposure to the indicated concentration of acetophenone for 5 days, normalized to the control condition (exposure to DPG for 5 days). ORs identified as activated in none of the concentrations, at 10% alone, or at 2 or more concentrations by acute (two-hour) Act-Seq are shown separately.
P. Correlation between environment-dependent changes in ES scores (relative to their mean across all OSN subtypes) after 24 hours and 2 weeks, for individual OSN subtypes.
Q. Mean change in z-scored expression of 73 functional genes (relative to the mean change observed across all OSN subtypes and show separately for genes associated with GEPHigh and GEPLow), at various timepoints after a shift to envA, for OSN subtypes categorized based on the sign and significance of their two-week ES score changes.
R. Mean ES scores for OSN subtypes from mice exposed to acetophenone or DPG for 2h and housed in home cages for 22h. Acetophenone-induced changes in the ES scores were not significantly different for acetophenone-responsive OSN subtypes compared to those that did not respond to acetophenone (Mann-Whitney U test, p = 0.214).
S. Mean expression of Nr4a1 and Fos normalized by the total number of transcripts per cell for OSN subtypes, as a function of the ES scores (binned by steps of 50) for each OSN subtype in mice housed in the home cage, compared to that observed in acetophenone-responsive OSN subtypes following two-hour exposure to acetophenone.
Figure S7. Odor environments determine acute functional odor responses in the olfactory bulb, related to Figure 7. A. Max-normalized mean fluorescence for individual sessions from a single mouse (mouse 1 in B.), imaged for five weeks across environments (rows). Scale bars = 200 μm.
B. Pairwise enhanced correlation coefficients for the entire FOV for all imaging sessions from each mouse (after registering FOVs across sessions, see Methods).
C. Percent of odor-responsive glomeruli (see Methods), for the three indicated odors at two separate concentrations. Each circle indicates a single mouse.
D. Percent of environment-sensitive glomeruli (of the glomeruli responsive to each odor) whose odor responses significantly differed (FDR ≤ 0.01 via permutation test), when mice were housed in home1 compared to environment A (envA). Odors were tested at two concentrations; pentanal-high corresponds to the concentration used in Fig. 7B–E. Each point represents a single mouse.
E. Fold-change in the number of glomeruli that differ significantly when comparing odor responses between home1/envA and home1/home2; each circle indicates a single mouse. A larger number of glomeruli change significantly across environments (see also F.).
F. Percent of glomeruli whose difference in odor response between home1/envA has the opposite sign as that observed between envA/home2, for all glomeruli significantly different between home1/envA (dots refer to individual mice); upon return to the home cage after experiencing envA, the odor responses of nearly all glomeruli are at least partially reverted.
G. Percent of odor-responsive glomeruli for the mice used for decoding analysis (mouse 5 and 6 in B.). Each circle indicates an odor, and colors correspond to those in Fig. 7H.
H. Percent of environment-sensitive glomeruli (of the glomeruli responsive to each odor, colored as in Fig. 7H) whose odor responses significantly differed (FDR ≤ 0.01 via permutation test), when mice were housed in home1 compared to envA.
I. Absolute changes in pairwise correlations for mean odor responses (see Fig. 7H–I). Observed values and distribution of means after permuting environment labels across 10,000 shuffles are shown for each mouse (p < 1 × 10−4 for each mouse).
J. Similar to Fig. 7K. Classification accuracy for each of the test × train pairs (120 × 119 pairs), colored by whether training and test data are from either the same or different environments, or from data with shuffled environment labels (and sorted independently for each of the three conditions).
Data Availability Statement
Single-cell RNA-seq data have been deposited at GEO and are publicly available as of the date of publication. The accession number is listed in the key resources table.
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Biological samples | ||
| Mouse olfactory epithelium | This paper | N/A |
| Chemicals, peptides, and recombinant proteins | ||
| Pentanal | Sigma-Aldrich | Cat# 110132 |
| Pentanone | Sigma-Aldrich | Cat# 471194 |
| Ethyl butyrate | Sigma-Aldrich | Cat# W242713 |
| Heptanal | Sigma-Aldrich | Cat# W254002 |
| Hexanal | Sigma-Aldrich | Cat# 115606 |
| Propanal | Sigma-Aldrich | Cat# 538124 |
| Butanal | Sigma-Aldrich | Cat# W217018 |
| Butanone | Sigma-Aldrich | Cat# W217018 |
| Hexanone | Sigma-Aldrich | Cat# 103004 |
| Ethyl acetate | Sigma-Aldrich | Cat# 270989 |
| Propyl acetate | Sigma-Aldrich | Cat# 537438 |
| Butyl acetate | Sigma-Aldrich | Cat# 287725 |
| Benzaldehyde | Sigma-Aldrich | Cat# B1334 |
| Tiglaldehyde | Sigma-Aldrich | Cat# W340707 |
| Methyl tiglate | Thermo Fisher | Cat# T024825ML |
| Methyl valerate | Sigma-Aldrich | Cat# W275204 |
| Acetophonone | Sigma-Aldrich | Cat# A10701 |
| 2-Hydroxy acetophenone | Sigma-Aldrich | Cat# H18607 |
| 4-Methyl acetophenone | Sigma-Aldrich | Cat# W267708 |
| Methyl salicylate | Sigma-Aldrich | Cat# M6752 |
| Octanal | Sigma-Aldrich | Cat# O5608 |
| Mineral oil | Sigma-Aldrich | Cat# M5904 |
| Dipropylene glycol | Sigma-Aldrich | Cat# D215554 |
| Actinomycin D | Sigma-Aldrich | Cat# A9415 |
| Triptolide | Sigma-Aldrich | Cat# T3652 |
| Anisomycin | Sigma-Aldrich | Cat# A9789 |
| Papain | Worthington | Cat# LK003178 |
| DNase I | Worthington | Cat# LK003172 |
| EBSS | Worthington | Cat# LK003188 |
| Hibernate-A | Thermo Fisher | Cat# A1247501 |
| Heat-inactivated Fetal Bovine Serum | Thermo Fisher | Cat# 16140071 |
| Propidium iodide | Thermo Fisher | Cat# P1304MP |
| PBS | Thermo Fisher | Cat# 70011044 |
| Critical commercial assays | ||
| Chromium Next GEM Single Cell 3' Kit v3.1 | 10x genomics | Cat# 1000268 |
| Chromium Next GEM Chip G Single Cell Kit | 10x genomics | Cat# 1000120 |
| Dual Index Kit TT Set A | 10x genomics | Cat# 1000215 |
| Agilent High Sensitivity DNA kit | Agilent | Cat# 5067–4626 |
| KAPA Library Quantification Kit, illumina platform, Complete kit (Universal) | Roche | Cat# KK4824 |
| Deposited data | ||
| Raw and analyzed sequencing data | This paper | GEO: GSE173947 |
| Experimental models: Organisms/strains | ||
| Mouse: C57BL/6J | The Jackson Laboratory | JAX: 000664, RRID:IMSR_JAX:000664 |
| Mouse: OMP-IRES-GFP | The Jackson Laboratory | JAX: 006667, RRID:IMSR_JAX:006667 |
| Mouse: B6J.Cg-Gt(ROSA)26Sortm95.1(CAG-GCaMP6f)Hze/MwarJ | The Jackson Laboratory | JAX:028865, RRID:IMSR_JAX:028865 |
| Mouse: Arrb2tm1Rjl/J | The Jackson Laboratory | JAX:011130, RRID:IMSR_JAX:011130 |
| Mouse: B6;129P2-Olfr160tm11(Olfr545)Mom/MomJ (S50(M72 locus)) | Thomas Bozza | JAX:006714, RRID:IMSR_JAX:006714 |
| Mouse: B6;129P2-Olfr545tm3(Olfr160)Mom/MomJ (M72(S50 locus)) | Thomas Bozza | JAX:006715, RRID:IMSR_JAX:006715 |
| Mouse: Olfr17tm7Mom/MomJ | Stavros Lomvardas | JAX:006669, RRID:IMSR_JAX:006669 |
| Mouse: OMP-IRES-Cre | Richard Axel | MGI:3045687 |
| Mouse: OMP-ChR2(H134R)-Venus | Thomas Bozza and Dmitry Rinberg | Li et al, 2014 |
| Software and algorithms | ||
| Python 3.6–3.8 | Python | https://www.python.org/ |
| MATLAB (2014a, 2015a) | Mathworks | https://www.mathworks.com/ |
| ScanImage 5 | Vidirio Technologies | http://scanimage.vidriotechnologies.com/display/SIH/ScanImage+Home |
| Wavesurfer 0.775 | Howard Hughes Medical Institute Janelia Research Campus | https://wavesurfer.janelia.org/ |
| Suite2p | Howard Hughes Medical Institute Janelia Research Campus | https://github.com/mouseland/suite2p |
| Adobe Illustrator | Adobe | RRID:SCR_010279, https://www.adobe.com/products/illustrator.html |
| Cell Ranger (2.2.0 for v2 and v3 data and 4.0.0 for v3.1 data) | 10x genomics | https://support.10xgenomics.com/single-cell-gene-expression/software/overview/welcome |
| Scanpy 1.6.0 | Wolf et al. 2018 | https://github.com/theislab/scanpy |
| Python analysis scripts | This paper | Zenode: https://doi.org/10.5281/zenodo.5550454 GitHub: https://github.com/dattalab/Tsukahara_Brann_OSN |
| Other | ||
| Teensy 3.2 | Adafruit | Cat# 2756 |
| blue LED | Digikey | Cat# 1416–1052-6-ND |
All original code has been deposited at GitHub and Zenodo, and the links and DOI are listed in the key resources table.
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.