

Abstract
Charcot-Marie-Tooth disease type 4H (CMT4H) is an autosomal recessive inherited demyelinating neuropathy caused by an FYVE, RhoGEF, and a PH domain-containing protein 4 (FGD4) gene mutation. CMT4H is characterized by an early onset, slow progression, scoliosis, distal muscle atrophy, and foot deformities. We herein present sibling cases of CMT4H with a homozygous mutation in the FGD4 gene. Both patients exhibited cauda equina thickening on magnetic resonance imaging, which had not been reported among the previous CMT4H cases. This is the first report of CMT4H with a homozygous FGD4 c.1730G>A (p.Arg577Gln) mutation showing mild progression and cauda equina thickening.
Keywords: Charcot-Marie-Tooth disease type 4H, FGD4, autosomal recessive, cauda equina thickening
Introduction
Charcot-Marie-Tooth disease (CMT) is a group of hereditary peripheral neuropathies characterized by progressive motor and sensory polyneuropathy with or without autonomic involvement. CMT typically presents with distal dominant muscle weakness/atrophy and sensory disturbance within the first two decades of life and thereafter slowly deteriorates over decades. It also presents characteristic foot deformities (pes cavus or pes planus) or scoliosis (1). CMT is highly heterogeneous genetically, and more than 80 causative genes linked to CMT-like phenotypes have been identified (2). A subset of these causative genes encodes proteins that are important for maintaining the metabolic function or structure of the myelin or axon. CMT has been mainly classified into CMT1, CMT2, CMT3, CMT4, and CMTX based on electrophysiological and pathological findings, where disease symbols are expressed by an alphabetical designation based on the responsible gene mutations. CMT4 is defined as demyelinating neuropathy (median motor nerve conduction velocities <38 m/s) and with autosomal recessive inheritance neuropathy (1). In 2005, De Sandre-Giovannoli et al. reported a case of two families showing severe demyelinating neuropathy linked to chromosome 12p11.2- p13.1 as CMT type 4H (CMT4H) (3). Subsequently, the responsible mutations in the gene encoding FGD4, also known as the FGD1-related F-actin binding protein (frabin) were identified, in both families (4). CMT4H is clinically characterized by an early onset, slow progression, scoliosis, and foot deformities, in addition to motor or sensory symptoms (5).
Peripheral nerve thickening has been reported in patients with chronic inflammatory demyelinating polyradiculoneuropathy (CIDP) or hereditary motor sensory neuropathy (also known as CMT). Nerve thickening can be detected by ultrasonography or magnetic resonance imaging (MRI) and its clinical effectiveness has been reported (6-14). We herein report the first sibling cases of CMT4H with characteristic cauda equina thickening.
Case Reports
Patient 1
Patient 1 was a 62-year-old woman, who complained of difficulty in walking and hand manipulation. She had been born by normal delivery, and her developmental milestones had been confirmed at the appropriate times. A pedigree of the present case and the affected brother, with their parents' consanguinity, is shown in Fig. 1. The patient realized that her running was slower than that of her classmates when she was 7 years old. At 12 years of age, she began experiencing difficulty in dorsiflexing her ankle joints, and her gait disturbance slowly progressed. However, because she could walk without assistance, she did not visit a hospital. At 51 years of age, she was suspected of having a neurological disorder when she visited the former hospital and was referred to our department. Neurological examinations revealed distal dominant muscle weakness/atrophy, hypoesthesia, and gait disturbance. Nerve conduction studies showed that the bilateral median motor conduction velocity (MCV) was reduced to 8.0 m/s, and the compound muscle action potential (CMAP) responses of the bilateral tibial and peroneal motor nerves were not evoked. The bilateral median, ulnar, and sural sensory nerve action potentials (SNAPs) were also unevoked. A fluorescent in situ hybridization analysis using the patient's lymphocytes, to detect loci of the peripheral myelin protein 22 gene, revealed a negative result of duplication/deletion of the gene. The diagnostic procedure did not progress further for the following 11 years until she visited our department for reevaluation.
Figure 1.
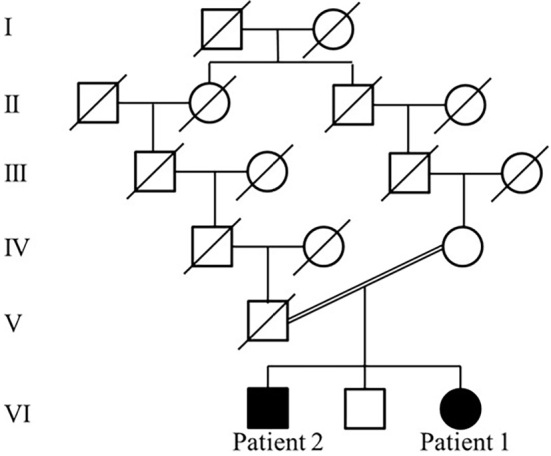
The pedigree of the present family carrying the FGD4 gene mutation is shown. There was consanguinity between the parents of Patients 1 and 2.
When she revisited our department, her mood, memory, and speech were normal. Her pupils were markedly miotic, with a diameter of 2.0 mm in the right eye and 1.5 mm in the left eye. The pupillary light reflex was not active in either eye, and the size of the pupils did not change even in a bright place. She had no blepharoptosis, oculomotor disturbance, facial weakness, bulbar symptoms, or any other cranial nerve disorder. Distal dominant muscle weakness and atrophy were observed in all limbs. Dorsiflexion of her ankle joints was most severely affected, thus resulting in a steppage gait. Hypoesthesia, hypalgesia, and decreased vibratory sensation were also observed in the distal limbs. The deep tendon reflexes showed generalized areflexia without the Babinski sign. Pes cavus foot deformity with hammer toes was also confirmed (Fig. 2A, B). Further, she had a history of hypertension and osteoarthritis of the bilateral hip joints and reported no use of illicit drugs.
Figure 2.

Photographs of characteristic foot deformities of patienst 1 and 2 are shown. Patient 1 showed pes cavus and hammer toes with distal atrophy of lower limbs (A and B). Patient 2 showed more severe foot deformities than Patient 1 (C and D).
The blood test results and chemistry panels were normal. The cerebrospinal fluid (CSF) test confirmed a mild elevation of protein level (88 mg/dL) with normal cell counts (1/mm3). Nerve conduction studies performed on the right upper and lower limbs revealed that the MCV was slower than the previous study; median and ulnar MCVs were 5.6 m/s and 6.7 m/s, and their amplitudes were 660 μV and 574 μV, respectively. No evoked CMAP responses in the tibial and peroneal nerves and no SNAP responses in the median, ulnar, and sural nerves were confirmed. Scoliosis was observed on the thoracic X-ray and computed tomography (CT) image. Skeletal muscle CT showed distal dominant muscle atrophy with fatty infiltration (Fig. 3). Cerebral MRI revealed neither abnormal parenchymal signal intensities nor thickening of the cranial nerves. Lumbar spinal MRI showed prominent thickening of the cauda equina (Fig. 4A-C). Contrast-enhanced lumbar spinal MRI was not performed.
Figure 3.

Skeletal muscle CT image of Patient 1 at the slices of the upper arm level (A), the forearm level (B), the thigh level (C) and the calf level (D) showed symmetrical distal dominant muscle atrophy with fatty degeneration.
Figure 4.

Lumbar spinal MRI of Patient 1 shows a prominent thickening of the cauda equina on T2-weighted sagittal (A), short tau inversion recovery (STIR) coronal (B) and T2-weighted axial (C) images. A lumbar spinal MRI of Patient 2 shows similar findings of cauda equina thickening with multiple disc herniation on T2-weighted sagittal (D), STIR coronal (E) and T2-weighted axial (F) images.
After obtaining written informed consent from the patient, targeted gene sequencing was performed using genomic DNA extracted from the patient's blood cells, and previously known 64 causative genes and 8 candidate genes for CMT were screened using a custom Ion AmpliSeq gene panel (15). A list of genes screened included AARS, APTX, ARHGEF10, BSCL7, DCAF8, DCTN1, DHH, DHTKD1, DNM2, DYNC1H1, EGR2, FBLN5, FBXO38, FGD4, FIG4, GALC, GAN, GARS, GDAP1, GJB1, GJB3, GNB4, HARS, HK1, HOXD10, HSPB1, HSPB3, HSPB8, IGHMBP2, INF2, KARS, KIF1A, LITAF, LMNA, LRSAM1, MARS, MED25, MFN2, MPZ, MTMR2, NDRG1, NEFL, PDK3, PLEKHG5, PMP22, PRPS1, PRX, RAB7A, REEP1, SACS, SBF1, SBF2, SETX, SH3TC2, SLC5A7, SLC12A6, SOX10, SURF1, TDP1, TFG, TRIM2, TRPV4, TTR, and YARS. An additional 8 candidate genes are targets of ongoing research project, and their names were not shown to protect research confidentiality. A genetic analysis revealed a homozygous missense mutation, c.1730G>A (p.Arg577Gln), in FGD4 (NM_139241.2), which is a causative gene for CMT4H. This mutation was also confirmed by direct sequencing of the polymerase chain reaction (PCR) products with the following forward and reverse primers: 5'-GAACATGGTTTGAGCAATGTAG-3' and 5'-TGTGTCCCTAGTTGATGACTTTAC-3' (Fig. 5).
Figure 5.

Direct sequencing of polymerase chain reaction products amplified from the exon 14 region of the FGD4 gene of patients 1 and 2 revealed a homozygous c.1730G>A (p.Arg577Gln) mutation. The arrow indicates the position of the missense mutation.
Patient 2
Patient 2 was a 68-year-old man, the elder brother of Patient 1. He was born by normal delivery, as the first child of his parents (Fig. 1). He visited our department because his sister was diagnosed with CMT and had not been previously examined by a neurologist. He noticed that his running was slower than that of his classmates at age 7. At 27 years of age, he was diagnosed with rheumatoid arthritis, and had been treated with sodium aurothiomalate for 3 years, but he discontinued it thereafter. He had mild difficulty with hand manipulation, but he regarded it to be due to rheumatoid arthritis. At 68 years of age, neurological examination clarified that he had a disease phenotype quite similar to that of his sister, although less severe than hers. He exhibited muscle weakness and atrophy, hypoesthesia, hypalgesia, and reduced vibratory sensation in the distal limbs. The deep tendon reflexes showed generalized areflexia without the Babinski sign. His foot deformities were similar to, but more severe than those of patient 1 (Fig. 2C, D). Miosis was not confirmed as the bilateral pupil diameter in a bright place was 3.0/3.0 mm, with a positive reaction to pupillary light reflex. In addition to rheumatoid arthritis, he also had a history of hypertension, appendicitis, and Meniere's disease.
Nerve conduction studies revealed that the right median and ulnar MCVs were reduced to 10.6 m/s and 11.7 m/s, and their amplitudes were 3.17 mV and 2.26 mV, respectively. CMAP responses were not evoked in the right tibial and peroneal nerves. Furthermore, SNAP responses were not confirmed in the right median, ulnar, and sural nerves. Mild scoliosis was observed on a thoracic X-ray image. The CSF test and skeletal muscle CT imaging were not performed. Lumbar spinal MRI showed prominent thickening of the cauda equina with multiple disc herniations (Fig. 4D, E, F). Contrast-enhanced lumbar spinal MRI was not performed. After obtaining written informed consent from the patient, a genetic analysis of the FGD4 gene was performed, and the same homozygous missense mutation was found as in Patient 1 (Fig. 5).
Discussion
The FGD4 gene encoding protein, frabin, which functions as a Rho guanosine diphosphate/guanosine triphosphate (GTP) nucleotide exchange factor, specifically acts on Cdc42, a member of the Rho family of small GTP-binding proteins (4,16,17). Rho GTPases play an important role in regulating the signal transduction pathways in eukaryotes to control actin cytoskeleton changes during cell migration, morphogenesis, polarization, and division (18). Delague et al. reported that in embryonic rat spinal motoneurons and rat RT4 schwannoma cells, the wild-type frabin colocalized with F-actin in neurite tips and growth cones and induced the formation of filopodia-like microspikes; the truncated forms of frabin also co-localized with F-actin but induced significantly fewer microspike formation than full-length frabin (4). These results suggested that the truncated forms of frabin are involved in a loss-of-function mechanism due to the FGD4 gene mutation (4). Horn et al. showed that FGD4-knockout mice reproduced the electrophysiological findings of demyelinating peripheral neuropathy (19).
CMT4H is an autosomal recessively inherited demyelinating peripheral neuropathy caused by FGD4 gene mutations. So far, at least 22 CMT4H families with FGD4 gene mutations have been reported (4,20-32). According to a review of the previous CMT4H cases, it is clinically characterized by early onset and slow progression, and most cases started their symptoms before 5 years old, ranging from birth to 2nd decade (4,20-32). The severity of distal muscle weakness, amyotrophy, and sensory involvement varies between affected individuals. Foot deformities were confirmed in 23 out of 25 patients (92%) and scoliosis were found in 13 out of 21 patients (62%) (4,20-29,31,32). Spinal syringomyelia, pupil asymmetry, multiple cranial nerve involvement, and cerebellar dysfunction have been reported in individual patients (21,26,28,30). Sural nerve biopsy was performed in 9 CMT4H cases, and the unique pathological feature of myelin outfoldings was confirmed in all cases. This pathological finding has also been reported in CMT types 1B and 4B (33). There were no reports mentioning the results of the CSF test.
The present sibling cases have a homozygous missense c.1730G>A mutation in the FGD4 gene, causing arginine to glutamine substitution at codon 577 of the frabin protein. This mutation has not been previously identified in Japanese CMT4H cases (25,30). Regarding the pathogenicity of this mutation, while a segregation analysis in this family could not be fully investigated, an identical mutation was reported in a Chinese CMT4 patient. This patient was a 10-year-old boy who began experiencing a weakness in the distal lower limbs due to polyneuropathy, at 6 years of age. A genetic analysis revealed that he carried heterozygous FGD4 gene mutations, c.338A> G and c.1730G>A, which were transmitted from his neurologically normal parents (29). The c.1730G>A mutation found in this boy indicates that this is a pathogenic mutation for CMT4H. Another finding to support the pathogenesis of the c.1730G>A mutation was a web-based prediction tool used to evaluate functional effects stemming from a DNA variation, PolyPhen-2. This confirmed that this mutation would be “probably damaging” to frabin protein function. Taken together, the c.1730G>A mutation found in our sibling CMT cases can be considered a disease-causing mutation.
An MRI finding of cauda equina thickening can be observed in cases with demyelinating peripheral neuropathy. A comparative MRI study focusing on the cauda equina, between CIDP and CMT1A, revealed that gadolinium enhancement of the cauda equina was seen in 11 of 16 CIDP patients, but in none of the 6 CMT1A patients, and nerve root thickening was seen in 3 of 16 CIDP patients, but in only 1 of the 6 CMT1A patients (34). Another study on the MRI findings of the cauda equina in seven CMT (type I, n=5; type II, n=2) and three Dejerine-Sottas disease (DSD) patients, revealed that MRI of CMT patients showed intradural nerve root thickening (n=2), signal abnormalities (n=2), and gadolinium enhancement (n=3), and two of three DSD patients' MRI showed intradural nerve enhancement. However, to date, there have been no previous reports of CMT4H cases with cauda equina thickening (14).
A CSF test of patient 1 revealed mild elevation of protein level. Although the level of CSF protein in CMT4H has not been reported, it has been reported that CSF protein elevation may occur in CMT1A (35). CMT cases complicated by autoimmune polyneuropathy had been reported, and these overlapping cases typically showed acute or subacute deterioration of symptoms (36). Immunotherapies including corticosteroids and/or intravenous immunoglobulin should be considered if CMT cases showed acute or subacute deterioration with nerve roots thickening and CSF protein elevation. Since acute or subacute deterioration had not been observed in our cases, overlapping autoimmune polyneuropathy was not supposed.
Patient 1 showed remarkable bilateral miosis, but Patient 2 showed no pupil abnormality. They showed no evidence of other autonomic dysfunction such as bladder dysfunction or orthostatic hypotension. It is speculated that miosis could be the hallmark of this disease, but the previous reports on CMT4 cases had not reported miosis as a characteristic feature except for a finding of asymmetric pupils in one CMT4 case (21). According to the reports of pupil abnormalities in CMTs, bilateral miosis was reported in a subset of CMT2B cases with a MPZ mutation (37). At present, the association between miosis and CMT4 is unclear.
In conclusion, we herein described the first sibling cases of CMT4H, carrying a homozygous c.1730G>A missense mutation in the FGD4 gene. In addition to previously reported characteristics of CMT4H, such as early onset, slow progression, distal muscle atrophy, scoliosis, and foot deformities, cauda equina thickening on MRI was observed in both cases. Cauda equina thickening may be a characteristic neuroradiological feature of CMT4H and thus warrants further investigation in the clinical identification of CMT4H cases.
The authors state that they have no Conflict of Interest (COI).
Financial Support
This study was supported by Grants-in-Aid for Scientific Research (C) from the Ministry of Education, Culture, Sports, Science and Technology, Japan [grant number 19K07813] (to Y.I.); Grants-in-Aid from the Research Committee of Ataxia, Health Labour Sciences Research Grant, The Ministry of Health, Labour and Welfare, Japan [grant number JPMH20FC1041] (to Y.I.).
References
- 1. Pareyson D, Marchesi C. Diagnosis, natural history, and management of Charcot-Marie-Tooth disease. Lancet Neurol 8: 654-667, 2009. [DOI] [PubMed] [Google Scholar]
- 2. Stojkovic T. Hereditary neuropathies: an update. Rev Neurol (Paris) 172: 775-778, 2016. [DOI] [PubMed] [Google Scholar]
- 3. De Sandre-Giovannoli A, Delague V, Hamadouche T, et al. Homozygosity mapping of autosomal recessive demyelinating Charcot-Marie-Tooth neuropathy (CMT4H) to a novel locus on chromosome 12p11.21-q13.11. J Med Genet 42: 260-265, 2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Delague V, Jacquier A, Hamadouche T, et al. Mutations in FGD4 encoding the Rho GDP/GTP exchange factor FRABIN cause autosomal recessive Charcot-Marie-Tooth type 4H. Am J Hum Genet 81: 1-16, 2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Delague V. Charcot-Marie-Tooth Neuropathy Type 4H - RETIRED CHAPTER, FOR HISTORICAL REFERENCE ONLY. In: GeneReviewsⓇ [Internet]. Adam MP, Ardinger HH, Pagon RA, et al. , Eds. University of Washington, Seattle, Seattle (WA), 2013: 1993-2020. [Google Scholar]
- 6. Shibuya K, Sugiyama A, Ito S, et al. Reconstruction magnetic resonance neurography in chronic inflammatory demyelinating polyneuropathy. Ann Neurol 77: 333-337, 2015. [DOI] [PubMed] [Google Scholar]
- 7. Sugimoto T, Ochi K, Hosomi N, et al. Ultrasonographic nerve enlargement of the median and ulnar nerves and the cervical nerve roots in patients with demyelinating Charcot-Marie-Tooth disease: distinction from patients with chronic inflammatory demyelinating polyneuropathy. J Neurol 260: 2580-2587, 2013. [DOI] [PubMed] [Google Scholar]
- 8. Zaidman CM, Harms MB, Pestronk A. Ultrasound of inherited vs. acquired demyelinating polyneuropathies. J Neurol 260: 3115-3121, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Grimm A, Rattay TW, Winter N, Axer H. Peripheral nerve ultrasound scoring systems: benchmarking and comparative analysis. J Neurol 264: 243-253, 2017. [DOI] [PubMed] [Google Scholar]
- 10. Shibuya K, Yoshida T, Misawa S, et al. Hidden Charcot-Marie-Tooth 1A as revealed by peripheral nerve imaging. Intern Med 58: 3157-3161, 2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Murata K, Morishita S, Nakamuro T, Sugata T, Takayanagi T. [A case report of the compression syndrome due to hypertrophic neuropathy]. Rinsho Shinkeigaku (Clin Neurol) 31: 213-215, 1991. (in Japanese). [PubMed] [Google Scholar]
- 12. Tachi N, Kozuka N, Ohya K, Chiba S, Naganuma M. MRI of peripheral nerves and pathology of sural nerves in hereditary motor and sensory neuropathy type III. Neuroradiology 37: 496-499, 1995. [DOI] [PubMed] [Google Scholar]
- 13. Friedman DP, Flanders AE, Tartaglino LM. Hypertrophic Charcot-Marie-Tooth disease: MR imaging findings. AJR Am J Roentgenol 163: 749-750, 1994. [DOI] [PubMed] [Google Scholar]
- 14. Cellerini M, Salti S, Desideri V, Marconi G. MR imaging of the cauda equina in hereditary motor sensory neuropathies: correlations with sural nerve biopsy. AJNR Am J Neuroradiol 21: 1793-1798, 2000. [PMC free article] [PubMed] [Google Scholar]
- 15. Yoshimura A, Yuan JH, Hashiguchi A, et al. Genetic profile and onset features of 1005 patients with Charcot-Marie-Tooth disease in Japan. J Neurol Neurosurg Psychiatry 90: 195-202, 2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Obaishi H, Nakanishi H, Mandai K, et al. Frabin, a novel FGD1-related actin filament-binding protein capable of changing cell shape and activating c-Jun N-terminal kinase. J Biol Chem 273: 18697-18700, 1998. [DOI] [PubMed] [Google Scholar]
- 17. Umikawa M, Obaishi H, Nakanishi H, et al. Association of frabin with the actin cytoskeleton is essential for microspike formation through activation of Cdc42 small G protein. J Biol Chem 274: 25197-25200, 1999. [DOI] [PubMed] [Google Scholar]
- 18. Etienne-Manneville S, Hall A. Rho GTPases in cell biology. Nature 420: 629-635, 2002. [DOI] [PubMed] [Google Scholar]
- 19. Horn M, Baumann R, Pereira JA, et al. Myelin is dependent on the Charcot-Marie-Tooth Type 4H disease culprit protein FRABIN/FGD4 in Schwann cells. Brain 135: 3567-3583, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Stendel C, Roos A, Deconinck T, et al. Peripheral nerve demyelination caused by a mutant Rho GTPase guanine nucleotide exchange factor, frabin/FGD4. Am J Hum Genet 81: 158-164, 2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Houlden H, Hammans S, Katifi H, Reilly MM. A novel Frabin (FGD4) nonsense mutation p.R275X associated with phenotypic variability in CMT4H. Neurology 72: 617-620, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Fabrizi GM, Taioli F, Cavallaro T, et al. Further evidence that mutations in FGD4/frabin cause Charcot-Marie-Tooth disease type 4H. Neurology 72: 1160-1164, 2009. [DOI] [PubMed] [Google Scholar]
- 23. Baudot C, Esteve C, Castro C, et al. Two novel missense mutations in FGD4/FRABIN cause Charcot-Marie-Tooth type 4H (CMT4H). J Peripher Nerv Syst 17: 141-146, 2012. [DOI] [PubMed] [Google Scholar]
- 24. Boubaker C, Hsairi-Guidara I, Castro C, et al. A novel mutation in FGD4/FRABIN causes Charcot Marie Tooth disease type 4H in patients from a consanguineous Tunisian family. Ann Hum Genet 77: 336-343, 2013. [DOI] [PubMed] [Google Scholar]
- 25. Hayashi M, Abe A, Murakami T, et al. Molecular analysis of the genes causing recessive demyelinating Charcot-Marie-Tooth disease in Japan. J Hum Genet 58: 273-278, 2013. [DOI] [PubMed] [Google Scholar]
- 26. Sivera R, Sevilla T, Vilchez JJ, et al. Charcot-Marie-Tooth disease: genetic and clinical spectrum in a Spanish clinical series. Neurology 81: 1617-1625, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Hyun YS, Lee J, Kim HJ, et al. Charcot-Marie-Tooth disease type 4H resulting from compound heterozygous mutations in FGD4 from nonconsanguineous Korean families. Ann Hum Genet 79: 460-469, 2015. [DOI] [PubMed] [Google Scholar]
- 28. Zimon M, Battaloglu E, Parman Y, et al. Unraveling the genetic landscape of autosomal recessive Charcot-Marie-Tooth neuropathies using a homozygosity mapping approach. Neurogenetics 16: 33-42, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Yan C, Yuan Z, Xu L, Jiang L, Gao F. [Peroneal myoatrophy type 4H FGD4 new gene mutation in one case and literature review]. Zhonghua Er Ke Za Zhi 54: 218-221, 2016. (in Chinese). [DOI] [PubMed] [Google Scholar]
- 30. Kondo D, Shinoda K, Yamashita KI, et al. A novel mutation in FGD4 causes Charcot-Marie-Tooth disease type 4H with cranial nerve involvement. Neuromuscul Disord 27: 959-961, 2017. [DOI] [PubMed] [Google Scholar]
- 31. Zis P, Reilly MM, Rao DG, Tomaselli P, Rossor AM, Hadjivassiliou M. A novel mutation in the FGD4 gene causing Charcot-Marie-Tooth disease. J Peripher Nerv Syst 22: 224-225, 2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Argente-Escrig H, Sánchez-Monteagudo A, Frasquet M, et al. A very mild phenotype of Charcot-Marie-Tooth disease type 4H caused by two novel mutations in FGD4. J Neurol Sci 402: 156-161, 2019. [DOI] [PubMed] [Google Scholar]
- 33. Wilmshurst JM, Ouvrier R. Hereditary peripheral neuropathies of childhood: an overview for clinicians. Neuromuscul Disord 21: 763-775, 2011. [DOI] [PubMed] [Google Scholar]
- 34. Midroni G, de Tilly LN, Gray B, Vajsar J. MRI of the cauda equina in CIDP: clinical correlations. J Neurol Sci 170: 36-44, 1999. [DOI] [PubMed] [Google Scholar]
- 35. Rossor AM, Evans MR, Reilly MM. A practical approach to the genetic neuropathies. Pract Neurol 15: 187-198, 2015. [DOI] [PubMed] [Google Scholar]
- 36. Ginsberg L, Malik O, Kenton AR, et al. Coexistent hereditary and inflammatory neuropathy. Brain 127: 193-202, 2004. [DOI] [PubMed] [Google Scholar]
- 37. Houlden H, Reilly MM, Smith S. Pupil abnormalities in 131 cases of genetically defined inherited peripheral neuropathy. Eye (Lond) 23: 966-974, 2009. [DOI] [PubMed] [Google Scholar]