

Profiling of DNMT3A variants revealed a large portion with dramatic loss of DNA methyltransferase activity and protein stability, which enabled the risk stratifi cation of malignancy development and revealed a molecular mechanism of regulated DNMT3A turnover.
Abstract
Clonal hematopoiesis is a prevalent age-related condition associated with a greatly increased risk of hematologic disease; mutations in DNA methyltransferase 3A (DNMT3A) are the most common driver of this state. DNMT3A variants occur across the gene with some particularly associated with malignancy, but the functional relevance and mechanisms of pathogenesis of the majority of mutations are unknown. Here, we systematically investigated the methyltransferase activity and protein stability of 253 disease-associated DNMT3A mutations, and found that 74% were loss-of-function mutations. Half of these variants exhibited reduced protein stability and, as a class, correlated with greater clonal expansion and acute myeloid leukemia development. We investigated the mechanisms underlying the instability using a CRISPR screen and uncovered regulated destruction of DNMT3A mediated by the DCAF8 E3 ubiquitin ligase adaptor. We establish a new paradigm to classify novel variants that has prognostic and potential therapeutic significance for patients with hematologic disease.
Significance:
DNMT3A has emerged as the most important epigenetic regulator and tumor suppressor in the hematopoietic system. Our study represents a systematic and high-throughput method to characterize the molecular impact of DNMT3A missense mutations and the discovery of a regulated destruction mechanism of DNMT3A offering new prognostic and future therapeutic avenues.
See related commentary by Ma and Will, p. 23.
This article is highlighted in the In This Issue feature, p. 1
Introduction
One trillion new blood cells are produced each day through a complex orchestrated hierarchy rooted in long-lived hematopoietic stem cells (HSC) in the bone marrow. Over time, all HSCs acquire somatic mutations, some of which confer a Darwinian fitness advantage, enabling them to produce a disproportionate number of progeny, a condition known as clonal hematopoiesis (CH). CH is increasingly prevalent with age and is associated with increased risk of myelodysplastic syndrome, hematologic malignancies, and all-cause mortality (1, 2).
Virtually all individuals from middle age on will have expanded HSC clones (3), but by the age of 70, about one fifth will have larger clones considered CH, comprising ≥4% of the peripheral blood (2% variant allele frequency, or VAF; ref. 4). Broadly, larger clones are associated with a higher risk of hematologic malignancies (1). Although mutations are randomly acquired throughout the genome, variants in a limited number of genes are recurrently observed (5). Understanding the mechanisms through which HSC clones expand and associate with malignancy development is therefore of significant interest (6).
DNA methyltransferase 3A (DNMT3A) is a critical tumor suppressor in the hematopoietic system and is the most frequently mutated gene in CH. Mutations in DNMT3A are spread across all three functional domains, namely, Pro-Trp-Trp-Pro (PWWP), ATRX-DNMT3A-DNMT3L (ADD), and the methyltransferase (MTase; Fig. 1A). The R882 hotspot mutation in the dimerization region of the MTase domain that accounts for around 60% of mutations in acute myeloid leukemia (AML) has been shown to act as a dominant negative, leading to reduced DNA methylation activity (7–10), despite R882-specific hypermethylation through alteration of flanking sequence preferences (11). Importantly, in CH, as well as in lymphoid malignancies, R882 is found in only around 20% of cases, and the impact of the other 80% of mutations, many of which are predicted to be pathogenic, is largely unknown. Mouse models have shown that homozygous ablation of Dnmt3a leads to focal hypomethylation, stem cell expansion, and predisposition to hematologic malignancies (12–14). Therefore, we hypothesized that at least some of the other missense mutations would also exhibit loss of DNA methylation activity.
Figure 1.

Methyltransferase activity analysis in 253 disease-associated DNMT3A missense mutations using the HOXA5-Snrpn-BFP methylation reporter. A, Schematic of missense mutations across DNMT3A in CH and cancer. Sites marked in orange represent tested mutations (Supplementary Table S1); the larger circles indicate recurrent mutations. Sites in gray represent mutations seen in the databases but not assayed here. B, Schematic of the methyltransferase activity assay using HOXA5-Snrpn-BFP methylation reporter. The Snrpn promoter region was linked to BFP and a bovine polyA signal and knocked in (KI) to the HOXA5 locus in reverse orientation in HEK293T cells. These cells have high BFP fluorescence, as shown in C in gray. When wild-type (WT) DNMT3A is introduced using lentiviral transduction, the methylation-sensitive HOXA5 regions promote DNA methylation, which leads to suppression of BFP expression (blue plot in C). C, Methyltransferase activity assay. The graph depicts blue fluorescence intensity in the DNMT3AWT-transduced cells as measured by flow cytometry on day 0 (gray) and day 15 (blue). D, The percentage of BFP-negative cells after transduction with DNMT3AWT or DNMT3AE756A constructs measured by flow cytometry over 18 days. E, Methyltransferase activity assay of 253 disease-associated DNMT3A missense mutations measured by flow cytometry. Lentiviral particles of each DNMT3A variant were infected in three biological replicates in HOXA5-Snrpn-BFP reporter cells. Activity was normalized to the percent BFP-negative percent cells in DNMT3AWT-transduced cells 15 days later. To visualize results, we defined and colored mutations based on their levels of methyltransferase activity. Methyltransferase activity of those mutations labeled were classified as having impaired methyltransferase activity (activity <0.5), whereas those in black were considered in the range of WT DNMT3A (activity >0.5). Data are presented as mean ± SEM. F, DNA methylation profile (WGBS) of cells transduced with the indicated DNMT3A mutants displayed on the UCSC genome browser. See also Supplementary Figs. S1–S3.
In addition to somatic mutations in CH and hematologic malignancies (15, 16), germline mutations in DNMT3A are clinically relevant: they can lead to Tatton–Brown–Rahman overgrowth syndrome (TBRS) in which patients exhibit obesity, excessive height, and intellectual disability (17, 18). Peripheral blood of one patient with TBRS with an R882 mutation was shown to exhibit DNA hypomethylation patterns relative to its healthy sibling (19), but other mutations, many of which overlap with those found in hematologic disorders, have not been characterized. Importantly, a patient with microcephaly is reported to have a mutation leading to hyperactive DNMT3A (20). Together, these findings suggest that the precise dosage of DNMT3A is physiologically critical, with heterozygous mutations leading to either an excess or a diminution of DNMT3A activity with potentially dire consequences.
Here, we sought to systematically profile DNMT3A variants found in patients to gain insights into the mechanism of pathogenesis, with a view toward risk stratification and identification of potential therapeutic strategies for hematologic malignancies.
Results
Methyltransferase Function of 253 Disease-Associated DNMT3A Variants
In order to profile methyltransferase activity of DNMT3A variants in a high-throughput manner, we first sought to establish a fluorescent reporter capable of faithfully representing functionality of DNMT3A variants. Our previous work had shown that DNA methylation at the HOXA5 promoter was a sensitive indicator of DNMT3A activity (21). Thus, we knocked in a Snrpn–blue fluorescence protein (BFP) reporter (22) to this locus in human embryonic kidney 293T (HEK293T) cells at a reverse orientation such that increased DNA methylation at HOXA5-Snrpn-BFP would lead to decreased BFP. This strategy can provide readout of methyltransferase functionality by flow cytometry (Fig. 1B). To test the fidelity of the assay, we transduced the cells with lentiviral vectors carrying DNMT3A wild-type (DNMT3AWT) and a known catalytically inactive variant DNMT3AE756A (23, 24). The proportion of BFP-negative cells in DNMT3AWT-transduced cells increased and plateaued at approximately 20% after 2 weeks (Fig. 1C and D). Conversely, the proportion of BFP-negative cells in DNMT3AE756A-transduced cells remained virtually unchanged over this period (Fig. 1D). Increased DNA methylation at the HOXA5-Snrpn reporter in cells transduced with WT-DNMT3A was verified (Supplementary Fig. S1A). These results confirmed that we could measure methyltransferase functionality of DNMT3A variants using the HOXA5-Snrpn-BFP reporter system.
We then selected 253 patient-associated DNMT3A variants, mostly missense mutations and some 3-bp in-frame deletions, distributed across three functional domains of the 912-residue protein (Fig. 1A; Supplementary Table S1). The variants were compiled from reports of mutations in CH (1, 2, 25), cancer, and TBRS (17, 18) and cover a large portion of reported mutations. These variants were cloned into a lentiviral vector, which was then transduced into the HOXA5-Snrpn-BFP reporter cells. The proportions of BFP-negative cells were monitored and normalized to DNMT3AWT 15 days later in order to measure the DNMT3A methyltransferase functionality. Across the protein, 74% (187 of 253) of the variants were classified as severe loss of function (LOF; Fig. 1E; Supplementary Figs. S1B–S1D, S2A, S3A, and S4A), with 85% (23 of 27) and 81% (114 of 140) of those in the PWWP and MTase domains, respectively, exhibiting little or no activity. In the ADD domain, a lower proportion (68%, 29 of 40) exhibited the same severity, whereas other variants had modest loss of activity.
To verify the observed reduction of methyltransferase activity, we introduced a number of variants into methylation-deficient mouse embryonic stem cells [Dnmt3a/b double knockout (DKO) mESC] with almost no background DNA methylation (∼5% of average DNA methylation of genome-wide CpGs; ref. 23; Supplementary Fig. S1E). We focused on frequently documented variants in CH, AML, and TBRS—a single amino acid in-frame Trp 297 deletion (W297Del) in the PWWP domain, a nonsynonymous Arg 749 missense variants (R749C), and the known catalytically inactive mutation Glu 756 (E756A) in the MTase domain as a control. After expression in DKO mESCs, the harvested cells were subjected to whole-genome bisulfite sequencing (WGBS). DNMT3AWT reestablished DNA methylation of about 60% of genome-wide CpGs. In contrast, DNMT3AW297Del, DNMT3AE756A, and the other DNMT3A variants failed to increase methylation (Fig. 1F; Supplementary Fig. S1F). Notably, DNMT3AE756A retained comparable protein expression to DNMT3AWT. DNMT3AW297Del and the other DNMT3A variants, on the other hand, had lower protein expression (Supplementary Fig. S1G). Western blot confirmed that DNMT3AW297Del expression was significantly reduced compared with DNMT3AWT (Fig. 2A), suggesting lack of methylation activity may be attributed to decreased protein levels.
Figure 2.

More than a third of missense mutations across DNMT3A decrease protein stability. A, Western blot analysis of GFP-DNMT3A fusion protein and GAPDH control expression measured in DKO embryonic stem cell after doxycycline induction. B, Schematic of protein stability assay using the DsRed-IRES-DNMT3A-GFP bicistronic vector. The vector produces a DNMT3A-GFP fusion protein, such that the level of GFP fluorescence allows inference of the amount of DNMT3A protein present. The fusion protein transcript is downstream of DsRed, following an internal ribosome entry site (IRES) so that DsRed serves as an internal control for differences in transcription efficiencies of the constructs. The construct is transfected into the HEK293T cells, and 48 hours later, green and red fluorescence is measured by flow cytometry (right panel). The ratio of the MFI of the green and red fluorescence is used to indicate the level of GFP-tagged DNMTA protein in the cells. The fluorescence ratios in cells transfected with DNMT3A-mutant constructs are normalized to that of the DNMT3A-WT–expressing constructs. C, The graph depicts protein stability of 253 DNMT3A missense mutations measured as described in B. To visualize results, we defined and colored mutations based on their levels of severity of impaired protein stability. Mutations marked in red indicate those with severely impaired protein stability (stability ratio <0.5), whereas those in black were considered comparable to DNMT3AWT (stability ratio >0.5). The mutation labeled in red is one identified above as unstable. Data are presented as mean ± SEM. D, Western blotting analysis and quantification of heterozygous mESC unstable mutants (DNMT3AW293Del, DNMT3AG681R, DNMT3AR732C) and stable mutants (DNMT3AE752A DNMT3AW856R) normalized to GAPDH. E, Quantification of RNA expression levels in mESC mutants described in D normalized to GAPDH. F, The distribution of variant allele frequencies of stable (gray data points) and unstable (red data points) variants plotted after controlling for study size, panel size, and mutation rate compared with maximum likelihood VAF distributions (red and gray lines) that assume a parameterized form for the distribution of fitness effects (29).
More Than a Third of DNMT3A Variants Lead to Protein Instability
Of the hundreds of identified DNMT3A mutations, none have been shown to result in high protein turnover. Furthermore, although algorithms can be used to predict whether amino acid changes are damaging protein function, they do not account for changes in protein stability. Therefore, we sought to systematically determine whether protein instability was a common molecular mechanism leading to loss of DNMT3A function. To investigate this, we used a bicistronic vector in which DNMT3A was fused to GFP as a sensor of DNMT3A protein level, linked to dsRed expression controlling for transfection efficiency (Fig. 2B). Protein stability of 253 variants was measured by the ratio of green to red fluorescence after transfection into HEK293T cells (Supplementary Figs. S2B, S3B, and S4B). Surprisingly, we found instability was a common feature, with 37% (94 of 253) of variants reducing protein stability (Fig. 2C), with a nonrandom distribution across DNMT3A.
The observations of protein instability were also verified in patient samples and mESCs at a heterozygous state in the Dnmt3a endogenous loci. Lymphoblastoid cell lines (LCL) generated from the peripheral blood of patients with AML carrying mutant DNMT3A and DNMT3A protein levels were examined. All DNMT3A variants shown to be stable above had similar amounts of protein, whereas one sample bearing a heterozygous unstable mutant (DNMT3AP307S) had lower DNMT3A (Supplementary Fig. S5A). Furthermore, Dnmt3a variants were introduced into the endogenous locus in mESCs using CRISPR/Cas9 genome editing. Single colonies bearing heterozygous knock-in of three unstable mutations (W293Del, G681R, and R732C) and two stable mutations (E752A and W856R), which correspond to W297, G685, R736, E756, and W860 in human DNMT3A, were validated by Sanger sequencing (Supplementary Fig. S5B). DNMT3A protein expression levels were significantly reduced in unstable mutants (DNMT3AW293Del, DNMT3AG681R, and DNMT3AR732C) and increased in stable mutants (DNMT3AE752A and DNMT3AW856R; Fig. 2D), whereas the RNA expression levels of these Dnmt3a mutants remained comparable (Fig. 2E). These data demonstrate that the reduced DNMT3A protein expression resulted from protein instability instead of defects in transcription. Together, data from the patient-derived LCLs and CRISPR-generated mESCs validate the finding of DNMT3A protein instability for multiple mutants in physiologically relevant heterozygous states.
To better understand whether particular features were associated with unstable and stable variants, we examined the structural distribution of the variants. Unstable PWWP domain (26) variants occurred across the entire domain (Supplementary Fig. S5C), in the hydrophobic core as well as in a region where intramolecular polar/charged interactions occur that likely promote stability. Similarly, several unstable MTase domain variants (e.g., R729W and R736C) are located at the intermolecular interface where DNMT3A interacts to form a highly active tetramer with DNMT3L (Supplementary Fig. S5D), a noncatalytic member of the DNMT3 family expressed during embryogenesis (27).
Overall, most DNMT3A variants leading to protein instability were located in the PWWP and MTase domains, with 57% (16 of 27) and 44% (62 of 140) respectively classified as unstable (Supplementary Figs. S2B and S3B). In the MTase domain, most unstable variants were located in the interior (Supplementary Fig. S5E). Others (e.g., P904L and E907V) were located at the intramolecular interface between the MTase and the ADD domains (Supplementary Fig. S5F), indicating that interdomain interactions may be critical for protein stability.
The MTase domain also contains mutations relatively resistant to instability, including the residues important for cofactor (S-adenosyl-L-methionine, or SAM) binding (27), target recognition, catalysis, and DNMT3A-3A homodimer formation (ref. 28; Supplementary Fig. S3B). These mutations likely lead to LOF through effects on catalysis rather than loss of stability.
Finally, fewer variants in the ADD domain (17.5%, 7 of 40) led to protein instability (Supplementary Fig. S4B). All seven variants impairing protein stability occurred at the cysteine residues and are involved in zinc coordination (Supplementary Fig. S5G), indicating the metal binding domains are important for overall protein structure and stability.
Together, these data establish that reduced DNMT3A stability is a common pathway to LOF. Domains involved in protein–protein interactions had a higher proportion of variants leading to instability. Mutations appearing in TBRS and CH were equally likely to result in protein instability, underscoring a remarkable dose sensitivity of DNMT3A for normal human physiology.
Unstable Variants Are Associated with Increased Clonal Expansion
Variants that lead to instability are expected to have around half the amount of WT DNMT3A protein and, collectively, may be expected to behave similarly at a physiologic level. Conversely, LOF variants that retain stability may exert effects to varying degrees via multiple mechanisms such as diminished SAM or DNA binding. Thus, we sought to determine whether variants that led to protein instability would exhibit any phenotypic correlations.
Recent work has shown that variants with cell-intrinsic fitness advantages are major driving forces shaping clonal hematopoiesis (29). To determine whether DNMT3A variants associated with protein instability displayed fitness advantages over stable variants, we estimated the distribution of fitness effects for the two classes of variants. We plotted VAF density histograms for the stable and unstable variants detected in four studies that targeted more than 99% of variants tested here (25, 30–32), normalizing by the study size and study-specific mutation rate (ref. 29; Fig. 2F). Performing a maximum likelihood fit to a family of stretched exponential distributions (29), we found that the unstable group of variants, as a class, had a higher proportion of variants in the mid to high fitness range compared with the stable group (Supplementary Fig. S6A). If destabilizing variants, on average, confer a larger fitness advantage compared with stable variants, we would expect to observe more unstable over stable variants after controlling for total mutation rate differences between the two classes. Indeed, we demonstrated a significant enrichment for unstable variants in data from four separate studies (Supplementary Fig. S6B), consistent with their having higher fitness effects, on average, than stable variants. Whereas destabilizing variants are predominantly in the moderate to high fitness class, expected of a “null” allele, stable variants have broader fitness effects, falling into both low and high fitness categories (Supplementary Fig. S6A). This is consistent with our expectation that individual stable variants exert a more varied impact, with some representing a partial LOF, and others, such as mutations like R882 that result in a dominant-negative effect (8, 9, 19), conferring an even higher fitness advantage (Supplementary Fig. S6C). These analyses again underscore the remarkable dose sensitivity of DNMT3A function.
To determine whether unstable variants also are associated with leukemia development, we examined VAFs of DNMT3A variants in 60 patients with pre-AML (average 6.3 years before AML diagnosis) and 192 patients of age- and sex-matched controls identified from a publicly available data set (33). Although no significant differences in clonal expansion were observed in the control group (Supplementary Fig. S6D), in the pre-AML group, greater expansion was observed in patients with unstable versus stable variants (Supplementary Fig. S6E). This suggests that the increased fitness effects of unstable variants associated with CH also translate to a higher risk of AML development.
DNMT3A Is Degraded through Proteasomal Machinery
Given the striking prevalence of variants leading to protein instability, we sought to examine the molecular consequences of such mutations in a murine model. One mutation leading to severe protein instability was a 3-bp deletion resulting in an in-frame deletion of W297, equivalent to W293 in the mouse. We used CRISPR/Cas9 editing in murine zygotes to re-create this variant, generating a Dnmt3aW293Del allele. To determine the severity of the W293Del variant compared with complete loss of DNMT3A, we intercrossed Dnmt3a+/− and Dnmt3aW293Del/+ mice to generate compound heterozygous Dnmt3aW293Del/− as well as homozygous mutant mice. Notably, both Dnmt3aW293Del/− and Dnmt3aW293Del/W293Del mice did not survive past postnatal day 24 (Fig. 3A), resembling the phenotype of Dnmt3a−/− mice (34), suggesting that the severity of the Dnmt3aW293Del variant is comparable to the null variant. We then examined whole bone marrow cells from Dnmt3aW293Del mice to determine whether they also exhibited a loss of DNMT3A. Indeed, DNMT3A was almost completely absent in homozygous mice, and about half the amount of WT protein was present in heterozygous mice (Fig. 3B), despite normal mRNA levels (Fig. 3C).
Figure 3.

DNMT3A is degraded through the ubiquitin-proteasome system. A, Survival after the birth of mice with the indicated genotypes. No Dnmt3a−/−, Dnmt3aW293Del/−, or Dnmt3aW293Del/W293Del mice survived beyond postnatal day 24. B, Western blot of DNMT3A and GAPDH protein expression in whole bone marrow cells of mice. C, Gene expression analysis of Dnmt3a in whole bone marrow measured by quantitative PCR and normalized for Gapdh mRNA expression. D, The stability ratio of the WT and W297Del mutant over the indicated times, calculated as described in Fig. 2C. E, Alterations in DNMT3A protein stability after administration of proteasome inhibitor (MG132), a CRL inhibitor (MLN4924), an autophagy inhibitor (chloroquine), and an unfolded protein response inhibitor (IREi). F, Stability ratio of MFI of DNMT3A-GFP versus MFI of DsRed before and after treatment with the proteasome inhibitor (MG132) as measured by flow cytometry 48 hours after transfection. Data are presented as mean ± SEM. *, P < 0.05; **, P < 0.01; ***, P < 0.001 using the unpaired t test. See also Supplementary Fig. S7.
The DNMT3AW297Del variant and its murine homolog showed similar severe protein instability; therefore, we sought to identify the responsible protein degradation pathway. Transfection of DNMT3A into HEK293T cells showed that after 12 hours, the amount of DNMT3AWT was constant, but DNMT3AW297Del was approximately half of WT and subsequently decreased (Fig. 3D). This confirmed that reduced DNMT3AW297Del was not due to a translational defect but rather to impaired stability. We then used the stability assay described above to assess the relevance of major degradation pathways, treating DNMT3AW297Del-transfected cells with inhibitors of autophagy, the unfolded protein response, the proteasome machinery, and Cullin-RING E3 ubiquitin ligases (CRL). Treatment with a proteasome inhibitor (MG132) rescued 2.5-fold expression of DNMT3AW297Del (Fig. 3E). The CRL inhibitor MLN4924 also led to a modest increase of mutant DNMT3AW297Del expression in HEK293T cells (Fig. 3E). These results suggest that DNMT3AW297Del is degraded through the proteasome and that CRLs may play a role in this process.
We next aimed to determine whether other unstable DNMT3A variants were degraded through the same mechanism. We treated cells expressing 20 of these variants distributed in the PWWP and MTase domains with inhibitors targeting multiple degradation pathways. Again, treatment with a proteasome inhibitor partially restored DNMT3A-mutant protein expression in cells with all 20 mutations (Fig. 3F). Notably, treatment with inhibitors of E1 ubiquitin-activating enzyme (MLN7243) conferred the highest rescue of DNMT3A variant expression (Supplementary Fig. S7A), indicating that DNMT3A is mainly degraded through ubiquitin-proteasome system. Treating cells expressing WT or DNMT3A variants with inhibitors of CRLs (MLN4924), autophagy inhibitors (i.e., chloroquine) or the unfolded protein response (i.e., IREi) did not rescue protein expression (Supplementary Fig. S7B–S7E). In addition, an HSP90 inhibitor further decreased protein stability (Supplementary Fig. S7F), suggesting a role for HSP90 in stabilizing DNMT3A during protein synthesis.
A Proteasome Inhibitor Partially Restores Patient Methylome and Transcriptome Patterns
The finding that many DNMT3A variants lead to protein instability offers the possibility of therapeutic intervention. Focusing on the W297Del mutation, we first aimed to confirm that DNMT3AW297Del is decreased in patient cells. We generated LCLs from the peripheral blood of a patient with TBRS and confirmed that the DNMT3A protein level was indeed reduced (Fig. 4A). Moreover, exposure to a proteasome inhibitor increased the level of DNMT3A in both WT and DNMT3AW297Del/+ LCLs (Fig. 4A). To further examine whether proteasome inhibitor treatment would restore DNA methylation and correct gene expression levels, we cultured the DNMT3AW297Del/+ LCL cells in the presence of a low dose of proteasome inhibitor (1 μmol/L MG132) for 30 days and then conducted WGBS and RNA sequencing (RNA-seq). Indeed, proteasome inhibitor treatment increased the average methylation in DNMT3AW297Del/+ LCLs from 59.4% to 63.5%, which is statistically identical to the 63.1% methylation ratio of genome-wide CpGs observed in WT LCLs (Fig. 4B). Proteasome inhibitor treatment also increased the average methylation in WT LCLs by 2%. We then examined 25 chromatin marks across 127 reference genomes from the Epigenome Roadmap Project (35) to identify genomic regions that were most responsive to remethylation in the presence of the proteasome inhibitors. We found that active enhancers, weak enhancers, heterochromatin, poised promoters, and regions with repressive polycomb complexes showed the most dynamic changes (Fig. 4C and D; Supplementary Fig. S8A–S8M), whereas regions close to the transcription start site and transcribed regions (Supplementary Fig. S8N–S8W) remained largely inert to proteasome inhibitor administration.
Figure 4.

Proteasome inhibitors partially restore the methylome and transcriptome in patient-derived cells. A, Western blotting and fold-change quantification of DNMT3A and GAPDH protein expression in LCL cells from WT or a patient with TBRS (W297del). DNMT3A was normalized to GAPDH. B, Box plots showing DNA methylation distribution analyzed by whole-genome bisulfite sequencing in the indicated LCL cells with and without proteasome inhibition through MG132 treatment. The bars represent methylation ratios between the first and third quartiles, with the median distribution, shown by a gap in the bars, of 66.7% (DNMT3AW297Del/+) and 80% (all other samples). The whiskers represent methylation in the first and fourth quartiles. The statistical values represent the Wilcox rank-sum test. C, Violin plots of DNA methylation distribution in 12,896 active enhancer regions as defined in the Roadmap Epigenomic Project. D, Violin plot of DNA methylation ratios in 10,973 heterochromatic regions as defined in the Roadmap Epigenomic Project. E, Gene expression heat map of the indicated cells and treatments. Cluster 1 genes were hypomethylated in DNMT3AW297Del/+ compared with WT LCLs and responsive to proteasome inhibitors. Cluster 2 genes were hypomethylated in DNMT3AW297Del/+ compared with WT LCLs but inert to proteasome inhibitors. See also Supplementary Fig. S8.
We also examined the correlation between DNA methylation changes and gene expression. We identified genes that were expressed in the LCL cells and also harbored at least 5 CpGs in their promoter region. Within this group, upregulated genes were separated into two clusters using unsupervised consensus clustering. Genes whose promoters were hypomethylated in DNMT3AW297Del/+ compared with WT LCLs showed higher expression in DNMT3AW297Del/+ LCLs, with one particular group of genes (cluster 1) particularly strongly correlated. Treatment with proteasome inhibitors rescued this phenotype with reduced expression of cluster 1 but not the more modestly affected cluster 2 genes (Fig. 4E). Genes on clusters 1 and 2 were generally found in diverse cellular pathways, although components associated with the plasma membrane were statistically enriched in the cluster 1 genes (Supplementary Table S2). Together, these data demonstrate that administration of proteasome inhibitor to cells harboring a DNMT3A variant leading to protein instability can at least partially restore the global methylome and transcriptome, opening up the possibility of therapeutic restoration of DNMT3A function in some contexts.
CRISPR Screening Links CUL4BDCAF8 Ubiquitination Complex to DNMT3A Degradation
We next sought to identify the specific ubiquitination complex involved in DNMT3A turnover using the unstable DNMT3AW297Del variant, using a CRISPR screen (36). We engineered HEK293T cells to constitutively overexpress Cas9 protein and a bicistronic DNMT3AW297Del stability reporter and then transduced the cells with single-guide RNA (sgRNA) libraries targeting ubiquitin ligases. Nine days later, we sorted for the cells with the highest (above the 95th percentile) and the lowest (below the 5th percentile) DNMT3A-GFP expression (Fig. 5A) and then examined which sgRNAs were enriched in the cells with the highest compared with lowest DNMT3A-GFP expression. Strikingly, we found that DCAF8, RBX1, and CUL4B were among the most enriched targets in the screen (Fig. 5B). To verify the results, we examined whether the substrate adaptor DCAF8 physically interacts with DNMT3A. We conducted immunoprecipitation for DCAF8 and then probed for interaction with DNMT3A after culture in the absence or presence of a E1 ubiquitin–activating enzyme inhibitor. We observed coimmunoprecipitation between DNMT3AW297Del and DCAF8 after 6 hours of inhibitor treatment, whereas weak interaction between WT DNMT3A and DCAF8 was also observed (Fig. 5C). This suggested that DCAF8 had higher binding affinity to the variant protein and confirmed the possibility that CUL4BDCAF8 could be the ubiquitin ligase for DNMT3A.
Figure 5.

Targeted CRISPR screening identifies CUL4BDCAF8 ubiquitin ligase complexes essential for DNMT3A protein degradation. A, Schematic of targeted CRISPR screening to identify ubiquitin ligases essential for DNMT3A protein degradation. HEK293T cells were engineered to constitutively overexpress Cas9 and the indicated bicistronic DNMT3AW297Del reporter and then infected with sgRNA libraries targeting ubiquitin ligases. Nine days after infection, we sorted both the top and bottom 5% of cells for DNMT3A-GFP expression. B, The graph depicts the gene enrichment score and P value in targeted CRISPR screening for ubiquitin ligases. DCAF8, RBX1, and CUL4B genes (red) were enriched and statistically significant. C, The image depicts DCAF8 immunoprecipitation in DNMT3A-WT or W297del-expressing cells after incubation for 6 hours with an inhibitor of E1 ubiquitin-activating enzymes (MLN7243), followed by blotting using GFP, DCAF8, DDB1, and GAPDH antibodies. D, DNMT3AW297del-expressing HEK293T cells treated with sgRNA targeting DCAF8, proteasome inhibitor, and E1 ubiquitin–activating enzyme, followed by Western blot analysis for DNMT3A-GFP, DCAF8, and GAPDH. E, Cycloheximide (CHX) treatment with 0, 2, 4, and 6 hours of DNMT3AWT and DNMT3AW297Del HEK293T cells with or without DCAF8-KO followed by Western blot for DNMT3A-GFP, DCAF8, and GAPDH. Inhibitor treatments of proteasome and E1 ubiquitin–activating enzyme in DNMT3AWT and DNMT3AW297Del HEK293 cells serve as rescue control of experiment. F, Western blot analysis of DNMT3A, DCAF8, and GAPDH in DCAF8-KO DNMT3AWT LCLs. G, The image depicts the levels of ubiquitination in DNMT3AWT and DNMT3AW297del HEK293T cells with or without DCAF8-KO. DNMT3A-GFP was pulled down by a GFP antibody, followed by Western blot analysis using ubiquitin and GFP antibody. H, Western blotting analysis of heterozygous mESC unstable mutants (DNMT3AW293Del and DNMT3AG681R) and stable mutants (DNMT3AE752A DNMT3AW856R) with or without Dcaf8-KO. I, Stability of variants before and after DCAF8-KO measured as described in Fig. 2C. J, Schematic of the RBX1–CUL4B–DCAF8 complex serving as the ubiquitin ligase for DNMT3A protein. ***, P < 0.001 using the unpaired t test.
We then sought to examine whether DNMT3AW297Del expression was rescued when DCAF8 was ablated. Indeed, knockout of DCAF8 with sgRNAs rescued DNMT3AW297Del level to a degree comparable to that of rescue by treatment with the proteosome inhibitor and E1 ubiquitin-activating enzymes (Fig. 5D). In addition, when we inhibited protein synthesis with cycloheximide treatment, we observed rapid disappearance of DNMT3A protein; DCAF8 knockout (KO) enabled sustained DNMT3A expression (Fig. 5E). Furthermore, knockout of DCAF8 in DNMT3AWT LCLs also increased DNMT3AWT expression (Fig. 5F). Together, these results indicate that DCAF8 plays a major role in degradation of DNMT3A, both WT and unstable variants.
We next sought to verify whether DCAF8 regulates DNMT3A through facilitating ubiquitination. We observed that DNMT3AW297Del showed high levels of ubiquitination relative to DNMT3AWT. Ubiquitination of both DNMT3AWT and DNMT3AW297Del was significantly reduced in DCAF8 KO cells (Fig. 5G). Moreover, in mESCs harboring heterozygous knock-in of the unstable DNMT3AW293Del and DNMT3AG681R variants, Dcaf8 ablation significantly rescued DNMT3A protein expression, whereas expression of stable DNMT3A variants (DNMT3AE752A and DNMT3AW856R) did not change (Fig. 5H). These results indicate that DCAF8 serves as a substrate adaptor for DNMT3A ubiquitination, and the ablation of DCAF8 prevents DNMT3A ubiquitination and degradation in a regulated manner. To determine whether DCAF8 also influenced stability of other unstable DNMT3A variants, we examined the turnover of the 20 selected unstable DNMT3A variants in DCAF8-KO HEK293T cells. DCAF8-KO modestly rescued protein stability of most DNMT3A variants (Fig. 5I). Importantly, the scaffold protein CUL4B was also enriched in the screen, as was RBX1, an E2-conjugating enzyme. These two components are both known to participate in a complex with the substrate adaptor DCAF8 (Fig. 5J). Together, these results suggest a new complex for regulation of DNMT3A degradation and stability, some components of which could serve as potential therapeutic targets for rescuing DNMT3A expression in patients with unstable variants.
Discussion
The work presented here, characterizing patient-associated variants across approximately one third of amino acids in DNMT3A, has revealed general principles of DNMT3A regulation and stability and has implications for the interpretation of novel variants across many disease types. Although the concept of reduced stability is not unknown, that such a large portion of variants exhibit this phenotype and can be classified together was unexpected. Furthermore, our work illustrates that when unstable variants can be classified together, their biological impact is consistent. Finally, our discovery of DNMT3A destruction mediated by the CUL4BDCAF8 E3 ligase unveils an important new regulatory pathway for this protein, whose biological role is remarkably dose-sensitive; this new pathway may offer new potential therapeutic avenues.
As large numbers of novel germline and somatic variants across the genome are identified by genome sequencing of patients, it is becoming increasingly important to infer their impact on protein function. Numerous algorithms have been designed to identify the likelihood of a particular mutation being deleterious, but none have taken into account loss of protein stability. Not only do we show, for the first time, that a large portion of missense variants lead to protein instability, but our analysis of their relevance with regard to activity and protein structure offers a framework to apply these principles to novel variants in other genes.
DNMT3A mutations leading to protein instability have clinical significance. We found a relationship between protein instability and CH as well as hematologic malignancies. Greater clonal expansion was observed in individuals with unstable compared with stable mutations in the clonal hematopoiesis and pre-AML cohorts, suggesting that DNMT3A mutations leading to protein instability may increase the risk of AML development. Although the stable dominant-negative R882 mutation has the greatest prognostic significance for development of AML, a comprehensive map of unstable mutations across DNMT3A may also have prognostic value. Because essentially all individuals will develop HSC clones bearing DNMT3A mutations during their lifetime (3), the functional implications of different variants are of broad interest.
Identification of DCAF8 as the substrate adaptor for DNMT3A protein degradation demonstrated a regulated turnover process that affects both mutant and WT protein, rather than general destruction of misfolded protein. We hypothesize that DCAF8 has some affinity for WT DNMT3A protein but that unstable mutations confer some degree of structural conformation change, further exposing potential degrons to DCAF8. This suggests a potential therapeutic target for patients with destabilizing mutations. A recent report showed that DNMT3A is normally recruited to intergenic regions by H3K36me2, but two destabilizing mutants of DNMT3A (W297Del and I310N) failed to bind to chromatin (37). We expect that CUL4BDCAF8 ubiquitin ligases may serve as a protein quality surveillance system for DNMT3A protein, eliminating protein that is not bound to chromatin. This would be consistent with other studies showing there is very little free cellular DNMT3A detected (38). Although our data clearly show that DCAF8 is a major regulator of DNMT3A, we cannot preclude the existence of others. Inhibition of E1-activating enzymes shows stronger rescue of protein stability across multiple DNMT3A mutants than DCAF8 depletion (Supplementary Fig. S7A; Fig. 5E), and treatment of DCAF8 KO cell lines with the E1 inhibitor further increases DNMT3A protein slightly (Fig. 5E). Together, these data suggest DCAF8 may not be the sole ubiquitin ligase responsible for DNMT3A degradation. Further work is needed to understand all the components and the context of DNMT3A protein quality control.
Finally, we showed that a proteasome inhibitor could partially restore the aberrant methylome and transcriptome in a patient-derived DNMT3A-mutant cell line. Several proteasome inhibitors have been approved by FDA for use in treating multiple myeloma (39, 40); therefore, the use of proteasome inhibitors, or conceivably DCAF8 inhibitors, could be a potential therapeutic avenue for a subset of patients with TBRS, CH, and hematologic disorders. As the proteasome inhibitor also increased WT DNMT3A protein, proteasome inhibition could be a potential therapeutic strategy even in patients with deletions or other mutations. More broadly, identification of effects on stability of variants in other genes could be similarly used to develop new therapeutic strategies. A caveat of proteasome inhibition is the broad effects; it is possible that the restoration of methylome and transcriptome patterns could be indirect. Given that DNMT3A mutations in patients are typically heterozygous (1, 2, 15), testing low-dose and long-term proteasome inhibition on patient-derived DNMT3Amutant/+ cells may represent possible outcomes.
Given the rapid pace with which mutations involved in human disease are now being identified, there is an increasing need to be able to anticipate the impact of new variants. We believe the approach presented here, of comprehensive characterization of a large fraction of novel variants to understand protein function, can serve as a paradigm to gain broad insights into other recurrently mutated genes.
Methods
Cell Lines, Cell Culture, and Lentiviral Particle Production
Peripheral blood mononuclear cells (PBMC) were isolated from whole blood by a density gradient centrifugation method using Ficoll. PBMCs (2 × 106) were incubated with concentrated supernatant from the EBV producer cell line B95-8 in a total of 200 μL RPMI 1640 medium (containing 10% FCS and 5% l-glutamine) for 30 minutes. The cells were then plated at 106 cells/well in a flat-bottomed 96-well plate, as well as 1 μg/mL cyclosporin A (Sandoz Pharmaceuticals). Cells were fed biweekly until LCLs were established. The use of human samples had institutional review board approval from Baylor College of Medicine (Houston, TX). Written informed consent was obtained from sample donors. All relevant ethical regulations on compliance were followed in this study.
Dnmt3a variants in previously established Dnmt3a-flagged mESC was generated using CRISPR/Cas9 genome editing. Then, 1 μg sgRNA (Synthego) was incubated with 1 μg Cas9 protein (PNA Bio) for 30 minutes at room temperature to obtain Cas9-sgRNA RNPs. Next, 1.2 μg single-stranded DNA donor template (Integrated DNA Technologies) containing desired variant sequence was added prior to electroporation using a Neon transfection system (Thermo Fisher Scientific), and 2 × 105 cells were electroporated using the optimized electroporation condition of 1,200 V, 20 ms, and two pulses. After electroporation, we collected single-cell colonies for each variant, and the sequence of single colonies was validated by Sanger sequencing.
HEK293T cells are commercially available and DKO mESCs were established from the Meissner laboratory (23). All cells used in this study were routinely tested to be Mycoplasma free (Mycoplasma Detection Kit; Minerva Biolabs). HEK293T cells were incubated in DMEM with 10% fetal bovine serum (FBS) and 1% penicillin/streptomycin at 5% CO2 in a 37°C incubator. DKO mESCs and their DNMT3A mutant–expressing derivatives were cultured in 12.5% Foundation ES serum, 1× Glutamax, 100 μmol/L β-mercaptoethanol, 1× nonessential amino acid and 1,000 U/mL leukemia inhibitory factor, and knockout DMEM.
HEK293T cells were transfected with Pinducer20-BSD DNMT3A-mutant plasmids using Lipofectamine 2000 (Life Technologies) and cotransfected with pMD2.G and psPAX2. Lentiviral particles were collected 48 hours and 72 hours after transfection. Then, 4× polyethylene glycol (PEG; 32% PEG6000, 0.4 mol/L NaCl, and 0.04 mol/L HEPES) was added to precipitate viral particles at 4°C overnight. Viral particles were then centrifuged at 1,500 × g for 45 minutes and resuspended in mESC medium. DKO mESCs were infected with Pinducer20 DNMT3A-mutant lentiviral particles and 4 μg/mL polybrene in the medium. Then, 4 μg/mL blasticidin (BSD) selection was conducted for 3 days in the DNMT3A mutant–infected mESCs, then recovered for 7 days, and conducted 4 μg/mL BSD for a further 7 days. DNMT3A-mutant mESCs were treated with 2 μg/mL doxycycline for 30 days and then collected for DNA and RNA extraction.
DNA Constructs
Full-length DNMT3A cDNA fused with a GFP sequence was cloned into pDONR223 using a Gateway cloning BP clonase II enzyme mix (Thermo Fisher Scientific). Pathogenic DNMT3A mutations listed in the Catalogue of Somatic Mutations in Cancer (COSMIC) database, and those causing TBRS and CH were selected and then modeled in pDONR-DNMT3A-GFP vectors using Quickchange II site-directed mutagenesis kits (Agilent Technologies). The full-length cDNA of every DNMT3A mutant in the pDONR vector used in subsequent experiments was verified by Sanger sequencing. For the DNA methylation assay, mutant DNMT3A pDONR vectors were further cloned into Pinducer20-BSD vectors using Gateway cloning LP clonase II enzyme mix as previously described (24). For DNMT3A protein stability assay, DNMT3A-mutant pDONR vectors were subsequently subcloned into the PsLenti-DsRed-IRES-DNMT3A-eGFP bicistronic vectors (41, 42) shown in Fig. 2B as described. DNMT3A-mutant pDONR vectors were first PCR amplified with 2× Phusion master mix, NotI and ClaI recognition sequence-containing primers using the following PCR program. Initially, PCR mix was heated to 95°C for 5 minutes for initial denaturation, kept at 95°C for 30 seconds for denaturation, and then annealed at 68°C for 30 seconds and extension at 72°C for 1 minute and 30 seconds. The cycle of elongation, annealing, and extension was performed 35 times, before a final elongation step at 72°C for 10 minutes. PCR products were run on 1.5% agarose gel electrophoresis and excised, and the amplified PCR product was extracted using a gel extraction kit (Qiagen). Gel-purified amplicons were then digested with NotI-HF and ClaI at least 4 hours. The digested amplicons were then ligated overnight at 16°C with double restriction enzyme digested and CIP-treated bicistronic vectors. Every DNMT3A mutant in the bicistronic vector used in protein stability assay was confirmed by Sanger sequencing.
HOXA5-Snrpn-BFP Methylation Reporter Analysis
DNA for the Snrpn promoter (22), BFP (tagBFP), woodchuck hepatitis virus posttranslational regulatory elements, bovine growth hormone polyadenylation signal, and HOXA5 homology arms were synthesized (Genscript). The linearized targeting vector was cotransfected with sgRNA-HOXA5 and Cas9-expressing vectors in HEK293T cells. Single BFP-expressing cells were then sorted into each well of 96-well plates, and then a functionally and PCR-verified single clone was chosen to conduct methylation reporter analysis. 5,000 HOXA5-Snrpn-BFP cells containing 10 μg/mL polybrene were seeded to each well in 96-well plates and then mixed with DNMT3A mutant-bearing lentiviral particles in three biological replicates. Then cells were centrifuged at 1,100 rpm for 90 minutes. DsRed, GFP, and BFP intensity were measured every 3 days using flow cytometry.
Protein Stability Assay
Bicistronic DNMT3A mutant reporter plasmids generated as previously described were transfected into HEK293T cells with four biological replicates in 96-well plates. Four controls, including untransfected, bicistronic DNMT3AWT, bicistronic DNMT3AW297Del, and bicistronic control (Addgene 92194), were always transfected in the same plate. Mean fluorescence intensity (MFI) of GFP and DsRed in the bicistronic DNMT3A mutant–transfected cells was measured 24 hours after transfection using flow cytometry. The stability ratio of DNMT3A protein was measured through MFI of GFP divided by MFI of DsRed and normalized with the bicistronic control. For experiments with various inhibitor treatments, including 10 μmol/L MG132 (a proteasome inhibitor; Sigma-Aldrich), 10 μmol/L MLN 4924 (a Cullin-RING E3 ligase inhibitor; Medchem), 50 μmol/L chloroquine (an autophagy inhibitor; Cell Signaling), 5 mmol/L 3-methyladenine (an autophagy inhibitor; Sigma-Aldrich), 50 μmol/L IREi (an unfolded protein response inhibitor; Sigma-Aldrich), 1 μmol/L 17-DMAG (HSP90i; Sigma-Aldrich), and 40 μmol/L VER155008 (HSP70i; Sigma-Aldrich), the inhibitors were added 24 hours after transfection, and MFI of fluorescence protein was measured 48 hours after transfection.
Generation of the Dnmt3aW293Del Murine Model
The sequence of sgRNA-293 has been listed in Supplementary Table S1 and was generated by in vitro transcription (MEGAshortscript T7 Transcription kit). Single-stranded DNA template (ssODN) carrying W293Del and a SacI restriction site was synthesized (Integrated DNA Technologies). Cas9 protein was purchased from PNA Bio. Then, 100 ng/μL Cas9 protein, 50 ng/μL sgRNA-293, and 100 ng/μL ssODN were diluted in nuclease-free PBS and further injected into fertilized C57BL/6 eggs. After culture, about 20 to 30 blastocysts were transferred into the uterus of pseudo-pregnant ICR females. Genotype identification of Dnmt3aW293Del mice was first PCR by genotyping primers, followed by SacI restriction enzyme digestion for more than 4 hours. Detailed blood phenotypes of this murine model were published (43).
qPCR
RNA was extracted from whole bone marrow cells of mice using a RNeasy micro kit (Qiagen). Then, 1 μg RNA of each sample was mixed with 1 μL oligo dTs and 1 μL 10 mmol/L dNTPs and ddH2O. RNA mixtures were heated to 65°C for 5 minutes and incubated on ice for 1 minute. Next, 4 μL 5× first-strand buffer, 1 μL 0.1 mol/L DTT, 1 μL RNase inhibitor, and 1 μL SuperscriptIII RT were added to the RNA mixture and incubated at 50°C for 60 minutes, and the reaction was inactivated by heating to 70°C for 15 minutes. Then, 0.5 μL cDNAs, 0.5 μL of 10 μmol/L forward and reverse primers, 3.5 μL ddH2O, and 5 μL 2× SsoAdvanced universal SYBR Green supermix were added to PCR tubes, and quantitative PCR was conducted as the following program. Samples were heat activated at 95°C for 3 minutes, then kept at 95°C for 10 seconds, 55°C for 10 seconds, and 72°C for 30 seconds and repeated from the second step for 40 cycles.
Modeling and Analysis of Clonal Hematopoiesis Mutations
We looked at studies that included ≥99% of the stabilizing and destabilizing variants (25, 30–32). Deletions were excluded due to uncertainties estimating site-specific mutation rates for deletions. Data were trimmed to exclude variants at VAFs below each study's estimated limit of reliable variant detection (as described in ref. 29, Supplemental Methods 1).
To enable comparison between studies, the densities were normalized by dividing by [number of individuals in the study × bin widths], and the densities were then rescaled by dividing by 2 μ, where μ is the study-specific haploid mutation rate summed across all the variants covered by the studies. Estimates for the distribution of fitness effects (s) were inferred by fixing Nτ (total number of HSCs × time in years between successive symmetric cell differentiation divisions) to ∼100,000 as inferred (29). The distribution of ages was assumed to be Gaussian with a mean of 60 years and a standard deviation of 15 years, which are the mean and standard deviation of participants in Coombs and colleagues (25), which contributed ∼85% of the data from these four studies. We parameterized the distribution of fitness effects using a family of stretched exponential distributions with a maximum s = smax. We then performed a maximum likelihood procedure, optimizing the shape (β) and scale (d) of the distribution as well as smax.
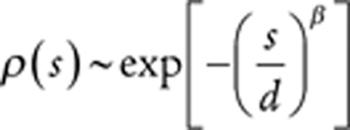 |
For Coombs and colleagues (25), we included only the individuals who were both chemotherapy naive and radiotherapy naive. For Desai and colleagues (31) and Young and colleagues (32), we included only the “control” participants. For the studies that reported replicate VAF measurements (Young and colleagues, refs. 30, 32), we required a variant to be detected in both replicate samples to be called, and the average of the replicate values was taken as the VAF at that time point. For studies that reported variants in participants from more than one time point (Young and colleagues, refs. 30, 32, and Desai and colleagues, ref. 31), we included only variants detected in the first blood sample for these studies.
RNA-seq and Analysis
RNA was extracted using a RNeasy micro kit (Qiagen) and quantified using Nanodrop. TruSeq stranded mRNA library preparation was based on the manufacturer's instruction (Illumina). Libraries were sequenced using Nextseq 500 sequencer. Paired-end RNA-seq reads were mapped to the human genome (hg19) using TopHat 2.0.10. The fragments per kilobase of exon per million fragments mapped values were calculated by cufflinks 2.2.1. To examine whether proteasome inhibitors can restore transcriptome in DNMT3AW297Del/+ LCLs, we selected the genes that matched with the criteria as follows: at least 5 CpGs that were detected in the promotor of this particular gene, and this gene has detectable expression in RNA-seq. Within these gene (11,401 genes), upregulated genes (fold-change >2, n = 1,208) and downregulated genes (fold-change <–2, n = 508) in the DNMT3AW297Del/+ versus DNMT3A+/+ sample were then identified. For visualization, we used unsupervised consensus clustering and separated genes into two clusters (clusters 1 and 2) within upregulated genes. We used the DAVID Functional Annotation Tools (https://david.ncifcrf.gov/tools.jsp) to perform the gene function enrichment analysis in cluster 1 and 2 gene sets and used all the detected genes as the background.
WGBS and Analysis
DNA (100 ng) was used for WGBS library preparation using a TruSeq DNA methylation kit as the manufacturer's instruction (Illumina). Libraries were sequenced using the Nextseq 500 sequencer. For each WGBS profile, we used BSMAP to trim the adaptor and low-quality sequence with the default threshold, as well as aligned bisulfite-treated reads to the human genome (hg19). Then the methylation ratio of each CpG covered with at least five reads was calculated by the module bsratio in BSMAP. Twenty-five chromatin states were defined using 12 epigenetic marks, including H3K4me1, H3K4me2, H3K4me3, H3K9ac, H3K27ac, H4K20me1, H3K79me2, H3K36me3, H3K9me3, H3K27me3, H2A.Z, and DNase, across all 127 reference epigenomes. All sequencing data are accessible at the NCBI GEO database, accession number GSE178798.
CRISPR-DNMT3A Stability Screen
The human BISON CRISPR-KO library, which contains 2,852 guide RNAs, targets 713 E1, E2, E3, deubiquitinases, and control genes (36). It was cloned into the pXPR003 as previously described (44) by the genome perturbation platform (Broad Institute). The lentivirus particles for the library were produced in a T-175 flask format. Briefly, 18 × 106 HEK293T cells were seeded in 25 mL DMEM supplemented with 10% FBS and penicillin/streptomycin/glutamine. The next day, a packaging mix was prepared: 40 μg psPAX2, 4 μg pVSV-G, and 32 μg of the library in 1 mL OptiMem (Invitrogen) and incubated for 5 minutes at room temperature. This mix was combined with 244 μL TransIT-LT1 (Mirus) in 5 mL OptiMem, incubated for 30 minutes at room temperature, and then applied to cells. Two days posttransfection, cell debris was removed by centrifugation. The lentivirus particles containing medium were collected and stored at −80°C before use.
HEK293T cells were engineered with constitutively expressing Cas9 and bicistronic DNMT3AW297Del reporter. Then, 2 × 106 engineered HEK293T cells were added with 10% (v/v) of the human BISON CRISPR-KO in 2 mL medium and spin-infected (2,400 rpm, 2 hours, 37°C). Twenty-four hours postinfection, sgRNA-infected cells were selected with 2 μg/mL puromycin for 2 days. On the ninth day postinfection, populations were separated using fluorescence-activated cell sorting. Two populations were collected (top 5% and lowest 5%) based on the eGFPDNMT3A to mCherry MFI ratio. Sorted cells were harvested by centrifugation and subjected to direct lysis buffer reactions (1 mmol/L CaCl2, 3 mmol/L MgCl2, 1 mmol/L EDTA, 1% Triton X-100, Tris pH 7.5, with freshly supplemented 0.2 mg/mL proteinase). The sgRNA sequence was amplified in a first PCR reaction with eight staggered forward primers. Then, 20 μL of direct lysed cells was mixed with 0.04 U Titanium Taq (Takara Bio 639210), 0.5 × Titanium Taq buffer, 800 μmol/L dNTP mix, 200 nmol/L P5-SBS3 forward primer, and 200 nmol/L SBS12-pXPR003 reverse primer in a 50-μL reaction. The samples were heat activated at 94°C for 5 minutes; kept at 94°C for 30 seconds, 58°C for 10 seconds, and 72°C for 30 seconds and repeated from the second step for 15 cycles; and heated at 72°C for 2 minutes. Then, 2 μL of the primary PCR product was used as the template for 15 cycles of the secondary PCR, where Illumina adapters and barcodes were added [0.04 U Titanium Taq (Takara Bio 639210), 1 × Titanium Taq buffer, 800 μmol/L dNTP mix, 200 nmol/L SBS3-Stagger-pXPR003 forward primer, 200 nmol/L P7-barcode-SBS12 reverse primer]. An equal amount of all samples was pooled and subjected to preparative agarose electrophoresis followed by gel purification (Qiagen). Eluted DNA was further purified by NaOAc and isopropanol precipitation. Amplified sgRNAs were quantified using the Illumina NextSeq platform. Read counts for all guides targeting the same gene were used to generate P values. The data analysis pipeline comprised the following steps: (i) Each sample was normalized to the total read number. (ii) For each guide, the ratio of reads in the stable versus unstable sorted gate was calculated, and guide RNAs were ranked. (iii) The ranks for each guide were summed for all replicates. (iv) The gene rank was determined as the median rank of the four guides targeting it. (v) P values were calculated, simulating a corresponding distribution over 100 iterations.
Immunoprecipitation
Cells from two 15-cm plates in each condition were harvested in 8 mL IP lysis buffer with protease/phosphatase inhibitor. Cell lysate then was sonicated and centrifuged at the highest speed at 4°C. Cell lysate was precleared with empty protein A Dynabeads and protein concentration was determined using a BCA kit (Thermo Scientific). Protein A Dynabeads were washed twice in IP lysis buffer and bound with anti-DCAF8 antibody (A301–556A; Bethyl Laboratories) at 4°C for 1 hour. Protein A Dynabeads with 4 μg anti-DCAF8 antibody in 2 mg cell lysate were then incubated at 4°C overnight. The beads were then washed three times with 0.5 mL IP lysis buffer and eluted with 40 μL freshly prepared 1× sample buffer by heating at 90°C for 10 minutes, followed by Western blotting.
Western Blotting
Whole bone marrow cells from different genotypes of mice, lymphoblastoid cell lines, and human cell lines were lysed using a cytobuster protein extraction buffer (Millipore) with protease inhibitor cocktail (GenDEPOT). Standard immunoblotting analysis was performed using anti-GFP (1:1,000, Novus, NB600–308), anti-DNMT3A antibody (1:1,000, Santa Cruz, H-295; 1:1,000, Abcam, ab16704), anti-DCAF8 antibody (1:500, Sigma-Aldrich, HPA027381), anti-DDB1 antibody (1:1,000, Bethyl Laboratories, A300–462A), and anti-GAPDH antibody (1:2,000, Millipore, MAB374).
Ubiquitination Assay
For detection of ubiquitination, 2 × 107 cells were harvested and lysed with 1 mL lysis buffer (2% SDS, 150 mmol/L NaCl, 10 mmol/L Tris-HCl, pH 8.0, with protease inhibitor) after 10 hours of MG132 treatment. Cell lysate was boiled for 10 minutes and sonicated. Then, 9 mL renaturing buffer (10 mmol/L Tris-HCl, pH 8.0; 150 mmol/L NaCl; 2 mmol/L EDTA; 1% Triton) was then added and incubated for 1 hour at 4°C. Clear cell lysate was obtained by centrifugation at the highest speed at 4°C. Protein A Dynabeads were washed twice in PBST and bound with 6 μg anti-GFP antibody (NB600–308; Novus) at room temperature for 30 minutes. Protein A Dynabeads with anti-GFP antibody were then incubated with precleared cell lysate at 4°C overnight. Beads were washed three times with 1 mL PBST and protein eluted with 40 μL freshly prepared 1× sample buffer by heating at 95°C for 10 minutes. Western blotting was performed using an antiubiquitination antibody (3936S; Cell Signaling Technology).
Authors' Disclosures
No disclosures were reported.
Supplementary Material
Acknowledgments
We thank Catherine Gillespie, Kelly Turner, and members of the Goodell lab for critical review. We also thank Manisha Manojkumar for her help with CRISPR screening experiments. This work was supported by the Cancer Prevention and Research Institute of Texas, a grant from the Edward P. Evans Foundation, the Samuel Waxman Cancer Research Foundation, the Welch Foundation (BE-1913), the American Cancer Society (RSG-18-043-01-LIB), the National Institutes of Health (DK092883, CA183252, CA125123, CA222736, HG007538, CA228140, HL134780, GM112003), CRUK Cambridge Centre, and UKRI. This project was supported by the Cytometry and Cell Sorting Core and the Genetically Engineered Mouse Core at BCM (CA125123, RR024574, HG006352).
The costs of publication of this article were defrayed in part by the payment of page charges. This article must therefore be hereby marked advertisement in accordance with 18 U.S.C. Section 1734 solely to indicate this fact.
Footnotes
Note: Supplementary data for this article are available at Cancer Discovery Online (http://cancerdiscovery.aacrjournals.org/).
Authors' Contributions
Y. Huang: Conceptualization, formal analysis, validation, investigation, visualization, methodology, writing–original draft. C. Chen: Formal analysis, validation, investigation, visualization. V. Sundaramurthy: Investigation. M. Słabicki: Investigation. D. Hao: Formal analysis, visualization. C.J. Watson: Investigation, visualization. A. Tovy: Methodology. J.M. Reyes: Formal analysis. O. Dakhova: Methodology. B.R. Crovetti: Investigation. C. Galonska: Methodology. M. Lee: Investigation. L. Brunetti: Investigation. Y. Zhou: Resources. K. Tatton-Brown: Resources. Y. Huang: Resources. X. Cheng: Resources. A. Meissner: Resources. P.J. Valk: Resources. L. Van Maldergem: Resources. M.A. Sanders: Resources. J.R. Blundell: Resources. W. Li: Resources, formal analysis. B.L. Ebert: Resources. M.A. Goodell: Conceptualization, resources, supervision, funding acquisition, writing–original draft, writing–review and editing.
References
- 1. Jaiswal S, Fontanillas P, Flannick J, Manning A, Grauman PV, Mar BGet al. Age-related clonal hematopoiesis associated with adverse outcomes. N Engl J Med 2014;371:2488–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Genovese G, Kahler AK, Handsaker RE, Lindberg J, Rose SA, Bakhoum SFet al. Clonal hematopoiesis and blood-cancer risk inferred from blood DNA sequence. N Engl J Med 2014;371:2477–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Crowgey EL, Mahajan N, Wong WH, Gopalakrishnapillai A, Barwe SP, Kolb EAet al. Error-corrected sequencing strategies enable comprehensive detection of leukemic mutations relevant for diagnosis and minimal residual disease monitoring. BMC Med Genomics 2020;13:32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Jaiswal S, Libby P. Clonal haematopoiesis: connecting ageing and inflammation in cardiovascular disease. Nat Rev Cardiol 2020;17:137–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Challen GA, Goodell MA. Clonal hematopoiesis: mechanisms driving dominance of stem cell clones. Blood 2020;136:1590–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Bowman RL, Busque L, Levine RL. Clonal hematopoiesis and evolution to hematopoietic malignancies. Cell Stem Cell 2018;22:157–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Holz-Schietinger C, Matje DM, Reich NO. Mutations in DNA methyltransferase (DNMT3A) observed in acute myeloid leukemia patients disrupt processive methylation. J Biol Chem 2012;287:30941–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Kim SJ, Zhao H, Hardikar S, Singh AK, Goodell MA, Chen T. A DNMT3A mutation common in AML exhibits dominant-negative effects in murine ES cells. Blood 2013;122:4086–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Russler-Germain DA, Spencer DH, Young MA, Lamprecht TL, Miller CA, Fulton Ret al. The R882H DNMT3A mutation associated with AML dominantly inhibits wild-type DNMT3A by blocking its ability to form active tetramers. Cancer Cell 2014;25:442–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Guryanova OA, Shank K, Spitzer B, Luciani L, Koche RP, Garrett-Bakelman FEet al. DNMT3A mutations promote anthracycline resistance in acute myeloid leukemia via impaired nucleosome remodeling. Nat Med 2016;22:1488–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Emperle M, Adam S, Kunert S, Dukatz M, Baude A, Plass Cet al. Mutations of R882 change flanking sequence preferences of the DNA methyltransferase DNMT3A and cellular methylation patterns. Nucleic Acids Res 2019;47:11355–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Challen GA, Sun D, Jeong M, Luo M, Jelinek J, Berg JSet al. Dnmt3a is essential for hematopoietic stem cell differentiation. Nat Genet 2012;44:23–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Jeong M, Park HJ, Celik H, Ostrander EL, Reyes JM, Guzman Aet al. Loss of Dnmt3a immortalizes hematopoietic stem cells in vivo. Cell Rep 2018;23:1–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Mayle A, Yang L, Rodriguez B, Zhou T, Chang E, Curry CVet al. Dnmt3a loss predisposes murine hematopoietic stem cells to malignant transformation. Blood 2015;125:629–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Ley TJ, Ding L, Walter MJ, McLellan MD, Lamprecht T, Larson DEet al. DNMT3A mutations in acute myeloid leukemia. N Engl J Med 2010;363:2424–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Yang L, Rau R, Goodell MA. DNMT3A in haematological malignancies. Nat Rev Cancer 2015;15:152–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Tatton-Brown K, Seal S, Ruark E, Harmer J, Ramsay E, Del Vecchio Duarte Set al. Mutations in the DNA methyltransferase gene DNMT3A cause an overgrowth syndrome with intellectual disability. Nat Genet 2014;46:385–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Tatton-Brown K, Zachariou A, Loveday C, Renwick A, Mahamdallie S, Aksglaede Let al. The Tatton-Brown-Rahman syndrome: a clinical study of 55 individuals with de novo constitutive DNMT3A variants. Wellcome Open Res 2018;3:46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Spencer DH, Russler-Germain DA, Ketkar S, Helton NM, Lamprecht TL, Fulton RSet al. CpG island hypermethylation mediated by DNMT3A is a consequence of AML progression. Cell 2017;168:801–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Heyn P, Logan CV, Fluteau A, Challis RC, Auchynnikava T, Martin CAet al. Gain-of-function DNMT3A mutations cause microcephalic dwarfism and hypermethylation of Polycomb-regulated regions. Nat Genet 2019;51:96–105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Huang YH, Su J, Lei Y, Brunetti L, Gundry MC, Zhang Xet al. DNA epigenome editing using CRISPR-Cas SunTag-directed DNMT3A. Genome Biol 2017;18:176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Stelzer Y, Shivalila CS, Soldner F, Markoulaki S, Jaenisch R. Tracing dynamic changes of DNA methylation at single-cell resolution. Cell 2015;163:218–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Galonska C, Charlton J, Mattei AL, Donaghey J, Clement K, Gu Het al. Genome-wide tracking of dCas9-methyltransferase footprints. Nat Commun 2018;9:597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Su J, Huang YH, Cui X, Wang X, Zhang X, Lei Yet al. Homeobox oncogene activation by pan-cancer DNA hypermethylation. Genome Biol 2018;19:108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Coombs CC, Zehir A, Devlin SM, Kishtagari A, Syed A, Jonsson Pet al. Therapy-related clonal hematopoiesis in patients with non-hematologic cancers is common and associated with adverse clinical outcomes. Cell Stem Cell 2017;21:374–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Wu H, Zeng H, Lam R, Tempel W, Amaya MF, Xu Cet al. Structural and histone binding ability characterizations of human PWWP domains. PLoS One 2011;6:e18919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Jia D, Jurkowska RZ, Zhang X, Jeltsch A, Cheng X. Structure of Dnmt3a bound to Dnmt3L suggests a model for de novo DNA methylation. Nature 2007;449:248–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Zhang ZM, Lu R, Wang P, Yu Y, Chen D, Gao Let al. Structural basis for DNMT3A-mediated de novo DNA methylation. Nature 2018;554:387–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Watson CJ, Papula AL, Poon GYP, Wong WH, Young AL, Druley TEet al. The evolutionary dynamics and fitness landscape of clonal hematopoiesis. Science 2020;367:1449–54. [DOI] [PubMed] [Google Scholar]
- 30. Young AL, Challen GA, Birmann BM, Druley TE. Clonal haematopoiesis harbouring AML-associated mutations is ubiquitous in healthy adults. Nat Commun 2016;7:12484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Desai P, Mencia-Trinchant N, Savenkov O, Simon MS, Cheang G, Lee Set al. Somatic mutations precede acute myeloid leukemia years before diagnosis. Nat Med 2018;24:1015–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Young AL, Tong RS, Birmann BM, Druley TE. Clonal hematopoiesis and risk of acute myeloid leukemia. Haematologica 2019;104:2410–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Abelson S, Collord G, Ng SWK, Weissbrod O, Mendelson Cohen N, Niemeyer Eet al. Prediction of acute myeloid leukaemia risk in healthy individuals. Nature 2018;559:400–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Okano M, Bell DW, Haber DA, Li E. DNA methyltransferases Dnmt3a and Dnmt3b are essential for de novo methylation and mammalian development. Cell 1999;99:247–57. [DOI] [PubMed] [Google Scholar]
- 35. Roadmap Epigenomics C, Kundaje A, Meuleman W, Ernst J, Bilenky M, Yen Aet al. Integrative analysis of 111 reference human epigenomes. Nature 2015;518:317–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Slabicki M, Kozicka Z, Petzold G, Li YD, Manojkumar M, Bunker RDet al. The CDK inhibitor CR8 acts as a molecular glue degrader that depletes cyclin K. Nature 2020;585:293–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Weinberg DN, Papillon-Cavanagh S, Chen H, Yue Y, Chen X, Rajagopalan KNet al. The histone mark H3K36me2 recruits DNMT3A and shapes the intergenic DNA methylation landscape. Nature 2019;573:281–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Sharma S, De Carvalho DD, Jeong S, Jones PA, Liang G. Nucleosomes containing methylated DNA stabilize DNA methyltransferases 3A/3B and ensure faithful epigenetic inheritance. PLoS Genet 2011;7:e1001286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Kouroukis TC, Baldassarre FG, Haynes AE, Imrie K, Reece DE, Cheung MC. Bortezomib in multiple myeloma: systematic review and clinical considerations. Curr Oncol 2014;21:e573–603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Dimopoulos M, Quach H, Mateos MV, Landgren O, Leleu X, Siegel Det al. Carfilzomib, dexamethasone, and daratumumab versus carfilzomib and dexamethasone for patients with relapsed or refractory multiple myeloma (CANDOR): results from a randomised, multicentre, open-label, phase 3 study. Lancet 2020;396:186–97. [DOI] [PubMed] [Google Scholar]
- 41. Yen HC, Xu Q, Chou DM, Zhao Z, Elledge SJ. Global protein stability profiling in mammalian cells. Science 2008;322:918–23. [DOI] [PubMed] [Google Scholar]
- 42. Rousseaux MW, de Haro M, Lasagna-Reeves CA, De Maio A, Park J, Jafar-Nejad Pet al. TRIM28 regulates the nuclear accumulation and toxicity of both alpha-synuclein and tau. Elife 2016;5:e19809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Tovy A, Rosas C, Gaikwad AS, Medrano G, Zhang L, Reyes JMet al. Perturbed hematopoiesis in individuals with germline DNMT3A overgrowth Tatton-Brown-Rahman syndrome. Haematologica 2021. June 3 [Epub ahead of print]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Doench JG, Fusi N, Sullender M, Hegde M, Vaimberg EW, Donovan KFet al. Optimized sgRNA design to maximize activity and minimize off-target effects of CRISPR-Cas9. Nat Biotechnol 2016;34:184–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.