

Abstract
Breakthroughs in molecular medicine have positioned the amyloid-β (Aβ) pathway at the center of Alzheimer’s disease (AD) pathophysiology. While the detailed molecular mechanisms of the pathway and the spatial-temporal dynamics leading to synaptic failure, neurodegeneration, and clinical onset are still under intense investigation, the established biochemical alterations of the Aβ cycle remain the core biological hallmark of AD and are promising targets for the development of disease-modifying therapies. Here, we systematically review and update the vast state-of-the-art literature of Aβ science with evidence from basic research studies to human genetic and multi-modal biomarker investigations, which supports a crucial role of Aβ pathway dyshomeostasis in AD pathophysiological dynamics. We discuss the evidence highlighting a differentiated interaction of distinct Aβ species with other AD-related biological mechanisms, such as tau-mediated, neuroimmune and inflammatory changes, as well as a neurochemical imbalance. Through the lens of the latest development of multimodal in vivo biomarkers of AD, this cross-disciplinary review examines the compelling hypothesis- and data-driven rationale for Aβ-targeting therapeutic strategies in development for the early treatment of AD.
Subject terms: Neuroscience, Diseases, Diagnostic markers
Introduction
Alzheimer’s disease (AD) is the primary cause of dementia, affecting ~45.0 million individuals worldwide and is ranked as the fifth leading cause of death globally [1]. In the United States alone, an estimated 5.8 million individuals live with AD dementia today, and this number is expected to grow to 13.8 million by 2050 [2, 3]. Similarly, in Western Europe, dementia affects ~2.5% of people aged 65–69 years, escalating to about 40% of those aged 90–94 years [4], and by 2050, there will likely be up to 18.9 million patients with dementia in Europe [5] and 36.5 million in East Asian countries [1].
To date, drugs approved for the treatment of AD are labeled for the disease’s clinical dementia stage and target the neurochemical systems underlying cognitive dysfunction and behavioral symptoms, with only short-term symptomatic effects. In the last 25 years, translational studies—including experimental animal and human neuropathological, genetic, and in vivo biomarker-based evidence—support a descriptive hypothetical model of AD pathophysiology characterized by the upstream brain accumulation of Aβ species and plaques, which precedes spreading of tau, neuronal loss and ultimately clinical manifestations by up to 20–30 years [6]. Such multi-dimensional evidence led to reshaping the conceptual framework of AD, into a clinical-biological construct along a continuum that spans preclinical, prodromal, and eventually dementia stages [6, 7].
This pathophysiological model has supported a considerable effort to develop therapeutic compounds targeting the Aβ pathway to slow AD progression in early clinical stages. More recently, several anti-Aβ therapeutic pipelines have been expanded to preclinical stages of AD, when the expected success rate of compounds with putative biological effects is higher [8]. While research and physician communities have raised theoretical and conceptual questions on the scientific appeal of Aβ-targeting therapeutic development due to the failures of AD drug clinical trials, anti-Aβ compounds are continually investigated with promising progress of several late-stage development agents towards regulatory approval steps. Moreover, thorough evaluation of disease relevance of a biological pathway—including sophisticated incorporation of latest biomarkers for target engagement, optimized dosing, and selection of participants and treatment response monitoring despite highly heterogenous populations and subsequent results—may help dispel the concern that negative clinical trials negate the true biological and pathophysiological validity of a complex entity such as the Aβ pathway in AD. Critical evaluation of the Aβ pathway in the sole context of clinical trials is a worthy topic for discussion and have been discussed frequently. Critical evaluation of evidence independent of clinical trial results of anti-Aβ drugs can provide the rationale and validation of the disease relevance of the Aβ pathway, especially as data from supporting non-clinical studies of the Aβ pathway continue to accrue.
In this evolving landscape, we present a systematic and cross-disciplinary state-of-art update of the translational literature based on genetic, epigenetic, and biological data that support the pathophysiological role of the Aβ pathway in the biological continuum of AD. We deliver a descriptive evidence-based overview without inferring any causal nexus between the Aβ pathophysiology and other established AD-related pathophysiological alterations occurring at different temporal scales. This multi-perspective endeavor describes an evidence-based state-of-the-art of the literature that points out a rationale for Aβ-targeting therapeutic strategies for the early treatment of AD and identifies knowledge gaps.
Early History of the Amyloid-β Pathway in AD
The Aβ is a 4 kDa fragment of the amyloid precursor protein (APP), a larger precursor molecule widely produced by brain neurons, vascular and blood cells (including platelets), and, to a lesser extent, astrocytes. Two subsequent proteolytic cleavages of APP by β-secretase (β-APP-cleaving enzyme-1 (BACE1)) at the ectodomain and γ-secretase at intra-membranous sites generate Aβ [9]. In 1984, Aβ and its amino acid sequence were reported for the first time as a primary constituent of meningovascular polymorphic deposits in patients with Down Syndrome; the full sequence of parenchymal Aβ plaque core was found to be identical to the peri-vascular component previously described except that the latter mainly extends to the 42nd residue [10]. Subsequently, the APP gene was sequenced, corroborating that Aβ is a by-product of the enzymatic processing of APP [11]. Eventually, dense Aβ aggregates were described as the main constituent of neocortical neuritic plaques, characterizing brain aging and constituting a pathological hallmark of AD along with tau neurofibrillary tangles (NTFs) [12].
Neuropathological studies, confirmed in vivo by recent quantitative neuroimaging investigations, indicate a spatial-temporal evolution of brain Aβ accumulation that occurs initially in cerebral regions with neuronal populations at high metabolic bio-energetic activity rates (such as association cortices) and spreads from neocortex to allocortex to brainstem, eventually reaching the cerebellum (see Fig. 1) [13]. During the 1990’s and early 2000’s, (i) mechanistic studies linking autosomal dominant AD genes, (ii) investigation of several genetic risk factors relating late‐onset AD to Aβ accumulation, and (iii) longitudinal biomarker-based studies conducted in individuals at risk led to draw the biological-clinical construct for AD including the evidence that Aβ pathophysiology occurs decades before the onset of clinical symptoms [14–16]. In addition, brain Aβ accumulation appears to be upstream to other pathomechanistic alterations of the biological continuum of AD, including the spreading of NTFs, and involvement of neuronal and synaptic loss (Fig. 2). The temporal and spatial evolution of these pathophysiological alterations underlies AD cognitive and functional decline across a clinical continuum, from preclinical to prodromal and dementia stages.
Fig. 1. Traditional neuropathological phases of amyloid-β deposition in Alzheimer’s disease dementia.

Red areas in Phase 1 depicts the cortical regions with the initial accumulation of amyloid-β during the early pre-clinical stage. Continued deposition in the same areas are shown in darker colors in the subsequent stages, with the new areas showing amyloid-β in red in each phase. Neocortical regions with the early accumulation of amyloid-β in phase 1 include association cortices. Additional accumulation is seen in allocortical regions and midbrain (phases 2 and 3), with the cerebellum and brain stem having amyloid-β accumulation in late phase clinical stages. The change to darker shading indicates the continuous accumulation of Aβ. Adapted with permission from ref. [13].
Fig. 2. Hypothetical biomarker evidence-driven model of AD pathophysiology.

Hypothetical model of dynamic biomarkers of the AD is expanded to explicate the preclinical phase. Aβ is identified by cerebrospinal fluid Aβ42 assay or PET amyloid imaging. Synaptic dysfunction evidenced by [18F]-fluorodeoxyglucose positron emission tomography (FDG-PET) or functional magnetic resonance imaging (fMRI), with a dashed yellow line to indicate that synaptic dysfunction may be detectable in carriers of the ε4 allele of the apolipoprotein E gene before detectable Aβ deposition. Neuronal injury is evidenced by cerebrospinal fluid tau or phospho-tau, and brain structure is documented by structural magnetic resonance imaging. Biomarkers change from normal to maximally abnormal (y-axis) as a function of disease stage (x-axis). The temporal trajectory of two key indicators used to stage the disease clinically, cognitive and behavioral measures, and clinical function are also illustrated. Neurofilament light chain (NfL) and neurogranin are newer and potentially more accurate markers of neuronal injury. Figure adapted with permission from ref. [391].
Experimental pathomechanistic and proof-of-concept studies indicate an imbalance between Aβ neuronal production and extracellular clearance of Aβ as the upstream event of Aβ dyshomeostasis, associated with protein misfolding, aggregation, and incipient extracellular accumulation in plaques [15, 17, 18]. While in early-onset AD (EOAD) such an imbalance is primarily due to genetic-driven deregulation of the amyloidogenic pathway with downstream overproduction of Aβ, in late‐onset cases of AD (LOAD) failure of proteostasis networks—mechanisms quality control, from protein synthesis to protein degradation—with insufficient cerebral Aβ clearance represents the key event in Aβ aggregation [19]. Such trickle-down effects comprise the initiating factor of brain Aβ accumulation as an early and central pathophysiological alteration within the AD biological continuum [7, 15, 17].
Genetic Evidence of the Role of the Aβ Pathway
Early-onset AD (EOAD)
Large-scale genetic analyses conducted in datasets of informative monogenic EOAD pedigrees identified highly penetrant mutations in the three genes—the APP gene and the presenilin 1 and 2 (PSEN1 and PSEN2) genes. These mutations are transmitted through autosomal dominant inheritance (i.e., autosomal dominant Alzheimer’s disease or ADAD). In mouse models of ADAD each monogenic mutation causes Aβ dyshomeostasis, with protein misfolding, aggregation, and accumulation in brain parenchymal Aβ plaques [15, 17, 20–22]. Such a linear pathomechanistic model (i.e., “one mutation-one misfolded protein”) led to the conceptualization of the “amyloid cascade” [20–22]. In humans, genetic EOAD accounts for around 1% of all AD cases, and most of the genetic forms are caused by mutations in the APP, PSEN1, and PSEN2 genes, with more than 300 different autosomal dominant mutations reported in these genes [23, 24].
The locus of the APP gene is on chromosome 21. Several genetic linkage studies and observational data indicate that individuals with Down syndrome, bearing APP gene triplication, develop cognitive impairment associated with AD biological signatures [25, 26]. Moreover, 25 genomic duplications encompassing APP were found to co-segregate with AD in families with autosomal dominant disease transmission [27, 28]. Most pathogenic mutations on the APP gene cluster around the proteolytic sites of the β- and γ-secretases with a downstream increase of the substrate affinity and either an overall increase of the total Aβ pool or shifts in Aβ peptides ratios. The latter is characterized by a relative increase of Aβ1-42 levels over the levels of Aβ1-40 and shorter species [25–28]. Such an imbalance is hypothesized to facilitate protein self-aggregation [29, 30].
The potential pathogenic role of the APP gene in humans is supported by the existence of a rare protective variant—APP A673T (or A2T)—next to the APP β-secretase site that reduces both APP cleavage and the production of amyloidogenic Aβ peptides [25–28]. The A673T rare variant is five times more common in non-demented older Icelandic individuals than in AD [31]. Notably, another novel variant of this gene—A673V—is linked to AD when the individual is homozygous for the gene, whereas the heterozygous state is unaffected, in line with a model of recessive Mendelian trait type of inheritance [32]. The opposite effects of APP A673V and APP A673T variants on amyloidogenesis indicate a distinct autosomal recessive pattern of inheritance [33]. PSEN1 accounts for most of the known AD-related mutations with the autosomal dominant transmission. Over 200 mutations involving this complex have been observed [34]. PSEN2 mutations are rare, with less than 40 mutations currently identified [31, 35]. In vitro studies and PSEN1/PSEN2 gene knockout mouse models show (i) reduced NOTCH signal due to a diminished cleavage, (ii) decreased formation of the APP Intracellular Domain fragment (AICD), and (iii) reduced processing of other γ-secretase substrates (see below for more information about the PSEN complex biology). These studies point at a genetically-driven γ-secretase loss of function [36]. Several pathogenic PSEN1/2 mutations induce a unique partial loss of function of APP γ-secretase-dependent cleavage, associated with a shift to Aβ1-42 position cleavage, and decrease both Aβ1-42 and Aβ1-40 production [37]. Unlike PSEN1, AD patients carrying PSEN2 mutations exhibit a wide range of age of onset, from 40 to 80 years. Mutations in PSEN2 have been reported in association with other diseases, including frontotemporal dementia (FTD), dementia with Lewy bodies (DLB), breast cancer, and dilated cardiomyopathy [38].
Late-onset AD (LOAD)
At present, no causal (autosomal dominant or recessive) genetic mutations are known in association with late-onset AD [39]. LOAD is hypothesized to be a multifactorial disease with a complex genetic background. Several critical genetic risk factors in AD susceptibility have been detected through large-scale genome-wide association studies (GWASes), with more than 50 susceptibility genes/loci associated with LOAD risk(see Table 1) [39]. Although GWASes do not uncover causative mechanisms, it is notable that many of these genes are linked to Aβ homeostasis, including its (i) expression (APP, PSEN1, PSEN2 and ADAM10), (ii) trafficking (APOE, CLU and SORL1), and (iii) degradation (PICALM, SORL1, CD33, BIN1, CD2AP, ABCA7, and RIN3 are associated with the endosomal-lysosomal system, and CLU and PTK2B are associated with the ubiquitin-proteasome pathway) [40].
Table 1.
Loci reaching genome-wide significance for association with sporadic late-onset AD.
| Locus | GWS locus or gene | original SNP and publication | Dataset | Functional information |
|---|---|---|---|---|
| 1 | APOE | rs429358 p.(Cys112Arg); ref. [380]. | Case–control | A multifactorial protein, known primarily for its role in lipid transport. Known to bind soluble Aβ. |
| rs7412 p.(Cys158Arg); ref. [380]. | ||||
| 3 | CLU | rs11136000; refs. [381, 382]. | GERAD EADI | Molecular chaperone. Role in immunity and cholesterol metabolism. Binds Aβ. |
| 7 | TREM2 | rs75932628 p.(Arg47His); refs.. [383, 384] | Mixed-cohorts | Receptor of the immunoglobulin superfamily, binds lipids and Aβ. Signals to affect multiple processes in myeloid cells including phagocytosis and cellular metabolism. |
| rs143332484 p.(Arg62His); ref. [385]. | IGAP | |||
| 15 | BIN1 | rs744373; ref. [386]. | CHARGE | Involved in endocytic recycling and Aβ production. also involved in membrane folding. |
| 21 | SORL1 | rs11218343; ref. [40]. | IGAP | Endocytic receptor involved in the uptake of lipo- proteins, APP processing and lysosomal targeting of Aβ. |
| Gene-wide; ref. [387] | ADES-FR | |||
| 22 | ABCA7 | rs3764650; ref. [388]. | GERAD+ | Transporter involved in cholesterol metabolism and phagocytic clearance of Aβ. |
| Gene-wide; ref. [389] | IGAP | |||
| 25 | ADAM10 | rs593742; refs. [383, 390]. | IGAP+ | Metalloprotease responsible for proteolytic processing of APP. |
| Combined UK Biobank | ||||
| and IGAP | ||||
| 36 | APP | rs63750847, p.(Ala673Thr); ref. [107]. | Icelandic, Finnish | APP. |
| and Swedish | ||||
| 37 | IGHG3 | rs77307099; ref. [384]. | ADSP | Immunoglobulin gene whose antibodies interact with Aβ. |
Datasets: Alzheimer’s disease sequencing project (ADSP); Psychiatric Genomics Consortium Alzheimer’s disease working group (PGC–ALZ); deCODE, a private corporation (https://www.decode.com); Genetic and Environmental Risk in AD (GERAD); International Genomics of Alzheimer’s Disease Consortium (IGAP); European AD Initiative (EADI); Cohorts for Heart and Aging Research in Genomic Epidemiology (CHARGE); Alzheimer’s Disease Exome Sequencing-France (ADES-FR).
Aβ amyloid beta, APP amyloid precursor protein. Table adapted from ref. [39].
In addition, pathway analyses indicate that polymorphisms in these genes may have a pleiotropic effect or may not be directly linked to the Aβ pathway but encode for proteins whose alterations are associated with impairment of Aβ homeostasis with a network-wise effect. Several genes related to LOAD play a role in the regulation of inflammatory and immune response pathways, endocytosis and cellular trafficking, cholesterol transport and lipid metabolism, post-translational modification—including ubiquitination, which is a crucial mechanism of cellular protein clearance; see Table 1 for details [39].
The association between APOE ε4 and the Aβ pathway
The apolipoprotein E (APOE) ε4 allele (locus on chromosome 19q13.2) is the first and most significant LOAD risk gene identified [41, 42]. A significant detrimental effect of APOE ε4 allele on EOAD pathophysiology has also been reported [43]. Age-related memory trajectories in APOE ε4 carriers may diverge from those of non-carriers before the age of 60 years despite ongoing normal clinical status as the presence of APOE ε4 correlates with an earlier decline [44]. Homozygosity for the APOE ε4 allele increases the risk of developing LOAD by 3- to 15-fold in a dose-dependent manner [45]. APOE has three major allelic variants, APOE ε2, APOE ε3, and APOE ε4, with the ε3 allele being the most common (77%) and ε2 allele the least common (8%) [46]. Human ApoE protein is a 34-kDa glycoprotein consisting of 299 amino acids. In the central nervous system (CNS), ApoE is abundantly expressed in astrocytes, microglia, vascular mural cells, and choroid plexus cells, and, to a lesser extent, in stressed neurons [45]. ApoE isoforms differentially modulate multiple brain intracellular signaling pathways, including lipid transport, synaptic homeostasis, glucose metabolism, and cerebrovascular function [45].
Clinical and neuropathological studies show a significant association between APOE genotype and Aβ metabolism and homeostasis [45, 47–49]. Brain tissue from AD patients shows that APOE ε4 is correlated with increased intraneuronal accumulation of misfolded Aβ, formation of neurotoxic Aβ species, and plaque parenchymal accumulation [45, 47–49]. Both neuroimaging and cerebrospinal fluid (CSF) biomarker studies indicate a consistent association of APOE ε4 with higher cerebral Aβ deposition in cognitively healthy elderly individuals and across the full clinical continuum of AD, i.e., in patients with subjective memory complaint, prodromal (or mild cognitive impairment (MCI)) and dementia [50–54].
The APOE ε4 effect is marked by earlier AD symptoms onset in cognitively healthy individuals with positive Aβ biomarkers [55] but with otherwise typical clinical progression. The impact of the APOE genotype on the risk of AD cognitive-functional decline is likely to be Aβ-mediated [56]. The effect of APOE ε4 on Aβ metabolism and aggregation appears to be most pronounced during the initiation phase of Aβ dyshomeostasis [57]. Increasing age exacerbates this effect, indicating a potential synergistic interaction between APOE and aging-related metabolic changes [58]. Investigation of the combined APOE ε4-age effect on Aβ accumulation has gained traction since it may help develop reliable predictive models of AD clinical trajectories in cognitively healthy at-risk individuals [45].
The link between the APOE ε4 allele and brain Aβ accumulation: experimental evidence
Studies in humans and transgenic mice support that a model in which brain levels of Aβ species aggregation and rates of Aβ plaque formation are ApoE isoform-dependent (ε4>ε3>ε2), allowing inference of a role for ApoE in modulating Aβ metabolism, aggregation, and deposition [45, 59]. Although the molecular dynamics underlying a direct effect of ApoE isoforms on amyloidogenic pathways are not elucidated yet, studies in vitro and in mouse models of AD indicate that ApoE modulates γ‑secretase activity and downstream Aβ production [60, 61].
ApoE upregulates APP transcription and Aβ production in human embryonic stem cells-derived and induced pluripotent stem cell (iPSC)-derived neurons in an isoform-dependent fashion (i.e., ApoE4 stimulating Aβ production more effectively than ApoE2 or ApoE3) [62]. Furthermore, Aβ secretion was significantly higher in iPSC-derived neurons carrying APOE ε4 than in those with APOE ε3, probably due to increased APP transcription or splicing [63, 64].
Preliminary in vivo evidence indicates that APP processing is not affected by ApoE isoforms [65]. By contrast, mouse models show that a primary mechanism for ApoE-mediated plaque formation to be effects of ApoE on aggregation dynamics rather than from isoforms themselves [66]. Some studies indicate that ApoE4 can facilitate the formation of Aβ fibrils by accelerating the initial seeding or nucleation of Aβ deposition [45, 67]. Astrocytic overexpression of ApoE4—but not ApoE3—was found to exacerbate Aβ seeding and increase brain Aβ half-life in a mouse model of aging [45, 67]. ApoE4 expression increased, whereas ApoE3 reduced, Aβ-related gliosis in the mouse brains, emphasizing the significant impact of ApoE4 on Aβ during the seeding stage that may occur by perturbing Aβ clearance and enhancing Aβ aggregation [68].
The major ApoE receptors are low-density lipoproteins (LDL) receptors (LDLRs), LDL receptor-related protein 1 (LRP1), and heparan sulfate proteoglycan (HSPG), and they mediate cellular uptake of Aβ and ApoE [69]. LDLR overexpression considerably decreases ApoE levels, demonstrating its role in ApoE catabolism [69–71]. Preliminary results indicate that overexpression of LDLR LRP1 mediates cellular Aβ uptake in neurons, astrocytes, and microglia [70, 72]. In addition, LRP1 deficiency exacerbated amyloid pathology in amyloid mouse models by suppressing cellular Aβ uptake and lysosomal degradation [73]. Finally, ApoE4 is assumed to exacerbate Aβ pathophysiology by mechanisms depending on neuronal LRP [74].
Potential protective role of APOE ε3 and APOE ε2
To better understand the potential protective role of APOE ε3 and APOE ε2, clinical observation of patient with a PSEN1 E280A variant provides insight. This rare variant was initially identified in the largest ADAD kindred to date [75]. This amino acid substitution is known to cause Aβ overproduction and subsequent early neurodegeneration, cognitive decline, and eventually dementia. Recently, a female carrier of this variant was identified who did not develop MCI until her seventies, i.e., three decades after the expected age of clinical disease onset [75–78]. Remarkably, a [11C]-PiB-PET scan revealed an unusually pronounced accumulation of cerebral amyloid plaques, much higher than that detected in other cognitively impaired young mutation carriers [79]. Whole-exome sequencing demonstrated that this carrier had two copies of APOE containing the rare Christchurch mutation R136S, a variant with known a protective effect likely due to a loss of normal ApoE function [79, 80]. This APOE ε3ch homozygosity was assumed to delay ADAD onset whereby the protective allele’s homozygosity promotes significant resilience to highly penetrant ADAD clinical onset, possibly mediated by mechanisms limiting tau spreading and pathology even in the presence of substantial accumulation of amyloid plaques. This effect may be associated with an altered affinity for HSPGs [79]. Therefore, the degree of affinity of ApoE for HSPGs might be a factor in triggering downstream neurodegeneration.
The APOE ε2 allele is associated with a lower risk of AD-related neurodegeneration [81, 82]. APOE ε2 carriers show a lower risk and delayed age of onset of AD compared with APOE ε3 homozygotes and APOE ε4 carriers [83]. Besides reduced AD-related pathological burden, greater cortical thickness and less age-related cognitive decline are associated with the protective effects of the APOE ε2 allele [81]. APOE ε2 was defined as an AD age-of-onset ‘decelerator’ since its variant rs7412 delayed age-of-onset by around 12 years [84].
Epigenetic, transcriptional, and post-translational alteration of APP and related genes
Epigenetic dysregulation—including histone modifications, DNA methylation, chromatin remodeling, and non-coding RNAs—is assumed to underlie aging-related functional decline which is itself a risk factor for several sporadic diseases, including cancer and AD [85, 86]. Human neuropathological and omics-based studies show that (i) APP mRNA is highly expressed in neurons, (ii) patterns of APP expression and the mechanisms of regulatory transcription change throughout the lifespan with an age gradient toward dysfunction, and (iii) APP expression is upregulated in AD brains [87–89]. DNA methylation changes in the AD brain are observed where DNA methylation of APP gene promoters differs from one brain region to another, with CpG island hypomethylation of the APP gene in AD brain tissue [90]. Differential DNA methylation is reported in other Aβ-related genes too. For example, the DSCAML1 enhancer region was recently shown to be hypomethylated in AD brain, which in turn, was correlated with the upregulated expression of nearby BACE1 genes [91]. In addition, histone acetylome changes in AD brain include differential H3K27-Ac peaks near MAPT encoding tau protein and hypoacetyl peaks downstream of APP and PSEN1/2 [92].
MicroRNAs (miRNAs) constitute a large family of small non-coding RNAs that exert an inhibitory effect on gene expression by destabilizing messenger RNAs and inhibiting the translation process [93]. Mouse models and human postmortem studies indicate that the deregulation of miRNA turnover has been linked to impairment of the Aβ pathway by either upregulation of the APP gene or increased activity of BACE1; for other miRNAs generally related to Aβ and AD, in mice and humans, a more detailed discussion can be found in other review articles [94].
As a transmembrane protein, APP is a glycosylated protein with constitutive cell surface insertion [95]. While phosphorylation of the C-terminal fragment of APP was previously shown to alter γ-secretase processing in vitro [96] and glycation of Aβ is important for aggregation (discussed in the following sections), it is not known what other post-translational modifications of APP influence its proteolytic processing in AD brain.
Transcriptomic response to Aβ in neuronal and glial cells
Technological advances in single-cell and single-nucleus RNA sequencing have significantly added to understanding cell type-specific changes in AD at cell level resolution in both neuronal and non-neuronal cells [97–100]. Recent data from single-cell analyses in AD mouse models and post-mortem brain from AD patients have highlighted the involvement and contribution of glial cells in AD and have led to the identification of glial subtypes that are associated with the disease, such as AD-associated microglia [101, 102] or astrocyte [103] subpopulations. The resolution offered by single-cell technologies provides an unprecedented opportunity to examine the molecular pathways and cellular processes that are associated with Aβ pathophysiology in a cell-type specific manner—particularly systematic cellular changes to the inflammatory response in microglia and astrocytes that reflect complex neuroimmune interactions in AD pathophysiology and novel disease risk genes [104].
APP Processing: Amyloidogenic and Non-amyloidogenic Pathways
APP cleavage and Aβ generation
Three main proteases—α-, β- and γ-secretases—are involved in APP processing through (1) the amyloidogenic pathway promoting Aβ production through sequential cleavage by β- and γ-secretases, and (2) the non-amyloidogenic pathway in which APP is cleaved in the middle, either generating soluble APPα directly by α-secretase or generating shorter Aβ species such as Aβ1-15 and Aβ1-16 by the sequential cleavage by β-secretase and α-secretase. The two pathways lead to different by-products with different intrinsic functional properties, putative physiological roles, and pathophysiological implications (Fig. 3) [15, 17, 18]. Besides secretase activity, APP trafficking due to the secretory pathway is another essential factor in APP metabolism. APP is first matured in the endoplasmic reticulum and the Golgi apparatus, then translocated to the cell surface. Alternatively, APP can enter the lysosomal pathway and undergo proteolytic degradation [105].
Fig. 3. Amyloidogenic vs non-amyloidogenic pathway.

Amyloid Precursor Protein (APP) is a single transmembrane protein. For the non-amyloidogenic pathway (left), APP is cleaved by A Disintegrin And Metalloprotease (ADAM) family proteases to yield the membrane-tethered C83 fragment and extracellularly released soluble APP alpha (sAPPα). In the amyloidogenic pathway (right), APP is first cleaved by β-secretase (β-APP-cleaving enzyme-1 or BACE1). CTF-β fragment is subsequently cleaved by γ-secretase composed of Presenilin 1 or 2, Nicastrin, PEN2 and APH-1. This proteolytic processing releases amyloid-β into the extracellular space. APP intracellular domain (AICD) from the initial β-secretase cleavage is released into intracellular space. Adapted with permission from ref. [392].
BACE1 is the β-secretase enzyme that cleaves the extracellular juxtamembrane region of APP (β-cleavage). Cleavage of APP by β-secretase liberates the soluble N-terminus of APP (sAPPβ) while the C-terminal fragment (CTF-β or C99) remains bound to the membrane. Two mutations at the β-secretase cleavage site of APP (the Swedish mutation KM/NL and an Italian variant A673V) are linked to EOAD, and are mechanistically linked to higher sAPPβ levels due to a putatively stronger affinity of BACE1 for the changed recognition motif in APP [32, 106]. Conversely, the APP variant A673T has been reported to protect against AD due to the lower affinity of BACE1 for the APP binding site [107]. High BACE1 enzymatic activity is found in human AD brain extracts, consistent with experimental evidence of neurons producing higher levels of Aβ in AD than ‘normal’ aging [108]. BACE1 is also accumulated in dystrophic neurites close to Aβ plaques, both in AD amyloidogenic mouse models and AD brains [109–111]. Inducing autophagy in human mutant neurons promotes retention of BACE1 in distal axons, leading to the enhanced β-cleavage of APP [112].
To produce Aβ, the CTF-β fragment produced by β-secretase cleavage of APP is subsequently cleaved by β-secretase, which then releases Aβ into the extracellular space and the AICD into the cytoplasm [108]. γ-secretase is an aspartyl-type protease membrane protein complex and consists of different several components. The catalytic elements of the membrane-embedded tetrameric γ-secretase complex are represented by presenilins 1 and 2, and intramembrane-cleaving proteases responsible for generating the Aβ carboxyl terminus from APP [113, 114]. Three other proteins accounting of the complex are (i) Nct and (ii) Aph1, thought to underlie formation of a stable, high-molecular-mass protein complex supporting the catalytic activity [115, 116], and (iii) Pen-2, hypothesized to regulate the endoproteolysis of presenilins to form a stable heterodimer that binds to the Nct/Aph1 complex [108]. Besides their function in the γ-secretase proteolytic activity, presenilins participate in fundamental cellular pathways, including cell differentiation, intracellular signaling (including anti-apoptosis) [117], and membrane trafficking [105, 118].
Presenilins play a critical role in maintaining cellular homeostasis and function by modulating membrane protein degradation, intracellular vesicle/protein trafficking, lysosomal activity, and autophagy [105, 110]. More than 90 type-I transmembrane proteins have been identified as substrates of the γ-secretase complex, with the most prominent substrate aside from APP being the NOTCH receptor. Processing of NOTCH by γ-secretase liberates the NOTCH intracellular domain, which translocates into the nucleus and regulates transcription of target genes involved in cell fate decisions during embryogenesis as well as adulthood. Abrogation of NOTCH receptor processing and signaling causes dramatic phenotypes in a variety of organisms [105, 110].
In a parallel competing non-amyloidogenic pathway, APP is cleaved either by α-secretase or η-secretase to release two additional variants of the APP ectodomain, namely sAPP-α and sAPP-η [119]. Juxtamembrane cleavage of APP by α-secretase precludes Aβ generation. In vitro studies have shown that several members of the ADAM (a disintegrin and metalloprotease) family of proteases—including isoforms 9, 10, and 17—display α-secretase activity [120]. In addition, recent evidence indicates that ADAM10 is the major α-secretase responsible for the ectodomain shedding of APP in the mouse brain and likely to be active in humans [112, 121]. The η-secretase pathway is an alternative rescue pathway when BACE1 is inhibited, causing a functional shift with increased Aη-α activity and subsequent lowering of neuronal activity by an unknown mechanism [113].
Physiological roles of APP
The expression of APP as a type I transmembrane protein is high in neurons, especially at the synaptic level. Although a full understanding of its biological function remains elusive, experimental evidence indicates a potential role in dendritic spines remodeling, molecular pathways of neurotransmission, and synaptic homeostasis [111, 122, 123]. Rescue experiments in APP KO mice show that sAPPα is sufficient to restore defects in spine density, long-term potentiation, and spatial learning [124, 125]. Most of the ectodomain shedding of APP is performed by α-secretase, which, as mentioned above, cleaves APP in the Aβ sequence, generating peptides mostly without aggregation or toxicity [126].
Although in vitro evidence suggests that soluble sAPPα has a higher impact on neural plasticity than sAPPβ [127], both peptides modulate basal synaptic transmission and short-term synaptic facilitation through binding to the GABAB receptor subunit 1a (GABABR1a) at the synapse [122]. The sushi domain of the GABABR1a binds to the full-length APP intracellularly [122], likely triggering a crucial mechanism for axonal trafficking of the complex and regulation of receptor exhibition at the presynaptic terminals. Delivery of the complex to the axonal cell surface diminishes the pool of APP available for BACE1 processing in endosomes and lowers Aβ production [122].
Aβ is an ancient neuropepetide, highly conserved across vertebrate taxa over at least 400 million years. The human Aβ sequence is shared by 60–70% of vertebrates [128], underscoring that this peptide has critical physiological functions. Aβ monomers, which are generated from the proteolytic processing of APP, can trigger or sustain intracellular signaling essential for neurotransmission, including the regulation of the excitation/inhibition balance, and synaptic vesicle trafficking [129–131]. In addition, Aβ monomers can initiate pathways mediated by the cyclic adenosine monophosphate response element-binding protein (CREB)-mediated transcription of the brain-derived neurotrophic factor (BDNF) axis, known to be involved in hippocampal neurogenesis, a key process for adult synaptic plasticity (i.e. a set of activity-dependent and adaptive structural/functional changes in synaptic strength or efficacy) [132, 133]. Loss of BDNF activation and decline of hippocampal neurogenesis have been observed in human AD dementia and MCI-AD patients, suggesting that hippocampal neurogenesis may be an early event in the synaptic failure characterizing AD [133]. Aβ released at the synaptic cleft has a critical role in sustaining neuronal bioenergetic levels essential for proper synaptic activity [134]. Experimental models of aging and AD indicate that Aβ-mediated molecular pathways are linked to lipid homeostasis and angiogenesis [135].
Aβ Clearance Mechanisms: a Focus on the Role of the Blood–Brain Barrier
The average fractional rates of Aβ production and clearance in cognitively healthy adults are estimated to be around 8% per hour, as assessed using stable isotope labelling kinetics (SILK) technology and measurements in the CSF [136]. It is hypothesized that small reductions in Aβ clearance from the brain are sufficient to cause Aβ accumulation since efficient clearance is vital for Aβ homeostasis and preventing its toxic accumulation in misfolded assemblies given continual APP processing and Aβ generation [136]. As with all other brain metabolites, the normal average Aβ turnover depends, in part, on bulk-flow via the CSF across the blood–brain barrier (BBB), the perivascular circulation, and the glia-lymphatic (glymphatic) system in the brain [136, 137]. Moreover, multiple molecular pathways and cellular machinery are involved in the clearance process beyond the CNS, with the BBB being of crucial importance in Aβ homeostasis and clearance dynamics. In physiological conditions, the BBB protects the CNS from exposure to toxic metabolites in the systemic circulation and maintains the highly regulated brain internal milieu. Conversely, BBB anatomical disruption and functional breakdown may be detrimental for Aβ homeostasis as a part of early pathophysiological alterations in AD individuals [138].
Aβ clearance through endothelial cells and pericytes
The core structure of the BBB is represented by endothelial cells connected by tight junctions, astrocytic end-feet, pericytes, and smooth muscle cells that ensure a selectively permeable system [139]. Soluble Aβ is transported across brain endothelial cells and transferred to the systemic blood stream mainly via LRP-1 [140] and ABC transporter sub-family A and B member 1 (ABCA1 and ABCB1 respectively) where ABCB1 on the abluminal side of the brain endothelium directly clears Aβ into systematic circulation in an ApoE-dependent fashion [139, 141].
Free Aβ can be transported from the circulation into the interstitium via receptors for advanced glycosylation end-products (RAGE). Soluble transporters (known as ‘sequestering agents’; including soluble forms of RAGE (sRAGE) and LRP (sLRP)) bind to soluble Aβ and inhibit its binding to RAGE, thereby preventing Aβ from entering the interstitium [139, 141]. Preliminary results indicate that, in AD, expression of the blood efflux transporters LRP1 and ABCB1 is decreased, whereas expression of the blood influx transporter RAGE is upregulated [139, 141].
Aβ clearance through intracellular and extracellular enzymatic degradation
There is preliminary evidence showing that intracellular Aβ can be degraded by proteasomes and Aβ-degrading enzymes (ADE) via the ubiquitin-proteasome pathway in neurons and the extracellular neprilysin–mediated pathway, respectively [142]. Mouse models of AD indicate that components of the ADE system can be impaired [139, 142] and that Aβ can inhibit the proteasome, through cross-pathways influences, including a lysosomal cathepsin B-mediated mechanism [143]. Therefore, experimental data suggest the existence of a self-reinforcing detrimental protein homeostasis cycle [143, 144].
The ADE encompasses the zinc metalloendopeptidase (NEP-1 and NEP-2, endothelin-converting enzyme (ECE)-1 and -2, angiotensin-converting enzyme (ACE)), thiol-dependent metalloendopeptdiase (insulin-degrading enzyme (IDE)), serine proteases (plasmin, myelin basic protein and acylpeptide hydrolase), cystein proteases (cathepsin B, D, and S), matrix metalloproteinase (MMP-9, MMP-2), Kallikrein-Related Peptidase 7 and others (GCPII, aminopeptidase A, mitochondrial peptidasome) [145, 146–148]. Many genes identified through GWASes and established as risk factors for AD are linked to Aβ degradation through the endosomal-lysosomal system (RIN3) or ubiquitin-proteasome pathway (CLU and PTK2B) [39, 40].
Aβ clearance via brain interstitial fluid (ISF) bulk-flow and CSF absorption
The perivascular drainage pathway has a significant role in ISF bulk-flow clearance of Aβ [19]. Failure of perivascular drainage of Aβ and increased Aβ deposition in arterial walls has two detrimental downstream effects: (a) microbleeds due to rupture of Aβ-laden arteries, namely cerebral amyloid angiopathy that has high comorbidity with AD, and (b) AD itself where the failure of elimination of ISF, Aβ, and other soluble metabolites from the brain alters homeostasis and the neuronal micro-environment, and is associated with synaptic decline and cognitive-functional impairment.
The glymphatic system was proposed as a CSF-ISF exchange system in absence of direct lymphatic access to the brain and with astrocytes as cellular links between brain parenchyma and the perivascular pathway, with eventual solute transport to the cerebrovenous network and meningeal lymphatic vessels [149]. While there is limited knowledge of the anatomy and function of the glymphatic system in humans, mouse models of aging and AD show that the glymphatic pathways represent a vital clearance system for driving the removal of soluble Aβ from the interstitium [149]. Several other glymphatic-related factors with implications for AD include expression and localization of aquaporin 4 (AQP4) channels on astrocytic endfeet, arterial pulsation, and diurnal glymphatic cycles corresponding to sleep-awake rhythms [150–152].
CSF absorption clearance of Aβ occurs via both circulatory and lymphatic systems. Such processes depends on CSF production by the choroid plexus, blood-CSF barrier structural integrity, relevant transporters, arachnoid villi resistance, and CSF lymphatic absorption, all of which decline with age [153]. In AD, the blood-CSF barrier structural integrity is affected and associated with aberrant Aβ clearance [154]. Both increased CSF outflow resistance at the arachnoid villi level and decreased lymphatic CSF absorption have been reported as brain aging alterations and primary risk factors for AD.
Aβ Biochemical Properties from Monomers through Higher Aggregation States, Including Plaques
After being generated as soluble monomers, Aβ is found in several different intermediate aggregation states, including dimers and trimers, soluble oligomers, and protofibrils, until it forms fibrils that accumulate in plaques, typically viewed as an AD neuropathological hallmark (Fig. 4). Understanding the biology and interlinked dynamics of these intermediate assemblies and their bio-activity, in either physiological and pathophysiological conditions, is essential from a diagnostic and therapeutic perspective.
Fig. 4. Amyloid-β aggregation species and evidence of reversible states: the amyloid-β cycle.
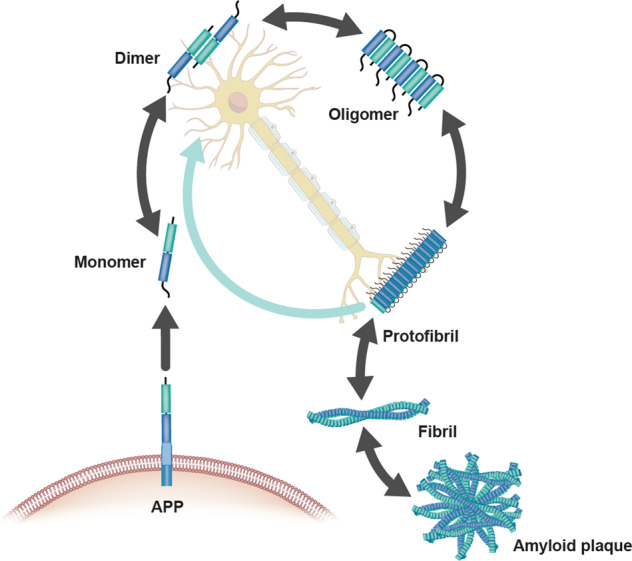
Aggregation species of Aβ can exist as monomers, dimers, oligomers, protofibril, fibril and amyloid plaques. These species exist in steady state where one form can convert to another in a bidirectional manner. The species are characterized by aggregate size, conformation state and solubility, with fibril and amyloid plaque being insoluble. Adapted with permission from ref. [108].
Monomers
As reported above, in physiological conditions, Aβ monomers are involved in neuronal cytoprotective pathways as well as intracellular signaling and synaptic functions [122, 123]. The molecular dynamics underlying the incipient Aβ monomer self-assembly are not known though some in vitro and animal models have provided plausible preliminary hypotheses.
Albeit observed only in vitro, the aggregation of Aβ involves a series of interconnected processes, which starts with a primary nucleation step leading to the formation of disordered oligomers that then convert into growth-competent nuclei [155]. These nuclei can then elongate into fibrillar assemblies, which catalyze the formation of new nuclei, in a feedback process known as secondary nucleation, responsible for the proliferation of the aggregates [156].
Aβ1-42 is less soluble than Aβ1-40 and thus more likely to form aggregates. In this regard, protein solubility has emerged as a critical aspect of protein homeostasis as proteins generally evolved to maintain the solubility required for their optimal function [157, 158]. A variety of aspects of Aβ homeostasis can affect Aβ aggregation. For example, glycation appears as a relevant early event that stimulats amyloid aggregation, followed by increased protease resistance and insolubility [159]. Proteins in amyloid deposits, like Aβ, are frequently glycated [160], suggesting a direct correlation between protein glycation and amyloidosis as well as a link to diabetes [161]. Advanced glycation end-products (AGEs)-modified Aβ peptide-nucleation can seed accelerated aggregation of soluble Aβ peptide versus non-modified seed material [162]. N-terminal truncations of Aβ are less soluble, more prone to aggregation and associated with enhanced toxicity [163], in particular pyroglutamylated variants form when N-terminal truncations expose a glutamate residue which is then transformed into pyroglutamate by the enzyme glutaminyl cyclase [164]. By contrast, the oxidation of methionine 35 increases the solubility of the C-terminal region of Aβ and reduces the aggregation propensity of the peptide [165].
Soluble oligomers
Soluble Aβ oligomers are biochemically defined as Aβ assemblies that are not pelleted from physiological fluids by high-speed centrifugation [166]. Generally, soluble protein misfolded oligomers of unrelated sequences share characteristic structural features with specific immunoreactivity, distinct from those of monomers and fibrils [167]. Soluble Aβ oligomers derived from human brains have molecular weight distributions corresponding to a mixture of dimers to dodecamers [168, 169]. Intracellular and secreted soluble dimeric and trimeric Aβ oligomers were observed in human-derived neurons, as well as APP transgenic mouse models [156, 170, 171]. Mass spectrometry studies have shown that brain-derived bioactive 7 kDa Aβ species are composed of a heterogeneous mixture of covalently cross-linked dimers of different Aβ fragments, which might represent the smallest synaptotoxic species [172, 173].
Robust evidence for the toxic potential of soluble Aβ oligomers derives from studies showing that soluble, low-number oligomers of naturally secreted human Aβ injected in rodent hippocampus can hinder the activity-dependent modulation of synaptic strength and long term depression (LTD) (i.e., synaptic plasticity) [172, 173]. In particular, different Aβ species—including soluble, low-number oligomers—can inhibit key electrophysiological and ultrastructural mechanisms of synaptic plasticity, such as long-term hippocampal potentiation (LTP), enhance LTD and lead to synaptic loss as assessed by the decrease of dendritic spine density [174]. With cell-derived Aβ oligomers, this inhibition occurs at low- to sub-nanomolar concentrations similar to those found in human CSF [175, 176]. Experimental models of AD showed that low-number Aβ oligomers obtained intracellularly from APP-expressing cultured cell lines, disrupt hippocampal LTP in brain slices and in vivo, impair memory of complex learned behavior in rats, and decrease dendritic spine density in organotypic hippocampal slice cultures [177]. Larger aggregates such as dodecamers also exhibit substantial neurotoxicity [178].
Aβ oligomers promote a rapid decrease in membrane expression of memory-related receptors, followed by abnormal spine morphology, reduction in spine density, and synaptic deterioration in cultures of hippocampal neurons [179]. Experiments performed with brain-derived oligomeric species provided a highly diversified picture, supporting the existence of a mixture of water-soluble Aβ species promoting synaptotoxicity [180]. Experimental data in AD extracts show that low molecular weight Aβ oligomers, which are the most aqueously diffusible, effectively mediated disruption of both neuronal structure (neurite integrity) and function (synaptic plasticity), suggesting that only a small pool of toxic Aβ oligomers displays bioactivity [180, 181].
Protofibrils
During the aggregation of monomeric Aβ to insoluble fibrils, several intermediate species are formed, including large soluble aggregates known as protofibrils, as described by Walsh and colleagues [182]. These protofibrils were defined as the soluble oligomeric species of synthetic Aβ peptides appearing as a peak in the void volume (>75 kDa) of a size exclusion chromatography with a Superdex G75 column [183, 184]. Such soluble Aβ species have been shown to induce electrophysiological changes, and neurotoxicity in rat cortical neurons [185]. Aβ protofibrils inhibit LTP-mediated synaptic plasticity in mouse hippocampus, thus impairing pivotal cognitive/behavioral functions such as spatial-temporal pattern separation and learning processes [186]. Aβ protofibrils can accumulate in glial cells, are associated with inflammatory responses, and are present in activated astrocytes in AD brains [187]. In cultured microglia in vitro, Aβ protofibrils are internalized by microglia more extensively than monomers [188]. They can further be released through microglia-derived microvesicles, possibly contributing to extracellular spread and neuroinflammation [189]. A peripheral immune response to the toxic Aβ protofibrils is suggested by the observation that the number of B cells producing auto-antibodies against Aβ protofibrils is significantly higher in AD patients than healthy controls [190].
Soluble protofibrils of various sizes have been identified in human brains and in brains from APP transgenic mice [191–193]. However, it is still unclear which particular aggregated soluble Aβ species confer toxicity. The detrimental agents may consist of high molecular weight and low molecular weight soluble Aβ aggregates with distinctive conformations.
An important model for the study of protofibrils is the Arctic APP mutant (APP E693G) which causes EOAD, and has been shown to specifically increase the rate of formation of these species [183, 184, 194]. In ArcSwe transgenic mice, a model with both the Swedish and the Arctic mutations and expressing abundant levels of protofibrils, cognitive deficits were shown to occur without plaques accumulation and concomitantly with the detection of early and widespread punctate (grain-like) intraneuronal Aβ-immunoreactive staining, as indicated by highly selective N-terminus 6E10 [epitope 1–16] and 3D6 [epitope 1–5] Aβ-antibodies. Such intraneuronal peptides are hypothesized to reflect intracellular non-fibrillar Aβ aggregates (protofibrils, given the underlying Artic mutation). Intraneuronal peptides predated parenchymal plaques accumulation [195]. Levels of Aβ protofibrils in the brain, but not of total Aβ, correlated with spatial learning, adding further evidence to the hypothesis of soluble protofibrils being the most toxic Aβ species [196]. The pool of soluble toxic Aβ assemblies consists of particles in the size range of 75–500 kDa [197]. Such species are selectively detected by the murine equivalent of BAN2401, mAb158, a protofibril-targeting antibody with low binding to monomers and insoluble Aβ fibrils [193, 198]. Importantly, mAb158 has been shown to significantly reduce protofibril levels in the brain and CSF from ArcSwe transgenic mice after chronic treatment [199].
Studies of AD patients with the Arctic mutation showed that they were, as expected, negative for fibrillar Aβ, as measured by the brain retention of the amyloid ligand Pittsburgh compound B ([11C]-PIB) with positron emission tomography (PET) [200]. A novel pathogenic APP mutation (E693del [Osaka]) was identified in Japanese pedigrees with AD, producing an Aβ variant—E22Δ—lacking Glu22 [201]. The E22Δ peptide variant was more resistant to proteolytic degradation and had the distinctive aggregation property of enhanced oligomerization (but no fibrillization) [201]. In vivo studies in rats demonstrated more effective hippocampal LTP inhibition by E22Δ peptide versus the wild-type Aβ peptides [201].
Taken together, and based on the current knowledge of underlying disease mechanisms, various soluble Aβ aggregates, and specifically, Aβ protofibrils, are particularly harmful and should be a compelling therapeutic target in AD.
Fibrils and plaques
Under physiological conditions, amyloidogenic proteins and peptides—such as Aβ—spontaneously aggregate into amyloid structures in a concentration-dependent manner. This phenomenon is general since, at the concentrations typically found in the cellular environment, proteins are metastable only in their native states [157, 158]. The conversion into the more stable amyloid state is prevented by the presence of high free energy barriers [157, 158]. In AD, specific brain micro-environmental conditions—including a vulnerable protein homeostasis system [202] and the abundance of a variety of poorly soluble proteins—appear to facilitate the formation of Aβ fibrils. Aβ fibrils form the characteristic cross-β-sheet structure of amyloid fibrils, in which Aβ peptides assemble into β-sheets with β-strands perpendicularly oriented to the long axis of the fibril and stabilized by hydrogen bonds [203–207].
Aβ fibrils are polymorphic with molecular structures that depend on the aggregation conditions [208]. Structurally distinct fibrils can have different levels of solubility, accumulation rates, and toxicity levels in neuronal cell cultures [206, 208]. Aβ fibrils, and to a lesser extent plaque, are associated with synaptic dysfunction in AD animal models and in AD patients. Fibrillar Aβ deposits are observed in the vicinity of disrupted neurites [209], of regions of decreased spine density, and in areas of neuronal loss [206, 210]. Moreover, primate models of AD show that microinjection of Aβ fibrillar assemblies in the cerebral cortex causes neurodegeneration, neurofibrillary pathology, and neuroinflammation [211]. These observations are consistent with the finding that Aβ fibril surfaces can catalyze the formation of Aβ oligomers [156], and Aβ oligomers have been observed surrounding Aβ fibrils [212].
Rates of recycling of the Aβ aggregation states
The interconversion of Aβ monomers, oligomers, protofibrils, and amyloid fibrils is implicated in AD pathogenesis [213]. By inspecting the nature of the amyloid fibrils structure, a continuous process of dissociation and re-association, resulting in the recycling of molecules within the fibril pool was observed [214]. Determining the kinetics of the individual association and dissociation reactions are challenging since the forward and reverse reactions to and from different Aβ aggregation states co-occur [155, 157, 213, 215]. Likewise, the heterogeneous set of oligomers consists mainly of unstable aggregations that can dissociate back to monomers but includes assembling species as well. Oligomers undergo repeated cycles of formation–dissociation before eventually turning into species that can grow into new fibrils [155].
Molecules making up Aβ1-40 fibrils recycle to a much greater extent than those of Aβ1-42. The rate constant for dissociation of molecules from the fibril is much higher for Aβ1-40 compared with Aβ1-42 [215]. Typically, the N-terminal region of Aβ contributes to improving fibrillar stability due to a gain of function mechanism at low pH, specifically at the pH range found within the endosomal and lysosomal pathways [216]. Along with pH, brain lipids play a critical function in destabilizing and rapidly re-solubilize mature Aβ fibers. This equilibrium is not reversed toward monomeric Aβ but, instead, toward soluble Aβ protofibrils [217]. A balance has been found between relatively inactive intermediate-sized Aβ aggregates and highly cytotoxic Aβ aggregates such as small oligomers and large protofibrils, which may have an impact on the role of amyloid plaques in the pathogenesis of cellular dysfunction in AD [181].
The Toxicity of the Aβ Pathway
Biomarker-based studies conducted in EOAD and LOAD have shown a temporal sequence between incipient Aβ pathophysiology, spreading of Aβ aggregation species and plaques through brain areas, and eventually increase of tau and neurodegeneration-based biological signatures [6, 8, 17, 54]. Although no causal effect has been established between Aβ pathophysiology and AD-related pathophysiological changes taking place at different temporal scales, a body of experimental and in-human studies indicates that Aβ aggregation species may exert a permissive/facilitating role on other pathophysiological pathways and/or unfold synergistically with them [8, 17, 76].
Aβ pathophysiology and tauopathy
The spatial-temporal relationship between the Aβ pathway and tau pathophysiology in AD, at both the molecular and macroscale, is critical to understanding AD pathogenesis and pathophysiological progression, and has gained momentum recently with the validation of several biomarkers charting different biological levels. The currently most accepted model indicates that Aβ pathophysiology may be an upstream pathophysiological event in AD and may function as a trigger/facilitator of downstream molecular pathways, including tau misfolding, tau-mediated toxicity, accumulation in tangles, and tau spreading that leads to cortical neurodegeneration (see Fig. 5) [218–222]. Genetic studies support biomarker-based observations and experimental studies which indicate a temporal Aβ–tau synergy where there is a pathophysiological sequence between aggregation of Aβ and tau-mediated toxicity [221].
Fig. 5. The evidence-driven experimental model of Aβ-tau synergy.

Accumulation of neurofibrillary tangles made up of tau (red) and amyloid plaques composed of amyloid-β (blue) coincides in the neocortical areas in the brain of Alzheimer’s disease subjects supporting amyloid-β dependent tau propagation across neocortical regions. Inter-neuronal spreading of tau (bottom) is enhanced in AD brains with both plaques and tangle build-up. Adapted with permission from ref. [221].
In a study of the Colombian ADAD kindred with PSEN1 E280A mutation carriers who were age- and sex-matched to mutation non-carriers, the onset of cortical Aβ deposition was around 15 years before dementia onset [223]. Notably, one mutation carrier exhibited tau-PET pathology in the medial temporal entorhinal-cortical area around 6 years before the estimated clinical symptoms onset, suggesting a 10-year gap between the development of Aβ pathology and tau-PET pathology [76]. Tau-PET pathology was not present in ADAD mutation carriers if Aβ cortical levels did not exceed the clinical disease threshold. Evidence suggests that the highest tau amounts detected by PET were found in those with the highest amyloid plaque pathology [76].
In sporadic AD, neuroimaging studies show that cortical tau-PET ligand retention is increased only in the presence of cortical Aβ accumulation and is associated with cortical thinning in AD [224]. Longitudinal studies show that a fast rate of antecedent Aβ accumulation predicts subsequent tau deposition in the inferior temporal cortex [225]. In the last 10 years, extensive research effort has been dedicated to understanding whether Aβ represents a trigger or a driver of AD, or both. Most of the studies report that tau markers, more than Aβ markers, significantly covary with neurodegeneration markers and long-term cognitive/functional outcome measures suggesting that Aβ pathophysiology triggers downstream pathways including tau-mediated toxicity and facilitates tau spreading [17, 214, 215]. These results, supported by experimental evidence (see below) suggest that AD is an Aβ-facilitated tauopathy leading to cognitive decline, MCI, and dementia. According to these PET-based investigations in both ADAD and LOAD, Aβ pathophysiology is likely to play a role in fostering the development of tau pathology.
Experimental models indicate that soluble forms of Aβ and tau synergize to exert synaptic toxicity independently of their assembly into plaques and tangles. Mouse models of AD show that modulation of tau seeding is associated with lower neurodegeneration rates and memory deficits without significant changes in the level of brain Aβ accumulation [226]. The triple transgenic mouse model (3xTg-AD) displays increasing extracellular Aβ accumulation in the neocortex and hippocampus before the seeding of tau into tangles [227].
Crossing familial AD-mutant APP mice with mutant MAPT transgenic mice leads to enhanced tau pathology and supports the occurrence of tangle-like alterations downstream of Aβ accumulation [220]. Injection of Aβ fibrils into P301L mutant tau transgenic mice’s brains triggers a five-fold rise in NFTs in cell bodies within the amygdala from where neurons project to the injection sites [222]. Crossing transgenic mice showing the spread of tau from the entorhinal cortex to other brain regions with APP/PS1 mice revealed that cortical amyloid deposition caused a dramatic increase in tau spreading to distal brain regions [228]. These findings support the hypothesis that cortical Aβ is permissive for the spread of tangles from the medial temporal lobe associated with cognitive decline in AD. According to the Braak neuropathological staging, such a pathophysiological model fits in the amyloid-independent progression of tau pathology [220].
Several findings deriving from mouse models converge toward an upstream role of Aβ on tau dyshomeostasis by facilitating/promoting tau conversion from a normal to a toxic state that can enhance Aβ toxicity via a feedback loop [228, 229]. Critical insights derive from in vitro studies. Tau hyperphosphorylation is promoted by synthetic Aβ oligomers and soluble extracts containing Aβ oligomers from AD brains (but not in non-AD brains) [230]. Treating healthy rat neurons in culture with soluble Aβ oligomers isolated from the AD cortex generated neuritic dystrophy and AD-type tau hyperphosphorylation. However, no dystrophy followed if tau expression was first knocked down [231]. Other similar studies suggested that Aβ, particularly soluble oligomers of Aβ1-42 [222], could trigger AD-type tau alterations, supporting the sequence that human genetics indicated. EOAD mutations in APP and PSEN1 promotes Aβ extracellular deposition, including Aβ plaques, in a human neural stem-cell-derived-3D culture system [232]. Cells expressing familial AD mutations exhibited high hyperphosphorylated tau levels in both the soma and neurites. In summary, there is extensive experimental evidence implying that inhibition of Aβ generation would be expected to decrease Aβ pathology and attenuates tauopathy [221].
Aβ pathophysiology and neuroinflammation
The spatiotemporal relationship between Aβ and glial cells, which are the critical orchestrators of neuroinflammation, is a rapidly expanding area of research to determine whether neuroinflammation can trigger and sustain incipient Aβ dyshomeostasis, or compensate for it, or carry out both in a stage-dependent manner. To date, most of the studies in vitro and in murine models of aging and AD support the notion that neuroinflammation is a key pathogenic event in AD etiology. The in-human exploration of neuroinflammatory mechanisms is still limited because of the early stage of development or the lack of clinical validation of relevant biomarkers.
Aβ species can interact with microglial and astrocytic pattern recognition receptors that initiate innate immunity where sustained microenvironment alterations—such as brain accumulation of Aβ—can trigger microglia “priming” [233]. Priming makes microglia susceptible to secondary inflammation stimulating factors, which can then amplify inflammatory reactions [233]. Two main phenotypical categories of microglia cells are present in the brain; resting (or quiescent) and activated. Activated microglia are typical pathophysiological features of AD and other neurodegenerative diseases [234–236].
Experimental AD models demonstrate that microglia surround plaques and fibrils, likely creating a physical barrier that can prevent their spreading and toxicity [237]. Microglia may contribute to Aβ clearance as well as limiting plaque growth and accumulation [238, 239]. Moreover, the dysregulation of microglia activity, including that from dystrophic microglia, may be a trigger and an aggravating factor of the seeding of aberrant protein aggregates in the brain [235, 236]. In AD mouse model, there is a transition from the resting to the activated states of microglia that may be the consequence of physiological stress, or Aβ triggered activation stimuli [240].
At a molecular level, inflammation is promoted by the presence of Aβ aggregates, including oligomers, protofibrils, and fibrils [241–244]. Microglia can bind to soluble Aβ oligomers, protofibrils, and insoluble Aβ fibrils through cell surface receptors, including the class A1 scavenger receptor (SCARA1), cell surface cluster of differentiation (CD) markers (CD36, CD14, CD47), α6β1 integrin, and Toll-like receptors [245–248]. Aβ species induce neuroinflammation and neurodegeneration by stimulating the microglia to release pro-inflammatory cytokines and interfering with the synthesis of anti-inflammatory cytokines such as transforming growth factor-beta1 (TGF-β1) [249–251]. TGF-β1 is a neurotrophic factor displaying both anti-inflammatory and neuroprotective actions stimulating Aβ clearance by microglia [252, 253]. TGF-β1 deficit exerts a key pro-inflammatory role in AD. A selective impairment of the TGF-β1 pathway is present in early AD, both in animal models and the human brain [242, 243, 254, 255]. Tumor necrosis factor-alpha (TNF-α) is a cytokine exerting a pivotal role in early pro-inflammatory processes in preclinical AD, as shown by both AD animal models and human longitudinal studies. In AD, TNF-α is chronically released by activated microglia, neurons, and astrocytes, and increased levels of extracellular Aβ stimulate its release [256–259]. TNF-α can stimulate γ-secretase activity, resulting in increased synthesis of Aβ peptides and a further increase in TNF-α release [249]. Animal studies highlight the association between TNF-α pathway blocking and histopathological marker reductions, such as Aβ plaques formation and microglial cell number decreases in the AD brain [260]. In humans, multiple studies detected elevated TNF-α levels in both MCI and AD dementia [260, 261].
In early AD pathogenesis, Aβ oligomers, protofibrils, and fibrils gather in the extracellular space and elicit a pathological cascade, eventually resulting in neuronal death [256–259]. Microglia eliminate these Aβ forms, as well as dying and dead cells through phagocytosis [262]. Aβ clearance can be stimulated by the release of numerous proteases participating in Aβ degradation [263]. In this regard, TREM2 modulates microglial functions by stimulating inflammatory cytokine production in response to Aβ plaques [264, 265]. The absence of TREM2 can enhance Aβ pathophysiology during early AD, which can be exacerbated by decreased phagocytic Aβ clearance in later disease stages [265], TREM2 variants reduce the Aβ phagocytic ability of microglia. TREM2 is the primary positive regulator of microglia phagocytosis, whereas CD33 is the negative regulator downstream to TREM2 [266, 267]. While additional in vivo studies will be necessary to clarify ApoE isoform-dependent function in cellular Aβ uptake and metabolism, there is evidence that microglial uptake of Aβ is facilitated by TREM2, ApoE, and CLU/ApoJ [268].
Along with microglia activation, hypertrophic reactive astrocytes can surround Aβ plaques as observed in human postmortem studies and in animal models [269, 270]. In AD, astrocytes release various pro-inflammatory molecules after exposure to Aβ (i.e., cytokines, interleukins (ILs), complement components, nitric oxide, and other cytotoxic compounds) and thus ultimately, amplify the neuroinflammatory response [260, 261, 271]. Human neuropathological studies conducted on AD brains report the presence of cytoplasmic inclusions of non-fibrillar Aβ in astrocytes, reflecting a phagocytic engulfment of extracellular Aβ deposits [260–262]. In addition, rodent models of AD indicate the astrocytes’ ability to take up and clear Aβ in individuals bearing cerebral fibrillar aggregates and diffuse plaques [260–262]. Conversely, compromise of astrocyte-mediated synaptic homeostasis is associated with increased Aβ plaque burden and synaptic terminal dystrophy [260–262]. This enhanced phagocytic activity may represent a compensatory mechanism to incipient increase in Aβ accumulation to neutralize its toxicity.
Aβ pathophysiology and the neurochemical systems in AD: the cholinergic system
There are complex and non-linear dynamics between Aβ homeostasis and the basal forebrain’s cholinergic system, one of the earliest brain anatomical structures to degenerate in AD. Both neuropathological and neuroimaging studies conducted in cognitively healthy older adults have reported correlations between increased BACE1 activity, Aβ accumulation with basal forebrain atrophy and loss of functional connectivity [272–276], and loss of projections to other cortical sub-cortical regions [277, 278]. Such an inverse correlation is likely to be aggravated by the presence of the APOE4 allele [279]. Furthermore, those progressing from MCI-to-dementia exhibited smaller baseline basal forebrain volumes and faster basal forebrain atrophy progression versus MCI-stable individuals [280]. These findings support previous evidence on the disruption of the cholinergic basal forebrain nuclei that may precede clinical onset [281].
Complex interactions exist at the molecular level. Muscarinic acetylcholine receptor agonists (mainly M1-type; to a lesser extent M3-type) can downregulate amyloidogenic and tau-generating pathways. M1 agonists may act as functional activators of protein kinase C (PKC) signaling, which, in turn, promotes a metabolic shift towards α-secretase activity by upregulating ADAM17 (also known as TNF-α-converting enzyme or TACE) [282]. Experiments in a mouse model of AD showed that the activation of α7 nicotinic receptors leads to downregulation of glycogen synthase kinase-3 (GSK3), a kinase involved in Aβ oligomer-induced inhibition of LTP as well as tau hyperphosphorylation [283, 284]. Possibly, α7 nicotinic activity and coupling of M1 to PKC lead to a downregulation of detrimental cell processes occurring in AD, such as GSK3-mediated tau hyperphosphorylation [285].
Aβ pathophysiology and the neurochemical systems in AD: the glutamatergic system
Glutamate excitotoxicity is considered one of the core molecular mechanisms of neurodegeneration in AD [286, 287]. The interaction between Aβ aggregates and glutamatergic neurotransmission is a possible critical event for the Aβ-induced disruption of excitatory synaptic transmission and plasticity associated with cognitive deficits [286, 287]. Aβ species can promote the dysregulation of N-methyl-D-aspartate (NMDA) and, to a lesser extent (α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid) (AMPA) ionotropic glutamate receptors (NMDARs and AMPARs) in the brain [286, 287].
Electrophysiological recordings on mouse hippocampal slices showed the ability of soluble Aβ oligomers to enhance the activation of NR2B/2A subunits of NMDARs while inhibiting glutamate uptake and recycling at the synapse [286, 288]. Consequently, a partial block of NMDA receptors coupled with a shift of the activation of NMDAR-dependent signaling cascades can take place, thus inducing LTD and downstream synaptic loss. The hippocampal overstimulation of Aβ oligomers is associated with decreased cell surface expression of NMDARs (downregulation via endocytosis) and alterations of dendritic spine density [286, 288, 289].
In AD, synaptic transmission and plasticity impairment is partially due to loss of AMPARs homeostasis with unbalanced trafficking and/or turnover [290]. AMPARs are the principal receptors mediating fast excitatory synaptic transmission in the mammalian brain [291]. Dynamic trafficking of AMPARs to and from synapses is a critical mechanism underlying the induction of synaptic plasticity. Overexpression of APP and high concentrations of soluble Aβ oligomers are associated with the downregulation of GluA1/2 subunits of AMPARs and downstream impairment of synaptic plasticity, spine loss, and memory deficits [292, 293]. As with the NMDARs, the mechanisms leading to AMPARs downregulation are not fully understood [294].
The spatial-temporal association between Aβ pathophysiology and brain networks damage
Multi-modal studies—conducted across the entire AD clinical continuum and combining molecular, structural and functional neuroimaging as well as fluid biological signatures—show a close spatial-temporal overlap between Aβ accumulation and distinct brain endophenotypes. The combination of amyloid-PET and volumetric/shape analysis MRI indicate that incipient higher rates of PET standardized update value ratios (SUVRs) are associated with hippocampal gray matter atrophy, an established biomarker of AD-type neurodegeneration, even in cognitively healthy individuals [6, 17, 295–297]. Such findings are consistent across studies investigating fluid biological signatures (CSF and or plasma Aβ species) and hippocampal volumes [298], experimental models of AD [227], human neuropathological data [6, 17, 18], and fluid biomarkers studies investigating dendritic proteins, like neurogranin, charting hippocampal disruption and synaptic dysfunction [299]. Hence, the overall evidence points toward hippocampal atrophy as a pathophysiological event observable as early as the incipient Aβ accumulation.
Selective brain structural damage—including at the hippocampal level—due to initial Aβ toxicity may occur downstream to ultrastructural changes that may underlie functional impairment [17]. In the limbic system [300, 301], the mesial temporal and superior parietal cortex [302, 303], activity change in the default-mode network (DMN) and the central executive (CEN) and the salience (SaN) networks [304, 305] is associated with worse cognitive trajectories in individuals displaying elevated Aβ burden [302, 303]. Early Aβ-associated reduction in DMN activity can take place before Aβ biomarkers (either PET or CSF) become positive, thus indicating a potential upstream toxic role of Aβ aggregation species in selectively vulnerable regions such those belonging to the DMN [306]. In prodromal stages of AD, loss of DMN functional connectivity is associated with neocortical and hippocampal gray matter volume loss, considered to reflect downstream neurodegeneration [302, 307]. As addressed above, whether this effect is necessarily tau-mediated or partially induced by Aβ species toxicity needs to be fully elucidated [308]. Eventually, lower DMN connectivity is associated with faster cortical shrinking, but only in those with elevated baseline Aβ-PET indexes [309]. This evidence in humans is supported by experimental models of aging and AD that point out the intrinsic bio-energetic vulnerability of the DMN neurons [300].
Multi-modal imaging studies show an increased Aβ-PET signal within the CEN and the SaN [300, 310, 311] throughout the biological continuum of AD and in aging. A spatial covariance between Aβ accumulation and connectivity and metabolism in the CEN and SaN (decreased) [300, 312] has been reported in AD [313–316]. The SaN plays a key role in the coordination of the DMN and the CEN, and whose functional impairment is associated with early learning and episodic memory deficits that characterize AD [317].
Discovery, Development, Validation, and Qualification of in vivo Biomarkers
CSF and blood-based biomarkers of Aβ: monomers
The three core AD CSF biomarkers Aβ42, total-tau (t-tau), and phosphorylated tau (p-tau) contribute diagnostically relevant information, especially during the early phases of the disease [318]. Low CSF Aβ1-42 concentrations display an average sensitivity greater than 90% for detecting cortical Aβ deposition of across all clinical stages of AD, including preclinical, prodromal, and dementia [319–322]. According to the current research diagnostic criteria, Aβ1-42 and tau (t-tau and p-tau) should be used in combination. The simultaneous presence of low Aβ1-42 and high t-tau and p-tau concentrations strongly suggests an AD diagnosis even at a prodromal stage, with a sensitivity of 90–95% and a specificity of about 90% [323]. The CSF tau/Aβ1-42 ratio represents a reliable tool for predicting cognitive decline in non-demented older adults and individuals with subjective cognitive decline, a risk factor for AD [324–326].
CSF Aβ1-42 has the potential to discriminate AD from frontotemporal lobar degeneration. Still, it shows significant overlap with other non-AD neurodegenerative diseases, specifically Lewy body disease, which is frequently characterized by concomitant Aβ pathology [299, 318]. The CSF matrix contains many different Aβ isoforms, of which Aβ1-40 is ~10 times more abundant than Aβ1-42 [327]. CSF levels of Aβ1-40 are unchanged in AD, but there is a reduction in the CSF Aβ1-42/Aβ1-40 ratio that is more marked than the decrease in Aβ1-42 alone. The CSF Aβ1-42/Aβ1-40 ratio improves diagnostic accuracy and has a better concordance with amyloid-PET positivity [299, 318, 328]. based on that CSF Aβ1-40 serves as a proxy for the ‘total’ Aβ production, thereby normalizing for differences in basal Aβ production between individuals [318, 328] or normalizing for between-individual differences in CSF dynamics or pre-analytical confounders affecting both Aβ1-42 and Aβ1-40. A marked reduction in CSF Aβ1-42 and the Aβ1-42/Aβ1-40 ratio has consistently been found in patients at different stages of AD [318, 329], and it supports the diagnostic differentiation between AD and non-AD clinical phenotypes.
From a biological standpoint, several factors—especially the APOE ε4 allele and sex—may influence the biomarker concentrations across large-scale populations [330]. However, it has been shown that the dependence of CSF Aβ1-42 levels on the APOE ε4 allele is due to carriers having more Aβ deposition; this association is not present in Aβ-negative young people [330]. From a methodological standpoint, pre-analytical and analytical factors may affect absolute levels of Aβ1-42 [331]. Unified protocols for CSF collection and handling have been published by the Alzheimer´s Association, such standardization will facilitate the introduction of globally accepted thresholds for CSF the AD biomarkers. Among the various recommendations and solutions put forward, the recent development of fully automated assays provides the basis for globally replicable and accepted cut-off points [299, 332]. Indeed, fully automated assays significantly minimize operator and lot-to-lot related variability (i.e., both intra-laboratory and inter-laboratory variability) [333].
CSF (and blood-based) biomarkers provide somewhat different information from Aβ-PET, with the latter showing the brain regional distribution of Aβ. In contrast, the former allows the simultaneous investigation of different pathophysiological mechanisms other than brain Aβ accumulation [299, 318, 331]. There is no consensus on whether CSF and PET biomarkers of Aβ accumulation become positive at the same preclinical disease stages. However, most studies show a small percentage of patients with abnormal CSF Aβ with negative Aβ-PET, and these progress to positive Aβ-PET, suggesting that CSF changes likely precede PET detection of cortical Aβ [334, 335]. A growing body of literature has demonstrated that the temporal dynamics of plasma Aβ mirror the CSF matrix [336].
Plasma biomarkers of brain Aβ accumulation
Blood-based biomarkers are expected to facilitate critical clinical solutions catalyzed by the global threat of AD. These biomarkers could be particularly suitable for the early screening and identification of individuals unlikely to develop AD-related pathophysiology and for increasing the probability that individuals with AD pathophysiology are being selected for further investigations using more specific, expensive and/or more invasive methods with reduced accessibility such as PET imaging or CSF assessment. The broad availability of blood-based biomarkers will facilitate a critical step towards a cost-, resource- and time-effective multi-step diagnostic work-up and accelerate the re-engineering of drug Research & Development (R&D) pipelines, from proof of pharmacology to clinical trial design.
The plasma Aβ1-42/Aβ1-40 ratio performs well in predicting the presence of brain amyloid as assessed through amyloid-PET across the AD clinical continuum. Moreover, reduced plasma Aβ1-42/Aβ1-40 is significantly associated with the overall increased risk for developing AD [336]. For measures of Aβ1-42/Aβ1-40 in blood, a variety of techniques have been employed, including single-molecule arrays (SiMoA) technology [337–339], immunoprecipitation coupled with mass spectrometry (IP-MS) [340], IP coupled with liquid chromatography-mass spectrometry [341], immune-magnetic reduction [342], and stable isotope labeling kinetics protocols [343, 344]. While these analytical methods provide similar positive results in MCI, dementia, and cognitively healthy individuals at risk for AD, IP-MS methods have higher concordance with brain amyloidosis than Simoa assays [344]. Some of the approaches, however, are not scalable and/or have high variability. Hence, like CSF, fully automated assays offer a viable solution for blood-based biomarkers of brain Aβ accumulation [345, 346].
Aβ oligomers in bodily fluids
With accumulating evidence of soluble Aβ oligomers and protofibrils being the critical toxic species within the Aβ pathway, the accurate detection and quantification of these species could prove useful for diagnostic and therapeutic context-of-use [166]. Different technologies are used for their detection/measurement, including enzyme-linked immunosorbent assay (ELISA), flow cytometry, nanoscale optical biosensors, amplified plasmonic exosome (APEX), single particle detection, nanoparticle-based detection, single-molecule fluorescence microscopy, a protein misfolding cyclic amplification assay method, and an assay based on the monoclonal antibody BAN50 [344, 347–351]. Most of these studies show a trend toward high CSF levels of Aβ oligomers in AD patients compared with healthy controls, but have proven controversial and, in some cases, with no clear discrimination between the groups, especially when involving prodromal AD. In many studies, oligomer concentrations were higher in MCI than AD or cognitively healthy individuals; there was significant overlap among concentrations in different populations [318, 352]. In some studies, patients with MCI who later converted to AD had increased Aβ oligomers CSF concentrations on a group level, but several samples had undetectable levels [353].
Assays to measure Aβ oligomers in plasma are under development [354–358]. A recently developed ELISA-Multimer Detection System (MDS), capable of differentiating multimers from their cellular monomers, detected higher plasma Aβ oligomers concentrations in AD versus healthy controls (HC) [359]. Before adapting MDS in clinical settings, further studies are needed to validate plasma Aβ oligomer concentration and use of the assay for screening patients, monitoring longitudinal changes across the course of AD, or determining the efficacy of Aβ-targeting drugs.
A study has been performed in human bodily fluids to assesses whether AD patients have higher levels of protofibrils compared with cognitively healthy controls. An enzyme-linked immunospot (ELISpot)-based investigation reported that AD patients display a significantly higher number of cells producing antibodies toward Aβ42 protofibrils compared to healthy controls [190]. Although the study did not directly assess plasma levels of protofibrils, it showed there is a specific immune response to the toxic Aβ protofibrils, which is significantly increased in AD patients [190].
PET radioligands of brain amyloid plaques
Molecular imaging in amyloid in AD is characterized by radiopharmaceuticals binding to aggregated insoluble fibrillary forms of cortical Aβ visualized via PET (Aβ-PET) [360]. Aβ neuroimaging allows (1) in vivo assessment of global and regional deposition of amyloid plaques, (2) exploration of the spatiotemporal relationships between brain Aβ accumulation (see Fig. 6), other pathomechanistic alterations of AD, and clinical outcomes, and (3) assessment of target engagement and treatment effect in anti-Aβ clinical trials as a quantifiable biomarker [361]. The FDA label for PET imaging emphasizes that a low Aβ-PET burden is incompatible with AD as the cause of the cognitive decline. Most older cognitively unimpaired or MCI individuals with low Aβ-PET burden will not develop or progress to AD in their lifetime [362]. Such a recommendation highlights the importance of employing a panel of biomarkers along with PET as prognostic indicators.
Fig. 6. Techniques of in vivo Alzheimer’s disease amyloid-β pathway staging, along the clinical continuum, based on molecular imaging and innovative algorithms.

Neocortical distribution of [18F]-florbetapir is shown in a composite representation according to Aβ stages. Early composite (positive in stage 1 in green; left), intermediate composite (positive in stage 2 in blue; middle) and late composite (positive in stage 3 in red; right) can allow global and regional assessment of amyloid plaque deposition. Adapted with permission from: ref. [393].
The first validated radiopharmaceutical developed for Aβ-PET was [11C]-PiB, a derivative of the amyloid-binding fluorescent dye thioflavin-T, which is a small molecule known to bind amyloid proteins aggregated into a cross β structure [363, 364]. [11C]-PiB has a short half-life, which limits its use to clinical centers with an on-site cyclotron and specialized radiochemistry expertise. Recently, the FDA has approved fluorine-18 [18F]-labeled compounds—[18F]-Florbetapir ([18F]-AV-45, AmyvidTM), [18F]-Florbetaben ([18F]-FBB, NeuraceqTM), [18F]-Flutemetamol ([18F]-FMT, VizamylTM)—that have a 110-min half-life, thus allowing for centralized production and regional distribution [365, 366].
Multi-center studies, systematic reviews, and meta-analyses of the PET radiotracers demonstrate substantial corroborating data for the capability Aβ-PET to differentiate AD patients from healthy controls (HC) and to predict the likelihood of progression to AD dementia in patients with MCI [367–369]. The results for sensitivity range from 89 to 97% by all study subgroups (HC versus AD dementia and HC versus MCI versus AD dementia individuals). The values ranged more widely, from 63% to 93%, for specificity [367–369]. Unlike neuroimaging of neurodegeneration and tau pathophysiology, the pattern of Aβ-PET deposition across the AD clinical spectrum (typical and atypical variants) does not show much regional differences [370].
Amyloid-PET imaging is primarily approved to be used as a binary visual reading approach (ordinal classification of positive or negative scans) to distinguish individuals with no/sparse Aβ plaques from those with moderate-to-frequent plaques. Recently, automatized pipelines that allow standardized quantitative measures have been developed. Quantitative studies enable regional investigation of brain Aβ deposition, allowing for tracking spatiotemporal evolution throughout the AD clinical continuum [13, 371]. These findings demonstrate a predictable regional sequence that may be used to stage an individual’s progress of in vivo cerebral amyloid pathology [371]. Regional Aβ staging based on amyloid-PET imaging has the potential to predict progression to cognitive impairment and dementia in individuals with preclinical and prodromal AD, with the most advanced amyloid stages able to identify high-risk groups of progression from MCI to dementia [371, 372]. For quantitative purposes, the three different tracers show considerable variability when measured using the typical SUVRs. To improve the comparability of the retention measurements across tracers and across centers, the Centiloid method has been proposed [360]. This method linearly scales the measure of a particular tracer from 0 to 100 scale, where “0” represents the average tracer retention in young controls, and “100” corresponds to the average racer retention in typical AD patients at the dementia stage [360].
Although radiopharmaceuticals target fibrillar Aβ, this does not represent a specific marker for a particular pool of Aβ, but for the global cerebral Aβ load [373]. For instance, AD patients with APP Arctic mutation or the Osaka variant show markedly low cerebral deposition of plaques as assessed through the Aβ-PET global SUVR [183, 200, 201]. On this basis, [11C]-PiB was tested for its ability to bind Aβ protofibrils and oligomers. Tritiated PiB ([3H]-PiB) bound strongly to Aβ1-42 fibrils and satisfactorily to protofibrils [374]. An earlier study also showed that PiB has three-fold less affinity for soluble forms than insoluble forms [375].
Concerning radiolabeled antibodies, recombinant antibody-based radioligand [124I]A3 could target soluble Aβ protofibrils [376, 377]. The radioligand di-scFv [124I]3D6-8D3 has a larger dynamic range and sensitivity for measuring more soluble forms of Aβ than [11C]-PiB [378]. Hence, antibody-based radioligands might visualize more subtle and earlier Aβ alterations than the conventional ones. There are several issues concerning this developing approach, including (i) technical difficulty to get antibodies into the brain in enough quantities to attain useful neuroimaging-based clinical information, (ii) the amount of soluble pools of Aβ is ~1% of all amount of Aβ in the brain and do not last long enough in the soluble form to allow imaging in the CNS, (iii) antibodies enter the brain very slowly, and maximal concentrations might take more than 1 day restricting its use to radiolabeling with very long T1/2 radioisotopes [379]. Future strategies to circumvent these physiological barriers include the use of nanoparticles, exosomes, or molecular chaperones that facilitate transport across the BBB.
Conclusions
Over the recent decades, translational and multi-disciplinary studies—from (epi)genetic, to biological, and to biomarker-based clinical investigations—have contributed to unveiling the biochemical, physiological, and pathophysiological features of the Aβ pathway, including its spatial and temporal dynamics throughout the AD continuum. All point to the Aβ pathway as a hallmark of disease pathophysiology rather than a passive readout of the disease process. As discussed above, anatomical and biomarker-based studies of familial and sporadic AD provide critical genetic and molecular evidence about the initiation of the Aβ pathway decades before the onset of the symptoms and upstream to other pathophysiological hallmarks of AD.
These advances in biology have culminated in the identification of tangible therapeutic molecular targets for AD in order to slow disease progression at the earliest possible clinical and preclinical stages. Progress in drug R&D has also been accelerated by the validation of Aβ biomarkers-based outcomes and endpoints and for different context(s)-of-use, including patient diagnosis for clinical trials, target engagement of drug candidates, and proof-of-mechanism. Implementation of biomarker-guided pipelines contributes to explaining why the first generation of compounds targeting Aβ aggregation species and with putative disease-modifying effect reached late-stage development and exhibited phase II and phase III failures. However, the field needs to fully uncover the physiological functions of the Aβ pathway, as well as the upstream molecular orchestrators of its dyshomeostasis in AD. Aβ homeostasis undergoes a complex interplay consisting of highly conserved feedback loops and interactions among an array of quality control mechanisms and protein clearance pathways across cells, tissues, and body systems. Understanding this hierarchical organization across tissues and body systems and its decline with aging and in an individual, genetically determined fashion will be essential to comprehensively target the Aβ cycle for preventive strategies. New multi-modal imaging integrative approaches coupled with molecular imaging and fluid biomarkers hold the potential to unravel the spatial and temporal coordinates the Aβ pathways dynamics and to map the critical genetic and biological factors influencing sub-population clinical and pathophysiological trajectories.
Within this conceptual framework, Aβ-oriented therapies will be more and more scaled to the disease stage and biological inter-individual differences for time-sensitive and effective pathway (a mechanism)-based preventive strategies for AD, aligned with the precision medicine paradigm.
Acknowledgements
Editorial support was provided by JD Cox, PhD of Mayville Medical Communications (funded by Eisai Inc).
Author contributions
HH conceptualized the article, performed a critical review of the literature, wrote and revised the manuscript. MC contributed to the writing and revision of the article and supervised artwork. JH, KB, CC, GP, SHK, VLV, PA, MV, TI, CLM, LL, and JLC supported the critical review of the literature, the writing, and/or the revision of the article. AV conceptualized the article, performed a critical review of the literature, wrote and revised the manuscript. All authors read and approved the final manuscript.
Competing interests
HH is an employee of Eisai Inc. HH serves as Senior Associate Editor for the Journal Alzheimer’s & Dementia and does not receive any fees or honoraria since May 2019; before May 2019 he had received lecture fees from Servier, Biogen and Roche, research grants from Pfizer, Avid, and MSD Avenir (paid to the institution), travel funding from Eisai, Functional Neuromodulation, Axovant, Eli Lilly and company, Takeda and Zinfandel, GE-Healthcare and Oryzon Genomics, consultancy fees from Qynapse, Jung Diagnostics, Cytox Ltd., Axovant, Anavex, Takeda and Zinfandel, GE Healthcare, Oryzon Genomics, and Functional Neuromodulation, and participated in scientific advisory boards of Functional Neuromodulation, Axovant, Eisai, Eli Lilly and company, Cytox Ltd., GE Healthcare, Takeda and Zinfandel, Oryzon Genomics and Roche Diagnostics. He is co-inventor in the following patents as a scientific expert and has received no royalties: • In Vitro Multiparameter Determination Method for The Diagnosis and Early Diagnosis of Neurodegenerative Disorders Patent Number: 8916388. • In Vitro Procedure for Diagnosis and Early Diagnosis of Neurodegenerative Diseases Patent Number: 8298784. • Neurodegenerative Markers for Psychiatric Conditions Publication Number: 20120196300. • In Vitro Multiparameter Determination Method for The Diagnosis and Early Diagnosis of Neurodegenerative Disorders Publication Number: 20100062463. • In Vitro Method for The Diagnosis and Early Diagnosis of Neurodegenerative Disorders Publication Number: 20100035286. • In Vitro Procedure for Diagnosis and Early Diagnosis of Neurodegenerative Diseases Publication Number: 20090263822. • In Vitro Method for The Diagnosis of Neurodegenerative Diseases Patent Number: 7547553. • CSF Diagnostic in Vitro Method for Diagnosis of Dementias and Neuroinflammatory Diseases Publication Number: 20080206797. • In Vitro Method for The Diagnosis of Neurodegenerative Diseases Publication Number: 20080199966. • Neurodegenerative Markers for Psychiatric Conditions Publication Number: 20080131921. • Method for diagnosis of dementias and neuroinflammatory diseases based on an increased level of procalcitonin in cerebrospinal fluid: Publication number: United States Patent 10921330. https://www.freepatentsonline.com/10921330.html. KB is supported by the Swedish Research Council (#2017-00915), the Alzheimer Drug Discovery Foundation (ADDF), USA (#RDAPB-201809-2016615), the Swedish Alzheimer Foundation (#AF-742881), Hjärnfonden, Sweden (#FO2017-0243), the Swedish state under the agreement between the Swedish government and the County Councils, the ALF-agreement (#ALFGBG-715986), and European Union Joint Program for Neurodegenerative Disorders (JPND2019-466-236). KB has served as a consultant, at advisory boards, or at data monitoring committees for Abcam, Axon, Biogen, Julius Clinical, Lilly, MagQu, Novartis, Roche Diagnostics, and Siemens Healthineers, and is a co-founder of Brain Biomarker Solutions in Gothenburg AB (BBS), which is a part of the GU Ventures Incubator Program. JLC has provided consultation to Acadia, Actinogen, Alkahest, Alzheon, Annovis, Avanir, Axsome, Biogen, Cassava, Cerecin, Cerevel, Cortexyme, Cytox, EIP Pharma, Eisai, Foresight, GemVax, Genentech, Green Valley, Grifols, Karuna, Merck, Novo Nordisk, Otsuka, Resverlogix, Roche, Samumed, Samus, Signant Health, Suven, and United Neuroscience pharmaceutical and assessment companies. Dr. Cummings has stock options in ADAMAS, AnnovisBio, MedAvante, BiOasis. Dr. Cummings owns the copyright of the Neuropsychiatric Inventory. Dr Cummings is supported by Keep Memory Alive (KMA); NIGMS grant P20GM109025; NINDS grant U01NS093334; and NIA grant R01AG053798. MC is an employee of Eisai Inc. AV is an employee of Eisai Inc. He does not receive any fees or honoraria since November 2019. Before November 2019 he had received lecture honoraria from Roche, MagQu LLC, and Servier.
Footnotes
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Contributor Information
Harald Hampel, Email: Harald_Hampel@eisai.com.
Andrea Vergallo, Email: Andrea_Vergallo@eisai.com.
References
- 1.Prince MJ, Wimo A, Guerchet MM, Ali GC, Wu Y-T, Prina M. World Alzheimer Report 2015 - The Global Impact of Dementia: An analysis of prevalence, incidence, cost and trends. London: Alzheimer’s Disease International, 2015. 84 p.
- 2.2020 Alzheimer’s disease facts and figures. Alzheimer’s & Dement. 2020;16:391–460. [DOI] [PubMed]
- 3.Hebert LE, Weuve J, Scherr PA, Evans DA. Alzheimer disease in the United States (2010-2050) estimated using the 2010 census. Neurology. 2013;80:1778–83. doi: 10.1212/WNL.0b013e31828726f5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Prince M, Knapp M, Guerchet M, McCrone P, Prina M, Comas-Herrera A et al. Dementia UK: Second Edition—Overview. Alzheimer’s Soc. 2014.
- 5.Canevelli M, Lacorte E, Cova I, Zaccaria V, Valletta M, Raganato R, et al. Estimating dementia cases amongst migrants living in Europe. Eur J Neurol. 2019;26:1191–9. doi: 10.1111/ene.13964. [DOI] [PubMed] [Google Scholar]
- 6.Dubois B, Feldman HH, Jacova C, Hampel H, Molinuevo JL, Blennow K, et al. Advancing research diagnostic criteria for Alzheimer’s disease: the IWG-2 criteria. Lancet Neurol. 2014;13:614–29. doi: 10.1016/S1474-4422(14)70090-0. [DOI] [PubMed] [Google Scholar]
- 7.Hampel H, Cummings J, Blennow K, Gao P, Jack CR, Vergallo A. Developing the ATX(N) classification for use across the Alzheimer disease continuum. Nat Rev Neurol. 2021; 10.1038/s41582-021-00520-w. [DOI] [PubMed]
- 8.Aisen PS, Vellas B, Hampel H. Moving towards early clinical trials for amyloid-targeted therapy in Alzheimer’s disease. Nat Rev Drug Discov. 2013;12:324. doi: 10.1038/nrd3842-c1. [DOI] [PubMed] [Google Scholar]
- 9.Blennow K, de Leon MJ, Zetterberg H. Alzheimer’s disease. Lancet. 2006;368:387–403. doi: 10.1016/S0140-6736(06)69113-7. [DOI] [PubMed] [Google Scholar]
- 10.Masters CL, Simms G, Weinman NA, Multhaup G, McDonald BL, Beyreuther K. Amyloid plaque core protein in Alzheimer disease and Down syndrome. Proc Natl Acad Sci USA. 1985;82:4245–9. doi: 10.1073/pnas.82.12.4245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kang J, Lemaire HG, Unterbeck A, Salbaum JM, Masters CL, Grzeschik KH, et al. The precursor of Alzheimer’s disease amyloid A4 protein resembles a cell-surface receptor. Nature. 1987;325:733–6. doi: 10.1038/325733a0. [DOI] [PubMed] [Google Scholar]
- 12.Cras P, Kawai M, Lowery D, Gonzalez-DeWhitt P, Greenberg B, Perry G. Senile plaque neurites in Alzheimer disease accumulate amyloid precursor protein. Proc Natl Acad Sci USA. 1991;88:7552–6. doi: 10.1073/pnas.88.17.7552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Thal DR, Rüb U, Orantes M, Braak H. Phases of A beta-deposition in the human brain and its relevance for the development of AD. Neurology. 2002;58:1791–1800. doi: 10.1212/wnl.58.12.1791. [DOI] [PubMed] [Google Scholar]
- 14.Hardy JA, Higgins GA. Alzheimer’s disease: the amyloid cascade hypothesis. Science. 1992;256:184–5. doi: 10.1126/science.1566067. [DOI] [PubMed] [Google Scholar]
- 15.Hardy J, Selkoe DJ. The amyloid hypothesis of Alzheimer’s disease: progress and problems on the road to therapeutics. Science. 2002;297:353–6. doi: 10.1126/science.1072994. [DOI] [PubMed] [Google Scholar]
- 16.Jansen WJ, Ossenkoppele R, Knol DL, Tijms BM, Scheltens P, Verhey FRJ, et al. Prevalence of cerebral amyloid pathology in persons without dementia: a meta-analysis. JAMA. 2015;313:1924–38. doi: 10.1001/jama.2015.4668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Jack CRJ, Bennett DA, Blennow K, Carrillo MC, Dunn B, Haeberlein SB, et al. NIA-AA research framework: toward a biological definition of Alzheimer’s disease. Alzheimers Dement. 2018;14:535–62. doi: 10.1016/j.jalz.2018.02.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Selkoe DJ, Hardy J. The amyloid hypothesis of Alzheimer’s disease at 25 years. EMBO Mol Med. 2016;8:595–608. doi: 10.15252/emmm.201606210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Mawuenyega KG, Sigurdson W, Ovod V, Munsell L, Kasten T, Morris JC, et al. Decreased clearance of CNS beta-amyloid in Alzheimer’s disease. Science. 2010;330:1774. doi: 10.1126/science.1197623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Levy-Lahad E, Wasco W, Poorkaj P, Romano DM, Oshima J, Pettingell WH, et al. Candidate gene for the chromosome 1 familial Alzheimer’s disease locus. Science. 1995;269:973–7. doi: 10.1126/science.7638622. [DOI] [PubMed] [Google Scholar]
- 21.Rogaev EI, Sherrington R, Rogaeva EA, Levesque G, Ikeda M, Liang Y, et al. Familial Alzheimer’s disease in kindreds with missense mutations in a gene on chromosome 1 related to the Alzheimer’s disease type 3 gene. Nature. 1995;376:775–8. doi: 10.1038/376775a0. [DOI] [PubMed] [Google Scholar]
- 22.Goate A, Chartier-Harlin MC, Mullan M, Brown J, Crawford F, Fidani L, et al. Segregation of a missense mutation in the amyloid precursor protein gene with familial Alzheimer’s disease. Nature. 1991;349:704–6. doi: 10.1038/349704a0. [DOI] [PubMed] [Google Scholar]
- 23.Jansen IE, Savage JE, Watanabe K, Bryois J, Williams DM, Steinberg S, et al. Genome-wide meta-analysis identifies new loci and functional pathways influencing Alzheimer’s disease risk. Nat Genet. 2019;51:404–13. doi: 10.1038/s41588-018-0311-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Pimenova AA, Raj T, Goate AM. Untangling genetic risk for Alzheimer’s disease. Biol Psychiatry. 2018;83:300–10. doi: 10.1016/j.biopsych.2017.05.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Wisniewski KE, Dalton AJ, McLachlan C, Wen GY, Wisniewski HM. Alzheimer’s disease in Down’s syndrome: clinicopathologic studies. Neurology. 1985;35:957–61. doi: 10.1212/wnl.35.7.957. [DOI] [PubMed] [Google Scholar]
- 26.St George-Hyslop PH, Tanzi RE, Polinsky RJ, Haines JL, Nee L, Watkins PC, et al. The genetic defect causing familial Alzheimer’s disease maps on chromosome 21. Science. 1987;235:885–90. doi: 10.1126/science.2880399. [DOI] [PubMed] [Google Scholar]
- 27.Cruts M, Theuns J, Van Broeckhoven C. Locus-specific mutation databases for neurodegenerative brain diseases. Hum Mutat. 2012;33:1340–4. doi: 10.1002/humu.22117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Hooli BV, Mohapatra G, Mattheisen M, Parrado AR, Roehr JT, Shen Y, et al. Role of common and rare APP DNA sequence variants in Alzheimer disease. Neurology. 2012;78:1250–7. doi: 10.1212/WNL.0b013e3182515972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Hartley D, Blumenthal T, Carrillo M, DiPaolo G, Esralew L, Gardiner K, et al. Down syndrome and Alzheimer’s disease: common pathways, common goals. Alzheimers Dement. 2015;11:700–9. doi: 10.1016/j.jalz.2014.10.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Cai XD, Golde TE, Younkin SG. Release of excess amyloid beta protein from a mutant amyloid beta protein precursor. Science. 1993;259:514–6. doi: 10.1126/science.8424174. [DOI] [PubMed] [Google Scholar]
- 31.Citron M, Teplow DB, Selkoe DJ. Generation of amyloid beta protein from its precursor is sequence specific. Neuron. 1995;14:661–70. doi: 10.1016/0896-6273(95)90323-2. [DOI] [PubMed] [Google Scholar]
- 32.Di Fede G, Catania M, Morbin M, Rossi G, Suardi S, Mazzoleni G, et al. A recessive mutation in the APP gene with dominant-negative effect on amyloidogenesis. Science. 2009;323:1473–7. doi: 10.1126/science.1168979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Di Fede G, Di Catania M, Morbin M, Giaccone G, Moro ML, Ghidoni R, et al. Good gene, bad gene: new APP variant may be both. Prog Neurobiol. 2012;99:281–92. doi: 10.1016/j.pneurobio.2012.06.004. [DOI] [PubMed] [Google Scholar]
- 34.Tanzi RE. The genetics of Alzheimer disease. Cold Spring Harb Perspect Med. 2012;2:a006296. [DOI] [PMC free article] [PubMed]
- 35.Maloney JA, Bainbridge T, Gustafson A, Zhang S, Kyauk R, Steiner P, et al. Molecular mechanisms of Alzheimer disease protection by the A673T allele of amyloid precursor protein. J Biol Chem. 2014;289:30990–31000. doi: 10.1074/jbc.M114.589069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Bentahir M, Nyabi O, Verhamme J, Tolia A, Horré K, Wiltfang J, et al. Presenilin clinical mutations can affect gamma-secretase activity by different mechanisms. J Neurochem. 2006;96:732–42. doi: 10.1111/j.1471-4159.2005.03578.x. [DOI] [PubMed] [Google Scholar]
- 37.Sun L, Zhou R, Yang G, Shi Y. Analysis of 138 pathogenic mutations in presenilin-1 on the in vitro production of Aβ42 and Aβ40 peptides by γ-secretase. Proc Natl Acad Sci USA. 2017;114:E476–85. doi: 10.1073/pnas.1618657114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Cai Y, An SSA, Kim S. Mutations in presenilin 2 and its implications in Alzheimer’s disease and other dementia-associated disorders. Clin Inter Aging. 2015;10:1163–72. doi: 10.2147/CIA.S85808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Sims R, Hill M, Williams J. The multiplex model of the genetics of Alzheimer’s disease. Nat Neurosci. 2020;23:311–22. doi: 10.1038/s41593-020-0599-5. [DOI] [PubMed] [Google Scholar]
- 40.Lambert J, Ibrahim-Verbaas CA, Harold D, Naj AC, Sims R, Bellenguez C, et al. Meta-analysis of 74,046 individuals identifies 11 new susceptibility loci for Alzheimer’s disease. Nat Genet. 2013;45:1452–8. doi: 10.1038/ng.2802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Poirier J, Davignon J, Bouthillier D, Kogan S, Bertrand P, Gauthier S. Apolipoprotein E polymorphism and Alzheimer’s disease. Lancet. 1993;342:697–9. doi: 10.1016/0140-6736(93)91705-q. [DOI] [PubMed] [Google Scholar]
- 42.Corder EH, Saunders AM, Strittmatter WJ, Schmechel DE, Gaskell PC, Small GW, et al. Gene dose of apolipoprotein E type 4 allele and the risk of Alzheimer’s disease in late onset families. Science. 1993;261:921–3. doi: 10.1126/science.8346443. [DOI] [PubMed] [Google Scholar]
- 43.Liu C-C, Liu C-C, Kanekiyo T, Xu H, Bu G. Apolipoprotein E and Alzheimer disease: risk, mechanisms and therapy. Nat Rev Neurol. 2013;9:106–18. doi: 10.1038/nrneurol.2012.263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Caselli RJ, Dueck AC, Osborne D, Sabbagh MN, Connor DJ, Ahern GL, et al. Longitudinal modeling of age-related memory decline and the APOE epsilon4 effect. N. Engl J Med. 2009;361:255–63. doi: 10.1056/NEJMoa0809437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Nyarko JNK, Quartey MO, Pennington PR, Heistad RM, Dea D, Poirier J, et al. Profiles of beta-amyloid peptides and key secretases in brain autopsy samples differ with sex and APOE epsilon4 status: impact for risk and progression of Alzheimer disease. Neuroscience. 2018;373:20–36. doi: 10.1016/j.neuroscience.2018.01.005. [DOI] [PubMed] [Google Scholar]
- 46.Mahley RW. Apolipoprotein E: cholesterol transport protein with expanding role in cell biology. Science. 1988;240:622–30. doi: 10.1126/science.3283935. [DOI] [PubMed] [Google Scholar]
- 47.Kok E, Haikonen S, Luoto T, Huhtala H, Goebeler S, Haapasalo H, et al. Apolipoprotein E-dependent accumulation of Alzheimer disease-related lesions begins in middle age. Ann Neurol. 2009;65:650–7. doi: 10.1002/ana.21696. [DOI] [PubMed] [Google Scholar]
- 48.Polvikoski T, Sulkava R, Haltia M, Kainulainen K, Vuorio A, Verkkoniemi A, et al. Apolipoprotein E, dementia, and cortical deposition of beta-amyloid protein. N. Engl J Med. 1995;333:1242–7. doi: 10.1056/NEJM199511093331902. [DOI] [PubMed] [Google Scholar]
- 49.Schmechel DE, Saunders AM, Strittmatter WJ, Crain BJ, Hulette CM, Joo SH, et al. Increased amyloid beta-peptide deposition in cerebral cortex as a consequence of apolipoprotein E genotype in late-onset Alzheimer disease. Proc Natl Acad Sci USA. 1993;90:9649–53. doi: 10.1073/pnas.90.20.9649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Sunderland T, Mirza N, Putnam KT, Linker G, Bhupali D, Durham R, et al. Cerebrospinal fluid beta-amyloid1-42 and tau in control subjects at risk for Alzheimer’s disease: the effect of APOE epsilon4 allele. Biol Psychiatry. 2004;56:670–6. doi: 10.1016/j.biopsych.2004.07.021. [DOI] [PubMed] [Google Scholar]
- 51.Morris JC, Roe CM, Xiong C, Fagan AM, Goate AM, Holtzman DM, et al. APOE predicts amyloid-beta but not tau Alzheimer pathology in cognitively normal aging. Ann Neurol. 2010;67:122–31. doi: 10.1002/ana.21843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Jack CR, Jr, Wiste HJ, Weigand SD, Knopman DS, Vemuri P, Mielke MM, et al. Age, sex, and APOE ε4 effects on memory, brain structure, and β-amyloid across the adult life span. JAMA Neurol. 2015;72:511–9. doi: 10.1001/jamaneurol.2014.4821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Lim YY, Ellis KA, Ames D, Darby D, Harrington K, Martins RN, et al. Aβ amyloid, cognition, and APOE genotype in healthy older adults. Alzheimers Dement. 2013;9:538–45. doi: 10.1016/j.jalz.2012.07.004. [DOI] [PubMed] [Google Scholar]
- 54.Sperling RA, Donohue MC, Raman R, Sun C-K, Yaari R, Holdridge K, et al. Association of factors with elevated amyloid burden in clinically normal older individuals. JAMA Neurol. 2020;77:735–45. doi: 10.1001/jamaneurol.2020.0387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Lim YY, Laws SM, Villemagne VL, Pietrzak RH, Porter T, Ames D, et al. Aβ-related memory decline in APOE ε4 noncarriers: implications for Alzheimer disease. Neurology. 2016;86:1635–42. doi: 10.1212/WNL.0000000000002604. [DOI] [PubMed] [Google Scholar]
- 56.Verghese PB, Castellano JM, Holtzman DM. Apolipoprotein E in Alzheimer’s disease and other neurological disorders. Lancet Neurol. 2011;10:241–52. doi: 10.1016/S1474-4422(10)70325-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Lim YY, Mormino EC. APOE genotype and early β-amyloid accumulation in older adults without dementia. Neurology. 2017;89:1028–34. doi: 10.1212/WNL.0000000000004336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Lim YY, Kalinowski P, Pietrzak RH, Laws SM, Burnham SC, Ames D, et al. Association of β-amyloid and apolipoprotein E ε4 with memory decline in preclinical Alzheimer disease. JAMA Neurol. 2018;75:488–94. doi: 10.1001/jamaneurol.2017.4325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Reiman EM, Chen K, Liu X, Bandy D, Yu M, Lee W, et al. Fibrillar amyloid-beta burden in cognitively normal people at 3 levels of genetic risk for Alzheimer’s disease. Proc Natl Acad Sci USA. 2009;106:6820–5. doi: 10.1073/pnas.0900345106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Riddell DR, Zhou H, Atchison K, Warwick HK, Atkinson PJ, Jefferson J, et al. Impact of apolipoprotein E (ApoE) polymorphism on brain ApoE levels. J Neurosci. 2008;28:11445–53. doi: 10.1523/JNEUROSCI.1972-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Sullivan PM, Han B, Liu F, Mace BE, Ervin JF, Wu S, et al. Reduced levels of human apoE4 protein in an animal model of cognitive impairment. Neurobiol Aging. 2011;32:791–801. doi: 10.1016/j.neurobiolaging.2009.05.011. [DOI] [PubMed] [Google Scholar]
- 62.Huang Y-WA, Zhou B, Wernig M, Südhof TC. ApoE2, ApoE3, and ApoE4 differentially stimulate APP transcription and Aβ secretion. Cell. 2017;168:e21. doi: 10.1016/j.cell.2016.12.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Lin Y-T, Seo J, Gao F, Feldman HM, Wen H-L, Penney J, et al. APOE4 causes widespread molecular and cellular alterations associated with Alzheimer’s disease phenotypes in human iPSC-derived brain cell types. Neuron. 2018;98:1294. doi: 10.1016/j.neuron.2018.06.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Wang C, Najm R, Xu Q, Jeong D-E, Walker D, Balestra ME, et al. Gain of toxic apolipoprotein E4 effects in human iPSC-derived neurons is ameliorated by a small-molecule structure corrector. Nat Med. 2018;24:647–57. doi: 10.1038/s41591-018-0004-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Castellano JM, Kim J, Stewart FR, Jiang H, DeMattos RB, Patterson BW, et al. Human apoE isoforms differentially regulate brain amyloid-β peptide clearance. Sci Transl Med. 2011;3:89ra57. doi: 10.1126/scitranslmed.3002156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Liao F, Li A, Xiong M, Bien-Ly N, Jiang H, Zhang Y, et al. Targeting of nonlipidated, aggregated apoE with antibodies inhibits amyloid accumulation. J Clin Investig. 2018;128:2144–55. doi: 10.1172/JCI96429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Yamazaki Y, Zhao N, Caulfield TR, Liu C-C, Bu G. Apolipoprotein E and Alzheimer disease: pathobiology and targeting strategies. Nat Rev Neurol. 2019;15:501–18. doi: 10.1038/s41582-019-0228-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Liu CC, Zhao N, Fu Y, Wang N, Linares C, Tsai C-W, et al. ApoE4 accelerates early seeding of amyloid pathology. Neuron. 2017;96:1024–32.e3. doi: 10.1016/j.neuron.2017.11.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Kanekiyo T, Xu H. Bu G. ApoE and Aβ in Alzheimer’s disease: accidental encounters or partners? Neuron. 2014;81:740–54. doi: 10.1016/j.neuron.2014.01.045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Kim J, Castellano JM, Jiang H, Basak JM, Parsadanian M, Pham V, et al. Overexpression of low-density lipoprotein receptor in the brain markedly inhibits amyloid deposition and increases extracellular A beta clearance. Neuron. 2009;64:632–44. doi: 10.1016/j.neuron.2009.11.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Holtzman DM, Herz J, Bu G. Apolipoprotein E and apolipoprotein E receptors: normal biology and roles in Alzheimer disease. Cold Spring Harb Perspect Med. 2012;2:a006312. doi: 10.1101/cshperspect.a006312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Laporte V, Lombard Y, Levy-Benezra R, Tranchant C, Poindron P, Warter J-M. Uptake of Abeta 1-40- and Abeta 1-42-coated yeast by microglial cells: a role for LRP. J Leukoc Biol. 2004;76:451–61. doi: 10.1189/jlb.1203620. [DOI] [PubMed] [Google Scholar]
- 73.Kanekiyo T, Cirrito JR, Liu C-C, Shinohara M, Li J, Schuler DR, et al. Neuronal clearance of amyloid-β by endocytic receptor LRP1. J Neurosci. 2013;33:19276–83. doi: 10.1523/JNEUROSCI.3487-13.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Tachibana M, Holm M-L, Liu C-C, Shinohara M, Aikawa T, Oue H, et al. APOE4-mediated amyloid-β pathology depends on its neuronal receptor LRP1. J Clin Invest. 2019;129:1272–7. doi: 10.1172/JCI124853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Lopera F, Ardilla A, Martínez A, Madrigal L, Arango-Viana JC, Lemere CA, et al. Clinical features of early-onset Alzheimer disease in a large kindred with an E280A presenilin-1 mutation. JAMA. 1997;277:793–9. [PubMed] [Google Scholar]
- 76.Quiroz YT, Sperling RA, Norton DJ, Baena A, Arboleda-Velasquez JF, Cosio D, et al. Association between amyloid and Tau accumulation in young adults with autosomal dominant Alzheimer disease. JAMA Neurol. 2018;75:548–56. doi: 10.1001/jamaneurol.2017.4907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Reiman EM, Quiroz YT, Fleisher AS, Chen K, Velez-Pardo C, Jimenez-Del-Rio M, et al. Brain imaging and fluid biomarker analysis in young adults at genetic risk for autosomal dominant Alzheimer’s disease in the presenilin 1 E280A kindred: a case-control study. Lancet Neurol. 2012;11:1048–56. doi: 10.1016/S1474-4422(12)70228-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.isher AS, Chen K, Quiroz YT, Jakimovich LJ, Gomez MG, Langois CM, et al. Florbetapir PET analysis of amyloid-β deposition in the presenilin 1 E280A autosomal dominant Alzheimer’s disease kindred: a cross-sectional study. Lancet Neurol. 2012;11:1057–65. doi: 10.1016/S1474-4422(12)70227-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Arboleda-Velasquez JF, Lopera F, O’Hare M, Delgado-Tirado S, Marino C, Chmielewska N, et al. Resistance to autosomal dominant Alzheimer’s disease in an APOE3 Christchurch homozygote: a case report. Nat Med. 2019;25:1680–3. doi: 10.1038/s41591-019-0611-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Wardell MR, Brennan SO, Janus ED, Fraser R, Carrell RW. Apolipoprotein E2-Christchurch (136 Arg—Ser). New variant of human apolipoprotein E in a patient with type III hyperlipoproteinemia. J Clin Invest. 1987;80:483–90. doi: 10.1172/JCI113096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Suri S, Heise V, Trachtenberg AJ, Mackay CE. The forgotten APOE allele: a review of the evidence and suggested mechanisms for the protective effect of APOE ɛ2. Neurosci Biobehav Rev. 2013;37:2878–86. doi: 10.1016/j.neubiorev.2013.10.010. [DOI] [PubMed] [Google Scholar]
- 82.Corder EH, Saunders AM, Risch NJ, Strittmatter WJ, Schmechel DE, Gaskell PCJ, et al. Protective effect of apolipoprotein E type 2 allele for late onset Alzheimer disease. Nat Genet. 1994;7:180–4. doi: 10.1038/ng0694-180. [DOI] [PubMed] [Google Scholar]
- 83.Farrer LA, Cupples LA, Haines JL, Hyman B, Kukull WA, Mayeux R, et al. Effects of age, sex, and ethnicity on the association between apolipoprotein E genotype and Alzheimer disease. A meta-analysis. APOE and Alzheimer Disease Meta Analysis Consortium. JAMA. 1997;278:1349–56. [PubMed] [Google Scholar]
- 84.Vélez JI, Lopera F, Sepulveda-Falla D, Patel HR, Johar AS, Chuah A, et al. APOE*E2 allele delays age of onset in PSEN1 E280A Alzheimer’s disease. Mol Psychiatry. 2016;21:916–24. doi: 10.1038/mp.2015.177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Hampel H, Lista S, Neri C, Vergallo A. Time for the systems-level integration of aging: Resilience enhancing strategies to prevent Alzheimer’s disease. Prog Neurobiol. 2019;181:101662. doi: 10.1016/j.pneurobio.2019.101662. [DOI] [PubMed] [Google Scholar]
- 86.Levine ME, Lu AT, Bennett DA, Horvath S. Epigenetic age of the pre-frontal cortex is associated with neuritic plaques, amyloid load, and Alzheimer’s disease related cognitive functioning. Aging. 2015;7:1198–211. doi: 10.18632/aging.100864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Schmechel DE, Goldgaber D, Burkhart DS, Gilbert JR, Gajdusek DC, Roses AD. Cellular localization of messenger RNA encoding amyloid-beta-protein in normal tissue and in Alzheimer disease. Alzheimer Dis Assoc Disord. 1998;2:96–111. doi: 10.1097/00002093-198802020-00002. [DOI] [PubMed] [Google Scholar]
- 88.Goedert M. Neuronal localization of amyloid beta protein precursor mRNA in normal human brain and in Alzheimer’s disease. EMBO J. 1987;6:3627–32. doi: 10.1002/j.1460-2075.1987.tb02694.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Lunnon K, Smith R, Hannon E, De Jager PL, Srivastava G, Volta M, et al. Methylomic profiling implicates cortical deregulation of ANK1 in Alzheimer’s disease. Nat Neurosci. 2014;17:1164–70. doi: 10.1038/nn.3782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.West RL, Lee JM, Maroun LE. Hypomethylation of the amyloid precursor protein gene in the brain of an Alzheimer’s disease patient. J Mol Neurosci. 1995;6:141–6. doi: 10.1007/BF02736773. [DOI] [PubMed] [Google Scholar]
- 91.Li P, Marshall L, Oh G, Jakubowski JL, Groot D, He Y, et al. Epigenetic dysregulation of enhancers in neurons is associated with Alzheimer’s disease pathology and cognitive symptoms. Nat Commun. 2019;10:2246. doi: 10.1038/s41467-019-10101-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Marzi SJ, Leung SK, Ribarska T, Hannon E, Smith AR, Pishva E, et al. A histone acetylome-wide association study of Alzheimer’s disease identifies disease-associated H3K27ac differences in the entorhinal cortex. Nat Neurosci. 2018;21:1618–27. doi: 10.1038/s41593-018-0253-7. [DOI] [PubMed] [Google Scholar]
- 93.Zhao Y, Alexandrov PN, Lukiw WJ. Anti-microRNAs as novel therapeutic agents in the clinical management of Alzheimer’s disease. Front Neurosci. 2016;10:59. doi: 10.3389/fnins.2016.00059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Amakiri N, Kubosumi A, Tran J, Reddy PH. Amyloid beta and microRNAs in Alzheimer’s disease. Front Neurosci. 2019;13:430. doi: 10.3389/fnins.2019.00430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Georgopoulou N, McLaughlin M, McFarlane I, Breen KC. The role of post-translational modification in beta-amyloid precursor protein processing. Biochem Soc Symp. 2001;67:23–36. doi: 10.1042/bss0670023. [DOI] [PubMed] [Google Scholar]
- 96.Vingtdeux V, Hamdane M, Gompel M, Bégard S, Drobecq H, Ghestem A, et al. Phosphorylation of amyloid precursor carboxy-terminal fragments enhances their processing by a gamma-secretase-dependent mechanism. Neurobiol Dis. 2005;20:625–37. doi: 10.1016/j.nbd.2005.05.004. [DOI] [PubMed] [Google Scholar]
- 97.Mathys H, Davila-Velderrain J, Peng Z, Gao F, Mohammadi S, Young JZ, et al. Single-cell transcriptomic analysis of Alzheimer’s disease. Nature. 2019;570:332–7. doi: 10.1038/s41586-019-1195-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Grubman A, Chew G, Ouyang JF, Sun G, Choo XY, McLean C, et al. A single-cell atlas of entorhinal cortex from individuals with Alzheimer’s disease reveals cell-type-specific gene expression regulation. Nat Neurosci. 2019;22:2087–97. doi: 10.1038/s41593-019-0539-4. [DOI] [PubMed] [Google Scholar]
- 99.Lau S-F, Cao H, Fu AKY, Ip NY. Single-nucleus transcriptome analysis reveals dysregulation of angiogenic endothelial cells and neuroprotective glia in Alzheimer’s disease. Proc Natl Acad Sci USA. 2020;117:25800–9. doi: 10.1073/pnas.2008762117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Zhou Y, Song WM, Andhey PS, Swain A, Levy T, Miller KR, et al. Human and mouse single-nucleus transcriptomics reveal TREM2-dependent and TREM2-independent cellular responses in Alzheimer’s disease. Nat Med. 2020;26:131–42. doi: 10.1038/s41591-019-0695-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Olah M, Menon V, Habib N, Taga MF, Ma Y, Yung CJ, et al. Single cell RNA sequencing of human microglia uncovers a subset associated with Alzheimer’s disease. Nat Commun. 2020;11:6129. doi: 10.1038/s41467-020-19737-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Alsema AM, Jiang Q, Kracht L, Gerrits E, Dubbelaar ML, Miedema A, et al. Profiling microglia from Alzheimer’s disease donors and non-demented elderly in acute human postmortem cortical tissue. Front Mol Neurosci. 2020;13:134. doi: 10.3389/fnmol.2020.00134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Habib N, McCabe C, Medina S, Varshavsky M, Kitsberg D, Dvir-Szternfeld R, et al. Disease-associated astrocytes in Alzheimer’s disease and aging. Nat Neurosci. 2020;23:701–6. doi: 10.1038/s41593-020-0624-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Sierksma A, Lu A, Mancuso R, Fattorelli N, Thrupp N, Salta E, et al. Novel Alzheimer risk genes determine the microglia response to amyloid-β but not to TAU pathology. EMBO Mol Med. 2020;12:e10606. doi: 10.15252/emmm.201910606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Nixon RA. Amyloid precursor protein and endosomal-lysosomal dysfunction in Alzheimer’s disease: inseparable partners in a multifactorial disease. FASEB J Publ Fed Am Soc Exp Biol. 2017;31:2729–43. doi: 10.1096/fj.201700359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Mullan M, Crawford F, Axelman K, Houlden H, Lilius L, Winblad B, et al. A pathogenic mutation for probable Alzheimer’s disease in the APP gene at the N-terminus of beta-amyloid. Nat Genet. 1992;1:345–7. doi: 10.1038/ng0892-345. [DOI] [PubMed] [Google Scholar]
- 107.Jonsson T, Atwal JK, Steinberg S, Snaedal J, Jonsson PV, Bjornsson S, et al. A mutation in APP protects against Alzheimer’s disease and age-related cognitive decline. Nature. 2012;488:96–99. doi: 10.1038/nature11283. [DOI] [PubMed] [Google Scholar]
- 108.Hampel H, Vassar R, De Strooper B, Hardy J, Willem M, Singh N, et al. The β-secretase BACE1 in Alzheimer’s disease. Biol Psychiatry. 2021;89:745–56. doi: 10.1016/j.biopsych.2020.02.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Zhao J, Fu Y, Yasvoina M, Shao P, Hitt B, O’Connor T, et al. Beta-site amyloid precursor protein cleaving enzyme 1 levels become elevated in neurons around amyloid plaques: implications for Alzheimer’s disease pathogenesis. J Neurosci. 2007;27:3639–49. doi: 10.1523/JNEUROSCI.4396-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Kandalepas PC, Sadleir KR, Eimer WA, Zhao J, Nicholson DA, Vassar R. The Alzheimer’s β-secretase BACE1 localizes to normal presynaptic terminals and to dystrophic presynaptic terminals surrounding amyloid plaques. Acta Neuropathol. 2013;126:329–52. doi: 10.1007/s00401-013-1152-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Sadleir KR, Kandalepas PC, Buggia-Prévot V, Nicholson DA, Thinakaran G, Vassar R. Presynaptic dystrophic neurites surrounding amyloid plaques are sites of microtubule disruption, BACE1 elevation, and increased Aβ generation in Alzheimer’s disease. Acta Neuropathol. 2016;132:235–56. doi: 10.1007/s00401-016-1558-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Feng T, Tammineni P, Agrawal C, Jeong YY, Cai Q. Autophagy-mediated regulation of BACE1 protein trafficking and degradation. J Biol Chem. 2017;292:1679–90. doi: 10.1074/jbc.M116.766584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.De Strooper B, Vassar R, Golde T. The secretases: enzymes with therapeutic potential in Alzheimer disease. Nat Rev Neurol. 2010;6:99–107. doi: 10.1038/nrneurol.2009.218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Kakuda N, Yamaguchi H, Akazawa K, Hata S, Suzuki T, Hatsuta H, et al. Secretase activity is associated with Braak Senile Plaque Stages. Am J Pathol. 2020;190:1323–31. doi: 10.1016/j.ajpath.2020.02.009. [DOI] [PubMed] [Google Scholar]
- 115.Serneels L, Van Biervliet J, Craessaerts K, Dejaegere T, Horré K, Van Houtvin T, et al. gamma-Secretase heterogeneity in the Aph1 subunit: relevance for Alzheimer’s disease. Science. 2009;324:639–42. doi: 10.1126/science.1171176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.De Strooper B. Aph-1, Pen-2, and Nicastrin with Presenilin generate an active gamma-secretase complex. Neuron. 2003;38:9–12. doi: 10.1016/s0896-6273(03)00205-8. [DOI] [PubMed] [Google Scholar]
- 117.Zhou Y, Zhang W, Easton R, Ray JW, Lampe P, Jiang Z, et al. Presenilin-1 protects against neuronal apoptosis caused by its interacting protein PAG. Neurobiol Dis. 2002;9:126–38. doi: 10.1006/nbdi.2001.0472. [DOI] [PubMed] [Google Scholar]
- 118.Voytyuk I, De Strooper B, Chávez-Gutiérrez L. Modulation of γ- and β-secretases as early prevention against Alzheimer’s disease. Biol Psychiatry. 2018;83:320–7. doi: 10.1016/j.biopsych.2017.08.001. [DOI] [PubMed] [Google Scholar]
- 119.Willem M, Tahirovic S, Busche MA, Ovsepian SV, Chafai M, Kootar S, et al. Secretase processing of APP inhibits neuronal activity in the hippocampus. Nature. 2015;526:443–7. doi: 10.1038/nature14864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Vassar R. ADAM10 prodomain mutations cause late-onset Alzheimer’s disease: not just the latest FAD. Neuron. 2013;80:250–3. doi: 10.1016/j.neuron.2013.09.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Kuhn PH, Wang H, Dislich B, Colombo A, Zeitschel U, Ellwart JW, et al. ADAM10 is the physiologically relevant, constitutive alpha-secretase of the amyloid precursor protein in primary neurons. EMBO J. 2010;29:3020–32. doi: 10.1038/emboj.2010.167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Rice HC, de Malmazet D, Schreurs A, Frere S, Van Molle I, Volkov AN, et al. Secreted amyloid-β precursor protein functions as a GABA(B)R1a ligand to modulate synaptic transmission. Science. 2019;363:eaao4827. doi: 10.1126/science.aao4827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Wilhelm BG, Mandad S, Truckenbrodt S, Kröhnert K, Schäfer C, Rammner B, et al. Composition of isolated synaptic boutons reveals the amounts of vesicle trafficking proteins. Science. 2014;344:1023–8. doi: 10.1126/science.1252884. [DOI] [PubMed] [Google Scholar]
- 124.Ring S, Weyer SW, Kilian SB, Waldron E, Pietrzik CU, Filippov MA, et al. The secreted beta-amyloid precursor protein ectodomain APPs alpha is sufficient to rescue the anatomical, behavioral, and electrophysiological abnormalities of APP-deficient mice. J Neurosci. 2007;27:7817–26. doi: 10.1523/JNEUROSCI.1026-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Hick M, Herrmann U, Weyer SW, Mallm J-P, Tschäpe J-A, Borgers M, et al. Acute function of secreted amyloid precursor protein fragment APPsα in synaptic plasticity. Acta Neuropathol. 2015;129:21–37. doi: 10.1007/s00401-014-1368-x. [DOI] [PubMed] [Google Scholar]
- 126.Suh J, Choi SH, Romano DM, Gannon MA, Lesinski AN, Kim DY, et al. ADAM10 missense mutations potentiate β-amyloid accumulation by impairing prodomain chaperone function. Neuron. 2013;80:385–401. doi: 10.1016/j.neuron.2013.08.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Turner PR, O’Connor K, Tate WP, Abraham WC. Roles of amyloid precursor protein and its fragments in regulating neural activity, plasticity and memory. Prog Neurobiol. 2003;70:1–32. doi: 10.1016/s0301-0082(03)00089-3. [DOI] [PubMed] [Google Scholar]
- 128.Tharp WG, Sarkar IN. Origins of amyloid-β. BMC Genomics. 2013;14:290. doi: 10.1186/1471-2164-14-290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Abramov E, Dolev I, Fogel H, Ciccotosto GD, Ruff E, Slutsky I. Amyloid-beta as a positive endogenous regulator of release probability at hippocampal synapses. Nat Neurosci. 2009;12:1567–76. doi: 10.1038/nn.2433. [DOI] [PubMed] [Google Scholar]
- 130.Kamenetz F, Tomita T, Hsieh H, Seabrook G, Borchelt D, Iwatsubo T, et al. APP processing and synaptic function. Neuron. 2003;37:925–37. doi: 10.1016/s0896-6273(03)00124-7. [DOI] [PubMed] [Google Scholar]
- 131.Hsieh H, Boehm J, Sato C, Iwatsubo T, Tomita T, Sisodia S, et al. AMPAR removal underlies Abeta-induced synaptic depression and dendritic spine loss. Neuron. 2006;52:831–43. doi: 10.1016/j.neuron.2006.10.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Zimbone S, Monaco I, Gianì F, Pandini G, Copani AG, Giuffrida ML et al. Amyloid beta monomers regulate cyclic adenosine monophosphate response element binding protein functions by activating type-1 insulin-like growth factor receptors in neuronal cells. Aging Cell. 2018;17. 10.1111/acel.12684. [DOI] [PMC free article] [PubMed]
- 133.Moreno-Jiménez EP, Flor-García M, Terreros-Roncal J, Rábano A, Cafini F, Pallas-Bazarra N, et al. Adult hippocampal neurogenesis is abundant in neurologically healthy subjects and drops sharply in patients with Alzheimer’s disease. Nat Med. 2019;25:554–60. doi: 10.1038/s41591-019-0375-9. [DOI] [PubMed] [Google Scholar]
- 134.Giuffrida ML, Tomasello MF, Pandini G, Caraci F, Battaglia G, Busceti C, et al. Monomeric ß-amyloid interacts with type-1 insulin-like growth factor receptors to provide energy supply to neurons. Front Cell Neurosci. 2015;9:297. doi: 10.3389/fncel.2015.00297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Morris GP, Clark IA, Vissel B. Questions concerning the role of amyloid-β in the definition, aetiology and diagnosis of Alzheimer’s disease. Acta Neuropathol. 2018;136:663–89. doi: 10.1007/s00401-018-1918-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Bateman RJ, Munsell LY, Morris JC, Swarm R, Yarasheski KE, Holtzman DM. Human amyloid-beta synthesis and clearance rates as measured in cerebrospinal fluid in vivo. Nat Med. 2006;12:856–61. doi: 10.1038/nm1438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Engelhardt B, Carare RO, Bechmann I, Flügel A, Laman JD, Weller ROVascular. glial, and lymphatic immune gateways of the central nervous system. Acta Neuropathol. 2016;132:317–38. doi: 10.1007/s00401-016-1606-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Blennow K, Wallin A. Clinical heterogeneity of probable Alzheimer’s disease. J Geriatr Psychiatry Neurol. 1992;5:106–13. doi: 10.1177/002383099200500208. [DOI] [PubMed] [Google Scholar]
- 139.Sweeney M, Sagare A, Zlokovic B. Blood–brain barrier breakdown in Alzheimer disease and other neurodegenerative disorders. Nat Rev Neurol. 2018;14:133–50. doi: 10.1038/nrneurol.2017.188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Yamada K, Hashimoto T, Yabuki C, Nagae Y, Tachikawa M, Strickland DK, et al. The low density lipoprotein receptor-related protein 1 mediates uptake of amyloid beta peptides in an in vitro model of the blood-brain barrier cells. J Biol Chem. 2008;283:34554–62. doi: 10.1074/jbc.M801487200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Zlokovic BV. The blood-brain barrier in health and chronic neurodegenerative disorders. Neuron. 2008;57:178–201. doi: 10.1016/j.neuron.2008.01.003. [DOI] [PubMed] [Google Scholar]
- 142.Iwata N, Tsubuki S, Takaki Y, Shirotani K, Lu B, Gerard NP, et al. Metabolic regulation of brain Abeta by neprilysin. Science. 2001;292:1550–2. doi: 10.1126/science.1059946. [DOI] [PubMed] [Google Scholar]
- 143.Farizatto KLG, Ikonne US, Almeida MF, Ferrari MFR, Bahr BA. Aβ42-mediated proteasome inhibition and associated tau pathology in hippocampus are governed by a lysosomal response involving cathepsin B: Evidence for protective crosstalk between protein clearance pathways. PLoS One. 2017;12:e0182895. doi: 10.1371/journal.pone.0182895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Kechko OI, Petrushanko IY, Brower CS, Adzhubei AA, Moskalev AA, Piatkov KI, et al. Beta-amyloid induces apoptosis of neuronal cells by inhibition of the Arg/N-end rule pathway proteolytic activity. Aging (Albany NY) 2019;11:6134–52. doi: 10.18632/aging.102177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Kidana K, Tatebe T, Ito K, Hara N, Kakita A, Saito T et al. Loss of kallikrein-related peptidase 7 exacerbates amyloid pathology in Alzheimer’s disease model mice. EMBO Mol Med 2018;10. 10.15252/emmm.201708184. [DOI] [PMC free article] [PubMed]
- 146.Eckman EA, Adams SK, Troendle FJ, Stodola BA, Kahn MA, Fauq AH, et al. Regulation of steady-state beta-amyloid levels in the brain by neprilysin and endothelin-converting enzyme but not angiotensin-converting enzyme. J Biol Chem. 2006;281:30471–8. doi: 10.1074/jbc.M605827200. [DOI] [PubMed] [Google Scholar]
- 147.Leissring MA, Farris W, Chang AY, Walsh DM, Wu X, Sun X, et al. Enhanced proteolysis of beta-amyloid in APP transgenic mice prevents plaque formation, secondary pathology, and premature death. Neuron. 2003;40:1087–93. doi: 10.1016/s0896-6273(03)00787-6. [DOI] [PubMed] [Google Scholar]
- 148.Zou L-B, Mouri A, Iwata N, Saido TC, Wang D, Wang M-W, et al. Inhibition of neprilysin by infusion of thiorphan into the hippocampus causes an accumulation of amyloid Beta and impairment of learning and memory. J Pharm Exp Ther. 2006;317:334–40. doi: 10.1124/jpet.105.095687. [DOI] [PubMed] [Google Scholar]
- 149.Rasmussen MK, Mestre H, Nedergaard M. The glymphatic pathway in neurological disorders. Lancet Neurol. 2018;17:1016–24. doi: 10.1016/S1474-4422(18)30318-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Reeves BC, Karimy JK, Kundishora AJ, Mestre H, Cerci HM, Matouk C, et al. Glymphatic system impairment in Alzheimer’s disease and idiopathic normal pressure hydrocephalus. Trends Mol Med. 2020;26:285–95. doi: 10.1016/j.molmed.2019.11.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Ju YE, Lucey BP, Holtzman DM Sleep. and Alzheimer disease pathology–a bidirectional relationship. Nat Rev Neurol. 2014;10:115–9. doi: 10.1038/nrneurol.2013.269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Zeppenfeld DM, Simon M, Haswell JD, D’Abreo D, Murchison C, Quinn JF, et al. Association of perivascular localization of aquaporin-4 with cognition and Alzheimer disease in aging brains. JAMA Neurol. 2017;74:91–99. doi: 10.1001/jamaneurol.2016.4370. [DOI] [PubMed] [Google Scholar]
- 153.Tarasoff-Conway JM, Carare RO, Osorio RS, Glodzik L, Butler T, Fieremans E, et al. Clearance systems in the brain-implications for Alzheimer disease. Nat Rev Neurol. 2015;11:457–70. doi: 10.1038/nrneurol.2015.119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Silverberg GD, Mayo M, Saul T, Rubenstein E, McGuire D. Alzheimer’s disease, normal-pressure hydrocephalus, and senescent changes in CSF circulatory physiology: a hypothesis. Lancet Neurol. 2003;2:506–11. doi: 10.1016/s1474-4422(03)00487-3. [DOI] [PubMed] [Google Scholar]
- 155.Michaels TCT, Šarić A, Curk S, Bernfur K, Arosio P, Meisl G, et al. Dynamics of oligomer populations formed during the aggregation of Alzheimer’s Aβ42 peptide. Nat Chem. 2020;12:445–51. doi: 10.1038/s41557-020-0452-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Cohen SIA, Linse S, Luheshi LM, Hellstrand E, White DA, Rajah L, et al. Proliferation of amyloid-β42 aggregates occurs through a secondary nucleation mechanism. Proc Natl Acad Sci USA. 2013;110:9758–63. doi: 10.1073/pnas.1218402110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Knowles TP, Vendruscolo M, Dobson CM. The amyloid state and its association with protein misfolding diseases. Nat Rev Mol Cell Biol. 2014;15:384–96. doi: 10.1038/nrm3810. [DOI] [PubMed] [Google Scholar]
- 158.Vecchi G, Sormanni P, Mannini B, Vandelli A, Tartaglia GG, Dobson CM, et al. Proteome-wide observation of the phenomenon of life on the edge of solubility. Proc Natl Acad Sci USA. 2020;117:1015–20. doi: 10.1073/pnas.1910444117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Iannuzzi C, Irace G, Sirangelo I. Differential effects of glycation on protein aggregation and amyloid formation. Front Mol Biosci. 2014;1:9. doi: 10.3389/fmolb.2014.00009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Halim A, Brinkmalm G, Rüetschi U, Westman-Brinkmalm A, Portelius E, Zetterberg H, et al. Site-specific characterization of threonine, serine, and tyrosine glycosylations of amyloid precursor protein/amyloid beta-peptides in human cerebrospinal fluid. Proc Natl Acad Sci USA. 2011;108:11848–53. doi: 10.1073/pnas.1102664108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Takeuchi M, Yamagishi S. Possible involvement of advanced glycation end-products (AGEs) in the pathogenesis of Alzheimer’s disease. Curr Pharm Des. 2008;14:973–8. doi: 10.2174/138161208784139693. [DOI] [PubMed] [Google Scholar]
- 162.Vitek MP, Bhattacharya K, Glendening JM, Stopa E, Vlassara H, Bucala R, et al. Advanced glycation end products contribute to amyloidosis in Alzheimer disease. Proc Natl Acad Sci USA. 1994;91:4766–70. doi: 10.1073/pnas.91.11.4766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Wildburger NC, Esparza TJ, LeDuc RD, Fellers RT, Thomas PM, Cairns NJ, et al. Diversity of amyloid-beta proteoforms in the Alzheimer’s disease brain. Sci Rep. 2017;7:9520. doi: 10.1038/s41598-017-10422-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Nussbaum JM, Schilling S, Cynis H, Silva A, Swanson E, Wangsanut T, et al. Prion-like behaviour and tau-dependent cytotoxicity of pyroglutamylated amyloid-β. Nature. 2012;485:651–5. doi: 10.1038/nature11060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Hou L, Shao H, Zhang Y, Li H, Menon NK, Neuhaus EB, et al. Solution NMR studies of the A beta(1-40) and A beta(1-42) peptides establish that the Met35 oxidation state affects the mechanism of amyloid formation. J Am Chem Soc. 2004;126:1992–2005. doi: 10.1021/ja036813f. [DOI] [PubMed] [Google Scholar]
- 166.Kulenkampff K, Wolf Perez A-M, Sormanni P, Habchi J, Vendruscolo M. Quantifying misfolded protein oligomers as drug targets and biomarkers in Alzheimer and Parkinson diseases. Nat Rev Chem. 2021;5:277–94. doi: 10.1038/s41570-021-00254-9. [DOI] [PubMed] [Google Scholar]
- 167.Kayed R, Head E, Thompson JL, McIntire TM, Milton SC, Cotman CW, et al. Common structure of soluble amyloid oligomers implies common mechanism of pathogenesis. Science. 2003;300:486–9. doi: 10.1126/science.1079469. [DOI] [PubMed] [Google Scholar]
- 168.Zahs KR, Ashe KH. β-Amyloid oligomers in aging and Alzheimer’s disease. Front Aging Neurosci. 2013;5:28. doi: 10.3389/fnagi.2013.00028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169.Lesné SE, Sherman MA, Grant M, Kuskowski M, Schneider JA, Bennett DA, et al. Brain amyloid-β oligomers in ageing and Alzheimer’s disease. Brain. 2013;136:1383–98. doi: 10.1093/brain/awt062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170.Podlisny MB, Ostaszewski BL, Squazzo SL, Koo EH, Rydell RE, Teplow DB, et al. Aggregation of secreted amyloid beta-protein into sodium dodecyl sulfate-stable oligomers in cell culture. J Biol Chem. 1995;270:9564–70. doi: 10.1074/jbc.270.16.9564. [DOI] [PubMed] [Google Scholar]
- 171.Walsh DM, Tseng BP, Rydel RE, Podlisny MB, Selkoe DJ. The oligomerization of amyloid beta-protein begins intracellularly in cells derived from human brain. Biochemistry. 2000;39:10831–9. doi: 10.1021/bi001048s. [DOI] [PubMed] [Google Scholar]
- 172.Shankar GM, Li S, Mehta TH, Garcia-Munoz A, Shepardson NE, Smith I, et al. Amyloid-beta protein dimers isolated directly from Alzheimer’s brains impair synaptic plasticity and memory. Nat Med. 2008;14:837–42. doi: 10.1038/nm1782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173.Brinkmalm G, Hong W, Wang Z, Liu W, O’Malley TT, Sun X, et al. Identification of neurotoxic cross-linked amyloid-β dimers in the Alzheimer’s brain. Brain. 2019;142:1441–57. doi: 10.1093/brain/awz066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174.Li S, Selkoe DJ. A mechanistic hypothesis for the impairment of synaptic plasticity by soluble Aβ oligomers from Alzheimer’s brain. J Neurochem. 2020;154:583–97. doi: 10.1111/jnc.15007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175.Walsh DM, Klyubin I, Fadeeva JV, Cullen WK, Anwyl R, Wolfe MS, et al. Naturally secreted oligomers of amyloid beta protein potently inhibit hippocampal long-term potentiation in vivo. Nature. 2002;416:535–9. doi: 10.1038/416535a. [DOI] [PubMed] [Google Scholar]
- 176.Townsend M, Shankar GM, Mehta T, Walsh DM, Selkoe DJ. Effects of secreted oligomers of amyloid beta-protein on hippocampal synaptic plasticity: a potent role for trimers. J Physiol. 2006;572:477–92. doi: 10.1113/jphysiol.2005.103754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177.Selkoe DJ. Soluble oligomers of the amyloid beta-protein impair synaptic plasticity and behavior. Behav Brain Res. 2008;192:106–13. doi: 10.1016/j.bbr.2008.02.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178.Bernstein SL, Dupuis NF, Lazo ND, Wyttenbach T, Condron MM, Bitan G, et al. Amyloid-β protein oligomerization and the importance of tetramers and dodecamers in the aetiology of Alzheimer’s disease. Nat Chem. 2009;1:326–31. doi: 10.1038/nchem.247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 179.Lacor PN, Buniel MC, Furlow PW, Clemente AS, Velasco PT, Wood M, et al. Abeta oligomer-induced aberrations in synapse composition, shape, and density provide a molecular basis for loss of connectivity in Alzheimer’s disease. J Neurosci. 2007;27:796–807. doi: 10.1523/JNEUROSCI.3501-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180.Hong W, Wang Z, Liu W, O’Malley TT, Jin M, Willem M, et al. Diffusible, highly bioactive oligomers represent a critical minority of soluble Aβ in Alzheimer’s disease brain. Acta Neuropathol. 2018;136:19–40. doi: 10.1007/s00401-018-1846-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181.Yang T, Li S, Xu H, Walsh DM, Selkoe DJ. Large soluble oligomers of amyloid β-protein from Alzheimer brain are far less neuroactive than the smaller oligomers to which they dissociate. J Neurosci. 2017;37:152–63. doi: 10.1523/JNEUROSCI.1698-16.2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 182.Walsh DM, Lomakin A, Benedek GB, Condron MM, Teplow DB. Amyloid beta-protein fibrillogenesis. Detection of a protofibrillar intermediate. J Biol Chem. 1997;272:22364–72. doi: 10.1074/jbc.272.35.22364. [DOI] [PubMed] [Google Scholar]
- 183.Nilsberth C, Westlind-Danielsson A, Eckman CB, Condron MM, Axelman K, Forsell C, et al. The ‘Arctic’ APP mutation (E693G) causes Alzheimer’s disease by enhanced Abeta protofibril formation. Nat Neurosci. 2001;4:887–93. doi: 10.1038/nn0901-887. [DOI] [PubMed] [Google Scholar]
- 184.Johansson AS, Berglind-Dehlin F, Karlsson G, Edwards K, Gellerfors P, Lannfelt L. Physiochemical characterization of the Alzheimer’s disease-related peptides A beta 1-42Arctic and A beta 1-42wt. FEBS J. 2006;273:2618–30. doi: 10.1111/j.1742-4658.2006.05263.x. [DOI] [PubMed] [Google Scholar]
- 185.Hartley DM, Walsh DM, Ye CP, Diehl T, Vasquez S, Vassilev PM, et al. Protofibrillar intermediates of amyloid beta-protein induce acute electrophysiological changes and progressive neurotoxicity in cortical neurons. J Neurosci. 1999;19:8876–84. doi: 10.1523/JNEUROSCI.19-20-08876.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 186.O’Nuallain B, Freir DB, Nicoll AJ, Risse E, Ferguson N, Herron CE, et al. Amyloid beta-protein dimers rapidly form stable synaptotoxic protofibrils. J Neurosci. 2010;30:14411–9. doi: 10.1523/JNEUROSCI.3537-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 187.Lasagna-Reeves CA, Kayed R. Astrocytes contain amyloid-β annular protofibrils in Alzheimer’s disease brains. FEBS Lett. 2011;585:3052–7. doi: 10.1016/j.febslet.2011.08.027. [DOI] [PubMed] [Google Scholar]
- 188.Gouwens LK, Makoni NJ, Rogers VA, Nichols MR. Amyloid-β42 protofibrils are internalized by microglia more extensively than monomers. Brain Res. 2016;1648:485–95. doi: 10.1016/j.brainres.2016.08.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 189.Gouwens LK, Ismail MS, Rogers VA, Zeller NT, Garrad EC, Amtashar FS, et al. Aβ42 protofibrils interact with and are trafficked through microglial-derived microvesicles. ACS Chem Neurosci. 2018;9:1416–25. doi: 10.1021/acschemneuro.8b00029. [DOI] [PubMed] [Google Scholar]
- 190.Söllvander S, Ekholm-Pettersson F, Brundin R-M, Westman G, Kilander L, Paulie S, et al. Increased number of plasma B cells producing autoantibodies against Aβ42 protofibrils in Alzheimer’s disease. J Alzheimers Dis. 2015;48:63–72. doi: 10.3233/JAD-150236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 191.McLean CA, Cherny RA, Fraser FW, Fuller SJ, Smith MJ, Beyreuther K, et al. Soluble pool of Abeta amyloid as a determinant of severity of neurodegeneration in Alzheimer’s disease. Ann Neurol. 1999;46:860–6. doi: 10.1002/1531-8249(199912)46:6<860::aid-ana8>3.0.co;2-m. [DOI] [PubMed] [Google Scholar]
- 192.Lesné S, Koh MT, Kotilinek L, Kayed R, Glabe CG, Yang A, et al. A specific amyloid-beta protein assembly in the brain impairs memory. Nature. 2006;440:352–7. doi: 10.1038/nature04533. [DOI] [PubMed] [Google Scholar]
- 193.Englund H, Sehlin D, Johansson A-S, Nilsson LNG, Gellerfors P, Paulie S, et al. Sensitive ELISA detection of amyloid-beta protofibrils in biological samples. J Neurochem. 2007;103:334–45. doi: 10.1111/j.1471-4159.2007.04759.x. [DOI] [PubMed] [Google Scholar]
- 194.Sahlin C, Lord A, Magnusson K, Englund H, Almeida CG, Greengard P, et al. The Arctic Alzheimer mutation favors intracellular amyloid-beta production by making amyloid precursor protein less available to alpha-secretase. J Neurochem. 2007;101:854–62. doi: 10.1111/j.1471-4159.2006.04443.x. [DOI] [PubMed] [Google Scholar]
- 195.Lord A, Kalimo H, Eckman C, Zhang XQ, Lannfelt L, Nilsson LN. The Arctic Alzheimer mutation facilitates early intraneuronal Abeta aggregation and senile plaque formation in transgenic mice. Neurobiol Aging. 2006;27:67–77. doi: 10.1016/j.neurobiolaging.2004.12.007. [DOI] [PubMed] [Google Scholar]
- 196.Lord A, Englund H, Söderberg L, Tucker S, Clausen F, Hillered L, et al. Amyloid-beta protofibril levels correlate with spatial learning in Arctic Alzheimer’s disease transgenic mice. FEBS J. 2009;276:995–1006. doi: 10.1111/j.1742-4658.2008.06836.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 197.Sehlin D, Englund H, Simu B, Karlsson M, Ingelsson M, Nikolajeff F, et al. Large aggregates are the major soluble Aβ species in AD brain fractionated with density gradient ultracentrifugation. PLoS One. 2012;7:e32014. doi: 10.1371/journal.pone.0032014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 198.Magnusson K, Sehlin D, Syvänen S, Svedberg MM, Philipson O, Söderberg L, et al. Specific uptake of an amyloid-β protofibril-binding antibody-tracer in AβPP transgenic mouse brain. J Alzheimers Dis. 2013;37:29–40. doi: 10.3233/JAD-130029. [DOI] [PubMed] [Google Scholar]
- 199.Tucker S, Möller C, Tegerstedt K, Lord A, Laudon H, Sjödahl J, et al. The murine version of BAN2401 (mAb158) selectively reduces amyloid-β protofibrils in brain and cerebrospinal fluid of tg-ArcSwe mice. J Alzheimers Dis. 2015;43:575–88. doi: 10.3233/JAD-140741. [DOI] [PubMed] [Google Scholar]
- 200.Schöll M, Wall A, Thordardottir S, Ferreira D, Bogdanovic N, Långström B, et al. Low PiB PET retention in presence of pathologic CSF biomarkers in Arctic APP mutation carriers. Neurology. 2012;79:229–36. doi: 10.1212/WNL.0b013e31825fdf18. [DOI] [PubMed] [Google Scholar]
- 201.Tomiyama T, Nagata T, Shimada H, Teraoka R, Fukushima A, Kanemitsu H, et al. A new amyloid beta variant favoring oligomerization in Alzheimer’s-type dementia. Ann Neurol. 2008;63:377–87. doi: 10.1002/ana.21321. [DOI] [PubMed] [Google Scholar]
- 202.Freer R, Sormanni P, Vecchi G, Ciryam P, Dobson CM, Vendruscolo M. A protein homeostasis signature in healthy brains recapitulates tissue vulnerability to Alzheimer’s disease. Sci Adv. 2016;2:e1600947. doi: 10.1126/sciadv.1600947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 203.Lu JX, Qiang W, Yau WM, Schwieters CD, Meredith SC, Tycko R. Molecular structure of β-amyloid fibrils in Alzheimer’s disease brain tissue. Cell. 2013;154:1257–68. doi: 10.1016/j.cell.2013.08.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 204.Agrawal N, Skelton AA. Structure and function of Alzheimer’s amyloid βeta proteins from monomer to fibrils: a mini review. Protein J. 2019;38:425–34. doi: 10.1007/s10930-019-09854-3. [DOI] [PubMed] [Google Scholar]
- 205.Paravastu AK, Leapman RD, Yau WM, Tycko R. Molecular structural basis for polymorphism in Alzheimer’s beta-amyloid fibrils. Proc Natl Acad Sci USA. 2008;105:18349–54. doi: 10.1073/pnas.0806270105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 206.Petkova AT, Leapman RD, Guo Z, Yau WM, Mattson MP, Tycko R. Self-propagating, molecular-level polymorphism in Alzheimer’s beta-amyloid fibrils. Science. 2005;307:262–5. doi: 10.1126/science.1105850. [DOI] [PubMed] [Google Scholar]
- 207.Knowles TP, Fitzpatrick AW, Meehan S, Mott HR, Vendruscolo M, Dobson CM, et al. Role of intermolecular forces in defining material properties of protein nanofibrils. Science. 2007;318:1900–3. doi: 10.1126/science.1150057. [DOI] [PubMed] [Google Scholar]
- 208.Xu G, Fromholt SE, Chakrabarty P, Zhu F, Liu X, Pace MC, et al. Diversity in Aβ deposit morphology and secondary proteome insolubility across models of Alzheimer-type amyloidosis. Acta Neuropathol Commun. 2020;8:43. doi: 10.1186/s40478-020-00911-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 209.Meyer-Luehmann M, Spires-Jones TL, Prada C, Garcia-Alloza M, de Calignon A, Rozkalne A, et al. Rapid appearance and local toxicity of amyloid-beta plaques in a mouse model of Alzheimer’s disease. Nature. 2008;451:720–4. doi: 10.1038/nature06616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 210.Knowles RB, Wyart C, Buldyrev SV, Cruz L, Urbanc B, Hasselmo ME, et al. Plaque-induced neurite abnormalities: implications for disruption of neural networks in Alzheimer’s disease. Proc Natl Acad Sci USA. 1999;96:5274–9. doi: 10.1073/pnas.96.9.5274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 211.Geula C, Wu CK, Saroff D, Lorenzo A, Yuan M, Yankner BA. Aging renders the brain vulnerable to amyloid beta-protein neurotoxicity. Nat Med. 1998;4:827–31. doi: 10.1038/nm0798-827. [DOI] [PubMed] [Google Scholar]
- 212.Koffie RM, Meyer-Luehmann M, Hashimoto T, Adams KW, Mielke ML, Garcia-Alloza M, et al. Oligomeric amyloid beta associates with postsynaptic densities and correlates with excitatory synapse loss near senile plaques. Proc Natl Acad Sci USA. 2009;106:4012–7. doi: 10.1073/pnas.0811698106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 213.Grüning CSR, Klinker S, Wolff M, Schneider M, Toksöz K, Klein AN, et al. The off-rate of monomers dissociating from amyloid-β protofibrils. J Biol Chem. 2013;288:37104–11. doi: 10.1074/jbc.M113.513432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 214.Carulla N, Caddy GL, Hall DR, Zurdo J, Gairí M, Feliz M, et al. Molecular recycling within amyloid fibrils. Nature. 2005;436:554–8. doi: 10.1038/nature03986. [DOI] [PubMed] [Google Scholar]
- 215.Sánchez L, Madurga S, Pukala T, Vilaseca M, López-Iglesias C, Robinson CV, et al. Aβ40 and Aβ42 amyloid fibrils exhibit distinct molecular recycling properties. J Am Chem Soc. 2011;133:6505–8. doi: 10.1021/ja1117123. [DOI] [PubMed] [Google Scholar]
- 216.Brännström K, Öhman A, Nilsson L, Pihl M, Sandblad L, Olofsson A. The N-terminal region of amyloid β controls the aggregation rate and fibril stability at low pH through a gain of function mechanism. J Am Chem Soc. 2014;136:10956–64. doi: 10.1021/ja503535m. [DOI] [PubMed] [Google Scholar]
- 217.Martins IC, Kuperstein I, Wilkinson H, Maes E, Vanbrabant M, Jonckheere W, et al. Lipids revert inert Abeta amyloid fibrils to neurotoxic protofibrils that affect learning in mice. EMBO J. 2008;27:224–33. doi: 10.1038/sj.emboj.7601953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 218.He Z, Guo JL, McBride JD, Narasimhan S, Kim H, Changolkar L, et al. Amyloid-β plaques enhance Alzheimer’s brain tau-seeded pathologies by facilitating neuritic plaque tau aggregation. Nat Med. 2018;24:29–38. doi: 10.1038/nm.4443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 219.Wang L, Benzinger TL, Su Y, Christensen J, Friedrichsen K, Aldea P, et al. Evaluation of Tau imaging in staging Alzheimer disease and revealing interactions between β-amyloid and tauopathy. JAMA Neurol. 2016;73:1070–7. doi: 10.1001/jamaneurol.2016.2078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 220.Lewis J, Dickson DW, Lin WL, Chisholm L, Corral A, Jones G, et al. Enhanced neurofibrillary degeneration in transgenic mice expressing mutant tau and APP. Science. 2001;293:1487–91. doi: 10.1126/science.1058189. [DOI] [PubMed] [Google Scholar]
- 221.Busche MA, Hyman BT. Synergy between amyloid-β and tau in Alzheimer’s disease. Nat Neurosci. 2020;23:1183–93. doi: 10.1038/s41593-020-0687-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 222.Götz J, Chen F, van Dorpe J, Nitsch RM. Formation of neurofibrillary tangles in P301l tau transgenic mice induced by Abeta 42 fibrils. Science. 2001;293:1491–5. doi: 10.1126/science.1062097. [DOI] [PubMed] [Google Scholar]
- 223.Lalli MA, Cox HC, Arcila ML, Cadavid L, Moreno S, Garcia G, et al. Origin of the PSEN1 E280A mutation causing early-onset Alzheimer’s disease. Alzheimers Dement. 2014;10:S277–83.e10. doi: 10.1016/j.jalz.2013.09.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 224.Jack CR, Wiste HJ, Botha H, Weigand SD, Therneau TM, Knopman DS, et al. The bivariate distribution of amyloid-β and tau: relationship with established neurocognitive clinical syndromes. Brain. 2019;142:3230–42. doi: 10.1093/brain/awz268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 225.Hanseeuw BJ, Betensky RA, Jacobs HIL, Schultz AP, Sepulcre J, Becker JA, et al. Association of amyloid and tau with cognition in preclinical Alzheimer disease: a longitudinal study. JAMA Neurol. 2019;76:915–24. doi: 10.1001/jamaneurol.2019.1424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 226.Roberson ED, Scearce-Levie K, Palop JJ, Yan F, Cheng IH, Wu T, et al. Reducing endogenous tau ameliorates amyloid beta-induced deficits in an Alzheimer’s disease mouse model. Science. 2007;316:750–4. doi: 10.1126/science.1141736. [DOI] [PubMed] [Google Scholar]
- 227.Oddo S, Caccamo A, Shepherd JD, Murphy MP, Golde TE, Kayed R, et al. Triple-transgenic model of Alzheimer’s disease with plaques and tangles: intracellular Abeta and synaptic dysfunction. Neuron. 2003;39:409–21. doi: 10.1016/s0896-6273(03)00434-3. [DOI] [PubMed] [Google Scholar]
- 228.Pooler AM, Polydoro M, Maury EA, Nicholls SB, Reddy SM, Wegmann S, et al. Amyloid accelerates tau propagation and toxicity in a model of early Alzheimer’s disease. Acta Neuropathol Commun. 2015;3:14. doi: 10.1186/s40478-015-0199-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 229.Bloom GS. Amyloid-β and tau: the trigger and bullet in Alzheimer disease pathogenesis. JAMA Neurol. 2014;71:505–8. doi: 10.1001/jamaneurol.2013.5847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 230.De Felice FG, Wu D, Lambert MP, Fernandez SJ, Velasco PT, Lacor PN, et al. Alzheimer’s disease-type neuronal tau hyperphosphorylation induced by A beta oligomers. Neurobiol Aging. 2008;29:1334–47. doi: 10.1016/j.neurobiolaging.2007.02.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 231.Jin M, Shepardson N, Yang T, Chen G, Walsh D, Selkoe DJ. Soluble amyloid beta-protein dimers isolated from Alzheimer cortex directly induce Tau hyperphosphorylation and neuritic degeneration. Proc Natl Acad Sci USA. 2011;108:5819–24. doi: 10.1073/pnas.1017033108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 232.Choi SH, Kim YH, Hebisch M, Sliwinski C, Lee S, D’Avanzo C, et al. A three-dimensional human neural cell culture model of Alzheimer’s disease. Nature. 2014;515:274–8. doi: 10.1038/nature13800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 233.Perry VH, Holmes C. Microglial priming in neurodegenerative disease. Nat Rev Neurol. 2014;10:217–24. doi: 10.1038/nrneurol.2014.38. [DOI] [PubMed] [Google Scholar]
- 234.Heneka MT, Carson MJ, El Khoury J, Landreth GE, Brosseron F, Feinstein DL, et al. Neuroinflammation in Alzheimer’s disease. Lancet Neurol. 2015;14:388–405. doi: 10.1016/S1474-4422(15)70016-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 235.Sarlus H, Heneka MT. Microglia in Alzheimer’s disease. J Clin Investig. 2017;127:3240–9. doi: 10.1172/JCI90606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 236.Wang S, Colonna M. Microglia in Alzheimer’s disease: a target for immunotherapy. J Leukoc Biol. 2019;106:219–27. doi: 10.1002/JLB.MR0818-319R. [DOI] [PubMed] [Google Scholar]
- 237.Condello C, Yuan P, Schain A, Grutzendler J. Microglia constitute a barrier that prevents neurotoxic protofibrillar Aβ42 hotspots around plaques. Nat Commun. 2015;6:6176. doi: 10.1038/ncomms7176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 238.Edwards FA. A unifying hypothesis for Alzheimer’s disease: from plaques to neurodegeneration. Trends Neurosci. 2019;42:310–22. doi: 10.1016/j.tins.2019.03.003. [DOI] [PubMed] [Google Scholar]
- 239.Streit WJ, Xue QS, Tischer J, Bechmann I. Microglial pathology. Acta Neuropathol Commun. 2014;2:142. doi: 10.1186/s40478-014-0142-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 240.Wang Y, Zhu T, Wang M, Zhang F, Zhang G, Zhao J, et al. Icariin attenuates M1 activation of microglia and Aβ plaque accumulation in the hippocampus and prefrontal cortex by up-regulating PPARγ in restraint/Isolation-stressed APP/PS1 mice. Front Neurosci. 2019;13:291. doi: 10.3389/fnins.2019.00291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 241.Forloni G, Balducci C. Alzheimer’s disease, oligomers, and inflammation. J Alzheimers Dis. 2018;62:1261–76. doi: 10.3233/JAD-170819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 242.Glass CK, Saijo K, Winner B, Marchetto MC, Gage FH. Mechanisms underlying inflammation in neurodegeneration. Cell. 2010;140:918–34. doi: 10.1016/j.cell.2010.02.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 243.Cameron B, Landreth GE. Inflammation, microglia, and Alzheimer’s disease. Neurobiol Dis. 2010;37:503–9. doi: 10.1016/j.nbd.2009.10.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 244.Yates SL, Kocsis-Angle J, Embury P, Brunden KR. Inflammatory responses to amyloid fibrils. Methods Enzymol. 199;309:723–33. [DOI] [PubMed]
- 245.Bamberger ME, Harris ME, McDonald DR, Husemann J, Landreth GE. A cell surface receptor complex for fibrillar beta-amyloid mediates microglial activation. J Neurosci. 2003;23:2665–74. doi: 10.1523/JNEUROSCI.23-07-02665.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 246.Liu Y, Walter S, Stagi M, Cherny D, Letiembre M, Schulz-Schaeffer W, et al. LPS receptor (CD14): a receptor for phagocytosis of Alzheimer’s amyloid peptide. Brain. 2005;128:1778–89. doi: 10.1093/brain/awh531. [DOI] [PubMed] [Google Scholar]
- 247.Paresce DM, Ghosh RN, Maxfield FR. Microglial cells internalize aggregates of the Alzheimer’s disease amyloid beta-protein via a scavenger receptor. Neuron. 1996;17:553–65. doi: 10.1016/s0896-6273(00)80187-7. [DOI] [PubMed] [Google Scholar]
- 248.Stewart CR, Stuart LM, Wilkinson K, van Gils JM, Deng J, Halle A, et al. CD36 ligands promote sterile inflammation through assembly of a Toll-like receptor 4 and 6 heterodimer. Nat Immunol. 2010;11:155–61. doi: 10.1038/ni.1836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 249.Sharma D, Kanneganti TD. The cell biology of inflammasomes: mechanisms of inflammasome activation and regulation. J Cell Biol. 2016;213:617–29. doi: 10.1083/jcb.201602089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 250.Merlini M, Rafalski VA, Ma K, Kim K-Y, Bushong EA, Rios Coronado PE, et al. Microglial G(i)-dependent dynamics regulate brain network hyperexcitability. Nat Neurosci. 2021;24:19–23. doi: 10.1038/s41593-020-00756-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 251.Torrisi SA, Geraci F, Tropea MR, Grasso M, Caruso G, Fidilio A, et al. Fluoxetine and vortioxetine reverse depressive-like phenotype and memory deficits induced by Aβ(1-42) oligomers in mice: a key role of transforming growth factor-β1. Front Pharm. 2019;10:693. doi: 10.3389/fphar.2019.00693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 252.von Bernhardi R, Cornejo F, Parada GE, Eugenín J. Role of TGFβ signaling in the pathogenesis of Alzheimer’s disease. Front Cell Neurosci. 2015;9:426. doi: 10.3389/fncel.2015.00426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 253.Chen JH, Ke KF, Lu JH, Qiu YH, Peng YP. Protection of TGF-β1 against neuroinflammation and neurodegeneration in Aβ1-42-induced Alzheimer’s disease model rats. PLoS One. 2015;10:e0116549. doi: 10.1371/journal.pone.0116549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 254.Tesseur I, et al. Deficiency in neuronal TGF-beta signaling promotes neurodegeneration and Alzheimer’s pathology. J Clin Investig. 2006;116:3060–9. doi: 10.1172/JCI27341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 255.Hopperton KE, Mohammad D, Trepanier MO, Giuliano V, Bazinet RP. Markers of microglia in post-mortem brain samples from patients with Alzheimer’s disease: a systematic review. Mol Psychiatry. 2018;23:177–98. doi: 10.1038/mp.2017.246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 256.Wang MM, Miao D, Cao XP, Tan L, Tan L. Innate immune activation in Alzheimer’s disease. Ann Transl Med. 2018;6:177. doi: 10.21037/atm.2018.04.20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 257.Cantarella G, Di Benedetto G, Puzzo D, Privitera L, Loreto C, Saccone S, et al. Neutralization of TNFSF10 ameliorates functional outcome in a murine model of Alzheimer’s disease. Brain. 2015;138:203–16. doi: 10.1093/brain/awu318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 258.Decourt B, Lahiri DK, Sabbagh MN. Targeting tumor necrosis factor alpha for Alzheimer’s disease. Curr Alzheimer Res. 2017;14:412–25. doi: 10.2174/1567205013666160930110551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 259.Liddelow SA, Guttenplan KA, Clarke LE, Bennett FC, Bohlen CJ, Schirmer L, et al. Neurotoxic reactive astrocytes are induced by activated microglia. Nature. 2017;541:481–7. doi: 10.1038/nature21029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 260.Van Eldik LJ, Carrillo MC, Cole PE, Feuerbach D, Greenberg BD, Hendrix JA, et al. The roles of inflammation and immune mechanisms in Alzheimer’s disease. Alzheimer’s Dement. 2016;2:99–109. doi: 10.1016/j.trci.2016.05.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 261.Brosseron F, Krauthausen M, Kummer M, Heneka MT. Body fluid cytokine levels in mild cognitive impairment and Alzheimer’s disease: a comparative overview. Mol Neurobiol. 2014;50:534–44. doi: 10.1007/s12035-014-8657-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 262.Salter MW, Stevens B. Microglia emerge as central players in brain disease. Nat Med. 2017;23:1018–27. doi: 10.1038/nm.4397. [DOI] [PubMed] [Google Scholar]
- 263.Nimmerjahn A, Kirchhoff F, Helmchen F. Resting microglial cells are highly dynamic surveillants of brain parenchyma in vivo. Science. 2005;308:1314–8. doi: 10.1126/science.1110647. [DOI] [PubMed] [Google Scholar]
- 264.Guerreiro R, Wojtas A, Bras J, Carrasquillo M, Rogaeva E, Majounie E, et al. TREM2 variants in Alzheimer’s disease. N. Engl J Med. 2013;368:117–27. doi: 10.1056/NEJMoa1211851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 265.Lee CYD, Daggett A, Gu X, Jiang L-L, Langfelder P, Li X, et al. Elevated TREM2 gene dosage reprograms microglia responsivity and ameliorates pathological phenotypes in Alzheimer’s disease models. Neuron. 2018;97:1032–48.e5. doi: 10.1016/j.neuron.2018.02.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 266.Griciuc A, Serrano-Pozo A, Parrado AR, Lesinski AN, Asselin CN, Mullin K, et al. Alzheimer’s disease risk gene CD33 inhibits microglial uptake of amyloid beta. Neuron. 2013;78:631–43. doi: 10.1016/j.neuron.2013.04.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 267.Griciuc A, Patel S, Federico AN, Choi SH, Innes BJ, Oram MK, et al. TREM2 acts downstream of CD33 in modulating microglial pathology in Alzheimer’s disease. Neuron. 2019;103:820–.e7. doi: 10.1016/j.neuron.2019.06.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 268.Yeh FL, Wang Y, Tom I, Gonzalez LC, Sheng M. TREM2 binds to apolipoproteins, including APOE and CLU/APOJ, and thereby facilitates uptake of amyloid-beta by microglia. Neuron. 2016;91:328–40. doi: 10.1016/j.neuron.2016.06.015. [DOI] [PubMed] [Google Scholar]
- 269.Olabarria M, Noristani HN, Verkhratsky A, Rodríguez JJ. Concomitant astroglial atrophy and astrogliosis in a triple transgenic animal model of Alzheimer’s disease. Glia. 2010;58:831–8. doi: 10.1002/glia.20967. [DOI] [PubMed] [Google Scholar]
- 270.Arranz AM, De, Strooper B. The role of astroglia in Alzheimer’s disease: pathophysiology and clinical implications. Lancet Neurol. 2019;18:406–14. doi: 10.1016/S1474-4422(18)30490-3. [DOI] [PubMed] [Google Scholar]
- 271.Wyss-Coray T, Loike JD, Brionne TC, Lu E, Anankov R, Yan F, et al. Adult mouse astrocytes degrade amyloid-beta in vitro and in situ. Nat Med. 2003;9:453–7. doi: 10.1038/nm838. [DOI] [PubMed] [Google Scholar]
- 272.Kerbler GM, Fripp J, Rowe CC, Villemagne VL, Salvado O, Rose S, et al. Basal forebrain atrophy correlates with amyloid β burden in Alzheimer’s disease. NeuroImage Clin. 2015;7:105–13. doi: 10.1016/j.nicl.2014.11.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 273.Grothe MJ, Heinsen H, Amaro E, Jr, Grinberg LT, Teipel SJ. Cognitive correlates of basal forebrain atrophy and associated cortical hypometabolism in mild cognitive impairment. Cereb Cortex. 2016;26:2411–26. doi: 10.1093/cercor/bhv062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 274.Chiesa PA, Cavedo E, Grothe MJ, Houot M, Teipel SJ, Potier M-C, et al. Relationship between basal forebrain resting-state functional connectivity and brain amyloid-β deposition in cognitively intact older adults with subjective memory complaints. Radiology. 2019;290:167–76. doi: 10.1148/radiol.2018180268. [DOI] [PubMed] [Google Scholar]
- 275.Teipel SJ, Cavedo E, Hampel H, Grothe MJ. Alzheimer’s Disease Neuroimaging Initiative; Alzheimer Precision Medicine Initiative (APMI). Basal Forebrain Volume, but Not Hippocampal Volume, is a Predictor of Global Cognitive Decline in Patients With Alzheimer’s Disease Treated With Cholinesterase Inhibitors. Front Neurol. 2018;9:642. doi: 10.3389/fneur.2018.00642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 276.Cavedo E, Grothe MJ, Colliot O, Lista S, Chupin M, Dormont D, et al. Reduced basal forebrain atrophy progression in a randomized Donepezil trial in prodromal Alzheimer’s disease. Sci Rep. 2017;7:11706. doi: 10.1038/s41598-017-09780-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 277.Grothe MJ, Ewers M, Krause B, Heinsen H, Teipel SJ. Alzheimer’s disease neuroimaging Initiative. Basal forebrain atrophy and cortical amyloid deposition in nondemented elderly subjects. Alzheimers Dement. 2014;10:S344–53. doi: 10.1016/j.jalz.2013.09.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 278.Beach TG, Honer WG, Hughes LH. Cholinergic fibre loss associated with diffuse plaques in the non-demented elderly: the preclinical stage of Alzheimer’s disease? Acta Neuropathol. 1997;93:146–53. doi: 10.1007/s004010050595. [DOI] [PubMed] [Google Scholar]
- 279.Lai MKP, Tsang SWY, Garcia-Alloza M, Minger SL, Nicoll JAR, Esiri MM, et al. Selective effects of the APOE epsilon4 allele on presynaptic cholinergic markers in the neocortex of Alzheimer’s disease. Neurobiol Dis. 2006;22:555–61. doi: 10.1016/j.nbd.2005.12.016. [DOI] [PubMed] [Google Scholar]
- 280.Grothe M, Heinsen H, Teipel S. Longitudinal measures of cholinergic forebrain atrophy in the transition from healthy aging to Alzheimer’s disease. Neurobiol Aging. 2013;34:1210–20. doi: 10.1016/j.neurobiolaging.2012.10.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 281.Hall AM, Moore RY, Lopez OL, Kuller L, Becker JT. Basal forebrain atrophy is a presymptomatic marker for Alzheimer’s disease. Alzheimers Dement. 2008;4:271–9. doi: 10.1016/j.jalz.2008.04.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 282.Cisse M, Braun U, Leitges M, Fisher A, Pages G, Checler F, et al. ERK1-independent α-secretase cut of β-amyloid precursor protein via M1 muscarinic receptors and PKCα/ε. Mol Cell Neurosci. 2011;47:223–32. doi: 10.1016/j.mcn.2011.04.008. [DOI] [PubMed] [Google Scholar]
- 283.Jaworski T, Dewachter I, Lechat B, Gees M, Kremer A, Demedts D, et al. GSK-3α/β kinases and amyloid production in vivo. Nature. 2011;480:E4–5. doi: 10.1038/nature10615. [DOI] [PubMed] [Google Scholar]
- 284.Chu J, Lauretti E, Praticò D. Caspase-3-dependent cleavage of Akt modulates tau phosphorylation via GSK3β kinase: implications for Alzheimer’s disease. Mol Psychiatry. 2017;22:1002–8. doi: 10.1038/mp.2016.214. [DOI] [PubMed] [Google Scholar]
- 285.Espada S, Rojo AI, Salinas M, Cuadrado A. The muscarinic M1 receptor activates Nrf2 through a signaling cascade that involves protein kinase C and inhibition of GSK-3beta: connecting neurotransmission with neuroprotection. J Neurochem. 2009;110:1107–19. doi: 10.1111/j.1471-4159.2009.06208.x. [DOI] [PubMed] [Google Scholar]
- 286.Snyder EM, Nong Y, Almeida CG, Paul S, Moran T, Choi EY, et al. Regulation of NMDA receptor trafficking by amyloid-beta. Nat Neurosci. 2005;8:1051–8. doi: 10.1038/nn1503. [DOI] [PubMed] [Google Scholar]
- 287.Busche MA, Eichhoff G, Adelsberger H, Abramowski D, Wiederhold K-H, Haass C, et al. Clusters of hyperactive neurons near amyloid plaques in a mouse model of Alzheimer’s disease. Science. 2008;321:1686–9. doi: 10.1126/science.1162844. [DOI] [PubMed] [Google Scholar]
- 288.Li S, Hong S, Shepardson NE, Walsh DM, Shankar GM, Selkoe D. Soluble oligomers of amyloid Beta protein facilitate hippocampal long-term depression by disrupting neuronal glutamate uptake. Neuron. 2009;62:788–801. doi: 10.1016/j.neuron.2009.05.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 289.Shankar GM, Bloodgood BL, Townsend M, Walsh DM, Selkoe DJ, Sabatini BL. Natural oligomers of the Alzheimer amyloid-beta protein induce reversible synapse loss by modulating an NMDA-type glutamate receptor-dependent signaling pathway. J Neurosci. 2007;27:2866–75. doi: 10.1523/JNEUROSCI.4970-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 290.Carter TL, Rissman RA, Mishizen-Eberz AJ, Wolfe BB, Hamilton RL, Gandy S, et al. Differential preservation of AMPA receptor subunits in the hippocampi of Alzheimer’s disease patients according to Braak stage. Exp Neurol. 2004;187:299–309. doi: 10.1016/j.expneurol.2003.12.010. [DOI] [PubMed] [Google Scholar]
- 291.Wei W, Nguyen LN, Kessels HW, Hagiwara H, Sisodia S, Malinow R. Amyloid beta from axons and dendrites reduces local spine number and plasticity. Nat Neurosci. 2010;13:190–6. doi: 10.1038/nn.2476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 292.Reinders NR, Pao Y, Renner MC, da Silva-Matos CM, Lodder TR, Malinow R, et al. Amyloid-β effects on synapses and memory require AMPA receptor subunit GluA3. Proc Natl Acad Sci USA. 2016;113:E6526–E6534. doi: 10.1073/pnas.1614249113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 293.Chang EH, Savage MJ, Flood DG, Thomas JM, Levy RB, Mahadomrongkul V, et al. AMPA receptor downscaling at the onset of Alzheimer’s disease pathology in double knockin mice. Proc Natl Acad Sci USA. 2006;103:3410–5. doi: 10.1073/pnas.0507313103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 294.Zhang Y, Guo O, Huo Y, Wang G, Man HY. Amyloid-β Induces AMPA Receptor Ubiquitination and Degradation in Primary Neurons and Human Brains of Alzheimer’s Disease. J Alzheimers Dis. 2018;62:1789–801. doi: 10.3233/JAD-170879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 295.Hadjichrysanthou C, Evans S, Bajaj S, Siakallis LC, McRae-McKee K, de Wolf F, et al. Alzheimer’s Disease Neuroimaging Initiative. The dynamics of biomarkers across the clinical spectrum of Alzheimer’s disease. Alzheimers Res Ther. 2020;12:74. doi: 10.1186/s13195-020-00636-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 296.Parker TD, Cash DM, Lane CAS, Lu K, Malone IB, Nicholas JM, et al. Hippocampal subfield volumes and pre-clinical Alzheimer’s disease in 408 cognitively normal adults born in 1946. PLoS One. 2019;14:e0224030. doi: 10.1371/journal.pone.0224030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 297.Chételat G, Villemagne VL, Pike KE, Ellis KA, Bourgeat P, Jones G, et al. Independent contribution of temporal beta-amyloid deposition to memory decline in the pre-dementia phase of Alzheimer’s disease. Brain. 2011;134:798–807. doi: 10.1093/brain/awq383. [DOI] [PubMed] [Google Scholar]
- 298.Wang L, Benzinger TL, Hassenstab J, Blazey T, Owen C, Liu J, et al. Spatially distinct atrophy is linked to β-amyloid and tau in preclinical Alzheimer disease. Neurology. 2015;84:1254–60. doi: 10.1212/WNL.0000000000001401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 299.Molinuevo JL, Ayton S, Batrla R, Bednar MM, Bittner T, Cummings J, et al. Current state of Alzheimer’s fluid biomarkers. Acta Neuropathol. 2018;136:821–53. doi: 10.1007/s00401-018-1932-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 300.Sheline YI, Raichle ME. Resting state functional connectivity in preclinical Alzheimer’s disease. Biol Psychiatry. 2013;74:340–7. doi: 10.1016/j.biopsych.2012.11.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 301.Agosta F, Pievani M, Geroldi C, Copetti M, Frisoni GB, Filippi M. Resting state fMRI in Alzheimer’s disease: beyond the default mode network. Neurobiol Aging. 2012;33:1564–78. doi: 10.1016/j.neurobiolaging.2011.06.007. [DOI] [PubMed] [Google Scholar]
- 302.Buckley RF, Schultz AP, Hedden T, Papp KV, Hanseeuw BJ, Marshall G, et al. Functional network integrity presages cognitive decline in preclinical Alzheimer disease. Neurology. 2017;89:29–37. doi: 10.1212/WNL.0000000000004059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 303.Sperling RA, Laviolette PS, O’Keefe K, O’Brien J, Rentz DM, Pihlajamaki M, et al. Amyloid deposition is associated with impaired default network function in older persons without dementia. Neuron. 2009;63:178–88. doi: 10.1016/j.neuron.2009.07.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 304.Mormino EC, Smiljic A, Hayenga AO, Onami SH, Greicius MD, Rabinovici GD, et al. Relationships between β-amyloid and functional connectivity in different components of the default mode network in aging. Cereb Cortex. 2011;21:2399–407. doi: 10.1093/cercor/bhr025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 305.Li Y, Yao Z, Yu Y, Zou Y, Fu Y, Hu B. Brain network alterations in individuals with and without mild cognitive impairment: parallel independent component analysis of AV1451 and AV45 positron emission tomography. BMC Psychiatry. 2019;19:165. doi: 10.1186/s12888-019-2149-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 306.Palmqvist S, Schöll M, Strandberg O, Mattsson N, Stomrud E, Zetterberg H, et al. Earliest accumulation of β-amyloid occurs within the default-mode network and concurrently affects brain connectivity. Nat Commun. 2017;8:1214. doi: 10.1038/s41467-017-01150-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 307.Chhatwal JP, Schultz AP, Johnson KA, Hedden T, Jaimes S, Benzinger TLS, et al. Preferential degradation of cognitive networks differentiates Alzheimer’s disease from ageing. Brain. 2018;141:1486–1500. doi: 10.1093/brain/awy053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 308.Pereira JB, Ossenkoppele R, Palmqvist S, Strandberg TO, Smith R, Westman E, et al. Amyloid and tau accumulate across distinct spatial networks and are differentially associated with brain connectivity. Elife. 2019;8. 10.7554/eLife.50830. [DOI] [PMC free article] [PubMed]
- 309.Hampton OL, Buckley RF, Manning LK, Scott MR, Properzi MJ, Peña-Gómez C, et al. Resting-state functional connectivity and amyloid burden influence longitudinal cortical thinning in the default mode network in preclinical Alzheimer’s disease. NeuroImage Clin. 2020;28:102407. doi: 10.1016/j.nicl.2020.102407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 310.Myers N, Pasquini L, Göttler J, Grimmer T, Koch K, Ortner M, et al. Within-patient correspondence of amyloid-β and intrinsic network connectivity in Alzheimer’s disease. Brain. 2014;137:2052–64. doi: 10.1093/brain/awu103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 311.Grothe MJ, Teipel SJ. Spatial patterns of atrophy, hypometabolism, and amyloid deposition in Alzheimer’s disease correspond to dissociable functional brain networks. Hum Brain Mapp. 2016;37:35–53. doi: 10.1002/hbm.23018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 312.Lin C, Ly M, Karim HT, Wei W, Snitz BE, Klunk WE, et al. The effect of amyloid deposition on longitudinal resting-state functional connectivity in cognitively normal older adults. Alzheimers Res Ther. 2020;12:7. doi: 10.1186/s13195-019-0573-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 313.Andrews-Hann JR, Snyder AZ, Vincent JL, Lustig C, Head D, Raichle ME, et al. Disruption of large-scale brain systems in advanced aging. Neuron. 2007;56:924–35. doi: 10.1016/j.neuron.2007.10.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 314.Brier MR, Thomas JB, Snyder AZ, Benzinger TL, Zhang D, Raichle ME, et al. Loss of intranetwork and internetwork resting state functional connections with Alzheimer’s disease progression. J Neurosci. 2012;32:8890–9. doi: 10.1523/JNEUROSCI.5698-11.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 315.Zhou J, Greicius MD, Gennatas ED, Growdon ME, Jang JY, Rabinovici GD, et al. Divergent network connectivity changes in behavioural variant frontotemporal dementia and Alzheimer’s disease. Brain. 2010;133:1352–67. doi: 10.1093/brain/awq075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 316.He X, Qin W, Liu Y, Zhang X, Duan Y, Song J, et al. Abnormal salience network in normal aging and in amnestic mild cognitive impairment and Alzheimer’s disease. Hum Brain Mapp. 2014;35:3446–64. doi: 10.1002/hbm.22414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 317.Lim YY, Baker JE, Bruns L, Jr., Mills A, Fowler CJ, Fripp J, et al. Deficits in learning are greater than memory dysfunction in preclinical Alzheimer’s disease. Alzheimer’s Dement. 2020;16:e045901. [Google Scholar]
- 318.Olsson B, Lautner R, Andreasson U, Öhrfelt A, Portelius E, Bjerke M, et al. CSF and blood biomarkers for the diagnosis of Alzheimer’s disease: a systematic review and meta-analysis. Lancet Neurol. 2016;15:673–84. doi: 10.1016/S1474-4422(16)00070-3. [DOI] [PubMed] [Google Scholar]
- 319.Toledo JB, Bjerke M, Da X, Landau SM, Foster NL, Jagust W, et al. Nonlinear association between cerebrospinal fluid and florbetapir F-18 β-amyloid measures across the spectrum of Alzheimer disease. JAMA Neurol. 2015;72:571–81. doi: 10.1001/jamaneurol.2014.4829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 320.Janelidze S, Pannee J, Mikulskis A, Chiao P, Zetterberg H, Blennow K, et al. Concordance between different amyloid immunoassays and visual amyloid positron emission tomographic assessment. JAMA Neurol. 2017;74:1492–501. doi: 10.1001/jamaneurol.2017.2814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 321.Shaw LM, Vanderstichele H, Knapik-Czajka M, Clark CM, Aisen PS, Petersen RC, et al. Cerebrospinal fluid biomarker signature in Alzheimer’s disease neuroimaging initiative subjects. Ann Neurol. 2009;65:403–13. doi: 10.1002/ana.21610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 322.Mattsson N, Zetterberg H, Hansson O, Andreasen N, Parnetti L, Jonsson M, et al. CSF biomarkers and incipient Alzheimer disease in patients with mild cognitive impairment. JAMA. 2009;302:385–93. doi: 10.1001/jama.2009.1064. [DOI] [PubMed] [Google Scholar]
- 323.Blennow K, Hampel H, Weiner M, Zetterberg H. Cerebrospinal fluid and plasma biomarkers in Alzheimer disease. Nat Rev Neurol. 2010;6:131–44. doi: 10.1038/nrneurol.2010.4. [DOI] [PubMed] [Google Scholar]
- 324.Fagan AM, Roe CM, Xiong C, Mintun MA, Morris JC, Holtzman DM. Cerebrospinal fluid tau/beta-amyloid(42) ratio as a prediction of cognitive decline in nondemented older adults. Arch Neurol. 2007;64:343–9. doi: 10.1001/archneur.64.3.noc60123. [DOI] [PubMed] [Google Scholar]
- 325.Buckley RF, Maruff P, Ames D, Bourgeat P, Martins RN, Masters CL, et al. Subjective memory decline predicts greater rates of clinical progression in preclinical Alzheimer’s disease. Alzheimers Dement. 2016;12:796–804. doi: 10.1016/j.jalz.2015.12.013. [DOI] [PubMed] [Google Scholar]
- 326.van Harten AC, Mielke MM, Swenson-Dravis DM, Hagen CE, Edwards KK, Roberts RO, et al. Subjective cognitive decline and risk of MCI: The Mayo Clinic Study of Aging. Neurology. 2018;91:e300–e312. doi: 10.1212/WNL.0000000000005863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 327.Blennow K, Mattsson N, Scholl M, Hansson O, Zetterberg H. Amyloid biomarkers in Alzheimer’s disease. Trends Pharm Sci. 2015;36:297–309. doi: 10.1016/j.tips.2015.03.002. [DOI] [PubMed] [Google Scholar]
- 328.Lewczuk P, Lelental N, Spitzer P, Maler JM, Kornhuber J. Amyloid-β 42/40 cerebrospinal fluid concentration ratio in the diagnostics of Alzheimer’s disease: validation of two novel assays. J Alzheimers Dis. 2015;43:183–91. doi: 10.3233/JAD-140771. [DOI] [PubMed] [Google Scholar]
- 329.Paterson RW, Slattery CF, Poole T, Nicholas JM, Magdalinou NK, Toombs J, et al. Cerebrospinal fluid in the differential diagnosis of Alzheimer’s disease: clinical utility of an extended panel of biomarkers in a specialist cognitive clinic. Alzheimers Res Ther. 2018;10:32. doi: 10.1186/s13195-018-0361-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 330.Lautner R, Palmqvist S, Mattsson N, Andreasson U, Wallin A, Pålsson E, et al. Apolipoprotein E genotype and the diagnostic accuracy of cerebrospinal fluid biomarkers for Alzheimer disease. JAMA Psychiatry. 2014;71:1183–91. doi: 10.1001/jamapsychiatry.2014.1060. [DOI] [PubMed] [Google Scholar]
- 331.Hampel H, Shaw LM, Aisen P, Chen C, Lleó A, Iwatsubo T. et al. State-of-the-art of lumbar puncture and its place in the journey of patients with Alzheimer’s disease. Alzheimers Dement. 2021;1–19. 10.1002/alz.12372. [DOI] [PMC free article] [PubMed]
- 332.Kaplow J, Vandijck M, Gray J, Kanekiyo M, Huyck E, Traynham CJ, et al. Concordance of Lumipulse cerebrospinal fluid t-tau/Aβ42 ratio with amyloid PET status. Alzheimers Dement. 2020;16:144–52. doi: 10.1002/alz.12000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 333.Vanderstichele H, Bibl M, Engelborghs S, Le Bastard N, Lewczuk P, Molinuevo JL, et al. Standardization of preanalytical aspects of cerebrospinal fluid biomarker testing for Alzheimer’s disease diagnosis: a consensus paper from the Alzheimer’s Biomarkers Standardization Initiative. Alzheimers Dement. 2012;8:65–73. doi: 10.1016/j.jalz.2011.07.004. [DOI] [PubMed] [Google Scholar]
- 334.Mattsson N, Insel PS, Donohue M, Landau S, Jagust WJ, Shaw LM, et al. Independent information from cerebrospinal fluid amyloid-β and florbetapir imaging in Alzheimer’s disease. Brain. 2015;138:772–83. doi: 10.1093/brain/awu367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 335.Palmqvist S, Mattsson N, Hansson O. Alzheimer’s disease neuroimaging initiative. Cerebrospinal fluid analysis detects cerebral amyloid-β accumulation earlier than positron emission tomography. Brain. 2016;139:1226–36. doi: 10.1093/brain/aww015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 336.Hampel H, O’Bryant SE, Molinuevo JL, Zetterberg H, Masters CL, Lista S, et al. Blood-based biomarkers for Alzheimer disease: mapping the road to the clinic. Nat Rev Neurol. 2018;14:639–52. doi: 10.1038/s41582-018-0079-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 337.Zetterberg H, Wilson D, Andreasson U, Minthon L, Blennow K, Randall J, et al. Plasma tau levels in Alzheimer’s disease. Alzheimers Res Ther. 2013;5:9. doi: 10.1186/alzrt163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 338.Vergallo A, Mégret L, Lista S, Cavedo E, Zetterberg H, Blennow K, et al. Plasma amyloid β 40/42 ratio predicts cerebral amyloidosis in cognitively normal individuals at risk for Alzheimer’s disease. Alzheimers Dement. 2019;15:764–75. doi: 10.1016/j.jalz.2019.03.009. [DOI] [PubMed] [Google Scholar]
- 339.Janelidze S, Stomrud E, Palmqvist S, Zetterberg H, van Westen D, Jeromin A, et al. Plasma β-amyloid in Alzheimer’s disease and vascular disease. Sci Rep. 2016;6:26801. doi: 10.1038/srep26801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 340.Nakamura A, Kaneko N, Villemagne VL, Kato T, Doecke J, Doré V, et al. High performance plasma amyloid-β biomarkers for Alzheimer’s disease. Nature. 2018;554:249–54. doi: 10.1038/nature25456. [DOI] [PubMed] [Google Scholar]
- 341.Schindler SE, Bollinger JG, Ovod V, Mawuenyega KG, Li Y, Gordon BA, et al. High-precision plasma β-amyloid 42/40 predicts current and future brain amyloidosis. Neurology. 2019;93:e1647–59. doi: 10.1212/WNL.0000000000008081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 342.Teunissen CE, Chiu M-J, Yang C-C, Yang S-Y, Scheltens P, Zetterberg H, et al. Plasma amyloid-β (Aβ42) correlates with cerebrospinal fluid Aβ42 in Alzheimer’s disease. J Alzheimers Dis. 2018;62:1857–63. doi: 10.3233/JAD-170784. [DOI] [PubMed] [Google Scholar]
- 343.Keshavan A, Pannee J, Karikari TK, Rodriguez JL, Ashton NJ, Nicholas JM, et al. Population-based blood screening for preclinical Alzheimer’s disease in a British birth cohort at age 70. Brain. 2021;144:434–49. doi: 10.1093/brain/awaa403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 344.Lim CZJ, Zhang Y, Chen Y, Zhao H, Stephenson MC, Ho NRY, et al. Subtyping of circulating exosome-bound amyloid β reflects brain plaque deposition. Nat Commun. 2019;10:1144. doi: 10.1038/s41467-019-09030-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 345.Palmqvist S, Janelidze S, Stomrud E, Zetterberg H, Karl J, Zink K, et al. Performance of fully automated plasma assays as screening tests for Alzheimer Disease-related β-amyloid status. JAMA Neurol. 2019;76:1060–9. doi: 10.1001/jamaneurol.2019.1632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 346.Yamashita K, Hasegawa T, Iino T, Miura M, Watanabe T, Watanabe S, et al. P075-prediction of amyloid pathology by the plasma Aβ(1-42)/Aβ(1-40) ratio measured with fully automated immunoassay system (HISCL™ SERIES) J Prev Alz Dis. 2019;6:S89. [Google Scholar]
- 347.Fukumoto H, Tokuda T, Kasai T, Ishigami N, Hidaka H, Kondo M, et al. High-molecular-weight beta-amyloid oligomers are elevated in cerebrospinal fluid of Alzheimer patients. FASEB J Publ Fed Am Soc Exp Biol. 2010;24:2716–26. doi: 10.1096/fj.09-150359. [DOI] [PubMed] [Google Scholar]
- 348.Savage MJ, Kalinina J, Wolfe A, Tugusheva K, Korn R, Cash-Mason T, et al. A sensitive aβ oligomer assay discriminates Alzheimer’s and aged control cerebrospinal fluid. J Neurosci. 2014;34:2884–97. doi: 10.1523/JNEUROSCI.1675-13.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 349.Yang T, O’Malley TT, Kanmert D, Jerecic J, Zieske LR, Zetterberg H, et al. A highly sensitive novel immunoassay specifically detects low levels of soluble Aβ oligomers in human cerebrospinal fluid. Alzheimers Res Ther. 2015;7:14. doi: 10.1186/s13195-015-0100-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 350.Santos AN, Ewers M, Minthon L, Simm A, Silber R-E, Blennow K, et al. Amyloid-β oligomers in cerebrospinal fluid are associated with cognitive decline in patients with Alzheimer’s disease. J Alzheimers Dis. 2012;29:171–6. doi: 10.3233/JAD-2012-111361. [DOI] [PubMed] [Google Scholar]
- 351.Horrocks MH, Lee SF, Gandhi S, Magdalinou NK, Chen SW, Devine MJ, et al. Single-molecule imaging of individual amyloid protein aggregates in human biofluids. ACS Chem Neurosci. 2016;7:399–406. doi: 10.1021/acschemneuro.5b00324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 352.Esparza TJ, Zhao H, Cirrito JR, Cairns NJ, Bateman RJ, Holtzman DM, et al. Amyloid-β oligomerization in Alzheimer dementia versus high-pathology controls. Ann Neurol. 2013;73:104–19. doi: 10.1002/ana.23748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 353.Hölttä M, Hansson O, Andreasson U, Hertze J, Minthon L, Nägga K, et al. Evaluating amyloid-β oligomers in cerebrospinal fluid as a biomarker for Alzheimer’s disease. PLoS One. 2013;8:e66381. doi: 10.1371/journal.pone.0066381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 354.Zhou L, Chan KH, Chu LW, Kwan JSC, Song YQ, Chen LH, et al. Plasma amyloid-β oligomers level is a biomarker for Alzheimer’s disease diagnosis. Biochem Biophys Res Commun. 2012;423:697–702. doi: 10.1016/j.bbrc.2012.06.017. [DOI] [PubMed] [Google Scholar]
- 355.Xia W, Yang T, Shankar G, Smith IM, Shen Y, Walsh DM, et al. A specific enzyme-linked immunosorbent assay for measuring beta-amyloid protein oligomers in human plasma and brain tissue of patients with Alzheimer disease. Arch Neurol. 2009;66:190–9. doi: 10.1001/archneurol.2008.565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 356.Santos AN, Simm A, Holthoff V, Boehm G. A method for the detection of amyloid-beta1-40:amyloid-beta1-42 and amyloid-beta oligomers in blood using magnetic beads in combination with Flow cytometry and its application in the diagnostics of Alzheimer’s disease. J Alzheimers Dis. 2008;14:127–31. doi: 10.3233/jad-2008-14201. [DOI] [PubMed] [Google Scholar]
- 357.An SS, Lim KT, Oh HJ, Lee BS, Zukic E, Ju YR, et al. Differentiating blood samples from scrapie infected and non-infected hamsters by detecting disease-associated prion proteins using Multimer Detection System. Biochem Biophys Res Commun. 2010;392:505–9. doi: 10.1016/j.bbrc.2010.01.053. [DOI] [PubMed] [Google Scholar]
- 358.Lim K, Kim SY, Lee B, Segarra C, Kang S, Ju Y, et al. Magnetic microparticle-based multimer detection system for the detection of prion oligomers in sheep. Int J Nanomed. 2015;10:241–50. doi: 10.2147/IJN.S88377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 359.Wang MJ, Yi S, Han J-Y, Park SY, Jang J-W, Chun IK, et al. Oligomeric forms of amyloid-β protein in plasma as a potential blood-based biomarker for Alzheimer’s disease. Alzheimers Res Ther. 2017;9:98. doi: 10.1186/s13195-017-0324-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 360.Klunk WE, Koeppe RA, Price JC, Benzinger TL, Devous MDS, Jagust WJ, et al. The Centiloid Project: standardizing quantitative amyloid plaque estimation by PET. Alzheimers Dement. 2015;11:1–4. doi: 10.1016/j.jalz.2014.07.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 361.Kadir A, Marutle A, Gonzalez D, Schöll M, Almkvist O, Mousavi M, et al. Positron emission tomography imaging and clinical progression in relation to molecular pathology in the first Pittsburgh Compound B positron emission tomography patient with Alzheimer’s disease. Brain. 2011;134:301–17. doi: 10.1093/brain/awq349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 362.Brookmeyer R, Abdalla N. Estimation of lifetime risks of Alzheimer’s disease dementia using biomarkers for preclinical disease. Alzheimers Dement. 2018;14:981–8. doi: 10.1016/j.jalz.2018.03.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 363.Klunk WE, Engler H, Nordberg A, Wang Y, Blomqvist G, Holt DP, et al. Imaging brain amyloid in Alzheimer’s disease with Pittsburgh Compound-B. Ann Neurol. 2004;55:306–19. doi: 10.1002/ana.20009. [DOI] [PubMed] [Google Scholar]
- 364.Rowe CC, Ng S, Ackermann U, Gong SJ, Pike K, Savage G, et al. Imaging beta-amyloid burden in aging and dementia. Neurology. 2007;68:1718–25. doi: 10.1212/01.wnl.0000261919.22630.ea. [DOI] [PubMed] [Google Scholar]
- 365.Rowe CC, Villemagne VL. Brain amyloid imaging. J Nucl Med Technol. 2013;41:11–8. doi: 10.2967/jnumed.110.076315. [DOI] [PubMed] [Google Scholar]
- 366.Leuzy A, Zimmer ER, Heurling K, Rosa-Neto P, Gauthier S. Use of amyloid PET across the spectrum of Alzheimer’s disease: clinical utility and associated ethical issues. Amyloid. 2014;21:143–8. doi: 10.3109/13506129.2014.926267. [DOI] [PubMed] [Google Scholar]
- 367.Wolk DA, Zhang Z, Boudhar S, Clark CM, Pontecorvo MJ, Arnold SE. Amyloid imaging in Alzheimer’s disease: comparison of florbetapir and Pittsburgh compound-B positron emission tomography. J Neurol Neurosurg Psychiatry. 2012;83:923–6. doi: 10.1136/jnnp-2012-302548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 368.Landau SM, Thomas BA, Thurfjell L, Schmidt M, Margolin R, Mintun M, et al. Amyloid PET imaging in Alzheimer’s disease: a comparison of three radiotracers. Eur J Nucl Med Mol Imaging. 2014;41:1398–407. doi: 10.1007/s00259-014-2753-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 369.Morris E, Chalkidou A, Hammers A, Peacock J, Summers J, Keevil S. Diagnostic accuracy of (18)F amyloid PET tracers for the diagnosis of Alzheimer’s disease: a systematic review and meta-analysis. Eur J Nucl Med Mol Imaging. 2016;43:374–85. doi: 10.1007/s00259-015-3228-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 370.Graff-Radford J, Yong KXX, Apostolova LG, Bouwman FH, Carrillo M, Dickerson BC, et al. New insights into atypical Alzheimer’s disease in the era of biomarkers. Lancet Neurol. 2021;20:222–34. doi: 10.1016/S1474-4422(20)30440-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 371.Grothe MJ, Barthel H, Sepulcre J, Dyrba M, Sabri O, Teipel SJ. Alzheimer’s Disease Neuroimaging Initiative. In vivo staging of regional amyloid deposition. Neurology. 2017;89:2031–8. doi: 10.1212/WNL.0000000000004643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 372.Teipel SJ, Dyrba M, Chiesa PA, Sakr F, Jelistratova I, Lista S, et al. In vivo staging of regional amyloid deposition predicts functional conversion in the preclinical and prodromal phases of Alzheimer’s disease. Neurobiol Aging. 2020;93:98–108. doi: 10.1016/j.neurobiolaging.2020.03.011. [DOI] [PubMed] [Google Scholar]
- 373.Clark CM, Pontecorvo MJ, Beach TG, Bedell BJ, Coleman RE, Doraiswamy PM, et al. Cerebral PET with florbetapir compared with neuropathology at autopsy for detection of neuritic amyloid-β plaques: a prospective cohort study. Lancet Neurol. 2012;11:669–78. doi: 10.1016/S1474-4422(12)70142-4. [DOI] [PubMed] [Google Scholar]
- 374.Yamin G, Teplow DB. Pittsburgh Compound-B (PiB) binds amyloid β-protein protofibrils. J Neurochem. 2017;140:210–5. doi: 10.1111/jnc.13887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 375.Maezawa I, Hong H-S, Liu R, Wu C-Y, Cheng RH, Kung M-P, et al. Congo red and thioflavin-T analogs detect Abeta oligomers. J Neurochem. 2008;104:457–68. doi: 10.1111/j.1471-4159.2007.04972.x. [DOI] [PubMed] [Google Scholar]
- 376.Syvänen S, Fang XT, Hultqvist G, Meier SR, Lannfelt L, Sehlin D. A bispecific Tribody PET radioligand for visualization of amyloid-beta protofibrils—a new concept for neuroimaging. Neuroimage. 2017;148:55–63. doi: 10.1016/j.neuroimage.2017.01.004. [DOI] [PubMed] [Google Scholar]
- 377.van Dongen GA, Poot AJ, Vugts DJ. PET imaging with radiolabeled antibodies and tyrosine kinase inhibitors: immuno-PET and TKI-PET. Tumour Biol. 2012;33:607–15. doi: 10.1007/s13277-012-0316-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 378.Fang XT, Hultqvist G, Meier SR, Antoni G, Sehlin D, Syvänen S. High detection sensitivity with antibody-based PET radioligand for amyloid beta in brain. Neuroimage. 2019;184:881–8. doi: 10.1016/j.neuroimage.2018.10.011. [DOI] [PubMed] [Google Scholar]
- 379.Sehlin D, Syvänen S. MINC faculty. Engineered antibodies: new possibilities for brain PET? Eur J Nucl Med Mol Imaging. 2019;46:2848–58. doi: 10.1007/s00259-019-04426-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 380.Saunders AM, Schmader K, Breitner JC, Benson MD, Brown WT, Goldfarb L, et al. Apolipoprotein E epsilon 4 allele distributions in late-onset Alzheimer’s disease and in other amyloid-forming diseases. Lancet. 1993;342:710–1. doi: 10.1016/0140-6736(93)91709-u. [DOI] [PubMed] [Google Scholar]
- 381.Harold D, Abraham R, Hollingworth P, Sims R, Gerrish A, Hamshere ML, et al. Genome-wide association study identifies variants at CLU and PICALM associated with Alzheimer’s disease. Nat Genet. 2009;41:1088–93. doi: 10.1038/ng.440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 382.Lambert JC, Heath S, Even G, Campion D, Sleegers K, Hiltunen M, et al. Genome-wide association study identifies variants at CLU and CR1 associated with Alzheimer’s disease. Nat Genet. 2009;41:1094–9. doi: 10.1038/ng.439. [DOI] [PubMed] [Google Scholar]
- 383.Kunkle BW, Grenier-Boley B, Sims R, Bis JC, Damotte V, Naj AC, et al. Genetic meta-analysis of diagnosed Alzheimer’s disease identifies new risk loci and implicates Aβ, tau, immunity and lipid processing. Nat Genet. 2019;51:414–30. doi: 10.1038/s41588-019-0358-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 384.Bis JC, Jian X, Kunkle BW, Chen Y, Hamilton-Nelson KL, Bush WS, et al. Whole exome sequencing study identifies novel rare and common Alzheimer’s-Associated variants involved in immune response and transcriptional regulation. Mol Psychiatry. 2020;25:1859–75. doi: 10.1038/s41380-018-0112-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 385.Sims R, van der Lee SJ, Naj AC, Bellenguez C, Badarinarayan N, Jakobsdottir J, et al. Rare coding variants in PLCG2, ABI3, and TREM2 implicate microglial-mediated innate immunity in Alzheimer’s disease. Nat Genet. 2017;49:1373–84. doi: 10.1038/ng.3916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 386.Seshadri S, Fitzpatrick AL, Ikram MA, DeStefano AL, Gudnason V, Boada M, et al. CHARGE Consortium; GERAD1 Consortium; EADI1 Consortium. Genome-wide analysis of genetic loci associated with Alzheimer disease. JAMA. 2010;303:1832–40. doi: 10.1001/jama.2010.574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 387.Bellenguez C, Charbonnier C, Grenier-Boley B, Quenez O, Le Guennec K, Nicolas G, et al. CNR MAJ collaborators. Contribution to Alzheimer’s disease risk of rare variants in TREM2, SORL1, and ABCA7 in 1779 cases and 1273 controls. Neurobiol Aging. 2017;59:220.e1–9. doi: 10.1016/j.neurobiolaging.2017.07.001. [DOI] [PubMed] [Google Scholar]
- 388.Hollingworth P, Harold D, Sims R, Gerrish A, Lambert JC, Carrasquillo MM, et al. Common variants at ABCA7, MS4A6A/MS4A4E, EPHA1, CD33 and CD2AP are associated with Alzheimer’s disease. Nat Genet. 2011;43:429–35. doi: 10.1038/ng.803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 389.Steinberg S, Stefansson H, Jonsson T, Johannsdottir H, Ingason A, Helgason H, et al. Loss-of-function variants in ABCA7 confer risk of Alzheimer’s disease. Nat Genet. 2015;47:445–7. doi: 10.1038/ng.3246. [DOI] [PubMed] [Google Scholar]
- 390.Marioni RE, Harris SE, Zhang Q, McRae AF, Hagenaars SP, Hill WD, et al. GWAS on family history of Alzheimer’s disease. Transl Psychiatry. 2018;8:99. doi: 10.1038/s41398-018-0150-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 391.Sperling RA, et al. Toward defining the preclinical stages of Alzheimer’s disease: recommendations from the National Institute on Aging-Alzheimer’s Association workgroups on diagnostic guidelines for Alzheimer’s disease. Alzheimers Dement. 2011;7:280–292. doi: 10.1016/j.jalz.2011.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 392.LaFerla FM, Green KN, Oddo S, et al. Intracellular amyloid-beta in Alzheimer’s disease. Nat Rev Neurosci. 2007;8:499–509. doi: 10.1038/nrn2168. [DOI] [PubMed] [Google Scholar]
- 393.Mattsson N, Palmqvist S, Stomrud E, Vogel J, Hansson O. Staging β-amyloid pathology with amyloid positron emission tomography. JAMA Neurol. 2019;76:1319–29. doi: 10.1001/jamaneurol.2019.2214. [DOI] [PMC free article] [PubMed] [Google Scholar]