

Abstract
Plasmodium and other vector-borne pathogens have evolved mechanisms to hijack the mammalian fibrinolytic system to facilitate infection of the human host and the invertebrate vector. Plasmin, the effector protease of fibrinolysis, maintains homeostasis in the blood vasculature by degrading the fibrin that forms blood clots. Plasmin also degrades proteins from extracellular matrices, the complement system, and immunoglobulins. Here, we review some of the mechanisms by which vector-borne pathogens interact with components of the fibrinolytic system and co-opt its functions to facilitate transmission and infection in the host and the vector. Further, we discuss innovative strategies beyond conventional therapeutics that could be developed to target the interaction of vector-borne pathogens with the fibrinolytic proteins and prevent their transmission.
Keywords: Fibrinolysis, Plasmin, Vector-borne Diseases, Malaria
Vector-Borne Diseases and Global Burden
Despite the progress in improving global public health systems, vector-borne diseases continue to emerge and re-emerge, accounting for more than 17% of all infectious diseases and over 700,000 deaths annually across the globe i. Vector-borne diseases are caused by parasites, bacteria or viruses that can transmit between various hosts, including humans and insects. A complex set of demographic, environmental and socio-economic factors define the distribution and dissemination of these diseases across the world, with tropical and subtropical regions experiencing the highest burden. The increase in global travel and trade, unplanned urbanization and climate change have led to outbreaks of these diseases in new regions and countries i.
During infection, vector-borne pathogens must overcome host and vector (see Glossary) physical barriers such as epithelial surfaces and extracellular matrices (ECM), that in combination with innate and adaptive immune responses provide protection against invasive microorganisms [1, 2]. However, vector-borne pathogens have evolved to persist in the host and the vector, resulting in acute diseases and chronic infections. Their adaptations include a myriad of molecular mechanisms, from antigenic hypervariability and the release of effector proteins that suppress the host immune system to the utilization of humoral factors to facilitate invasion [3]. Blood feeding is an absolute requirement for the reproduction of hematophagous vectors and for transmission of their pathogens. Consequently, both organisms have adapted to exploit and counteract host hemostatic and immune responses. For example, the saliva of hematophagous vectors contains multiple molecules that modulate inflammatory responses, coagulation, complement attack, and fibrinolysis [4]. In addition, various vector-borne pathogens co-opt molecules from the vector or the host, like the mammalian fibrinolytic proteins, used to breakdown fibrin, ECM and complement proteins during invasion and immune evasion [5].
In this review, we highlight the role of the fibrinolytic system during infection by various vector-borne diseases, with emphasis on Plasmodium infection of the mosquito and the vertebrate hosts.
Overview of the Fibrinolytic System
When a blood vessel is injured, clotting factors are activated in a series of complex biochemical reactions that culminate with the cleavage of fibrinogen by thrombin, inducing the spontaneous polymerization of fibrin that together with platelets form a hemostatic plug (clot) to prevent excessive bleeding. Fibrinolysis is the enzymatic breakdown of fibrin by the serine protease plasmin, an essential process that sustains plasma homeostasis by preventing excessive fibrin and clots from occluding blood vessels [6]. Plasmin is formed from its inactive zymogen plasminogen through proteolysis by the serine proteases tissue-type plasminogen activator (tPA) and urokinase-type plasminogen activator (uPA) [7]. Plasminogen circulates in the blood at a concentration of ~200 μg/ml, whereas tPA and uPA circulate at ~5 ng/ml and ~8 ng/ml, respectively [8]. Plasminogen activation is regulated by plasminogen activator inhibitor 1 (PAI-1), a serine protease inhibitor (serpin) that inhibits tPA and uPA by binding irreversibly to their active site, thus inhibiting plasminogen activation [9]. In addition, the protease inhibitors α2-antiplasmin and α2-macroglobulin regulate fibrinolysis by inhibiting free plasmin [10] (Figure 1A).
Figure 1. The fibrinolytic system and its roles in humans and pathogens.
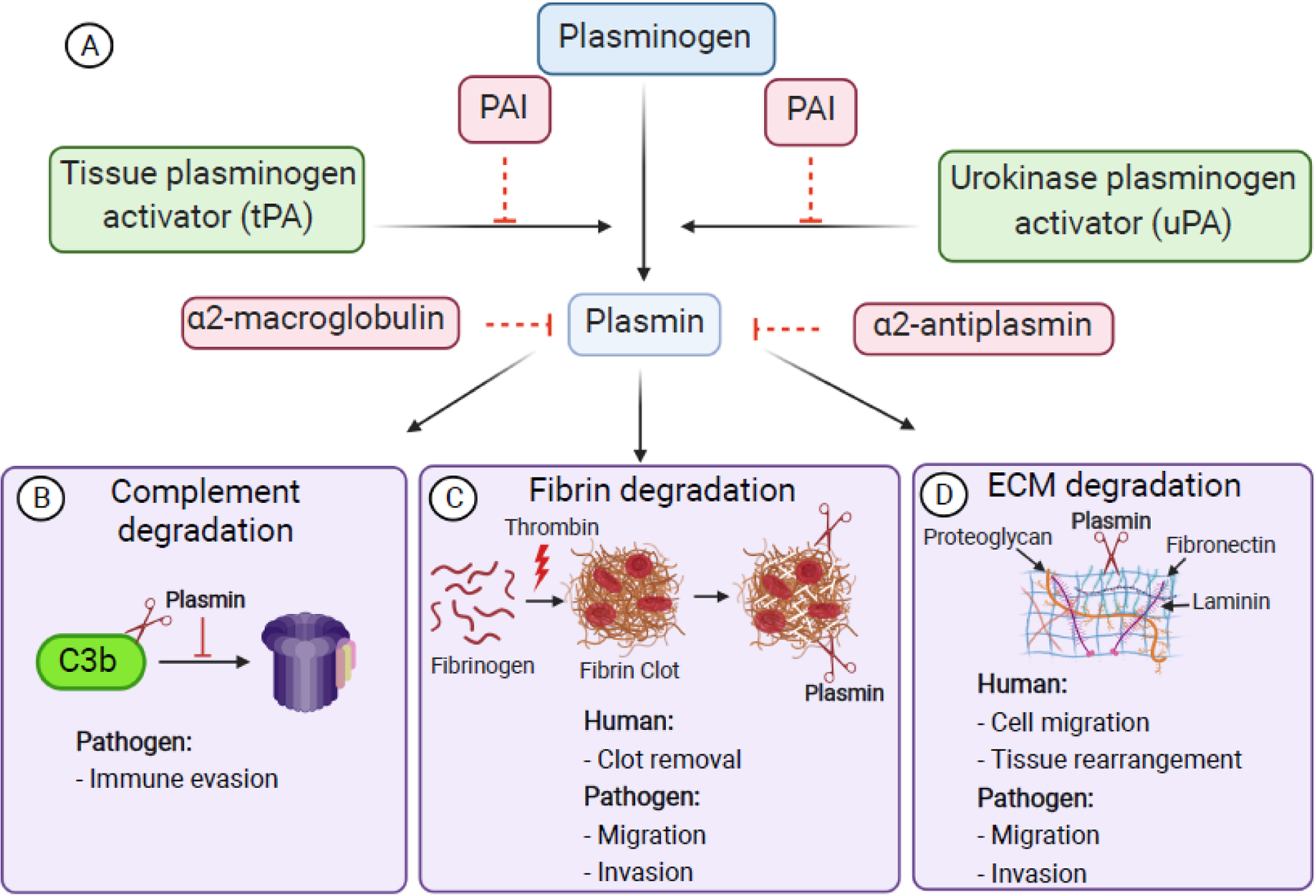
A. Plasminogen is activated into the serine protease plasmin by the tissue-type plasminogen activator (tPA) and urokinase-type plasminogen activator (uPA). Both tPA and uPA are inhibited by plasminogen activator inhibitor-1 (PAI-1). Plasmin is inhibited by α2- antiplasmin and α2-macroglobulin. B. Pathogens evade immunity by co-opting plasmin to degrade C3b and prevent the formation of the terminal complement complex (TCC). C. During coagulation, thrombin converts fibrinogen into insoluble fibrin which polymerizes to form a clot. Plasmin degrades fibrin, thus facilitating clot removal to maintain homeostasis. D. Plasmin degrades ECM proteins, like fibronectin, laminin, and proteoglycans, thus facilitating cell migration and tissue rearrangement. Pathogens co-opt plasmin to facilitate migration through fibrin networks (C) and ECM (D). Figure created with BioRender.com.
The formation of fibrin clots at injury sites not only prevents excessive bleeding, but also provides protection against microbial infections by trapping pathogens within the clot [11]. Therefore, fibrin formation presents a potential bottleneck to pathogen infectivity, and the understanding of how pathogens circumvent this obstacle is imperative. Plasmin can also degrade immune factors (complement), ECM proteins (laminin, fibronectin, vitronectin, proteoglycans) and activate latent collagenases, leading to enhanced cell migration during processes like tissue rearrangement and cancer metastasis [12–14] (Figure 1).
The Fibrinolytic System and Vector-borne Diseases
The fact that plasmin can degrade a variety of structural and immune molecules makes plasminogen an ideal binding partner for invasive pathogens. Surface-bound plasminogen can be activated to plasmin by either pathogen- or host-derived plasminogen activators (Table 1). Pathogens express several plasminogen receptors, including “moonlight” proteins like enolase and glyceraldehyde 3-phosphate dehydrogenase (GAPDH) that are secreted through non-classical pathways and perform critical roles in the organism’s development besides binding plasminogen (Table 1). Plasminogen binds to receptors via its Kringle domains which have high affinity for C-terminal or internal lysine residues in the receptor. Lysine-dependent binding to fibrin or to a receptor on a cell surface facilitates plasminogen activation while protecting plasmin from inhibition by α2-antiplasmin and α2-macroglobulin [15, 16]. Together, this represents a conserved mechanism whereby pathogens co-opt plasminogen to facilitate dissemination within the host.
Table 1.
The Role of the Fibrinolytic System in Infectious Diseases
| Group | Infectious species | Receptor(s) | Activator(s) | Plasmin role |
|---|---|---|---|---|
| Viruses | Dengue Virus (DENV) [40, 42, 83] | DENV Env. | Unknown | Enhances mosquito midgut infection. |
| Bacteria | Borelia burgdorferi [46, 48, 51, 56–58, 60, 61, 64] | OspA OspC ErpP, ErpA, ErpC FHBP CspA Enolase |
uPA | ECM protein degradation and activation of matrix metalloproteases. |
| Protozoa | Leishmania mexicana [32–34] | Enolase LACK |
tPA | Enhances parasite virulence and migration within skin lesions. |
| Trypanosoma cruzi [35] | Unknown | uPA | Parasite survival within triatomine midgut, potentially by degrading immune molecules and digesting other proteins for nutrient supply. | |
| Plasmodium spp. [28, 29] | Enolase | tPA, uPA | Fibrin degradation facilitates gamete migration for successful fertilization and traversal of sporozoites through host physical barriers. | |
| Helminths | Schistosoma spp. [84–86] | Enolase GAPDH Actin |
tPA | Prevents clot formation for survival in blood vessels. |
The multiple roles of plasmin during Plasmodium infection
Malaria is caused by parasites of the genus Plasmodium, of which Plasmodium falciparum is responsible for most malaria-related deaths. The infection begins when an infected Anopheles mosquito injects sporozoites in the dermis while probing in search of a blood vessel. Sporozoites migrate within the ECM of the dermis to locate and invade a blood vessel to enter the blood circulation. Sporozoites exit the circulation by traversing the liver tissue to invade hepatocytes, where the parasite undergoes asexual schizogony to generate thousands of merozoites. Merozoites are released in the blood circulation where they invade red blood cells (RBCs) and develop into the intraerythrocytic stages known as ring, trophozoite and schizont, which are responsible for the clinical symptoms of malaria. Merozoites produced by schizogony employ a combination of endogenous and host-derived proteases to degrade the parasitophorous vacuole and the RBC membranes, ensuing egress and the infection of more RBCs. Severe malaria infections caused by P. falciparum are often associated with disseminated intravascular coagulation and increased fibrin levels in plasma, presumably caused by increased or decreased expression of pro-coagulation factors and fibrinolytic factors, respectively [17]. For example, a study in India showed that patients with complicated malaria had higher PAI-1 and lower tPA levels [18]. Similarly, a field study involving Zambian children aged <6 years old reported an association between severe malaria and elevated levels of plasma PAI-1 [19]. Interestingly, infection of PAI-1 deficient mice with the non-lethal P. chabaudi resulted in a significant delay in parasite clearance, suggesting that plasminogen activation enhances parasite infectivity [20]. However, earlier studies showed that the infectivity of the lethal Plasmodium vinckei vinckei was not affected in mice deficient in plasminogen, tPA, uPA and the uPA receptor [21]. Although the evidence in the rodent models is somewhat contrasting, possibly due to the differences in the experimental settings, the evidence in P. falciparum suggests that the fibrinolytic system has a role in parasite development.
P. falciparum rings and trophozoites can internalize plasminogen, which is hydrolyzed by the parasite into angiostatin, an angiogenesis inhibitor [22] (Figure 2A, Key Figure). Supernatants from infected erythrocytes pre-incubated with plasminogen strongly inhibited (72%) angiogenesis in endothelial cells cultured in vitro. Angiostatin binds to endothelial cells and induces apoptosis [23]; therefore, the elevation of angiostatin has been associated with cerebral malaria [24]. Whether angiostatin directly affects parasite infectivity during the intraerythrocytic cycle remains to be determined. In addition, uPA binds to trophozoite-infected RBCs, and its depletion from the culture medium inhibits merozoite release. This suggests that uPA is critical for parasite egress, presumably by activating plasminogen [25]. Since uPA binding to infected erythrocytes is stage specific and there is no known uPA receptor on the erythrocyte surface, it is reasonable to speculate that the receptor is a parasite protein exported to the erythrocyte surface.
Figure 2, Key Figure. Vector-borne pathogens co-opt the fibrinolytic system for invasion and transmission.

A. The role of the fibrinolytic proteins in malaria. A1. uPA binding on mature trophozoites is essential for merozoite egress. A2. Plasmodium gametes recruit host tPA to activate surface-bound plasminogen into plasmin. It is proposed that plasmin degrades fibrin networks in the midgut blood bolus and thus facilitate gamete migration and fertilization. A3. tPA activates plasminogen into plasmin at the sporozoite surface and plasmin facilitates sporozoite migration in the dermis and the liver by degrading ECM proteins. A4. tPA activates plasminogen to plasmin on infected RBCs. Plasmin facilitates parasite complement evasion by degrading C3b on the infected RBC surface, thereby preventing the formation of the terminal complement complex (TCC) and parasite lysis. B1. Plasminogen-deficient mice restrict the formation of leishmaniasis lesions suggesting the role of plasmin for parasite dissemination. C1. T. cruzi epimastigotes enhance plasminogen activation by tPA, which might have a role in parasite traversal and survival (*). D1. Plasmin enhances DENV infection of Ae. aegypti mosquitoes. It is proposed that plasmin on the surface of DENV facilitates the degradation of the glycocalyx matrix and enhances internalization of the virus by the midgut epithelial cells. E1. Borrelia spp. plasminogen activation by tPA and uPA facilitates systemic bacteria dissemination in mice, and dissemination from the tick gut to the salivary glands. E2. Plasmin enhances Borrelia spp. infection of the brain and heart by disrupting the tissue through the degradation of tight junction proteins. Figure created with BioRender.com.
Recently, Reiss et al. [26] studied the role of plasmin during complement evasion in the erythrocytic stage of P. falciparum (Figure 2A). They showed that tPA activates plasminogen on the surface of the infected RBC, and that plasmin specifically degraded C3b bound to the infected RBC. C3b is a central molecule required for further activation of the human complement system and for the recruitment of the Terminal Complement Complex (TCC) that mediates pathogen lysis. Culturing P. falciparum in plasminogen-depleted serum increased TCC formation on infected RBCs, whereas plasminogen supplementation significantly reduced TCC formation, showing the importance of plasminogen in halting the complement cascade [26]. Additionally, depletion of plasminogen in serum reduced parasite growth, implying that plasminogen was required for complement evasion [26]. Interestingly, schizonts and trophozoites partially activated plasminogen in the absence of exogeneous tPA or uPA, suggesting that P. falciparum asexual stages might have an intrinsic ability to activate plasminogen [26].
Plasmodium parasites are transmitted by Anopheles mosquitoes. After the mosquito ingests blood from a malaria infected person, Plasmodium gametocytes transform into male and female gametes that mate within the midgut blood bolus to form zygotes. The zygotes develop into motile ookinetes that invade the midgut epithelium and form oocysts on the basal wall of the midgut. Each oocyst produces thousands of sporozoites that are released into the hemocoel where they invade the mosquito salivary glands [27]. A study by Ghosh et al. [28] demonstrated that plasminogen binds to the surface of Plasmodium ookinetes and that depletion of plasminogen from the infectious blood meal strongly reduced oocyst formation in the mosquito. This phenotype was rescued by supplementing the blood meal with plasminogen, whereas supplementation with a catalytically inactive plasminogen failed to restore oocyst formation, demonstrating the significance of plasmin activity for mosquito infection. In addition, enolase was identified as a plasminogen receptor on the ookinete surface [28]. Bioinformatic analysis showed that an internal plasminogen-binding lysine motif conserved among enolases of multiple pathogens is also conserved in Plasmodium enolase (DKSLVK) [28]. The synthetic peptide DKSLVK inhibited the binding of plasminogen to ookinetes and mosquito infection by P. berghei, showing that the interaction with parasite enolase is via the plasminogen Kringle domains [28].
Our laboratory has further characterized the role of plasminogen during Plasmodium invasion of the mosquito midgut (Figure 2A). Immunofluorescence assays showed that tPA and uPA bind to the surface of P. falciparum gametes, zygotes and ookinetes where they activate plasminogen. Enzymatic assays using live female gametes showed that the activation of parasite-bound plasminogen was only observed in the presence of tPA, suggesting that the mosquito stages do not produce an endogenous plasminogen activator [29]. Importantly, the inhibition of plasminogen activation by supplementing the infectious blood meal with PAI-1 strongly reduced ookinete formation in mosquitoes, showing that plasmin is required for the development of gametes and zygotes [29]. This inhibition was only observed in vivo in the mosquito blood meal, where the coagulation cascade is partially active, resulting in the polymerization of fibrin after blood feeding. Fibrin, the main substrate for plasmin, increases the blood bolus viscosity, which hampers parasite development, presumably by imposing a barrier for gamete migration [29]. We propose that Plasmodium parasites use surface plasmin to cleave fibrin polymers to facilitate motility within the blood bolus.
Plasmodium sporozoites, the infectious stage of the parasite, must migrate through the ECM of the dermis to find and penetrate a blood vessel. The blood circulation carries the sporozoites to the liver where they traverse Kupffer cells and the space of Disse to infect the hepatocytes. Similar to gametes, sporozoites bind plasminogen, tPA and uPA, and plasmin activity on the sporozoite surface is only detected in the presence of tPA [29]. Inhibition of plasminogen activation by PAI-1 significantly reduced P. berghei sporozoite migration in the mouse dermis as well as in the liver tissue, resulting in a significant reduction of infectivity. PAI-1 also inhibited sporozoite migration in Matrigel, a reconstituted basement membrane composed of ECM proteins [29]. These findings suggest that plasminogen enhances sporozoite infectivity by facilitating traversal through ECMs, presumably by degrading ECM proteins(Figure 2A). Taken together, data demonstrates that Plasmodium utilizes plasminogen for infection of the mosquito vector and the vertebrate host by facilitating migration and evasion of the complement system [29].
Plasminogen facilitates the dissemination of Leishmania mexicana in skin lesions
Leishmaniasis is caused by obligate intracellular protozoans of the genus Leishmania and is transmitted by female phlebotomine sand flies. During their life cycle, Leishmania parasites alternate between two forms: extracellular promastigotes that develop within the gut lumen of the female sandfly vector and intracellular amastigotes which inhabit and multiply within the macrophages of vertebrate host [30]. Previous studies show that culture-derived promastigotes and lesion-derived amastigotes of Leishmania mexicana bind plasminogen and utilize tPA to aactivate plasminogen[31]. Studies by Maldonado et al. [32] showed that the distribution of L. mexicana amastigotes injected intradermally on plasminogen-deficient mice was restricted to isolated foci within the skin lesions, instead of the scattered pattern observed in the lesions of wild type mice. This data suggests that plasmin promotes Leishmania migration in the skin by degrading the fibrin and ECM deposited during wound healing [32] Plasmin(ogen) could enhance L. mexicana dissemination in the skin (Figure 2B). The same study showed the presence of parasites only in the skin lesions and the draining popliteal lymph node in both wild type mice and plasminogen-deficient mice, indicating that plasminogen does not facilitate L. mexicana dissemination to other organs. Leishmania amastigotes are transmitted to sandflies after ingestion of an infectious blood meal. In the sandfly midgut lumen, amastigotes egress from macrophages to transform into motile promastigotes that must migrate within the blood bolus to traverse the peritrophic matrix. Different from other vector-borne pathogens, Leishmania promastigotes do not cross the sandfly midgut epithelium. It is possible that the acquired host plasminogen might enhance sandfly infection by facilitating promastigote motility through the degradation of fibrin and traversal of the peritrophic matrix.
Enolase has been identified as a plasminogen receptor in L. mexicana and binds to plasminogen via an enolase internal lysine motif (249AYDAERKMY257), similar to the plasminogen binding site of Streptococcus pneumoniae enolase. Plasminogen binding to promastigotes was partially inhibited by anti-enolase antibodies, suggesting that the parasite might have additional plasminogen receptors [33]. Indeed, the Leishmania homolog of receptors for activated C-kinase (LACK) binds plasminogen and facilitates its activation [34]. Binding of plasminogen to LACK was inhibited by the lysine analog ε-aminocaproic acid (ε-ACA), suggesting a lysine-dependent interaction. Like enolase, LACK has an internal lysine motif (260VYDLESKAV268) that may act as the plasminogen binding site. LACK and enolase could synergize to enhance plasminogen activation on the parasite surface and increase its invasiveness [34].
Trypanosomes bind and activate plasminogen
Trypanosoma parasites can be classified in two subgroups, salivarian or stercorarian, based on the site of development in the vector (foregut or hindgut, respectively) and their transmission route. Trypanosoma cruzi, which causes Chagas disease, is a stercorarian parasite that develops in the hindgut of the triatomine insect vector. Metacyclic trypomastigotes, the infectious stage, are released in the insect feces as it defecates while feeding on blood. Trypomastigotes invade the cells underlying the mucosal membrane (macrophages, fibroblasts, and epithelial cells) near the bite site to differentiate into intracellular amastigotes that multiply and differentiate into trypomastigotes that cause the clinical manifestations of Chagas disease. Trypomastigotes ingested by the triatomine bug transform into epimastigotes and then into metacyclic trypomastigotes in the midgut of the bug, completing the parasite life cycle. Plasminogen has been shown to bind to T. cruzi epimastigotes and metacyclic trypomastigotes in a lysine-dependent mechanism [35, 36], and it is activated by tPA on both parasitic forms (Figure 2C). Ligand-blotting analysis of proteins extracted from epimastigote microsomal fractions revealed five soluble and one hydrophobic protein that bind plasminogen. However, the identity of these putative plasminogen receptors is still unknown [36].
T. evansi, a salivarian parasite that infects horses, cows, pigs and other mammals, also binds plasminogen and plasmin on their surface. Interestingly, treatment with the lysine analogue ε-ACA partially inhibited binding of plasminogen to the parasite, whereas plasmin binding was completely inhibited [37]. It is hypothesized that plasminogen binding to T. evansi might also occur via a lysine-independent mechanism. This may be possible due to the conformational changes induced by the activation of plasminogen, which exclude the amino hexyl and the single weak lysine binding sites from the binding motif [38]. T. evansi did not activate plasminogen in the absence or presence of tPA, suggesting that the parasite might utilize uPA to activate plasminogen, a hypothesis that remains to be tested. Several proteins with molecular masses between ~18 and ~70 kDa exhibited plasminogen binding properties [38], thus warranting further experimental studies to identify plasminogen receptors in Trypanosoma. Plasminogen has been hypothesized to play multiple roles in Trypanosoma, including parasite dissemination through the degradation of ECM and fibrin, as well as immune evasion through the degradation of complement proteins and immunoglobulins [37].
Plasmin facilitates Dengue virus infection of the mosquito midgut epithelium
Dengue virus (DENV) is a flavivirus transmitted by the mosquito vectors Aedes aegypti and Aedes albopictus. DENV ingested from an infected human invades the mosquito midgut epithelium, where it multiplies and disseminates to other tissues, including the salivary glands from where its transmitted during subsequent biting [39]. DENV has been shown to induce a hyper-fibrinolytic state in plasma that could be partially explained by the formation of DENV-induced plasminogen autoantibodies that enhance plasminogen activation [40, 41]. Such hyperactivation of fibrinolysis could prevent the formation of fibrin in the mosquito blood bolus and reduce its viscosity, which might have significant implications during DENV transmission from humans to mosquitoes. A recent report showed that feeding Ae. aegypti mosquitoes with pig blood supplemented with human plasmin induced an increase in DENV infection [42]. On the contrary, plasmin supplementation of in vitro mammalian (BHK-21) or Ae. albopictus (C6/36) cell cultures did not change DENV infection levels, suggesting that the increase of infection is specific to the mosquito midgut environment. Further, feeding DENV to mosquitoes in blood supplemented with plasmin did not increase the midgut infection rate but significantly increased the number of infection foci per mosquito midgut. This data suggests that plasmin increases the onset of DENV infection in the midgut, which ultimately results in higher infection rate and DENV dissemination to other organs [42]. Feeding mosquitoes with FITC-labelled dextran molecules together with DENV and plasmin significantly increased the number of fluorescent dextran foci in the midgut epithelium. Since dextran molecules are internalized by a similar clathrin-dependent endocytosis mechanism as DENV, this result suggests that recruitment of plasmin by DENV facilitates proteolysis and increases DENV internalization [42]. The authors proposed a model where DENV utilizes plasmin to degrade the midgut glycocalyx [43], thus exposing the midgut epithelial cells and facilitating virus internalization (Figure 2D).
Additionally, Ae. aegypti possesses a Kazal-type serine protease inhibitor (AaTI), which is expressed in the midgut and inhibits plasmin [44, 45]. Using a chromogenic in vitro assay, Ramesh et al. [42] showed that recombinant AaTI can potently inhibit the proteolytic activity of plasmin. Knockdown of AaTI expression by intrathoracic injection of AaTI dsRNA significantly increased the infection rate and the number of foci per midgut suggesting that AaTI inhibits DENV infection. Further, the authors developed a mutant rAaTI that lost its inhibitory activity against plasmin by substituting the P1 residue from arginine to alanine. Infecting mosquitoes with blood supplemented with wild type rAaTI increased DENV infection, whereas infections with mutant rAaTI remained unaltered. These results suggest that rAaTI inhibits plasmin proteolysis and reduces DENV infection of the mosquito midgut thus emphasizing the transmission-blocking potential of this molecule [42, 45]. Moreover, the authors propose that administration of AaTI to patients might restrict vascular leakage induced by fibrinolysis [42].
The more, the better: Tick-borne pathogens deploy multiple plasminogen receptors for infection of the tick vector and the host
Lyme disease is caused by the spirochete Borrelia burgdorferi and is transmitted by infected Ixodes ticks while blood feeding on the skin of a human. The spirochete traverses the dermis and enters the blood circulation to invade distant organs like the central and peripheral nervous systems, the heart and the joints. In vitro studies show that plasminogen and plasmin bind to the surface of B. burgdorferi through lysine-dependent interactions and that plasminogen activation by uPA and tPA is enhanced 4 to 5-fold in the presence of spirochetes [46]. Incubating spirochetes with human serum, which contains high concentrations of the plasmin inhibitor α2-antiplasmin, did not affect the plasmin enzymatic activity, showing that plasmin bound to B. burgdorferi is protected from serum-derived inhibitors [46].
Studies in endothelial cells showed that plasmin on the spirochete surface degrades the ECM glycoprotein fibronectin, thus enhancing the spirochete penetration of the endothelium [47–51]. Furthermore, in vivo studies with plasminogen-deficient mice showed that plasmin enhances spirochetemia in mice and is crucial for B. burgdorferi dissemination from the tick gut to the salivary glands, thus facilitating its transmission [52] (Figure 2E). By using a murine model of relapsing fever, Gebbia et al. [53] showed that plasminogen deficiency reduces B. burgdorferi invasion of the central nervous system (CNS) and the heart. They propose that plasmin on the surface of Borrelia could interact with endothelial plasminogen receptors to anchor the bacterium to the tissue. Such interaction can facilitate the degradation of proteins in the tight junctions of the blood brain barrier (BBB) leading to increased dissemination of Borrelia in the brain [53] (Figure 2E). In support of this hypothesis, Grab et al. [54] studied the migration of B. burgdorferi using an in vitro model of the BBB consisting of cultured human brain microvascular endothelial cells (BMEC), the BBB functional unit. B. burgdorferi traversal of the BMEC barrier induced the expression of plasminogen activators and its receptors, which could promote the focal degradation of BBB tight junctions and spirochete dissemination into the CNS [54]. In addition, plasmin enhances invasion of the heart tissue by B. hermsii [52, 53, 55].
Several plasminogen receptors have been characterized for Borrelia. Immuno-blot studies identified B. burgdorferi outer-surface protein A (OspA) as a predominant plasminogen receptor, and the interaction is facilitated by lysine residues [46]. However, in vivo immunofluorescence studies show the loss of OspA expression in most spirochetes 72 hours after attachment of the tick to a mouse, suggesting that OspA might not serve as a plasminogen receptor in the vector or in the vertebrate host [52]. Outer surface protein C (OspC), another lipoprotein of the same family, is expressed on the spirochete surface during development in the tick and is a virulence factor required for mammalian infection [56]. OspC strongly binds to plasminogen and fibrin and delays the formation of fibrin clots, presumably by facilitating plasminogen activation and fibrin degradation [57, 58]. Further, by using B. burgdorferi invasive and non-invasive strains with specific OspC polymorphisms, Lagal et al. showed that Borrelia invasiveness positively correlates with the plasminogen-binding capacity of the bacterium [59]. Furthermore, three members of the Erp family of outer-surface lipoproteins, ErpP, ErpA and ErpC, were shown to bind to plasminogen via a lysine dependent mechanism and support plasminogen activation by uPA [60]. Some plasminogen receptors like Erp proteins, factor H binding protein A and complement regulator-acquiring surface proteins (CspA), can simultaneously bind to other ligands, like ECM proteins and the complement regulator factor H, enhancing the degradation of matrices and evasion of complement [61]. Enolase is also exported to the outer-membrane surface of B. burgdorferi where it binds plasminogen in a lysine-dependent manner [62–64].
Tick saliva produces a vast variety of biomolecules that can regulate hemostasis, inflammation, immunity and vasoconstriction [65]. For example, longistatin from the hard tick Haemaphysalis longicornis plays a vital role during blood feeding by facilitating the formation of a blood pool beneath the host skin. Longistatin hydrolyzes the α, β and γ chains of fibrinogen and therefore delays the formation of fibrin clots. In addition, longistatin binds to fibrin and activates plasminogen bound to fibrin clots at similar levels tPA. Longistatin might have a dual role as a plasminogen activator and as an anticoagulant, thus facilitating blood feeding and survival of the tick. Recently, ixonnexin, a member of the basic-tail family of salivary proteins from I. scapularis, has been shown to enhance fibrinolysis [66]. Ixonnexin binds to plasminogen and tPA, forming a ternary complex that facilitates plasminogen activation, thus revealing the importance of another potent pro-fibrinolytic enzyme present in tick saliva. As a novel fibrinolysis modulator, ixonnexin can be further studied to define its role during Borrelia infection and as a therapeutic agent [66]. Serpin 19, a serine protease inhibitor from Amblyomma americanum tick saliva, can inhibit plasmin and thrombin-triggered plasma clotting [67]. These studies show the diverse mechanisms by which proteins from the vector saliva can interact with the fibrinolytic system and modulate hemostasis to facilitate blood ingestion and digestion, and inadvertently, enhance pathogen transmission.
Novel interventions to disrupt the parasite interaction with the fibrinolytic system
Growing evidence shows that multiple vector-borne pathogens share similar mechanisms to co-opt the fibrinolytic system to infect their hosts and vectors. However, no interventions have been developed to disrupt these interactions and prevent pathogen transmission and pathogenesis. Current therapeutics to modulate fibrinolysis in specific clinical disorders, like alpha 2-plasmin inhibitor deficiency and cancer, mostly rely on the use of compounds or peptides that inhibit the activation of plasminogen or plasmin activity [68]. These strategies are not suited as prophylaxis to prevent the transmission of vector-borne diseases, since they may alter the homeostasis between fibrinolysis and coagulation in otherwise healthy individuals. Therefore, novel approaches will be required to prevent the interaction of the pathogen with the fibrinolytic system. One conventional approach is to target the pathogen receptors for plasminogen and/or its activators by developing antibodies or small molecules that disrupt their interaction. This might be challenging due to the high amino acid conservation between some of the pathogen receptors and the human homologues, as is the case for enolase and GAPDH. However, a recent study used epitope mapping to identify Plasmodium GAPDH antibodies that inhibit sporozoite infectivity and do not cross-react with human GAPDH [69].
Another approach involves the recruitment of the mosquito and its microbiome as allies to block the parasite interaction with plasmin. Recent studies have cleverly transformed some of the mosquito midgut bacteria to secrete molecules that hamper parasite development in the midgut lumen, a technology known as paratransgenesis. In two elegant studies Wang et al. [70, 71] engineered Pantoea agglomerans and Serratia AS1, two Anopheles commensal bacteria, to secrete the Plasmodium enolase lysine motif peptide (DKSLVK) in the blood bolus where it binds to the plasminogen Kringle domain, thereby blocking its interaction with the parasite. Expression of the peptide by both bacteria strongly inhibited P. falciparum midgut infection. Of relevance, they found that Serratia AS1 can be transmitted vertically and horizontally between mosquitoes and can infect multiple Anopheles species, two important traits required for successful bacteria dissemination within the mosquito population [71].
An alternative approach is to engineer the mosquito to secrete molecules that inhibit the interaction between the parasite and the fibrinolytic proteins. Dong et al. [72] recently developed An. gambiae mosquitoes secreting multiple anti-plasmodial molecules into the midgut lumen, including the Plasmodium DKSLVK peptide. These mosquitoes significantly inhibited the formation of P. falciparum oocysts. Another molecule that could be employed in paratransgenesis or mosquito transgenesis interventions is PAI-1. We have shown that PAI-1 can inhibit parasite infection of the mosquito midgut and sporozoite motility in the mammalian dermis [28, 29]. Transgenic mosquitoes can be engineered to inhibit plasminogen activation by expressing human PAI in the midgut and/or the saliva, thereby targeting the parasite midgut stages and the sporozoite. Alternatively, the mosquito microbiome could be engineered to secret PAI-1. Similar approaches can be adapted to other vector-borne pathogens that co-opt the fibrinolytic system for transmission.
Concluding Remarks
An increasing body of evidence indicates that different components of the mammalian fibrinolytic system play crucial roles during pathogen development in the vector and during transmission to the host, a concept known as host exploitation. The ability of pathogens to utilize host proteins for their survival is presumably a consequence of within-host pathogen evolution to maximize transmission. Therefore, it is likely that other plasma proteins (not related to fibrinolysis) are also crucial for the development and transmission of vector-borne pathogens. Likewise, hematophagous vectors interact with host factors during blood feeding, so it will be relevant to investigate how the interplay between vectors and host factors shape survival and vector competence. For example, recently Pereira-filho et al. [73] showed that the midgut epithelium of A. aegypti mosquitoes acquire the human complement regulator factor H to degrade C3b and to evade complement attack. Similarly, several factors with anticomplement activities have been shown to be secreted by ticks [74]. It is not yet established if plasminogen can be acquired for a similar role in the gut of mosquitoes, sandflies and other vectors. Additionally, plasmin could also facilitate the digestion of the blood meal by degrading the fibrin that forms within minutes of blood ingestion [29], facilitating nutrient acquisition and perhaps enhancing pathogen transmission. These studies and many others highlight critical interactions between vectors and host humoral factors that could be targeted by novel vector control strategies.
Additionally, vector factors from saliva also influence pathogen transmission, a phenomen known as saliva assisted transmission (SAT). A plethora of vector molecules contained in saliva have been shown to be crucial for the pathogens’ infectivity to the vertebrate host [65, 75]. A good example is sialostatin that suppresses the immune response by inhibiting the JAK–STAT pathway and IFNβ production and enhances B. burgdorferi infection [76]. Components of the vector saliva can also influence pathogen infectivity by inducing anti-homeostatic responses in the host, such as metalloproteases and longistatin, which have anticoagulant activity [77].
Vector-borne pathogens have evolved similar mechanisms to interact with the fibrinolytic system, like binding plasminogen in a lysine-dependent manner and utilizing tPA and/or uPA to activate plasminogen. Therefore, these mechanisms could be targeted by interventions that are effective against multiple vector-borne pathogens, including transgenic mosquitoes and paratransgenesis. These are complementary strategies that rely on the genetic modification of the vector or the vector microbiome to express molecules that inhibit activation of fibrinolysis. Transgenic mosquitoes require the engineering of individual vectors species, which could be challenging for diseases like malaria that is transmitted by more than 40 anopheline species. Paratransgenesis approaches might overcome this limitation by engineering a bacterium that is common to multiple vectors and targets multiple pathogens. Several studies have shown the effectiveness of these strategies in blocking malaria transmission under laboratory settings, although testing and further implementation in the field is limited. The progress made in the establishment of policies and facilities for semi-field studies [78–80], the development of gene drive technologies [80] and the discovery of commensal bacteria that can be transmitted transstadially and transovarially [81, 82] are significant milestones towards this goal.
Thus, a more detailed understanding of the molecular interactions between vectors, vector-borne pathogens, and plasma proteins, including the fibrinolytic proteins, may provide new targets to develop novel interventions to prevent transmission of multiple vector-borne pathogens (see Outstanding Questions).
Outstanding Questions.
How do pathogens utilize plasmin (i.e. substrates) to enhance survival, dissemination, and produce a chronic infection?
Which other pathogen receptors bind the fibrinolytic proteins?
Why do some pathogens produce multiple plasminogen receptors? Are different receptors used in different stages of the pathogen’s life cycle?
Do vector-borne pathogens produce endogenous plasminogen activators? If so, what is the identity of such activators?
Could novel interventions be developed to disrupt the interaction of the pathogen with the fibrinolytic system without altering the fibrinolytic balance of the human?
Highlights.
Plasmin, the effector protease of the human fibrinolytic system, degrades fibrin, extracellular matrix proteins, complement proteins and immunoglobulins.
Plasmodium and other vector-borne pathogens co-opt plasmin to facilitate tissue invasion and dissemination, and to evade immune responses in the human host and the arthropod vector.
Evolutionary distant pathogens share conserved mechanisms for the acquisition and activation of plasmin.
Novel strategies, including paratransgenesis and transgenic mosquitoes, could be developed to block plasminogen activation by Plasmodium parasites, thereby targeting crucial developmental stages required for malaria transmission. If successful, these approaches can be adapted to impair the transmission of other vector-borne diseases.
Acknowledgements
This study was supported by the NIH Distinguished Scholars Program, and the Intramural Research Program of the Division of Intramural Research AI001250-01, National Institutes of Allergy and Infectious Diseases, National Institutes of Health. Z.R.P acknowledges the Malaria Research Program Fellowship.
Glossary
- Extracellular matrices
The non-cellular component within tissues and organs that provides structural support for cells and can also regulate biochemical and biophysical cell signaling
- Fibrin
An insoluble fibrous protein formed when thrombin cleaves fibrinogen during blood coagulation. Fibrin polymerizes to form a mesh that, together with platelets, form a hemostatic plug (clot) at the injury site
- Fibrinolysis
The enzymatic degradation of fibrin in blood clots to prevent the obstruction of blood vessels and maintain homeostasis
- Hematophagous
Living organism that feeds on blood
- Kringle domains
Autonomous protein domains that fold into big loops stabilized by three disulfide linkages. They are involved in protein-protein interactions and are found in some proteins from the coagulation and the fibrinolytic systems, such as Plg and tPA
- Merozoite
Daughter cell produced by schizogony. Merozoites invade red blood cells to for rings
- Paratransgenesis
Genetic engineering of vector symbionts to produce anti-pathogen effector molecules
- Phlebotomine
Sandfly arthropods of the genus Phlebotomus that suck blood from mammals and other animals and transmit sandfly fever and Leishmania
- Plasmin
Serine protease responsible for degradation of fibrin clots
- Plasminogen
The inactive precursor of plasmin
- Ring
Early stage of the malaria parasite that forms after merozoites invade the red blood cell. Rings precede the trophozoite stage
- Schizogony
Asexual reproduction by multiple segmentation
- Schizont
The stage of the malaria parasite that proceeds the trophozoite. Schizonts produce daughter cells known as merozoites through schizogony
- Serine proteases
Enzymes that cleave peptide bonds of target proteins and have a nucleophilic serine at the active sites
- Serine Protease Inhibitor (serpin)
A superfamily of proteins that irreversibly inhibit serine proteases to regulate proteolytic cascades
- Tissue-type plasminogen activator (tPA)
Serine protease that activates plasminogen on fibrin clots and cell surfaces
- Trophozoite
Amoeboid feeding stage of the malaria parasite. The trophozoite develops inside the red blood cell and proceeds the ring stage. Trophozoites are metabolically active and responsible for the digestion of the hemoglobin within the red blood cell
- Urokinase-type plasminogen activator (uPA)
Serine protease that activates plasminogen on cell surfaces
- Vector
Living organism that transmits pathogens between humans or between animals and humans
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Declaration of Interests
The authors declare no competing interests.
Resources
References
- 1.Sperandio B, et al. (2015) Mucosal physical and chemical innate barriers: Lessons from microbial evasion strategies. Semin Immunol 27, 111–118 [DOI] [PubMed] [Google Scholar]
- 2.Kobayashi SD, et al. (2018) Neutrophils and Bacterial Immune Evasion. J Innate Immun 10, 432–441 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Finlay BB and McFadden G (2006) Anti-immunology: Evasion of the host immune system by bacterial and viral pathogens. Cell 124, 767–782 [DOI] [PubMed] [Google Scholar]
- 4.Arca B and Ribeiro JM (2018) Saliva of hematophagous insects: a multifaceted toolkit. Curr Opin Insect Sci 29, 102–109 [DOI] [PubMed] [Google Scholar]
- 5.Ayon-Nunez DA, et al. (2018) Plasminogen-binding proteins as an evasion mechanism of the host’s innate immunity in infectious diseases. Bioscience Rep 38, BSR20180705 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Chandler WL (2019) Fibrinolytic Testing. Transfusion Medicine and Hemostasis: Clinical and Laboratory Aspects, 3rd Edition, 861–863 [Google Scholar]
- 7.Ghosh AK and Vaughan DE (2012) PAI-1 in tissue fibrosis. J Cell Physiol 227, 493–507 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Collen D and Lijnen HR (1986) Chapter 8 Fibrinolysis and thrombolysis. In New Comprehensive Biochemistry (Neuberger A and van Deenen LLM, eds), pp. 243–258, Elsevier [Google Scholar]
- 9.Gong LH, et al. (2015) Crystal Structure of the Michaelis Complex between Tissue-type Plasminogen Activator and Plasminogen Activators Inhibitor-1. J Biol Chem 290, 25795–25804 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Schaller J and Gerber SS (2011) The plasmin-antiplasmin system: structural and functional aspects. Cell Mol Life Sci 68, 785–801 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Behl T, et al. (2017) Role of altered coagulation-fibrinolytic system in the pathophysiology of diabetic retinopathy. Vasc Pharmacol 92, 1–5 [DOI] [PubMed] [Google Scholar]
- 12.Alexander CM and Werb Z (1989) Proteinases and extracellular matrix remodeling. Curr Opin Cell Biol 1, 974–982 [DOI] [PubMed] [Google Scholar]
- 13.Bhattacharya S, et al. (2012) Bacterial Plasminogen Receptors Utilize Host Plasminogen System for Effective Invasion and Dissemination. J Biomed Biotechnol 2012, 482096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Loof TG, et al. (2014) The role of coagulation/fibrinolysis during Streptococcus pyogenes infection. Front Cell Infect Mi 4, 128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Law RH, et al. (2012) The X-ray crystal structure of full-length human plasminogen. Cell Rep 1, 185–190 [DOI] [PubMed] [Google Scholar]
- 16.Sasaki T, et al. (1986) Identification of the plasminogen-binding site of human alpha 2-plasmin inhibitor. J Biochem 99, 1699–1705 [DOI] [PubMed] [Google Scholar]
- 17.Francischetti IM, et al. (2008) Blood coagulation, inflammation, and malaria. Microcirculation 15, 81–107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Mohanty D, et al. (1997) Fibrinolysis, inhibitors of blood coagulation, and monocyte derived coagulant activity in acute malaria. Am J Hematol 54, 23–29 [DOI] [PubMed] [Google Scholar]
- 19.Thuma PE, et al. (2011) Distinct clinical and immunologic profiles in severe malarial anemia and cerebral malaria in Zambia. J Infect Dis 203, 211–219 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Krucken J, et al. (2005) Testosterone suppresses protective responses of the liver to blood-stage malaria. Infect Immun 73, 436–443 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Rosenthal PJ, et al. (1998) Plasminogen activators are not required in the erythrocytic life cycle of malaria parasites. Mol Biochem Parasitol 97, 253–257 [DOI] [PubMed] [Google Scholar]
- 22.Melo PM, et al. (2014) Plasmodium falciparum proteases hydrolyze plasminogen, generating angiostatin-like fragments. Mol Biochem Parasitol 193, 45–54 [DOI] [PubMed] [Google Scholar]
- 23.Claesson-Welsh L, et al. (1998) Angiostatin induces endothelial cell apoptosis and activation of focal adhesion kinase independently of the integrin-binding motif RGD. Proc Natl Acad Sci U S A 95, 5579–5583 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Deininger MH, et al. (2003) Angiogenic proteins in brains of patients who died with cerebral malaria. J Neuroimmunol 142, 101–111 [DOI] [PubMed] [Google Scholar]
- 25.Roggwiller E, et al. (1997) Host urokinase-type plasminogen activator participates in the release of malaria merozoites from infected erythrocytes. Mol Biochem Parasitol 86, 49–59 [DOI] [PubMed] [Google Scholar]
- 26.Reiss T, et al. (2021) Acquisition of human plasminogen facilitates complement evasion by the malaria parasite Plasmodium falciparum. Eur J Immunol 51, 490–493 [DOI] [PubMed] [Google Scholar]
- 27.Meibalan E and Marti M (2017) Biology of Malaria Transmission. Cold Spring Harb Perspect Med 7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ghosh AK, et al. (2011) Plasmodium ookinetes coopt mammalian plasminogen to invade the mosquito midgut. Proc Natl Acad Sci U S A 108, 17153–17158 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Alves ESTL, et al. (2021) The fibrinolytic system enables the onset of Plasmodium infection in the mosquito vector and the mammalian host. Sci Adv 7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Handman E (1999) Cell biology of Leishmania. Adv Parasitol 44, 1–39 [DOI] [PubMed] [Google Scholar]
- 31.Avilan L, et al. (2000) Interaction of Leishmania mexicana promastigotes with the plasminogen-plasmin system. Mol Biochem Parasit 110, 183–193 [DOI] [PubMed] [Google Scholar]
- 32.Maldonado J, et al. (2006) A study of cutaneous lesions caused by Leishmania mexicana in plasminogen-deficient mice. Exp Mol Pathol 80, 289–294 [DOI] [PubMed] [Google Scholar]
- 33.Vanegas G, et al. (2007) Enolase as a plasminogen binding protein in Leishmania mexicana. Parasitol Res 101, 1511–1516 [DOI] [PubMed] [Google Scholar]
- 34.Gómez-Arreaza A, et al. (2011) Leishmania mexicana: LACK (Leishmania homolog of receptors for activated C-kinase) is a plasminogen binding protein. Exp Parasitol 127, 752–761 [DOI] [PubMed] [Google Scholar]
- 35.Rojas M, et al. (2008) Characteristics of plasminogen binding to Trypanosoma cruzi epimastigotes. Acta Trop 107, 54–58 [DOI] [PubMed] [Google Scholar]
- 36.Almeida L, et al. (2004) Plasminogen interaction with Trypanosoma cruzi. Mem I Oswaldo Cruz 99, 63–67 [DOI] [PubMed] [Google Scholar]
- 37.Acosta H, et al. (2016) Interaction of Trypanosoma evansi with the plasminogen-plasmin system. Veterinary Parasitology 226, 189–197 [DOI] [PubMed] [Google Scholar]
- 38.Christensen U (1984) The AH-site of plasminogen and two C-terminal fragments. A weak lysine-binding site preferring ligands not carrying a free carboxylate function. Biochem J 223, 413–421 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Salazar MI, et al. (2007) Dengue virus type 2: replication and tropisms in orally infected Aedes aegypti mosquitoes. Bmc Microbiol 7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Huang YH, et al. (2001) Activation of coagulation and fibrinolysis during dengue virus infection. J Med Virol 63, 247–251 [DOI] [PubMed] [Google Scholar]
- 41.Chuang YC, et al. (2011) Dengue Virus-Induced Autoantibodies Bind to Plasminogen and Enhance Its Activation. Journal of Immunology 187, 6483–6490 [DOI] [PubMed] [Google Scholar]
- 42.Ramesh K, et al. (2019) Increased Mosquito Midgut Infection by Dengue Virus Recruitment of Plasmin Is Blocked by an Endogenous Kazal-type Inhibitor. Iscience 21, 564–576 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Becker BF, et al. (2015) Degradation of the endothelial glycocalyx in clinical settings: searching for the sheddases. Brit J Clin Pharmaco 80, 389–402 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Rimphanitchayakit V and Tassanakajon A (2010) Structure and function of invertebrate Kazal-type serine proteinase inhibitors. Dev Comp Immunol 34, 377–386 [DOI] [PubMed] [Google Scholar]
- 45.Watanabe RMO, et al. (2010) A novel trypsin Kazal-type inhibitor from Aedes aegypti with thrombin coagulant inhibitory activity. Biochimie 92, 933–939 [DOI] [PubMed] [Google Scholar]
- 46.Fuchs H, et al. (1994) The Outer Surface Protein-a of the Spirochete Borrelia burgdorferi Is a Plasmin(Ogen) Receptor. P Natl Acad Sci USA 91, 12594–12598 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Fuchs H, et al. (1996) Borrelia burgdorferi induces secretion of pro-urokinase-type plasminogen activator by human monocytes. Infect Immun 64, 4307–4312 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Coleman JL, et al. (1995) Borrelia burgdorferi Binds Plasminogen, Resulting in Enhanced Penetration of Endothelial Monolayers. Infection and Immunity 63, 2478–2484 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Hu LT, et al. (1995) Binding of human plasminogen to Borrelia burgdorferi. Infect Immun 63, 3491–3496 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Klempner MS, et al. (1995) Binding of human plasminogen and urokinase-type plasminogen activator to the Lyme disease spirochete, Borrelia burgdorferi. J Infect Dis 171, 1258–1265 [DOI] [PubMed] [Google Scholar]
- 51.Coleman JL, et al. (1999) Plasmin-coated Borrelia burgdorferi degrades soluble and insoluble components of the mammalian extracellular matrix. Infection and Immunity 67, 3929–3936 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Coleman JL, et al. (1997) Plasminogen is required for efficient dissemination of B. burgdorferi in ticks and for enhancement of spirochetemia in mice. Cell 89, 1111–1119 [DOI] [PubMed] [Google Scholar]
- 53.Gebbia JA, et al. (1999) The plasminogen activation system enhances brain and heart invasion in murine relapsing fever borreliosis. J Clin Invest 103, 81–87 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Grab DJ, et al. (2005) Borrelia burgdorferi, host-derived proteases, and the blood-brain barrier. Infect Immun 73, 1014–1022 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Nordstrand A, et al. (2001) Delayed invasion of the kidney and brain by Borrelia crocidurae in plasminogen-deficient mice. Infection and Immunity 69, 5832–5839 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Grimm D, et al. (2004) Outer-surface protein C of the Lyme disease spirochete: a protein induced in ticks for infection of mammals. Proc Natl Acad Sci U S A 101, 3142–3147 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Onder O, et al. (2012) OspC is potent plasminogen receptor on surface of Borrelia burgdorferi. J Biol Chem 287, 16860–16868 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Bierwagen P, et al. (2019) Borrelia outer surface protein C is capable of human fibrinogen binding. FEBS J 286, 2415–2428 [DOI] [PubMed] [Google Scholar]
- 59.Lagal V, et al. (2006) Borrelia burgdorferi sensu stricto invasiveness is correlated with OspC-plasminogen affinity. Microbes Infect 8, 645–652 [DOI] [PubMed] [Google Scholar]
- 60.Brissette CA, et al. (2009) Borrelia burgdorferi infection-associated surface proteins ErpP, ErpA, and ErpC bind human plasminogen. Infect Immun 77, 300–306 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Rottgerding F and Kraiczy P (2020) Immune Evasion Strategies of Relapsing Fever Spirochetes. Front Immunol 11, 1560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Toledo A, et al. (2012) The Enolase of Borrelia burgdorferi Is a Plasminogen Receptor Released in Outer Membrane Vesicles. Infection and Immunity 80, 359–368 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Nogueira SV, et al. (2012) A Surface Enolase Participates in Borrelia burgdorferi-Plasminogen Interaction and Contributes to Pathogen Survival within Feeding Ticks. Infection and Immunity 80, 82–90 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Floden AM, et al. (2011) Borrelia burgdorferi Enolase Is a Surface-Exposed Plasminogen Binding Protein. Plos One 6, e27502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Simo L, et al. (2017) The Essential Role of Tick Salivary Glands and Saliva in Tick Feeding and Pathogen Transmission. Front Cell Infect Microbiol 7, 281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Assumpcao TC, et al. (2018) Ixonnexin from Tick Saliva Promotes Fibrinolysis by Interacting with Plasminogen and Tissue-Type Plasminogen Activator, and Prevents Arterial Thrombosis. Sci Rep-Uk 8, 4806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Kim TK, et al. (2015) Conserved Amblyomma americanum tick Serpin19, an inhibitor of blood clotting factors Xa and XIa, trypsin and plasmin, has anti-haemostatic functions. Int J Parasitol 45, 613–627 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Lin HL, et al. (2020) Therapeutics targeting the fibrinolytic system. Exp Mol Med 52, 367–379 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Cha SJ, et al. (2018) Identification of Plasmodium GAPDH epitopes for generation of antibodies that inhibit malaria infection. Life Sci Alliance 1, e201800111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Wang S, et al. (2012) Fighting malaria with engineered symbiotic bacteria from vector mosquitoes. Proc Natl Acad Sci U S A 109, 12734–12739 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Wang SB, et al. (2017) Driving mosquito refractoriness to Plasmodium falciparum with engineered symbiotic bacteria. Science 357, 1399–1402 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Dong YM, et al. (2020) Versatile transgenic multistage effector-gene combinations for Plasmodium falciparum suppression in Anopheles. Science Advances 6, eaay5898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Pereira-Filho AA, et al. (2020) The gut anti-complement activity of Aedes aegypti: Investigating new ways to control the major human arboviruses vector in the Americas. Insect Biochem Mol Biol 120, 103338. [DOI] [PubMed] [Google Scholar]
- 74.Kazimirova M and Stibraniova I (2013) Tick salivary compounds: their role in modulation of host defences and pathogen transmission. Front Cell Infect Mi 3, 43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Fontaine A, et al. (2011) Implication of haematophagous arthropod salivary proteins in host-vector interactions. Parasit Vectors 4, 187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Lieskovska J, et al. (2015) Tick salivary cystatin sialostatin L2 suppresses IFN responses in mouse dendritic cells. Parasite Immunol 37, 70–78 [DOI] [PubMed] [Google Scholar]
- 77.Pham M, et al. (2021) Changing the Recipe: Pathogen Directed Changes in Tick Saliva Components. Int J Environ Res Public Health 18 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Ritchie SA, et al. (2011) A secure semi-field system for the study of Aedes aegypti. PLoS Negl Trop Dis 5, e988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Niang A, et al. (2019) Semi-field and indoor setups to study malaria mosquito swarming behavior. Parasit Vectors 12, 446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Devos Y, et al. (2021) Risk management recommendations for environmental releases of gene drive modified insects. Biotechnol Adv, 107807. [DOI] [PubMed] [Google Scholar]
- 81.Huang W, et al. (2020) Use of Microbiota to Fight Mosquito-Borne Disease. Front Genet 11, 196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Elston KM, et al. (2021) Engineering insects from the endosymbiont out. Trends Microbiol S0966–842X(21)00126–8 [DOI] [PubMed] [Google Scholar]
- 83.Monroy V and Ruiz BH (2000) Participation of the Dengue virus in the fibrinolytic process. Virus Genes 21, 197–208 [DOI] [PubMed] [Google Scholar]
- 84.Pirovich DB, et al. (2020) Schistosoma mansoni glyceraldehyde-3-phosphate dehydrogenase enhances formation of the blood-clot lysis protein plasmin. Biol Open 9, bio050385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Ramajo-Hernandez A, et al. (2007) Schistosoma bovis: plasminogen binding in adults and the identification of plasminogen-binding proteins from the worm tegument. Exp Parasitol 115, 83–91 [DOI] [PubMed] [Google Scholar]
- 86.Figueiredo BC, et al. (2015) Schistosomes Enhance Plasminogen Activation: The Role of Tegumental Enolase. PLoS Pathog 11, e1005335. [DOI] [PMC free article] [PubMed] [Google Scholar]