

Abstract
Despite advances, the roles of genetic variants from the APOE-harboring 19q13.32 region in Alzheimer’s disease (AD) remain controversial. We leverage a comprehensive approach to gain insights into a more homogeneous genetic architecture of AD in this region. We use a sample of 2,673 AD-affected and 16,246 unaffected subjects from four studies and validate our main findings in the landmark Alzheimer’s Disease Genetics Consortium cohort (3,662 AD-cases and 1,541 controls). We report the remarkably high excesses of the AD risk for carriers of the ε4 allele who also carry minor alleles of rs2075650 (TOMM40) and rs12721046 (APOC1) polymorphisms compared to carriers of their major alleles. The exceptionally high 4.37-fold (p=1.34×10−3) excess was particularly identified for the minor allele homozygotes. The beneficial and adverse variants were significantly depleted and enriched, respectively, in the AD-affected families. This study provides compelling evidence for the definitive roles of the APOE-TOMM40-APOC1 variants in the AD risk.
Keywords: Alzheimer’s disease, apolipoprotein E polymorphism, haplotypes, linkage disequilibrium
1. INTRODUCTION
The autosomal dominant (familial) form of early-onset Alzheimer’s disease is considered to be caused by mutations in the APP, PSEN1, and PSEN2 genes that support its deterministic mechanism (Lanoiselee, et al., 2017,Levy-Lahad, et al., 1995,Rogaev, et al., 1995,Sherrington, et al., 1995). Unlike the early form, late-onset Alzheimer’s disease, which is prevalent after approximately 60–65 years, herein referred to as AD, is a polygenic heterogeneous disorder which risk is influenced by a complex interplay of various endogenous (e.g., genetics, physiology) and exogenous (e.g., environmental exposures, social milieu) factors, and their interactions (Escott-Price, et al., 2017,Finch and Kulminski, 2019,Sweeney, et al., 2019). The complexity of such influences and their uncertainty forces the view of a sporadic origin of AD.
A vivid example is the potential role of the APOE ε4 allele in AD pathogenesis. This well-studied variant is known as the strongest individual genetic risk factor of AD in various populations (Raichlen and Alexander, 2014). Nevertheless, even this variant is not considered a causative factor of AD (Belloy, et al., 2019) and can presumably contribute through multiple mechanisms (Yamazaki, et al., 2019,Zhao, et al., 2018). Furthermore, despite a quarter-century of APOE-AD research, neither the role of the APOE gene nor its interplay with the other genes in the neighboring region is evident because of uncertainty about how to treat genetic variants from this region (Belloy, et al., 2019,Lutz, et al., 2016,Roses, et al., 2010,Zhou, et al., 2019). The research driven by the medical genetics hypothesis of one gene, one function, one phenotype assumes the existence of genes with causal variants (Jansen, et al., 2019,Lambert, et al., 2013). This hypothesis may not apply, however, in the genetics of complex traits (A. M. Kulminski, et al., 2020a,Visscher, et al., 2017). Then, alternative explanations of genetic predisposition to complex traits, --such as, for example, the roles of common/rare variants with small effects, structural diversity of the human genome, intricate genetic architectures of complex traits (Eichler, et al., 2010,Gibson, 2012)--, are required. Prior research also supports the roles of haplotypes with variants from APOE and other genes in AD (Lescai, et al., 2011,Linnertz, et al., 2014,Zhou, et al., 2019).
The complexity of the AD pathogenesis is further augmented by an inherently heterogeneous genetic architecture of AD. This heterogeneity is supported by an elusive role of natural selection in driving molecular mechanisms of complex traits characteristic of post-reproductive life such as AD (Nesse and Williams, 1994). Indeed, following the famous essay by Theodosius Dobzhansky (1973), evolutionary medicine suggests potential mechanisms of such age-related traits. These mechanisms represent side-effects of natural selection rather than its direct role in the pathogenesis of a disease in post-reproductive life. For example, evolutionary medicine discusses mechanisms such as mismatch of disease and the environment, trade-offs between reproductive success and health, the cost of organism defenses, etc. (Nesse, et al., 2012). These mechanisms are inherent sources of heterogeneity in genetic predisposition to age-related traits such as AD. The role of natural selection is further challenged by increased human life span (Oeppen and Vaupel, 2002) and environmental changes during recent centuries (Corella and Ordovas, 2014,Crespi, et al., 2010,Kulminski, 2013,Vijg and Suh, 2005).
The evolutionary implications support non-trivial contributions of genetic variants to complex traits in a heterogeneous manner. This heterogeneity can be dissected by identifying the context in the genetic contributions that increases accuracy of the estimates of AD risks. Thus, consistently with the 2018 NIA-Alzheimer’s-Association framework (Jack, et al., 2018,Knopman, et al., 2018,Silverberg, et al., 2018) and personalized medicine (Schork, 2015), gaining insights into mechanisms of AD pathogenesis requires thorough approaches in dissecting heterogeneity in predisposition to AD (Kulminski, et al., 2018).
Here, we adopt a comprehensive approach leveraging the analyses of differences in linkage disequilibrium (LD) structures in AD-affected and unaffected subjects, called molecular signatures, complemented by the analyses of allele frequencies and associations, to gain insights into a more homogeneous genetic architecture of AD in the APOE 19q13.32 region. For the main analysis, we use a pooled sample of 2,673 AD-affected and 16,246 AD-unaffected subjects of European ancestry from the Framingham Heart Study (FHS), the Cardiovascular Health Study (CHS), the Health and Retirement Study (HRS), and the National Institute on Aging (NIA) Late-Onset Alzheimer Disease Family Study (LOADFS). The main findings were validated using an independent sample of 3,662 AD-affected and 1,541 AD-unaffected subjects from the landmark Alzheimer’s Disease Genetics Consortium (ADGC) initiative. Our primary research objective is to examine in detail the roles of variants from APOE and the neighboring TOMM40 and APOC1 genes in the AD risks.
2. METHODS
2.1. Study cohorts and phenotypes
The main (discovery) analysis used data from four independent studies: FHS, comprised of the original (FHS_C1) and offspring (FHS_C2) cohorts (Cupples, et al., 2009), CHS (Fried, et al., 1991), HRS (Juster and Suzman, 1995), and the NIA LOADFS (Lee, et al., 2008). In LOADFS and FHS, AD was defined based on diagnoses made according to National Institute of Neurological and Communicative Disorders and Stroke and the Alzheimer’s disease and Related Disorders Association. A diagnosis of AD in HRS and CHS was defined based on ICD-9:331.0x codes. Individuals with AD constituted the case group, N=2,673, and those without AD constituted the non-case group, N=16,246 (Supplemental Table S1).
The main findings from the discovery stage were validated using an independent sample from the NIA Alzheimer’s Disease Centers (ADCs) cohort, which is a part of the Alzheimer’s Disease Genetics Consortium (ADGC) initiative (Naj, et al., 2011). This sample consisted of autopsy-confirmed and clinically-confirmed AD-affected (N=3,662) and cognitively normal (N=1,541) subjects who were ascertained by the clinical and neuropathology cores of the NIA-funded ADCs.
We also used data from the FHS 3rd generation cohort (FHS_C3) and Coronary Artery Risk Development in Young Adults cohort (CARDIA) to examine the differences in proportions of the selected compound genotypes in younger and older AD-unaffected subjects.
All analyses focused on individuals who identified themselves as of European ancestry.
2.2. Genotypes
Genotype data were available from the same customized Illumina iSelect array in the FHS and CHS cohorts, Affymetrix 500K in the FHS, Illumina HumanCNV370v1 chip in the CHS, Illumina HumanOmni 2.5 Quad chip in the HRS, and Illumina Human 610Quadv1_B Beadchip in LOADFS.
The BCAM-NECTIN2-TOMM40-APOE-APOC1 (19q13.32) region was represented by 32 SNPs, which were in moderate LD (r2<0.8) and directly genotyped in at least two studies. We excluded individuals with >5% missingness. To facilitate cross-platform comparisons, we selected directly genotyped target SNPs or their proxies (r2>0.8 in the 1000 Genomes Project, CEU population) using all available arrays for each study. Non-genotyped SNPs were imputed (IMPUTE2 (Howie, et al., 2009)) according to the 1000 Genomes Project Phase I integrated variant set release (SHAPEIT2) in the NCBI build 37 (hg19) coordinate. Only SNPs with high imputation quality (info>0.8) were retained for the analyses (Supplemental Table S2). Genotype data for the ADCs cohort included in ADGC was available from Human660W-Quad_v1_A array.
2.3. LD analysis
LD was characterized by the correlation coefficient r using a haplotype-based method, as argued in (Kulminski, et al., 2018,A. M. Kulminski, et al., 2020b). The significance of the LD estimates was evaluated using chi-square statistics, defined as χ2=r2n, where n=2N is the number of gametes and N is the sample size (Lewontin, 1988). Given the potential loss of power because of inferring haplotypes from genotypes, we used a more conservative estimate, with N instead of n.
We employed a LD contrast test (Zaykin, et al., 2006) to characterize the significance of the differences in pair-wise estimates of LD between affected (r1) and unaffected (r0) subjects using Z2 = 2(r1 – r0)2 statistics. We used a permutation procedure by shuffling the labels for the affected and unaffected subjects to obtain an empirical distribution of Z2 under the null hypothesis r1 = r0. The distribution of r1 – r0 was then tested for normality using Q-Q (quantile-quantile) plots and Shapiro-Wilk tests. Since the permutation distribution of r1 – r0 is approximately normal, we used the sample mean and standard deviation s of the permutation distribution of r1 – r0 to calculate where r is the estimate of the correlation coefficient obtained using the original (non-permuted) labels. To obtain p-values, we then compared z2 to a chi-squared distribution with one degree of freedom. This parametric procedure allows one to compute accurate significance levels using far fewer permutations since one only needs to estimate the parameters of a normal distribution. In our analysis, we used 1,000 permutations to ensure robustness as the results were stable after 200 permutations.
Adopting a conservative Bonferroni correction for multiple testing, the locus-wide significance level for the LD estimates is p=0.05/32=1.6×10−3, whereas for the difference in LD between the AD affected and unaffected subjects is p=0.05/496(=32×31/2)=10−4. Asymptotically valid confidence intervals were constructed using asymptotic variance adapted from (Wellek and Ziegler, 2009).
2.4. Statistical analysis
We evaluated effect sizes (beta) and odds ratios (ORs) for AD risk for carriers of compound genotypes constructed from rs429358, rs2075650, and rs12721046 SNPs using the base R function glm for logistic regression. The models were adjusted for age, sex, and study composition, defined by field centers (CHS), cohorts (FHS and HRS), and three ADC centers (ADGC). No other adjustments have been made.
We employed a fixed-effects model meta-analysis with inverse-variance weighting. The combined effect size was estimated as , and the variance of this effect-size was , where is the effect size in the study i and wi is the reciprocal of the variance of . To produce the p-values for the meta-analysis, we adopted a Wald test with null hypothesis given by the test statistic .
2.5. Familial history of AD in LOADFS
LOADFS did not provide information on the complete history of AD in families. We used the reported data on the AD affection status in LOADFS families to define the AD-affected families. Herein, the families whose members were affected by AD were referred to as having a familial history of AD. Consequently, the families whose members were not affected by AD were referred to as not having a familial history of AD.
3. RESULTS
3.1. The APOE ε4- and ε2-specific molecular signatures of AD
We evaluated molecular signatures of AD as differences in LD between 32 SNPs representing the APOE region (Supplemental Table S2) in AD-affected (cases) and unaffected (non-cases) subjects (Δr=rcases–rnon-cases) who do not have either the ε4 (Figure 1A) or ε2 (Figure 1B) allele in the pooled sample of the LOADFS, HRS, CHS, and two older cohorts of the FHS, the original (FHS_C1) and offspring (FHS_C2) cohorts (see Methods). Figure 1 shows a substantially more heterogeneous AD signature associated with the ε4 allele (Figure 1B) compared to that associated with the ε2 allele (Figure 1A). Specifically, the ε2-negative signature is characterized by locus-wide significance (p≤10−4) for Δr for 173 SNP pairs compared to six pairs in the ε4-negative signature (Supplemental Table S3).
Fig. 1. Molecular signatures of Alzheimer’s Disease (AD) in (A) ε4-negative and (B) ε2-negative samples.
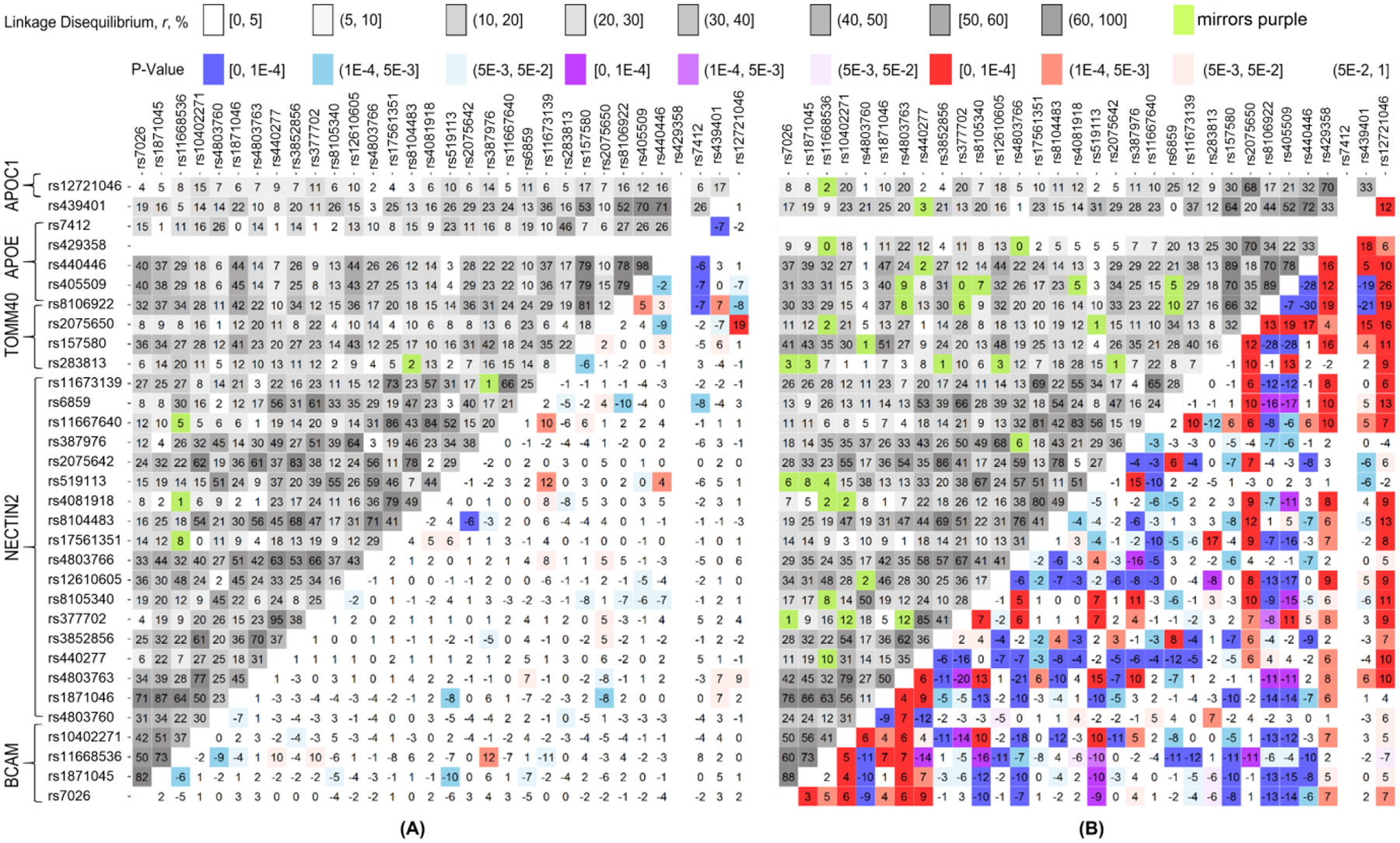
Upper-left triangle: Linkage disequilibrium (LD) pattern (r, %) in the pooled sample, non-cases. Lower-right triangle: heat map for Δr=rcases–rnon-cases representing the molecular signature of AD. Red denotes rcases>rnon-cases and blue denotes rcases<rnon-cases. Purple and green show the estimates with opposite signs of rcases and rnon-cases. For convenience, positive sign of rnon-cases has been selected. The legend on the top shows color-coded p-values and grey-coded LD. Numerical estimates are given in Supplemental Table S3.
We verified that the signs of Δr for the significant differences were consistent in the independent samples of the LOADFS and non-LOADFS (HRS, CHS, and FHS) studies, which is regarded as replication (Marigorta, et al., 2018). Specifically, directions of the significant differences Δr were the same for all six SNP pairs in the ε4-negative sample and a vast majority of SNP pairs, 160 of 173 (93%), in the ε2-negative sample (Supplemental Table S3). Furthermore, the signs were also consistent for a vast majority of Δr which attained suggestive significance (5×10−3≤p<10−4) i.e., for 16 of 17 (94%) SNP pairs in the ε4-negative sample and 57 of 66 (86%) SNP pairs in the ε2-negative sample (Supplemental Table S4).
We found that the difference in LD between unfavorable AD-affected non-ε2 sample (rε2negative-AD) and the most favorable AD- and ε4-negative sample (rε4negative-noAD) was locus-wide significant (p≤10−4) for 250 SNP pairs (Supplemental Table S3). Top LD difference between these samples rε2negative-AD – rε4negative-noAD =77.6% (p<10−100) was for rs2075650 (TOMM40) and rs12721046 (APOC1) SNPs (Figure 2, ε2-negative red vs ε4-negative green). The most favorable sample was characterized by negligible LD, rε4negative-noAD=7% (p=3.02×10−25). This SNP pair was well separated from the other pairs, followed by the rs2075650 and rs405509 (APOE) pair with the 2-fold smaller difference in LD of 39% (p<10−100). The further analysis focuses on the rs429358, rs2075650, and rs12721046 triple.
Fig. 2. Linkage disequilibrium (r) between rs2075650 (TOMM40) and rs12721046 (APOC1) in the ε2- and ε4-negative samples.
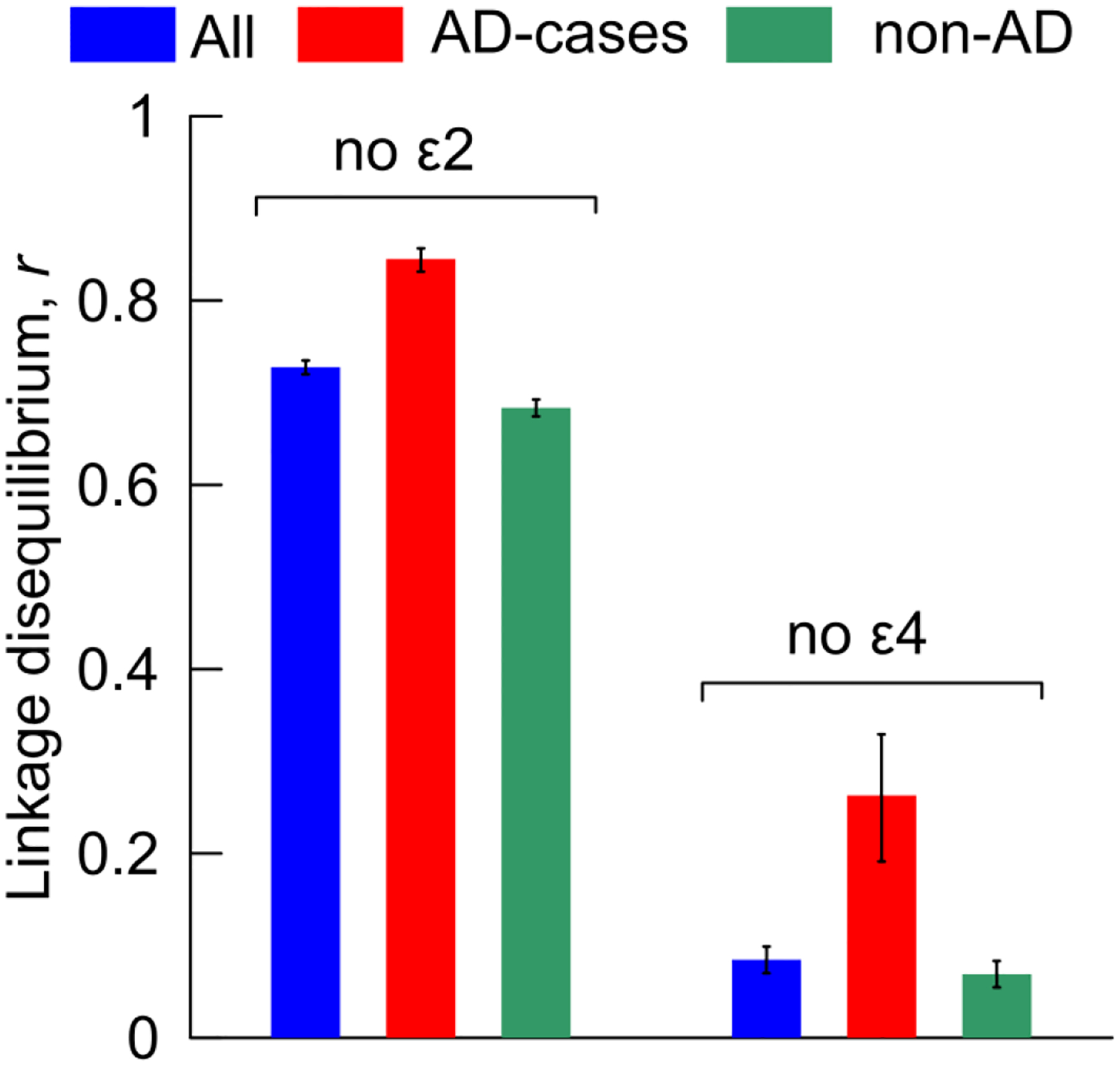
Blue, red, and green denote pooled samples from all studies of (all) AD-cases and non-cases combined, AD-cases, and (non-AD) AD-non-cases, respectively. Vertical lines show standard errors. Numerical estimates are given in Supplemental Table S3.
3.2. Proportions of compound genotypes in the AD-affected and unaffected subjects
Proportions of carriers of compound genotypes constructed from rs429358, rs2075650, and rs12721046 were evaluated in the samples without exclusion of the ε2 allele as this exclusion did not make a difference. Of the 27 (=3×3×3) possible compound genotypes for three bi-allelic SNPs, there were 13 genotypes with more than 10 AD-affected or unaffected subjects, which we focus on thereafter (Table 1 and Supplemental Table S5), except explicitly noted. The most common genotypes in the pooled samples of LOADFS, HRS, CHS, and FHS studies, cases and non-cases combined and separately, were complete major allele homozygote for the three SNPs (rs429358_TT, rs2075650_AA, and rs12721046_GG, herein denoted as TT/AA/GG), followed by the complete heterozygote (Tc/Ag/Ga).
Table 1.
Proportions of the most frequent 13 compound genotypes in the pooled sample.
| ID | MA coding | Genotype | All | AD cases | Non-cases | Δf, % | OR | p value | |||
|---|---|---|---|---|---|---|---|---|---|---|---|
| N | % | N | % | N | % | ||||||
| 1 | 000 | TT/AA/GG | 10,359 | 60.3 | 731 | 33.0 | 9,628 | 64.4 | 31.3 | 0.27 | 4.10E-174 |
| 2 | 001 | TT/AA/Ga | 851 | 5.0 | 45 | 2.0 | 806 | 5.4 | 3.4 | 0.36 | 1.14E-11 |
| 4 | 010 | TT/Ag/GG | 782 | 4.6 | 59 | 2.7 | 723 | 4.8 | 2.2 | 0.54 | 4.98E-06 |
| 5 | 011 | TT/Ag/Ga | 138 | 0.8 | 18 | 0.8 | 120 | 0.8 | 0.0 | 1.01 | 9.57E-01 |
| 10 | 100 | Tc/AA/GG | 950 | 5.5 | 225 | 10.2 | 725 | 4.8 | −5.3 | 2.22 | 1.68E-24 |
| 11 | 101 | Tc/AA/Ga | 216 | 1.3 | 51 | 2.3 | 165 | 1.1 | −1.2 | 2.12 | 2.22E-06 |
| 13 | 110 | Tc/Ag/GG | 123 | 0.7 | 37 | 1.7 | 86 | 0.6 | −1.1 | 2.94 | 1.13E-08 |
| 14 | 111 | Tc/Ag/Ga | 3,000 | 17.5 | 718 | 32.4 | 2,282 | 15.3 | −17.2 | 2.67 | 7.19E-88 |
| 15 | 112 | Tc/Ag/aa | 104 | 0.6 | 21 | 0.9 | 83 | 0.6 | −0.4 | 1.72 | 2.58E-02 |
| 17 | 121 | Tc/gg/Ga | 107 | 0.6 | 35 | 1.6 | 72 | 0.5 | −1.1 | 3.32 | 8.36E-10 |
| 19 | 200 | cc/AA/GG | 27 | 0.2 | 11 | 0.5 | 16 | 0.1 | −0.4 | 4.67 | 1.54E-05 |
| 23 | 211 | cc/Ag/Ga | 146 | 0.9 | 77 | 3.5 | 69 | 0.5 | −3.0 | 7.78 | 3.28E-47 |
| 27 | 222 | cc/gg/aa | 265 | 1.5 | 155 | 7.0 | 110 | 0.7 | −6.3 | 10.17 | 2.04E-110 |
The pooled sample includes National Institute on Aging Late Onset Alzheimer’s disease Family Study (LOADFS), Health and Retirement Study (HRS), Cardiovascular Health Study (CHS), Framingham Heart Study (FHS) original cohort, and FHS offspring cohort.
ID corresponds to that in the extended Supplementary Table S5.
MA coding: the number of minor alleles in each SNP ordered as rs429358, rs207650, and rs12721046.
Genotype: actual genotypes of rs429358, rs207650, and rs12721046, in that order; upper/lower case denotes major/minor allele.
All: Alzheimer’s disease (AD) cases and non-cases combined.
Δf = fnon-cases – fcases is the difference of proportions of a given compound genotype in AD non-cases and cases.
OR is odds ratio defined as (fcases/fnon-cases)×(1 – fnon-cases)/(1 – fcases).
Bonferroni-adjusted significant effects (the two proportion z-test), p≤0.05/13=3.8×10−3, characterized by the differences in proportions Δf=fnon-cases–fcases or odds ratios (ORs), were observed for 11 of 13 compound genotypes (Table 1). Of them, there were three beneficial effects for non-carriers of the ε4 allele, TT/AA/GG, TT/AA/Ga, and TT/Ag/GG. The strongest adverse effect (OR=10.17, p=2.04×10−110) was observed for carriers of the complete minor allele homozygote (cc/gg/aa).
3.3. Proportions of compound genotypes in AD-unaffected subjects and familial history of AD
We found that proportions of the selected 13 compound genotypes in the older population of N=14,633 AD-unaffected subjects from the pooled sample of LOADFS, HRS, CHS, and FHS (55 years and older; 78.7±8.8 years; mean age [MA] and standard deviation [SD]) resembled those in a younger population of N=5,914 subjects (younger than 55 years; MA±SD=37.8±10.1 years) from the FHS_C3 and CARDIA cohorts (Supplemental Table S6). Specifically, the differences between these proportions were at most 1.5%. None of them attained a Bonferroni-adjusted level of significance p=3.8×10−3. As the younger sample has been under negligible survival selection, these proportions represent unbiased estimates in a general population.
In contrast, proportions of compound genotypes in LOADFS sample without AD (Supplemental Table S7) were substantially different from these unbiased estimates (Supplemental Tables S5, non-cases, and S6). We observed significant depletion of the TT/AA/GG genotype and enrichment of Tc/AA/GG, Tc/Ag/Ga, and cc/gg/aa genotypes in LOADFS AD-unaffected subjects from families with (MA±SD=64.7±10.6 years) and without (MA±SD=76.2±8.9 years) history of AD (Supplemental Table S8). The proportions of genotypes in subjects without familial history of AD resembled those in the general (unbiased) population (Figure 3; Table 1 and Supplemental Table S8). We found that the LOADFS AD-unaffected subjects from families with a history of AD are relatively young (MA±SD= 64.7±10.6 years), and that they are substantially younger than the AD-affected subjects from the same families (MA±SD=81.0±7.6 years). Thus, they may merely not be old enough to develop AD yet. The observed differences in proportions indicate the clustering of the adverse compound genotypes in families with a history of AD. In the pooled sample of LOADFS, HRS, CHS, and FHS studies, this effect is diluted, and the exclusion of subjects with familial history of AD did not make a difference.
Fig. 3. Proportions of the beneficial (A) and adverse (B and C) compound genotypes in the Alzheimer’s disease (AD) unaffected subjects.
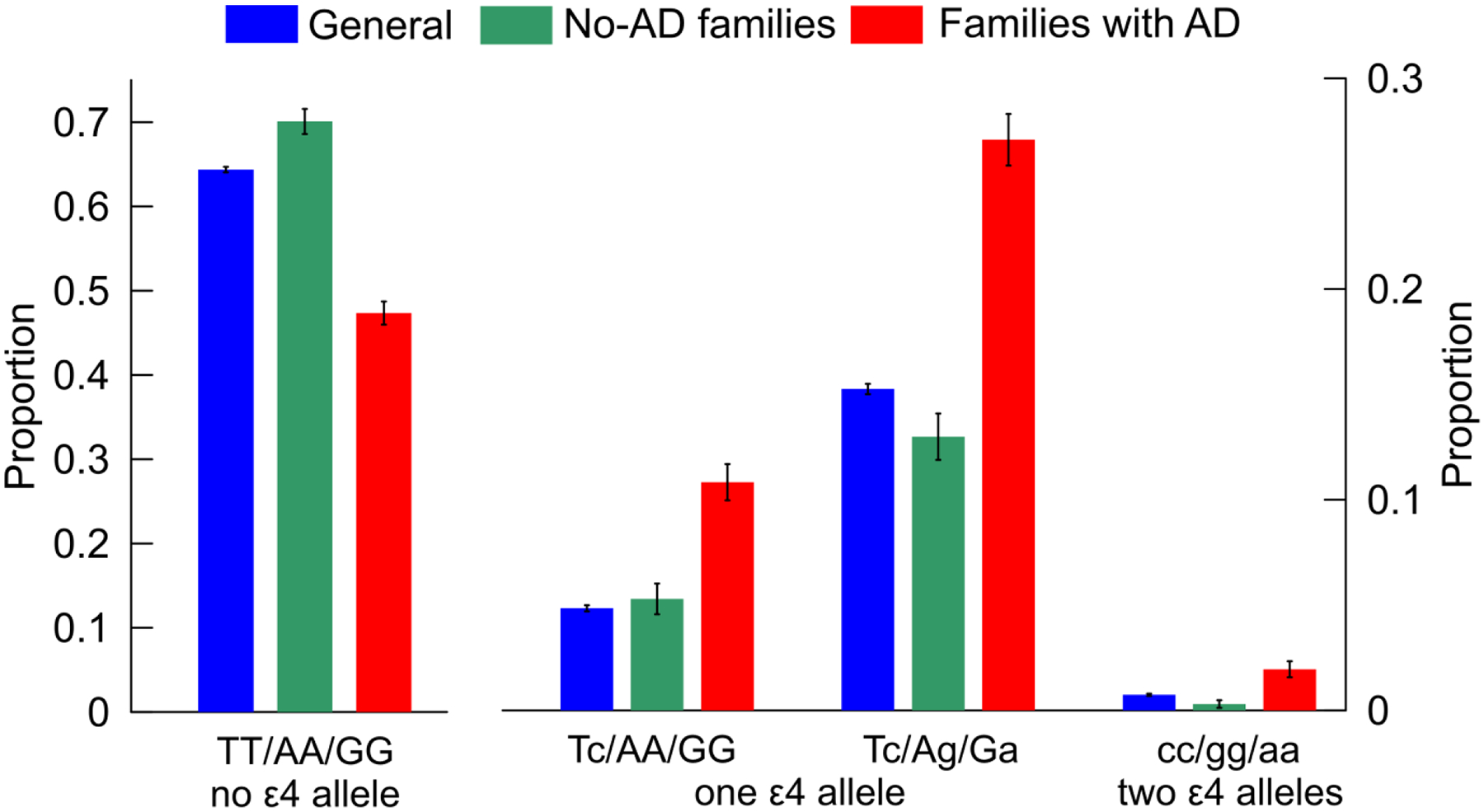
The labels on the x-axis show compound genotypes constructed from SNPs ordered as rs429358, rs2075650, or rs12721046 comprising samples with: (A) no ε4 allele, (B) one copy of ε4 allele, and (C) two copies of ε4 allele. Blue (“general”): the pooled sample of LOADFS, HRS, FHS, and CHS (data are in Supplemental Table S5, non-cases). Samples from the LOADFS from families: (green, “no-AD families”) without a history of AD and (red, “families with AD”) with history of AD (data are in Supplemental Table S8). Vertical lines show standard errors.
3.4. Compound-genotype-specific risks of AD
The regression analyses were not adjusted for family structure because the clustering of compound genotypes in families (see above Section) indicated meaningful biological effect. Subjects carrying a minor allele of rs429358, rs2075650, or rs12721046 SNP were at higher risks of AD measured by odds ratio (OR) (Supplemental Table S9). The smallest risks in groups of no, one, and two ε4 alleles were for TT/AA/GG, Tc/AA/GG, and cc/AA/GG carriers, respectively, i.e., for non-carriers of minor alleles of rs2075650 and rs12721046. Thus, carriers of minor alleles of rs2075650 and rs12721046 were at higher AD risk in each ε4 group (Figure 4, left panel, Supplemental Tables S9 and S10). The ε4 carriers of minor alleles of rs2075650 and rs12721046 were also at higher AD risk than non-carriers of these alleles (Figure 4, right panel, Supplemental Table S11). The risk for carriers of one ε4 allele who have at least one minor allele of rs2075650 and rs12721046 (Figure 4, 111 or 1XY) resembled that for carriers of two ε4 alleles who do not have minor alleles of these SNPs (Figure 4, 200).
Fig. 4. Odds ratios (ORs) for Alzheimer’s disease (AD) for selected compound genotypes.
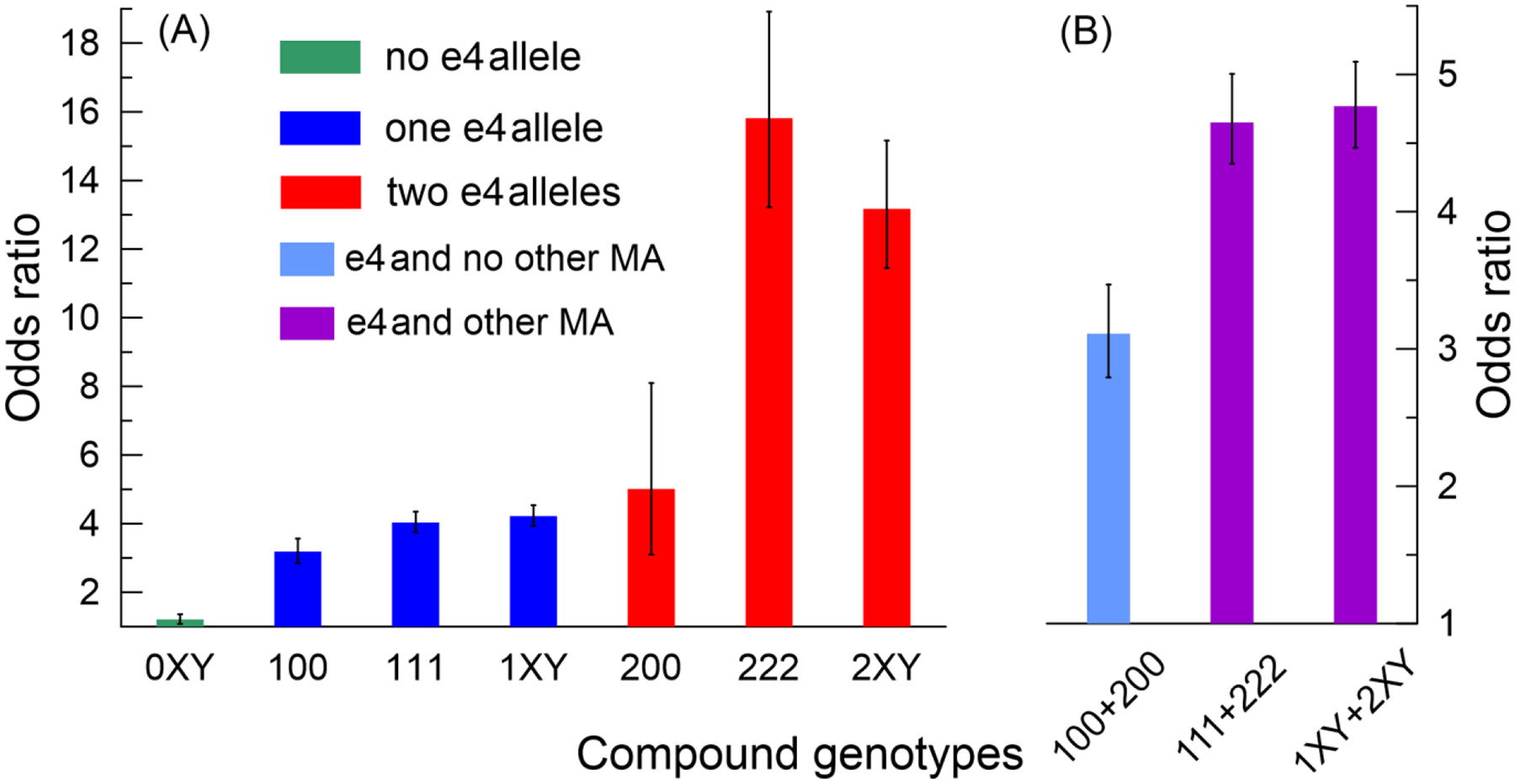
(A) green, blue, and red denote ORs in samples with no, one, and two copies of the ε4 allele, respectively. (B) ORs for the ε4 allele carriers who do not carry (light blue) and carry (purple) minor alleles (MAs) of rs2075650 and rs12721046 SNPs. Numbers in the labels on the x-axes show the number of MAs for SNPs ordered as rs429358, rs2075650, or rs12721046. Symbols “X” and “Y” denote aggregated compound genotypes; these symbols take values of 0, 1, or 2 but not simultaneously 0. Bars show the estimates of ORs from the models with the major allele homozygous genotype (TT/AA/GG) as a reference. Numerical estimates for ORs for: (i) 100, 111, 200, and 222 are in Supplemental Table S9, (ii) 0XY, 1XY, and 2XY are in Supplemental Table S10, and (iii) 100+200, 111+222, and 1XY+2XY are in Supplemental Table S11. Vertical lines show standard errors.
3.5. Validation in ADGC: Definitive roles of the ε4-bearing compound genotypes in the AD risks
Next, we characterized the excess of the AD risks for the ε4 carriers who carry and do not carry minor alleles of rs2075650 and rs12721046. Table 2 and Supplemental Table S12 show that carrying minor alleles of rs2075650 and rs12721046 substantially increased the risks of AD for carriers of the ε4 allele in the pooled sample. These findings were validated in an independent ADGC sample of the AD-affected (N=3,662, MA±SD=79.7±7.7 years) and unaffected (N=1,541, MA±SD=75.8±9.5 years) subjects. A meta-analysis of these results showed that carriers of one copy of the ε4 allele who also carry one minor allele of rs2075650 and rs12721046 (complete heterozygote, Tc/Ag/Ga, 111) were under 1.59-fold (p=8.46×10−7) higher risk of AD than those who do not carry these minor alleles (Tc/AA/GG [100] genotype). Carriers of two copies of the ε4 allele who have two minor alleles of rs2075650 and rs12721046 (complete minor allele homozygote, cc/gg/aa [222]) were under 4.37-fold (p=1.34×10−3) higher risk of AD compared to non-carriers of minor alleles of rs2075650 and rs12721046 (cc/AA/GG [200] genotype). Overall, the risk of AD for carriers of the ε4 allele who carry minor alleles of rs2075650 and rs12721046 compared to those who do not have them was 1.89-fold higher (p=4.69×10−13; Table 2, 1XY+2XY). Excluding carriers of the ε2 allele did not explain the observed excesses (Supplemental Table S12).
Table 2.
Excess of the Alzheimer’s disease (AD) risk for the ε4 carriers who carry and do not carry minor alleles of rs2075650 and rs12721046.
| MA coding | Sample | Ntotal | NAD | Beta | SE | Odds ratio | p-value |
|---|---|---|---|---|---|---|---|
| 100 | Reference | ||||||
| 111 | Pooled | 3000 | 718 | 0.28 | 0.12 | 1.32 | 2.51E-02 |
| 111 | ADGC | 1437 | 1230 | 0.71 | 0.14 | 2.03 | 8.29E-07 |
| 111 | Meta | 4437 | 1948 | 0.46 | 0.09 | 1.59 | 8.46E-07 |
| 1XY | Pooled | 3558 | 863 | 0.32 | 0.12 | 1.38 | 7.73E-03 |
| 1XY | ADGC | 1733 | 1463 | 0.61 | 0.14 | 1.85 | 8.39E-06 |
| 1XY | Meta | 5291 | 2326 | 0.45 | 0.09 | 1.57 | 7.81E-07 |
| 200 | Reference | ||||||
| 222 | Pooled | 265 | 155 | 1.40 | 0.63 | 4.05 | 2.73E-02 |
| 222 | ADGC | 310 | 299 | 1.56 | 0.67 | 4.76 | 1.96E-02 |
| 222 | Meta | 575 | 454 | 1.48 | 0.46 | 4.37 | 1.34E-03 |
| 2XY | Pooled | 468 | 257 | 1.23 | 0.56 | 3.42 | 2.77E-02 |
| 2XY | ADGC | 559 | 535 | 1.25 | 0.63 | 3.49 | 4.75E-02 |
| 2XY | Meta | 1027 | 792 | 1.24 | 0.42 | 3.45 | 3.05E-03 |
| 100+200 | Reference | ||||||
| 111+222 | Pooled | 3265 | 873 | 0.41 | 0.12 | 1.51 | 6.95E-04 |
| 111+222 | ADGC | 1747 | 1529 | 0.86 | 0.14 | 2.37 | 4.56E-10 |
| 111+222 | Meta | 5012 | 2402 | 0.61 | 0.09 | 1.84 | 2.68E-11 |
| 1XY+2XY | Pooled | 4026 | 1120 | 0.48 | 0.12 | 1.61 | 5.69E-05 |
| 1XY+2XY | ADGC | 2292 | 1998 | 0.84 | 0.13 | 2.32 | 2.08E-10 |
| 1XY+2XY | Meta | 6318 | 3118 | 0.64 | 0.09 | 1.89 | 4.69E-13 |
Column MA coding shows compound genotypes coded by the number of minor alleles (0, 1, or 2) in each SNP ordered as rs429358, rs2075650 and rs12721046. Symbols “X” and “Y” denote aggregated compound genotypes; these symbols take values of 0, 1, or 2 but not simultaneously 0.
Column Sample shows the results for the pooled sample and the AD Genetics Consortium (ADGC) sample, and the results from the meta-analysis of the pooled and ADGC samples. The pooled sample includes the NIA Late Onset Alzheimer’s disease Family Study, the Health and Retirement Study, the Cardiovascular Health Study, and the Framingham Heart Study original and offspring cohorts. Ntotal is the total number of subjects; NAD is the number of AD cases; SE is the standard error.
4. DISCUSSION
This article advances the understanding of the contribution of genetic variants from the APOE-harboring 19q13.32 region to AD risk, emphasizing the APOE ε2 and ε4 alleles, along three lines. First, we show that LD structures in this region differ in the AD affected and unaffected subjects in an ε2/ε4-dependent manner, and that the molecular signature of AD is substantially more heterogeneous in the ε2-negative sample than in the ε4-negative sample. This finding extends previous qualitative observations of the differences in LD structures in AD affected and unaffected subjects (Takei, et al., 2009,Yu, et al., 2007,Zhou, et al., 2019) and rigorous quantitative characterizations of such differences in the APOE ε2/ε4 non-stratified populations (Kulminski, et al., 2018). These signatures show that AD is associated with polygenic profiles rather than with individual alleles in this region, and that the architecture of these profiles is more affected by the ε4 allele than the ε2 allele. The difference in the ε2- and ε4-based molecular signatures of AD supports the independence of the ε2- and ε4-based genetic mechanisms of protection against AD and predisposition to AD, respectively. Accordingly, different explanations for the ε2-related protective effect and the ε4-related adverse effect is required contributing, thus, to a central question in AD research on elucidating a spectrum of APOE function (Belloy, et al., 2019).
Second, we provided compelling evidence on the non-independent role of the ε4 allele in AD, and identified the leading role of the compound genotype comprised of rs429358 (the ε4-coding SNP), rs2075650 (TOMM40), and rs12721046 (APOC1) in AD. These findings are supported by the differences in (i) LD between rs2075650 and rs12721046 SNPs in the ε2-negative and ε4-negative samples (Figure 2), (ii) proportions of compound genotypes in the AD-affected and unaffected subjects (Table 1), and (iii) the AD risks for carriers of the ε4 allele who carry and do not carry minor alleles of rs2075650 and rs12721046 (Figure 4). The leading role of this triple of SNPs in AD is also supported by the highly significant difference in joint variations of this triple in AD-affected and unaffected subjects accessed in (Alexander M. Kulminski, et al., 2020) via an alternative metric.
The definitive roles of the ε4-bearing compound genotypes comprised of alleles from this triple of SNPs are supported by the high excess of the AD risk for carriers of the ε4 allele who carry minor alleles of rs2075650 and rs12721046 compared to those who do not have them (Table 2). These findings were validated in the landmark NIA-funded ADGC initiative study. For example, meta-analysis showed the remarkably high 4.37-fold (p=1.34×10−3) excess of the AD risk for carriers of two ε4 alleles who carry two minor alleles of rs2075650 and rs12721046 (cc/gg/aa) compared to those who do not have them (cc/AA/GG). We propose considering the cc/gg/aa genotype as a more specific exceptionally high-risk genetic profile of AD.
Third, we show that the beneficial and adverse compound genotypes are clustered in families with a history of AD (Figure 3), regardless of whether these families are contrasted by older (MA=78.7 years) or younger (MA=37.8 years) general population. Enrichment of the adverse compound genotypes and depletion of the beneficial genotype in families with a history of AD compared to the younger population, which was not under noticeable survival selection, provides compelling support on the clustering of such genotypes due to their transmittance through generations. This finding is supported by the significant difference of LD structures between AD-affected and the younger population, and the lack of such difference between older AD-unaffected and the younger population (A. M. Kulminski, et al., 2020b). These findings raise a fundamental issue of driving forces of the AD-related compound genotypes. These forces should be related to recent evolutionary selection and be indirectly relevant to AD. Clustering of adverse/beneficial compound genotypes in families suggests that such driving forces should be associated with the AD-specific familial “exposures” such as ancestry, lifestyle, toxins, familial AD-related risk factors, etc. Accordingly, our findings call for comprehensive studies of potential mediating or moderating roles of various AD-related factors, especially those in AD-affected families, rather than just routinely considering them as adjustable covariates in the models. The importance of such studies is that they can help in identifying modifiable AD-related factors amenable to preventive interventions (Finch and Kulminski, 2019).
Despite the rigor of this study, we acknowledge its limitations. First, our study inherits intrinsic limitations pertinent to gathering AD-related information in large-scale studies that can affect the quality of AD diagnoses. Second, unlike model organisms and human monozygotic twins, human cohorts include subjects with a different genetic background. These individual-level genetic differences, complemented by potential ancestral differences between sub-populations, contribute to genetic heterogeneity. In large-scale studies, however, these limitations are partly offset by the size of the studied cohorts.
Thus, this study provides compelling evidence for the definitive roles of the APOE-TOMM40-APOC1 variants in the AD risk that aligns with the complex role of the APOE region in AD pathogenesis reported in previous studies (Babenko, et al., 2018,Crenshaw, et al., 2013,Lescai, et al., 2011,Linnertz, et al., 2014,Lutz, et al., 2016,Zhou, et al., 2019). Our findings support the independence of the mechanisms of the ε2-based protection against AD and the ε4-based predisposition to AD. Clustering of adverse/beneficial compound genotypes in families supports the role of haplotypes in AD. The complex structure of the ε4-based molecular signature of AD shows that other SNPs (or their proxies not examined in this work) may be involved in compound genotypes or haplotypes in a heterogeneous manner. Then, AD can be further tailored to even more homogeneous genetic profiles, consistently with the idea of personalized AD medicine. A better understanding of the potential roles of variants in the APOE region is critical for gaining insights into the biological mechanisms of AD and for defining more specific genetic profiles for assessment of subjects at an exceptionally high risk of AD.
Supplementary Material
Highlights.
The ε2 and ε4 alleles are associated with different molecular signatures of AD
AD is associated with polygenic profiles rather than with individual APOE alleles
Complex architecture of these profiles is more affected by the ε4 than ε2 allele
The ε4-bearing haplotype, but not ε4 allele alone, confers the strongest AD risk
Acknowledgments
This article was prepared using a data obtained through dbGaP (accession numbers phs000007.v28 [FHS], phs000287.v6 [CHS], phs000428.v2 [HRS], phs000168.v2 [LOADFS], phs000285.v.3 [CARDIA], and phs000372.v1 [ADGC]) and the University of Michigan. Phenotypic HRS data are available publicly and through restricted access from http://hrsonline.isr.umich.edu/index.php?p=data. The authors thank Arseniy P. Yashkin for help in the preparation of phenotypes in HRS. See extended acknowledgment in the Supplemental Acknowledgement file.
Funding Sources
This research was supported by Grants R01 AG047310, R01 AG061853, R01 AG065477, and R01 AG070488 from the NIA. The funders had no role in study design, data collection, and analysis, decision to publish, or preparation of the manuscript. The content is solely the authors’ responsibility and does not necessarily represent the official views of the National Institutes of Health.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Supplemental Information includes
Supplementary Acknowledgement text and 12 Supplementary Tables.
References:
- Babenko VN, Afonnikov DA, Ignatieva EV, Klimov AV, Gusev FE, Rogaev EI 2018. Haplotype analysis of APOE intragenic SNPs. BMC Neurosci 19(Suppl 1), 16. doi: 10.1186/s12868-018-0413-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Belloy ME, Napolioni V, Greicius MD 2019. A Quarter Century of APOE and Alzheimer’s Disease: Progress to Date and the Path Forward. Neuron 101(5), 820–38. doi: 10.1016/j.neuron.2019.01.056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Corella D, Ordovas JM 2014. Aging and cardiovascular diseases: the role of gene-diet interactions. Ageing research reviews 18, 53–73. doi: 10.1016/j.arr.2014.08.002. [DOI] [PubMed] [Google Scholar]
- Crenshaw DG, Gottschalk WK, Lutz MW, Grossman I, Saunders AM, Burke JR, Welsh-Bohmer KA, Brannan SK, Burns DK, Roses AD 2013. Using genetics to enable studies on the prevention of Alzheimer’s disease. Clin Pharmacol Ther 93(2), 177–85. doi: 10.1038/clpt.2012.222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crespi B, Stead P, Elliot M 2010. Evolution in health and medicine Sackler colloquium: Comparative genomics of autism and schizophrenia. Proc Natl Acad Sci U S A 107 Suppl 1, 1736–41. doi: 10.1073/pnas.0906080106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cupples LA, Heard-Costa N, Lee M, Atwood LD 2009. Genetics Analysis Workshop 16 Problem 2: the Framingham Heart Study data. BMC Proc 3 Suppl 7, S3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dobzhansky T 1973. Nothing in biology makes sense except in the light of evolution. pp 125–9.
- Eichler EE, Flint J, Gibson G, Kong A, Leal SM, Moore JH, Nadeau JH 2010. Missing heritability and strategies for finding the underlying causes of complex disease. Nat Rev Genet 11(6), 446–50. doi: 10.1038/nrg2809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Escott-Price V, Shoai M, Pither R, Williams J, Hardy J 2017. Polygenic score prediction captures nearly all common genetic risk for Alzheimer’s disease. Neurobiol Aging 49, 214 e7–e11. doi: 10.1016/j.neurobiolaging.2016.07.018. [DOI] [PubMed] [Google Scholar]
- Finch CE, Kulminski AM 2019. The Alzheimer’s Disease Exposome. Alzheimers Dement 15(9), 1123–32. doi: 10.1016/j.jalz.2019.06.3914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fried LP, Borhani NO, Enright P, Furberg CD, Gardin JM, Kronmal RA, Kuller LH, Manolio TA, Mittelmark MB, Newman A, et al. 1991. The Cardiovascular Health Study: design and rationale. Annals of epidemiology 1(3), 263–76. [DOI] [PubMed] [Google Scholar]
- Gibson G 2012. Rare and common variants: twenty arguments. Nat Rev Genet 13(2), 135–45. doi: 10.1038/nrg3118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Howie BN, Donnelly P, Marchini J 2009. A flexible and accurate genotype imputation method for the next generation of genome-wide association studies. PLoS Genet 5(6), e1000529. doi: 10.1371/journal.pgen.1000529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jack CR Jr., Bennett DA, Blennow K, Carrillo MC, Dunn B, Haeberlein SB, Holtzman DM, Jagust W, Jessen F, Karlawish J, Liu E, Molinuevo JL, Montine T, Phelps C, Rankin KP, Rowe CC, Scheltens P, Siemers E, Snyder HM, Sperling R, Contributors. 2018. NIA-AA Research Framework: Toward a biological definition of Alzheimer’s disease. Alzheimers Dement 14(4), 535–62. doi: 10.1016/j.jalz.2018.02.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jansen IE, Savage JE, Watanabe K, Bryois J, Williams DM, Steinberg S, Sealock J, Karlsson IK, Hagg S, Athanasiu L, Voyle N, Proitsi P, Witoelar A, Stringer S, Aarsland D, Almdahl IS, Andersen F, Bergh S, Bettella F, Bjornsson S, Braekhus A, Brathen G, de Leeuw C, Desikan RS, Djurovic S, Dumitrescu L, Fladby T, Hohman TJ, Jonsson PV, Kiddle SJ, Rongve A, Saltvedt I, Sando SB, Selbaek G, Shoai M, Skene NG, Snaedal J, Stordal E, Ulstein ID, Wang Y, White LR, Hardy J, Hjerling-Leffler J, Sullivan PF, van der Flier WM, Dobson R, Davis LK, Stefansson H, Stefansson K, Pedersen NL, Ripke S, Andreassen OA, Posthuma D 2019. Genome-wide meta-analysis identifies new loci and functional pathways influencing Alzheimer’s disease risk. Nat Genet 51(3), 404–13. doi: 10.1038/s41588-018-0311-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Juster FT, Suzman R 1995. An overview of the health and retirement study. Journal of Human Resources 30, S7–S56. [Google Scholar]
- Knopman DS, Haeberlein SB, Carrillo MC, Hendrix JA, Kerchner G, Margolin R, Maruff P, Miller DS, Tong G, Tome MB, Murray ME, Nelson PT, Sano M, Mattsson N, Sultzer DL, Montine TJ, Jack CR Jr., Kolb H, Petersen RC, Vemuri P, Canniere MZ, Schneider JA, Resnick SM, Romano G, van Harten AC, Wolk DA, Bain LJ, Siemers E 2018. The National Institute on Aging and the Alzheimer’s Association Research Framework for Alzheimer’s disease: Perspectives from the Research Roundtable. Alzheimers Dement 14(4), 563–75. doi: 10.1016/j.jalz.2018.03.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kulminski AM 2013. Unraveling genetic origin of aging-related traits: evolving concepts. Rejuvenation research 16(4), 304–12. doi: 10.1089/rej.2013.1441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kulminski AM, Huang J, Wang J, He L, Loika Y, Culminskaya I 2018. Apolipoprotein E region molecular signatures of Alzheimer’s disease. Aging Cell 17(4), e12779. doi: 10.1111/acel.12779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kulminski AM, Loika Y, Nazarian A, Culminskaya I 2020a. Quantitative and Qualitative Role of Antagonistic Heterogeneity in Genetics of Blood Lipids. J Gerontol A Biol Sci Med Sci 75(10), 1811–9. doi: 10.1093/gerona/glz225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kulminski AM, Philipp I, Loika Y, He L, Culminskaya I 2020. Haplotype architecture of the Alzheimer’s risk in the APOE region via co - skewness. Alzheimer’s & Dementia: Diagnosis, Assessment & Disease Monitoring 12(1), e12129. doi: 10.1002/dad2.12129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kulminski AM, Shu L, Loika Y, He L, Nazarian A, Arbeev K, Ukraintseva S, Yashin A, Culminskaya I 2020b. Genetic and regulatory architecture of Alzheimer’s disease in the APOE region. Alzheimer’s & Dementia: Diagnosis, Assessment & Disease Monitoring. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lambert JC, Ibrahim-Verbaas CA, Harold D, Naj AC, Sims R, Bellenguez C, DeStafano AL, Bis JC, Beecham GW, Grenier-Boley B, Russo G, Thorton-Wells TA, Jones N, Smith AV, Chouraki V, Thomas C, Ikram MA, Zelenika D, Vardarajan BN, Kamatani Y, Lin CF, Gerrish A, Schmidt H, Kunkle B, Dunstan ML, Ruiz A, Bihoreau MT, Choi SH, Reitz C, Pasquier F, Cruchaga C, Craig D, Amin N, Berr C, Lopez OL, De Jager PL, Deramecourt V, Johnston JA, Evans D, Lovestone S, Letenneur L, Moron FJ, Rubinsztein DC, Eiriksdottir G, Sleegers K, Goate AM, Fievet N, Huentelman MW, Gill M, Brown K, Kamboh MI, Keller L, Barberger-Gateau P, McGuiness B, Larson EB, Green R, Myers AJ, Dufouil C, Todd S, Wallon D, Love S, Rogaeva E, Gallacher J, St George-Hyslop P, Clarimon J, Lleo A, Bayer A, Tsuang DW, Yu L, Tsolaki M, Bossu P, Spalletta G, Proitsi P, Collinge J, Sorbi S, Sanchez-Garcia F, Fox NC, Hardy J, Deniz Naranjo MC, Bosco P, Clarke R, Brayne C, Galimberti D, Mancuso M, Matthews F, European Alzheimer’s Disease, I., Genetic, Environmental Risk in Alzheimer’s, D., Alzheimer’s Disease Genetic, C., Cohorts for, H., Aging Research in Genomic, E., Moebus S, Mecocci P, Del Zompo M, Maier W, Hampel H, Pilotto A, Bullido M, Panza F, Caffarra P, Nacmias B, Gilbert JR, Mayhaus M, Lannefelt L, Hakonarson H, Pichler S, Carrasquillo MM, Ingelsson M, Beekly D, Alvarez V, Zou F, Valladares O, Younkin SG, Coto E, Hamilton-Nelson KL, Gu W, Razquin C, Pastor P, Mateo I, Owen MJ, Faber KM, Jonsson PV, Combarros O, O’Donovan MC, Cantwell LB, Soininen H, Blacker D, Mead S, Mosley TH Jr., Bennett DA, Harris TB, Fratiglioni L, Holmes C, de Bruijn RF, Passmore P, Montine TJ, Bettens K, Rotter JI, Brice A, Morgan K, Foroud TM, Kukull WA, Hannequin D, Powell JF, Nalls MA, Ritchie K, Lunetta KL, Kauwe JS, Boerwinkle E, Riemenschneider M, Boada M, Hiltuenen M, Martin ER, Schmidt R, Rujescu D, Wang LS, Dartigues JF, Mayeux R, Tzourio C, Hofman A, Nothen MM, Graff C, Psaty BM, Jones L, Haines JL, Holmans PA, Lathrop M, Pericak-Vance MA, Launer LJ, Farrer LA, van Duijn CM, Van Broeckhoven C, Moskvina V, Seshadri S, Williams J, Schellenberg GD, Amouyel P 2013. Meta-analysis of 74,046 individuals identifies 11 new susceptibility loci for Alzheimer’s disease. Nat Genet 45(12), 1452–8. doi: 10.1038/ng.2802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lanoiselee HM, Nicolas G, Wallon D, Rovelet-Lecrux A, Lacour M, Rousseau S, Richard AC, Pasquier F, Rollin-Sillaire A, Martinaud O, Quillard-Muraine M, de la Sayette V, Boutoleau-Bretonniere C, Etcharry-Bouyx F, Chauvire V, Sarazin M, le Ber I, Epelbaum S, Jonveaux T, Rouaud O, Ceccaldi M, Felician O, Godefroy O, Formaglio M, Croisile B, Auriacombe S, Chamard L, Vincent JL, Sauvee M, Marelli-Tosi C, Gabelle A, Ozsancak C, Pariente J, Paquet C, Hannequin D, Campion D, collaborators of the, C.N.R.M.A.J.p. 2017. APP, PSEN1, and PSEN2 mutations in early-onset Alzheimer disease: A genetic screening study of familial and sporadic cases. PLoS Med 14(3), e1002270. doi: 10.1371/journal.pmed.1002270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee JH, Cheng R, Graff-Radford N, Foroud T, Mayeux R, National Institute on Aging Late-Onset Alzheimer’s Disease Family Study, G. 2008. Analyses of the National Institute on Aging Late-Onset Alzheimer’s Disease Family Study: implication of additional loci. Arch Neurol 65(11), 1518–26. doi: 10.1001/archneur.65.11.1518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lescai F, Chiamenti AM, Codemo A, Pirazzini C, D’Agostino G, Ruaro C, Ghidoni R, Benussi L, Galimberti D, Esposito F, Marchegiani F, Cardelli M, Olivieri F, Nacmias B, Sorbi S, Tagliavini F, Albani D, Martinelli Boneschi F, Binetti G, Santoro A, Forloni G, Scarpini E, Crepaldi G, Gabelli C, Franceschi C 2011. An APOE haplotype associated with decreased epsilon4 expression increases the risk of late onset Alzheimer’s disease. J Alzheimers Dis 24(2), 235–45. doi: 10.3233/JAD-2011-101764. [DOI] [PubMed] [Google Scholar]
- Levy-Lahad E, Wasco W, Poorkaj P, Romano DM, Oshima J, Pettingell WH, Yu CE, Jondro PD, Schmidt SD, Wang K, et al. 1995. Candidate gene for the chromosome 1 familial Alzheimer’s disease locus. Science 269(5226), 973–7. [DOI] [PubMed] [Google Scholar]
- Lewontin RC 1988. On measures of gametic disequilibrium. Genetics 120(3), 849–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Linnertz C, Anderson L, Gottschalk W, Crenshaw D, Lutz MW, Allen J, Saith S, Mihovilovic M, Burke JR, Welsh-Bohmer KA, Roses AD, Chiba-Falek O 2014. The cis-regulatory effect of an Alzheimer’s disease-associated poly-T locus on expression of TOMM40 and apolipoprotein E genes. Alzheimers Dement 10(5), 541–51. doi: 10.1016/j.jalz.2013.08.280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lutz MW, Crenshaw D, Welsh-Bohmer KA, Burns DK, Roses AD 2016. New Genetic Approaches to AD: Lessons from APOE-TOMM40 Phylogenetics. Curr Neurol Neurosci Rep 16(5), 48. doi: 10.1007/s11910-016-0643-8. [DOI] [PubMed] [Google Scholar]
- Marigorta UM, Rodriguez JA, Gibson G, Navarro A 2018. Replicability and Prediction: Lessons and Challenges from GWAS. Trends Genet 34(7), 504–17. doi: 10.1016/j.tig.2018.03.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Naj AC, Jun G, Beecham GW, Wang LS, Vardarajan BN, Buros J, Gallins PJ, Buxbaum JD, Jarvik GP, Crane PK, Larson EB, Bird TD, Boeve BF, Graff-Radford NR, De Jager PL, Evans D, Schneider JA, Carrasquillo MM, Ertekin-Taner N, Younkin SG, Cruchaga C, Kauwe JS, Nowotny P, Kramer P, Hardy J, Huentelman MJ, Myers AJ, Barmada MM, Demirci FY, Baldwin CT, Green RC, Rogaeva E, St George-Hyslop P, Arnold SE, Barber R, Beach T, Bigio EH, Bowen JD, Boxer A, Burke JR, Cairns NJ, Carlson CS, Carney RM, Carroll SL, Chui HC, Clark DG, Corneveaux J, Cotman CW, Cummings JL, DeCarli C, DeKosky ST, Diaz-Arrastia R, Dick M, Dickson DW, Ellis WG, Faber KM, Fallon KB, Farlow MR, Ferris S, Frosch MP, Galasko DR, Ganguli M, Gearing M, Geschwind DH, Ghetti B, Gilbert JR, Gilman S, Giordani B, Glass JD, Growdon JH, Hamilton RL, Harrell LE, Head E, Honig LS, Hulette CM, Hyman BT, Jicha GA, Jin LW, Johnson N, Karlawish J, Karydas A, Kaye JA, Kim R, Koo EH, Kowall NW, Lah JJ, Levey AI, Lieberman AP, Lopez OL, Mack WJ, Marson DC, Martiniuk F, Mash DC, Masliah E, McCormick WC, McCurry SM, McDavid AN, McKee AC, Mesulam M, Miller BL, Miller CA, Miller JW, Parisi JE, Perl DP, Peskind E, Petersen RC, Poon WW, Quinn JF, Rajbhandary RA, Raskind M, Reisberg B, Ringman JM, Roberson ED, Rosenberg RN, Sano M, Schneider LS, Seeley W, Shelanski ML, Slifer MA, Smith CD, Sonnen JA, Spina S, Stern RA, Tanzi RE, Trojanowski JQ, Troncoso JC, Van Deerlin VM, Vinters HV, Vonsattel JP, Weintraub S, Welsh-Bohmer KA, Williamson J, Woltjer RL, Cantwell LB, Dombroski BA, Beekly D, Lunetta KL, Martin ER, Kamboh MI, Saykin AJ, Reiman EM, Bennett DA, Morris JC, Montine TJ, Goate AM, Blacker D, Tsuang DW, Hakonarson H, Kukull WA, Foroud TM, Haines JL, Mayeux R, Pericak-Vance MA, Farrer LA, Schellenberg GD 2011. Common variants at MS4A4/MS4A6E, CD2AP, CD33 and EPHA1 are associated with late-onset Alzheimer’s disease. Nat Genet 43(5), 436–41. doi: 10.1038/ng.801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nesse RM, Ganten D, Gregory TR, Omenn GS 2012. Evolutionary molecular medicine. J Mol Med (Berl) 90(5), 509–22. doi: 10.1007/s00109-012-0889-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nesse RM, Williams GC 1994. Why we get sick : the new science of Darwinian medicine. 1st ed. Times Books, New York. [Google Scholar]
- Oeppen J, Vaupel JW 2002. Demography. Broken limits to life expectancy. Science 296(5570), 1029–31. doi: 10.1126/science.1069675. [DOI] [PubMed] [Google Scholar]
- Raichlen DA, Alexander GE 2014. Exercise, APOE genotype, and the evolution of the human lifespan. Trends Neurosci 37(5), 247–55. doi: 10.1016/j.tins.2014.03.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rogaev EI, Sherrington R, Rogaeva EA, Levesque G, Ikeda M, Liang Y, Chi H, Lin C, Holman K, Tsuda T, et al. 1995. Familial Alzheimer’s disease in kindreds with missense mutations in a gene on chromosome 1 related to the Alzheimer’s disease type 3 gene. Nature 376(6543), 775–8. doi: 10.1038/376775a0. [DOI] [PubMed] [Google Scholar]
- Roses AD, Lutz MW, Amrine-Madsen H, Saunders AM, Crenshaw DG, Sundseth SS, Huentelman MJ, Welsh-Bohmer KA, Reiman EM 2010. A TOMM40 variable-length polymorphism predicts the age of late-onset Alzheimer’s disease. Pharmacogenomics J 10(5), 375–84. doi: 10.1038/tpj.2009.69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schork NJ 2015. Personalized medicine: Time for one-person trials. Nature 520(7549), 609–11. doi: 10.1038/520609a. [DOI] [PubMed] [Google Scholar]
- Sherrington R, Rogaev EI, Liang Y, Rogaeva EA, Levesque G, Ikeda M, Chi H, Lin C, Li G, Holman K, Tsuda T, Mar L, Foncin JF, Bruni AC, Montesi MP, Sorbi S, Rainero I, Pinessi L, Nee L, Chumakov I, Pollen D, Brookes A, Sanseau P, Polinsky RJ, Wasco W, Da Silva HA, Haines JL, Perkicak-Vance MA, Tanzi RE, Roses AD, Fraser PE, Rommens JM, St George-Hyslop PH 1995. Cloning of a gene bearing missense mutations in early-onset familial Alzheimer’s disease. Nature 375(6534), 754–60. doi: 10.1038/375754a0. [DOI] [PubMed] [Google Scholar]
- Silverberg N, Elliott C, Ryan L, Masliah E, Hodes R 2018. NIA commentary on the NIA-AA Research Framework: Towards a biological definition of Alzheimer’s disease. Alzheimers Dement 14(4), 576–8. doi: 10.1016/j.jalz.2018.03.004. [DOI] [PubMed] [Google Scholar]
- Sweeney MD, Montagne A, Sagare AP, Nation DA, Schneider LS, Chui HC, Harrington MG, Pa J, Law M, Wang DJJ, Jacobs RE, Doubal FN, Ramirez J, Black SE, Nedergaard M, Benveniste H, Dichgans M, Iadecola C, Love S, Bath PM, Markus HS, Salman RA, Allan SM, Quinn TJ, Kalaria RN, Werring DJ, Carare RO, Touyz RM, Williams SCR, Moskowitz MA, Katusic ZS, Lutz SE, Lazarov O, Minshall RD, Rehman J, Davis TP, Wellington CL, Gonzalez HM, Yuan C, Lockhart SN, Hughes TM, Chen CLH, Sachdev P, O’Brien JT, Skoog I, Pantoni L, Gustafson DR, Biessels GJ, Wallin A, Smith EE, Mok V, Wong A, Passmore P, Barkof F, Muller M, Breteler MMB, Roman GC, Hamel E, Seshadri S, Gottesman RF, van Buchem MA, Arvanitakis Z, Schneider JA, Drewes LR, Hachinski V, Finch CE, Toga AW, Wardlaw JM, Zlokovic BV 2019. Vascular dysfunction-The disregarded partner of Alzheimer’s disease. Alzheimers Dement 15(1), 158–67. doi: 10.1016/j.jalz.2018.07.222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takei N, Miyashita A, Tsukie T, Arai H, Asada T, Imagawa M, Shoji M, Higuchi S, Urakami K, Kimura H, Kakita A, Takahashi H, Tsuji S, Kanazawa I, Ihara Y, Odani S, Kuwano R, Japanese Genetic Study Consortium for Alzheimer, D. 2009. Genetic association study on in and around the APOE in late-onset Alzheimer disease in Japanese. Genomics 93(5), 441–8. doi: 10.1016/j.ygeno.2009.01.003. [DOI] [PubMed] [Google Scholar]
- Vijg J, Suh Y 2005. Genetics of longevity and aging. Annu Rev Med 56, 193–212. doi: 10.1146/annurev.med.56.082103.104617. [DOI] [PubMed] [Google Scholar]
- Visscher PM, Wray NR, Zhang Q, Sklar P, McCarthy MI, Brown MA, Yang J 2017. 10 Years of GWAS Discovery: Biology, Function, and Translation. Am J Hum Genet 101(1), 5–22. doi: 10.1016/j.ajhg.2017.06.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wellek S, Ziegler A 2009. A genotype-based approach to assessing the association between single nucleotide polymorphisms. Hum Hered 67(2), 128–39. doi: 10.1159/000179560. [DOI] [PubMed] [Google Scholar]
- Yamazaki Y, Zhao N, Caulfield TR, Liu CC, Bu G 2019. Apolipoprotein E and Alzheimer disease: pathobiology and targeting strategies. Nat Rev Neurol 15(9), 501–18. doi: 10.1038/s41582-019-0228-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu CE, Seltman H, Peskind ER, Galloway N, Zhou PX, Rosenthal E, Wijsman EM, Tsuang DW, Devlin B, Schellenberg GD 2007. Comprehensive analysis of APOE and selected proximate markers for late-onset Alzheimer’s disease: patterns of linkage disequilibrium and disease/marker association. Genomics 89(6), 655–65. doi: 10.1016/j.ygeno.2007.02.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zaykin DV, Meng Z, Ehm MG 2006. Contrasting linkage-disequilibrium patterns between cases and controls as a novel association-mapping method. Am J Hum Genet 78(5), 737–46. doi: 10.1086/503710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao N, Liu CC, Qiao W, Bu G 2018. Apolipoprotein E, Receptors, and Modulation of Alzheimer’s Disease. Biol Psychiatry 83(4), 347–57. doi: 10.1016/j.biopsych.2017.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou X, Chen Y, Mok KY, Kwok TCY, Mok VCT, Guo Q, Ip FC, Chen Y, Mullapudi N, Alzheimer’s Disease Neuroimaging I, Giusti-Rodriguez P, Sullivan PF, Hardy J, Fu AKY, Li Y, Ip NY 2019. Non-coding variability at the APOE locus contributes to the Alzheimer’s risk. Nat Commun 10(1), 3310. doi: 10.1038/s41467-019-10945-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.